


Abstract
Biobank projects are generating genomic data for many thousands of individuals. Computational methods are needed to handle these massive data sets, including genetic ancestry (GA) inference tools. Current methods for GA inference do not scale to biobank-size genomic datasets. We present Rye—a new algorithm for GA inference at biobank scale. We compared the accuracy and runtime performance of Rye to the widely used RFMix, ADMIXTURE and iAdmix programs and applied it to a dataset of 488221 genome-wide variant samples from the UK Biobank. Rye infers GA based on principal component analysis of genomic variant samples from ancestral reference populations and query individuals. The algorithm's accuracy is powered by Metropolis-Hastings optimization and its speed is provided by non-negative least squares regression. Rye produces highly accurate GA estimates for three-way admixed populations—African, European and Native American—compared to RFMix and ADMIXTURE (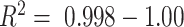 ), and shows 50× runtime improvement compared to ADMIXTURE on the UK Biobank dataset. Rye analysis of UK Biobank samples demonstrates how it can be used to infer GA at both continental and subcontinental levels. We discuss user consideration and options for the use of Rye; the program and its documentation are distributed on the GitHub repository: https://github.com/healthdisparities/rye.
), and shows 50× runtime improvement compared to ADMIXTURE on the UK Biobank dataset. Rye analysis of UK Biobank samples demonstrates how it can be used to infer GA at both continental and subcontinental levels. We discuss user consideration and options for the use of Rye; the program and its documentation are distributed on the GitHub repository: https://github.com/healthdisparities/rye.
INTRODUCTION
Genetic ancestry (GA) refers to genetic similarities indicating the geographic origins of common ancestors (1,2). The GA of modern humans reflects recurrent historical patterns of migration, followed by geographical and reproductive isolation, and subsequent admixture whereby previously isolated populations come back together (3–5). GA is a characteristic of the genome, and it can be inferred based on correlated allele frequency differences that accumulate owing to the action of evolutionary forces on ancestral populations (6). GA can be characterized objectively and with precision, as a categorical or continuous variable, at the genome-wide or local level, and at different levels of scale, e.g. continental versus subcontinental ancestry. In this way, GA is distinct from the socially ascribed and more subjective categories of race and ethnicity (7,8).
Studies of GA have been widely used to illuminate the complex evolutionary history of our species (9–16). GA inference can also be used to help understand how genetic variation within and between populations contributes to health and disease. For example, the characterization of GA is crucial for the application of genome-wide association studies and polygenic risk prediction to globally diverse populations (17–19). GA can be used to help decompose genetic and environmental contributions to health disparities since it is characterized independently of the social dimensions of race and ethnicity (7,8).
There are numerous programs available for GA inference (6), including tools that characterize genome-wide ancestry (20,21) and local ancestry (22), along with applications for fine-scale ancestry and admixture (23). Current methods for GA inference produce accurate and reliable results, but they do not scale well to increasingly large genomic datasets. Biobanks projects, such as the UK Biobank (24) and the NIH All of Us project (25), are generating genome-wide variant datasets for hundreds of thousands of individuals, and similarly ambitious biobank projects are underway around the world (26). Biobanks promise to revolutionize precision medicine, but their potential will not be fully realized without the development of the computational methods needed to analyze such massive datasets. GA inference methods that scale to biobank-size genomic data, while retaining the accuracy of previous generation methods, are urgently needed.
We present the program Rye as one solution to this challenge. Rye provides for rapid and accurate genome-wide GA inference on biobank-size genomic datasets, and it can be used to infer GA at continental and subcontinental levels. Rye and its source code are made freely available; it is well documented, easy to install and use, relies on standard genomic variant formats, and works well with limited computational resources.
MATERIALS AND METHODS
Algorithm overview
The Rye algorithm infers genome-wide ancestry fractions for individual genomic variant samples by comparing principal component (PC) vectors of global reference population individuals with PC vectors of query individuals (Figure 1). Reference populations are grouped into user-defined ancestry groups, which can be assigned at varying levels of biogeographic and genetic relatedness (e.g. continental or subcontinental groups). Principal component analysis (PCA) is run on a combined variant dataset of reference and query individual samples to yield PC vectors for all individuals. PC vectors representative of each ancestry group are computed via Markov chain Monte carlo (MCMC) optimization of reference group-mean PC vectors. Finally, the optimized ancestry-representative PC vectors are compared with PC vectors of query individuals, using non-negative least squares (NNLS), to generate ancestry estimates for all individual samples, expressed as fractions of each user-defined ancestry group.
Figure 1.

Overview of the Rye algorithm. Rye utilizes eigenvectors (PC vectors) and eigenvalues generated by PCA of reference and query individual genome-wide variant samples (left panel). Ancestry group-representative PC vectors are weighted via Metropolis-Hastings optimization of ancestry group-mean PC vectors. Non-negative least squares regression (NNLS) is used to estimate GA fractions via comparison of query individual PC vectors and the weighted ancestry group representative PC vectors.
Algorithm workflow
A schematic illustrating the details of the Rye algorithm is shown in Supplementary Figure S1. Genetic ancestry inference with Rye is performed on a user-supplied genomic variant file that includes reference population samples and query individual samples. Ancestry inference with Rye proceeds via the following steps:
PCA is run on the genomic variant file to yield eigenvectors (i.e. vectors of PC values) for all reference and query individuals. PC vectors are scaled from 0 to 1 for downstream calculations.
Reference individuals are grouped into user-defined ancestry groups, and mean scaled PC vectors are calculated for each ancestry group.
Ancestry-representative PC vectors are computed from ancestry group mean PC vectors using a nested Metropolis-Hastings optimization procedure (see next section for details).
- Optimized ancestry-representative PC vectors are used with non-negative least squares (NNLS) regression to estimate ancestry group fraction values for query individuals, with the constraint that ancestry group fraction values must sum to one. The NNLS equation below shows an example for
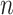 PC values and
PC values and 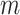 reference ancestry groups, yielding
reference ancestry groups, yielding 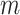 ancestry fractions (
ancestry fractions (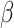 ) for a query individual PC vector.
) for a query individual PC vector.
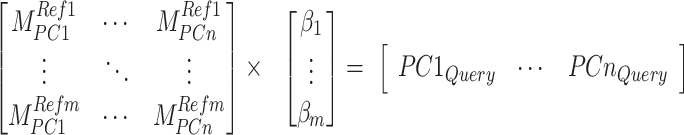
(1)
Metropolis-hastings optimization
A nested Metropolis-Hastings procedure is used to optimize two parameter sets for the ancestry-representative PC vectors: PC weights and shrinkage values. PC weights are used to scale the contribution of each PC to ancestry assignment, and shrinkage values are used to scale the values of each PC. PC weights are the same for all ancestry groups, whereas shrinkage values are specific to each ancestry group. PC weights are initialized using the fraction of variance explained (i.e. the eigenvalues) for each PC from the PCA, and shrinkage values are initialized uniformly. The shrinkage values are used to ensure that outlier individuals, i.e. individuals with extreme PC values compared to their ancestry group, do not bias the ancestry estimation results. The shrinkage values are used to shrink the ancestry group representative PC values towards a value of 0.5. Optimized ancestry-representative PC vectors are calculated as:
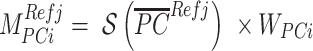 |
(2) |
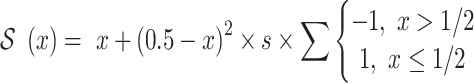 |
(3) |
where 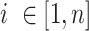 PCs,
PCs, 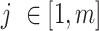 ancestry groups,
ancestry groups,  is the maximum-normalized weight for
is the maximum-normalized weight for 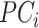 , and
, and 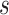 is the shrinkage value for
is the shrinkage value for 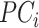 and ancestry group
and ancestry group 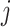 .
.
Optimization of the weight and shrinkage parameters is done using the Metropolis-Hasting algorithm, executed in a nested manner across 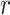 rounds,
rounds, 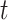 attempts within each round, and
attempts within each round, and 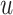 iterations within each attempt (Supplementary Figure S2). Rounds are executed sequentially, and attempts are launched independently within each round. Within each attempt, the Metropolis-Hastings algorithm is used to iterate over weight and shrinkage parameter values, probabilistically selecting the optimal combination of values with which to proceed after each iteration. The optimization criterion is based on NNLS prediction of group-specific ancestry values for reference individuals, as shown in equation 1 with reference individuals treated as query individuals. Reference individuals are expected to show ancestry values that maximally correspond to their group membership.
iterations within each attempt (Supplementary Figure S2). Rounds are executed sequentially, and attempts are launched independently within each round. Within each attempt, the Metropolis-Hastings algorithm is used to iterate over weight and shrinkage parameter values, probabilistically selecting the optimal combination of values with which to proceed after each iteration. The optimization criterion is based on NNLS prediction of group-specific ancestry values for reference individuals, as shown in equation 1 with reference individuals treated as query individuals. Reference individuals are expected to show ancestry values that maximally correspond to their group membership.
Algorithm testing and validation
Rye was used to estimate ancestry fractions—African, European, and Native American—for three-way admixed individuals from the Americas. Rye ancestry estimates were compared against ancestry estimates obtained with the widely used RFMix (22), ADMIXTURE (20) and iAdmix programs (21). Genomic variant data were taken from the 1000 Genomes Project (1KGP) whole genome sequence data (27), and previously published set of Native American genome-wide genotypes (10). A total of 1686 reference individuals were taken from African, European, and Native American populations, and 504 query individuals were taken from Admixed American populations (Supplementary Table S1). Genomic variant data from global reference samples (n = 2190) and UK Biobank genomic variant data (n = 488221) were merged and harmonized as previously described (28–30), yielding a final merged and LD pruned dataset of 490411 samples and 171880 variants. Variants were merged by identifying the set of variants common to both datasets, with strand flips and variant identifier inconsistencies corrected as needed. The initial merged and harmonized variant data set was filtered for variants with >1% missingness and <0.1% minor allele frequency among samples. The resulting data set was tested for Hardy-Weinberg equilibrium (cutoff of 1 × 10−30) and LD pruning was performed using the –indep-pairwise command with window size = 50 SNPs, step = 10 SNPs, and pairwise threshold <0.1 using PLINK.
Algorithm accuracy was measured by comparing observed ancestry fractions for three-way admixed individuals calculated with Rye to expected ancestry fractions calculated with RFMix and ADMIXTURE using Pearson correlation (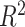 ) and residual sum of squares (
) and residual sum of squares (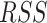 ) error. Sensitivity of the algorithm to reference sequence selection was measured using jackknife resampling with 10% of reference sequences removed in each of 10 replicates. Runtime performance was measured on a 40-core (Intel Xeon), 512GB RAM system, running on Red Hat Enterprise Linux Server release 6.10 (Santiago).
) error. Sensitivity of the algorithm to reference sequence selection was measured using jackknife resampling with 10% of reference sequences removed in each of 10 replicates. Runtime performance was measured on a 40-core (Intel Xeon), 512GB RAM system, running on Red Hat Enterprise Linux Server release 6.10 (Santiago).
UK biobank
Rye was used to estimate ancestry fractions for seven regional ancestry groups—African, Central Asian, East Asian, European, Middle Eastern, Native American and South Asian—on 488221 participants from the UK Biobank (UKBB). UKBB participants’ genome-wide genotypes were characterized using the UKBB Axiom Array or United Kingdom BiLEVE Array as previously described (24,31). UKBB participant genome-wide genotypes were merged and harmonized with genomic variant data from global reference populations characterized as part of the 1KGP (27) and the Human Genome Diversity Project (HGDP) (32) as previously described (33,34). Reference populations were grouped into seven regional ancestry groups based on their genetic and geographic affinity. Rye ancestry estimates were compared to participants’ self-identified ethnicity (Field 21000: Ethnic background https://biobank.ctsu.ox.ac.uk/crystal/field.cgi?id=21000), and Rye runtime performance was compared to ADMIXTURE.
PCA, RFmix, ADMIXTURE and iAdmix
PCA was run using the FastPCA program (35) implemented in PLINK v2 (36), using the command plink –pca, with data from the first 20 PCs retained for ancestry inference with Rye. PCA run on the merged and harmonized variant dataset samples took 1 hour 30 minutes, with 40 threads on a ∼512GB RAM computational server. RFMix was run with 22 threads for 12 generations in the ‘PopPhased’ mode with a minimum node size of five and the ‘‘-use-reference-panels-in-EM’’ for two rounds of expectation maximization (EM) (22). RFmix ancestry assignments were made for chromosomal regions where the RFMix ancestral certainty was at least 95%. ADMIXTURE v.1.30 was run with 40 threads in the supervised mode using default settings, with K = 3 for the admixed American populations across K = 3–20 for the UKBB (20). iAdmix was run with default settings, where the plink genotype files were supplied along with the HapMap3 allele frequency file derived from African, Asian and European reference populations (21).
RESULTS AND DISCUSSION
Accuracy and runtime performance
Rye was used to characterize the genetic ancestry of three-way admixed individuals from the Americas (Supplementary Table S1), and the observed Rye results were compared to expected results obtained from the widely used RFMix, ADMIXTURE and iAdmix programs. Rye is run using a nested optimization approach across a specified number of rounds, attempts, and iterations. Fractions of African, European, and Native American ancestry estimated for Rye and RFMix are highly correlated at both the low and high end of the numbers of rounds and iterations (Figure 2A). Similar high correlations can be seen when Rye ancestry fractions are compared to GA estimates inferred with the ADMIXTURE and iAdmix programs (Supplementary Figure S3). Higher numbers of rounds and iterations yield more accurate results, but the increase in accuracy with increasing rounds and iterations is marginal (Figure 2B). Increasing the number of rounds and iterations entails a marked decrease in runtime performance (Figure 2C). Runtime increases three-orders of magnitude at the highest numbers of rounds and iterations; nevertheless, the longest runtime is just over 20 min.
Figure 2.

Accuracy and runtime performance. (A) GA estimates—African (blue), European (orange), and Native American (red)—are compared for Rye (y-axis) and RFMix (x-axis) for n = 2190 Admixed American and reference individuals. (B) Accuracy of Rye measured by residual sum of squares (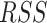 ) and
) and 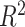 across a range of optimization rounds and iterations. (C) Runtime performance of Rye across a range of optimization rounds and iterations.
across a range of optimization rounds and iterations. (C) Runtime performance of Rye across a range of optimization rounds and iterations.
We assessed the sensitivity of Rye performance to changes in ancestry group reference samples. Jacknife resampling was used to remove 10% of reference samples across 10 replicates, and this procedure was repeated across multiple rounds and iterations (Supplementary Figure S4). Rye is relatively insensitive to changes in the composition of ancestry group reference samples. High accuracy (low RSS) is achieved even at the lowest numbers of rounds and iterations. Increasing the number rounds and iterations leads to marginal improvements in accuracy, comparable to what is seen when the full reference sample sets are used.
Biobank scale performance
The scalability of Rye was evaluated using the UK Biobank (UKBB); genetic ancestry estimates were computed for 488221 participants. The genetic relationship among UKBB participants and reference samples from seven regional ancestry groups, computed using PCA, are shown in Figure 3A. UKBB participants’ self-identified ethnicity are mapped onto their genetic relationships in Figure 3B. Genetic ancestry fractions for each of the seven regional ancestry groups are shown for six ethnic groups (Figure 3C). The Chinese and White ethnic groups shown the most homogenous ancestry patterns, East Asian and European respectively, whereas the Mixed and Other groups are highly diverse. The Asian ethnic group shows mostly South Asian ancestry, followed by East Asian and Middle Eastern components. The Black ethnic group shows most African ancestry followed by European and Middle Eastern components. The ancestry estimates are consistent with participants’ self-identified ethnic backgrounds, which is a second level of ethnic identity beneath the ethnic group designation (https://biobank.ctsu.ox.ac.uk/crystal/field.cgi?id=21000).
Figure 3.

GA inference on the UK Biobank (UKBB). (A) PCA of UKBB participants (gray) and ancestry group reference samples (colored as shown). (B) PCA of UKBB participants labeled by self-identified ethnicity (colored as shown). (C) Ancestry and admixture patterns for UKBB participants, organized by self-identified ethnicity groups. Ancestry fractions (colored as shown) are indicated for each individual. The White ethnic group is not shown to scale owing to its large size; all other groups are scaled based on the number of participants. (D) Runtime comparison for ADMIXTURE and Rye, decomposed into model building (the optimization step for Rye) and GA projection steps.
Rye can also be used for subcontinental GA inference via the delineation of more fine-scale reference ancestry groups (Figure 4). When Rye is run in this way, it reveals clearly distinct ancestry components that constitute broader East Asian, South Asian, African, and European ancestry groups. For example, UKBB participants that identify Pakistani, Indian, and Bangladeshi ethnic backgrounds, within the broader Asian ethnic group, show a gradient of distinct ancestry patterns. Similar results can be seen for African and European ancestry. African reference populations show distinct fine-scale ancestry patterns, and admixed New World African populations show largely similar African ancestry but distinct non-African admixture patterns. Rye clearly distinguishes northeastern, northwestern, southeastern, and southwestern ancestry components within Europe. As was seen for continental ancestry inference, the European subcontinental ancestry components inferred with Rye are significantly concordant with results from ADMIXTURE (Supplementary Figure S5). Rye infers greater Northwest European ancestry in the UKBB British sample, consistent with the demographics of the participants, whereas ADMIXTURE infers more Southeast European ancestry in the UKBB sample.
Figure 4.

Fine-scale GA inference with Rye. Results for UKBB, 1KGP and Native American reference and query individuals are shown. GA estimates for (A) East Asian, (B) South Asian, (C) African and (D) European query individuals from UKBB are shown along with 1KGP and Native American reference populations.
The runtime performance of Rye on the 488221 UKBB participants was compared to ADMIXTURE (Figure 3C). The runtime for both programs was decomposed into model building and projection phases. For Rye, model building corresponds to optimization phase and projection corresponds to the NNLS ancestry fraction calculation. Optimization was performed at the high end of Rye options, with 200 rounds and 200 iterations, to yield a conservative runtime estimate. Overall, Rye is >50× faster than ADMIXTURE: 2687 se for Rye (∼45 min) compared to 136031 s for ADMIXTURE (∼38 h). Model building is ∼2× faster in ADMIXTURE, but this can be attributed to the large number of rounds and iterations used for Rye optimization, which can be substantially reduced without appreciable loss of accuracy. RFMix runs prohibitively slow on a dataset of this size, and therefore its runtime performance could not be directly compared to Rye. It is estimated that RFMix would take more than two years to characterize a dataset of this size on the system used here.
User considerations and options
Documentation and instructions for running Rye are provided on the GitHub repository: https://github.com/healthdisparities/rye. Starting with a merged reference and query genotype file, users need to run PCA and provide Rye with the output eigenvalue and eigenvector files. Samples in the eigenvector output file should include population labels in the first column and sample identifiers in the second column. Rye also requires a population to ancestry group mapping file. These three files are the only required arguments for Rye: –eigenval –eigenvec –pop2group. Other important arguments for Rye include the number of rounds, attempts, and iterations. Higher numbers for each yield more accurate ancestry estimates at the cost of slower runtime (Figure 2B). The default settings are set towards the upper end for these values, yielding the most accurate ancestry estimates. Users with large datasets or limited computational resources may considering reducing the value of these parameters. An order of magnitude time savings can be achieved in this way with little loss of accuracy (Figure 2A).
The choice of individual reference samples to be used for each ancestry group is an important consideration when using Rye. The optimization criteria for the Metropolis-Hastings algorithm assumes that reference individuals will have close to 100% ancestry for each reference group. Accordingly, the use of individuals with distinct ancestry, or admixed individuals, within the same ancestry group could impact the accuracy of ancestry estimates. Users are advised to select a subset of individuals from any given reference population, or closely related group of reference populations, that have highly similar and coherent ancestry patterns. It is not always possible to depend on population labels to choose a coherent reference individual sample set.
Limitations and future directions
Rye is currently designed to run in supervised mode with user-specified reference ancestry groups. This reflects its intended use for population biobank data from cosmopolitan countries where the admixture (ancestry) components are generally known. This design allows users to infer ancestry across multiple levels of scale, e.g. continental and subcontinental, as demonstrated via the application to the UK Biobank data (see Figures 3 and 4). Given this potential limitation, compared to ancestry inference programs that can run in both supervised and unsupervised mode, we plan to incorporate an unsupervised mode in future development plans.
Rye can infer GA at both the continental and subcontinental levels using data from individual (unlinked) genomic variants. Information provided by haplotypes of linked variants could, in principle, provide for even greater resolution of fine-scale ancestry inference. Our future development efforts for biobank-scale ancestry inference also include haplotype-informed methods for fine-scale analysis.
DATA AVAILABILITY
The Rye program, its source code and documentation are freely distributed on the GitHub repository: https://github.com/healthdisparities/rye.
Supplementary Material
ACKNOWLEDGEMENTS
This study was made possible by the United Kingdom Biobank application number 65206.
Contributor Information
Andrew B Conley, National Institute on Minority Health and Health Disparities, National Institutes of Health, Bethesda, MD, USA; IHRC-Georgia Tech Applied Bioinformatics Laboratory, Atlanta, GA, USA; PanAmerican Bioinformatics Institute, Valle del Cauca, Cali, Colombia.
Lavanya Rishishwar, National Institute on Minority Health and Health Disparities, National Institutes of Health, Bethesda, MD, USA; IHRC-Georgia Tech Applied Bioinformatics Laboratory, Atlanta, GA, USA; PanAmerican Bioinformatics Institute, Valle del Cauca, Cali, Colombia; School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA, USA.
Maria Ahmad, National Institute on Minority Health and Health Disparities, National Institutes of Health, Bethesda, MD, USA.
Shivam Sharma, National Institute on Minority Health and Health Disparities, National Institutes of Health, Bethesda, MD, USA; IHRC-Georgia Tech Applied Bioinformatics Laboratory, Atlanta, GA, USA; School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA, USA.
Emily T Norris, National Institute on Minority Health and Health Disparities, National Institutes of Health, Bethesda, MD, USA; IHRC-Georgia Tech Applied Bioinformatics Laboratory, Atlanta, GA, USA; PanAmerican Bioinformatics Institute, Valle del Cauca, Cali, Colombia.
I King Jordan, IHRC-Georgia Tech Applied Bioinformatics Laboratory, Atlanta, GA, USA; PanAmerican Bioinformatics Institute, Valle del Cauca, Cali, Colombia; School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA, USA.
Leonardo Mariño-Ramírez, National Institute on Minority Health and Health Disparities, National Institutes of Health, Bethesda, MD, USA; PanAmerican Bioinformatics Institute, Valle del Cauca, Cali, Colombia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
A.B.C., L.R., E.T.N., M.A., S.S. and I.K.J. were supported by the IHRC-Georgia Tech Applied Bioinformatics Laboratory [RF383]; L.M.R. was supported by the National Institutes of Health (NIH) Distinguished Scholars Program (DSP) and the Division of Intramural Research (DIR) of the National Institute on Minority Health and Health Disparities (NIMHD) at NIH [1ZIAMD000016 and 1ZIAMD000018]. Funding for open access charge: IHRC [RF383]; NIMHD [1ZIAMD000016 and 1ZIAMD000018].
Conflict of interest statement. None declared.
REFERENCES
- 1. Mathieson I., Scally A.. What is ancestry?. PLoS Genet. 2020; 16:e1008624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Royal C.D., Novembre J., Fullerton S.M., Goldstein D.B., Long J.C., Bamshad M.J., Clark A.G.. Inferring genetic ancestry: opportunities, challenges, and implications. Am. J. Hum. Genet. 2010; 86:661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wohns A.W., Wong Y., Jeffery B., Akbari A., Mallick S., Pinhasi R., Patterson N., Reich D., Kelleher J., McVean G.. A unified genealogy of modern and ancient genomes. Science. 2022; 375:eabi8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nielsen R., Akey J.M., Jakobsson M., Pritchard J.K., Tishkoff S., Willerslev E.. Tracing the peopling of the world through genomics. Nature. 2017; 541:302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hellenthal G., Busby G.B.J., Band G., Wilson J.F., Capelli C., Falush D., Myers S.. A genetic atlas of human admixture history. Science. 2014; 343:747–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schraiber J.G., Akey J.M.. Methods and models for unravelling human evolutionary history. Nat. Rev. Genet. 2015; 16:727–740. [DOI] [PubMed] [Google Scholar]
- 7. Yudell M., Roberts D., DeSalle R., Tishkoff S.. SCIENCE AND SOCIETY. Taking race out of human genetics. Science. 2016; 351:564–565. [DOI] [PubMed] [Google Scholar]
- 8. Borrell L.N., Elhawary J.R., Fuentes-Afflick E., Witonsky J., Bhakta N., Wu A.H.B., Bibbins-Domingo K., Rodriguez-Santana J.R., Lenoir M.A., Gavin J.R. 3rdet al.. Race and genetic ancestry in medicine - a time for reckoning with racism. N. Engl. J. Med. 2021; 384:474–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tishkoff S.A., Reed F.A., Friedlaender F.R., Ehret C., Ranciaro A., Froment A., Hirbo J.B., Awomoyi A.A., Bodo J.M., Doumbo O.et al.. The genetic structure and history of Africans and African Americans. Science. 2009; 324:1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reich D., Patterson N., Campbell D., Tandon A., Mazieres S., Ray N., Parra M.V., Rojas W., Duque C., Mesa N.et al.. Reconstructing Native American population history. Nature. 2012; 488:370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Novembre J., Johnson T., Bryc K., Kutalik Z., Boyko A.R., Auton A., Indap A., King K.S., Bergmann S., Nelson M.R.et al.. Genes mirror geography within Europe. Nature. 2008; 456:98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L.et al.. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008; 319:1100–1104. [DOI] [PubMed] [Google Scholar]
- 13. Ioannidis A.G., Blanco-Portillo J., Sandoval K., Hagelberg E., Barberena-Jonas C., Hill A.V.S., Rodriguez-Rodriguez J.E., Fox K., Robson K., Haoa-Cardinali S.et al.. Paths and timings of the peopling of Polynesia inferred from genomic networks. Nature. 2021; 597:522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Homburger J.R., Moreno-Estrada A., Gignoux C.R., Nelson D., Sanchez E., Ortiz-Tello P., Pons-Estel B.A., Acevedo-Vasquez E., Miranda P., Langefeld C.D.et al.. Genomic insights into the ancestry and demographic history of South America. PLos Genet. 2015; 11:e1005602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bryc K., Durand E.Y., Macpherson J.M., Reich D., Mountain J.L.. The genetic ancestry of African Americans, Latinos, and European Americans across the United States. Am. J. Hum. Genet. 2015; 96:37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baharian S., Barakatt M., Gignoux C.R., Shringarpure S., Errington J., Blot W.J., Bustamante C.D., Kenny E.E., Williams S.M., Aldrich M.C.et al.. The great migration and African-American genomic diversity. PLoS Genet. 2016; 12:e1006059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martin A.R., Kanai M., Kamatani Y., Okada Y., Neale B.M., Daly M.J.. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 2019; 51:584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Atkinson E.G., Maihofer A.X., Kanai M., Martin A.R., Karczewski K.J., Santoro M.L., Ulirsch J.C., Kamatani Y., Okada Y., Finucane H.K.et al.. Tractor uses local ancestry to enable the inclusion of admixed individuals in GWAS and to boost power. Nat. Genet. 2021; 53:195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Simonin-Wilmer I., Orozco-Del-Pino P., Bishop D.T., Iles M.M., Robles-Espinoza C.D.. An overview of strategies for detecting genotype-phenotype associations across ancestrally diverse populations. Front. Genet. 2021; 12:703901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alexander D.H., Novembre J., Lange K.. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009; 19:1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bansal V., Libiger O.. Fast individual ancestry inference from DNA sequence data leveraging allele frequencies for multiple populations. BMC Bioinf. 2015; 16:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maples B.K., Gravel S., Kenny E.E., Bustamante C.D.. RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am. J. Hum. Genet. 2013; 93:278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lawson D.J., Hellenthal G., Myers S., Falush D. Inference of population structure using dense haplotype data. PLoS Genet. 2012; 8:e1002453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J.et al.. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018; 562:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. All of Us Research Program, I. Denny J.C., Rutter J.L., Goldstein D.B., Philippakis A., Smoller J.W., Jenkins G., Dishman E.. The “all of us” research program. N. Engl. J. Med. 2019; 381:668–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abul-Husn N.S., Kenny E.E.. Personalized medicine and the power of electronic health records. Cell. 2019; 177:58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Genomes Project, C. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A.et al.. A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nagar S.D., Conley A.B., Chande A.T., Rishishwar L., Sharma S., Marino-Ramirez L., Aguinaga-Romero G., Gonzalez-Andrade F., Jordan I.K.. Genetic ancestry and ethnic identity in Ecuador. HGG Adv. 2021; 2:100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jordan I.K., Rishishwar L., Conley A.B.. Native American admixture recapitulates population-specific migration and settlement of the continental United States. PLos Genet. 2019; 15:e1008225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Conley A.B., Rishishwar L., Norris E.T., Valderrama-Aguirre A., Marino-Ramirez L., Medina-Rivas M.A., Jordan I.K.. A comparative analysis of genetic ancestry and admixture in the Colombian populations of Choco and Medellin. G3 (Bethesda). 2017; 7:3435–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Welsh S., Peakman T., Sheard S., Almond R.. Comparison of DNA quantification methodology used in the DNA extraction protocol for the UK Biobank cohort. BMC Genomics. 2017; 18:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bergstrom A., McCarthy S.A., Hui R., Almarri M.A., Ayub Q., Danecek P., Chen Y., Felkel S., Hallast P., Kamm J.et al.. Insights into human genetic variation and population history from 929 diverse genomes. Science. 2020; 367:eaay5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nagar S.D., Napoles A.M., Jordan I.K., Marino-Ramirez L.. Socioeconomic deprivation and genetic ancestry interact to modify type 2 diabetes ethnic disparities in the United Kingdom. EClinicalMedicine. 2021; 37:100960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nagar S.D., Conley A.B., Sharma S., Rishishwar L., Jordan I.K., Marino-Ramirez L.. Comparing genetic and socioenvironmental contributions to ethnic differences in C-reactive protein. Front. Genet. 2021; 12:738485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Galinsky K.J., Bhatia G., Loh P.R., Georgiev S., Mukherjee S., Patterson N.J., Price A.L.. Fast principal-component analysis reveals convergent evolution of ADH1B in Europe and East Asia. Am. J. Hum. Genet. 2016; 98:456–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J.. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015; 4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Rye program, its source code and documentation are freely distributed on the GitHub repository: https://github.com/healthdisparities/rye.