

Abstract
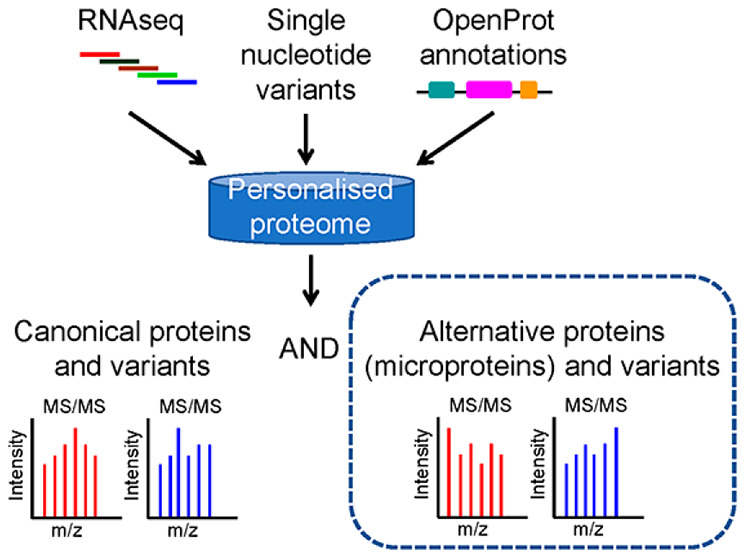
Proteomic diversity in biological samples can be characterized by mass spectrometry (MS)-based proteomics using customized protein databases generated from sets of transcripts previously detected by RNA-seq. This diversity has only been increased by the recent discovery that many translated alternative open reading frames rest unannotated at unsuspected locations of mRNAs and ncRNAs. These novel protein products, termed alternative proteins, have been left out of all previous custom database generation tools. Consequently, genetic variations that impact alternative open reading frames and variant peptides from their translated proteins are not detectable with current computational workflows. To fill this gap, we present OpenCustomDB, a bioinformatics tool that uses sample-specific RNaseq data to identify genomic variants in canonical and alternative open reading frames, allowing for more than one coding region per transcript. In a test reanalysis of a cohort of 16 patients with acute myeloid leukemia, 5666 peptides from alternative proteins were detected, including 201 variant peptides. We also observed that a significant fraction of peptide-spectrum matches previously assigned to peptides from canonical proteins got better scores when reassigned to peptides from alternative proteins. Custom protein libraries that include sample-specific sequence variations of all possible open reading frames are promising contributions to the development of proteomics and precision medicine. The raw and processed proteomics data presented in this study can be found in PRIDE repository with accession number PXD029240.
Keywords: proteogenomics, alternative proteins, alternative ORFs, database, variants, precision medicine, multicoding
Introduction
The inability to detect variants of canonical proteins represents a major drawback when using reference protein databases for shotgun mass spectrometry-based proteomics to perform large-scale proteomic profiles of biological samples. In addition, several thousands of functional ORFs have been discovered in regions of the transcriptome that were not expected to be protein-coding, thus are not annotated in conventional databases. These novel or alternative ORFs (altORFs) are present within 5′- and 3′-UTRs, overlap a known coding sequence (CDS) in a frameshifted reading frame, or are present in transcripts expressed from noncoding RNA genes or from pseudogenes and automatically annotated as noncoding.1 These are particularly major issues in the field of precision medicine, which, among other things, aims at associating each biological sample with a specific proteomic profile. Proteogenomics is an increasingly popular solution to this problem, enabling the construction of customized protein databases using sample-specific genomic or transcriptomic data. For this approach, two main types of computational tools have been developed. In the first group, variants discovered in RNA-seq data are added to an existing reference protein database.2−6 Here, variations may include single or multiple nucleotide variations, indels, frameshifts, novel alternative splice forms, and gene fusions according to the proteogenomic workflow. These tools do not enable the detection of variants of novel proteins. The second group of computational tools enables the detection of novel proteins and their variants, increasing the depth of proteomic profiling of a biological sample.7,8 These proteogenomics workflows insert a 3-frame or a 6-frame translation step of transcripts or genomes to annotate possible novel proteins, resulting in very large databases and therefore dramatically increasing both computing time and the challenge of false positive identifications.9 To address this problem, different experimental or computational strategies are incorporated to reduce the size of the customized database.7,8,10,11 However, these proteogenomics workflows are primarily designed to discover novel peptides and a limited number of variant types, rather than to enable precision medicine studies on a routine basis. Finally, none of these tools allows for the possibility of more than one ORF in the same reading frame or in different reading frames in a single RNA or in different reading frames in a single RNA.
OpenProt is a proteogenomic resource that provides functional annotation of any ORF with a minimum size of 30 codons in the transcriptome (NCBI RefSeq and Ensembl) of several species without a priori regarding the coding biotype of the transcripts, the number of ORFs in each transcript, and the reading frame12 Therefore, OpenProt allows several ORFs within the same transcripts, in agreement with experimental observations of a significant number of dual coding (overlapping ORFs) and polycistronic (at least two nonoverlapping ORFs) genes in mammals.13,14 Proteins annotated by OpenProt in human include (1) 134,477 canonical proteins that are already annotated in UniProt, NCBI RefSeq or Ensembl; (2) 68,612 novel isoforms that are novel proteins with a significant identity with a canonical protein from the same gene; and (3) 488,956 alternative proteins (altProts) that are novel proteins with no significant identity with any canonical proteins.
To build RNaseq-based custom protein databases using OpenProt annotations for precision medicine, we created OpenCustomDB, available both as a Python package and as a web application. In contrast to other proteogenomic workflows, OpenCustomDB-derived protein databases allow the identification of canonical proteins, novel isoforms of canonical proteins, and altProts encoded in the transcriptome of a sample of interest, and of their variants (Figure 1). To test this new proteogenomic workflow, we used OpenCustomDB on leukemic cells from 16 patients with acute myeloid leukemia.15 Acute myeloid leukemia is among the most lethal of all hematologic cancers that affect both children and adults. From a total of 91,372 unique detected peptides, 5666 are from alternative proteins, including 201 variant peptides.
Figure 1.

OpenCustomDB enables the construction of customized protein sequence databases integrating noncanonical proteins, including genetic variants. (A) Compared to other strategies, OpenCustomDB uses OpenProt annotations to integrate proteins that are not included in conventional databases. (B) OpenCustomDB workflow. OpenProt annotations include all ORFs longer than 30 codons and allow more than one ORF per transcript. Therefore, transcripts annotated as mRNAs may contain 1 or more ORFs in addition to the annotated coding sequence (CDS). (C) Representative composition of a customized database generated using data from patient 05H143. OpenCustomDB was run with (OpenCustomDB, green columns) or without (canonical proteins only, gray columns) the integration of altProts and novel isoforms. The number of protein entries is indicated. The size of the database with altProts and novel isoforms was limited to a total of 100,000 entries. A total of 16 patients were included in this study and all databases are shown in supplementary Figure 1. (D) Transcript sequence localization of ORFs encoding noncanonical proteins from the patient database 05H143.
EXPERIMENTAL PROCEDURES
Human AML Samples from the Quebec Leukemia Cell Bank
AML specimens were purified from bone marrow or leukapheresis samples by Ficoll density centrifugation and cryopreserved in liquid nitrogen (DMSO 10%). All samples were collected, prepared, and cryopreserved by the Quebec Leukemia Cell Bank (BCLQ, http://bclq.org/), which is certified by the Canadian Tissue Repository Network (CTRNet). The technical and clinical characteristics have previously been described.15 AML samples were thawed (1 min in 37 °C water bath) and resuspended in 48 mL of 4 °C PBS. Two million cells (1 mL) were pelleted and resuspended in 1 mL of Trizol for RNA sequencing, while 5 million were pelleted and snap frozen in liquid nitrogen for mass spectrometry analyses.
RNA Sequencing
RNA extraction, library preparation, and sequencing were performed as previously described.15 RNA-Seq reads were trimmed for sequencing adapters and low quality 3′ bases using TrimGalore version 0.6.4 (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). The reads were then aligned to the reference genome GRCh38.p12 using STAR version 2.7.3a16 running with default parameters except for ‘–outSAMprimaryFlag: AllBestScore,–outFilterMismatchNmax: 5, –alignSJoverhangMin 10, –alignMatesGapMax 200 000, –alignIntronMax 200 000, –alignSJstitchMismatchNmax “5-1 5 5”,–bamRemoveDuplicatesType UniqueIdenticalNotMulti’. Transcript expression was quantified in transcripts per million (tpm) with kallisto version 0.46.017 running with default parameters.
Variant Calling and Integration in the Transcripts
Variants were called from genomic DNA. Variant calling files (VCF) were generated from BAM files with FreeBayes version 1.3.1 with the setting “–min-alternate-count” set to 5. SNPs and Indels with FreeBayes quality of less than 20 were filtered out with an internal Python script. Then, OpenCustomDB uses OpenVar18 to predict the impact of variations on the primary structure of the corresponding canonical proteins, novel isoforms, and altProts and inserts the variations at their correct positions in the transcripts.
OpenCustomDB Inputs
OpenCustomDB has two mandatory inputs: a VCF file that can be generated with any variant caller such as FreeBayes, and a transcript expression file.
OpenCustomDB also uses optional inputs:
By default, the maximum number of proteins in the custom database is set at 100,000 and the number of transcripts is limited by this value. This value can be modified.
An additional list of transcripts may be included to recover proteins from transcripts that do not reach the threshold enforced by the maximum number of proteins.
An additional list of transcripts to be removed may be uploaded to exclude specific proteins.
Customised Database Generation
First, a VCF file is used by the variant annotator OpenVar to annotate variants in canonical and noncanonical proteins. After this step, all protein sequences specific to the sample transcriptome are obtained, including cases where, for example, a single nucleotide variation caused a change in amino acid in the protein sequence. Then, the Kallisto quantified transcripts are ranked from the highest to the lowest expression level, and all OpenProt-annotated proteins that are associated with these transcripts (Figure 1B) are added to the customized database until a maximum number of proteins is reached (100,000 by default). When a protein variant is added to the database, the corresponding wild-type protein with no variation is also included in the database (to account for the possibility of heterozygosity).
Search and Postprocessing
MS/MS spectra were searched against sample-specific customized databases using MaxQuant v1.6.14.0 without false discovery rate (FDR) filtering.19 All other parameters were set by default. All peptide-spectrum matches (PSMs) were rescored using a combination of the spectral intensity predictor MS2PIP,20 HPLC retention time predictor DeepLC21 and the postprocessing tool Percolator22 within MS2Rescore23 as previously described.24 Percolator was run with MS2Rescore to compute PSM scores as well as q-values.22,25 The PSMs were selected by applying a FDR < 1%, and the unicity of nonvariant and variant peptides from novel isoforms and altProts was checked against Ensembl and UniProt. Peptides were classified into 6 categories: derived from canonical proteins, alternative proteins, novel isoforms, each with their corresponding variants. The Percolator Fido protein inference tool was used to obtain proteins groups. The protein groups were selected applying FDR < 1%. Identified protein numbers were calculated from Percolator Fido results with the following rules: (1) one canonical protein was counted for each gene; (2) variant proteins were computed only if the corresponding peptides showing the variation were identified in the protein group; (3) altProts were computed if at least one unique peptide was identified.
Cell Lysis, Protein Extraction, In-Gel Digestion
Cells were lysed with 0.5% w/v CHAPS (final concentration) containing a protease inhibitor cocktail (Sigma, cat#P8340-5ML) for 60 min with tumbling at 4 °C. BCA dosage was done following by reduction in 10 mm DTT and alkylation in 50 mM iodoacetamide. SDS-Page, in-gel digestion, peptide extraction and desalting were performed as previously described26 with minor modifications. Mini-PROTEAN TGX Precast Gels were used, and a ratio of 1:10 (w/w) of Trypsin Gold (Promega) were used for tryptic digestion.
LC-MS/MS Analysis
The peptides were separated with a Dionex Ultimate 3000 nanoHPLC system. Here, 10 μL of sample (1.5 μg) in 1% (v/v) formic acid were loaded with a constant flow of 4 μL/min onto an Acclaim PepMap100 C18 column (0.3 mm id × 5 mm, Dionex Corporation). After trap enrichment, the peptides were eluted in an EasySpray PepMap C18 nano column (75 μm × 50 cm, Dionex Corporation) with a linear gradient of 5–35% solvent B (90% acetonitrile with 0.1% formic acid) with a constant flow of 200 nL/min for 240 min. The HPLC system was coupled to an OrbiTrap Q-Exactive mass spectrometer (Thermo Fisher Scientific Inc.) via an EasySpray source. The spray voltage was set to 2.0 kV and the column temperature was set to 40 °C. Full scan MS survey spectra (m/z 350–1600) in profile mode were acquired in the Orbitrap with a resolution of 70,000 after accumulation of 1,000,000 ions. The 10 most intense peptide ions from the Orbitrap survey scan were fragmented by high-energy collision dissociation (normalized collision energy 25% and resolution of 17,500) after the accumulation of 50,000 ions. Maximal filling times were 250 ms for the full scans and 60 ms for the MS/MS scans. Precursor ion charge state screening was enabled and all unassigned charge states as well as singly, 7 and 8 charged species were rejected. The dynamic exclusion list was restricted to a maximum of 500 entries with a maximum retention period of 40 s and a relative mass window of 10 ppm. The lock mass option was enabled for survey scans to improve mass accuracy. Data were acquired using the Xcalibur software.
Availability and Implementation
OpenCustomDB is an open-source pipeline written in Python. It is also available as a web application https://www.openprot.org/opencustomdb.
Results
The translation of proteins unannotated in conventional protein sequence databases, including novel isoforms of canonical proteins and altProts has been largely reported using MS-based proteomics and ribosome profiling strategies. However, there are no tools for the annotation and detection of variants of these novel proteins. OpenCustomDB fills this gap and enables the detection of a deeper proteome.
OpenCustomDB: Design and Implementation
OpenCustomDB uses RNA-seq data to generate customized protein sequence databases (Figure 1B). It takes advantage of the OpenProt annotation that includes altProts, predicted novel isoforms of canonical proteins, and canonical proteins.12 A VCF (Variant Calling File) is generated with the OpenVar genomic variant annotator, which integrates altORFs.18 A transcript expression file is used as a second input to control database size by either adjusting the number of transcripts according to their level of expression (i.e., threshold value) or by establishing a maximum number of protein entries. With the default parameters, the maximum number of protein entries is 100,000 and the Transcripts Per Kilobase Million or TPM threshold is adjusted accordingly. Finally, specific transcripts can also be added or removed. Transcripts must have a TPM > 0 to be considered in the analysis, even if the maximum number of protein entries has not been reached.
We used RNA-seq data from cells from 16 patients with acute myeloblastic leukemia.15 For example, the resulting customized database for patient 05H143 contains a significant number of novel proteins and variants compared to the standard proteogenomic database containing only canonical proteins and their variants (Figure 1C). Here, the default parameters were used and 13,382 transcripts with TPM > 5.38 were selected to limit the database to 100,000 protein entries. The conventional database with canonical proteins and their variants contains a total of 13,716 protein entries (11,600 canonical proteins and 2116 variants). The custom database contains 78,083 altProts and novel isoforms, and 8201 variants, in addition to the 13,716 canonical proteins including their variants. The altORFs encoding the predicted altProts are mainly located within mRNAs (Figure 1D). Among them, the majority overlap the canonical CDS in a different reading frame or localize in 3′-UTRs. Similar results were obtained for 15 other patients (Supplementary Figure 1). The RNaseq approach included the selection of poly-A+, thus is necessarily biased for mRNAs and poly-A+ ncRNAs.
Peptide Level Analysis
We used MaxQuant19 followed by a combination of the MS2Rescore spectral intensity predictor and the Percolator postprocessing tool24 for proteomics analyses. This workflow is particularly effective in the context of proteogenomics with large databases.21 and is therefore well suited for our analyses with custom databases containing 100,000 protein entries. Here, we use the OpenProt assignment rule where a peptide that can be assigned to both a canonical protein and an altProt is always assigned to the canonical protein. In addition to reference and variant peptides from canonical proteins, a significant number of reference and variant peptides from altProts and novel isoforms were also identified in each patient, confirming the validity of our proteogenomic approach for the discovery of novel proteins (Table 1). Variant peptides are listed in Supplementary Table 1.
Table 1. Number of Unique Reference and Variant Peptides in Canonical Proteins, Alternative Proteins, and Novel Isoforms in 16 Patients with Acute Myeloid Leukemiaa.
| Peptides
derived from canonical proteins |
Peptides
derived from alternative proteins |
Peptides
derived from novel isoforms |
||||
|---|---|---|---|---|---|---|
| Patients | Total | Variants | Total | Variants | Total | Variants |
| 05H143 | 45895 | 143 | 607 | 26 | 21 | 4 |
| 05H149 | 27771 | 73 | 463 | 11 | 10 | 9 |
| 07H063 | 45504 | 145 | 612 | 14 | 23 | 2 |
| 07H122 | 46882 | 181 | 545 | 14 | 12 | 3 |
| 07H141 | 33691 | 111 | 486 | 18 | 13 | 2 |
| 08H039 | 31784 | 91 | 384 | 7 | 8 | 2 |
| 08H053 | 33801 | 93 | 397 | 10 | 7 | 6 |
| 11H008 | 25517 | 61 | 252 | 8 | 9 | 1 |
| 11H035 | 32586 | 95 | 467 | 10 | 13 | 3 |
| 12H172 | 43334 | 129 | 527 | 28 | 17 | 5 |
| 15H013 | 32250 | 32 | 342 | 3 | 14 | 2 |
| 15H023 | 40593 | 135 | 404 | 10 | 18 | 1 |
| 15H063 | 31231 | 94 | 322 | 4 | 13 | 3 |
| 15H080 | 43293 | 153 | 449 | 14 | 25 | 2 |
| 16H123 | 35204 | 103 | 471 | 19 | 13 | 3 |
| 16H145 | 46346 | 183 | 612 | 15 | 25 | 3 |
Peptide level FDR 1%.
These identifications of peptides derived from altProts and novel isoforms may result from the replacement of doubtful identifications of peptides originating from canonical proteins with more confident identifications of peptides from altProts, from the successful assignment of PSMs typically unassigned with a conventional database, or from both processes.
To address this question, we first compared statistical confidence measures of PSMs obtained using a customized database with conventional proteins only to those obtained using a customized database containing altProts and novel isoforms in addition to canonical proteins. We determined that 6493 spectra were matched to canonical peptides with a custom database containing canonical proteins only, but the majority (63.5%) did not pass the PSM-level FDR threshold of 1%. In contrast, in the analysis using a custom database containing canonical proteins, altProts, and novel isoforms, these same 6493 spectra were matched to altProts or novel isoforms, all of which passed the PSM-level FDR threshold of 1% (Figure 2A).
Figure 2.

Many PSMs typically assigned to peptides from canonical proteins can be assigned to peptides from altProts and novel isoforms with better statistical confidence measures. (A) The left and right panels show the Percolator score distributions for the same set of 6493 spectra from two analyses of the same dataset. On the left, only canonical proteins were present in the database at the peptide-spectrum matching step. On the right, altProts and novel isoforms were included. The dashed line represents the q-value at which PSMs on the right are excluded when enforcing an FDR below 1%. (B) Percolator-derived peptide posterior error probability (log10) for the same 6493 spectra from A associated either to peptide from canonical proteins (custom database with canonical proteins only; grey) or to peptides from altProts and novel isoforms (custom database with canonical proteins, altProts and novel isoforms; green), as indicated.
This indicates that peptides from novel proteins are much better explanations for these spectra than the poorly matched peptides from canonical proteins. Examples of MS2 spectra randomly selected are provided in Supplementary Figure 3. The distribution of posterior error probability scores associated to theses 6493 spectra assigned to peptides from canonical proteins was shifted toward the distribution for the decoy proteins compared to the same spectra assigned to peptides from altProts and novel isoforms. This highlights the fact that a significant fraction of PSMs is not accurate when using a database with canonical proteins only and obtain more confident statistical measures with a database containing altProts and novel isoforms in addition to canonical proteins (Figure 2B).
Second, we analyzed the PSMs that could not be assigned at all using a customized database containing canonical proteins only but were assigned to peptides from altProts and novel isoforms using a custom database containing canonical proteins, altProts, and novel isoforms. These represent the data acquired at the MS/MS that go unnoticed when analyzed conventionally, but are explained by the presence of altProts and novel isoforms. We identified 41,480 of those PSMs, 19.13% of which passed the FDR of 1% (i.e., 7939) (Figure 3A). The distribution of posterior error probability values for these PSMs compared to the decoys confirmed that most are very unlikely to be false-positive PSMs (Figure 3B).
Figure 3.
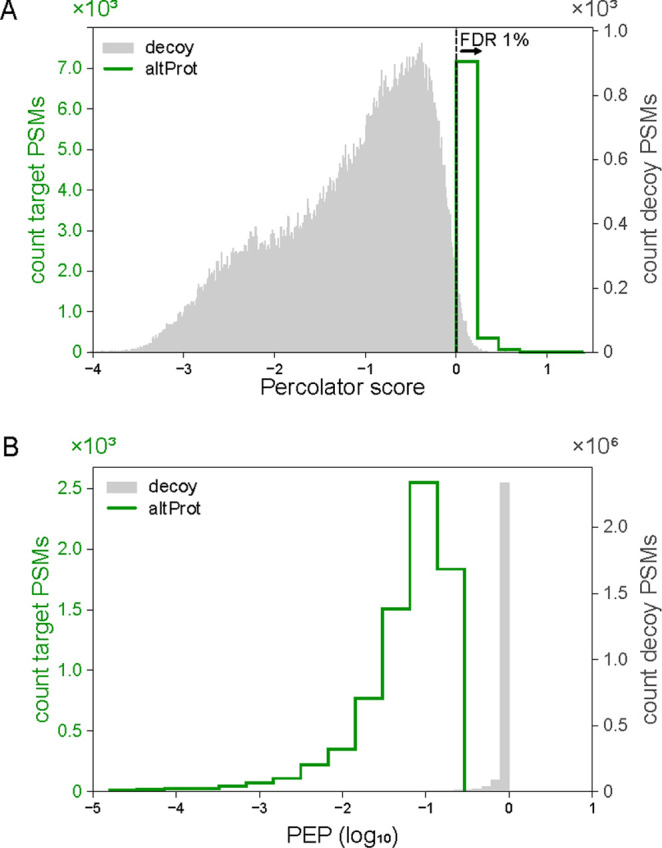
Statistical measures of spectra assigned using OpenCustomDB. (A) Percolator score distribution of 7939 spectra unassigned in the analysis using the database excluding altProts and novel isoforms, but confidently assigned to peptides from altProts or novel isoforms when they are included in the database. The dashed line represents the q-value at which PSMs on the right are excluded when enforcing an FDR below 1%. (B) Percolator peptide posterior error probability (log10) for the PSMs shown in (A).
Protein level analysis
We used the Percolator Fido inference software tool to assemble the identified peptides into a list of proteins. The number of canonical proteins detected in each patient ranged between 3202 and 5004, including 826 variants (Table 2). The analysis identified a total of 434 altProts, including 9 variants and 30 novel Isoforms, including 19 variants (Table 2).
Table 2. Number of Reference and Variant Protein Groups in 16 Patients with Acute Myeloid Leukemiaa.
| Canonical
proteins |
Alternative
proteins |
Novel
isoforms |
||||
|---|---|---|---|---|---|---|
| Patients | Total | Variants | Total | Variants | Total | Variants |
| 05H143 | 4380 | 76 | 53 | 2 | 21 | 1 |
| 05H149 | 3202 | 41 | 23 | 0 | 44 | 9 |
| 07H063 | 4975 | 92 | 53 | 0 | 7 | 3 |
| 07H122 | 5004 | 105 | 48 | 0 | 13 | 2 |
| 07H141 | 3627 | 56 | 42 | 1 | 9 | 2 |
| 08H039 | 3941 | 51 | 50 | 2 | 12 | 2 |
| 08H053 | 4105 | 47 | 32 | 1 | 8 | 2 |
| 11H008 | 3548 | 34 | 20 | 0 | 7 | 1 |
| 11H035 | 3999 | 54 | 40 | 1 | 10 | 1 |
| 12H172 | 4572 | 74 | 40 | 2 | 9 | 3 |
| 15H013 | 3894 | 46 | 35 | 1 | 8 | 1 |
| 15H023 | 4452 | 72 | 34 | 1 | 10 | 1 |
| 15H063 | 4014 | 41 | 30 | 1 | 13 | 3 |
| 15H080 | 4703 | 79 | 32 | 1 | 15 | 2 |
| 16H123 | 3889 | 51 | 33 | 0 | 6 | 2 |
| 16H145 | 4510 | 91 | 40 | 0 | 9 | 3 |
FDR 1% at the protein level.
Among the noncanonical proteins identified in the largest number of patients, 9 altProts and 2 novel isoforms, including 1 variant, were detected across 9 to 15 patients (Table 3).
Table 3. Several altProts and Novel Isoforms Are Detected in Many Patients.
| Accession | Gene | Protein type | RNA biotype | Number of patients | Location |
|---|---|---|---|---|---|
| IP_260057 | JPT1 | AltProt | mRNA | 15 | 3′UTR |
| IP_078777 | ABCB10 | AltProt | mRNA | 14 | CDS |
| IP_196754 | AHNAK | AltProt | mRNA | 13 | CDS |
| IP_295104 | DESI1 | AltProt | mRNA | 13 | 3′UTR |
| IP_105341 | PBRM1 | AltProt | mRNA | 12 | 3′UTR |
| IP_595290 | ASS1P1 | AltProt | ncRNA | 11 | N/A |
| IP_120014 | PPM1K | AltProt | mRNA | 11 | 3′UTR |
| II_772356 | NDUFA9 | Novel isoform | ncRNA | 11 | N/A |
| IP_580779 | TRIM74, STAG3L3, STAG3L1, STAG3L2 | AltProt | ncRNA, miscRNA | 10 | N/A |
| IP_065105 | SSBP3 | AltProt | mRNA | 9 | CDS, 5′UTR |
| II_726022@Ala112Thr | RPL13 | Novel isoform | ncRNA | 9 | N/A |
The corresponding altORFs are in UTRs and ncRNAs, confirming previous observations that regions of the transcriptome previously believed to be noncoding can actually encode proteins.14,27,28 As a result of the identification of novel proteins, OpenCustomDB typically increases the number of genes identified for each patient (supplementary Figure 2).
Discussion
With the rise of personalized medicine comes increasing interest in proteogenomics, a set of analytical methods where variant proteins predicted from a personal genome or transcriptome information are added to a protein search database prior analysis of the raw data. However, existing approaches that rely on canonical annotations ignore the many altProts that have been shown to be expressed.12,29 The integration of altProts and their variants could help determine personalized proteomes with better resolution. We built OpenCustomDB, a new variant annotator that relies on OpenProt annotations and enables the generation of customized databases from RNaseq data. We showed that the analysis of proteomics data with OpenCustomDB helps identify altProts and their variants. Furthermore, we demonstrated that previously unassigned spectra may be successfully assigned to peptides derived from altProts and that spectra matched with low confidence to canonical peptides may have better scores when matched to alternative peptides.
Proteogenomic databases customized using sample-specific RNaseq data and noncanonical annotations such as OpenProt can be powerful tools for identifying novel peptides and variants that are not included in generic protein sequence databases. However, this promise of proteogenomics is offset by the fact that such databases are much larger than conventional proteomic databases because they include predicted and therefore potentially spurious proteins, as it is not known whether they are expressed. This typically results in fewer PSMs at fixed FDRs, more erroneous identifications, and a considerable increase in computing costs.9,10,30 OpenCustomDB allows users to control the size of the database by adjusting the transcript expression level threshold or by removing specific transcripts. The user may also choose to completely remove altProts from the database to generate conventional proteogenomic databases that integrate only canonical proteins. This flexibility is intended to give control to the user, who can thus customize the database according to the objectives of the study.
Protein databases assembled using OpenCustomDB enable not only the identification of peptides from canonical proteins and alternative proteins with variants specific to each sample but also a general improvement of PSM assignments. This is because spectra assigned to peptides of canonical proteins with low confidence are reassigned with better scores to peptides of alternative proteins. This observation is particularly important given the central role of accurate PSM assignment in shotgun MS-based proteomics and database search.31 In fact, a list of candidate PSMs is typically returned for each spectrum and sorted according to their calculated scores. The peptide with the highest PSM score is then selected as the best match. When altProts are included in customized databases, a fraction of spectra receives higher PSM scores with peptides derived from altProts; hence, OpenCustomDB prevents some inaccuracies in peptide identifications and possible downstream erroneous conclusions about the presence of the corresponding proteins.
While OpenCutsomDB presents a useful first step in the inclusion of the multicoding aspect of many human genes into proteogenomic investigations, there remain some drawbacks that will need addressing in future works. For example, OpenProt annotations currently do not consider open reading frames smaller than 30 codons. This is a problem because many proteins shorter than this length are already known to be expressed and play important functional roles; current OpenProt annotation does not allow the discovery of new proteins in this class.
Another drawback comes from the filter based on transcript expression. This filter serves to keep the database in a manageable size range for a more effective use of the target-decoy strategy, but it may be inadequate for the detection of some proteins whose abundance does not correlate well with the abundance of their respective transcripts. As new machine learning strategies are applied to the problem of peptide spectrum matching task, the size of the protein library may become less of a hindrance.32
Conclusions
We have built OpenCustomDB to generate customized protein databases from sample-specific RNaseq data. In contrast to previous tools, OpenCustomDB-derived databases enable the identification of genetic variants in canonical and noncanonical ORFs and the detection of the corresponding protein products. Continued work in this field is increasingly important because new proteins and their disease-associated variants can have a great impact in precision medicine and proteomics. OpenCustomDB is a contribution toward this goal by providing a convenient and easy-to-use tool for researchers to better decipher the true diversity proteomes.
Acknowledgments
Computing resources from Digital Research Alliance of Canada are gratefully acknowledged. We thank Raphaelle Lambert and Jennifer Huber at the genomics facility for RNA-seq, Patrick Gendron, Eric Audemard, and Genevieve Boucher at the bioinformatic platform for assistance with RNA-seq analysis, the Institute for Research in Immunology and Cancer of the Université de Montréal. We acknowledge the work of Claude Rondeau and all members of the Quebec Leukemia Cell Bank. This work was supported by a grant from the Canadian Institutes of Health Research (CIHR, PJT-175322) to X.R. M.A.B., a Canada research chair to X.R., a grant from the Canadian Cancer Society (705604) to C.P. and P.T., a grant from the Leukemia and Lymphoma Society of Canada to C.P. and a grant from The Oncopole to C.P., P.T., and X.R. M.A.B. is a Junior 1 research fellow from the Fonds de Recherche du Québec–Santé. G.E. is supported by postdoctoral fellowships from the Institute for Research in Immunology and Cancer of the Université de Montréal, the Fonds de Recherche du Québec–Santé, and the Cole Foundation. Operation of the mp2 supercomputer is funded by the Canada Foundation of Innovation (CFI), le ministère de l’Économie, de la science et de l’innovation du Québec (MESI), and les Fonds de Recherche du Québec.
Data Availability Statement
The OpenCustomDB codebase is open source and hosted on GitHub at https://github.com/MAB-Lab/OpenCustomDB. The raw and processed proteomics data presented in this study can be found in PRIDE repository with accession number PXD029240.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.3c00054.
Supplementary Figure 1: Customised protein sequence databases that integrate reference and variant proteins. For each patient, both the composition (left) and the localization of predicted coding sequences (right) are shown. OpenCustomDB was run with (green bars) or without (gray bars) integration of altProts and novel isoforms. The size of the databases with altProts and novel isoforms was limited to a total of 100,000 entries by imposing the corresponding TPM threshold. Supplementary Figure 2: Number of genes coding for proteins identified with OpenCustomDB libraries with and without alternative proteins. For each patient, the number of genes identified using customized protein sequence databases integrating reference and variant proteins with alternative proteins (green) and without (canonical library only, gray). Despite the difference in size between the two databases for each patient, there is a notable overlap in identified genes. Supplementary Figure 3: Examples of MS2 spectra assigned to a reference protein with a canonical protein database and reassigned to an alternative protein with a database containing canonical proteins, altProts, and novel isoforms. (A) MS/MS spectrum confidently mapped to either a peptide unique to the reference protein ENSP0000378967 (A) or Q96QH2 (B) using a database containing canonical proteins only (bottom spectrum), or to a peptide unique to the novel isoform II_726022 (A) or IP_643011 (B) using a database containing canonical proteins, altProts, and novel isoforms (top spectrum). Peaks are represented by their mass over charge ratios (m/z) and their intensity relative to the highest (relative intensity). The y ions are colored in red, the b ions in blue and the unannotated peaks appear in gray. (PDF)
Supplementary Table 1: list of variant peptides in canonical and alternative proteins. (XLSX)
The authors declare no competing financial interest.
Supplementary Material
References
- Brunet M. A.; Leblanc S.; Roucou X. Reconsidering proteomic diversity with functional investigation of small ORFs and alternative ORFs. Exp. Cell Res. 2020, 393, 112057. 10.1016/j.yexcr.2020.112057. [DOI] [PubMed] [Google Scholar]
- Cesnik A. J.; Miller R. M.; Ibrahim K.; Lu L.; Millikin R. J.; Shortreed M. R.; Frey B. L.; Smith L. M. Spritz: A Proteogenomic Database Engine. J. Proteome Res. 2021, 20, 1826–1834. 10.1021/acs.jproteome.0c00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheynkman G. M.; Johnson J. E.; Jagtap P. D.; Shortreed M. R.; Onsongo G.; Frey B. L.; Griffin T. J.; Smith L. M. Using Galaxy-P to leverage RNA-Seq for the discovery of novel protein variations. BMC Genomics 2014, 15, 703. 10.1186/1471-2164-15-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggles K. V.; Tang Z.; Wang X.; Grover H.; Askenazi M.; Teubl J.; Cao Z.; McLellan M. D.; Clauser K. R.; Tabb P. M.; et al. An analysis of the sensitivity of proteogenomic mapping of somatic mutations and novel splicing events in cancer. Mol. Cell Proteomics 2016, 15, 1060–1071. 10.1074/mcp.M115.056226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Slebos R. J.; Wang D.; Halvey P. J.; Tabb D. L.; Liebler D. C.; Zhang B. Protein identification using customized protein sequence databases derived from RNA-seq data. J. Proteome Res. 2012, 11, 1009–1017. 10.1021/pr200766z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Zhang B. CustomProDB: An R package to generate customized protein databases from RNA-Seq data for proteomics search. Bioinformatics 2013, 29, 3235–3237. 10.1093/bioinformatics/btt543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Orre L. M.; Johansson H. J.; Huss M.; Boekel J.; Vesterlund M.; Fernandez-Woodbridge A.; Branca R. M. M.; Lehtiö J. Discovery of coding regions in the human genome by integrated proteogenomics analysis workflow. Nat. Commun. 2018, 9, 903. 10.1038/s41467-018-03311-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj S. H.; Waddell N.; Madugundu A. K.; Wood S.; Jones A.; Mandyam R. A.; Nones K.; Pearson J. V.; Grimmond S. M. PGTools: A software suite for proteogenomic data analysis and visualization. J. Proteome Res. 2015, 14, 2255–2266. 10.1021/acs.jproteome.5b00029. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii A. I. Proteogenomics: Concepts, applications and computational strategies. Nat. Methods 2014, 11, 1114–1125. 10.1038/nmeth.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Joh Y. S.; Kim H.; Paek E.; Lee S.-W.; Hwang K.-B. Evaluating the effect of database inflation in proteogenomic search on sensitive and reliable peptide identification. BMC Genomics 2016, 17 (Suppl 13), 1031. 10.1186/s12864-016-3327-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Wang X.; Cho J.-H.; Shaw T. I.; Wu Z.; Bai B.; Wang H.; Zhou S.; Beach T. G.; Wu G.; et al. JUMPg: An integrative proteogenomics pipeline identifying unannotated proteins in human brain and cancer cells. J. Proteome Res. 2016, 15, 2309–2320. 10.1021/acs.jproteome.6b00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet M. A.; Lucier J.-F.; Levesque M.; Leblanc S.; Jacques J. F.; Al-Saedi H. R. H.; Guilloy N.; Grenier F.; Avino M.; Fournier I.; et al. OpenProt 2021: Deeper functional annotation of the coding potential of eukaryotic genomes. Nucleic Acids Res. 2021, 49 (D1), D380–D388. 10.1093/nar/gkaa1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Brunner A. D.; Cogan J. Z.; Nunez J. K.; Fields A. P.; Adamson B.; Itzhak D. N.; Li J. Y.; Mann M.; Leonetti M. D.; et al. Pervasive functional translation of noncanonical human open reading frames. Science 2020, 367, 140–146. 10.1126/science.aay0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet M. A.; Levesque S. A.; Hunting D. J.; Cohen A. A.; Roucou X. Recognition of the polycistronic nature of human genes is critical to understanding the genotype-phenotype relationship. Genome Res. 2018, 28, 609–624. 10.1101/gr.230938.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehx G.; Larouche J.-D.; Durette C.; Laverdure J.-P.; Hesnard L.; Vincent K.; Hardy M.-P.; Thériault C.; Rulleau C.; Lanoix J.; Bonneil E.; et al. Atypical acute myeloid leukemia-specific transcripts generate shared and immunogenic MHC class-I-associated epitopes. Immunity 2021, 54, 737–752. 10.1016/j.immuni.2021.03.001. [DOI] [PubMed] [Google Scholar]
- Dobin A.; Davis C. A.; Schlesinger F.; Drenkow J.; Zaleski C.; Jha S.; Batut P.; Chaisson M.; Gingeras T. R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray N. L.; Pimentel H.; Melsted P.; Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- Brunet M. A.; Leblanc S.; Roucou X. Openvar: functional annotation of variants in non-canonical open reading frames. Cell Biosci. 2022, 12 (1), 130. 10.1186/s13578-022-00871-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J.; Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Silva A. S. C.; Bouwmeester R.; Martens L.; Degroeve S. Accurate peptide fragmentation predictions allow data driven approaches to replace and improve upon proteomics search engine scoring functions. Bioinformatics 2019, 35, 5243–5248. 10.1093/bioinformatics/btz383. [DOI] [PubMed] [Google Scholar]
- Bouwmeester R.; Gabriels R.; Hulstaert N.; Martens L.; Degroeve S. DeepLC can predict retention times for peptides that carry as yet unseen modifications. Nat. Methods 2021, 18, 1363–1369. 10.1038/s41592-021-01301-5. [DOI] [PubMed] [Google Scholar]
- The M.; MacCross M. J.; Noble W. S.; Käel L. Fast and Accurate Protein False Discovery Rates on Large-Scale Proteomics Data Sets with Percolator 3.0. J. Am. Soc. Mass Spectrom. 2016, 27, 1719–1727. 10.1007/s13361-016-1460-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Declercq A.; Bouwmeester R.; Hirschler A.; Carapito C.; Degroeve S.; Martens L.; Gabriels R. MS2Rescore: data-driven rescoring dramatically boosts immunopeptide identification rates. Mol. Cell Proteomics 2022, 21, 100266. 10.1016/j.mcpro.2022.100266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbruggen S.; Gessulat S.; Gabriels R.; Matsaroki A.; Van de Voorde H.; Kuster B.; Degroeve S.; Martens L.; Van Criekinge W.; Wilhem M.; et al. Spectral prediction features as a solution for the search space size problem in proteogenomics. Mol. Cell Proteomics 2021, 20, 100076. 10.1016/j.mcpro.2021.100076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kall L.; Canterbury J. D; Weston J.; Noble W. S.; MacCoss M. J Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 2007, 4, 923–925. 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- Chauvin A.; Boisvert F. M. Proteomics analysis of colorectal cancer cells. Methods Mol. Biol. 2018, 1765, 155–166. 10.1007/978-1-4939-7765-9_9. [DOI] [PubMed] [Google Scholar]
- Landry C. R.; Zhong X.; Nielly-Thibault L.; Roucou X. Found in translation: Functions and evolution of a recently discovered alternative proteome. Curr. Opin Struct Biol. 2015, 32, 74–80. 10.1016/j.sbi.2015.02.017. [DOI] [PubMed] [Google Scholar]
- Ruiz-Orera J.; Messeguer X.; Subirana J. A.; Alba M. N. Long non-coding RNAs as a source of new peptides. ELife 2014, 3, e.03523 10.7554/eLife.03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samandi S.; Roy A. V.; Delcourt V.; Lucier J.-F.; Gagnon J.; Beaudoin M. C.; Vanderperre B.; Breton M.-A.; motard J.; Jacques J.-F.; et al. Deep transcriptome annotation enables the discovery and functional characterization of cryptic small proteins. Elife 2017, 6, e27860 10.7554/eLife.27860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakeley P.; Overton I. M.; Hubbard S. J. Addressing statistical biases in nucleotide-derived protein databases for proteogenomic search strategies. J. Proteome Res. 2012, 11, 5221–5234. 10.1021/pr300411q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granholm V.; Käll L. Quality assessments of peptide-spectrum matches in sotgun proteomics. Proteomics 2011, 11, 1086–1093. 10.1002/pmic.201000432. [DOI] [PubMed] [Google Scholar]
- Degroeve S.; Gabriels R.; Velghe K.; Bouwmeester R.; Tichshenko N.; Martens L. ionbot: a novel, innovative and sensitive machine learning approach to LC-MS/MS peptide identification. bioRxiv 2022, 10.1101/2021.07.02.450686. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The OpenCustomDB codebase is open source and hosted on GitHub at https://github.com/MAB-Lab/OpenCustomDB. The raw and processed proteomics data presented in this study can be found in PRIDE repository with accession number PXD029240.