

Abstract
An X-ray structure of a CLICK chemistry-based BET PROTAC bound to BRD2(BD2) inspired synthesis of JQ1 derived heterocyclic amides. This effort led to the discovery of potent BET inhibitors displaying overall improved profiles when compared to JQ1 and birabresib. A thiadiazole derived 1q (SJ1461) displayed excellent BRD4 and BRD2 affinity and high potency in the panel of acute leukaemia and medulloblastoma cell lines. A structure of 1q co-crystalised with BRD4-BD1 revealed polar interactions with the AZ/BC loops, in particular with Asn140 and Tyr139, rationalising the observed affinity improvements. In addition, exploration of pharmacokinetic properties of this class of compounds suggest that the heterocyclic amide moiety improves drug-like features. Our study led to the discovery of potent and orally bioavailable BET inhibitor 1q (SJ1461) as a promising candidate for further development.
Keywords: BET inhibitors, JQ1, amides
Graphical Abstract
Introduction
Histones are positively charged proteins that enable cells to pack and protect DNA molecules via interactions of lysine residues with acidic phosphate moieties of DNA creating chromatin.[1] [2] Stability of this complex can be affected by acetylation of the ε-amino functionality of lysine, a transformation that eliminates basicity of this group and prevents its involvement in the ionic bond formation with the phosphates.[3] [4] [5] A consequence of the acetylation is a loss of DNA-histone contacts and formation of a complex with weak interactions which consequently promotes gene expression. One of the major mechanisms of chromatin activation involves recognition of N-acetyl lysine residue at the N-terminal tails of histones by the Bromo- and Extra-Terminal domain (BET) family of proteins. [6] [7] [8] Four members of this family have been described to date: BRD2, BRD3, BRD4 and BRDT. [9] [10] [11] [12] [13] [14] The structure of BET proteins comprises of two bromodomains (BD1 and BD2), an extraterminal domain (ET) and a C-terminal domain (CTD). [15] The BDs are hydrophobic regions created by four α-helices (Z, A, B and C) and two loops (ZA and BC) and can recognise N-acetyl residue in histones and other proteins. [16] The ET domain is involved in recruitment of various components of the transcriptional complexes while CTD, present only in BRD4 and BRDT, interact with positive elongation factor (p-TEFb). [17] [18] BET proteins are involved in various pathophysiological processes, but particularly important is their role in cancer development. [19] [20] [21] In recent years a range of BET inhibitors have been described in the literature (Figure 1). [22] [23] [24] [25]
Figure 1.

Selected BET inhibitors reported in the literature
The first amongst them was diazepine derivative 1a known as JQ1, now widely used as a tool in studying BET biology. [26] JQ1 displaces BRD4 from chromatin and induces differentiation, G1 cell cycle arrest and apoptosis in cancer cells such as human-derived NUT midline carcinoma. [27] However, the compound’s short half-life in vivo prevented its progression into clinical studies. [28] On the other hand, its amide derivative known as birabresib, 1c, entered clinical trial demonstrating dose-proportional exposure, favourable safety profile and activity in NMC. [29] Our interest in BET inhibitors sparked from an X-ray structure of a CLICK chemistry-based BET PROTAC bound to BRD2(BD2) revealing novel ligand-protein interactions. This observation followed by an optimization effort described here led to the discovery of potent BET inhibitors displaying overall improved profiles when compared to JQ1 and birabresib.
2. Results and discussion
2.1. Background of the study
Proteolysis-targeting chimeras (PROTACs) have received considerable attention in recent years as a promising novel paradigm in drug discovery. [30] [31] [32] [33] [34] [35] [36] [37] [38] Our interest in developing novel BET-PROTACs led us to the discovery of a series of CLICK-based degraders such as 7 that displayed significantly increased affinity in BRD2 TR-FRET assay compared to parent JQ1 1a and related PROTACs such as dBET1 8. [39] [40] [41]
This finding suggests that the side chain of the click PROTAC forms additional interactions resulting in improved BRD affinity. Indeed, the crystal structure of CLICK-PROTAC 7 in complex with BRD2/BD2 that we previously generated (PDB: 6WWB) showed several additional interactions with the protein compared to JQ1, which included a water-bridged hydrogen bond network between NH-amide/Asn429 and CO-amide/Leu381, and hydrogen bonding between the amide triazole/His433 (Figure 3A). [39] The electron density around the thalidomide part of ligand 7 was weak and its role is not clear. The crystal structure generally revealed that the network of H-bonds created by 7 is similar to 1a (JQ1) in the corresponding region except JQ1 is lacking an interaction with Leu381 (Figure 3B) (PDB:3ONI). [26] Similarly to 1a, 1c (birabresib) in complex with related BRD4 (PDB: 5WMD) adopts an analogous conformation with the key interactions involving the amide functionality (Figure 3C). [42] Here the H-bonding network is more complex and includes water-mediated H-bonds with Leu92, Asn140, and with a non-conserved Asp144 in lieu of His433 in BRD2. The phenol moiety of 1c that is projecting towards the solvent may be important for balancing other properties of this ligand. All three ligands maintain H-bonding with the conserved Tyr386 via a water molecule. These results suggest the critical role of the triazole moiety in improving the affinity of the PROTAC ligand towards BRD2/BD2.
Figure 3.

Comparison of ligand binding modes of BET family members. Crystal structure of BRD2/BD2 in complex with (A) 7 (magenta, PDB: 6WWB) and (B) JQ1, 1a, (yellow, PDB: 3ONI). (C) Crystal structure of birabresib, 1c, bound to BRD4/BD1 (purple, PDB: 5WMD). Hydrogen bonds are shown as dotted lines.
2.2. Chemistry
To explore and further leverage these novel interactions, we synthesised several JQ1 amide derivatives and compared their properties with the parent inhibitor as well as with a more potent birabresib 1c (Table 1). Our focus was on substituents that can probe the subpocket occupied by the 1,2,3-triazole in 7. Consequently, majority of compounds contained heterocyclic substituents with the potential to create H-bonding interactions. All amides were synthesised from the appropriate acid 9 and amine using either HATU or DEPBT as coupling reagent in presence of DIPEA as base and in DMF as solvent, Scheme 1. The amides were isolated by column chromatography in 20–95% yields.
Table 1.
Potency of the JQ1 amide derivativesa
| compound | 1, NHR | IC50 [μM]b | IC50 [μM]b | ||
|---|---|---|---|---|---|
| BRD4 (BD1) BRD4(BD2) | BRD2 (BD1)BRD2(BD2) | ||||
|
| |||||
|
1a
(JQ1) |
− | 0.0482 | 0.0132 | 0.0285 | 0.0095 |
|
1c
(birabresib) |
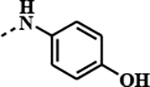
|
0.1810 | 0.0098 | 0.0587 | 0.0032 |
| 1d |
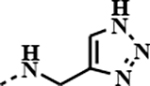
|
0.0790 | 0.0013 | 0.0086 | 0.0018 |
| 1e |
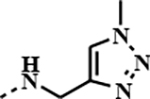
|
1.4127 | 0.0261 | 0.6396 | 0.1616 |
| 1f |
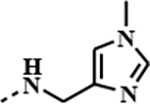
|
0.0823 | 0.0006 | 0.0176 | 0.0030 |
| 1g |
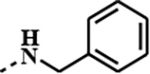
|
0.7251 | 0.0003 | 0.0217 | 0.0002 |
| 1h |
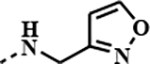
|
0.0057 | 0.0002 | 0.0031 | 0.0001 |
| 1i |
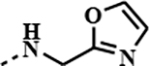
|
0.0104 | 0.0003 | 0.0092 | 0.0001 |
| 1j |
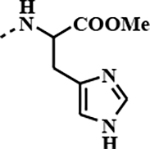
|
0.0346 | 0.0065 | 0.0321 | 0.0096 |
| 1k |
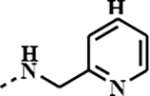
|
0.0405 | 0.0007 | 0.0163 | 0.0003 |
| 1l |
|
0.1058 | 0.0004 | 0.1467 | 0.0003 |
| 1m |
|
0.0028 | 0.0001 | 0.0034 | 0.0001 |
| 1n |
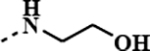
|
0.0212 | 0.0033 | 0.0222 | 0.0025 |
| 1o |
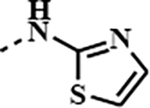
|
0.5138 | 0.0016 | 0.0109 | 0.0026 |
| 1p |
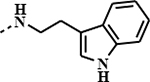
|
0.7401 | 0.0600 | 0.2529 | 0.0505 |
|
1q
(SJ1461) |
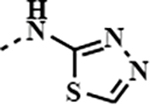
|
0.0065 | 0.0002 | 0.0016 | 0.0001 |
See Supporting Information for SD values.
TR-FRET assay.
Scheme 1.

Synthesis of amide derivatives
2.3. BRD2/BRD4 binding assay
Binding affinity of the synthesised amides were determined in TR-FRET assay against BRD4 and BRD2 proteins, as shown in Table 1. For both proteins BD2 seems to be more sensitive towards the ligands with several compounds showing sub-nanomolar activity and better potency than JQ1 1a or birabresib 1c. In general, majority of compounds possessing a heterocyclic ring within the amide moiety showed higher affinity to BRD2 than the parent JQ1 1a or birabresib 1c. Interestingly, glycine ester derivative 1m (IC50 BRD2/BD1 3.4nM), also showed a high affinity in this assay, possibly having the ester group engaged with the protein in a similar, bioisosteric fashion to five membered heterocycles. [43] [44] This is well exemplified by comparing the activities of 1m (IC50 BRD2/BD1 3.4nM) and oxazole derivative 1i (IC50 BRD2/BD1 9.2nM). Derivatives with the benzyl type of structure, 1d-f, 1h, 1i and 1k, were less potent than the most active thiadiazole 1q (IC50 BRD2/BD1 1.6nM) with the shorter amide chain. Notably, structurally similar to 1q, thiazole derivative 1o (IC50 BRD2/BD1 10.9nM) demonstrated lower affinity suggesting significance of an additional nitrogen or perhaps better positioning for the H-bonding interacting group. Surprisingly, we did not observe a difference between 1k (IC50 BRD2/BD1 16.3nM) and the N-deleted analogue 1g (IC50 BRD2/BD1 21.7nM). This may suggest that positioning of heteroatom in the amide moiety is important for the affinity. A similar trend was observed in the BRD4 assay. Derivative 1q (IC50 BRD4/BD1 6.5nM) was amongst the most active compounds with isoxazole 1h (IC50 BRD4/BD1 5.7nM) having comparable activity and glycine derivative 1m (IC50 BRD4/BD1 2.8nM) being even slightly more potent.
Overall, several of our most active compounds described above performed better in both BRD4 and BRD2 binding assays than JQ1 1a or birabresib 1c (Table 1).
2.4. Cell-based assay
Encouraging initial results prompted further profiling of the synthesised amides in cancer cell viability assays. The cell line selection was influenced by our general focus on high risk paediatric cancers such as acute leukaemia (MV4–11, NALM-16, MOLM-13) and medulloblastoma (HDMB03, D283), the two leading causes of childhood cancer death. (Table 2). Cells were treated with compounds over 72 hours, and their viability was assessed using the CellTiter-Glo assay kit (Promega). The IC50 values were determined by the proprietary software Robust Investigation of Screening Experiments (RISE), developed in house on the Pipeline Pilot platform (Biovia, v. 17.2.0). [45] Medulloblastoma cells HDMB03 showed high sensitivity towards 1i (IC50 1.0nM) and 1q (IC50 6.6nM), which were very potent in biochemical assays as well. Compound 1i was ~90-fold and ~130-fold more toxic than JQ1 1a (IC50 90.3nM) and birabresib 1c (IC50 134.3.3nM), respectively. It is also worth noting that heterocyclic amides 1o (IC50 4.6nM) and 1d (IC50 3.8nM) showed >20-fold better potency than 1a or 1c. Leukaemia cells NALM16 displayed similar sensitivity towards 1i (IC50 0.8nM) and 1q (IC50 3.6nM), while birabresib 1c (IC50 14.0nM) demonstrated ~15-fold lower potency than 1i. Additional heterocyclic amides such as 1d (IC50 7.3nM) or 1o (IC50 6.4nM) also showed significant potency against this cell line supporting generally observed trend for the JQ1 heterocyclic amide derivatives. A similar tendency was observed in other cell lines, although, with less significant differences in activity of the most active amides 1i/1q and JQ1 1a/birabresib 1c (Table 2). Glycine derivative 1m showed differences between the results obtained in two assay formats. Specifically, 1m showed considerably higher affinity in TR-FRET assay (IC50 BRD2/BD1 3.4nM, IC50 BRD4/BD1 2.8nM) compared to its activity in cell-based assays (IC50 9.9–359.2nM). Similar trends were obtained with other two amino acid amide analogues 1l and 1j. The observed lower potency in the cell-based assays might indicate susceptibility of the amino acid derive amides to hydrolytic transformations affecting activity of these compounds.
Table 2.
Cell lines studies of the amide derivativesa
| amide | HDMB03, IC50, uM |
D283, IC50, uM |
NALM16, IC50, uM |
MOLM-13, IC50, uM |
MV-4–11, IC50, uM |
|---|---|---|---|---|---|
|
| |||||
|
1a
(JQ1) |
0.0903 |
0.0609 |
0.0189 |
0.0247 |
0.099 |
|
1c
(birabresib) |
0.1343 |
0.0669 |
0.0140 |
0.0112 |
0.022 |
| 1d | 0.0038 | 0.9645 | 0.0073 | 0.0035 | 0.253 |
| 1e | 0.0407 | 0.2275 | 0.0145 | 0.0110 | 0.059 |
| 1f | 0.3589 | 0.7892 | 0.0938 | 0.0526 | 0.143 |
| 1g | 0.6190 | 0.5763 | 0.4524 | 0.3857 | 0.729 |
| 1h | 0.0173 | 0.0252 | 0.0121 | 0.0147 | 0.032 |
| 1i | 0.0010 | 0.0187 | 0.0008 | 0.0022 | 0.018 |
| 1j | 1.2443 | 2.9340 | 0.7914 | 0.2002 | 1.138 |
| 1k | 0.2630 | 0.2280 | 0.0148 | 0.0089 | 0.444 |
| 1l | 9.3408 | >8.8486 | >8.8486 | 2.4027 | 3.055 |
| 1m | 0.3592 | 0.3374 | 0.0164 | 0.0099 | 0.297 |
| 1n | 0.0427 | 0.4132 | 0.0071 | 0.0088 | 0.033 |
| 1o | 0.0046 | 0.1493 | 0.0064 | 0.0220 | 0.040 |
| 1p | 0.2546 | 0.7492 | 0.1723 | 0.3189 | 0.182 |
|
1q
(SJ1461) |
0.0066 |
0.0562 |
0.0036 |
0.0102 |
0.020 |
See supporting information for full data
Based on overall properties compound 1q was selected as a lead compound and progressed to BROMOscan™ profiling, where it showed high affinity for all BET isoforms (Figure S2). Furthermore, the lead compound 1q effect on MYC was assessed in MV4–11 cell line by immunoblotting. In this study 1q showed a very similar profile to JQ1 and birabresib. After 6 hours incubation at 100 nM concentration of 1q MYC protein abundance were reduced to around 50%, while at 1 μM concentration of 1q MYC protein levels were down to 22% (Figure S3/4).
2.5. Structural studies
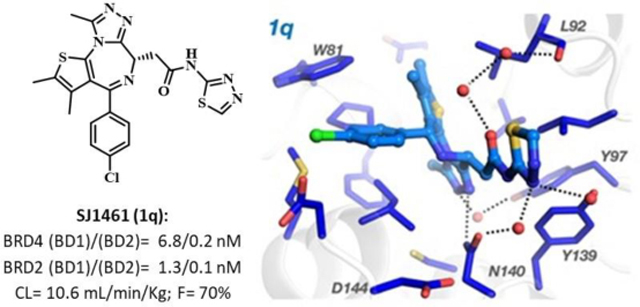
Given the promising activity of JQ-derived heterocyclic amide analogues we set to explore interactions of these derivatives with BRD proteins in more detail. We solved the X-ray crystal structures of two of our compounds, 1k and 1q, bound to BRD4/BD1 (Figure 4a–b). At resolutions of 1.05 Å with 1k (PDB: 7RN2) and 1.18 Å with 1q (PDB: 7RMD) the electron density for placement of the ligand is unambiguous (Figure S5). Analysis of the binding mode shows that both 1k and 1q occupy the same position in the active site of BRD4/BD1 as JQ1 1a and birabresib 1c, Figure 4c–d. In all cases, the conserved diazepine fragment is located deep in the pocket and H-bonds with Tyr97 through a water molecule. The amide substituents, pyridine in 1k and thiadiazole in 1q, are oriented in the same area of the binding pocket as the ester moiety in JQ1 1a or the amide fragment of birabresib 1c and interact with ZA and BC loop residues. Like 1a/1c, 1k and 1q H-bond directly with the amide NH moiety of Asn140 in the BC loop. Although 1k and 1q both contain a triazole functionality, the amide substituents are oriented orthogonally to one another and thus interact with the CO moiety of Asn140 in different ways. The conserved water bridging the H-bond with Asn140-CO binds to the secondary amide in 1k but interacts with the thiadiazole nitrogen of 1q. This allows the pyridine nitrogen of 1k to interact with a second water creating an additional H-bond with Asn140-CO like in JQ1 1a and birabresib 1c. This second water-mediated H-bond involves Asp144 as well, which 1q lacks. Yet, 1q contains a unique interaction where the thiadiazole interacts with Tyr139 in the BC loop, again through a water-mediated H-bond. Another water-mediated H-bond bridges the amide carbonyl in 1k to the backbone oxygen of Leu92 in the ZA loop and is also observed in birabresib 1c. This interaction requires two waters in 1q and is missing in JQ1 1a. The variability in water placement between 1q, 1a, and 1c/1k is most likely a consequence of the orientation of the analogues. This suggests that the amide group is critical for high affinity but also demonstrates that additional important H-bonding interactions are possible by the amide substituent. It is also worth noting that all these additional contacts are created on the rim of the BD binding pocket towards the solvent. Therefore, this provides opportunity to further derivatise the amide moiety to optimise, for example, physico-chemical properties without diminishing binding affinity.
Figure 4.

High-resolution structures of JQ1 derivatives bound to BRD4(BD1). (A) 1k interacts with Asn140, Asp144, and Leu92 (PDB: 7RN2). (B) 1q interacts with Asn140 and Tyr139 (PDB: 7RMD). (C) JQ1 1a (PDB: 3MXF) interacts with Asn140 and Asp144. (D) birabresib 1c (PDB: 5WMD) interacts with Asn140, Asp144, and Leu92. Dotted lines represent hydrogen bonds and red spheres represent ordered water molecules. Ligand electron densities are shown in SI Fig S2.
2.6. Physico-chemical and ADME studies
Upon demonstrating the beneficial effect of the heterocyclic substituent for the anti-tumour cell activity of JQ1 derivatives, we sought to determine how this moiety influences their drug-like properties (Table 3, and Supporting Information). [46] Solubility for the compounds was established in phosphate buffer solution after 18h incubation period at room temperature (Table 3). [47] Concentration of dissolved compounds was measured by UV spectrometry. The solubility of heterocyclic amides was found to be generally greater than that of JQ1 1a (37.5μM) or birabresib 1c (23.4μM), with triazole 1e (80.8μM) being the most soluble. The solubility of pyridine derivative 1k (72.5μM), was comparable to 1e, whereas thiadizole derivative 1q (12.0μM), showed lower solubility than its derivatives, including JQ1 1a and birabresib 1c. Next, we evaluated permeability of the amides using parallel artificial membrane permeability (PAMPA) and Caco2 assays. [48] [49] PAMPA assay was performed in parallel with compounds dissolved in DMSO and further diluted with phosphate buffer solutions. Donor and acceptor plates were analysed by UV spectrometry after incubation period of 0.5h. The results outlined in Table 3 suggested that the presence of polar NH/OH bond slowed down permeation of compounds (1d, 2.1; 1n 20,8; 1j, 0; 1l, 0.5 ×10−6cm/s) through the nonpolar PAMPA membrane while the heterocycle ring seemed to have a balanced effect. Lead compound 1q (534.0 ×10−6cm/s) demonstrated good permeability, while the compounds with lipophilic amide groups showed the highest permeation (1a >1800, 1g 1797.1; 1p >1800 ×10−6cm/s). In Caco2 assay we observed low permeability for the above mentioned NH/OH derivatives while 1h (Papp 150.34nm/s), 1k (Papp 98.4 nm/s) and 1q (Papp 66.5× nm/s) performed better than others. Compared to the heterocyclic amides birabresib 1c (Papp 221.6 nm/s) showed better permeability in Caco 2 than PAMPA assay. Interestingly, whereas some of the analogues such as 1f and 1e were subjects of a high efflux, lead compound 1q displayed low efflux ratio in the Caco2 assay.
Table 3.
Physico-chemical and ADME properties of the JQ1 derived amidesa
| amide | Solubility (pH 7.4, μM) |
PAMPAb (10−6cm/s) |
Caco2 A/B (nm/s) |
Caco2 B/A (nm/s) |
Efflux Ratio |
PB (%) |
PSc (t1/2,h) |
MS (t1/2,h) |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| 1a | 37.5 | ULOQ | 64.0 | 46.7 | 0.7 | 99.2 | ULOQ | 0.4 |
| 1c | 23.4 | 315.1 | 221.6 | 485.4 | 2.2 | 97.3 | ULOQ | 1.7 |
| 1f | 65.7 | 0.0 | 9.2 | 311.2 | 34.0 | 98.5 | ULOQ | 0.5 |
| 1d | 57.4 | 2.1 | 2.5 | 28.0 | 11.2 | 94.8 | ULOQ | 0.6 |
| 1g | 34.1 | 1797.1 | 42.2 | 44.5 | 1.1 | 98.7 | ULOQ | 0.1 |
| 1e | 80.8 | 14.0 | 6.2 | 246.5 | 39.9 | 97.5 | ULOQ | 0.5 |
| 1h | 56.9 | 274.5 | 150.3 | 777.9 | 5.2 | 93.5 | ULOQ | 0.4 |
| 1k | 72.5 | 177.5 | 98.4 | 568.4 | 5.8 | 97.2 | ULOQ | 0.1 |
| 1n | 76.1 | 20.8 | 7.0 | 83.3 | 11.9 | 86.9 | ULOQ | 3.2 |
| 1m | 63.3 | 231.3 | 63.8 | 307.3 | 4.8 | 91.0 | 15.8 | 1.1 |
| 1i | 62.0 | 97.2 | 30.4 | 297.4 | 9.8 | 94.1 | ULOQ | 5.8 |
| 1o | 12,41 | 544.8 | 19.1 | 41.1 | 2.2 | 98.2 | ULOQ | 1.1 |
| 1j | 68.4 | 0.0 | 7.7 | 14.2 | 1.8 | 92.7 | 24.7 | 4.7 |
| 1l | 66.4 | 0.5 | 4.5 | 7.8 | 1.7 | 85.6 | ULOQ | 11.3 |
| 1p | 1.9 | ULOQ | 5.7 | 13.8 | 2.4 | 99.6 | ULOQ | 0.3 |
| 1q | 12.0 | 534.0 | 66.5 | 180.1 | 2.7 | 97.2 | ULOQ | 3.2 |
See supporting information for full data; PAMPA: Parallel artificial membrane permeability assay; Caco2:_human colorectal adenocarcinoma cells permeability assay; PB: human plasma binding; PS: human plasma stability; MS: metabolic stability (human microsomes).
ULOQ on the PAMPA value indicates compound quantification was upper the limit of quantification (>1800 ×10–6cm/s).
ULOQ on the PS value indicates compound quantification was upper the limit of quantification (>100 h).
Plasma protein binding of a drug or a studied compound is important pharmacokinetic parameter as it can be often related to their efficacy. [50] Lower level of protein binding makes larger proportion of the drug/compound available to reach the biological target. Our experiments to determine this factor were performed using Rapid Equilibrium Dialysis (RED) Assay (Table 3). The results suggested high level of binding for all heterocyclic amides (>90%) including JQ1 1a and birabresib 1c. With the current set of compounds, we did not observe significant changes in this parameter with variations of the heterocyclic ring. Human plasma stability experiments were performed by incubating the compounds for up to 48h followed by LC-MS/MS analysis. Majority of the compounds showed excellent stability including derivative 1q (100%), Table 3. Finally, we investigated metabolic stability of the synthesised amides in human microsomes (Table 3). [51] Amongst the amide derivatives lead compound 1q (t1/2 3.2h) showed moderate metabolic stability while pyridine derivative 1k (t1/2 0.1h) and benzyl derivative 1g (t1/2 0.1h) were subjects of rapid metabolic transformation. Notably, the human microsome stability of lead compound 1q was also better than that of JQ1 1a (t1/2 0.4h) or birabresib 1c (t1/2 1.7h).
2.7. In vivo pharmacokinetic studies
The promising overall properties of 1q warranted its progression into in vivo pharmacokinetic studies. [52] This was carried out in female CD1 mice following a single intravenous (at 1mg/kg) and oral administration (at 3mg/kg and at 10mg/kg). Blood samples were collected over a time course and they were analysed by LC-MS/MS method. The critical pharmacokinetic parameters are outlined in Table 4.
Table 4.
In vivo pharmacokinetic studies for 1q
| Route | Dose (mg/kg) |
Tmax (h) |
C0/Cmax (ng/mL) |
AUClast (hr*ng/mL) |
T1/2 (hr) |
CL/CL_F (mL/min/kg) |
Vss/Vz_F (L/kg) |
F (%) |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| IV | 1 | NA | 1931.3 | 1567.1 | 1.5 | 10.6 | 0.8 | NA |
| PO | 3 | 1.00 | 1555.3 | 3275.4 | 3.9 | 15.2 | 5.2 | 70 |
| PO | 10 | 0.50 | 3155.6 | 9473.0 | 3.9 | 17.5 | 6.0 | 60 |
Tmax: the time to achieve the highest concentration of drug in the blood
Cmax: maximal concentration of drug
AUC: area under the curve
T1/2: elimination half-life
CL: clearance
Vss: volume of distribution
F: bioavailability
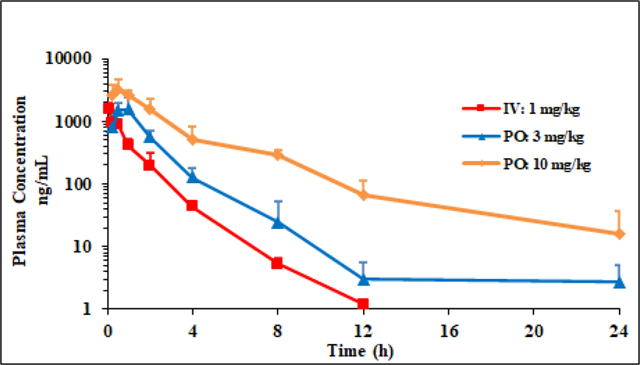
Compound 1q showed low plasma clearance (Cl=10.6 mL/min/kg) and volume of distribution typical of non-basic compounds (Vss=0.8 L/Kg), with elimination half-life of 1.5h. Subsequent P.O. experiments performed with 3mg/kg and 10mg/kg doses suggested good absorption (Tmax 1hr and 0.5hr, respectively) and high bioavailability (Table 4).
3. Conclusion
A follow up on an observation from our CLICK-derived BET-PROTACs program led to the discovery of JQ1-derived amides with overall properties comparable or better to those of JQ1 and currently clinically studied birabresib. Biochemical assay demonstrated >20-fold better potency of thiadiazole derived 1q (SJ1461) towards BRD4/BRD2, while investigation in a panel of cancer cell lines suggested particular sensitivity of HDMB03 towards oxazole 1i (~130- fold more potent than birabresib 1c). Initially studied pharmacokinetic parameters of the amides are comparable to those of birabresib 1c but the drawback for 1c is the potentially toxicophoric p-aminophenol as constitutional part of its structure. Analysis of the crystal structure of BRD4(BD1)-1q/1k provided an insight into the protein-ligand interactions revealing novel contacts that can be exploited for improving activity of JQ1 derivatives. The heterocyclic moiety was also shown to project towards the solvent implicitly suggesting potential route to optimise physicochemical properties of the studied compounds. The overall in vitro/vivo properties of SJ1461 makes this compound promising candidate for further exploration.
4. Experimental
All experimental procedures are delineated in Supporting Information.
Supplementary Material
Figure 2.

JQ1 derived PROTAC compounds
Highlights.
A PROTAC X-ray structure inspired synthesis of JQ1-based heteroaromatic amides.
Best derivatives showed improved overall profile compared to JQ1 and birabresib.
X-ray structure of SJ1461-BRD4 complex provided a rationale for improved affinity.
SJ1461 is being evaluated as a potential clinical candidate.
Acknowledgment
This work was supported by ALSAC and R35GM142772 (to MF), and R35GM140837 (to WCKP). Crystallographic data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. SER-CAT is supported by its member institutions, and equipment grants (S10_RR25528, S10_RR028976 and S10_OD027000) from the National Institutes of Health. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. This research was also supported by the Science fund of the Republic of Serbia: program, Diaspora; Grant no: 6463913; Acronym DeSyHPRO and by the Ministry of Education, Science and Technological Development, Republic of Serbia, Grant Agreement with University of Belgrade-Faculty of Pharmacy No: 451-03-9/2021-14/200161.
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supporting Information
The Supporting Information, experimental procedures and spectral data, is available.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hnilica LS, Structure and Biological Functions of Histones, 1st edition, CRC Press, Boca Raton, 2017. [Google Scholar]
- [2].Shen C, ed., Histones: Class, Structure and Function, 1st edition, Nova Science Pub Inc, New York, 2012. [Google Scholar]
- [3].Lee CY, Grant PA, Chapter 1–1 - Role of Histone Acetylation and Acetyltransferases in Gene Regulation, in: McCullough SD, Dolinoy DC (Eds.), Toxicoepigenetics, Academic Press, 2019: pp. 3–30. 10.1016/B978-0-12-812433-8.00001-0. [DOI] [Google Scholar]
- [4].Verdone L, Agricola E, Caserta M, Di Mauro E, Histone acetylation in gene regulation, Brief. in Func. Genomics. 5 (2006) 209–221. 10.1093/bfgp/ell028. [DOI] [PubMed] [Google Scholar]
- [5].Struhl K, Histone acetylation and transcriptional regulatory mechanisms, Genes Dev. 12 (1998) 599–606. [DOI] [PubMed] [Google Scholar]
- [6].Romero FA, Taylor AM, Crawford TD, Tsui V, Cote A, Magnuson S, Disrupting Acetyl-Lysine Recognition: Progress in the Development of Bromodomain Inhibitors, J. Med. Chem. 59 (2016) 1271–1298. 10.1021/acs.jmedchem.5b01514. [DOI] [PubMed] [Google Scholar]
- [7].Umehara T, Nakamura Y, Jang MK, Nakano K, Tanaka A, Ozato K, Padmanabhan B, Yokoyama S, Structural Basis for Acetylated Histone H4 Recognition by the Human BRD2 Bromodomain, J. Biol. Chem. 285 (2010) 7610–7618. 10.1074/jbc.M109.062422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mujtaba S, Zeng L, Zhou M-M, Structure and acetyl-lysine recognition of the bromodomain, Oncogene. 26 (2007) 5521–5527. 10.1038/sj.onc.1210618. [DOI] [PubMed] [Google Scholar]
- [9].Schwalm MP, Knapp S, BET bromodomain inhibitors, Curr. Opin. Chem. Biol. 68 (2022) 102148. 10.1016/j.cbpa.2022.102148. [DOI] [PubMed] [Google Scholar]
- [10].Stathis A, Bertoni F, BET Proteins as Targets for Anticancer Treatment, Cancer Discov. 8 (2018) 24–36. 10.1158/2159-8290.CD-17-0605. [DOI] [PubMed] [Google Scholar]
- [11].Padmanabhan B, Mathur S, Manjula R, Tripathi S, Bromodomain and extra-terminal (BET) family proteins: New therapeutic targets in major diseases, J, Biosci. 41 (2016) 295–311. 10.1007/s12038-016-9600-6. [DOI] [PubMed] [Google Scholar]
- [12].Sahai V, Redig AJ, Collier KA, Eckerdt FD, Munshi HG, Targeting BET bromodomain proteins in solid tumors, Oncotarget. 7 (2016) 53997–54009. 10.18632/oncotarget.9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Taniguchi Y, The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins, Int. J. Mol. Sci. 17 (2016) 1849. 10.3390/ijms17111849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ali HA, Li Y, Bilal AHM, Qin T, Yuan Z, Zhao W, A Comprehensive Review of BET Protein Biochemistry, Physiology, and Pathological Roles, Front. Pharmacol. 13 (2022) article 818891. https://www.frontiersin.org/articles/10.3389/fphar.2022.818891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Werner MT, Wang H, Hamagami N, Hsu SC, Yano JA, Stonestrom AJ, Behera V, Zong Y, Mackay JP, Blobel GA, Comparative structure-function analysis of bromodomain and extraterminal motif (BET) proteins in a gene-complementation system, J. Biol. Chem. 295 (2020) 1898–1914. 10.1074/jbc.RA119.010679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ferri E, Petosa C, McKenna CE, Bromodomains: Structure, function and pharmacology of inhibition, Biochem. Pharmacol. 106 (2016) 1–18. 10.1016/j.bcp.2015.12.005. [DOI] [PubMed] [Google Scholar]
- [17].Aiyer S, Swapna GVT, Ma L-C, Liu G, Hao J, Chalmers G, Jacobs BC, Montelione GT, Roth MJ, A common binding motif in the ET domain of BRD3 forms polymorphic structural interfaces with host and viral proteins, Structure. 29 (2021) 886–898.e6. 10.1016/j.str.2021.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Crowe BL, Larue RC, Yuan C, Hess S, Kvaratskhelia M, Foster MP, Structure of the Brd4 ET domain bound to a C-terminal motif from γ-retroviral integrases reveals a conserved mechanism of interaction, Proc. Natl. Acad. Sci. U.S.A. 113 (2016) 2086–2091. 10.1073/pnas.1516813113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shorstova T, Foulkes WD, Witcher M, Achieving clinical success with BET inhibitors as anti-cancer agents, Br. J Cancer. 124 (2021) 1478–1490. 10.1038/s41416-021-01321-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sarnik J, Popławski T, Tokarz P, BET Proteins as Attractive Targets for Cancer Therapeutics, Int. J. Mol. Sci. 22 (2021) 11102. 10.3390/ijms222011102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Trojer P, Targeting BET Bromodomains in Cancer, Annu. Rev. Cancer Biol. 6 (2022) 313–336. 10.1146/annurev-cancerbio-070120-103531. [DOI] [Google Scholar]
- [22].Schwalm MP, Knapp S, BET bromodomain inhibitors, Curr. Opin. Chem. Biol. 68 (2022) 102148. 10.1016/j.cbpa.2022.102148. [DOI] [PubMed] [Google Scholar]
- [23].Feng L, Wang G, Chen Y, He G, Liu B, Liu J, Chiang C-M, Ouyang L, Dual-target inhibitors of bromodomain and extra-terminal proteins in cancer: A review from medicinal chemistry perspectives, Med. Res. Rev. 42 (2022) 710–743. 10.1002/med.21859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen J, Tang P, Wang Y, Wang J, Yang C, Li Y, Yang G, Wu F, Zhang J, Ouyang L, Targeting Bromodomain-Selective Inhibitors of BET Proteins in Drug Discovery and Development, J. Med. Chem. 65 (2022) 5184–5211. 10.1021/acs.jmedchem.1c01835. [DOI] [PubMed] [Google Scholar]
- [25].Fu Y, Zhang Y, Sun H, Progress in the development of domain selective inhibitors of the bromo and extra terminal domain family (BET) proteins, Eur. J. Med. Chem. 226 (2021) 113853. 10.1016/j.ejmech.2021.113853. [DOI] [PubMed] [Google Scholar]
- [26].Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE, Selective inhibition of BET bromodomains, Nature. 468 (2010) 1067–1073. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jiang G, Deng W, Liu Y, Wang C, General mechanism of JQ1 in inhibiting various types of cancer, Mol. Med. Rep. 21 (2020) 1021–1034. 10.3892/mmr.2020.10927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li F, MacKenzie KR, Jain P, Santini C, Young DW, Matzuk MM, Metabolism of JQ1, an inhibitor of bromodomain and extra terminal bromodomain proteins, in human and mouse liver microsomes, Biol. Reprod. 103 (2020) 427–436. 10.1093/biolre/ioaa043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lewin J, Soria J-C, Stathis A, Delord J-P, Peters S, Awada A, Aftimos PG, Bekradda M, Rezai K, Zeng Z, Hussain A, Perez S, Siu LL, Massard C, Phase Ib Trial With Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients With Selected Advanced Solid Tumors, J. Clin. Oncol. 36 (2018) 3007–3014. 10.1200/JCO.2018.78.2292. [DOI] [PubMed] [Google Scholar]
- [30].Li K, Crews CM, PROTACs: past, present and future, Chem. Soc. Rev. 51 (2022) 5214–5236. 10.1039/D2CS00193D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bekes M, Langley DR, Crews CM, PROTAC targeted protein degraders: the past is prologue, Nat. Rev. Drug Discov. 21 (2022) 181–200. 10.1038/s41573-021-00371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Nemec V, Schwalm MP, Müller S, Knapp S, PROTAC degraders as chemical probes for studying target biology and target validation, Chem. Soc. Rev. 51 (2022) 7971–7993. 10.1039/D2CS00478J. [DOI] [PubMed] [Google Scholar]
- [33].Sun X, Gao H, Yang Y, He M, Wu Y, Song Y, Tong Y, Rao Y, PROTACs: great opportunities for academia and industry, Sig. Transduct. Target Ther. 4 (2019) 1–33. 10.1038/s41392-019-0101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wurz RP, Dellamaggiore K, Dou H, Javier N, Lo M-C, McCarter JD, Mohl D, Sastri C, Lipford JR, Cee VJ, A “Click Chemistry Platform” for the Rapid Synthesis of Bispecific Molecules for Inducing Protein Degradation, J. Med. Chem. 61 (2018) 453–461. 10.1021/acs.jmedchem.6b01781. [DOI] [PubMed] [Google Scholar]
- [35].Huang Y, Yokoe H, Kaiho-Soma A, Takahashi K, Hirasawa Y, Morita H, Ohtake F, Kanoh N, Design, Synthesis, and Evaluation of Trivalent PROTACs Having a Functionalization Site with Controlled Orientation, Bioconjugate Chem. 33 (2022) 142–151. 10.1021/acs.bioconjchem.1c00490. [DOI] [PubMed] [Google Scholar]
- [36].Ohoka N, Tsuji G, Shoda T, Fujisato T, Kurihara M, Demizu Y, Naito M, Development of Small Molecule Chimeras That Recruit AhR E3 Ligase to Target Proteins, ACS Chem. Biol. 14 (2019) 2822–2832. 10.1021/acschembio.9b00704. [DOI] [PubMed] [Google Scholar]
- [37].Henning NJ, Manford AG, Spradlin JN, Brittain SM, Zhang E, McKenna JM, Tallarico JA, Schirle M, Rape M, Nomura DK, Discovery of a Covalent FEM1B Recruiter for Targeted Protein Degradation Applications, J. Am. Chem. Soc. 144 (2022) 701–708. 10.1021/jacs.1c03980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chan K-H, Zengerle M, Testa A, Ciulli A, Impact of Target Warhead and Linkage Vector on Inducing Protein Degradation: Comparison of Bromodomain and Extra-Terminal (BET) Degraders Derived from Triazolodiazepine (JQ1) and Tetrahydroquinoline (I-BET726) BET Inhibitor Scaffolds, J. Med. Chem. 61 (2018) 504–513. 10.1021/acs.jmedchem.6b01912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Min J, Mayasundari A, Keramatnia F, Jonchere B, Yang SW, Jarusiewicz J, Actis M, Das S, Young B, Slavish J, Yang L, Li Y, Fu X, Garrett SH, Yun M-K, Li Z, Nithianantham S, Chai S, Chen T, Shelat A, Lee RE, Nishiguchi G, White SW, Roussel MF, Potts PR, Fischer M, Rankovic Z, Phenyl-Glutarimides: Alternative Cereblon Binders for the Design of PROTACs, Angew. Chem. Int. Ed. 133 (2021) 26663–26670. 10.1002/ange.202108848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE, Selective Target Protein Degradation via Phthalimide Conjugation, Science. 348 (2015) 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Xue G, Wang K, Zhou D, Zhong H, Pan Z, Light-Induced Protein Degradation with Photocaged PROTACs, J. Am. Chem. Soc. 141 (2019) 18370–18374. 10.1021/jacs.9b06422. [DOI] [PubMed] [Google Scholar]
- [42].Ozer HG, El-Gamal D, Powell B, Hing ZA, Blachly JS, Harrington B, Mitchell S, Grieselhuber NR, Williams K, Lai T-H, Alinari L, Baiocchi RA, Brinton L, Baskin E, Cannon M, Beaver L, Goettl VM, Lucas DM, Woyach JA, Sampath D, Lehman AM, Yu L, Zhang J, Ma Y, Zhang Y, Spevak W, Shi S, Severson P, Shellooe R, Carias H, Tsang G, Dong K, Ewing T, Marimuthu A, Tantoy C, Walters J, Sanftner L, Rezaei H, Nespi M, Matusow B, Habets G, Ibrahim P, Zhang C, Mathé EA, Bollag G, Byrd JC, Lapalombella R, BRD4 Profiling Identifies Critical Chronic Lymphocytic Leukemia Oncogenic Circuits and Reveals Sensitivity to PLX51107, a Novel Structurally Distinct BET Inhibitor, Cancer Discov. 8 (2018) 458–477. 10.1158/2159-8290.CD-17-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Meanwell NA, Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design, J. Med. Chem. 54 (2011) 2529–2591. 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- [44].Hoshi A, Sakamoto T, Takayama J, Xuan M, Okazaki M, Hartman TL, Buckheit RW, Pannecouque C, Cushman M, Systematic evaluation of methyl ester bioisosteres in the context of developing alkenyldiarylmethanes (ADAMs) as non-nucleoside reverse transcriptase inhibitors (NNRTIs) for anti-HIV-1 chemotherapy, Bioorg. Med. Chem. 24 (2016) 3006–3022. 10.1016/j.bmc.2016.05.010. [DOI] [PubMed] [Google Scholar]
- [45].Ward E, DeSantis C, Robbins A, Kohler B, Jemal A, Childhood and adolescent cancer statistics, 2014, CA Cancer J. Clin. 64 (2014) 83–103. 10.3322/caac.21219. [DOI] [PubMed] [Google Scholar]
- [46].Di L, Kerns E, Drug-Like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization, 2nd edition, Academic Press, Amsterdam; Boston, 2016. [Google Scholar]
- [47].Di L, Fish PV, Mano T, Bridging solubility between drug discovery and development, Drug Discov. 17 (2012) 486–495. 10.1016/j.drudis.2011.11.007. [DOI] [PubMed] [Google Scholar]
- [48].Ottaviani G, Martel S, Carrupt P-A, Parallel Artificial Membrane Permeability Assay: A New Membrane for the Fast Prediction of Passive Human Skin Permeability, J. Med. Chem. 49 (2006) 3948–3954. 10.1021/jm060230+. [DOI] [PubMed] [Google Scholar]
- [49].Lipinski CA, Lombardo F, Dominy BW, Feeney PJ, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Deliv. Rev. 64 (2012) 4–17. 10.1016/j.addr.2012.09.019. [DOI] [PubMed] [Google Scholar]
- [50].Bohnert T, Gan L-S, Plasma protein binding: from discovery to development, J. Pharm. Sci. 102 (2013) 2953–2994. 10.1002/jps.23614. [DOI] [PubMed] [Google Scholar]
- [51].Baranczewski P, Stańczak A, Sundberg K, Svensson R, Wallin A, Jansson J, Garberg P, Postlind H, Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development, Pharmacol. Rep. 58 (2006) 453–472. [PubMed] [Google Scholar]
- [52].Pelkonen O, Turpeinen M, Raunio H, In vivo-in vitro-in silico pharmacokinetic modelling in drug development: current status and future directions, Clin. Pharmacokinet. 50 (2011) 483–491. 10.2165/11592400-000000000-00000. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.