

SUMMARY
Suppressive regulatory T cell (Treg) differentiation is controlled by diverse immunometabolic signaling pathways and intracellular metabolites. Here we show that cell-permeable α-ketoglutarate (αKG) alters the DNA methylation profile of naive CD4 T cells activated under Treg polarizing conditions, markedly attenuating FoxP3+ Treg differentiation and increasing inflammatory cytokines. Adoptive transfer of these T cells into tumor-bearing mice results in enhanced tumor infiltration, decreased FoxP3 expression, and delayed tumor growth. Mechanistically, αKG leads to an energetic state that is reprogrammed toward a mitochondrial metabolism, with increased oxidative phosphorylation and expression of mitochondrial complex enzymes. Furthermore, carbons from ectopic αKG are directly utilized in the generation of fatty acids, associated with lipidome remodeling and increased triacylglyceride stores. Notably, inhibition of either mitochondrial complex II or DGAT2-mediated triacylglyceride synthesis restores Treg differentiation and decreases the αKG-induced inflammatory phenotype. Thus, we identify a crosstalk between αKG, mitochondrial metabolism and triacylglyceride synthesis that controls Treg fate.
In brief
Matias et al. show that naive T cell differentiation to a suppressive Treg fate is attenuated by ectopic α-ketoglutarate. Alterations in the cell’s DNA methylation profile, associated with increased oxidative phosphorylation and lipidome-wide remodeling, results in an inflammatory Th1-like phenotype. Inhibition of triacylglyceride synthesis restores Treg differentiation, decreasing inflammatory gene expression.
Graphical Abstract
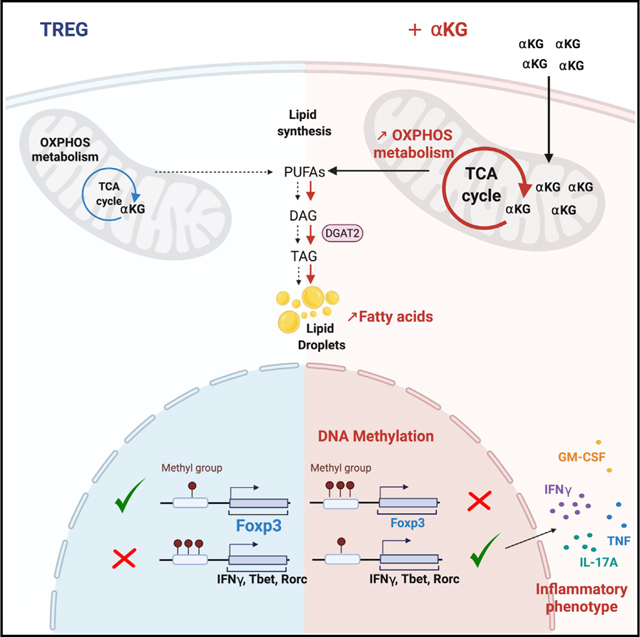
INTRODUCTION
The potential of a T cell to respond to infections, tumor antigens, and even auto-antigens is dependent on a massive augmentation of cellular resources, with the energetic profile of the cell intricately related to the harnessing of these resources (Almeida et al., 2016; Geltink et al., 2018; Makowski et al., 2020; Yong et al., 2017a). The energetic state of the cell is also linked to the extracellular environment; activation of the T cell receptor (TCR) leads to an increase in the transcription and translation of nutrient transporters, resulting in an augmented uptake of the corresponding nutrients (Cretenet et al., 2016; Hukelmann et al., 2016; Macintyre et al., 2014; Sinclair et al., 2013). Indeed, incorporation of glucose- and glutamine-derived carbons into nucleotides promotes optimal T cell proliferation (Buck et al., 2017; Carr et al., 2010; Clerc et al., 2019; Klysz et al., 2015; Loftus and Finlay, 2016; Wang et al., 2011).
Nutrient resources do not merely provide energy and substrates for nucleotide synthesis; their utilization regulates the specificity of the T cell response to a foreign stimulus. This regulation is especially critical in the context of CD4 T lymphocytes where effector cells are highly glycolytic, and even lipogenic, while suppressive regulatory T cells (Tregs) display a mixed metabolism with increased levels of lipid oxidation (Buck et al., 2017). Genetic deletion of nutrient transporters that are responsible for the uptake of glucose, leucine or glutamine, inhibits CD4 T cell differentiation to an effector (Teff) but not Treg fate (Macintyre et al., 2014; Nakaya et al., 2014; Sinclair et al., 2013). The balance between Th1 effector cells and Tregs is also regulated by the extracellular metabolic environment. As compared to Th1 cells, Tregs are less impacted by reduced glycolytic capacity (Beier et al., 2015; Gerriets et al., 2015) and decreased extracellular glutamine (Cham et al., 2008; Chang et al., 2013; Klysz et al., 2015; Macintyre et al., 2014; Metzler et al., 2016; Nakaya et al., 2014). Conversely, increasing glycolysis enhances T effector function (Buck et al., 2017; Chang et al., 2013; Peng et al., 2016). This balance in Teff/Treg differentiation is critical in the tumor environment where the competition between T cells and tumor cells for limiting amounts of nutrients has a negative impact on the former.
Interestingly, mitochondrial metabolism has been found to play a role in the function of both Treg and Th1 cells-–mitochondrial integrity as well as complex III activity of the electron transport chain (ETC) are required for Treg suppressive activity (Beier et al., 2015; Field et al., 2019; Miska et al., 2019; Weinberg et al., 2019) whereas complex II activity is a requirement for terminal Th1 function (Bailis et al., 2019). Consistent with a role for OXPHOS in Th1 activity, the catalytic conversion of glutamine to alpha-ketoglutarate (αKG), which directly fuels the tricarboxylic acid cycle (TCA), is critical for Th1 cell differentiation (Johnson et al., 2018) and ectopic αKG rescues Th1 differentiation under glutamine-deprived conditions (Klysz et al., 2015). However, it is not known whether alterations in OXPHOS and recruitment of ETC complexes regulate the potential of a naive T cell to differentiate to a Th1 as compared to a Treg fate.
Here, we show that an augmented OXPHOS, achieved by directly increasing the αKG TCA cycle intermediate, attenuated Treg differentiation and induced the secretion of inflammatory cytokines. Conversely, Th1 polarization was significantly augmented by αKG. The attenuation of Treg differentiation was associated with an altered epigenetic profile, defined here as cytosine DNA methylation (5mC) in CpG (5mCpG) regions. Consistent with these data, ERBB2-targeted chimeric antigen receptor (CAR) T cells activated in Treg-polarizing conditions in the presence of αKG delayed the growth of an ERBB2-expressing fibrosarcoma. Mechanistically, inhibition of succinate dehydrogenase, the bridging enzyme between the TCA cycle and the ETC, enforced Treg differentiation. Furthermore, membrane-permeable αKG was utilized in the de novo generation of fatty acids. This lipidome-wide remodeling was critical as inhibiting DGAT2-mediated triacylglyceride (TAG) synthesis restored Treg differentiation. Thus, αKG alters both OXPHOS and lipid homeostasis, modifying the balance between Th1 and Treg differentiation.
RESULTS
Increasing OXPHOS is associated with an attenuated Treg polarization and an augmented Th1 differentiation
Oxidative phosphorylation is critical for both Treg and Th1 differentiation and function (Bailis et al., 2019; Beier et al., 2015; Chapman et al., 2018; He et al., 2017; Howie et al., 2017; Layman et al., 2017; Weinberg et al., 2019) but the alterations in OXPHOS and recruitment of ETC complexes in naive CD4 T cells activated under Treg as compared to Th1 polarization conditions are not clear (Bailis et al., 2019; Field et al., 2019; Geltink et al., 2018; Shin et al., 2020; Weinberg et al., 2019). We found that the basal oxygen consumption rate (OCR, correlating with OXPHOS) as well as spare respiratory capacity (SRC) were significantly higher in Th1- than Treg-polarizing conditions, with polarization evaluated on the basis of Tbet and FoxP3 expression, respectively (p < 0.01 and p < 0.05; Figures 1A and S1A). These data are in accord with a recent study reporting a lower OXPHOS and glycolysis in activated thymic Tregs (tTreg) as well as TGFβ-induced Treg cells as compared to Th0 and Th1 polarized cells (Priyadharshini et al., 2018). The lower OXPHOS in Treg cells (as compared to Th1) was associated with a decreased extracellular acidification rate (ECAR; Figure S1B). Furthermore, ATP-linked OCR was higher in Th1- than Treg-polarized cells, 131.4 ± 15.5 and 79.3 ± 8.3, respectively (p < 0.01; Figure S1B). Thus, by day 4 of polarization, CD4 T cells polarized to a Treg fate exhibited a lower energetic profile than cells polarized to a Th1 fate.
Figure 1. αKG-induced increases in OXPHOS are associated with decreased Treg polarization and augmented Th1 differentiation.

(A) OCR was monitored at day 4 of Th1 and Treg polarization following sequential injection of oligomycin, FCCP and rotenone/antimycin A (arrows; left panel). The differentiation status of Th1- and Treg-polarized cells was monitored by Tbet and FoxP3 expression and representative histograms are shown (n = 7). Mean basal OCR levels ± SEM (n = 7 independent experiments) and SRC (n = 6) are presented.
(B) The direct impact of αKG on cell metabolism was evaluated by injecting αKG into the XFe96 flux analyzer at day 2 of Treg polarization and fold change in basal OCR as well as OCR/ECAR energy plots ± SEM are shown (n = 8).
(C) OCR of CD4 T cells activated under Treg-polarizing conditions ± αKG (day 4) are presented (n = 1 of 10). Quantification of basal OCR ± SEM (n = 8) and SRC (n = 8) are shown. The percentage of the ATP production rate derived from mitochondrial and glycolytic pathways are presented as means ± SEM (n = 7).
(D) FoxP3 expression was evaluated following stimulation of naive CD4 T cells under Treg-polarizing conditions in the absence (control) or presence of αKG (day 4). Representative plots and means ± SEM are presented (right, n = 42).
(E) IFNγ expression by naive CD4 T cells stimulated under Th1-polarizing conditions was evaluated by flow cytometry and means ± SEM are presented (n = 20). Statistical analyses were evaluated by paired (A and C) and unpaired (D and E) 2-tailed t tests.
Based on these data, it was of interest to determine the consequences of increasing OXPHOS during Treg polarization. To that end, we first tested the impact of membrane-permeable αKG, a key intermediate in the TCA cycle that has recently been shown to increase mitochondrial function in hematopoietic stem cells (Gonzalez-Menendez et al., 2021). Notably, αKG directly increased the OCR of naive CD4 T cells activated under Treg polarizing conditions, within minutes of its entry into the cell. The immediate effect was specific to OCR as ECAR was not altered (p < 0.001 and NS, respectively; Figure 1B). The impact of αKG was further amplified during differentiation, resulting in significant increases in basal respiration and SRC following activation in Treg-polarizing conditions for 4 days (p < 0.001; Figure 1C). This change in the cell’s energetic activity was associated with an increased ATP-linked OCR (p < 0.01; Figure S1C) and a metabolic reprogramming, such that there was a shift in the rate of ATP production toward mitochondrial respiration (p < 0.05, Figure 1C).
We therefore evaluated whether the αKG-mediated increase in OXPHOS impacted the cell’s differentiation state. Notably, the presence of αKG significantly diminished the potential of naive T cells stimulated under Treg polarization conditions to upregulate FoxP3, a transcription factor serving as a master regulator of Treg differentiation; the percentage of FoxP3+ T cells decreased from 60 ± 2% to 28 ± 2% (p < 0.0001; Figure 1D). This decrease in Treg polarization was associated with 3.4-fold lower Foxp3 mRNA levels (p < 0.05) and an attenuation of RNASeq reads in the Foxp3 gene (Figure S1D). Moreover, Treg differentiation was reduced even when T cells were initially activated in control Treg polarizing conditions and OXPHOS was increased by adding αKG 24h postactivation (from 68 ± 8% to 35 ± 9%; p < 0.05; Figures 1B and S1E). Importantly though, the αKG-mediated inhibition of FoxP3 induction was specific to Treg polarizing conditions because when isolated tTreg were activated in the presence of αKG, FoxP3 expression was not altered (78 ± 4% and 75 ± 4%, respectively; Figure S1F).
As the potential of a CD4 T cell to generate a suppressive Treg phenotype is often inversely related to its potential to adopt an effector Th1 phenotype (Delgoffe et al., 2011; Klysz et al., 2015; Zeng et al., 2013), we assessed whether αKG-induced OXPHOS modulates the balance between Treg and Th1 differentiation. Indeed, following activation in Th1 conditions, the percentage of CD4 T cells producing IFNγ, increased from 25 ± 2 to 53 ± 3% (p < 0.0001; Figure 1E). This increase was regulated at the transcriptional level with a 16-fold augmentation in IFNγ mRNA levels (p < 0.01) and a marked increase in RNASeq reads of the Ifng gene (Figure S1G). Thus, αKG-induced OXPHOS is associated with an increased potential of CD4 T cells to produce IFNγ following stimulation in Th1-polarizing conditions. Together, these data reveal the importance of αKG in regulating the balance in the polarization of naive CD4 T cells to a Th1 versus a Treg fate.
αKG alters the epigenetic profile of differentiating CD4 T cells, reprogramming Tregs toward an inflammatory phenotype
As αKG decreased the potential of CD4 T cells to differentiate into Tregs, we evaluated the consequences of this metabolite under conditions of Treg polarization. Interestingly, the potential of these cells to produce IFNγ was significantly augmented by αKG, increasing from 0.5 ± 0.1% to 5.8 ± 0.6% (p < 0.0001; Figure 2A), correlating with significantly augmented mRNA and protein level of the Th1 master transcription factor, T-bet (Figures 2B and S2A). Furthermore, these CD4 T cells harbored higher levels of granzyme B (GzmB) transcripts (Figure S2B), a marker of CD4 cytotoxicity (Takeuchi and Saito, 2017). It is also notable that the increased intracellular IFNγ levels (Figure 2A) paralleled the rise in Ifng transcripts (Figure S2C). While neither Tnf nor Csf2 (GM-CSF) transcripts were increased, secretion of TNF and GM-CSF as well as IFNγ and IL-17A were markedly elevated (p < 0.01 to p < 0.0001; Figure 2C). Together, these data highlight the inflammatory nature of these CD4 T cells, despite their activation under Treg-polarizing conditions.
Figure 2. αKG induces an inflammatory profile in CD4 T cells activated under Treg-polarizing conditions.

(A) IFNγ expression was assessed following activation under Treg polarizing conditions ±αKG (day 4, n = 40).
(B) T-bet protein levels were evaluated as a function of mean fluorescent intensity (MFI, n = 8).
(C) IFNγ, IL-17A, TNF, and GM-CSF levels were evaluated at day 3 of Treg polarization (n = 5).
(D)DNA methylation in a CpG context (5mCpG) was evaluated by nanopore sequencing in the indicated conditions and the percentage of methylation is presented as boxplots with the number of reads in the 6 samples ranging from 1,260,564 to 6,145,824 reads (triplicates, 2 independent experiments).
(E) Distribution of the methylation status of differentially methylated regions (DMR) in the Ifng, Tbet, Rorc and Foxp3 loci are presented. Quantifications of means ± SEM are shown (A–C). Statistical analyses were performed by an unpaired 2-tailed t test.
αKG has been shown to alter DNA and histone methylation states (Chisolm et al., 2017), including the specific methylation of the Foxp3 gene locus (Xu et al., 2017). We therefore used nanopore sequencing to detect 5mC modifications at CpG positions and found that the global methylation of CD4 T cells polarized in Treg conditions was decreased by the presence of αKG (p < 0.05) with methylation levels significantly lower than that detected in tTreg (Figure 2D). Moreover, methylated regions differed in multiple regions (Tables S1 and S2) including genes associated with a Th1 phenotype—Ifng, Tbx2 (Tbet), and Rorc (Figure 2E). Conversely, αKG-treated conditions were associated with a higher methylation in the promoter and CNS1 regions of Foxp3, differential methylation in CNS2 and CNS3 was less pronounced (Figure 2E).
Our data showing that the inflammatory state induced by αKG was associated with epigenetic changes suggested that the status of these cells might be maintained in the absence of αKG. We therefore evaluated the in vivo fate of CD4 T cells that underwent Treg polarization in the presence of αKG. Following a 4-day Treg polarization, T cells were adoptively transferred into RAG2−/− recipients and then evaluated 6 days later. While the percentage of recovered CD4 T cells was not altered by αKG, FoxP3 expression was significantly reduced and the percentage of IFNγ-secreting cells was increased by 3-fold to 31.7 ± 2.9% (p < 0.05; Figure S2D). Furthermore, in a competitive adoptive transfer setting, αKG did not decrease the level of T cell engraftment/proliferation but was similarly associated with a lower FoxP3 expression and a higher level of IFNγ-secretion (Figure S2E). Thus, the αKG-induced alteration in Treg differentiation was maintained following in vivo adoptive transfer.
Tumor infiltration of ERBB2-CAR T cells activated under Treg-polarizing conditions is enhanced by short-term αKG-induced OXPHOS augmentation
The data presented above suggested that αKG might alter the phenotype and persistence of antitumor T cells in an in vivo setting. This is particularly critical since it has been shown that the intratumoral presence of Tregs negatively impacts the potential of effector T cells to eradicate tumor (Darrasse-Jèze et al., 2009) and the “hostile” environment of the tumor consumes high levels of nutrients (Yong et al., 2017b). Furthermore, αKG increased the ATP production rate from mitochondrial metabolism, potentially making them less reliant on glucose and alleviating competition with tumor cells (Chang et al., 2015). To test this hypothesis, we tested the impact of αKG on the effector function of transgenic murine T cells harboring a chimeric antigen receptor (CAR) directed against the ERBB2/HER2 tumor antigen (ERBB2-CAR) (Figure 3A). Efficacy was evaluated against an ERBB2-expressing 24JK fibrosarcoma (24JK-ERB) in RAG2−/− mice (Yong et al., 2016). We found that similarly to polyclonal T cells, αKG treatment of transgenic ERBB2-CAR CD4 T cells resulted in a 2-fold increase in IFNγ secretion following activation in Th1 conditions and a decrease in FoxP3+ cells upon activation in Treg conditions (Figure S3A). Both non-polarized and Th1-polarized ERBB2-CAR CD4 T cells inhibited the growth of 24JK-ERB (p < 0.05 and p < 0.01, respectively) and inhibition of tumor growth was maintained following activation in the presence of αKG (Figure S3B).
Figure 3. ERBB2-CAR T cells polarized ex vivo in the presence of αKG maintain an in vivo inflammatory profile following adoptive transfer into tumor-bearing mice, delaying tumor growth.

(A) Schematic of the experimental setup evaluating the impact of ERBB2-CAR T cells in mice bearing the ERBB2+24JK fibrosarcoma (24JK-ERB).
(B) Representative plots and quantifications ± SEM of CD4+, FoxP3+ CD4+, and IFNγ+CD4+ T cells in lymph nodes (LN) of tumor-bearing mice.
(C) Representative plots and quantifications ± SEM of intratumoral CD4 T cell subsets (n = 6–8).
(D) Tumor area was monitored at days 17 and 30 following T cell injection (n = 3–4 per group). Statistical differences were determined by a one-way ANOVA and Tukey test for multiple comparisons (A–C) and by paired one-tailed t test (D).
Naive ERBB2-CAR CD4 T cells were then activated under Th1 or Treg polarizing conditions in the absence or presence of αKG and adoptively transferred into 24JK-ERB tumor-bearing mice. At day-20-posttumor inoculation, the percentages of adoptively transferred ERBB2-CAR CD4 T cells in lymph nodes (LN) were lower following ex vivo Treg polarization than following either Th1 polarization or Treg polarization in the presence of αKG (Figures 3B and S3C). Notably though, T cell infiltration into the tumor was markedly enhanced by αKG, increasing to levels equivalent to those detected for Th1-polarized cells (52 ± 6% versus 23 ± 4%, p = 0.002; Figures 3C and S3C). Furthermore, αKG markedly diminished the percentages of FoxP3+ ERBB2-CAR T cells in both LNs and tumors (p < 0.001) and the potential of these cells to secrete IFNγ was augmented (p < 0.01; Figure 3B). Thus, increasing OXPHOS during the ex vivo activation period alters the in vivo intratumoral infiltration and persistence of adoptively transferred T cells. Importantly, these characteristics were associated with a delayed tumor growth following adoptive transfer of either Th1 or αKG-treated Treg-polarized ERBB2-CART. In contrast, tumor size increased significantly following transfer of ERBB2-CAR Treg (p < 0.01, Figure 3D). Thus, αKG-induced changes in the metabolism and epigenetic state of CD4 T cells polarized in Treg conditions were associated with enhanced antitumor cytotoxicity in a solid tumor model.
Gene ontology analysis of αKG-reprogrammed T cells reveals a broad induction of mitochondrial pathway genes
While we found that αKG altered the methylation status of loci regulating T cell inflammation and FoxP3 expression, the contributions of αKG to Treg differentiation are not completely understood. Indeed, previous studies showed that TET demethylases, dependent on αKG for the conversion of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) (Lio and Rao, 2019; Rose et al., 2011; Tahiliani et al., 2009), are required for Treg stability and immune homeostasis (Nakatsukasa et al., 2019; Yang et al., 2015; Yue et al., 2019; Yue et al., 2016), but inhibiting αKG generation skewed naive T cells from a Th17 toward a Treg fate (Xu et al., 2017).
To gain further insights into the mechanisms via which αKG enhances Th1 differentiation while negatively regulating Treg differentiation, RNASeq analyses were performed. In accord with the alteration in T cell fate under Treg but not Th1 conditions, αKG-mediated changes in gene expression were more robust in Treg conditions (Figure 4A). There were substantially more downregulated as well as upregulated genes in Treg as compared to Th1 conditions (237 genes in Th1 conditions and 1,639 genes in Treg conditions with an adjusted p < 0.01; Tables S3 and S4). In Treg conditions, upregulation of transcription factors such as T-bet (Tbx21, padj = 1.1×10−56) were expected based on our initial qRT-PCR analyses (Figure S2A); but other transcription factors such as Irf1, Irf8, and Stat1 were also highly upregulated (padj < 5×10−7). These data suggest increased signaling through interferon regulatory factors in these cells (Figure 4B) while housekeeping control genes were unaffected (Rpl11, Ube3c; data not shown). Most importantly, evaluation of nonredundant biological processes (Liao et al., 2019) revealed the importance of mitochondrial processes among the most highly upregulated biological processes (558 protein-encoding upregulated genes; Figure 4C, 4D and Table S5). αKG-mediated increases in OXPHOS were therefore associated with a broad induction of genes involved in mitochondrial processes.
Figure 4. Mitochondrial and respiratory ETC genes are markedly upregulated by αKG in Treg-polarizing conditions.

(A) Volcano plot representations of differential expression analyses in naïve CD4 T cells activated in Th1- and Treg-polarizing conditions ± αKG (day 4).
(B) Alterations in transcription factor genes in the indicated conditions are shown. Each row represents an independent sample and statistical significance is indicated in the top bar. Transcripts that are differentially expressed (FDR < 0.05 and baseMean expression > 100) are colored.
(C) The top GO biological processes for genes upregulated following activation under Treg polarizing conditions ± αKG (558), the enrichment ratio for each GO term, and FDRs are presented. Full datasets for upregulated genes can be found at http://www.webgestalt.org/results/1628071504/#.
D) GSEA enrichment plots of mitochondrial respiratory chain complex assembly and the respiratory electron transport chain. The green curve corresponds to the enrichment score and the barcode plot indicates the position of the genes in each gene set.
αKG-induced increases in SRC and mitochondrial complex II activity are associated with attenuated Treg differentiation
To determine whether an αKG-induced increase in mitochondrial respiratory gene transcripts was associated with an increase in TCA cycling, we quantified these metabolites by mass spectrometry. As expected, the marked increase in αKG levels, from the ectopic addition of mM quantities (p < 0.0001), was associated with significant increases in other TCA cycle intermediates (Figure 5A). Furthermore, ectopic αKG was directly utilized to generate TCA cycle intermediates as monitored by the tracing of heavy carbons from glutamine and glucose (13C5 and 13C6, respectively; Figure S4A). Under control conditions, carbons from heavy glutamine contributed significantly to the generation of αKG, detected in 85.8% of all molecules (Figures 5B and S4B). In agreement with a previous report on human T cells (Clerc et al., 2019), carbons from glucose played a more minor role, incorporated in only 39.3% of all αKG molecules (Figure S4C). Notably though, the presence of αKG markedly reduced the contribution of both glutamine and glucose carbons in all measured TCA cycle intermediates (i.e., 80.3% to 39.7% of glutamine carbons in malate). In this condition, the non-labeled (C0) TCA metabolites were increased, representing synthesis of these intermediates from ectopic αKG (Figures 5B and S4C). The enhanced level of TCA cycling was also associated with increased pyruvate and a decreased generation of intracellular lactate (Figures 5A and 5B). As expected from these data, as well as the increased OXPHOS and ATP production rate (Figures 1C and S1C), the ATP/AMP ratio in these cells was increased (p < 0.05; Figure 5C).
Figure 5. Treg differentiation is negatively regulated by αKG-induced mitochondrial complex II activity.

(A) The peak area of αKG, succinate, malate, citrate, 2-hydroxyglutarate (2HG), pyruvate, and lactate were evaluated by mass spectrometry (MS) in the indicated conditions and the αKG/succinate peak area ratio ± SEM is presented.
(B) The peak area ± SEM of each metabolite and percentage incorporation of the carbon isotopologues from [13C5]glutamine into TCA cycle intermediates are presented.
(C) The ATP/AMP peak area ratio ± SEM was evaluated by MS.
(D–F) (D) The impact of malonate (Mal, 10 mM) on Treg polarization was evaluated as a function of FoxP3 expression at day 4 and quantifications ± SEM are shown (n = 19). (E) IFNγ expression was evaluated by flow cytometry and quantifications ± SEM are shown (n = 12). (F) IFNγ, IL-17A, TNF, and GM-CSF secretion was evaluated by CBA following Treg-polarization in the indicated conditions (day 4, n = 5). n = 2 independent experiments of technical triplicates (panels A–C).
Significance was determined by an unpaired 2-tailed t test (A–C) or a one-way ANOVA and Tukey multiple comparison test (D–F).
The increased ratio of Αkg/succinate in these cells highlighted the altered metabolism in these cells (Figure 5A). Conditions that modulate the αKG/succinate ratio have been shown to regulate the balance between stem cell self-renewal and differentiation (Carey et al., 2015; TeSlaa et al., 2016). As such, we monitored the impact of ectopic succinate (Succ) on differentiation. The addition of succinate partially attenuated the negative impact of αKG, increasing the percentages of FoxP3+ cells from 27 ± 4% and 38 ± 5% (Figure S5A). While this difference was not significant, succinate has both cytosolic and mitochondrial effects (King et al., 2006; Koivunen et al., 2007; Selak et al., 2005). We specifically inhibited succinate oxidation with dimethyl malonate (Mal), to assess whether the mitochondrial effects of succinate were involved in Treg differentiation. Malonate impairs the oxidation of succinate to fumarate without altering prolyl hydroxylase (PHD) activity in the cytoplasm (Mills et al., 2016) (Figure S5B). Notably, under these conditions, malonate not only restored the differentiation of FoxP3+ T cells in the presence of αKG (p < 0.0001) but also significantly augmented FoxP3 levels under control conditions (control, 64 ± 3%; αKG, 32 ± 2%; Mal, 75 ± 2%; Mal + αKG, 66 ± 2%; p < 0.01; Figure 5D). Furthermore, in the presence of αKG, malonate massively reduced the production of inflammatory cytokines including IFNγ, IL-17A, and TNF (p < 0.01; Figures 5E and 5F).
To corroborate the potential of malonate to rescue the αKG-mediated inhibition of Treg differentiation, we tested the capacity of another mitochondrial-respiratory complex II inhibitor, 3-nitropropionic acid (3-NP). Like malonate, 3-NP significantly increased the differentiation of FoxP3+ T cells in the presence of αKG (p < 0.05; Figure S5C) and massively reduced the percentage of cells secreting IFNγ (p < 0.001; Figure S5D). These data were surprising in light of previous research showing that inhibition of OXPHOS by oligomycin, an ATP synthase inhibitor of the ETC, attenuates Treg differentiation (Raud et al., 2018). However, in Th17-polarizing conditions, oligomycin has recently been reported to enhance Treg differentiation (Shin et al., 2020). Our results show that agents which completely blocked OXPHOS—oligomycin, rotenone, and antimycin A (Figure S5E)—significantly attenuated FoxP3 expression under Treg-polarizing conditions (Figure S5F), as previously reported (Ozay et al., 2018; Raud et al., 2018). Notably though, in contrast with these agents, 3-NP—rescuing Treg differentiation in the presence of αKG —did not block basal respiration but rather attenuated the cell’s SRC (Figure S5E). These data suggest that a basal level of OXPHOS is required for optimal Treg differentiation, while a metabolic environment with high levels of SRC leads to an attenuated differentiation. This hypothesis is supported by our finding that αKG treatment increased all mitochondrial complexes with CII and CIV, showing significantly increased expression (Figure S5G), while malonate decreased expression to levels that were not significantly different from control Treg conditions. Thus, mitochondrial function may serve as a Goldilocks principle for Treg differentiation, with attenuated FoxP3 expression occurring under conditions of both low basal respiration and enhanced SRC.
αKG-mediated inhibition of Treg differentiation is associated with lipidome remodeling
While genes involved in mitochondrial processes represented the GO category that was the most highly upregulated in response to αKG, the GO categories that were the most highly downregulated were associated with lipid processes (Figure 6A; Table S6). Furthermore, generation of citrate, through the direct reductive carboxylation of αKG, can contribute to fatty acid synthesis (Du et al., 2016; Fendt et al., 2013; Metallo et al., 2011; Mullen et al., 2014). Reductive carboxylation generates a citrate molecule with five 13C carbons (m+5, during the first turn of the cycle), whereas citrate is generally synthesized through oxidative metabolism resulting in 4 13C carbons (m+4). Notably, αKG treatment resulted in a 4-fold increase in the relative incorporation of m+5/m+4 13C into citrate (p < 0.0001; Figure 6B). We therefore hypothesized that the αKG-mediated alterations in mitochondrial metabolism were associated with changes in lipid homeostasis. Notably, increasing intracellular αKG resulted in a 2-fold increase in total cellular lipids in T cells activated in Treg-polarizing conditions (p < 0.0001; Figure 6C). Because plasma membranes contain more than 90% of lipids species, we wondered whether αKG altered membrane composition. We focused on cholesterol, a hallmark of membrane dynamics (Needham and Nunn, 1990; Sezgin et al., 2017), in part because of a significant decrease in the ATP-binding cassette transporters Abca1 and Abcg1 (Figure S6A), responsible for cholesterol efflux (Yvan-Charvet et al., 2010). However, a similar relative abundance of cholesterol in the lipid membrane was observed in our lipidomic analyses in the absence or presence of αKG (Figure 6D). Moreover, cholesterol supplementation was not sufficient to rescue the αKG-mediated attenuation of Treg differentiation (Figure S6B). Indeed, we found that cholesterol homeostasis in these cells, in the context of low levels of cholesterol efflux genes, was likely compensated by decreased expression of genes involved in cholesterol biosynthesis (Figure S6C). Several other major membrane lipid species, such as phosphatidylcholine (PC), phosphatidylinositol (PI) and phosphatidylserine (PS) were also not significantly altered in αKG-treated Treg (Figure 6D).
Figure 6. Lipidome remodeling in Treg-polarized cells in response to αKG is associated with dramatic increases in storage and mitochondrial lipids.

(A) The top GO terms for non-redundant biological processes are presented for downregulated genes (676) following Treg polarizing conditions ± αKG as in Figure 4.
(B) The contribution of reductive carboxylation to citrate generation was evaluated as a fraction of the m+5/m+4 carbons from [13C5]glutamine (n = 2 with technical triplicates).
(C) Fold change in lipids in CD4 T cells undergoing Treg polarization in the presence (red) of αKG (control Treg conditions (blue) are arbitrarily presented as “1”). Each point represents an individual triplicate sample from 4 biological experiments (black, gray, pink, blue).
(D) The abundance of the different membrane-lipid classes was assessed by lipidomic analysis and mean levels ± SEM are presented. Abbreviations: PC, phosphatidylcholines; PC O-, alkyl-ether-linked phosphatidylcholines; PE, phosphatidylethanolamines; PE O-, ether-linked phosphatidylethanolamines; PI, phosphatidylinositols; PS, phosphatidylserines; SL, sphingolipids; Chol, cholesterol; DAG, Diacylglycerol; PA, phosphatidate.
(E) Percent incorporation of carbon isotopologues from [13C6]glucose, [13C5]glutamine, and [13C5]dimethyl-αKG into C16:0 palmitic acid is shown in the indicated conditions (n = 2 technical replicates).
(F) Percent incorporation of carbon isotopologues from [13C6]glucose into C16:0, C16:1, C18:0, and C18:1 FAs is shown for Th1 and Treg polarizing conditions.
(G) Quantification of membrane packing was evaluated by C-Laurdan spectral microscopy and is presented as Generalized Polarization (GP; n = 3, 129–132 cells).
(H) Total and relative abundance of mitochondrial lipids (cardiolipin and phosphatidylglycerol) are presented.
(I) Total (pmol) and relative abundance of storage lipids (TAGs, triacylglyceride and CEs, cholesterol esters) are presented. Quantifications ± SEM are shown (B–I).
Significance was determined by unpaired 2-tailed t tests.
To determine whether αKG treatment altered the incorporation of carbons from glucose, glutamine and αKG itself into total fatty acids, we profiled the tracing of their uniformly labeled 13C isotopologues. MS evaluation showed that the FAs of most abundance were C16:0, C16:1, C18:0, and C18:1. Irrespective of exogenous αKG, glucose carbons were incorporated into fatty acids at significantly higher rates than glutamine carbons; for C16:0, mean incorporation was 19.9% and 3.1%, respectively (Figure 6E). These differences were also maintained in C16:1, C18:0 and C18:1 FAs (Figure S6D). Interestingly, the utilization of glucose carbons in the de novo synthesis of FAs was higher in Th1- than Treg-polarized cells at day 3–4 of differentiation, highlighting differences in FA synthesis (Figure 6F). Notably though, the presence of 13C5-membrane permeable dimethyl ketoglutarate directly altered FA synthesis; these carbons were incorporated at high levels into C16:0, C18:0, and C18:1 FAs, with a range of 8.7%–22.9%. This incorporation was dramatically elevated as compared to that detected for glutamine carbons, with a range of only 0.8%–5.5% (Figures 6E and S6D). These data reveal the importance of exogenous membrane permeable αKG in directly providing carbons for the synthesis of FAs, thereby altering fatty acid metabolism.
Quantitative shotgun lipidomics also revealed highly significant changes in the unsaturation state of phospholipids; decreased levels of saturated and mono-unsaturated lipids and conversely, increased polyunsaturated lipids (p < 0.0001; Figure S6E). Furthermore, other phospholipids were significantly altered by αKG-mediated T cell reprogramming including phosphatidylethanolamine (PE), sphingolipids (SL), and alkyl-etherlinked phosphatidylcholine (PC O-), a plasmalogen whose orientation of the polar head group differs with respect to the membrane surface from diacylphosphatidylcholine (Han and Gross, 1990) (p < 0.0001; Figure 6D).
These robust lipidomic findings led us to evaluate changes in the physical properties of the membrane in the polarized CD4 T cells. Using the polarity Di-4-AN(F)EPPTEA probe and spectral imaging (Sezgin et al., 2015a; Sezgin et al., 2019), we determined that αKG treatment significantly increased membrane packing, quantified by the dimensionless parameter generalized polarization (GP), which reports on the fluorescence emission shift of the membrane sensitive fluorophore (p < 0.0001, Figures 6G and S6F). This small yet significant change in GP is consistent with the broad lipid remodeling in response to αKG treatment. While incorporation of polyunsaturated fatty acids (PUFAs) into phospholipids generally increases fluidity (Ernst et al., 2018; Levental et al., 2016; Sezgin et al., 2015a; Sezgin et al., 2017), these data strongly suggest that complex compensatory changes between the lipidome-wide remodeling and high levels of PUFAs (Levental et al., 2020) contributed to increased membrane packing and rigidity.
Mitochondrial lipids and storage lipids are massively increased following Treg polarization in the presence of ectopic αKG
Mitochondrial function is closely linked to mitochondrial biogenesis, a process that requires the generation of mitochondrial membranes. Specifically, the presence of the two nonbilayer-forming phospholipids in the mitochondrial inner membrane, PE and cardiolipin (CL) (Kojima et al., 2019; Pennington et al., 2019), have been shown to reflect the close association between lipid metabolism and mitochondrial function (Schenkel and Bakovic, 2014). Moreover, CL biosynthesis has itself been linked to efficient mitochondrial metabolism (Gohil et al., 2004). Notably, the αKG-mediated induction of OXPHOS under Treg-polarizing conditions (Figures 1B and 1C) was associated with a dramatic increase in both CL and phosphatidylglycerol (PG); the proportion of mitochondrial lipids increased from 80 ± 10 to 166 ± 23 pmol/106 cells (Figure 6H, left panel), accounting for 1.4 ± 0.04% and 1.7 ± 0.06% of total membrane lipids, respectively (p < 0.01; Figure 6H, right panel). Thus, this lipid remodeling may stabilize a larger pool of mitochondrial membranes, thereby promoting mitochondrial respiration.
Mitochondrial lipid biosynthesis often parallels lipid storage in lipid droplets (Jarc and Petan, 2020; Pernes et al., 2019). Indeed, fatty acids are not only components of plasma membrane lipids but are also present in the cells, where they are stored as triacylglycerides (TAGs). Such TAGs are a major source of stored energy for the cell, presenting a dynamic pool of fatty acids that can be rapidly mobilized in response to cellular stress and energy requirements (Howie et al., 2019; Jarc and Petan, 2019). Importantly, αKG dramatically increased both the absolute levels of storage lipids, from 117 ± 14 to 795 ± 79 pmol/106 cells, representing an increase from 2.3 ± 0.3% to 7.7 ± 0.3% of total lipids (p < 0.0001; Figure 6I). These data, together with the critical contribution of ectopic αKG in FA synthesis, highlight an important role for lipid metabolism in increasing the energetic state of a naive CD4 T cell activated in the presence of αKG.
DGAT2-mediated generation of triacylglycerides inhibits Treg differentiation
TAG synthesis is catalyzed by the acyl-CoA:diacylglycerol acyltransferase (DGAT) enzymes DGAT1 and DGAT2. Both catalyze the same reaction, condensing diacylglycerol and fatty acyl-CoA to form TAGs (Cases et al., 2001; Harris et al., 2011; Lardizabal et al., 2001) (Figure 7A). Based on the dramatic increase in TAGs under conditions where Treg differentiation was attenuated, we were interested in specifically evaluating the role of these enzymes in Treg differentiation. While DGATs are increased in tissue-resident Tregs and in pathological conditions (Burzyn et al., 2013; Graham et al., 2019; Miragaia et al., 2019; Panduro et al., 2016; Peligero-Cruz et al., 2020), their roles in Treg function are likely to be tissue- and subset-specific. Inhibition of DGAT1 has been reported to impair FoxP3 expression (Howie et al., 2019) but conversely, DGAT1 has also been found to inhibit Treg differentiation and exacerbate autoimmune encephalitis (Graham et al., 2019). In accord with the latter study by Graham and colleagues (Graham et al., 2019), we found that Dgat1 RNA levels were significantly higher in Th1 than Treg (by day 4 of polarization). Furthermore, they were significantly upregulated by αKG in the latter (padj = 0.1; Figure S7A and data not shown). However, specific inhibition of DGAT1 by the Α−925200 inhibitor (Zhao et al., 2008) did not restore Treg differentiation in the presence of αKG (Figure S7B).
Figure 7. DGAT2-mediated generation of triacylglycerides inhibits Treg differentiation.

(A) Schematic representation of the classical pathway leading to the generation of triacylglycerides (TAG).
(B) The impact of the DGAT2 inhibitor (PF-06424439) on Treg polarization was evaluated as a function of FoxP3 expression at day 4 (n = 5).
(C) IFNγ, IL-17A, TNF, and GM-CSF secretion was quantified in the indicated conditions (day 3, n = 4).
(D) Lipid droplet quantification was evaluated by Nile Red staining and representative histograms and quantifications are presented at day 4 (levels in control Treg conditions were arbitrarily set at “1”). Quantifications ± SEM are shown (B–D). Significance was determined by one-way ANOVA and Tukey multiple comparison tests.
We therefore evaluated the role of DGAT2 in Treg differentiation. While both DGAT1 and DGAT2 catalyze the esterification of fatty acyl-CoA to diacylglycerol, they are thought to have a bias for exogenously derived and endogenously synthesized FAs, respectively (Bhatt-Wessel et al., 2018). Dgat2 RNA levels were not significantly altered by αKG (Figure S7C) but notably, inhibition of DGAT2 activity with the DGAT2-specific inhibitor PF-06424439 (Futatsugi et al., 2015) resulted in an increased generation of FoxP3+ Tregs, increasing from a mean of 37.2% to 68.2% (p < 0.001; Figure 7B). Furthermore, DGAT2 inhibition markedly decreased secretion of inflammatory cytokines, including IFNγ, IL-17, TNF and GM-CSF (p < 0.01–0.0001; Figure 7C). As expected, this impact of DGAT2 inhibition was associated with a significant decrease in the formation of lipid droplets, as monitored by Nile-Red staining (p < 0.01, Figure 7D). Thus, inhibition of DGAT2 activity, in the context of αKG-induced alterations in lipid homeostasis, resulted in augmented Treg differentiation.
DISCUSSION
Many recent studies have highlighted the critical roles that metabolic pathways play in T cell differentiation. However, the role of oxidative phosphorylation in controlling Treg differentiation is still not clear. Induced Tregs have been reported to depend on OXPHOS and lipid oxidation (Beier et al., 2015; Michalek et al., 2011), but recent studies have also found that Treg differentiation and function are not impacted by conditions wherein OXPHOS and/or FAO are inhibited (Field et al., 2019; Raud et al., 2018; Saravia et al., 2020; Tarasenko et al., 2017). To evaluate the role of OXPHOS in Treg differentiation, we utilized αKG, a metabolite that serves to replenish TCA cycle intermediates. Notably, αKG markedly increased OXPHOS and dramatically attenuated Treg generation. Furthermore, ectopic αKG augmented IFNγ secretion under both Th1- and Treg-polarizing conditions. While αKG altered the epigenetic profile of genes that are critical in both Th1 and Treg differentiation, with increased methylation in the Foxp3 locus and decreased methylation in Tbx21 and Ifng loci, it is significant that Treg polarization was rescued by mitochondrial complex II inhibitors alone. Furthermore, using transcriptomic, metabolomic, and lipidomic approaches, we found that membrane-permeable αKG, but not glutamine-derived αKG, contributed to fatty acid synthesis and massively increased triacylglycerol stores. The latter served as a negative regulator of Treg differentiation; inhibition of DGAT2, catalyzing the generation of TAG, restored Treg differentiation in the presence of αKG and inhibited the αKG-induced secretion of high levels of inflammatory cytokines.
Tregs have been found to use fatty acids for OXPHOS and promote lipid storage (Howie et al., 2017; Howie et al., 2019; Michalek et al., 2011), but it is interesting to note that inhibiting mitochondrial enzymes such as FABP5, resulting in loss of mitochondrial cristae structure and metabolism, promotes Treg suppressive activity (Field et al., 2019). Consistent with these data, the loss of COX10, a subunit of complex IV of the mitochondrial respiratory chain, inhibits T effector but not Treg differentiation (Tarasenko et al., 2017). Indeed, we and others have found that TGF-β-induced Treg exhibit a much lower basal state of OXPHOS than Th1 or Th17 cells (Priyadharshini et al., 2018; Tarasenko et al., 2017; Figure 1). While other groups have reported high levels of OXPHOS in Treg (Angelin et al., 2017; Gerriets et al., 2015; Michalek et al., 2011), the apparent discrepancies are likely due to the activation/ polarization protocols that are used and the distinct types of Treg subsets that were being evaluated (Koch et al., 2009; Sun et al., 2018). The vast majority of the published data point to the importance of a glutamine-linked metabolism in Teff but not Treg differentiation.
The importance of extracellular nutrients in regulating the plasticity of T lymphocytes has been highlighted by the tumor microenvironment. It appears that only five amino acids, of which glutamine is one, are depleted in the core of the tumor as compared to the periphery (Lee et al., 2019; Pan et al., 2016). Moreover, low levels of glutamine in the tumor core may decrease T cell recruitment into these regions (Byun et al., 2020; Matias et al., 2020). In this regard, it is interesting to note that in our studies, αKG pretreatment of ERBB2-directed CAR-T cells activated under Treg-polarizing conditions resulted in a significantly increased tumor infiltration and in vivo IFNγ secretion in mice harboring an ERBB2+ tumor. Furthermore, in the context of translational applications, we found that similarly to its impact on murine T cell differentiation, αKG significantly decreased human Treg differentiation (Figure S7D). These data support the development of new therapies aimed at altering the metabolic state of an adoptively transferred antitumor T cell such that it can optimally function in a tumor microenvironment characterized by a low availability of nutrients.
T cell metabolism has elegantly been shown to integrate transcriptional profiles with epigenetic changes, modulating the differentiation state of the cell (Geltink et al., 2018; Patel and Powell, 2017; Yong et al., 2017a). αKG specifically alters the epigenetic state of CD4 T cells by targeting DNA and histone methylation (Chisolm et al., 2017; Xu et al., 2017). This effect is modulated at least in part by αKG’s role as a required cofactor for TET dioxygenases (Lio and Rao, 2019; Rose et al., 2011; Tahiliani et al., 2009) whose demethylation of the Foxp3 locus is required for its subsequent expression (Nakatsukasa et al., 2019; Yang et al., 2015; Yue et al., 2019; Yue et al., 2016). However, decreasing the conversion of glutamate to αKG was also found to enhance FoxP3 expression, reducing the generation of the competitive 2-hydroxyglutarate (2-HG) TET inhibitor (Xu et al., 2017). In our study, high intracellular αKG levels were associated with a significant increase in intracellular 2-HG levels and augmented methylation in the Foxp3 locus. Conversely, methylation in several DMR including Tbx21, Ifng, and Rorc was decreased. These epigentic changes can also be due to alterations in the αkg/succinate ratio, found to regulate cell fate through histone and DNA demethylation (Carey et al., 2015; Liu et al., 2017; Mills et al., 2016; TeSlaa et al., 2016; Tischler et al., 2019). Indeed, our data reveal a critical role for malonate, inhibiting succinate oxidation, in restoring Treg differentiation. Furthermore, 3-NP, inhibiting mitochondrial complex II activity, rescued Treg differentiation and downregulated expression of IFNγ. Together, these data highlight a crosstalk between the αKG/succinate ratio, mitochondrial activity, and DNA methylation in controlling Treg differentiation.
Extensive research has focused on the contributions of fatty acid oxidation to lipid metabolism and cell fate decisions but there has been a paucity of research on the role of nonoxidative lipid pathways in regulating lymphoid cell fate (Lee et al., 2018; Pernes et al., 2019). While the role of lipidome remodeling in T lymphocyte fate has not yet been evaluated, recent studies have highlighted the importance of the cell’s lipidome in mesenchymal stem cell differentiation (Levental et al., 2017), myelopoiesis (Mitroulis et al., 2018), stem cell pluripotency (Wu et al., 2019), and lifespan (Schmeisser et al., 2019). Our study showed that during CD4 T cell differentiation, glucose provides carbons for fatty acid synthesis. This de novo FA synthesis, which is rare in non-transformed cells but occurring in immune cells (Berod et al., 2014; Qian et al., 2018), may regulate T cell differentiation. Indeed, incorporation of glucose carbons into FAs was higher in Th1 than Treg polarizing conditions. More importantly, in the conditions used here, carbons from ectopic membrane-permeable αKG, but not glutamine, were extensively used in FA synthesis; this may be due to potential differences in the activity (Parker et al., 2021) and/or localization of membrane-permeable as compared to endogenous αKG. It will be of much interest to study how the synthesis of lipids from carbons derived from esterified and nonesterified αKG impacts T cell differentiation.
Previous studies support a role for αKG in modulating lipid metabolism but the specific effects are not completely understood. High αKG levels have been associated with both obesity (Rodríguez-Gallego et al., 2015) and the prevention of weight gain in high-fat experimental models (Nagaoka et al., 2020; Radzki et al., 2009; Sliwa et al., 2009). In our study, the αKG-mediated increase in oxidative phosphorylation was coupled to a 2-fold increase in mitochondrial phospholipids (CL and PG, p < 0.01) and a > 5-fold increase in TAGs, or storage lipids. These storage lipids, representing a highly reduced form of carbon, serve as an energy reserve that allows cells to meet increased cellular demands (Aon et al., 2014). While DGAT1-mediated TAG synthesis was found to promote Treg differentiation in one study (Howie et al., 2019), other research has found that it inhibits Treg differentiation by sequestering retinol (Graham et al., 2019). Moreover, research from the latter group revealed high levels of DGAT1 in multiple sclerosis (MS) patients and as such, are evaluating DGAT1 inhibitors as a novel means of increasing Treg function in these patients (Graham et al., 2019). Notably, we found that inhibiting DGAT2, decreasing the generation of lipid droplets, significantly increased the generation of FoxP3+ Tregs in the presence of αKG.
Altogether, our study identifies the pleiotropic αKG metabolite as a negative checkpoint regulator in T cell function, increasing IFNγ secretion by CD4 T cells as well as CD4 CAR-T, irrespective of whether they are activated in Treg or Th1 conditions. These findings suggest that the Goldilocks principle can be applied to Treg polarization; attenuated basal OXPHOS and augmented SRC both mitigate differentiation. αKG-mediated attenuation of Treg differentiation can be counteracted at one of two related metabolic checkpoints—ETC complex II and DGAT2-induced TAG generation—opening new axes for the development of novel strategies aimed at targeting autoimmune disorders. Conversely, activation of these pathways can promote the generation of antitumor T lymphocytes that may function more optimally in a nutrient-poor tumor microenvironment.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contacts, Naomi Taylor (taylorn4@mail.nih.gov) and Valérie Dardalhon (vdardalhon@igmm.cnrs.fr).
Materials availability
This study did not generate new unique reagents.
Data and code availability
RNΑ-Seq data generated in this study are available at gene expression omnibus under accession number: GSE156827: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE156827
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Ly5.1, Ly5.2 C57BL/6J (obtained from Charles River), or transgenic mice harboring the ERBB2-CAR transgene (ERBB2-CAR Tg / C57BL/6 J background) downstream of the Vav promoter (Yong et al., 2015) were crossed with FoxP3-GFP reporter mice (Wang et al., 2008). Mice were housed in ventilated racks (with controlled temperature /hygrometry) in a conventional, pathogen-free facility at the Institut de Génétique Moléculaire de Montpellier. Animal care and experiments were approved by the local and national animal facility institutional review boards in accordance with French national and ARRIVE guidelines. All experiments were performed with adult (male and female) mice between the ages of 6–12 weeks of age. For in vivo tumor experiments, mice were randomized into treatment groups and flow cytometry analyses were performed in a blinded manner.
Naive CD4+ T cell isolation and ex vivo differentiation assays
Murine CD4+ T cells were purified using the MACS CD4+ T cell negative selection kit (Miltenyi Biotec) and naive CD4+ T cells from female and male adult C57BL6/J, FoxP3-GFP, or ERBB2-CAR transgenic mice were sorted on the basis of a CD4+CD8−CD62L+CD44−GFP−CD25− expression profile on a FACSAria flow cytometer (BD Biosciences). Thymic Tregs (tTregs) were sorted from FoxP3-GFP reporter mice based on a CD4+CD8−CD62L+CD44−GFP+ profile. T cell activation (0.5–1e6 cells/well/ml) was performed on non-tissue-treated 24-well plates (ThermoFisher) using plate-bound α-CD3 (clone 145–2C11, 1 μg/ml) and α-CD28 (clone PV-1, 1 μg/ml) monoclonal antibodies in RPMI 1640 medium (Life Technologies) supplemented with 10% FCS (PAN-Biotech, Eurobio), 1% penicillin/streptomycin (GIBCO-Life technologies) and β-mercaptoethanol (50 μM). For Th1 and Treg-polarizing conditions, IL-12 (15 ng/ml) and α-IL-4 mAb (5 μg/ml) or hTGF-β (3 ng/ml) and rhIL-2 (100 U/ml), respectively, were added to the cultures. When indicated, cell permeable αKG (dimethyl ketoglutarate, 3.5 mM; Sigma), malonate (dimethyl malonate, 10mM; Sigma), succinate (diethyl succinate, 3.5 mM; Sigma), 3-NP (3-Nitropropionic acid, 62.5 μM; Sigma), oligomycin (250 nM; Sigma), rotenone (25 nM; Sigma) and antimycin A (250 nM; Sigma), water-soluble cholesterol (50 mM, Sigma), Α−922500 (20 μM, Sigma) (Zhao et al., 2008) and PF-06424439 (40 μM, Sigma) (Futatsugi et al., 2015) were added. Cells were split 2–3 days later with medium supplemented with rhIL-2 (100U/mL) and drugs were added at their original or half-dose concentrations. Cells were maintained in a standard tissue culture incubator containing atmospheric O2 and 5% CO2.
Human CD4+ T cells were isolated from healthy adult donors (leukoreduction filters). All experiments using primary human cells were conducted in accordance with the Declaration of Helsinki and IRB approval to the French Blood Bank (Etablissement Français du Sang). 12 PB samples were used in this study. Samples did not contain any identifiers including sex, race, or ethnic origin. All samples were divided such that they were used in all experimental groups. As such, there was no need to use any specific criteria to allocate biological samples to experimental groups. T lymphocytes were purified by negative-selection using Rosette tetramers (StemCell Technologies, Inc. Inc) and the purity was monitored by flow cytometry. For sorting of human CD4+ naive T cells, CD4+ T cells were sorted on the basis of a CD4+CD8−CD45RA+CD45RO−CCR7+CD62L+CD127+CD25− phenotype on a BD FACSAria flow cytometer. For Treg-polarizing conditions, hTGF-β (3 ng/ml) and rhIL-2 (100 U/ml) were added to the cultures. T cell activation was performed using plate-bound α-CD3 (clone OKT3, Biolegend) and α-CD28 (clone 9.3) mAbs at a concentration of 1 mg/ml in RPMI medium 1640 (Life Technologies) supplemented with 10% FCS and 1% penicillin/streptomycin (GIBCO, ThermoFisher).
Adoptive T cell transfers and CAR-T tumor model
To evaluate the in vivo fate of Tregs generated in the absence or presence of αKG, they were retro-orbitally injected 4 days following polarization into RAG2−/− recipients (1e6). In other experiments, naive T cells from CD45.1 and CD45.2 mice were polarized in the presence or absence of αKG as previously described and adoptively transferred in a competitive setting (0.5e6 of each population). RAG2−/− recipients were sacrificed 6 days later and T cell phenotypes in dissociated lymph nodes were analyzed by flow cytometry.
The murine 24JK fibrosarcoma cell line expressing the human ERBB2/Her2 antigen was generated as described (Yong et al., 2016). The 24JK fibrosarcoma cell line was thawed 5–7 days before the injection, amplified in FCS-supplemented RPMI and tested for the presence of pathogens (Test IMPACT™ I, IDEXX BioResearch). 24JK-ERB cells (1e6) were subcutaneously injected into RAG2−/− mice and allowed to establish over a 7-day period. On day 7, 3–5e6 in vitro ERBB2-CAR T cells, activated as indicated, were injected retro-orbitally into tumor-bearing mice. Tumor development was initially evaluated twice a week and daily upon tumor progression. For endpoint analyses, tumors were excised, enzymatically digested in T cell culture medium (as above) using collagenase IV (1 mg/ml, Sigma) and DNASE I (500 μg/ml, Roche), incubated for 25 minutes at 37°C, and processed into single cell suspensions for FACS analysis. Lymph nodes and spleens of tumor bearing mice were isolated and dissociated into single cell suspensions in PBS supplemented with 2% FCS and analyzed by flow cytometry.
METHOD DETAILS
Flow cytometry
Expression of the CD4, CD62L, CD44, CD25, CD3, CD45.1, CD45.2, surface markers was evaluated on 2e5 − 1e6 cells using the appropriate fluorochrome-conjugated monoclonal antibodies at a 1:200 dilution (mAb), in a total volume of 50–100 μls as previously described. Cells were incubated in the dark for 20 minutes in PBS containing 2% FBS at 4C and then washed once in the same medium at 300 g for 5 min prior to evaluation.
Lipid droplets were assessed by staining 2e5 cells with Nile Red (1ug/ml, 100 μl, Thermofisher). Incubations were performed in the dark for 20 minutes in PBS + 2% FBS at 37°C.
For intracellular cytokine staining, 2e5 − 2e6 cells were stimulated with phorbol 12-myristate 13-acetate (PMA) (100 ng/ml) and ionomycin (1 μg/ml) in the presence of brefeldin A (10 μg/ml; all from Sigma) in complete RPMI media for 3.5–4h at 37°C (total volume of 200–2000 μl). For staining of intracellular cytokines and transcription factors, cells were incubated with fixable viability dye prior to fixation and permeabilization (intracellular staining kit from ThermoFisher or BD Biosciences) as per the manufacturer’s instructions. Cells were then washed with the permeabilization buffer and incubated with either FoxP3-PeCy7 and T-Bet-PE mAbs (3h) and/or IFNγ-APC (30 min).
Cytokine production (IFNγ, TNF, IL-17A, GM-CSF) was also assessed by cytometric bead array (CBA, BD Biosciences) using cell culture supernatants collected at day 2–3 of polarization. Supernatants (10 μl) were incubated with capture beads cocktail (PE) for 1 hour at room temperature and detection was performed as per the manufacturer’s instructions on a FACSCanto flow cytometer. A minimum of 300 events was recorded for each bead. Data analysis was performed using FCAPArray Software.
Cell sorting was performed on a FACSARIA high-speed cell sorter and analyses were performed on FACS-Canto II or LSRII-Fortessa cytometers (BD Biosciences). A minimum of 10,000 events were recorded for each staining. Data analyses were performed using FlowJo software (Tree Star, Ashland, OR).
Nanopore sequencing for evaluation of DNA methylation
T cells (from male mice) were polarized in the indicated conditions and DNA (3 μg) from each sample (2 biological replicates) was enriched in immune identity genes using cas9 guided RNPs (Key Resources Table, Table S7), as previously published (Gilpatrick et al., 2020) and characterized (Goldsmith et al., 2021a; Goldsmith et al., 2021b). TracrRNA and crRNA were purchased from Integrated DNA Technologies (IDT). Samples were barcoded and multiplexed using the Nanopore Ligation Sequencing kit (SQK-LSK109) and Native barcode expansion kit according to manufacturer’s instructions (Oxford Nanopore Technology, Oxford UK) without PCR amplification. Sequencing of native DNA was conducted on a MinION sequencer on ONT flow cells with protein pore R9.4 1D chemistry for 48h. Fast5 files were basecalled with Megalodon (using the Rerio ONT model “res_dna_r941_min_modbases_5mC_5hmC_v001.cfg”) and aligned with minimap2 to the mm10 mouse genome. The methylation status of each CpG site was determined by Megalodon after demultiplexing with Deepbinner (v.0.2.0) (Liu et al., 2021; Wick et al., 2018).
KEY RESOURCES TABLE
| Reagent or resource | Source | Identifier |
|---|---|---|
|
| ||
| Antibodies and Flow cytometry reagents | ||
|
| ||
| Anti-mCD25- eF780 (clone PC61.5) | eBioscience/ThermoFisher | Cat# 47-0251.82 RRID: AB_1272179 |
| Anti-mCD25- Pe-Cy7 (clone PC61.5) | BD Biosciences | Cat#552880 RRID: AB_394509 |
| Anti-mCD25- PE (clone PC61) | BD Biosciences | Cat# 553866, RRID:AB_395101 |
| Anti-mCD4- BV711 (clone RM4-5) | BD Biosciences | Cat#563726 RRID: AB_2738389 |
| Anti-mCD4- PE (clone RM4-5) | BD Biosciences | Cat#553049 RRID: AB_394585 |
| Anti-mCD4- PcP Cy5.5 (clone RM4-5) | BD Biosciences | Cat#550954 RRID: AB_393977 |
| Anti-mCD4- V450 (clone RM4-5) | BD Biosciences | Cat#560468 RRID: AB_1645271 |
| Anti-mCD4- BV650 (clone RM4-5) | BD Biosciences | Cat#563647 RRID: AB_2716859 |
| Anti-mCD44-eF780 (clone IM7) | eBioscience/ThermoFisher | Cat# 47-0441-82 RRID: AB_1272244 |
| Anti-mCD44- Pe-Cy7 (clone IM7) | eBioscience/ThermoFisher | Cat#25-0441-82 RRID: AB_469623 |
| Anti-mCD44- V450 (clone IM7) | BD Biosciences | Cat#560451 RRID: AB_1645273 |
| Anti-mCD44- V500 (clone IM7) | BD Biosciences | Cat#560780 RRID: AB_1937316 |
| Anti-mCD45- APC-eF780 (clone 30-F11) | eBioscience/ThermoFisher | Cat#47-0451-80 RRID: AB_1548790 |
| Anti-mCD45.2- BV711 (clone 104) | BD Biosciences | Cat#563685 RRID: AB_2738374 |
| Anti-mCD45.2- FITC (clone 104) | BD Biosciences | Cat#553772 RRID: AB_395041 |
| Anti-mCD45.2- PE (clone 104) | BD Biosciences | Cat# 560695 RRID: AB_1727493 |
| Anti-mCD62L- APC (clone MEL-14) | BD Biosciences | Cat# 553152 RRID: AB_398533 |
| Anti-mCD62L- PE (clone MEL-14) | BD Biosciences | Cat# 553151 RRID: AB_394666 |
| Anti-mCD8α-PE (clone 53-6.7) | BD Biosciences | Cat# 553033 RRID: AB_394571 |
| Anti-mCD8α- FITC (clone 53-6.7) | BD Biosciences | Cat# 553031 RRID: AB_394569 |
| Anti-mCD8α- APC (clone 53-6.7) | BD Biosciences | Cat#553035 RRID: AB_398527 |
| Anti-mCD8α- APC-R700 (clone 53-6.7) | BD Biosciences | Cat#564983 RRID: AB_2739032 |
| Anti-mCD8α- APC-eF780 (clone 53-6.7) | eBioscience/ThermoFisher | Cat# 47-0081-82 RRID: AB_1272185 |
| Anti-mCD8α- PcP Cy 5.5 (clone 53-6.7) | BD Biosciences | Cat#551162 RRID: AB_394081 |
| Anti-mTbet- PE (clone 4B10) | eBioscience/ThermoFisher | Cat# 12-5825-82 RRID: AB_925761 |
| Anti-mTbet- PercpCy5.5 (clone 4B10) | eBioscience/ThermoFisher | Cat# 45-5825-82 RRID: AB_953657 |
| Anti-mFoxp3- PC7 (clone FJK-16S) | eBioscience/ThermoFisher | Cat# 25-5773-82 RRID: AB_891552 |
| Anti-mIFNγ- APC (clone XMG1.2) | eBioscience/ThermoFisher | Cat#17-7311-82 RRID: AB_469504 |
| Anti-mTCRβ- AF594 (clone H57-597) | Biolegend | Cat# 109238 RRID: AB_2563324 |
| Anti-hCD4- PcP Cy5.5 (clone OKT4) | eBioscience/ThermoFisher | Cat# 45-004842 RRID:AB_10804390 |
| Anti-hCD25- FITC (clone B1,49,9) | Beckman Coulter | Cat# IM0478U RRID: AB_130985 |
| Anti-hCD45RΑ- BV711 (Hl100) | BD Biosciences | Cat# 563733 RRID: AB_2738392 |
| Anti-hCD45RO- BV650 (clone UCHL1) | BD Biosciences | Cat# 563750 RRID: AB_2744412 |
| Anti-hCD62L- PE (clone DREG56) | Beckman Coulter | Cat# IM2214U RRID: |
| Anti-hCD62L- PE (clone DREG56) | BD Biosciences | Cat# 555544 RRID: AB_395928 |
| Anti-hCD62L- PE (clone 555544) | BD Biosciences | Cat# 555544 RRID: AB_395928 |
| Anti-hCD127- Pc7 (clone R34.34) | Beckman Coulter | Cat# A64618 RRID: AB_2833031 |
| Anti-hCD127- BV786 (clone HIL-7R-M21) | BD Biosciences | Cat# 563324 RRID: AB_2738138 |
| Anti-hFoxp3- APC (clone PCH101) | eBioscience/ThermoFisher | Cat# 17-4776-41 RRID: AB_1603281 |
| Anti-hCCR7-PeCy7 (Clone 3D12) | BD Biosciences | Cat# 557648, RRID:AB_396765 |
| InVivoMab anti-mouse CD16/CD32 (2.4G2) | BioXcell | Cat# BE0307 RRID: AB_2736987 |
| InVivoMAb anti-mouse IL-4 (clone 11B11) | BioXcell | Cat# BE0045 RRID: AB_1107707 |
| InVivoMAb anti-mouse CD3ε (clone 145-2C11) | BioXcell | Cat# BE0001-1 RRID: AB_1107634 |
| InVivoMAb anti-mouse CD28 (clone PV1) | BioXcell | Cat# BE0015-5 RRID: AB_1107628 |
| InVivoMAb anti-human CD3 (clone OKT3) | BioXcell | Cat# BE0001-2 RRID: AB_1107632 |
| InVivoMAb anti-human CD28 (clone 9.3) | BioXcell | Cat# BE0248 RRID: AB_2687729 |
| Live/Dead v506 | eBioscience/ThermoFisher | Cat#65-0866-14 |
| Live/Dead eF780 | eBioscience/ThermoFisher | Cat#65-0865-18 |
| Nile Red | Thermofisher Scientific | Cat#N1142 |
|
| ||
| Biological Samples | ||
|
| ||
| Human peripheral blood | EFS | N/A |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Dimethyl α-ketoglutarate | Sigma-Aldrich | Cat#349631 |
| Dimethyl-[13C5]-α-Ketoglutarate | RTI International /NIH | N/A |
| Diethylsuccinate | Sigma-Aldrich | Cat#112402 |
| Cholesterol-Water Soluble | Sigma-Aldrich | Cat#C4951 |
| Dimethyl malonate 98% | Sigma-Aldrich | Cat#13644 |
| 3-Nitropropionic acid | Sigma-Aldrich | Cat# N5636 |
| PF-06424439 | Sigma-Aldrich | Cat#PZ0233 |
| A922500 | Sigma-Aldrich | Cat# A1737 |
| 13C5-Glutamine | Cambridge Isotope Laboratories | Cat#CNLM-1275-H-0.1 |
| 13C6-Glucose | Sigma-Aldrich | Cat#389374 |
| Di-4-AN(F)EPPTEA | Gift from Dr. Leslie Loew and then from Potentiometric Probes | Potentiometric Probes |
| Oligomycin | Sigma-Aldrich | Cat#O4876 |
| FCCP | Sigma-Aldrich | Cat#C2920 |
| Rotenone | Sigma-Aldrich | Cat#R8875 |
| Antimycin A | Sigma-Aldrich | Cat#A8674 |
| DMSO | Sigma-Aldrich | Cat#D2650 |
| Phorbol 12-myristate 13-acetate | Sigma-Aldrich | Cat#P8139 |
| Ionomycin | Sigma-Aldrich | Cat#I0634 |
| Brefeldin A | Sigma-Aldrich | Cat#B7651 |
| Murine recombinant IL12 | R&D Systems | Cat#419-ML |
| Human recombinant TGF-β1 | R&D Systems | Cat#240-B |
| ProLeukin IL-2 « Clinical grade reagent » | https://base-donnees-publique.medicaments.gouv.fr/affichageDoc.php?specid=61637983&typedoc=N | N/A |
| DNASE I | Roche (Sigma-Aldrich) | Cat#10104159001 |
| Collagenase IV | Roche (Sigma-Aldrich) | Cat#C4-28 |
| RPMI 1640 medium | Eurobio scientific | Cat#CM1RPM08-01 |
| Dulbecco’s phosphate buffered saline (PBS) | Eurobio scientific | Cat#CS1PBS01-01 |
| RPMI glutamax | Life Technologies/ThermoFisher | Cat#61870036 |
| RPMI w/o glutamine and glucose | Pan Biotech | Cat#P04-17550 |
| Penicillin/streptomycin | GIBCO-Life technologies | Cat#15140-122 |
| β-mercaptoethanol | Sigma-Aldrich | Cat#M3148 |
| FCS Eurobio scientific | Eurobio scientific | Cat# CVFSVF00-01 |
| FCS Pan Biotech | Pan Biotech | Cat# P30-5500 |
| Seahorse XF RPMI Assay Medium | Agilent Technologies | Cat#103576-100 |
| Seahorse XF Calibrant Solution | Agilent Technologies | Cat#100840-000 |
| Agilent XFe96 Extracellular Flux Assay cartridges | Agilent Technologies | Cat#102416-100 |
| Poly-D-lysine | Merck-Sigma Aldrich | Cat# Α−003-E |
| Ultima Gold liquid scintillation cocktail | Perkin Elmer | Cat#6013329 |
|
| ||
| Commercial Assays | ||
|
| ||
| CD4+ T Cell Isolation Kit, mouse | Miltenyi Biotec | Cat#130-104-454 |
| RosetteSep Human CD4+ T Cell Enrichment Cocktail |
StemCell Technologies Inc | Cat# 15062 |
| RNeasy Mini kit | QIAGEN | Cat# 74104 |
| QuantiTect™ Reverse Transcription Kit | QIAGEN | Cat# 205311 |
| QuantiTect SYBR green PCR Master mix | Roche | Cat# 04887352001 |
| Qubit® RNA HS Assay Kit | Thermo Fisher Scientific | Cat# Q32852 |
| Agilent RNA 6000 Nano kit | Agilent Technologies | Cat# 5067-1512 |
| Agilent High Sensitivity DNA Kit | Agilent Technologies | Cat# 5067-4626 |
| ERCC ExFold RNA Spike-In Mix 1 | Life Technologies | Cat# 4456740 |
| TruSeq® Stranded mRNA Library Prep for NeoPrepTM kit | Illumina | Cat# NP-202-1001 |
| Quant-iT PicoGreen dsDNA Assay Kit | Thermo Fisher Scientific | Cat# P7589 |
| Nanopore Ligation Sequencing kit | Oxford Nanopore Technology | Cat#SQK-LSK109 |
| Native barcode expansion kit | Oxford Nanopore Technology | Cat#EXP-NBD104 |
| CBA Flex Set mIFNγ | BD Biosciences | Cat#558296 RRID: AB_2869141 |
| BD CBA Flex Set mTNF | BD Biosciences | Cat#558299 RRID: AB_2869144 |
| BD CBA Flex Set mIL-17A | BD Biosciences | Cat#560283 RRID: AB_2869329 |
| BD CBA Flex Set mGM-CSF | BD Biosciences | Cat#558347 RRID: AB_2869171 |
| Mouse/Rat Soluble Protein Master Buffer Kit | BD Biosciences | Cat#558266 RRID: AB_2869126 |
| Fix/perm kit: BD Cytofix:Cytoperm Fixation: Permeabilization Kit | BD Biosciences | Cat#554714 RRID: AB_2869008 |
| Fix/perm kit: eBioscience FOXP3 /Transcription Factor staining buffer set |
eBioscience/ThermoFisher | Cat# 00-5523-00 |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| 24JK-ERB | P. Darcy/ M. Kershaw | (Yong et al., 2016) |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| C57BL/6J | Charles River | Cat# 632C57BL/6J |
| Ly5.1-C57BL/6J | Charles River | Cat# 494C57BL/6J-Ly5.1 |
| FoxP3.GFP reporter mouse line | B. Malissen | (Wang et al., 2008) |
| RAG2 KO- C57BL/6J | E. Verhoeyen | (Hao and Rajewsky, 2001) |
| ERBB2-CAR Tg | P. Darcy/ M. Kershaw | (Yong et al., 2015) |
|
| ||
| Oligonucleotides | ||
|
| ||
| FoxP3 (foxp3) Forward 5′- GGCCCTTCTCCAGGACAGΑ- 3′ |
IDT | Custom order |
| FoxP3 (foxp3) Reverse 5′- GCTGATCATGGCTGGGTTGT- 3′ |
IDT | Custom order |
| IFNγ (IFNγ) Forward 5′- TGGCTCTGCAGGATTTTCATG- 3′ |
IDT | Custom order |
| IFNγ (IFNγ Reverse 5′- TCAAGTGGCATAGATGTGGAAGAΑ- 3′ |
IDT | Custom order |
| Tbet (tbx21) Forward 5′-CAACAACCCCTTTGCCAAAG −3′ |
IDT | Custom order |
| Tbet (tbx21) Reverse 5′-TCCCCCAAGCAGTTGACAGT −3′ |
IDT | Custom order |
| GM-CSF (csf2) Forward 5′-TTTACTTTTCCTGGGCATTG-3′ |
IDT | Custom order |
| GM-CSF (csf2) Reverse 5′-TAGCTGGCTGTCATGTTCAA-3′ |
IDT | Custom order |
| TNF (tnf) Forward 5′-CATCTTCTCAAAATTCGAGTGACAΑ−3′ |
IDT | Custom order |
| TNF (tnf) Reverse 5′-GGGAGTAGACAAGGTACAACCC-3′ |
IDT | Custom order |
| GZMB (gzmb) Forward 5′-TGCTGACCTTGTCTCTGGCC-3′ |
IDT | Custom order |
| GZMB (gzmb) Reverse 5′-TAGTCTGGGTGGGGAATGCΑ−3′ |
IDT | Custom order |
| HPRT (hprt) Forward 5′-CTGGTGAAAAGGACCTCTCG-3′ |
IDT | Custom order |
| HPRT (hprt) Reverse 5′-TGAAGTACTCATTATAGTCAAGGGCΑ−3′ |
IDT | Custom order |
| ABCG1 (abcg1) Forward 5′-CTTTCCTACTCTGTACCCGAGG-3′ |
IDT | Custom order |
| ABCG1 (abcg1) Reverse 5′- CGGGGCATTCCATTGATAAGG-3′ |
IDT | Custom order |
| ABCA1 (abca1) Forward 5′- AAAACCGCAGACATCCTTCAG-3′ |
IDT | Custom order |
| ABCA1 (abca1) Reverse 5′- CATACCGAAACTCGTTCACCC-3′ |
IDT | Custom order |
| Cas9-crRNA guides: see Table S7 | IDT | Custom order |
|
| ||
| Software and Algorithms | ||
|
| ||
| FlowJo (version 10) | BD Biosciences | https://www.flowjo.com/ |
| Prism (version 8) | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Wave (version 2) | Agilent Technologies | https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-software/seahorse-wave-desktop-software-740897 |
| Agilent ATP Assay Report Generator | Agilent Technologies | https://www.agilent.com/en/products/cell-analysis/xf-real-time-atp-rate-assay-report-generator?productURL=https%3A%2F%2Fwww.agilent.com%2Fen%2Fproduct%2Fcell-analysis%2Freal-time-cell-metabolic-analysis%2Fxf-software%2Fseahorse-xf-real-time-atp-rate-assay-report-generators-740902 |
| Xcalibur 2.2 | ThermoFisher | https://assets.thermofisher.com/TFS-Assets/CMD/Product-Guides/Xcalibur-2-2-SP1-Release-Notes.pdf |
| LCquan 2.7 | ThermoFisher | https://assets.thermofisher.com/TFS-Assets/CMD/manuals/LCquan-Admin-2-7.pdf |
| TraceFinder 4.1 | ThermoFisher | https://www.thermofisher.com/fr/fr/home/industrial/mass-spectrometry/liquid-chromatography-massspectrometry-lc-ms/lc-ms-software/lc-ms-data-acquisition-software/tracefinder-software.html |
| FCAP Array V3.0.1 | BD Biosciences | Cat#655100 https://www.bdbiosciences.com/en-us/products/instruments/software-informatics/instrument-software/fcap-array-software-v3-0.652099 |
| WebGestalt (WEΒ-based GEne SeT AnaLysis Toolkit) |
WebGestalt | http://www.webgestalt.org |
| GSEA | Broad Institute | https://www.gsea-msigdb.org/gsea/index.jsp |
| STAR (version 2.5.2a) | Encode/Stanford University | https://github.com/alexdobin/STAR/releases/tag/2.5.2a |
| BioConductor | (Huber et al., 2015) | https://www.bioconductor.org |
| Megalodon | (Liu et al., 2021) | https://github.com/nanoporetech/megalodon |
| ImageJ (with custom GP plugin) | NIH | https://imagej.nih.gov/ij/download.html |
Data processing and statistical analyses were performed using R/Bioconductor (R version 4.0.3). Bed files containing modified cytosine information were transformed into a BSseq object for differential methylation analysis with dispersion shrinkage for sequencing data (DSS) as previously described (Gigante et al., 2019; Park and Wu, 2016). DSS tests for differential methylation at single CpG-sites were evaluated using a Wald test on the coefficients of a beta-binomial regression of count data with an ‘arcsine’ link function. For DSS, a multifactor model of experimental conditions (i.e., tTregs, polarized Tregs, and Tregs polarized in the presence of αKG) was used to account for the biological replicates as well as on- and off-target regions, regardless of coverage. Differentially methylated regions (DMRs) were defined as those loci with at least 3 CpG sites within a distance of less than 50 bp, and with changes in > 50% of all CpG sites exhibiting p < 0.05. DMRs were plotted using the plotDMRs function of the dmrseq package.
RNaseq analysis
T cells (5e6 cells) from triplicates of 2 independent experiments were lysed in TRIzol reagent (ThermoFisher). Total RNA was isolated as per the manufacturer’s instructions and resuspended in RNase free water. RNA was quantified with Qubit® RNA HS Assay Kit (Thermo Fisher Scientific) and RNA integrity was verified with Agilent RNA 6000 Nano kit on the 2100 Bioanalyzer (Agilent Technologies) according to the manufacturers’ protocol. RNΑ-Seq libraries were prepared from 65 ng of total RNA with ERCC ExFold RNA Spike-In Mix 1 (Life Technologies) of 1 μL of 1:5000 (v/v) dilution with TruSeq® Stranded mRNA Library Prep for NeoPrepTM kit as per the NeoPrep Library Preparation System Guide on the instrument (Illumina). The libraries were diluted 1:5 (v/v), with 1 μL used to check the expected size (~300 bp) and the samples’ purity were verified with the Agilent High Sensitivity DNA Kit as manufacturer’s recommendation (Agilent Technologies). One μL of the diluted libraries were used to quantify as recommended in the Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific) with standard range from 0 to 14 ng/ well. Pooled libraries of 1 nM were denatured and diluted as per the Standard Normalization Method for the NextSeq® System (Illumina). Final loading concentration of 1.7 pM and 1% (v/v) PhiX was used with the NextSeq® 500/550 High Output Kit v2 (150 cycles). Illumina Samples were sequenced paired-end with 75 cycles of each read and single index of 6 cycles.
Reads were aligned to mm10 by STAR (version 2.5.2a) (Dobin et al., 2013). NCBI’s genome-build GRCm38.p5 (12–2016) was used as the gene model. ERCC spike-in controls were added for normalization. The alignments to transcriptome were quantified using RSEM (version 1.2.31) (Li and Dewey, 2011). Differential expression was evaluated using the DESeq2 package (Love et al., 2014) and analyzed using the WebGestalt (WEΒ-based Gene SeT AnaLysis Toolkit) functional enrichment analysis web tool (Liao et al., 2019).
Gene expression analysis
Total RNA was isolated at the indicated time points and cDNA was synthetized using the RNeasy Mini kit and then reverse-transcribed to produce cDNA using oligonucleotide priming with the QuantiTect™ Reverse Transcription Kit (both QIAGEN) as per the manufacturer’s instructions. Quantitative PCR of cDNAs was performed using the Quantitect SYBR green PCR Master mix (Roche) with 10 ng of cDNA (by NanoDrop) and 0.5 μM primers in a final volume of 10μl. Primer sequences are detailed in the table below. Each sample was amplified in triplicate. Amplification of cDNAs was performed using the LightCycler 480 (Roche). Cycling conditions included a denaturation step for 5 min at 95°C, followed by 40 cycles of denaturation (95°C for 15 s), annealing (63°C for 30 s) and extension (72°C for 30 s). After amplification, melting curve analysis was performed with denaturation at 95°C for 5 s and continuous fluorescence measurements from 65°C to 97°C at 0.11°C/s. Each sample was amplified in triplicate.
Data were analyzed by LightCycler® 480 Software (Version 1.5) and Microsoft Excel. Relative expression was calculated by normalization to HPRT as indicated (delta-Ct). Real-time PCR CT values were analyzed using the 2(-Delta Delta C(T)) method to calculate fold expression (ddCt).
Evaluation of mitochondrial complexes
Cells (1e6) were lysed in a RIPA or NP-40 lysis buffer. After a 20-min incubation on ice, extracts were centrifuged, and supernatants were harvested. Extracts were resolved on SDS–polyacrylamide pre-cast electrophoresis gels (8.5 to 12%) and transferred electrophoretically onto polyvinylidene difluoride (PVDF) membranes. PVDF membranes were incubated with the OXPHOS rodent monoclonal antibody cocktail (1/250, Thermo Fisher) diluted in PBST-1% milk in for 45 min-1 hour at RT and with horseradish peroxidase–conjugated anti-mouse secondary Ab (1/5000). Immunoreactive proteins were visualized using enhanced chemiluminescence (ECL, Amersham) according to the manufacturer’s instructions. Loading control levels were assessed following amido black staining and proteins were quantified with ImageJ software and normalized to actin levels.
Lipid analyses
Mass spectrometry-based lipid analysis was performed at Lipotype GmbH (Dresden, Germany) as previously described (Sampaio et al., 2011) and triplicate samples (1×106) were evaluated. Briefly, lipids were extracted using a two-step chloroform/methanol procedure (Ejsing et al., 2009). Samples were analyzed by direct infusion on a QExactive mass spectrometer (Thermo Scientific) equipped with a TriVersa NanoMate ion source (Advion Biosciences) as described (Mitroulis et al., 2018).
Data were analyzed as previously described, using an in-house lipid identification software based on LipidXplorer (Levental et al., 2020; Wang and Han, 2016). Analyses were only performed on samples with a signal-to-noise ratio > 5 and a 5-fold higher signal intensity than in corresponding blank samples. Lipidomic analysis yielded a list of > 600 individual lipid species. Quantities were determined by first transforming all detected lipids into mol% as a function of total lipids. Molar percentages of membrane lipids were determined after removing TAG and CE from the analysis. Storage lipids were analyzed as a function of TAGs and CE. Class composition was further evaluated in the datasets as well as the number of carbons and level of unsaturation in individual species.
Spectral imaging and generalized polarization
Spectral imaging was performed as previously described (Levental and Levental, 2015; Levental et al., 2016; Levental et al., 2017; Sezgin et al., 2012; Sezgin et al., 2015b). Following staining of cells with 1 μg/ml Di-4-AN(F)EPPTEA for 5 min at room temperature, cells were evaluated by confocal based spectral imaging (Sezgin et al., 2015b). Di-4-AN(F)EPPTEA was excited with 488 nm laser and emissions were collected with a spectral channel in the spectral region 410–691 nm. For GP calculations, intensities at 560 nm and 650 nm wavelengths were used as ordered and disordered signal ( and ), respectively. GP was calculated as:
GP analysis was performed with custom GP plugin for ImageJ (Sezgin et al., 2015b).
Mass spectrometry (LC-MS)
For the profiling of intracellular metabolites, cells (1e6) were treated as previously described (Clerc et al., 2019; Oburoglu et al., 2014). Briefly, at day 3 of activation/polarization, cells were collected, washed and incubated in RPMI medium without glucose or glutamine and supplemented with uniformly labeled [U-13C6]glucose or [U-13C5-15N2]glutamine (Cambridge Isotope Laboratories, Inc) at the same concentration of the complete/original media (11mM and 2mM respectively), respectively, for 24 hours and all cultures were performed in triplicate. Cells were then rapidly collected, washed in ice-cold PBS and metabolites were extracted in a 50% methanol / 30% acetonitrile/ 20% water solution. Samples were resuspended in extraction solution at a concentration of 2e6 cells/ml. Cell extracts and media were centrifuged at 16,000 g for 10min at 4°C and supernatants were analyzed by HPLC-MS. Samples were separated on a Thermo Scientific Accela liquid chromatography system coupled with a guard column (Hichrom) and a ZIC-pHILIC column (Merck). Separations were performed using a gradient elution in a solution of 20mM ammonium carbonate, with 0.1% ammonium hydroxide and acetonitrile. Metabolites were detected using a Thermo Scientific Exactive Orbitrap mass spectrometer with electrospray ionization, operating in a polarity switching mode. Raw data were analyzed with Xcalibur 2.2 and LCquan 2.7 Thermo scientific software.
Tracing of metabolites into fatty acids
T cells polarized in the indicated conditions were incubated in RPMI medium without glucose or glutamine and supplemented with uniformly labeled [U-13C6]glucose, [U-13C5-15N2]glutamine, or [U-13C5]dimethyl alpha ketoglutarate (generated by RTI international and confirmed by GC-MS) for 24h. Lipids corresponding to total fatty acids were extracted according to Bligh and Dyer (Bligh and Dyer, 1959) in dichloromethane/water/methanol 2% acetic acid (2.5/2.5/2, v/v/v), in the presence of glyceryl trinonadecanoate as an internal standard (4μg). After centrifugation during 6min at 2500 rpm, the lipid extract was hydrolysed in KOH (0.5 M in methanol, 1mL) at 55°C for 30 min. A second extraction was performed with dichloromethane/water/methanol (2.5/2/1.5, v/v/v). The lipid extract was evaporated to dryness and dissolved in ethyl acetate (10 μL). Derivatization was performed with PentafluoroBenzylBrommide in ACN (1%) and Diisopropylethylamine in ACN (1%) (1/1, v/v). FAME (1 μL) were analyzed on a gas chromatography Thermo Trace 1310 with a mass spectrometer Thermo TSQ 8000 EVO. Chemical ionization was used as ionization, helium as carrier gas, and a HP-5MS column (30 m x 0.25 mm, 0.25 μm) was used for separation. Oven temperature was programmed from 180°C to 220C at a rate of 8°C/min, from 220°C to 280°C at a rate of 2°C/min. And 10°C/min to 310°C. A full scan method was used to detected compounds. The majority of lipids detected were C16, C16:1, C18:0 and C18:1 FAs and thus, data are presented for these species.
Extracellular flux analysis
OCR and ECAR were measured using the XFe96 Extracellular Flux Analyzer (Seahorse Biosciences, Agilent). Agilent XFe96 Extracellular Flux Assay cartridges were hydrated overnight in pure water, and in the morning, water was replaced by 200 μL of Seahorse Calibrant and incubated at least 1h at 37°C (non-CO2). T cells (1.7–2e5)/well) were harvested at day 2 or 4 following TCR activation, resuspended in 50 μL Agilent XF media (buffered RPMI pH7.4) in the presence of glucose (11 mM) and L-glutamine (2 mM), and seeded in 96-well Seahorse XF96 cell culture microplates (Seahorse Biosciences, Agilent) coated with poly-D-lysine (0.1mg/ml, Millipore- 1 hour coating performed at room temperature). T cells were incubated at 37°C (non-CO2) for 30 minutes and the total volume of each well was set to 180 μL by adding 130 μL of XF RPMI medium with or without drugs/metabolites depending on the tested conditions. Metabolites or inhibitors were added/injected as indicated. αKG, as well as other tested metabolites/drugs, were added to the pH-adjusted Agilent XF media at a 10X concentration (35mM) 24h prior to utilization and buffered with NaOH at that time. Prior to use, the 10X αKG solution was warmed to 37°C and the pH was re-checked.
OCR and ECAR parameters were monitored in basal conditions and in response to oligomycin (1 μM), FCCP (1 μM), rotenone (100 nM) and antimycin A (1 μM; Sigma). The following parameters were calculated as followed: Baseline respiration, baseline cellular OCR – non-mitochondrial respiration (post injection of Antimycin A/Rotenone); OCR-linked ATP, basal respiration rate minus the rate measured after the addition of oligomycin; and SRC (Spare Respiration Capacity), basal respiration rate minus the rate measured after addition of FCCP. ATP Rate Assays were performed as per the manufacturer’s instructions (XF Real-Time ATP Rate Assay Kit) and data were analyzed either with the « Agilent ATP Assay Report Generator », as previously described (Passalacqua et al., 2019), or using Agilent-based equations.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are represented as individual values or means. Error bars represent the standard error of the mean (SEM). Data were analyzed with GraphPad Software versions 5 and 8 (GraphPad Prism), and p values were calculated using a one-way ANOVA (Tukey’s post hoc test), unpaired or paired 2-tailed t tests as indicated. Tests were paired or unpaired as indicated and a normal distribution was used in all experiments. All statistical tests are indicated in the figure legends. Significance is indicated as * p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 as evaluated by GraphPad. Schematics presented in Figures 7A and S4B and the graphical abstract were created using BioRender.
Supplementary Material
α-ketoglutarate (αKG) induces an inflammatory phenotype in Treg-polarized cells
Tumor infiltration of Treg-polarized CAR-T is enhanced by ex vivo αKG treatment
αKG carbons contribute to de novo lipid biosynthesis and lipidome remodeling
DGAT2-mediated inhibition of triacylgleride synthesis restores Treg differentiation
ACKNOWLEDGMENTS
We thank all members of our laboratories for discussions and scientific critique. We are indebted to Ünal Coskun, Michal Grzybek, Alessandra Palladini, and Triantafyllos Chavakis for all their expertise and assistance with lipidomics analyses. We are grateful to Myriam Boyer-Clavel and Stéphanie Viala of the imaging facility MRI, member of the national infrastructure France-BioImaging infrastructure supported by the French National Research Agency (ANR-10-INBS-04, «Investments for the future»). We are grateful to Amal Makrini of the SERANAD bioinformatics platform for her assistance with analyses. M.I.M. was funded by the French Ministry of Health and a fellowship from ARC and C.S.Y. by an Australian Postgraduate Award, Cancer Therapeutics Australia and ARC. C.G. was supported by the ERICAN program of MSD Avenir (J.C.M.). S.T. is supported by funding from Cancer Research UK (C596/A17196 and A23982). We are grateful to MetaboHUΒ-MetaToul (Toulouse, France) and MetaboHUΒ-ANR-11-INBS-0010 for lipid tracing experiments. This research was in part supported by the NIH Intramural Research Program of NIAID (S.A.M.) and NCI (N.T.). This work was supported by generous funding from the ANR research grants (CHIC-20-CE14-0049, NutriDiff, and PolarAttack), FRM, ARC, Sidaction, ANRS, and the French laboratory consortiums EpiGenMed and GR-Ex.
Footnotes
DECLARATION OF INTERESTS
C.M., S.K., V.D., and N.T. are inventors on patents describing the use of ligands for detection of and modulation of metabolite transporters (N.T. gave up her rights), licensed to METAFORΑ-biosystems.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109911.
REFERENCES
- Almeida L, Lochner M, Berod L, and Sparwasser T. (2016). Metabolic pathways in T cell activation and lineage differentiation. Semin. Immunol 28, 514–524. [DOI] [PubMed] [Google Scholar]
- Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ 3rd, Kopinski PK, Wang L, et al. (2017). Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 25, 1282–1293.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aon MA, Bhatt N, and Cortassa SC (2014). Mitochondrial and cellular mechanisms for managing lipid excess. Front. Physiol 5, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailis W, Shyer JA, Zhao J, Canaveras JCG, Al Khazal FJ, Qu R, Steach HR, Bielecki P, Khan O, Jackson R, et al. (2019). Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 571, 403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, Koike MA, Hancock SA, Bhatti TR, Han R, et al. (2015). Essential role of mitochondrial energy metabolism in Foxp3+ T-regulatory cell function and allograft survival. FASEB J. 29, 2315–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, Sandouk A, Hesse C, Castro CN, Bä hre, H., et al. (2014). De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med 20, 1327–1333. [DOI] [PubMed] [Google Scholar]
- Bhatt-Wessel B, Jordan TW, Miller JH, and Peng L. (2018). Role of DGAT enzymes in triacylglycerol metabolism. Arch. Biochem. Biophys 655, 1–11. [DOI] [PubMed] [Google Scholar]
- Bligh EG, and Dyer WJ (1959). A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol 37, 911–917. [DOI] [PubMed] [Google Scholar]
- Buck MD, Sowell RT, Kaech SM, and Pearce EL (2017). Metabolic Instruction of Immunity. Cell 169, 570–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C, and Mathis D. (2013). A special population of regulatory T cells potentiates muscle repair. Cell 155, 1282–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun JK, Park M, Lee S, Yun JW, Lee J, Kim JS, Cho SJ, Jeon HJ, Lee IK, Choi YK, and Park KG (2020). Inhibition of Glutamine Utilization Synergizes with Immune Checkpoint Inhibitor to Promote Antitumor Immunity. Mol. Cell 80, 592–606.e8. [DOI] [PubMed] [Google Scholar]
- Carey BW, Finley LW, Cross JR, Allis CD, and Thompson CB (2015). Intracellular a-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, Turay AM, and Frauwirth KA (2010). Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol 185, 1037–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cases S, Stone SJ, Zhou P, Yen E, Tow B, Lardizabal KD, Voelker T, and Farese RV Jr. (2001). Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J. Biol. Chem 276, 38870–38876. [DOI] [PubMed] [Google Scholar]
- Cham CM, Driessens G, O’Keefe JP, and Gajewski TF (2008). Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur. J. Immunol 38, 2438–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, et al. (2013). Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, et al. (2015). Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162, 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman NM, Zeng H, Nguyen TM, Wang Y, Vogel P, Dhungana Y, Liu X, Neale G, Locasale JW, and Chi H. (2018). mTOR coordinates transcriptional programs and mitochondrial metabolism of activated Treg subsets to protect tissue homeostasis. Nat. Commun 9, 2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisolm DA, Savic D, Moore AJ, Ballesteros-Tato A, León B, Crossman DK, Murre C, Myers RM, and Weinmann AS (2017). CCCTC-binding factor translates interleukin 2- and α-ketoglutarate-sensitive metabolic changes in T cells into context-dependent gene programs. Immunity 47, 251–267.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerc I, Moussa DA, Vahlas Z, Tardito S, Oburoglu L, Hope TJ, Sitbon M, Dardalhon V, Mongellaz C, and Taylor N. (2019). Entry of glucose- and glutamine-derived carbons into the citric acid cycle supports early steps of HIV-1 infection in CD4 T cells. Nat. Metab 1, 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cretenet G, Clerc I, Matias M, Loisel S, Craveiro M, Oburoglu L, Kinet S, Mongellaz C, Dardalhon V, and Taylor N. (2016). Cell surface Glut1 levels distinguish human CD4 and CD8 T lymphocyte subsets with distinct effector functions. Sci. Rep 6, 24129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrasse-Jèze G, Bergot AS, Durgeau A, Billiard F, Salomon BL, Cohen JL, Bellier B, Podsypanina K, and Klatzmann D. (2009). Tumor emergence is sensed by self-specific CD44hi memory Tregs that create a dominant tolerogenic environment for tumors in mice. J. Clin. Invest 119, 2648–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, and Powell JD (2011). The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol 12, 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNΑ-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Yanagida A, Knight K, Engel AL, Vo AH, Jankowski C, Sadilek M, Tran VT, Manson MA, Ramakrishnan A, et al. (2016). Reductive carboxylation is a major metabolic pathway in the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 113, 14710–14715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejsing CS, Sampaio JL, Surendranath V, Duchoslav E, Ekroos K, Klemm RW, Simons K, and Shevchenko A. (2009). Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc. Natl. Acad. Sci. USA 106, 2136–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst R, Ballweg S, and Levental I. (2018). Cellular mechanisms of physicochemical membrane homeostasis. Curr. Opin. Cell Biol 53, 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendt SM, Bell EL, Keibler MA, Olenchock BA, Mayers JR, Wasylenko TM, Vokes NI, Guarente L, Vander Heiden MG, and Stephanopoulos G. (2013). Reductive glutamine metabolism is a function of the α-ketoglutarate to citrate ratio in cells. Nat. Commun 4, 2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field CS, Baixauli F, Kyle RL, Puleston DJ, Cameron AM, Sanin DE, Hippen KL, Loschi M, Thangavelu G, Corrado M, et al. (2019). Mitochondrial integrity regulated by lipid metabolism is a cell-intrinsic checkpoint for Treg suppressive function. Cell Metab 31, 422–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- for Treg Futatsugi K., Kung DW, Orr ST, Cabral S, Hepworth D, Aspnes G, Bader S, Bian J, Boehm M, Carpino PA, et al. (2015). Discovery and optimization of imidazopyridine-based inhibitors of diacylglycerol acyltransferase 2 (DGAT2). J. Med. Chem 58, 7173–7185. [DOI] [PubMed] [Google Scholar]
- Geltink RIK, Kyle RL, and Pearce EL (2018). Unraveling the complex interplay between T cell metabolism and function. Annu. Rev. Immunol 36, 461–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, et al. (2015). Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Invest 125, 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigante S, Gouil Q, Lucattini A, Keniry A, Beck T, Tinning M, Gordon L, Woodruff C, Speed TP, Blewitt ME, and Ritchie ME (2019). Using long-read sequencing to detect imprinted DNA methylation. Nucleic Acids Res. 47, e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpatrick T, Lee I, Graham JE, Raimondeau E, Bowen R, Heron A, Downs B, Sukumar S, Sedlazeck FJ, and Timp W. (2020). Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat. Biotechnol 38, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohil VM, Hayes P, Matsuyama S, Schä gger H, Schlame M, and Greenberg ML (2004). Cardiolipin biosynthesis and mitochondrial respiratory chain function are interdependent. J. Biol. Chem 279, 42612–42618. [DOI] [PubMed] [Google Scholar]
- Goldsmith C, Cohen D, Dubois A, Martinez MG, Petitjean K, Corlu A, Testoni B, Hernandez-Vargas H, and Chemin I. (2021a). Cas9-targeted nanopore sequencing reveals epigenetic heterogeneity after de novo assembly of native full-length hepatitis B virus genomes. Microb. Genom. 7 10.1099/mgen.0.000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith C, Rodríguez-Aguilera JR, El-Rifai I, Jarretier-Yuste A, Hervieu V, Raineteau O, Saintigny P, Chagoya de Sánchez V, Dante R, Ichim G, and Hernandez-Vargas H. (2021b). Low biological fluctuation of mitochondrial CpG and non-CpG methylation at the single-molecule level. Sci. Rep 11, 8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Menendez P, Romano M, Yan H, Deshmukh R, Papoin J, Oburoglu L, Daumur M, Dumé AS, Phadke I, Mongellaz C, et al. (2021). An IDH1-vitamin C crosstalk drives human erythroid development by inhibiting pro-oxidant mitochondrial metabolism. J. Cel. Rep 34, 108723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham KL, Werner BJ, Moyer KM, Patton AK, Krois CR, Yoo HS, Tverskoy M, LaJevic M, Napoli JL, Sobel RA, et al. (2019). DGAT1 inhibits retinol-dependent regulatory T cell formation and mediates autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 116, 3126–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han XL, and Gross RW (1990). Plasmenylcholine and phosphatidylcholine membrane bilayers possess distinct conformational motifs. Biochemistry 29, 4992–4996. [DOI] [PubMed] [Google Scholar]
- Hao Z, and Rajewsky K. (2001). Homeostasis of peripheral B cells in the absence of B cell influx from the bone marrow. J. Exp. Med 194, 1151–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CA, Haas JT, Streeper RS, Stone SJ, Kumari M, Yang K, Han X, Brownell N, Gross RW, Zechner R, and Farese RV Jr. (2011). DGAT enzymes are required for triacylglycerol synthesis and lipid droplets in adipocytes. J. Lipid Res 52, 657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He N, Fan W, Henriquez B, Yu RT, Atkins AR, Liddle C, Zheng Y, Downes M, and Evans RM (2017). Metabolic control of regulatory T cell (Treg) survival and function by Lkb1. Proc. Natl. Acad. Sci. USA 114, 12542–12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie D, Cobbold SP, Adams E, Ten Bokum A, Necula AS, Zhang W, Huang H, Roberts DJ, Thomas B, Hester SS, et al. (2017). Foxp3 drives oxidative phosphorylation and protection from lipotoxicity. JCI Insight 2, e89160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie D, Ten Bokum A, Cobbold SP, Yu Z, Kessler BM, and Waldmann H. (2019). A novel role for triglyceride metabolism in Foxp3 expression. Front. Immunol 10, 1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hukelmann JL, Anderson KE, Sinclair LV, Grzes KM, Murillo AB, Hawkins PT, Stephens LR, Lamond AI, and Cantrell DA (2016). The cytotoxic T cell proteome and its shaping by the kinase mTOR. Nat. Immunol 17, 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarc E, and Petan T. (2019). Lipid droplets and the management of cellular stress. Yale J. Biol. Med 92, 435–452. [PMC free article] [PubMed] [Google Scholar]
- Jarc E, and Petan T. (2020). A twist of FATe: Lipid droplets and inflammatory lipid mediators. Biochimie 169, 69–87. [DOI] [PubMed] [Google Scholar]
- Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, Maseda D, Liberti MV, Paz K, Kishton RJ, et al. (2018). Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. J. Cell 175, 1780–1795 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King A, Selak MA, and Gottlieb E. (2006). Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene 25, 4675–4682. [DOI] [PubMed] [Google Scholar]
- Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, Mongellaz C, Floess S, Fritz V, Matias MI, et al. (2015). Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal 8, ra97. [DOI] [PubMed] [Google Scholar]
- Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, and Campbell DJ (2009). The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol 10, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen P, Hirsilä M, Remes AM, Hassinen IE, Kivirikko KI, and Myllyharju J. (2007). Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J. Biol. Chem 282, 4524–4532. [DOI] [PubMed] [Google Scholar]
- Kojima R, Kakimoto Y, Furuta S, Itoh K, Sesaki H, Endo T, and Tamura Y. (2019). Maintenance of cardiolipin and crista structure requires cooperative functions of mitochondrial dynamics and phospholipid transport. Cell Rep. 26, 518–528.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardizabal KD, Mai JT, Wagner NW, Wyrick A, Voelker T, and Hawkins DJ (2001). DGAT2 is a new diacylglycerol acyltransferase gene family: purification, cloning, and expression in insect cells of two polypeptides from Mortierella ramanniana with diacylglycerol acyltransferase activity. J. Biol. Chem 276, 38862–38869. [DOI] [PubMed] [Google Scholar]
- Layman AAK, Deng G, O’Leary CE, Tadros S, Thomas RM, Dybas JM, Moser EK, Wells AD, Doliba NM, and Oliver PM (2017). Ndfip1 restricts mTORC1 signalling and glycolysis in regulatory T cells to prevent autoinflammatory disease. Nat. Commun 8, 15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MKS, Al-Sharea A, Dragoljevic D, and Murphy AJ (2018). Hand of FATe: lipid metabolism in hematopoietic stem cells. Curr. Opin. Lipidol 29, 240–245. [DOI] [PubMed] [Google Scholar]
- Lee SW, Zhang Y, Jung M, Cruz N, Alas B, and Commisso C. (2019). EGFR-Pak signaling selectively regulates glutamine deprivation-induced macropinocytosis. Dev. Cell 50, 381–392.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental KR, and Levental I. (2015). Giant plasma membrane vesicles: models for understanding membrane organization. Curr. Top. Membr 75, 25–57. [DOI] [PubMed] [Google Scholar]
- Levental KR, Lorent JH, Lin X, Skinkle AD, Surma MA, Stockenbojer EA, Gorfe AA, and Levental I. (2016). Polyunsaturated lipids regulate membrane domain stability by tuning membrane order. Biophys. J 110, 1800–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental KR, Surma MA, Skinkle AD, Lorent JH, Zhou Y, Klose C, Chang JT, Hancock JF, and Levental I. (2017). u-3 polyunsaturated fatty acids direct differentiation of the membrane phenotype in mesenchymal stem cells to potentiate osteogenesis. Sci. Adv 3, eaao1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental KR, Malmberg E, Symons JL, Fan YY, Chapkin RS, Ernst R, and Levental I. (2020). Lipidomic and biophysical homeostasis of mammalian membranes counteracts dietary lipid perturbations to maintain cellular fitness. Nat. Commun 11, 1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNΑ-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Wang J, Jaehnig EJ, Shi Z, and Zhang B. (2019). WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 47 (W1), W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lio CJ, and Rao A. (2019). TET enzymes and 5hmC in adaptive and innate immune systems. Front. Immunol 10, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, Di Conza G, Cheng WC, Chou CH, Vavakova M, et al. (2017). a-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol 18, 985–994. [DOI] [PubMed] [Google Scholar]
- Liu Y, Rosikiewicz R, Pan Z, Jillette N, Taghbalout A, Foox J, Mason C, Carroll M, Cheng A, and Li S. (2021). DNA methylation calling tools for Oxford Nanopore sequencing: a survey and human epigenome-wide evaluation. bioRxiv. 10.1101/2021.05.05.442849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftus RM, and Finlay DK (2016). Immunometabolism: Cellular metabolism turns immune regulator. J. Biol. Chem 291, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNΑ-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, and Rathmell JC (2014). The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 20, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makowski L, Chaib M, and Rathmell JC (2020). Immunometabolism: From basic mechanisms to translation. Immunol. Rev 295, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matias MI, Dardalhon V, and Taylor N. (2020). Targeting glutamine metabolism and PD-L1: A novel anti-tumor pas de deux. Mol. Cell 80, 555–557. [DOI] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. (2011). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzler B, Gfeller P, and Guinet E. (2016). Restricting glutamine or glutamine-dependent purine and pyrimidine syntheses promotes human T Cells with high Foxp3 expression and regulatory properties. J. Immunol 196, 3618–3630. [DOI] [PubMed] [Google Scholar]
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, and Rathmell JC (2011). Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol 186, 3299–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Däbritz JHM, Gottlieb E, Latorre I, et al. (2016). Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167, 457–470.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miragaia RJ, Gomes T, Chomka A, Jardine L, Riedel A, Hegazy AN, Whibley N, Tucci A, Chen X, Lindeman I, et al. (2019). Single-cell transcriptomics of regulatory T cells reveals trajectories of tissue adaptation. Immunity 50, 493–504.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska J, Lee-Chang C, Rashidi A, Muroski ME, Chang AL, Lopez-Rosas A, Zhang P, Panek WK, Cordero A, Han Y, et al. (2019). HIF-1α is a metabolic switch between glycolytic-driven migration and oxidative phosphorylation-driven immunosuppression of Tregs in glioblastoma. Cell Rep. 27, 226–237.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, Eugster A, Troullinaki M, Palladini A, Kourtzelis I, et al. (2018). Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell 172, 147–161.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, Hu Z, Shi X, Jiang L, Boroughs LK, Kovacs Z, Boriack R, Rakheja D, Sullivan LB, Linehan WM, et al. (2014). Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 7, 1679–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka K, Mulla J, Cao K, Cheng Z, Liu D, Mueller W, Bay A, Hilldebrand G, Lu S, and Huang CK (2020). The metabolite, alpha-ketoglutarate inhibits non-alcoholic fatty liver disease progression by targeting lipid metabolism. Liver Res. 4, 94–100. [Google Scholar]
- Nakatsukasa H, Oda M, Yin J, Chikuma S, Ito M, Koga-Iizuka M, Someya K, Kitagawa Y, Ohkura N, Sakaguchi S, et al. (2019). Loss of TET proteins in regulatory T cells promotes abnormal proliferation, Foxp3 destabilization and IL-17 expression. Int. Immunol 31, 335–347. [DOI] [PubMed] [Google Scholar]
- Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, Blonska M, Lin X, and Sun SC (2014). Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 40, 692–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham D, and Nunn RS (1990). Elastic deformation and failure of lipid bilayer membranes containing cholesterol. Biophys. J 58, 997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oburoglu L, Tardito S, Fritz V, de Barros SC, Merida P, Craveiro M, Mamede J, Cretenet G, Mongellaz C, An X, et al. (2014). Glucose and glutamine metabolism regulate human hematopoietic stem cell lineage specification. Cell Stem Cell 15, 169–184. [DOI] [PubMed] [Google Scholar]
- Ozay EI, Sherman HL, Mello V, Trombley G, Lerman A, Tew GN, Yadava N, and Minter LM (2018). Rotenone treatment reveals a role for electron transport complex I in the subcellular localization of key transcriptional regulators during t helper cell differentiation. Front. Immunol 9, 1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan M, Reid MA, Lowman XH, Kulkarni RP, Tran TQ, Liu X, Yang Y, Hernandez-Davies JE, Rosales KK, Li H, et al. (2016). Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell Biol 18, 1090–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panduro M, Benoist C, and Mathis D. (2016). Tissue Tregs. Annu. Rev. Immunol 34, 609–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y, and Wu H. (2016). Differential methylation analysis for BS-seq data under general experimental design. Bioinformatics 32, 1446–1453. [DOI] [PubMed] [Google Scholar]
- Parker SJ, Encarnación-Rosado J, Hollinshead KER, Hollinshead DM, Ash LJ, Rossi JAK, Lin EY, Sohn ASW, Philips MR, Jones DR, and Kimmelman AC (2021). Spontaneous hydrolysis and spurious metabolic properties of a-ketoglutarate esters. Nat. Commun 12, 4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passalacqua KD, Lu J, Goodfellow I, Kolawole AO, Arche JR, Maddox RJ, Carnahan KE, O’Riordan MXD, and Wobus CE (2019). Glycolysis is an intrinsic factor for optimal replication of a norovirus. MBio 10. 10.1128/mBio.02175-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel CH, and Powell JD (2017). Targeting T cell metabolism to regulate T cell activation, differentiation and function in disease. Curr. Opin. Immunol 46, 82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peligero-Cruz C, Givony T, Sebé-Pedrós A, Dobes J, Kadouri N, Nevo S, Roncato F, Alon R, Goldfarb Y, and Abramson J. (2020). IL18 signaling promotes homing of mature Tregs into the thymus. eLife 9. 10.7554/eLife.58213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, and Li MO (2016). Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354, 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington ER, Funai K, Brown DA, and Shaikh SR (2019). The role of cardiolipin concentration and acyl chain composition on mitochondrial inner membrane molecular organization and function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1864, 1039–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernes G, Flynn MC, Lancaster GI, and Murphy AJ (2019). Fat for fuel: Lipid metabolism in haematopoiesis. Clin. Transl. Immunology 8, e1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priyadharshini B, Loschi M, Newton RH, Zhang JW, Finn KK, Gerriets VA, Huynh A, Rathmell JC, Blazar BR, and Turka LA (2018). Cutting edge: TGF-β and phosphatidylinositol 3-kinase signals modulate distinct metabolism of regulatory T cell subsets. J. Immunol 201, 2215–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Yang Z, Mao E, and Chen E. (2018). Regulation of fatty acid synthesis in immune cells. Scand. J. Immunol 88, e12713. [DOI] [PubMed] [Google Scholar]
- Radzki RP, Bieńko M, and Pierzynowski SG (2009). Effect of dietary alpha-ketoglutarate on blood lipid profile during hypercholesterolaemia in rats. Scand. J. Clin. Lab. Invest 69, 175–180. [DOI] [PubMed] [Google Scholar]
- Raud B, Roy DG, Divakaruni AS, Tarasenko TN, Franke R, Ma EH, Samborska B, Hsieh WY, Wong AH, Stüve P, et al. (2018). Etomoxir actions on regulatory and memory T cells are independent of Cpt1a-mediated fatty acid oxidation. Cell Metab. 28, 504–515.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Gallego E, Guirro M, Riera-Borrull M, Hernández-Aguilera A, Mariné -Casadó R, Fernández-Arroyo S, Beltrán-Debón R, Sabench F, Hernández M, del Castillo D, et al. (2015). Mapping of the circulating metabolome reveals α-ketoglutarate as a predictor of morbid obesity-associated non-alcoholic fatty liver disease. Int. J. Obes 39, 279–287. [DOI] [PubMed] [Google Scholar]
- Rose NR, McDonough MA, King ON, Kawamura A, and Schofield CJ (2011). Inhibition of 2-oxoglutarate dependent oxygenases. Chem. Soc. Rev 40, 4364–4397. [DOI] [PubMed] [Google Scholar]
- Sampaio JL, Gerl MJ, Klose C, Ejsing CS, Beug H, Simons K, and Shevchenko A. (2011). Membrane lipidome of an epithelial cell line. Proc. Natl. Acad. Sci. USA 108, 1903–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saravia J, Zeng H, Dhungana Y, Bastardo Blanco D, Nguyen TM, Chapman NM, Wang Y, Kanneganti A, Liu S, Raynor JL, et al. (2020). Homeostasis and transitional activation of regulatory T cells require c-Myc. Sci. Adv 6, eaaw6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkel LC, and Bakovic M. (2014). Formation and regulation of mitochondrial membranes. Int. J. Cell Biol 2014, 709828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser S, Li S, Bouchard B, Ruiz M, Des Rosiers C, and Roy R. (2019). Muscle-specific lipid hydrolysis prolongs lifespan through global lipidomic remodeling. Cell Rep. 29, 4540–4552.e8. [DOI] [PubMed] [Google Scholar]
- Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, and Gottlieb E. (2005). Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7, 77–85. [DOI] [PubMed] [Google Scholar]
- Sezgin E, Kaiser HJ, Baumgart T, Schwille P, Simons K, and Levental I. (2012). Elucidating membrane structure and protein behavior using giant plasma membrane vesicles. Nat. Protoc 7, 1042–1051. [DOI] [PubMed] [Google Scholar]
- Sezgin E, Gutmann T, Buhl T, Dirkx R, Grzybek M, Coskun Ü, Solimena M, Simons K, Levental I, and Schwille P. (2015a). Adaptive lipid packing and bioactivity in membrane domains. PLoS ONE 10, e0123930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sezgin E, Waithe D, Bernardino de la Serna J, and Eggeling C. (2015b). Spectral imaging to measure heterogeneity in membrane lipid packing. Chem-PhysChem 16, 1387–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sezgin E, Levental I, Mayor S, and Eggeling C. (2017). The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol 18, 361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sezgin E, Schneider F, Galiani S, Urbanci c, I., Waithe D, Lagerholm BC, and Eggeling C. (2019). Measuring nanoscale diffusion dynamics in cellular membranes with super-resolution STED-FCS. Nat. Protoc 14, 1054–1083. [DOI] [PubMed] [Google Scholar]
- Shin B, Benavides GA, Geng J, Koralov SB, Hu H, Darley-Usmar VM, and Harrington LE (2020). Mitochondrial oxidative phosphorylation regulates the fate decision between pathogenic Th17 and regulatory T cells. Cell Reports 30, 1898–1909 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, and Cantrell DA (2013). Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol 14, 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliwa E, Dobrowolski P, Tatara MR, Piersiak T, Siwicki A, Rokita E, and Pierzynowski SG (2009). Alpha-ketoglutarate protects the liver of piglets exposed during prenatal life to chronic excess of dexamethasone from metabolic and structural changes. J. Anim. Physiol. Anim. Nutr (Berl.) 93, 192–202. [DOI] [PubMed] [Google Scholar]
- Sun IH, Oh MH, Zhao L, Patel CH, Arwood ML, Xu W, Tam AJ, Blosser RL, Wen J, and Powell JD (2018). mTOR complex 1 signaling regulates the generation and function of central and effector Foxp3+ regulatory T cells. J. Immunol 201, 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, and Rao A. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi A, and Saito T. (2017). CD4 CTL, a cytotoxic subset of CD4+ T cells, their differentiation and function. Front. Immunol 8, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasenko TN, Pacheco SE, Koenig MK, Gomez-Rodriguez J, Kapnick SM, Diaz F, Zerfas PM, Barca E, Sudderth J, DeBerardinis RJ, et al. (2017). Cytochrome c oxidase activity is a metabolic checkpoint that regulates cell fate decisions during T cell activation and differentiation. Cell Metab. 25, 1254–1268.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TeSlaa T, Chaikovsky AC, Lipchina I, Escobar SL, Hochedlinger K, Huang J, Graeber TG, Braas D, and Teitell MA (2016). α-Ketoglutarate accelerates the initial differentiation of primed human pluripotent stem cells. Cell Metab. 24, 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischler J, Gruhn WH, Reid J, Allgeyer E, Buettner F, Marr C, Theis F, Simons BD, Wernisch L, and Surani MA (2019). Metabolic regulation of pluripotency and germ cell fate through α-ketoglutarate. EMBO J. 38. 10.15252/embj.201899518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, and Han X. (2016). advanced shotgun lipidomics for characterization of altered lipid patterns in neurodegenerative diseases and brain injury. Methods Mol. Biol 1303, 405–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kissenpfennig A, Mingueneau M, Richelme S, Perrin P, Chevrier S, Genton C, Lucas B, DiSanto JP, Acha-Orbea H, et al. (2008). Th2 lymphoproliferative disorder of LatY136F mutant mice unfolds independently of TCR-MHC engagement and is insensitive to the action of Foxp3+ regulatory T cells. J. Immunol 180, 1565–1575. [DOI] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, and Green DR (2011). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martıńez-Reyes I, Gao P, Helmin KA, Abdala-Valencia H, Sena LA, et al. (2019). Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 565, 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick RR, Judd LM, and Holt KE (2018). Deepbinner: demultiplexing barcoded Oxford Nanopore reads with deep convolutional neural networks. PLoS Comput. Biol 14, e1006583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Chen K, Xing G, Li L, Ma B, Hu Z, Duan L, and Liu X. (2019). Phospholipid remodeling is critical for stem cell pluripotency by facilitating mesenchymal-to-epithelial transition. Sci. Adv 5, eaax7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Stewart KM, Wang X, Liu K, Xie M, Ryu JK, Li K, Ma T, Wang H, Ni L, et al. (2017). Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature 548, 228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Qu C, Zhou Y, Konkel JE, Shi S, Liu Y, Chen C, Liu S, Liu D, Chen Y, et al. (2015). Hydrogen sulfide promotes Tet1- and Tet2-mediated Foxp3 demethylation to drive regulatory T cell differentiation and maintain immune homeostasis. Immunity 43, 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong CS, Westwood JA, Schrö der, Papenfuss AT, von Scheidt B, Moeller M, Devaud C, Darcy PK, and Kershaw (2015). Expression of a chimeric antigen receptor in multiple leukocyte lineages in transgenic mice. PLoS ONE 10, e0140543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong CS, John LB, Devaud C, Prince MH, Johnstone RW, Trapani JA, Darcy PK, and Kershaw MH (2016). A role for multiple chimeric antigen receptor-expressing leukocytes in antigen-specific responses to cancer. Oncotarget 7, 34582–34598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong CS, Abba Moussa D, Cretenet G, Kinet S, Dardalhon V, and Taylor N. (2017a). Metabolic orchestration of T lineage differentiation and function. FEBS Lett. 591, 3104–3118. [DOI] [PubMed] [Google Scholar]
- Yong CSM, Dardalhon V, Devaud C, Taylor N, Darcy PK, and Kershaw MH (2017b). CAR T-cell therapy of solid tumors. Immunol. Cell Biol 95, 356–363. [DOI] [PubMed] [Google Scholar]
- Yue X, Trifari S, Äijö T, Tsagaratou A, Pastor WA, Zepeda-Martínez JA, Lio CW, Li X, Huang Y, Vijayanand P, et al. (2016). Control of Foxp3 stability through modulation of TET activity. J. Exp. Med 213, 377–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue X, Lio CJ, Samaniego-Castruita D, Li X, and Rao A. (2019). Loss of TET2 and TET3 in regulatory T cells unleashes effector function. Nat. Commun 10, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvan-Charvet L, Pagler TA, Seimon TA, Thorp E, Welch CL, Witztum JL, Tabas I, and Tall AR (2010). ABCA1 and ABCG1 protect against oxidative stress-induced macrophage apoptosis during efferocytosis. Circ. Res 106, 1861–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Yang K, Cloer C, Neale G, Vogel P, and Chi H. (2013). mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 499, 485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Souers AJ, Voorbach M, Falls HD, Droz B, Brodjian S, Lau YY, Iyengar RR, Gao J, Judd AS, et al. (2008). Validation of diacyl glycerolacyltransferase I as a novel target for the treatment of obesity and dyslipidemia using a potent and selective small molecule inhibitor. J. Med. Chem 51, 380–383. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNΑ-Seq data generated in this study are available at gene expression omnibus under accession number: GSE156827: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE156827
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.