

Abstract
Rieske-type non-heme iron oxygenases/oxidases catalyze a wide range of transformations. Their applications in bioremediation or biocatalysis face two key barriers: the need of expensive NAD(P)H as a reductant and a proper reductase to mediate the electron transfer from NAD(P)H to the oxygenases. To bypass the need of both the reductase and NAD(P)H, using Rieske-type oxygenase (Stc2) catalyzed oxidative demethylation as the model system, we report Stc2 photocatalysis using eosin Y/sulfite as the photosensitizer/sacrificial reagent pair. In a flow-chemistry setting to separate the photo-reduction half-reaction and oxidation half-reaction, Stc2 photo-biocatalysis outperforms the Stc2-NAD(P)H-reductase (GbcB) system. In addition, in a few other selected Rieske enzymes (NdmA, CntA, and GbcA), and a flavin-dependent enzyme (iodotyrosine deiodinase, IYD), the eosin Y/sodium sulfite photo-reduction pair could also serve as the NAD(P)H-reductase surrogate to support catalysis, which implies the potential applicability of this photo-reduction system to other redox enzymes.
Keywords: N-demethylation, Rieske-type oxygenase, photo-biocatalysis, eosin Y and sulfite, flow-chemistry, O2 delivery
Graphical Abstract
Introduction
Rieske-type oxygenases are a class of non-heme iron enzymes widely distributed in nature.1–5 The most well-characterized Rieske-type reactions are those involved in the degradation of aromatic compounds.6–7 In recent years, several Rieske-type oxygenases in natural product biosynthetic pathways were reported,2 including RedG/McpG-catalyzed cyclization in prodiginine biosynthesis,8 arylamine oxidation in pyrrolnitrin biosynthesis,9 the oxidation of chlorophyll a to chlorophyll b by chlorophyllide a oxygenase,10 hydroxylation of salvigenin by flavone-8-hydroxylase,11 steroid metabolism,12 guaiacol O-demethylation,13 several steps in the biosynthesis of shellfish toxin saxitoxin,14 paralytic shellfish toxin and azomycin biosynthesis.15–16
A soil bacterium Sinorhizobium meliloti 1021 uses N,N-dimethylproline (stachydrine 1) as a carbon and nitrogen source.17 In the stachydrine catabolic operon (Stc2-Stc3-Stc4, Scheme 1a), Stc2 is a Rieske-type oxygenase and Stc3/Stc4 pair are the Stc2 reductase pair. In many Rieske-type oxygenases, the reductase and ferredoxin are fused into one-protein.18 Our recent crystallographic work19 indicates that Stc2 is a trimer arranged in a head-to-tail fashion (Scheme 1c), similar to other Rieske oxygenases.4 In each Stc2 monomer, the distance between the mononuclear iron and the [2Fe-2S] cluster is ∼46 Å, while the mononuclear iron site in one subunit and the Rieske-type [2Fe-2S] cluster in an adjacent subunit are linked together via Glu201 at a distant of ∼12 Å (Scheme 1d).19 In Rieske oxygenases, cooperation between subunits is needed for the efficient electron transfer and catalysis. Genetic studies suggest that Stc3 and Stc4 are the Stc2 reductases and the Stc2-Stc3-Stc4 complex catalyze the N-demethylation of stachydrine to N-methylproline.17, 19 In this process, NAD(P)H and stachydrine each provide two electrons in an overall four-electron O2 reduction process.
Scheme 1. The Stc2-catalyzed stachydrine N-demethylation reaction.
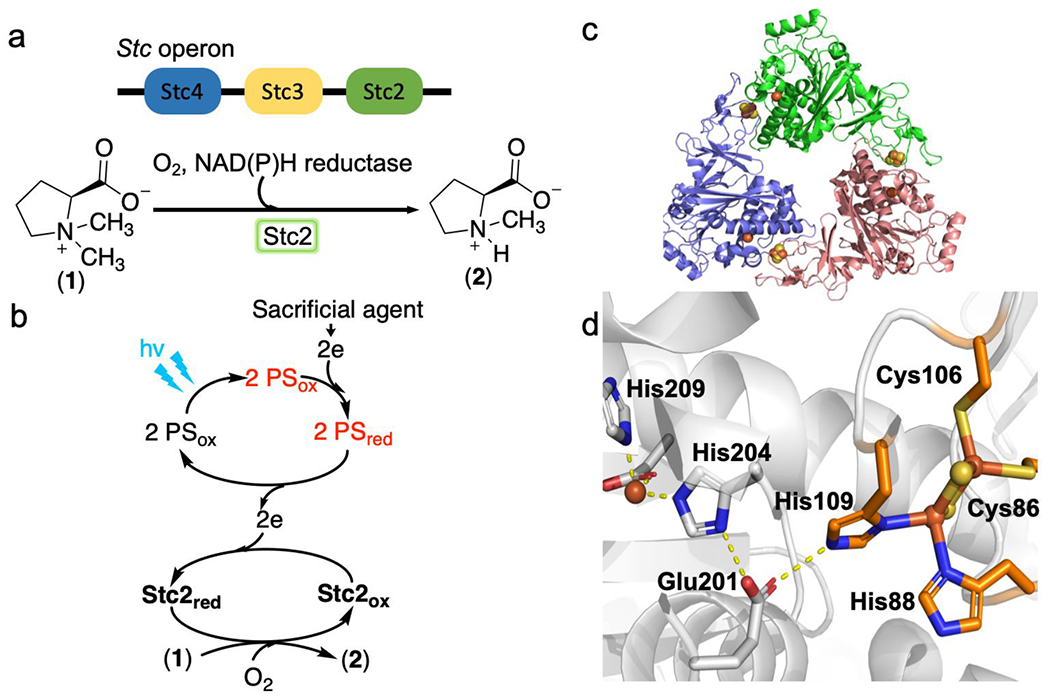
a), The Stc operon from S. meliloti 1021 and the proposed Stc2-catalyzed N-demethylation reaction using its biological reductases Stc3 and Stc4. b), Stc2-catalyzed N-demethylation reaction using a photo-reduction system with the help of photo-sensitizers (PS). c), The overall structure of Stc2 trimer. d), Stc2 active site, including its mononuclear non-heme iron site, the Rieske-type [2Fe-2S]2+ cluster on an adjacent subunit, and Glu201 used to link them.
In our prior report using dithionite as the reductant, a very low Stc2 demethylase activity was observed (one turnover in hours of incubation time),19 and the low activity might be attributed to a combination of several factors. First, dithionite can compete with the reduced Stc2 for reacting with O2, an oxygen dilemma faced by many oxygenase/oxidases.20–21 Second, Stc2 and dithionite may not have proper interactions, which limits the electron transfer efficiency. To overcome this challenge, in IspG- and IspH-based catalysis reported by us, methylviologen was used as an electron transfer mediator to facilitate the reduction of their [4Fe-4S] clusters by dithionite.22–23 Third, studies in some other enzyme systems suggested that the dithionite reaction products may inhibit enzymatic activity.24 In addition, the Rieske oxygenase reaction mechanism and the related electron transfer and protonation processes remain unclear.
There is a growing interest in the application of oxidase/oxygenase-catalyzed reactions in bioremediation or in the production of value-added products.25–28 Besides obtaining enzymes with a sufficient level of activity, to be economically competitive, two additional issues need to be addressed: 1) Rieske-enzymes require an appropriate reductase or reductase pair to achieve optimal catalytic performance;6, 18 and 2) NAD(P)H is expensive.29 In P450s or flavin-dependent oxygenases/oxidases, at least four methods have been explored to address these challenges. First, NAD(P)H is regenerated by an enzymatic system (e.g., formate dehydrogenase using formate as the sacrificial reagent).30 Second, NADP+ could be reduced electrochemically or photochemically.29, 31 Third, in whole-cell-based biocatalysis, a cell permeable photosensitizer is used to support photo-reduction to regenerate NAD(P)H, reduce the reductases, or directly provide electrons for oxygenases.32 Fourth, it has been reported recently that photo-activated NADH could directly deliver electrons to reduce iron-sulfur clusters,33 which eliminates the need of a reductase system.
Our prior Stc2-stachydrine co-crystallization efforts suggested that there may be other means to achieve non-NAD(P)H based reduction of Stc2.19 Our X-ray crystallographic analysis revealed a Stc2-proline complex, instead of the Stc2-stachydrine complex, attributed to a successful stachydrine demethylation reaction where X-ray radiation exposure during the synchrotron data-collection process provided the electrons needed for Stc2-catalyzed N-demethylation reaction.19 This unexpected result motivated us to explore NAD(P)H-reductase free Stc2 photo-biocatalysis that we present here. After screening a list of photo-sensitizers, sacrificial reagents, and buffer systems, we identified the eosin Y/sodium sulfite photo-reduction pair that bypasses the need of both NAD(P)H and a reductase to support the Stc2-catalyzed stachydrine N-demethylation reaction (Scheme 1b). We further demonstrate that under flow-chemistry settings, the performance of the Stc2-eosin Y/sodium sulfite photo-biocatalytic process outperforms the Stc2-NAD(P)H-reductase system. Finally, we illustrate that in addition to Stc2-catalysis, the eosin Y/sodium sulfite pair could also serve as the photo-reduction system in several other Rieske enzymes and a flavin enzyme.
RESULTS AND DISCUSSION
Stc2 was overexpressed and purified following our previously reported protocol.19 The purified Stc2 had 1.9 ± 0.08 iron and 1.1 ± 0.1 sulfide per protein monomer (Figure S1). Ru-complexes tris-(bipyridine)Ruthenium(II) ([Ru(bpy)3]2+) has been widely used in both synthetic organic chemistry and enzymatic catalysis.34–36 To examine the feasibility of Stc2 photo-biocatalysis, we first examined [Ru(bpy)3]2+ as the photo-sensitizer. Under an anaerobic condition, we mixed 100 μM of Stc2 and 10 μM [Ru(bpy)3]2+ in the presence of 20 mM ascorbate as the sacrificial reagent. The above mixture was illuminated under a 430 nm LED light with a power density of 9.76 mW/cm2 in an anaerobic chamber. The UV-visible spectrum of the solution was monitored over time (Figure 1b and Figure S2). The absorbances associated with the oxidized Stc2 Rieske [2Fe-2S]2+ cluster at 460 nm and 550 nm decreased within the first 15 min of illumination without further changes beyond this time point. The resulting Stc2 UV-visible spectrum closely matches that of dithionite reduced Stc2, implying the reduction of Stc2 iron-sulfur cluster (Figure S3a).
Figure 1. Stc2-catalysis using a [Ru(bpy)3]2+-ascorbate pair as a photo-reduction system.
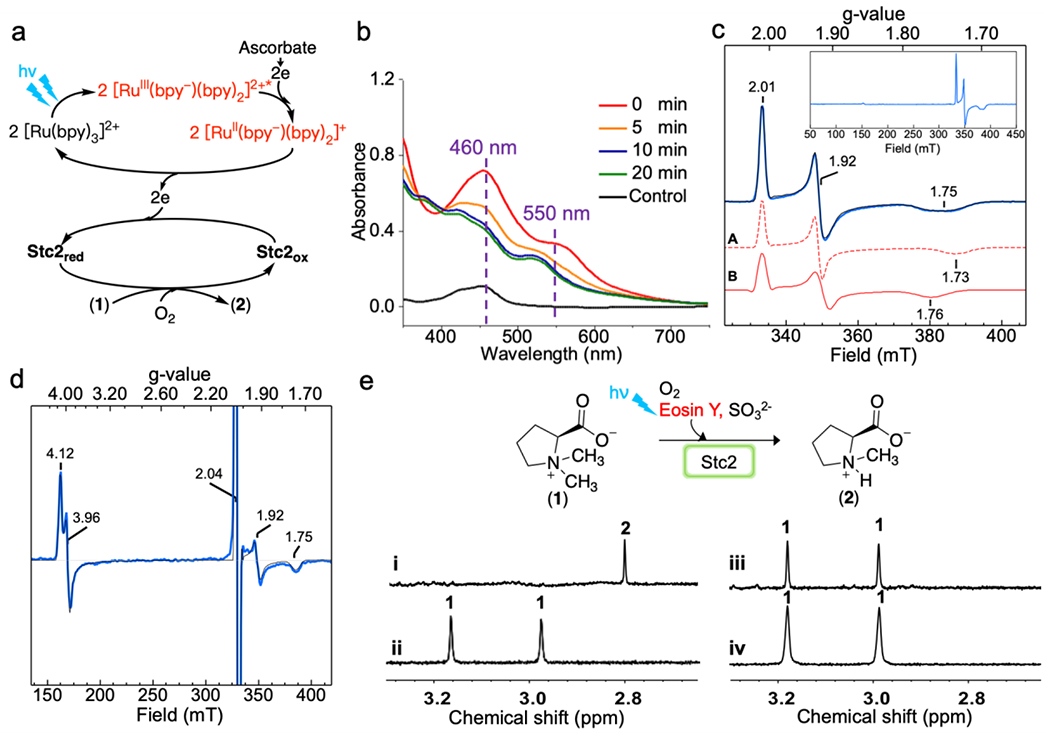
a), Scheme of Stc2-catalyzed N-demethylation reaction with [Ru(bpy)3]2+ as the photo-sensitizer (PS). b), Stc2 reduction time-course monitored by UV-visible spectroscopy. The control used is the [Ru(bpy)3]2+ solution. c), EPR spectrum of reduced Stc2, which contains two species, A and B (red traces). Experimental data (in blue) are overlaid with simulation (black trace). The insert is the full EPR spectrum, showing no other species. d), EPR spectrum of NO treated reduced Stc2. The features near g = 4 correspond to {S = 3/2}7 species, with E/D = 0.015. The signal at g = 2.04 is DNIC, and the g = 1.92 and 1.75 signals are from [2Fe-2S]+. e), 1H-NMR spectra of Stc2-catalyzed stachydrine N-demethylation reaction [N-methyl group signals of the substrate (1) and product (2) in 1H-NMR spectrum], under single turnover condition (trace i); under dark (trace ii); in the absence of Stc2 (trace iii); and under multiple turnover conditions (trace iv) using [Ru(bpy)3]2+ as the photo-sensitizer and ascorbate as the sacrficial reagent.
To provide further support for the successful reduction of the Rieske [2Fe-2S]2+ cluster in Stc2 to its [2Fe-2S]+ state by the [Ru(bpy)3]2+/ascorbate pair under LED light illumination, we characterized the above Stc2 samples using EPR spectroscopy. Specifically, a mixture containing 500 μM Stc2, 500 μM stachydrine, 25 μM [Ru(bpy)3]2+, and 1.5 mM ascorbate in 20 mM Tris-HCl, pH 8.0 buffer, was placed under the same 430 nm LED light source for 1 hour. The reduced Stc2 sample was divided in two. One was frozen directly for EPR analysis without further treatment and the other was treated with NO before EPR analysis. The EPR spectrum of the reduced Stc2 sample (black trace, Figure 1c) was best fit by quantitative simulations using a combination of two species (two red traces, Figure 1c), with g values of 2.01,1.92, 1.73 for species A and g values of 2.01, 1.92, 1.76 for species B. Both species are consistent with the properties of a reduced Rieske [2Fe-2S]+ cluster with 2His-2Cys coordination.37 Non-heme Fe(II) centers are typically EPR silent. To detect the ferrous active site, half of the reduced Stc2 sample was treated with NO (Figure 1d). NO is commonly used in EPR characterization of non-heme iron enzymes as an O2 surrogate,38 and when bound, forms an EPR-accessible {S = 3/2}7 complex (using the Enemark-Feltham notation).39 The EPR spectrum shows a majority of species near g = 4, typical of {S = 3/2}7 species.40–41 The splitting of the two features at 4.12 and 3.96 (with E/D = 0.015) reflects the asymmetry of the complex. Additionally, the signal near g = 2.04 is from the dinitrosyl iron complex (DNIC), which is a common degradation product of [2Fe-2S]1+ in the presence of NO.42–43 The signals near g =1.92 and 1.75 are from the reduced [2Fe-2S]+.44–45 Results from these EPR spectra are consistent with the reduction of the Rieske iron-sulfur cluster to its [2Fe-2S]+ state and the presence of mononuclear non-heme iron center in the Fe2+ state in the reduced Stc2.
After demonstrating the successful reduction of Stc2 by the [Ru(bpy)3]2+/ascorbate pair under LED light illumination, we conducted the stachydrine N-demethylation reaction under both single-turnover and multiple turnover conditions and monitored the reaction by 1H-NMR spectroscopy (Figure 1e). Upon mixing the photo-reduced Stc2 protein-substrate mixture with O2-saturated buffer under single turnover conditions (200 μM Stc2, 200 μM stachydrine, 20 μM [Ru(bpy)3]2+, and 20 mM sodium ascorbate in 20 mM Tris-HCl, pH 8.0), mono-methyl proline was observed (~ 2.82 ppm for the N-methyl group in trace i, Figure 1e), suggesting that the reaction is complete in mixing time (within minutes). In contrast, the reaction required hours to achieve one turnover when dithionite was used as the chemical reductant in our prior report.19 Therefore, the photo-reduction system indeed behaves differently from the chemical reduction system.
To provide further evidence to support that the N-demethylation reaction is due to Stc2 photocatalysis, we conducted two control experiments: a dark reaction and another reaction in the absence of Stc2. The dark reaction mixture contained all of the components except that the mixture was incubated under dark for 30 min. and then mixed with O2 containing buffer in a dark room. Within 5 min., the mixture was quenched by chloroform treatment to denature the Stc2 protein. The aqueous layer was then analyzed by 1H-NMR spectroscopy. No N-demethylation of stachydrine was observed under dark conditions (trace ii, Figure 1e). For the second control experiment, the reaction was conducted under the same conditions as the trace ii reaction in Figure 1e, except that Stc2 protein was excluded. In the absence of Stc2 (trace iii, Figure 1e), the two the N-methyl group signals (~ 3.17 ppm and ~ 2.97 ppm) from stachydrine were observed, while, no signal from the mono-methyl proline (~ 2.82 ppm) was detected. The results in these reactions (trace i – iii, Figure 1e) suggest that the photo-reduced Stc2 indeed is capable of catalyzing stachydrine N-demethylation reaction.
After demonstrating that the [Ru(bpy)3]2+/ascorbate pair could serve as the photo-reduction system to support Stc2-catalysis under single-turnover conditions, we next explored the reaction progress under multiple-turnover conditions. In the multiple turnover reaction, the mixture (2 mM strachydrine, 20 μM Stc2, 100 μM [Ru(bpy)3]2+, and 20 mM of sodium ascorbate in 20 mM Tris-HCl, pH 8.0 buffer) were stirred in an O2 atmosphere while illuminated by a mercury-vapor lamp (125 W with a power density of 25-40 mW/cm2) or a LED lamp. After 1 hour, the reaction mixture was quenched and analyzed by 1H-NMR (trace iv in Figure 1e). Surprisingly, the 1H-NMR spectrum does not show detectable amounts of product formation when the Ru[Ru(bpy)3]2+/ascorbate pair was used under multiple-turnover conditions (trace iv in Figure 1e) .
The single turnover results (Figure 1e) indicate that [Ru(bpy)3]2+/ascorbate pair serves as a photo-reductant for Stc2, and the reduced Stc2 is catalytically active. However, the system performs poorly under multiple-turnover conditions. This discrepancy may be due to several reasons:46 1) the lifetime of the photo-sensitizer activated state may be too short, resulting in poor protein reduction efficiency; 2) after electron-transfer from the photo-sensitizer to the enzyme, the back-electron transfer from the reduced Rieske iron-sulfur cluster [2Fe-2S]+ to [Ru(bpy)3]2+ might be fast, which could also result in a poor Stc2 reduction efficiency; and, 3) the sacrificial reagents may not be appropriate, which competes with the reduced enzyme to react with O2.20–21 To further improve the performance of Stc2 photo-biocatalysis, we screened a list of photo-sensitizers, sacrificial reagents, and buffer systems under both single and multiple turnover conditions, with special emphasis on catalysis under multiple turnover conditions.
The screening comprised varying three main factors (Table S1 for the complete list):31, 36
Photo-sensitizers. We evaluated inorganic nanoparticles and organic dyes, e.g., TiO2, Zinc porphyrin, [Ru(bpy)3]2+, eosin Y, rose B, and rhodamine B.
Sacrificial reductants. We examined the efficacy of commonly used sacrificial reagents, including ethylenediaminetetraacetic acid (EDTA), tris(hydroxymethyl)aminomethane (Tris), ascorbate, formic acid, dithionite, methionine, sodium sulfite, 2(N-morpholino)ethanesulfonic acid (MES), 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), and 3-(N-morpholino)propanesulfonic acid (MOPS).47–48 We have also examined another reagent that had not been previously examined, sodium sulfite.
Electron mediators. We also tested redox mediators commonly used in photo-reduction systems, e.g., methyl viologen, TiO2, CdSe/ZnS quantum dots, Ir(ppy)3, Ir(pCF3-ppy)3, [Ir(dtbbpy)(ppy)2]PF6, [Ir{dF(CF3)ppy}2(dtbpy)]PF6, eosin Y, 5-carboxyeosin, and 5-maleido-eosin Y.
Three representative 1H-NMR spectra for each variable listed above are shown in Figure 2 and more details are in Table S1 and Figure S4–6. Due to the difficulties faced in the [Ru(bpy)3]2+/ascorbate pair in achieving multiple-turnover, in these screening efforts, we focused on Stc2 photo-biocatalysis under multiple-turnover assay conditions. As shown in Figure 2, among the combinations we screened, the eosin Y/sodium sulfite pair has the best performance (trace iv, Figure 2). Further addition of titanium dioxide mediator only leads to a slightly better performance. Stc2 reduction progress using the eosin Y/sodium sulfite pair mediated photo-reduction was then monitored (Figure 3b). Using only 0.3 μM eosin Y, which is 0.3% relative to the Stc2 concentration, Stc2 was fully reduced after 40 min. In the presence of an excess amount of sodium sulfite, with 0.15%, 0.3%, 0.42%, and 0.6% eosin Y load relative to Stc2 concentration, we have measured the Stc2 reduction rate. Reduction progress was monitored by UV-visible spectroscopy at various time points. Reduction kinetics are calculated based on the decrease in 460 nm and 550 nm absorption features associated with the oxidized [2Fe-2S]2+ cluster in Stc2 (Figure 3b, 3c, and Figure S7). While [Ru(bpy)3]2+ in Figure 1 was present in 5-10% molar equivalents of Stc2 concentration, as little as ~0.3% molar equivalents of eosin Y relative to Stc2 could catalyze complete photo-reduction Stc2 (Figure 3b) and analysis of the reduction traces give a Stc2 photo reduction 2nd order reduction rate constant k of ~0.17 min−1•μM−1, which confirms the eosin Y/sulfite pair’s efficacy as a Stc2 photo-reduction system.
Figure 2. 1H-NMR Screening of the Stc2 photo-reduction system components under multiple turnover conditions.
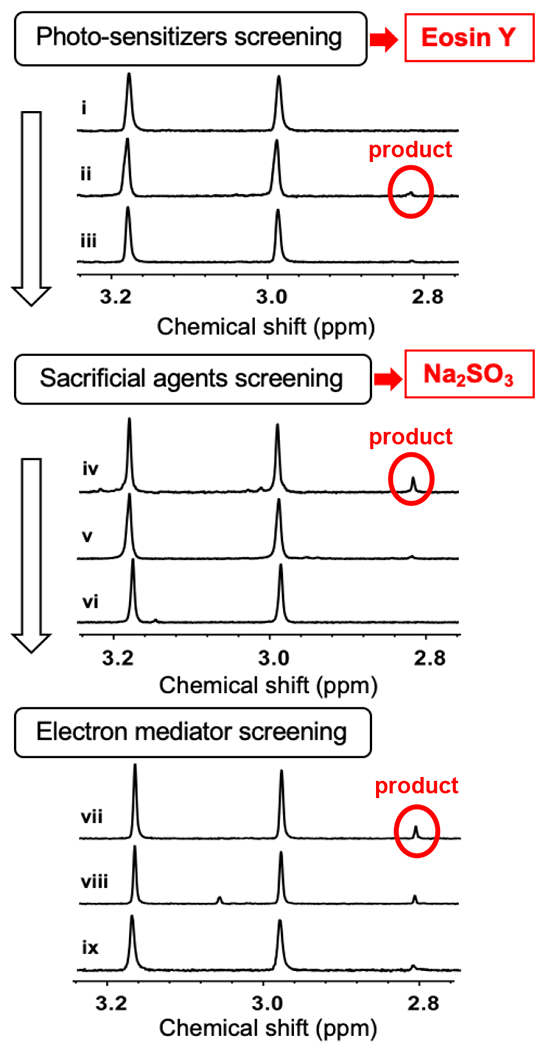
Photo sensitizer, sacrificial reagent, and electron mediators were screened in a sequential manner. First, photo-sensitizers were screened using sodium ascorbate as a sacrificial reagent in Tris-buffer (e.g., zinc porphyrin in trace i, eosin Y in trace ii, [Ru(bpy)3]2+ in trace iii). Second, with eosin Y as the photosensitizer, sacrificial reagents were screened (e.g., Na2SO3 in trace iv; Na2S2O4 in v; and formic acid in vi). Third, using eosin Y/Na2SO3 pair, additional electron mediators were screened (e.g., titanium dioxide in trace vii; CdSe quantum dots in viii; and methyl viologen in ix). More complete list is in Table S1.
Figure 3. Eosin Y/sodium sulfite photo-reduction system for Stc2 reaction.
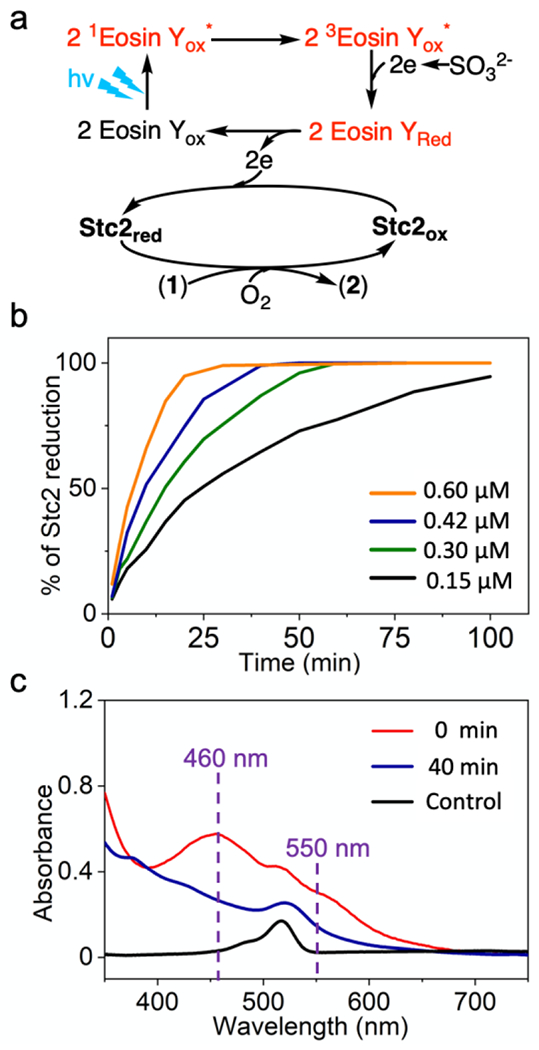
a), Scheme of Stc2-catalyzed N- demethylation reaction with eosin Y as the photo-sensitizer and sulfite as the sacrificial reagent. b), Time course of Stc2 reduction under various eosin concentrations. The reaction mixture contained 2 mM stachydrine, 100 μM Stc2, 0.15 – 0.6 μM of eosin Y, and 20 mM sodium sulfite in 20 mM Tris, pH 8.0 buffer. c), Stc2 reduction time-course monitored by UV-visible spectroscopy. The UV spectra were collected at 0 min (red trace) and 40 min (dark blue trace). The same mixture without Stc2 is set up as control (black trace, with 0.3 μM of eosin Y) to show the change of UV-visible spectra during the photoreduction process.
After the quantitative analysis of the Stc2 reduction rate under various eosin Y concentrations (Figure 3), we re-evaluated Stc2 catalysis under multiple-turnover conditions. At a Stc2:eosin molar ratio of 1:0.5, a turnover number of ~20-30 was observed (Figure 4b and Figure S8). In these multiple turnover reactions, to quantitatively measure the concentrations of stachydrine and N-methylproline, a known concentration of ethylviologen was added into the NMR samples to serve as an internal standard. Achieving a turnover number of ~20-30 under multiple turnover conditions is a significant improvement in comparison to the results in our prior report, where dithioniate was used as the reductant and one turnover could take a few hours.19
Figure 4. Further optimization of Stc2 photo-biocatalysis and a comparative characterization between the Stc2-photo-biocatalysis and the Stc2-NAD(P)H-reductase system.
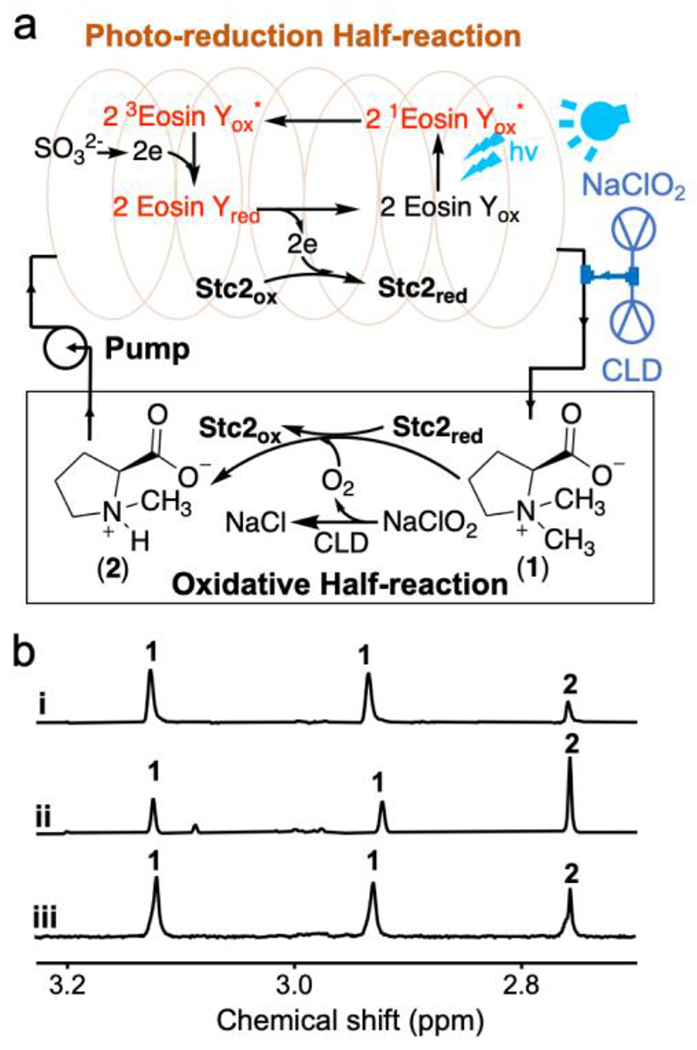
a), Flow-chemistry setting in Stc2-catalysis by separating the photo-reduction half-reaction and the oxidation half-reaction. To address the O2 delivery challenge in flow-chemistry, chlorite dismutase (CLD) mediated in situ O2 production was further introduced into Stc2-catalysis. b), a representative 1H-NMR spectrum [the methyl group signals for substrate (1) and product (2) in 1H-NMR] of Stc2 reaction under the multiple turnover reaction condition using eosin Y/Na2SO3 photo-reduction system (trace i) in batch reaction; under an optimized eosin Y concentration flow-chemistry setting, which further improved the Stc2 turnover number to ~65-70 (trace ii); and Stc2 reaction using GbcB as the reductase and NADH as the reductant (trace iii).
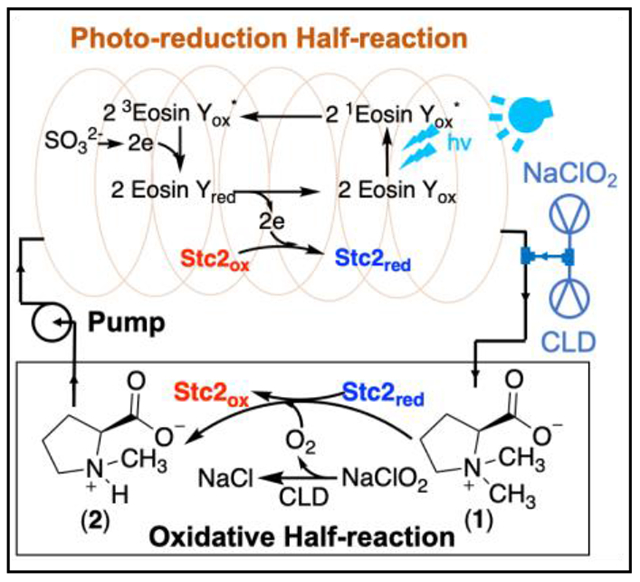
While the [Ru(bpy)3]2+/ascorbate pair reported in Figure 1 supports Stc2-catalysis under single-turnover conditions, its performance is poor under multiple-turnover conditions. In the prior sections, we suggested possible attenuating factors:46 1) the lifetime of the activated state of the photo-sensitizer; 2) the reduction rate of the activated photo-sensitizer by the sacrificial reagent, 3) the electron transfer between [Ru(bpy)3]+ and Stc2; 4) the competition between various reductants and reduced Stc2 for reacting with O2 (the O2 dilemma referred in literature).20–21 Screening of photo-sensitizers, sacrificial reagents, and buffer systems under both single and multiple turnover conditions possibly addressed reasons 1 – 3 and improved the turnover numbers (e.g., Stc2 reaction under batch reaction conditions shown in trace i, Figure 4b). In addition, it has been reported in literature that the photo-oxidation product of sulfite is sulfate,49 which may not affect the enzyme activity. However, the O2 dilemma has not been directly addressed yet. One potential solution is to separate the photo-reduction half-reaction and the oxidation half-reaction. To address this need, we incorporated flow-chemistry50 into Stc2 photo-biocatalysis (Figure 4a).
The general setting is shown in Figure 4a. The system was split into two halves: light reaction (Stc2 photoreduction) and dark-reaction (Stc2 catalysis). The reaction mixture is circulated between the two halves driven by a peristaltic pump. The reaction mixture (20 μM Stc2, 2 mM stachydrine, 20 μM eosin Y, and 20 mM sodium sulfite in 20 mM Tris-HCl, pH 8.0) was placed in the reservoir. The solution was pumped into the PFA tube, which was illuminated by either the LED light or by the white light. For the peristaltic pump used in this experiment, its maximal capacity is 6 mL/min. The volume of the PFA tube exposed to light is 3 mL. Therefore, the photoreduction half-reaction time is ~ 30 seconds. Based on the photoreduction rate constant of 0.17 min−1•μM−1 determined previously (Figure 3), a residence time of 30 seconds in the PFA tube is long enough to reduce most Stc2 for catalysis.
To apply flow-chemistry in Stc2-catalysis, another challenge is in the oxidation half-reaction, where O2 (gas) is the oxidant. In gas-liquid reactions in synthetic organic chemistry under flow-chemistry settings, the gas is delivered into microreactors to produce segmented flow (also called slug flow or Taylor flow). The slug-flow setting could significantly increase gas-liquid interface to improve the reaction performance.51 Unfortunately, when we applied this method to deliver O2 or air into the Stc2 reaction mixture, foaming of the Stc2 reaction mixture caused protein inactivation. To address the O2 delivery issue in photo-biocatalytic flow-chemistry setting, we decided to generate O2 in situ by taking advantage of the reaction catalyzed by chlorite dismutase (CLD).52 CLD catalyzes the decomposition of chlorite to chloride and O2 with kcat as high as 2 × 104 s−1. Such a high kcat for chlorite dismutase ensures that when ClO2− solution is introduced into a CLD-containing solution, molecular oxygen (O2) will be produced in situ almost instantaneously. In this new design, the mixture is reduced in the PFA tube under LED light illumination in the photo-reduction half, and chlorite solution and CLD are introduced to the reduced solution through another pump to generate O2 in situ to initiate the oxidation half reaction. In this setting, the photo-reduction and oxidation half reactions were separated (Figure 4a). For example, in the Figure 4b trace ii experiment, the CLD/chlorite ratio used is 1:1200 and the chlorite delivery rate is from 7.2 μmol/h to 2.0 μmol/h, which matches with the Stc2 circulation rate in the reaction mixture to minimize the production of O2 in excess. Under these conditions, we can successfully further improve the turnover numbers to 65 – 70 in at least three replicates (1H-NMR of the reaction in trace ii, Figure 4b). In the current setting, the turnover rate and turnover numbers are limited by the peristaltic pump-controlled solution circulation rate. Based on the results from our current settings, by adjusting the photo-reduction time (tube length to minimize photo-bleaching) and the peristaltic pump-controlled circulation rate, the performance of this photo-biocatalytic system has the potential to be further optimized.
For mononuclear non-heme iron enzymes, it is common to have self-inactivation after certain number of turnovers. Due to this reason, a total turnover number of ~100 or less is common.53–54 This is certainly true for Stc2 too and it is also possible that the turnover number for Stc2 in the flow-chemistry setting reported in Figure 4b is close to the maximal capacity already. To provide a benchmark for quantitative evaluation of the performance of this photo-reduction system, we further characterized Stc2-catalysis using an NAD(P)H-reductase system. Because the expression of the two Stc2 reductases (Stc3 and Stc4) is problematic, we turned our focus to another Rieske-enzyme, glycine betaine N-demethylase, which also catalyzes an oxidative demethylation reaction.55 In contrast to the Stc2 system, the glycine betaine N-demethylase has only one reductase and has been successfully overexpressed. We cloned and overexpressed glycine betaine N-demethylase reductase (GbcB) from P. aeruginosa. We then examined the Stc2 reaction under multiple turnover conditions using NADH-GbcB as the reduction system. The quantitative analysis of the NMR result in trace iii, Figure 4b indicate that the turnover number for this enzymatic reduction system is ~35-40. The results (trace ii vs trace iii, Figure 4b) demonstrate that using the eosin Y/sodium sulfite pair photo-catalytic system in flow-chemistry setting, Stc2 photo-biocatalysis is at a level comparable or outperforms the NAD(P)H-reductase system.
With the success in Stc2 photo-biocatalysis, the other question is whether eosin Y/sodium sulfite pair can be applied to other enzymes. We further examined this photo-reduction system in a few other Rieske enzymes, including hydroxylation reaction catalyzed by carnitine monooxygenase (CntA),56–57 caffeine N-demethylation catalyzed by NdmA,58–59 and glycine betaine N-demethylation by GbcA.55 Beside iron-sulfur clusters, flavin is another common cofactor for redox-chemistries or for electron-transfer processes. Therefore, we also selected a flavin-dependent enzyme, iodotyrosine deiodinase (IYD),60–62 to test the potential application of the eosin Y/sodium sulfite pair in flavin-enzyme photo-biocatalysis. These genes were synthesized by Genscript. The codon-optimized genes were sub-cloned into IBA-pASK-3plus vector. For the Rieske enzymes, their overexpression and purification protocols were similar to those used in Stc2 overexpression (Supplementary method and Figure S9). For IYD, the codon optimized gene was sub-cloned into pET28a-(+) vector, and overexpressed in E. coli with supplementation of 0.1 mM of riboflavin in the culture media (Supplementary method and Figure S9), and then purified by Ni-NTA affinity chromatography. We tested the activity of these purified proteins using the eosin Y/sodium sulfite pair under multiple turnover conditions in batch reactions in a manner similar to Stc2 (Figure 2). In batch conditions, the eosin Y/sodium sulfite pair could support these photo-biocatalytic transformations (Figure 5). For the NdmA reaction, the reactions were analyzed by HPLC (Figure 5a). To further confirm the identities of the products, the corresponding fractions were collected and then characterized by high resolution mass spectrometry (Figure S10–11). Consistent with literature reports,58–59, 63–64 the eosin Y/sulfite pair does support NdmA-catalysis. For the CntA-catalysis, GbcA-catalysis and IYD-reaction, we directly monitored the reaction by 1H-NMR because the methyl group in CntA, GbcA substrates and the aromatic hydrogens in the IYD substrate have distinct chemical shifts and do not overlap with the buffer or protein signals. 1H-NMR clearly support the success of CntA- (Figure 5b and Figure S12), GbcA- (Figure 5c and Figure S13) and IYD-catalysis (Figure 5d and Figure S14). The reaction product assignments were further confirmed by spiking the NMR samples with the product standards.
Figure 5. Additional examples of eosin Y/sodium sulfite pair supported photo-biocatalytic transformations.
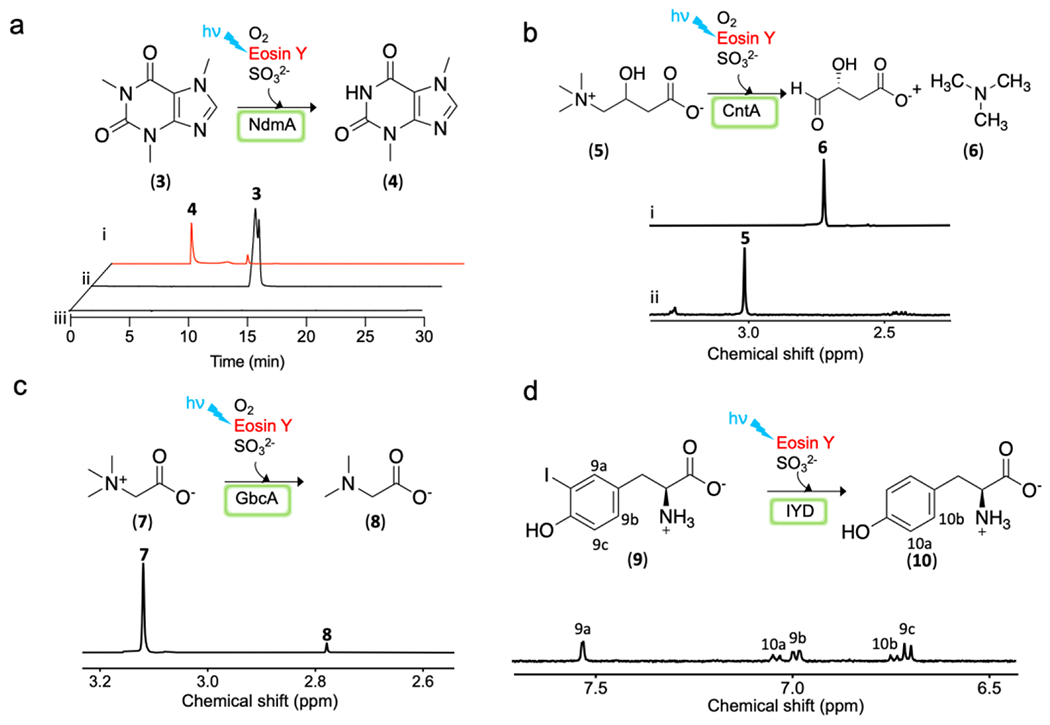
a), Reaction scheme and HPLC profile of NdmA reaction, under multiple turnover condition (trace i), a control without protein (trace ii) and the 2nd control without substrate (trace iii). b), Reaction scheme and 1H-NMR analysis of CntA reaction under single turnover condition (trace i) and a control without protein (trace ii) [methyl signals for the substrate (5) and the product (6) in 1H-NMR spectrum]. c), Reaction scheme and 1H-NMR analysis of GbcA reaction [methyl signals for the substrate (7) and the product (8) in 1H-NMR spectrum]. d), Reaction scheme and 1H-NMR analysis of IYD reaction (aromatic hydrogen signals for the substrate (9) and product (10)].
For Rieske oxygenases, especially for those that are involved in the degradation of aromatic compounds,6–7 they do have some level of flexibility in accepting alternative substrates. S. meliloti 1021 is a soil bacterium, using N,N-dimethylproline (stachydrine 1) as a carbon and nitrogen source.17 In order to make use of stachydrine as a carbon and nitrogen source, Stc2 might have evolved to improve the substrate specificity. After the establishment of the Stc2 photo-biocatalytic system under multiple turnover conditions, we have also selected a few substrate analogs for characterization to test Stc2 substrate flexibility. Besides stachydrine, Stc2 does accept glycine betaine as an alternative substrate, producing di-methylglycine as the product (Figure S15). When histidine betaine (trimethylhistidine or hercynine) was used as the substrate, no detectable activity was observed. For the other Rieske enzymes listed in Figure 5, none of their substrates could be accepted by Stc2 as an alternative substrate either. Therefore, consistent with the capacity of S. meliloti 1021 to use stachydrine as the carbon and nitrogen source, Stc2 might have been evolved to increase its specificity for stachydrine, which limits Stc2’s capacity to use alternative substrates.
CONCLUSIONS
In this work, using Stc2-catalyzed stachydrine N-demethylation as the model system, we describe a NAD(P)H- and reductase-free photo-reduction system (eosin Y-sulfite pair). Under multiple turnover conditions, the performance of this photo-reduction system is comparable to the native NAD(P)H-reductase system. By further introducing the flow-chemistry setting to separate the photo-reduction half-reaction and the oxidation half-reaction, we further improved the performance of Stc2 photo-biocatalystic system. In our flow-chemistry setting, there might be a small amount of unreacted O2 left in the photo-reduction half-reaction. However, Stc2-eosin Y/sodium sulfite already works better than the batch reaction and the Stc2-NAD(P)H-reductase system. These results highly suggest that the flow-chemistry setting could partially address the O2 delivery issue. In a few other selected Rieske enzymes (NdmA, CntA, GbcA), and flavin-dependent enzyme (IYD), the eosin Y/sodium sulfite pair could also serve as a surrogate for their reductases or reductants. Recently, cell permeable photosensitizers have been examined in supporting Rieske-type oxygenase activity in whole-cell systems.65 Eosin Y is cell permeable and sulfite is a common metabolite. Therefore, it may also worth exploring this photo-reduction system in whole-cell catalysis. With the growing interest in applying oxidase/oxygenase catalyzed reactions in bioremediation and in biocatalysis for the production of value-added products, the eosin Y-sulfite pair photo-reduction might be useful in these applications.
EXPERIMENTAL SECTION
Materials:
Reagents and photosensitizers were purchased from Sigma-Aldrich. Stachydrine was synthesized based on the previously published protocol.19 Proton nuclear magnetic resonance spectra were recorded using an Agilent 500 NMR (500 MHz VNMRS). Chelex-100 resin was purchased from Bio-Rad laboratory. The Iron content was quantified by MP AES atomic emission spectroscopy (Agilent).
Cell growth and purification of Stc2.
Stc2 gene sequence was codon optimized for Escherichia coli by Genscript and sub-cloned into the IBA-pASK-3plus vector, with EcoRI and XhoI restriction sites. Competent cell pDB1281/BL21 was transformed with the plasmid and a single colony was inoculated into 50 mL LB media supplemented with 50 μg/mL kanamycin and 100 μg/mL ampicillin resistance at 37 °C overnight. Then, 10 mL of seed culture was used to inoculate 1 L of LB media supplemented with 50 μg/mL kanamycin and 100 μg/mL ampicillin at 37 °C. When OD600 nm reached 0.2, arabinose was added to 5g/L final concentration. At OD600 nm = 0.6 ~ 0.8, protein overexpression was induced with anhydrous tetracycline to 0.5 mg/L final concentration. The protein was overexpressed at 25 °C for 14 hours. The cell was collected by centrifugation. The protein purification process was performed inside an anaerobic glove box (Coy Laboratory Products). 9 g of Stc2 cells were suspended in 50 mL of anaerobic lysis buffer (100 mM Tris-HCl, 150 mM NaCl, pH 8.0). 45 mg of lysozyme was added into the cell suspension, which was incubated on ice for 30 min with shaking. Before sonication, sodium ascorbate and ferrous ammonium sulfate were added to 0.4 mM and 0.2 mM (respectively) final concentrations. The cell suspension was disrupted by sonication (505 Sonic Dismembrator). Supernatant and cell debris were separated by centrifugation at 19500 rpm, 4 °C for 45 min. The supernatant was loaded to 20 mL of strep-tag resin and shaken on ice for 40 min. The flow through was collected and the resin was washed by lysis buffer and the target protein was eluted by lysis buffer supplemented with 2.5 mM desthiobiotin. The Stc2-containing fractions were combined and concentrated by ultrafiltration with 10 kDa membrane. The concentrated protein was frozen by liquid nitrogen and stored at −80 °C freezer. The purity of protein purified is analyzed by SDS-PAGE analysis.
Iron content quantification of Stc2.
20 μL of 0.90 mM Stc2 was diluted by adding 980 μL 2 % (w/w) HNO3. The protein precipitates were removed by centrifugation at 16000 rpm for 10 min. Iron standards (0.125 ppm to 2 pm) were made by diluting 1000 ppm iron stock solution by 2 % (w/w) HNO3 and used to make a calibration curve. The iron content was analyzed by atomic emission spectroscopy. The iron content was calculated based on the protein concentration, iron concentration, and sample dilution factor.
Stc2 sulfur quantification of Stc2.
A previously reported protocol was followed.66 200 μL of 90 μM Stc2 was added to a 2 mL plastic tube with stir bar. 600 μL of 1% zinc acetate was added and 30 μL of 12 % NaOH was added immediately with stirring until the solution was well mixed. After the solution was sat for 1 hour, 150 μL of 0.1 % N,N-dimethyl-p-phenylenediamine solution was added to the mixture and stir gently. Then, 30 μL of 23 mM FeCl3 was added. The precipitates were removed by centrifuge at 15500 rpm for 20 min. A670 nm, A710 nm, and A750 nm were collected and A670 nm was used for analysis, with ε670 nm = 34.6 cm−1•mM−1. Based on this analysis, the labile sulfur content was quantified as 1.1 ± 0.1.
Stc2 single turnover reaction using sodium dithionite as the reductant.
A 0.5 mL reaction with 200 μM Stc2, 200 μM stachydrine, 400 μM sodium dithionite in 20 mM Tris-HCl, pH 8.0 buffer was mixed in the anaerobic gloves box and incubated on ice for 10 min. Then, the reaction mixture was removed out of the anaerobic box and connected to an oxygen balloon. The reaction was stirred in air at room temperature for 2 hours. The protein precipitated was quenched by adding 300 μL chloroform and vortex for 5 min. The protein precipitates were removed by centrifugation at 15500 rpm for 10 min. The water layer was collected and lyophilized. The sample was dissolved in 300 μL of D2O. The product formation was analyzed by 1H-NMR.
We used ethyl viologen as an internal standard to normalize sample concentrations. Turnover numbers could be calculated by the relative intensities of the signals for N-methyl groups in staychydrine vs mono-methyl proline. This is possible because the methyl group signals for stachydrine and mono-methyl proline are well-separated from all of the other signals of the reaction mixture.
Stc2 single turnover reaction using [Ru(bpy)3]2+-sodium ascorbate photo-reduction system.
A 0.25 mL reaction mixture contained 400 μM Stc2, 400 μM stachydrine, 40 μM [Ru(bpy)3]2+, and 40 mM sodium ascorbate in 20 mM Tris-HCl, pH 8.0 buffer in the anaerobic gloves box and incubated on ice for 10 min. The sample was placed under 430 nm LED light with a power density of 9.76 mW/cm2 in the anaerobic box for 1 hour. Then, the reduced mixture was taken out of the gloves box and mixed with 0.25 mL of oxygenated 20 mM Tris-HCl, pH 8.0 buffer. The reaction was stirred at the room temperature for 5 min. The protein was quenched and precipitated by adding 300 μL chloroform and vortexed for 5 min. The protein precipitates were removed by centrifugation at 15500 rpm for 10 min. The aqueous layer was collected and lyophilized. The sample was dissolved in 300 μL of D2O. The product formation was analyzed by 1H-NMR.
Screening of photo-sensitizers of Stc2-reaction under multiple turnover conditions.
To identify the suitable photosensitizers with improved turnover number for Stc2, multiple photo-sensitizers were tested. One typical reaction was composed of 0.25 mL mixture with 40 μM Stc2, 4 mM stachydrine, 40 mM sodium ascorbate, 200 μM photosensitizers ([Ru(bpy)3]2+, eosin Y, rhodamine B, rose bengal or Zinc- porphyrin) or 1 mg of solid form of photosensitizers (TiO2, or mesoporous graphitic carbon nitride (mpg-CN) in 20 mM Tris-HCl, pH 8.0 buffer. The mixture was incubated on ice for 10 min in the anaerobic gloves box. Then, the reaction mixture was transferred out of the gloves box and quickly mixed with 0.25 mL oxygenated, 20 mM Tris-HCl, pH 8.0 buffer. The reaction mixture was stirred at room temperature for 1 hour under the bright mercury-vapor lamp light (125 W, 25-40 mW/cm2.). Ethyl viologen was added to 1 mM final concentration as an internal standard. The reaction was quenched by adding 300 μL chloroform and vortex for 5 min. The protein precipitates were removed by centrifugation at 15,500 rpm for 10 min. The water layer was collected and lyophilized. The sample was dissolved in 300 μL of D2O. The product formation was analyzed by 1H-NMR.
Screening of sacrificial reagents of Stc2-reaction under multiple turnover conditions.
One typical reaction was composed of 0.25 mL mixture with 40 μM Stc2, 4 mM stachydrine, 40 mM sacrificial reagents (sodium ascorbate, Na2SO3, methionine, Na2S2O4, diethyldithiocarbamate, triethanolamine, EDTA, MES, HEPES, fomic acid, NaHPO3 and Na2S), 200 μM eosin Y in 20 mM Tris-HCl, pH 8.0 buffer. The mixture was incubated on ice for 10 min in the anaerobic gloves box. Then, the reaction mixture was transferred out of the glove box and quickly mixed with 0.25 mL oxygenated 20 mM Tris-HCl, pH 8.0 buffer. The reaction mixture was stirred at room temperature for 1 hour under the bright mercury-vapor lamp light. Ethyl viologen was added to 1 mM final concentration as an internal standard and the reaction was quenched by adding 300 μL chloroform and vortex for 5 min. The protein precipitates were removed by centrifugation at 15,500 rpm for 10 min. The water layer was collected and lyophilized. The sample was dissolved in 300 μL of D2O. The product formation was analyzed by 1H-NMR.
Screening of electron mediators of Stc2 reaction under multiple turnover conditions.
One typical reaction was composed of 0.25 mL mixture with 40 μM Stc2, 4 mM stachydrine, 40 mM Na2SO3, 4 mM methyl violgen or 0.1 mg of other electron mediators (TiO2, CdSe/ZnS quantum dots, Ir(ppy)3, Ir(p-CF3-ppy)3, Ir(dtbbpy)(ppy)2 and (Ir[dF(CF3)ppy]2(dtbpy))PF6), 200 μM eosin Y in 20 mM Tris-HCl, pH 8.0 buffer. The mixture was incubated on ice for 10 min in the anaerobic glove box. Then, the reaction mixture was transferred out of the gloves box and quickly mixed with 0.25 mL oxygenated 20 mM Tris-HCl, pH 8.0 buffer. The reaction mixture was stirred at room temperature for 1 hour under the bright mercury-vapor lamp light. Ethyl viologen was added to 1 mM final concentration as an internal standard. The reaction was quenched by adding 300 μL chloroform and vortex for 5 min. The protein precipitates were removed by centrifugation at 15,500 rpm for 10 min. The water layer was collected and lyophilized. The sample was dissolved in 300 μL of D2O. The product formation was analyzed by 1H-NMR.
Screening of substrate selectivity of Stc2 reaction under multiple turnover conditions.
One typical reaction was composed of 0.25 mL mixture with 300 μM Stc2, 3 mM substrates (glycine betaine, histidine betaine, L-carnitine, caffeine, 3-iodo-L-tyrosine), 40 mM Na2SO3, 150 μM eosin Y in 20 mM Tris-HCl, pH 8.0 buffer. The mixture was incubated on ice for 10 min in the anaerobic gloves box. Then, the reaction mixture was transferred out of the gloves box and quickly mixed with 0.25 mL oxygenated 20 mM Tris-HCl, pH 8.0 buffer. The reaction mixture was stirred at room temperature for 1 hour under the bright mercury-vapor lamp light. Ethyl viologen was added to 1 mM final concentration as an internal standard. The reaction was quenched by adding 10 μL 6 M hydrochloric acid and vortex for 5 min. The protein precipitates were removed by centrifugation at 15,500 rpm for 10 min. The water layer was collected and lyophilized. The sample was dissolved in 300 μL of D2O. The product formation was analyzed by 1H-NMR.
Stc2 flow-chemistry.
A 5 mL reaction mixture with 20 μM Stc2, 2 mM stachydrine, 20 μM eosin Y and 20 mM Na2SO3 in 20 mM Tris-HCl, pH 8.0 buffer was placed in a reservoir as the starting solution. Set up the flow system as shown in Figure 4b. The reaction mixture was circulated between the two halves driven by a peristaltic pump. The solution was pumped into a 3mL PFA tube, which was illuminated by either the LED light or by the white light. The peristaltic pump used in this experiment, operated at a flow rate of 6 mL/min. Therefore, the photoreduction half-reaction time is ~ 30 seconds before exiting the PFA tube. 5μM CLD in 20 mM Tris-HCl, pH 8.0 buffer and 1.25 mM NaClO2 in 20 mM Tris-HCl, pH 8.0 buffer were prepared separately and transferred into a sealed 10 mL syringe for delivering. The CLD/chlorite ratio is: 1:1200 and the chlorite delivery rate is from 7.2 μmol/h to 2.0 μmol/h which matches with the Stc2 circulation rate in the reaction mixture. The CLD solution was mixed with the sodium chlorite solution first and then mixed with the reduced Stc2 reaction mixture. In some reactions, we supplement some additional eosin Y to compensate the loss of eosin Y due to photo-bleaching. At the end of the reaction, the mixture was lyophilized and dissolved in 1 mL D2O and analyzed by 1H-NMR.
Stc2 reaction under multiple turnover conditions using NADH-GbcB as the reduction system.
A 0.25 mL mixture with 40 μM Stc2, 4 mM stachydrine, 40μM GbcB, 40μM FAD and 6mM NADH in 20 mM Tris-HCl, pH 8.0 buffer. The mixture was incubated on ice for 10 min in the anaerobic gloves box. Then, the reaction mixture was transferred out of the gloves box and quickly mixed with 0.25 mL oxygenated 20 mM Tris-HCl, pH 8.0 buffer. The reaction mixture was stirred at room temperature for 1 hour. Ethyl viologen was added to 1 mM final concentration as an internal standard and the reaction was quenched by adding 300 μL chloroform and vortex for 5 min. The protein precipitates were removed by centrifugation at 15,500 rpm for 10 min. The water layer was collected and lyophilized. The sample was dissolved in 300 μL of D2O. The product formation was analyzed by 1H-NMR.
Stc2 photo-reduction kinetics using [Ru(bpy)3]2+ and sodium ascorbate as the photo-reduction system.
1 mL reaction mixture with 100 μM Stc2, 2 mM stachydrine, 20 mM sodium ascorbate, 10 μM [Ru(bpy)3]2+ in 20 mM Tris-HCl buffer was mixed in the anaerobic box and sealed in the air-sealed quartz cuvette. The reaction mixture was incubated under the 430 nm LED light with a power density of 9.76 /cm2. The UV scan at different time points from 350 nm to 700 nm was collected by Cary UV-vis spectrometer.
Stc2 photo-reduction kinetics using eosin Y and Na2SO3.
1 mL reaction mixtures contained 2 mM of stachydrine, 100 μM Stc2, 20 mM sodium sulfite, and eosin Y as the photo-sensitizer at four different concentrations (0.15 μM, 0.3 μM, 0.42 μM, and 0.6 μM). These reaction mixtures were placed under the mercury lamp’s white light in an air-sealed quartz cuvette. The UV scan at different time points from 350 nm to 700 nm was collected by Cary UV-vis instrumentation. The reduction progress was moni-tored by collecting UV-visible spectra at various time points. Reaction progress reflected as a percentage of total Stc2 reduction by monitoring the Electronic Absorption band at 460 nm which is associated with the oxidized [2Fe-2S]2+ cluster in Stc2. Kinetic data were fit using the KinTek Explorer software to directly simulate the processed data and to fit the data to single-exponential curves (Figure 3b, 3c, and Figure S7).
Eosin Y concentration dependence in Stc2 multiple turnover reaction.
After demonstrating that eosin Y could serve as a photo-sensitizer for Stc2 reduction, the ratio between Stc2 and eosin Y was optimized to improve the total turnover number. One typical 0.25 mL reaction was composed of 40 μM Stc2, 40 mM Na2SO3, various concentrations of eosin Y (0.4 μM, 2 μM, 4 μM, 10 μM, 20 μM, 40 μM, 120 μM, 200 μM and 400 μM) in 20 mM Tris-HCl, pH 8.0 buffer. The mixture was incubated on ice for 10 min in the anaerobic gloves box. Then, the reaction mixture was transferred out of the gloves box and quickly mixed with 0.25 mL oxygenated 20 mM Tris-HCl, pH 8.0 buffer. The reaction mixture was stirred at room temperature for 1 hour under the bright mercury-vapor lamp light. The reaction was quenched by adding 300 μL chloroform and vortex for 5 min. The protein precipitates were removed by centrifugation at 15,500 rpm for 10 min. The aqueous layer was collected and lyophilized. The sample was dissolved in 300 μL of D2O. The product formation was analyzed by 1H-NMR.
Preparation of NO-Stc2 sample for EPR analysis.
Standardization of PROLI-NONOate stock solutions: All samples were prepared in a Coy Laboratories anaerobic chamber. First, PROLI-NONOate (Cayman Chemical Company) stock solutions were prepared and standardized following a previously established methodology.67 Briefly, approximately 10 mg PROLI-NONOate were dissolved in 1 mL of deoxygenated 10 mM NaOH on the same day of EPR sample preparation. The NO delivery equivalents of the PROLI-NONOate stock solution were standardized by the reaction of the generated NO with FeIIEDTA at pH 7. A 50 mM stock solution of Fe(NH4)(SO4)2•6H2O was prepared fresh and mixed 1:1 (v/v) with 100 mM EDTA at pH 7. 10 μL PROLI-NONOate solution was mixed with 490 μL of 50 mM HEPES pH 7.0, followed approx. 60 s later by the addition of 500 μL of the EDTA stock solution. The resultant mixture was quantified by measuring the absorbance at 440 nm of the FeIIEDTA-NO complex (ϵ440nm = 900 M−1•cm−1). With the stock solution of PROLI-NONOate standardized, an appropriate volume of stock PROLI-NONOate was diluted in 10 mM NaOH to prepare 1 mL of 5 mM NO(g) equivalents solution. This solution was used for the preparation of the EPR samples.
Stc2 EPR sample preparation: An 1 mL reaction mixture with 0.5 mM Stc2 was mixed with 0.5 mM stachydrine, 25 μM [Ru(bpy)3]2+ and 1.5 mM ascorbate in anaerobic 20 mM Tris-HCl, pH 8.0 buffer was set up in the anaerobic gloves box and incubated under the light for 1 hour. Then the sample was equally split into two portions. One portion was directly aliquoted into an EPR tube for EPR spectroscopy directly. Another portion was treated with PROLI-NONOate to 0.6 mM final concentration. Then the sample was aliquoted to another EPR tube for EPR spectroscopy. The samples were frozen and stored in the liquid nitrogen until the EPR measurement.
EPR Spectroscopy.
X-Band EPR spectra were collected on a Bruker ELEXYS-II E500 EPR spectrometer equipped with a helium continuous flow cryostat model ESR 900 (Oxford Instruments, Inc.). The temperature was controlled with an Oxford Instruments ITC 503 Digital Cryogenic temperature controller. Unless otherwise noted, all spectra were recorded using the following standard parameters: modulation frequency, 100 kHz; modulation amplitude, 10 G; conversion time, 100 ms; time constant 3.6 ms; microwave frequency 9.38 GHz. Microwave power and sample temperature were adjusted to ensure spectra were non-saturating. EPR signals were quantified relative to a 2.78 mM CuIIEDTA standard at pH 4.0 in 10% (v/v) glycerol. The CuII of known concentration was from a commercially-certified AAS standard solution in 2% nitric acid (Sigma Aldrich).
Quantitative spectral simulations were performed using the software SpinCount developed by M.P. Hendrich.68 The simulations were calculated with least-squares fitting using the spin Hamiltonian:
Where D and E are the axial and rhombic zero-field splitting parameters, and B represents the applied magnetic field.
Supplementary Material
ACKNOWLEDGMENT
We thank Prof. Aaron Beeler and his students Sara El-Arid and Jason Lenihan for the help for setting up the flow-chemistry. Prof. Yisong Guo provided the CLD plasmid and protein. We thank the Clare Boothe Luce Award for supporting Jessica C. Murray working on the project.
Funding Sources
This work is supported in part by grants from the National Institute of Health (GM140040 to P. Liu, R35-GM136294 to S.J. Elliott), the National Science Foundation (CHE-2004109 to P. Liu), and the National Natural Science Foundation of China (21931005, 21720102002, and 21673140 to J.Chen).
Footnotes
The authors declare no competing financial interest.
Supporting Information. Supporting material, including protein purification & experimental methods, and supporting data are available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Knapp M; Mendoza J; Bridwell-Rabb J, An aerobic route for C-H bond functionalization: The Rieske non-heme iron oxygenases. 2021; pp 413–424. [Google Scholar]
- 2.Perry C; de los Santos ELC; Alkhalaf LM; Challis GL, Rieske non-heme iron-dependent oxygenases catalyse diverse reactions in natural product biosynthesis. Nat. Prod. Rep 2018, 35 (7), 622–632. [DOI] [PubMed] [Google Scholar]
- 3.Barry SM; Challis GL, Mechanism and Catalytic Diversity of Rieske Non-Heme Iron-Dependent Oxygenases. ACS Catal. 2013, 3 (10), 2362–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferraro DJ; Gakhar L; Ramaswamy S, Rieske business: structure–function of Rieske non-heme oxygenases. Biochem. Biophys. Res. Commun 2005, 338 (1), 175–190. [DOI] [PubMed] [Google Scholar]
- 5.Kovaleva EG; Lipscomb JD, Versatility of biological non-heme Fe(II) centers in oxygen activation reactions. Nat. Chem. Biol 2008, 4 (3), 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kweon O; Kim SJ; Baek S; Chae JC; Adjei MD; Baek DH; Kim YC; Cerniglia CE, A new classification system for bacterial Rieske non-heme iron aromatic ring-hydroxylating oxygenases. BMC Biochem. 2008, 9, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kincannon WM; Zahn M; Clare R; Lusty Beech J; Romberg A; Larson J; Bothner B; Beckham GT; McGeehan JE; DuBois JL, Biochemical and structural characterization of an aromatic ring-hydroxylating dioxygenase for terephthalic acid catabolism. Proc. Natl. Acad. Sci. USA 2022, 119 (13), e2121426119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sydor PK; Barry SM; Odulate OM; Barona-Gomez F; Haynes SW; Corre C; Song LJ; Challis GL, Regio- and stereodivergent antibiotic oxidative carbocyclizations catalysed by Rieske oxygenase-like enzymes. Nat. Chem 2011, 3 (5), 388–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JK; Simurdiak M; Zhao HM, Reconstitution and characterization of aminopyrrolnitrin oxygenase, a Rieske N-oxygenase that catalyzes unusual arylamine oxidation. J. Biol. Inorg. Chem 2005, 280 (44), 36719–36728. [DOI] [PubMed] [Google Scholar]
- 10.Satoh S; Tanaka A, Identification of chlorophyllide a oxygenase in the Prochlorococcus genome by a comparative genomic approach. Plant Cell Physiol. 2006, 47 (12), 1622–1629. [DOI] [PubMed] [Google Scholar]
- 11.Berim A; Park JJ; Gang DR, Unexpected roles for ancient proteins: flavone 8-hydroxylase in sweet basil trichomes is a Rieske-type, PAO-family oxygenase. Plant J. 2014, 80 (3), 385–395. [DOI] [PubMed] [Google Scholar]
- 12.Penfield JS; Worrall LJ; Strynadka NC; Eltis LD, Substrate specificities and conformational flexibility of 3-ketosteroid 9α-hydroxylases. J. Biol. Chem 2014, 289 (37), 25523–25536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bleem A; Kuatsjah E; Presley GN; Hinchen DJ; Zahn M; Garcia DC; Michener WE; König G; Tornesakis K; Allemann MN; Giannone RJ; McGeehan JE; Beckham GT; Michener JK, Discovery, characterization, and metabolic engineering of Rieske non-heme iron monooxygenases for guaiacol O-demethylation. Chem Catalysis 2022, 2, 1–23. [Google Scholar]
- 14.Lukowski AL; Ellinwood DC; Hinze ME; DeLuca RJ; Du Bois J; Hall S; Narayan ARH, C-H hydroxylation in paralytic shellfish toxin biosynthesis. J. Am. Chem. Soc 2018, 140 (37), 11863–11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lukowski AL; Liu J; Bridwell-Rabb J; Narayan ARH, Structural basis for divergent C-H hydroxylation selectivity in two Rieske oxygenases. Nat. Commun 2020, 11 (1), 2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hedges JB; Ryan KS, In vitro reconstitution of the biosynthetic pathway to the nitroimidazole antibiotic azomycin. Angew. Chem. Int. Ed 2019, 58 (34), 11647–11651. [DOI] [PubMed] [Google Scholar]
- 17.Burnet MW; Goldmann A; Message B; Drong R; El Amrani A; Loreau O; Slightom J; Tepfer D, The stachydrine catabolism region in Sinorhizobium meliloti encodes a multi-enzyme complex similar to the xenobiotic degrading systems in other bacteria. Gene 2000, 244 (1-2), 151–161. [DOI] [PubMed] [Google Scholar]
- 18.Friemann R; Lee K; Brown EN; Gibson DT; Eklund H; Ramaswamy S, Structures of the multicomponent Rieske non-heme iron toluene 2,3-dioxygenase enzyme system. Acta Crystallogr. D Biol. Crystallogr 2009, 65 (Pt 1), 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daughtry KD; Xiao Y; Stoner-Ma D; Cho E; Orville AM; Liu P; Allen KN, Quaternary ammonium oxidative demethylation: X-ray crystallographic, resonance raman, and UV–Visible spectroscopic analysis of a Rieske-type demethylase. J. Am. Chem. Soc 2012, 134 (5), 2823–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang W; Hollmann F, Nonconventional regeneration of redox enzymes - a practical approach for organic synthesis? Chem. Commun 2018, 54 (53), 7281–7289. [DOI] [PubMed] [Google Scholar]
- 21.Hoschek A; Buhler B; Schmid A, Overcoming the gas-liquid mass transfer of oxygen by coupling photosynthetic water oxidation with biocatalytic oxyfunctionalization. Angew. Chem. Int. Ed 2017, 56 (47), 15146–15149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiao Y; Zahariou G; Sanakis Y; Liu P, IspG enzyme activity in the deoxyxylulose phosphate pathway: roles of the iron-sulfur cluster. Biochemistry 2009, 48 (44), 10483–5. [DOI] [PubMed] [Google Scholar]
- 23.Xiao Y; Chu L; Sanakis Y; Liu P, Revisiting the IspH catalytic system in the deoxyxylulose phosphate pathway: achieving high activity. J. Am. Chem. Soc 2009, 131 (29), 9931–3. [DOI] [PubMed] [Google Scholar]
- 24.Martini MA; Rudiger O; Breuer N; Noring B; DeBeer S; Rodriguez-Macia P; Birrell JA, The nonphysiological reductant sodium dithionite and [FeFe] hydrogenase: Influence on the enzyme mechanism. J. Am. Chem. Soc 2021, 143 (43), 18159–18171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cirino PC; Arnold FH, Protein engineering of oxygenases for biocatalysis. Curr. Opin. Chem. Biol 2002, 6 (2), 130–5. [DOI] [PubMed] [Google Scholar]
- 26.Wu S; Snajdrova R; Moore JC; Baldenius K; Bornscheuer UT, Biocatalysis: Enzymatic synthesis for industrial applications. Angew. Chem. Int. Ed 2021, 60 (1), 88–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pyser JB; Chakrabarty S; Romero EO; Narayan ARH, State-of-the-Art Biocatalysis. ACS Cent. Sci 2021, 7 (7), 1105–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gibson DT; Parales RE, Aromatic hydrocarbon dioxygenases in environmental biotechnology. Curr. Opin. Biotechnol 2000, 11 (3), 236–43. [DOI] [PubMed] [Google Scholar]
- 29.Immanuel S; Sivasubramanian R; Gul R; Dar MA, Recent progress and perspectives on electrochemical regeneration of reduced nicotinamide adenine dinucleotide (NADH). Chem. Asian J 2020, 15 (24), 4256–4270. [DOI] [PubMed] [Google Scholar]
- 30.Uppada V; Bhaduri S; Noronha SB, Cofactor regeneration - an important aspect of biocatalysis. Curr. Sci 2014, 106 (7), 946–957. [Google Scholar]
- 31.Ozgen FF; Runda ME; Schmidt S, Photo-biocatalytic Cascades: Combining Chemical and Enzymatic Transformations Fueled by Light. ChemBiochem 2021, 22 (5), 790–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park JH; Lee SH; Cha GS; Choi DS; Nam DH; Lee JH; Lee JK; Yun CH; Jeong KJ; Park CB, Cofactor-free light-driven whole-cell cytochrome P450 catalysis. Angew. Chem. Int. Ed 2015, 54 (3), 969–73. [DOI] [PubMed] [Google Scholar]
- 33.Shanmugam M; Quareshy M; Cameron AD; Bugg TDH; Chen Y, Light-activated electron transfer and catalytic mechanism of carnitine oxidation by Rieske-Type oxygenase from human microbiota. Angew. Chem. Int. Ed 2021, 60 (9), 4529–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Douglas JJ; Sevrin MJ; Stephenson CRJ, Visible light photocatalysis: applications and new disconnections in the synthesis of pharmaceutical agents. Org. Process Res. Dev 2016, 20 (7), 1134–1147. [Google Scholar]
- 35.Arias-Rotondo DM; McCusker JK, An overview of the physical and photophysical properties of [Ru(bpy)3]2+. In Visible light photocatalysis in organic chemistry, Stephenson CRJ; Yoon TP; MacMillan WC, Eds. Wiley-VCH: 2019; pp 1–24. [Google Scholar]
- 36.Schmermund L; Jurkas V; Ozgen FF; Barone GD; Buchsenschutz HC; Winkler CK; Schmidt S; Kourist R; Kroutil W, Photo-biocatalysis: Biotransformations in the presence of light. ACS Catal. 2019, 9 (5), 4115–4144. [Google Scholar]
- 37.Stegmaier K; Blinn CM; Bechtel DF; Greth C; Auerbach H; Müller CS; Jakob V; Reijerse EJ; Netz DJA; Schünemann V; Pierik AJ, Apd1 and Aim32 are prototypes of bishistidinyl-coordinated Non-Rieske [2Fe–2S] proteins. J. Am. Chem. Soc 2019, 141 (14), 5753–5765. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y; Pavlosky MA; Brown CA; Westre TE; Hedman B; Hodgson KO; Solomon EI, Spectroscopic and theoretical description of the electronic structure of the S= 3/2 nitrosyl complex of non-heme iron enzymes. J. Am. Chem. Soc 1992, 114 (23), 9189–9191. [Google Scholar]
- 39.Enemark JH; Feltham RD, Principles of structure, bonding, and reactivity for metal nitrosyl complexes. Coord. Chem. Rev 1974, 13 (4), 339–406. [Google Scholar]
- 40.Dubichev AG; Parfenov NN; Chaplygina NM; Borovik AS; Dreizin RS; Zolotarskaya EE; Tikhonenko TI, Physicochemical properties of human adenovirus type 6 DNA. Voprosy Virusologii 1978, (1), 47–51. [PubMed] [Google Scholar]
- 41.Sardar S; Weitz A; Hendrich MP; Pierce BS, Outer-sphere tyrosine 159 within the 3-mercaptopropionic acid dioxygenase S-H-Y motif gates substrate-coordination denticity at the non-heme iron active site. Biochemistry 2019, 58 (51), 5135–5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tonzetich ZJ; Do LH; Lippard SJ, Dinitrosyl iron complexes relevant to Rieske cluster nitrosylation. J. Am. Chem. Soc 2009, 131 (23), 7964–7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Truzzi DR; Medeiros NM; Augusto O; Ford PC, Dinitrosyl iron complexes (DNICs). From spontaneous assembly to biological roles. Inorg. Chem 2021, 60 (21), 15835–15845. [DOI] [PubMed] [Google Scholar]
- 44.Tinberg CE; Tonzetich ZJ; Wang H; Do LH; Yoda Y; Cramer SP; Lippard SJ, Characterization of iron dinitrosyl species formed in the reaction of nitric oxide with a biological Rieske center. J. Am. Chem. Soc 2010, 132 (51), 18168–18176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tarasev M; Pinto A; Kim D; Elliott SJ; Ballou DP, The “bridging” aspartate 178 in phthalate dioxygenase facilitates interactions between the Rieske center and the iron (II)− mononuclear center. Biochemistry 2006, 45 (34), 10208–10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adam D; Bosche L; Castaneda-Losada L; Winkler M; Apfel UP; Happe T, Sunlight-dependent hydrogen production by photosensitizer/hydrogenase systems. ChemSusChem 2017, 10 (5), 894–902. [DOI] [PubMed] [Google Scholar]
- 47.Goncalves LCP; Mansouri HR; PourMehdi S; Abdellah M; Fadiga BS; Bastos EL; Sa J; Mihovilovic MD; Rudroffe F, Boosting photobioredox catalysis by morpholine electron donors under aerobic conditions. Catal. Sci. Technol 2019, 9 (10), 2682–2688. [Google Scholar]
- 48.Lee SH; Choi DS; Kuk SK; Park CB, Photobiocatalysis: activating redox enzymes by direct or indirect transfer of photoinduced electrons. Angew. Chem. Int. Ed 2018, 57 (27), 7958–7985. [DOI] [PubMed] [Google Scholar]
- 49.Soliman MA; Zakaria M, Kinetics of photolysis and photocatalytic oxidation of ammonium sulfite for hydrogen production. Journal of King Saud University– Engineering Sciences 2021, online ASAP, 10.1016/j.jksues.2021.11.006. [DOI] [Google Scholar]
- 50.Chanquia SH; Valotta A; Gruber-Woelfler H; Kara S, Photobiocatalysis in continuous flow. Front. Catal 2022, 1, 816538. [Google Scholar]
- 51.Straathof NJW; Noel T, Accelerating visible-light photoredox catalysis in continuous-flow reactors. In Visible light photocatalysis in organic chemistry, Stephenson CRJ; Yoon TP; MacMillan WC, Eds. Wiley-VCH: 2019; pp 389–414. [Google Scholar]
- 52.Streit BR; Blanc B; Lukat-Rodgers GS; Rodgers KR; DuBois JL, How active-site protonation state influences the reactivity and ligation of the heme in chlorite dismutase. J. Am. Chem. Soc 2010, 132 (16), 5711–5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mantri M; Zhang ZH; McDonough MA; Schofield CJ, Autocatalysed oxidative modifications to 2-oxoglutarate dependent oxygenases. FEBS J. 2012, 279 (9), 1563–1575. [DOI] [PubMed] [Google Scholar]
- 54.Liu PH; Mehn MP; Yan F; Zhao ZB; Que L; Liu HW, Oxygenase activity in the self-hydroxylation of (S)-2-hydroxypropylphosphonic acid epoxidase involved in fosfomycin biosynthesis. J. Am. Chem. Soc 2004, 126 (33), 10306–10312. [DOI] [PubMed] [Google Scholar]
- 55.Shao Y-H; Guo L-Z; Zhang Y-Q; Yu H; Zhao B-S; Pang H-Q; Lu W-D, Glycine betaine monooxygenase, an unusual Rieske-type oxygenase system, catalyzes the oxidative N-demethylation of glycine betaine in Chromohalobacter salexigens DSM 3043. Appl. Environ. Microbiol 2018, 84 (13), e00377–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu YJ; Jameson E; Crosatti M; Schafer H; Rajakumar K; Bugg TDH; Chen Y, Carnitine metabolism to trimethylamine by an unusual Rieske-type oxygenase from human microbiota. Proc. Natl. Acad. Sci. USA 2014, 111 (11), 4268–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Massmig M; Reijerse E; Krausze J; Laurich C; Lubitz W; Jahn D; Moser J, Carnitine metabolism in the human gut: characterization of the two-component carnitine monooxygenase CntAB fromAcinetobacter baumannii. J. Biol. Chem 2020, 295 (37), 13065–13078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim JH; Kim BH; Brooks S; Kang SY; Summers RM; Song HK, Structural and mechanistic insights into caffeine degradation by the bacterial N-demethylase complex. J. Mol. Biol 2019, 431 (19), 3647–3661. [DOI] [PubMed] [Google Scholar]
- 59.Summers RM; Louie TM; Yu CL; Gakhar L; Louie KC; Subramanian M, Novel, highly specific N-demethylases enable bacteria to live on caffeine and related purine alkaloids. J. Bacteriol 2012, 194 (8), 2041–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun ZD; Su Q; Rokita SE, The distribution and mechanism of iodotyrosine deiodinase defied expectations. Arch. Biochem. Biophys 2017, 632, 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bobyk KD; Ballou DP; Rokita SE, Rapid kinetics of dehalogenation promoted by iodotyrosine deiodinase from human thyroid. Biochemistry 2015, 54 (29), 4487–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pimviriyakul P; Chaiyen P, Flavin-dependent dehalogenases. In Flavin-dependent enzymes: mechanisms, structures and applications (The Enzymes serial vol. 47), Chaiyen P; F. T, Eds. 2020; pp 365–398. [DOI] [PubMed] [Google Scholar]
- 63.Herman PL; Behrens M; Chakraborty S; Chrastil BM; Barycki J; Weeks DP, A three-component dicamba O-demethylase from Pseudomonas maltophilia, strain DI-6: gene isolation, characterization, and heterologous expression. J. Biol. Chem 2005, 280 (26), 24759–67. [DOI] [PubMed] [Google Scholar]
- 64.Dumitru R; Jiang WZ; Weeks DP; Wilson MA, Crystal structure of dicamba monooxygenase: a Rieske nonheme oxygenase that catalyzes oxidative demethylation. J. Mol. Biol 2009, 392 (2), 498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Feyza Ozgen F; Runda ME; Burek BO; Wied P; Bloh JZ; Kourist R; Schmidt S, Artificial light-harvesting complexes enable rieske oxygenase catalyzed hydroxylations in non-photosynthetic cells. Angew. Chem. Int. Ed 2020, 59 (10), 3982–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Beinert H, Semi-micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron-sulfur proteins. Anal. Biochem 1983, 131 (2), 373–378. [DOI] [PubMed] [Google Scholar]
- 67.Caranto JD; Weitz A; Hendrich MP; Kurtz DM, The Nitric Oxide Reductase Mechanism of a Flavo-Diiron Protein: Identification of Active-Site Intermediates and Products. J. Am. Chem. Soc 2014, 136 (22), 7981–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Petasis DT; Hendrich MP, Quantitative interpretation of multifrequency multimode EPR spectra of metal containing proteins, enzymes, and biomimetic complexes. Methods Enzymol. 2015, 563, 171–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.