

Abstract
Despite the success of PD-1 blockade in melanoma and other cancers, effective treatment strategies to overcome resistance to cancer immunotherapy are lacking1,2. We identified the innate immune kinase TANK-binding kinase 1 (TBK1)3 as a candidate immune evasion gene in a pooled genetic screen4. Using a suite of genetic and pharmacologic tools across multiple experimental model systems, we confirm a role for TBK1 as an immune evasion gene. Targeting TBK1 enhances response to PD-1 blockade by lowering the cytotoxicity threshold to effector cytokines (TNFα/IFNγ). TBK1 inhibition in combination with PD-1 blockade also demonstrated efficacy using patient-derived tumour models, with concordant findings in matched patient-derived organotypic tumour spheroids (PDOTS) and matched patient-derived organoids (PDOs). Tumour cells lacking TBK1 are primed to undergo RIPK- and caspase-dependent cell death in response to TNFα/IFNγ in a JAK/STAT-dependent manner. Taken together, our results demonstrate that targeting TBK1 is a novel and effective strategy to overcome resistance to cancer immunotherapy.
Keywords: TBK1, PD-1, immunotherapy, tumour microenvironment, organotypic tumour spheroids, 3D microfluidic culture, cytokines
Cancer immunotherapy with immune checkpoint blockade (ICB) has transformed the treatment of advanced melanoma and other cancers, although overcoming resistance remains a central challenge1,2. There are currently no approved therapies for patients with innate or acquired resistance to ICB. Clinical trials evaluating novel immune modulatory agents in combination with anti-PD-1/PD-L1 therapies to overcome primary resistance are already underway5. Recently, the results of two phase III, placebo-controlled, randomized clinical trials comparing novel, promising combination strategies were reported, neither showing a survival benefit compared with single-agent PD-1 blockade6,7, prompting renewed focus on the pre-clinical and early-phase clinical development of combination strategies.
Approaches to unbiased target identification include loss-of-function genetic screens using CRISPR-Cas9 genome editing, which have successfully nominated novel targets to enhance anti-tumour immune responses4,8. Pooled in vivo and in vitro CRISPR-Cas9 based screening have nominated several tumour intrinsic drivers of resistance to immunotherapy4,8–11, but therapeutic applications of these findings remain limited.
TANK-binding kinase 1 (TBK1) is a multi-functional serine/threonine kinase with an established role coordinating innate immune responses to viruses and other invading pathogens12. TBK1 integrates upstream signals from pattern recognition receptors and cytosolic nucleic acid sensors to regulate the activation of interferon regulatory factor 3 (IRF3) and consequent induction of type I interferons (IFN-α/β) and interferon-stimulated genes (ISGs) critical to the host immune response3. Activation of cytosolic nucleic acid sensing pathways has emerged as a promising strategy to stimulate innate anti-tumour immune responses to inflame immunologically ‘cold’ tumours13, thus it is surprising that TBK1 has been nominated as a candidate immune evasion gene4,9–11 and that disrupting TBK1 signaling has shown early promise enhancing response to ICB in murine tumour models14,15. Given these seemingly contradictory findings, the precise role of TBK1 in influencing sensitivity to cancer immunotherapy remains unclear.
Here, we show that genetic deletion of TBK1 sensitizes tumours to immune attack and demonstrate that pharmacologic inhibition of TBK1 can overcome resistance to PD-1 blockade using established murine tumour models and novel patient-derived tumour models. Targeting TBK1 lowers the cytotoxicity threshold following exposure to immune cell-derived effector cytokines thereby sensitizing resistant tumours to ICB.
TBK1 loss sensitizes tumours to ICB
In a previous in vivo CRISPR screen4, Tbk1-targeting single-guide RNAs (sgRNAs) were significantly depleted from B16 melanoma tumours in immunocompetent mice following PD-1 blockade (Fig. 1a), suggesting more effective tumour control of cells lacking TBK1. In contrast, the sgRNAs targeting the homologous innate immune signaling kinase IKKε (Ikbke) were not enriched (Extended Data Fig. 1a), suggesting specificity for TBK1. To determine if deletion of Tbk1 enhanced response to PD-1 blockade, we generated B16 murine melanoma cells lacking Tbk1 by CRISPR-Cas9 knockout using two different sgRNAs and confirmed loss of TBK1 protein expression (Extended Data Fig. 1b). Tbk1-null and control sgRNA B16 cells grew at comparable rates in culture and when implanted in immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (Fig. 1b, Extended Data Fig. 1c–d). Tumour growth and survival was comparable in immunocompetent wild-type (WT) mice bearing control and Tbk1-null B16 tumours, whereas anti-PD-1 treatment resulted in improved tumour shrinkage and increased survival in mice bearing Tbk1-null B16 tumours compared to mice bearing control sgRNA B16 tumours (Fig. 1c, Extended Data Fig. 1e). These results confirm that Tbk1-null B16 tumours demonstrate normal growth compared to control sgRNA B16 tumours and are more sensitive to cancer immunotherapy with PD-1 blockade in vivo.
Figure 1|. TBK1 loss sensitizes tumours to PD-1 blockade.
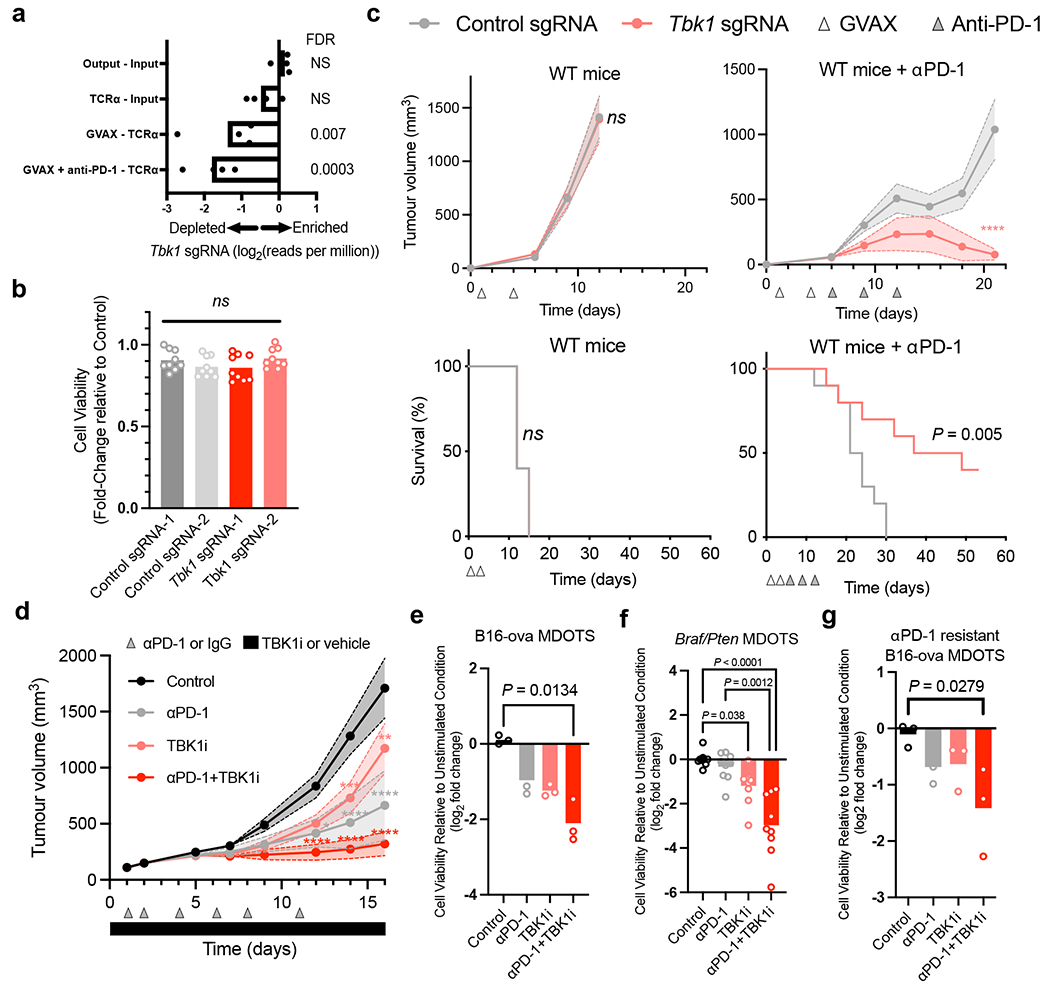
a, Relative depletion of Tbk1 sgRNAs from a pool of sgRNAs targeting 2,368 genes expressed by Cas9-expressing B16 melanoma cells (n = 4 independent guides targeting each gene; false discovery rate (FDR) was calculated using the STARS algorithm v1.3, as previously described 6,7). b, Viability of Tbk1-null and control B16 tumour cells following 3 days of in vitro culture. Means (bars) and individual values (open circles) are shown (n = 9, across 3 independent experiments. One-way ANOVA with Tukey’s multiple comparisons test; ns, not significant. c, Tumour volume and survival analysis of control (grey), Tbk1-null (light red) B16 tumours in wild-type (WT) and WT αPD-1-treated C57BL/6 mice with overlapping survival curves for GVAX WT mice. Data in c represent two independent experiments with n = 5 animals per guide with two separate guides for the control group and two separate guides for each Tbk1-null group. Mean tumour volumes (solid circles) are shown +/− s.e.m. (shaded region). d, Tumour volume analysis of mice bearing B16-ova tumours treated with TBK1i (Compound 1, 40mg/kg daily by oral gavage), αPD-1 (200 mg i.p. tiw x 6 doses), or combination compared to control (IgG + vehicle); n=10 mice per treatment group. Mean tumour volumes (solid circles) are shown +/− s.e.m. (shaded region). 2-way ANOVA with Tukey’s multiple comparisons test ***P < 0.001; compared to control group. e-g, Viability assessment of (e) treatment-naïve B16-ova MDOTS (n=3 per treatment group), (f) treatment-naïve Braf/Pten (D4M.3A) MDOTS (n=9 per treatment group), (g) αPD-1 resistant B16-ova MDOTS (n=3 per treatment group); one-way ANOVA with Dunn’s multiple comparisons test. *P < 0.05; **P < 0.01; **** P < 0.0001; ns, not significant.
TBK1 inhibition enhances ICB response
TBK1 plays an important role in innate immune sensing3 and TBK1 inhibitors are being evaluated in the treatment of autoimmune and inflammatory diseases16. This raises the possibility that systemic inhibition of TBK1 may dampen inflammation and fail to recapitulate the sensitization mediated by tumour-specific TBK1 loss. Thus, we sought to determine if pharmacologic inhibition of TBK1 phenocopied the observations in TBK1-null B16 tumours. To this end, WT mice bearing B16 tumours expressing the model antigen ovalbumin (B16-ova) were treated with IgG or anti-PD-1 antibodies +/− a previously described small molecule TBK1 inhibitor (TBK1i)14. Improved tumour control was observed in mice treated with anti-PD-1 plus TBK1i compared to mice with single-agent treatments or control mice (Fig. 1d, Extended Data Fig. 1f–g), and was well tolerated without evidence of toxicity or diminished body weight (Extended Data Fig. 1h). Ex vivo profiling using murine-derived organotypic tumour spheroids (MDOTS)14 derived from untreated mice bearing B16-ova tumours confirmed enhanced response to anti-PD-1 plus TBK1i (Fig. 1e). Using CT26 MDOTS (partially responsive to PD-1 blockade +/− TBK1i)14 +/− anti-CD8a treatment, we demonstrated that CD8 T cell activity was required for the combinatorial effect of anti-PD-1 plus TBK1i, but not single-agent TBK1i (Extended Data Fig. 1i). Ex vivo profiling of MDOTS derived from anti-PD-1 resistant D4M.3A (Braf-mutant/Pten-null) tumours17 showed that TBK1i could overcome primary (intrinsic) resistance using a separate autochthonous murine melanoma model (Fig. 1f). A similar sensitizing effect of TBKi was observed in B16-ova MDOTS prepared from mice that developed acquired (secondary) resistance to PD-1 blockade in vivo (Fig. 1g). We also observed improved in vivo tumour control with combined TBK1i plus PD-L1 blockade in MC38 (responsive) and MB49 (partially responsive) syngeneic murine tumour models (Extended Data Fig. 1j–k). These findings demonstrate activity of TBK1i + anti-PD-1 in murine tumour models of primary (intrinsic) and secondary (acquired) resistance to PD-1 blockade.
TBK1i enhances ICB response in PDOTS
To explore TBK1 inhibition as a strategy to overcome intrinsic or acquired resistance to ICB in human cancer, we performed ex vivo profiling of patient-derived organotypic tumour spheroids (PDOTS)14,18 from explanted human tumours (Fig. 2a). PDOTS established from patients with melanoma and other cancers were cultured ex vivo with TBK1i (1μM) +/− anti-PD-1. Analysis of PDOTS (n=30) from patients with cutaneous melanoma (n=15), non-cutaneous melanoma (n=2) and other cancer types (n=13), revealed reduced tumour growth in response to TBK1i (30% response) and TBK1i plus PD-1 blockade (40% response) compared to single-agent PD-1 blockade (16.6% response) (Fig. 2b, Extended Data Fig. 2a, Supplementary Table 1). We did not observe an effect of IgG4 antibody control (Extended Data Fig. 2b), consistent with previous reports14,19. PDOTS from patients with immunotherapy-resistant metastatic cutaneous melanoma were sensitive to combined TBK1i + anti-PD-1 treatment and were unresponsive to ex vivo anti-PD-1 +/− anti-CTLA-4 treatment (Fig. 2c–d). Exceptional ex vivo response to TBK1i + PD-1 blockade was also observed in other cancer types, especially colorectal carcinoma (CRC) with evidence of microsatellite instability (MSI) (Fig. 2e–f). To our knowledge, these are the first data that demonstrate efficacy of a TBK1 inhibitor in PD-1 refractory patients using patient-derived tumour models.
Figure 2|. TBK1 inhibition enhances sensitivity to PD-1 blockade using PDOTS.
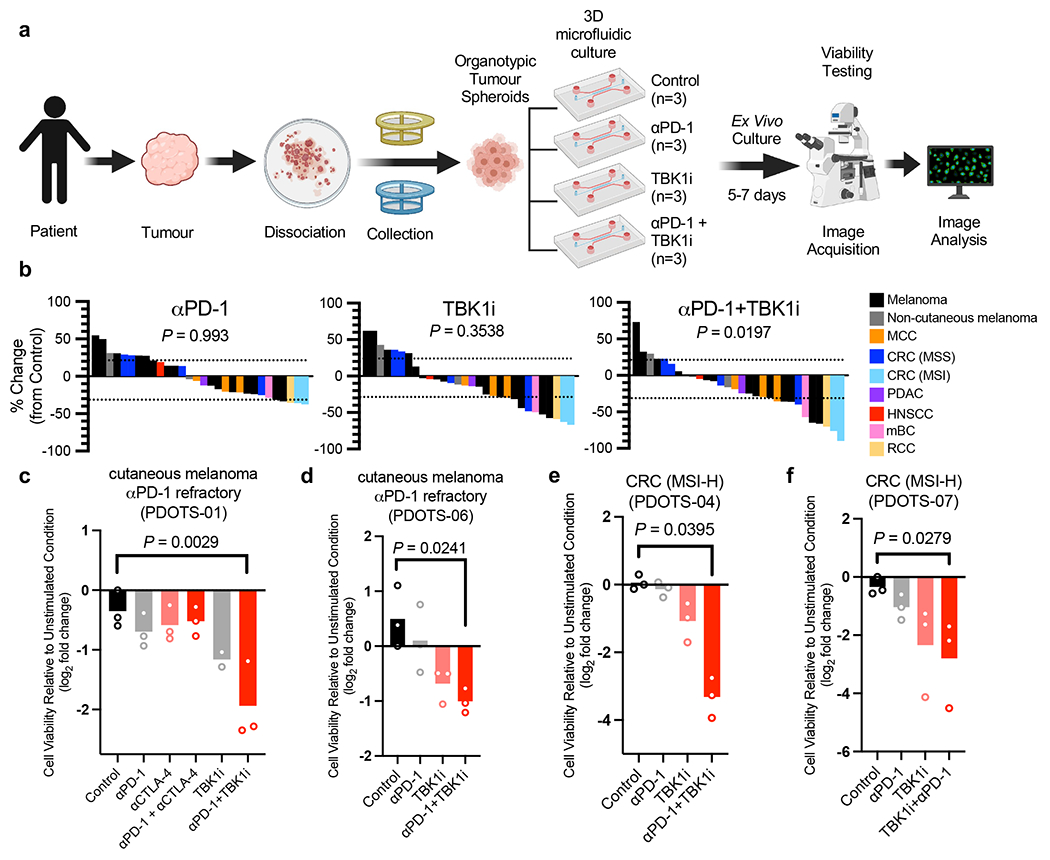
a, Scheme of PDOTS preparation. b, waterfall plots for PDOTS (n=30, indicated tumour types) treated with αPD-1 (250μg/mL pembrolizumab), TBK1i (1 μM), or combined αPD-1+TBK1i. Means (bars) for each specimen are shown. 1-way ANOVA (matched) with Dunnett’s multiple comparisons test compared to control. c-f, PDOTS viability assessment from patients with treatment-refractory melanoma (c-d) and treatment-naïve MSI-colon adenocarcinoma (e-f) with indicated treatments. Means (bars) and individual values (open circles) are shown (n = 3, biological replicates, one-way ANOVA with Dunn’s multiple comparisons test; *P < 0.05; **P < 0.01).
TBK1i and the tumour immune landscape
Tbk1 and IKKε (IKBKE) are widely expressed across lymphoid and myeloid cells in human melanoma20 (Extended Data Fig. 3a–b). Recent studies have demonstrated critical roles for TBK1 and/or IKKε in regulation of activity of numerous immune cell types, including T cells21, B cells22,23, dendritic cells24, and macrophages25,26. To examine the effect of TBK1 inhibition on the tumour immune microenvironment, we performed scRNA-seq on CD45+ cells (n=53,637) from B16-ova tumours from mice treated with anti-PD-1, TBK1i, or anti-PD-1 plus TBK1i, compared to isotype control (IgG) (Fig. 3a). We aggregated data from each treatment condition to perform clustering in order to create a stable set of clusters across conditions and then quantified changes in the relative abundance of populations between conditions (Fig. 3b; Extended Data Fig. 3b). As expected, treatment with anti-PD-1 expanded the populations of T and NK cells relative to the other treatment conditions (Fig. 3b, Extended Data Fig. 3c–d) with an increase in the proportion of terminal exhausted/effector CD8+ T cells (Extended Data Fig. 3e–g). In contrast, tumours from mice treated with TBK1i +/− anti-PD-1 demonstrated enrichment in early exhausted/effector CD8+ T cells with concomitant reduction in the abundance of terminal exhausted/effector CD8+ T cells (Extended Data Fig. 3f–g). In vitro treatment of T lymphocytes derived from murine spleens with TBK1i enhanced the production of cytokines and tumour necrosis factor-α (TNFα), interleukin-2 (IL-2), and interferon-γ (IFNγ) (Extended Data Fig. 3h–j), consistent with an enhanced effector function.
Figure 3|. TBK1 inhibition remodels the tumour immune microenvironment.
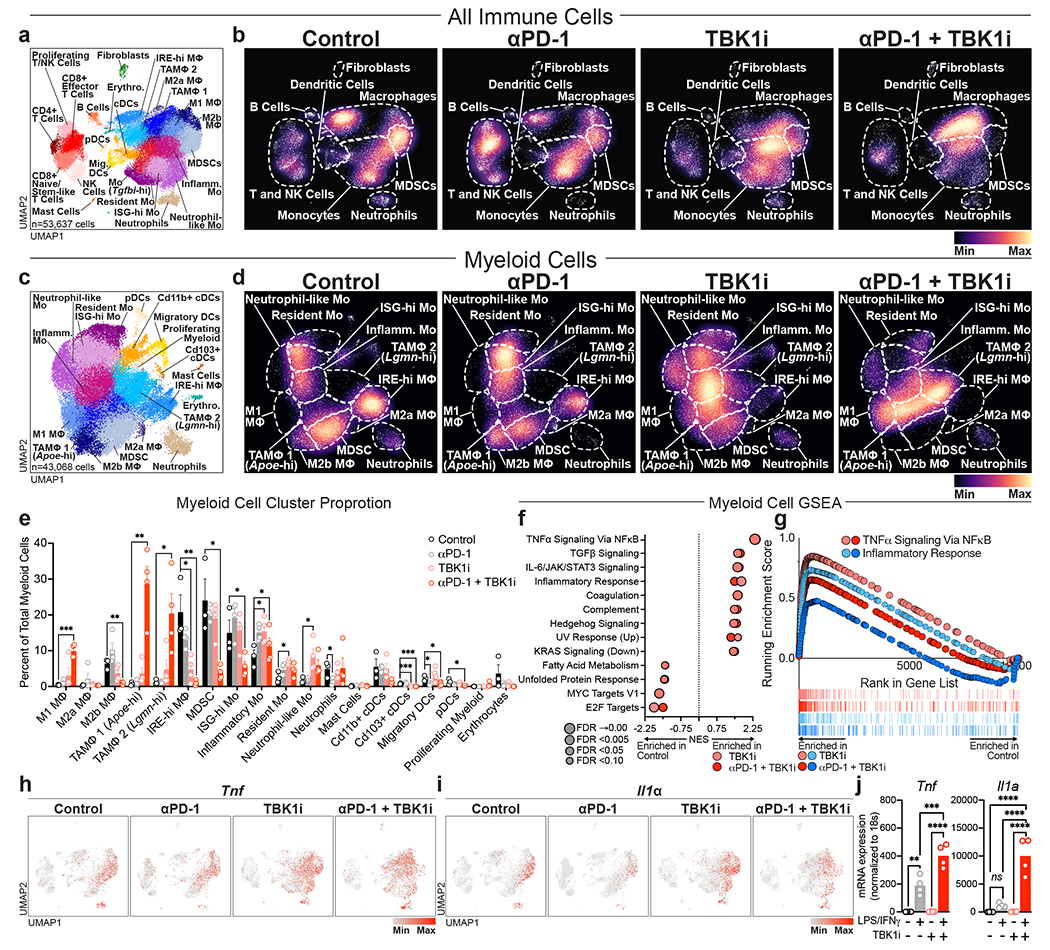
a, Uniform Manifold Approximation and Projection (UMAP) of all immune cells (n=53,637) with 25 unique populations identified among CD45+-enriched immune cells from single cell RNA sequencing on tumour-infiltrating leukocytes from B16-ova tumours from Control (Vehicle/IgG, n=3), αPD-1 (Vehicle/anti-PD-1, n=4), TBK1i (TBK1i/IgG n=4), and αPD-1 + TBK1i (TBK1i/anti-PD-1, n=4) treated tumours. b, Downsampled cell density projections by condition. c, UMAP of 43,068 cells and 19 unique populations identified among sub-clustered myeloid cells. d, Downsampled myeloid sub-cluster cell density projections by condition. e, Bar plots of proportional changes by unique cluster of myeloid sub-cluster immune cells by treatment. Means (bars) and individual values (circles) +/− s.e.m (error bars) are shown (n = 4 biologically independent samples, except for control, n = 3; multiple unpaired t-test, **P < 0.01; ***P < 0.001; **** P <.0001). f, Top enriched or decreased Hallmark gene signatures in the myeloid sub-cluster determined by GSEA Prerank on differentially expressed genes calculated by a logistic regression by condition. g, Mountain plots showing enrichment scores for the TNFα Signaling Via NFκB Hallmark gene set and Inflammatory Response gene set in the myeloid sub-cluster by condition. h-i, downsampled UMAP of all immune cells showing (h) Tnf gene expression and (i) Il1a gene expression by condition. j, gene expression (qRT-PCR) of Tnf and Il1a in bone marrow-derived macrophages (BMDMs) pre-treated with TBK1i (1 μM) for 24 hours prior to 2-hour stimulation with LPS (20 ng/mL) plus IFNγ (20ng/mL) versus PBS control. Means (bars) and individual values (open circles) are shown (n=4 biological replicates; 2-way ANOVA with Sidak’s multiple comparisons test, **P < 0.01; ***P < 0.001; **** P <.0001).
A marked expansion of myeloid cells was observed in tumours from mice treated with TBKi +/− anti-PD-1 (Fig. 3b). Sub-clustering of tumour-infiltrating myeloid cells revealed a marked increase in the abundance of several pro-inflammatory macrophage populations (e.g., M1 macrophages) with decreased abundance of certain immune suppressive myeloid populations, including myeloid-derived suppressor cells (MDSCs) (Fig. 3c–e). To gain additional insight into the impact of TBK1i +/− anti-PD-1 treatment on immune cell function, gene set enrichment analysis was performed. TBK1i +/− anti-PD-1 treatment was associated with enrichment for numerous gene sets associated with TNFα/NFкB signaling and inflammation (Fig. 3f–g). TNFα (Tnf) and IL-1α (Il1a) expression was largely observed in myeloid cell clusters which was further enhanced in tumours from mice treated with TBK1i +/− anti-PD-1 (Fig. 3h–i). TBK1i pre-treatment enhanced expression of Tnf and Il1a in bone marrow-derived macrophages (BMDMs) in response to lipopolysaccharide (LPS)/IFNγ challenge (Fig. 3j), confirming a direct effect of TBK1 inhibition on myeloid cell inflammatory responses. These findings demonstrate a tumour extrinsic impact of TBK1i with marked remodeling of the myeloid compartment in response to TBK1i +/− anti-PD-1 treatment and confirm that TBK1i is sufficient to enhance expression of inflammatory cytokines (e.g., IFNγ, TNFα) in the TME.
We next sought to determine if tumour-specific loss of TBK1 influenced the tumour immune microenvironment. Flow cytometric analysis of tumour-infiltrating immune cells from control and Tbk1-null B16 tumours implanted into wild-type mice and treated with anti-PD-1 treatment revealed no significant differences in CD8+ or CD4+ T cells, granzyme B+ CD8+ T cells, FoxP3+ regulatory T cells (Tregs), natural killer (NK) cells, or F4/80+ myeloid cells (Extended Data Fig. 4a). We next performed single-cell RNA sequencing (scRNA-seq) of CD45+ cells (n=31,810) from control and Tbk1-null B16 tumours following anti-PD-1 treatment and identified distinct lymphoid and myeloid cell clusters, as well as contaminating tumour cells (Extended Data Fig. 4b). Evaluation of immune cell states by scRNA-seq revealed limited immune remodeling in Tbk1-null B16 tumours with modest increases in CD8+ T cells and M1-like macrophages (Extended Data Fig. 4c–d). We confirmed expression of Tbk1 and IKKε (Ikbke) across lymphoid and myeloid cell types/states, with the highest expression in macrophages, MDSCs, and CD8+ T cells (Extended Data Fig. 4e–f). As expected, we observed loss of Tbk1 expression in tumour cells from Tbk1-null B16 tumours with intact expression of Ikbke (Extended Data Fig. 4f). These findings confirm that the enhanced efficacy of anti-PD-1 therapy in mice bearing Tbk1-null tumours is not dependent on significant remodeling of the immune compartment, consistent with a tumour-intrinsic role for TBK1 as an immune evasion gene.
TBK1 loss enhances TNFα/IFNγ sensitivity
IFNγ and TNFα are key effector cytokines that contribute to anti-tumour immune responses9,11,27–30, and genes associated with IFN and TNF signaling pathways contribute to immune evasion4,9,29. In a cohort of 203 patients with metastatic melanoma, elevated circulating levels of TNFα and IFNγ were observed in both responders (R) and non-responders (NR) 6 weeks after initiating ICB treatment, although levels remained elevated at 6 months in NR patients (Fig. 4a–b). Single cell RNA-seq data from melanoma patients treated with ICB20 confirmed higher expression of IFNG and TNF in NR versus R tumours (Fig. 4c). Expression of IFNG was largely restricted to the lymphoid compartment (highest expression in exhausted CD8 T cells), whereas TNF expression was enriched in macrophages/monocytes (Fig. 4d), consistent with findings in B16 tumours (Extended Data Fig. 5a). Importantly, levels of Tnf and Ifng were similar across immune, stromal, and tumour cell populations from control and Tbk1-null B16 tumours (Extended Data Fig. 5b). These results confirm upregulation of TNFα and IFNγ following ICB and demonstrate persistent cytokine elaboration in patients that are not responding to therapy.
Figure 4|. Loss of TBK1 sensitizes tumour cells to TNFα/IFNγ.
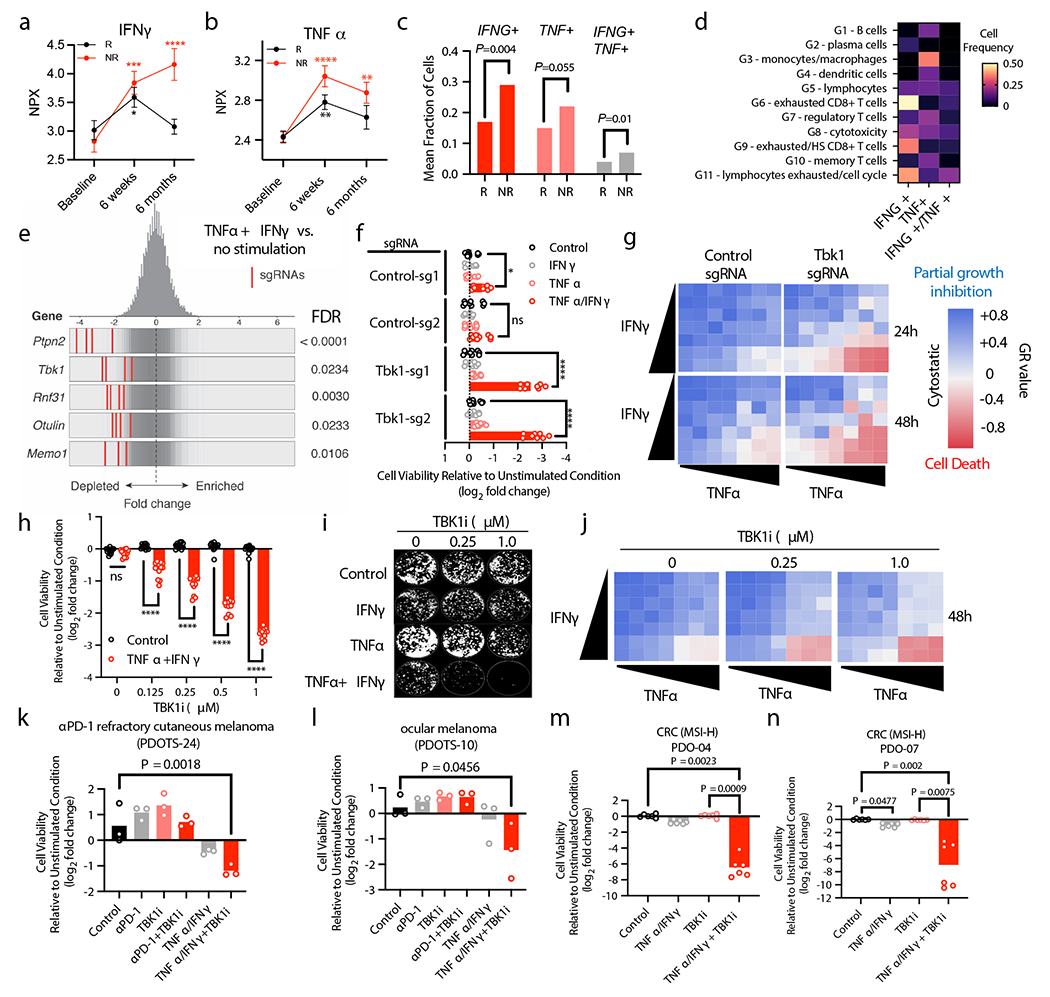
a-b, Plasma protein levels (NPX, normalized protein expression) from patients with metastatic melanoma responsive (R) or non-responsive (NR) to ICB at baseline (n=179), 6 weeks after starting ICB (n=173) and 6 months after starting ICB (n=151). Mean values (solid circles) +/− s.e.m. are shown (2-way ANOVA with Dunnett’s multiple comparisons test: **P < 0.01; ***P < 0.001; **** P <.0001). c-d, Mean fraction of CD45+ cells (c) and cell frequency across lineage-defined clusters (d) for cells expressing IFNG and TNF in patients with metastatic melanoma20. e, Frequency histograms of depletion (z-score) for all sgRNAs per target in a Cas9+ B16 control sgRNA cell line +/− in vitro stimulation with TNFα and IFNγ. f, Viability assessment of indicated B16 cell lines with indicated treatments (24 hours). Means (bars) and individual values (open circles) are shown (n=9, 3 independent experiments, 2-way ANOVA, Dunnett’s multiple comparisons test: * P <.05, **** P <.0001, ns, not significant). g, Heatmap of mean GR values (n=3) for cells treated with increasing concentrations of TNFα and IFNγ. h, Viability assessment of B16 cells with indicated treatments compared to unstimulated cells (24 hours). Means (bars) and individual values (open circles) are shown (n=12, 4 independent experiments, 2-way ANOVA, Dunnett’s multiple comparisons test: **** P <.0001, ns, not significant). i, Clonogenic assay of B16 cells (representative images shown; n=3). j, Heatmap of GR values for B16 cells treated with TBK1i (n=3) across TNFα/IFNγ concentrations. k-l, PDOTS viability assessment with indicated treatments. Means (bars) and individual values (open circles) are shown (n = 3, one-way ANOVA with Dunn’s multiple comparisons test). m-n, patient-derived organoids with indicated treatments. Means (bars) and individual values (open circles) are shown (n=6 biological replicates, 2 independent experiments: one-way ANOVA with Dunn’s multiple comparisons test).
Given the limited remodeling of the immune compartment in Tbk1-null B16 tumours and comparable expression of effector cytokines, we reasoned that B16 cells lacking TBK1 exhibited increased sensitivity to TNFα and IFNγ. In a whole-genome, in vitro pooled CRISPR screen, Tbk1 was amongst the top depleted sgRNAs in cells challenged with TNFα/IFNγ (Fig. 4e, Extended Data Fig. 6a), in line with in vivo CRISPR screening findings in B16 melanoma tumours (Fig. 1a)6. In vitro essentiality analysis confirmed that Tbk1 is not an essential gene (Extended Data Fig. 6b), consistent with our initial findings on the in vitro (Fig. 1b, Extended Data Fig. 1b) and in vivo (Fig. 1c, Extended Data Fig. 1c) growth characteristics of Tbk1-null B16 melanoma cells. Tbk1-null B16 cells exhibited marked sensitivity to combined TNFα/IFNγ treatment, but not with either cytokine alone (Fig. 4f). Using single cell clones, we demonstrate that the response to TNFα/IFNγ was influenced by extent of TBK1 deletion with dramatic reduction in cell viability in clones 3 and 4 (complete loss of TBK1 expression), whereas no effect was observed in clone 2 (intact TBK1 expression) and partial response was observed in clone 1 (+/− TBK1 expression) (Extended Data Fig. 6c–d). Normalized growth rate inhibition (GR) analysis31 across a range of both IFNγ and TNFα concentrations revealed partial growth inhibition with TNFα/IFNγ treatment in control B16 cells, whereas a marked cytotoxic response was observed exclusively in Tbk1-null B16 cells above threshold concentrations of TNFα and IFNγ (Fig. 4g).
TBK1i promotes TNFα/IFNγ cytotoxicity
To determine the effect of pharmacologic TBK1 inhibition on cell viability, parental B16 cells were treated with increasing concentrations of TBK1i +/− TNFα/IFNγ. TBK1i alone had no effect on cell viability at the doses evaluated (up to 1.0 μM), whereas TBK1i in combination with TNFα/IFNγ reduced cell viability in a dose-dependent manner in B16 cells (Fig. 4h). TBK1i also prevented B16 colony formation in the presence of TNFα/IFNγ, and to a lesser extent with TNFα alone (Fig. 4i). Activity of TBK1i in the presence of TNFα/IFNγ was confirmed using B16-ova cells (Extended Data Fig. 6a) and parental B16 tumour spheroids in 3D culture (Extended Data Fig. 6b). Comparable findings in B16 cells were observed using two additional TBK1 inhibitors, MRT6730732 and GSK861233 (Extended Data Fig. 6g–h) and the TBK1-targeted proteolysis targeting chimera (TBK1 PROTAC 3i)34 (Extended Data Fig. 6i).
GR analysis confirmed a dose-dependent effect of TBK1i in parental B16 melanoma cells at concentrations of TNFα and IFNγ required to induce cytotoxicity in Tbk1-null B16 cells (Fig. 4j). Dose response studies demonstrated that TBK1i (up to 1.0 μM) +/− IFNγ or TNFα alone did not affect growth or viability of control or Tbk1-null B16 cells, whereas TBK1i induced a dose-dependent cytotoxic response in cells co-treated with combined TNF𝛼/IFNγ, mirroring the cytotoxic response observed in Tbk1-null B16 cells treated with TNFα/IFNγ (Extended Data Fig. 7a). Further GR analysis of B16 cells confirmed enhanced TBK1i potency (half-maximal effect, GEC50) and overall efficacy (area over the GR curve, GRAOC) in cells treated with TNFα/IFNγ (Extended Data Fig. 7b–c).
To confirm these findings in other model systems, we explored tumour cell intrinsic sensitivity to TNFα/IFNγ using human melanoma cell lines, PDOTS, and patient-derived organoids. Similar to B16 cells, TBK1i sensitized A375 human melanoma cells to TNFα/IFNγ in a time- and dose-dependent manner (Extended Data Fig. 7d). Notably, A375 with acquired resistance to combined BRAF/MEK inhibition exhibited increased sensitivity to TBK1i compared to parental A375 cells (Extended Data Fig. 7e). Evaluation of PDOTS, including anti-PD-1 refractory cutaneous melanoma (Fig. 4k) and ocular melanoma (Fig. 4l), demonstrated that tumours poorly responsive to ICB could be sensitized to exogenous TNFα/IFNγ by co-administration of TBK1i. Lastly, matched patient-derived organoids (PDOs) from exceptional ex vivo responders to combined TBK1i + anti-PD-1 (PDOTS-04 and PDOTS-07; Fig. 2e–f, Extended Data Fig. 2), demonstrated dramatic sensitivity to TBK1i + TNFα/IFNγ (Fig. 4m–n). These results show in human and murine melanoma cells lines, as well as novel patient-derived tumour models including PDOTS and PDOs, that TBK1i treatment lowers the cytotoxic threshold to TNFα/IFNγ.
TBK1 restrains necroptosis
TBK1 restrains cell death signaling downstream of the TNF receptor (TNFR)35,36 by phosphorylating receptor interacting protein kinase 1 (RIPK1). Loss of TBK1 reduces this inhibitory phosphorylation thereby promoting RIPK1 activation leading to enhanced TNFR-complex II formation and subsequent caspase 8 cleavage and activation35. To determine the impact of TBK1 loss on RIPK1 activation and caspase cleavage, we performed immunoblotting on cell lysates from control and Tbk1-null B16 cells treated with TNFα/IFNγ. Increased levels of p-RIPK1 (S166/T189) were observed in Tbk1-null B16 within 3 hours of TNFα/IFNγ treatment, which preceded cleavage of caspase 8, caspase 3, and PARP, and c-FLIP degradation (Fig. 5a). To assess the requirement for RIPK and caspase signaling in response to TNFα/IFNγ, control and Tbk1-null B16 cells were pre-treated with Nec-1s (RIPK1 inhibitor) and/or zDEVD-fmk (caspase 3 inhibitor). Nec-1s and zDEVD-fmk each partially prevented loss of cell viability in Tbk1-null B16 cells following TNFα/IFNγ challenge, whereas combined RIPK1 and caspase 3 inhibitor was necessary to completely prevent cell death (Fig. 5b). Similar findings were observed using pan-caspase inhibitors (Q-VD-OPh and zVAD-fmk), and a caspase 8 selective inhibitor (z-IETD-fmk) (Extended Data Fig. 8a–c).
Figure 5|. IFNγ sensing is required for RIPK- and caspase-dependent death of Tbk1-null cells.
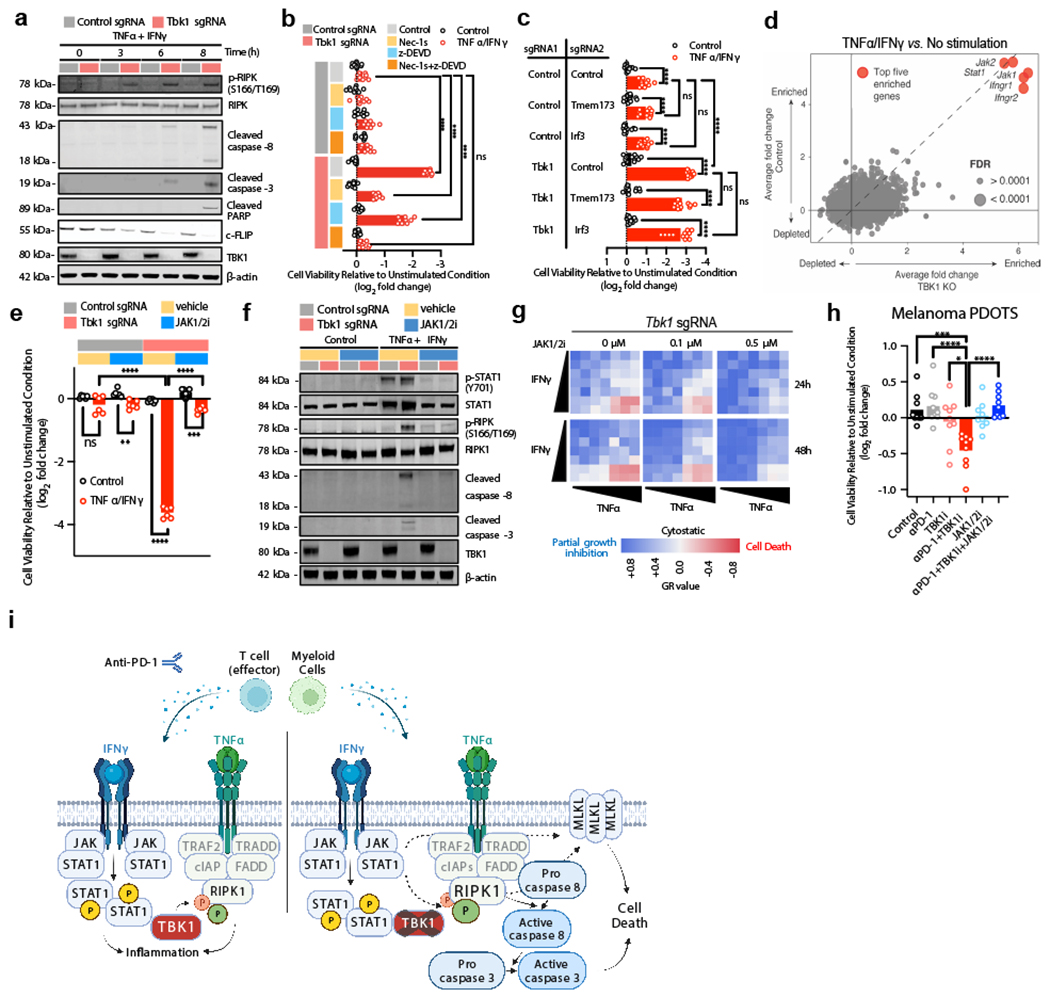
a, Western blot for indicated proteins in control sgRNA and Tbk1-null B16 cells treated with TNFα (160 ng/mL) and IFNγ (40 ng/mL) for the indicates times. b, Viability assessment of control and Tbk1-null B16 cells with indicated pre-treatments +/− TNFα/IFNγ. Means (bars) and individual values (open circles) are shown (n=9, 3 independent experiments; 2-way ANOVA, Dunnett’s multiple comparisons test; **** P < 0.0001; ns, not significant). c, Viability assessment of indicated B16 cells after 48 hours treatment with TNFα/IFNγ compared to unstimulated cells. Means (bars) and individual values (open circles) are shown (n=8, 2 independent experiments; 2-way ANOVA, Tukey’s multiple comparisons test; **** P < 0.0001; ns, not significant). d, Scatter plot depicting relative depletion and enrichment of sgRNAs targeting 19,674 genes in a Cas9+ B16 control and Tbk1 sgRNA cell line +/− in vitro stimulation with TNFα/IFNγ. e, cell viability assessment of indicated B16 cells pre-treated with ruxolitinib (0.5 μM) +/− TNFα/IFNγ. Means (bars) and individual values (open circles) are shown (n=6, 2 independent experiments; 2-way ANOVA, Dunnett’s multiple comparisons test: **P <.01, ***P < 0.001; **** P <.0001; ns, not significant). f, Western blot for indicated proteins in control sgRNA and Tbk1-null B16 cells pre-treated with vehicle or ruxolitinib (0.5 μM) followed by TNFα/IFNγ or PBS (control) for 8 hours. g, Heatmap of mean GR values (n=3) for Tbk1-null B16 cells treated with increasing concentrations of TNFα and IFNγ for 24 and 48 hours with 0, 0.1, and 0.5 μM ruxolitinib. h, Viability assessment of melanoma PDOTS with indicated treatments. Means (bars) and individual values (open circles) are shown (n=9, 3 independent specimens: one-way ANOVA with Dunn’s multiple comparisons test; *P <.05, ***P < 0.001; **** P <.0001). i, Scheme of TNFα/IFNγ-driven RIPK1- and caspase-dependent cell death in cells lacking TBK1.
Necroptosis is a form of death regulated by RIPK1 involving downstream activation of RIPK3 and the pseudokinase, mixed-lineage domain-like (MLKL)37. Consistent with our findings with RIPK1, treatment with small molecule inhibitors of RIPK3 (HS-1371)38 or MLKL (GW806742X)39 rescued Tbk1-null B16 cells from TNFα/IFNγ-induced cell death when combined with caspase inhibition (Extended Data Fig. 8d–e). Inhibition of RIPK1, RIPK3 or MLKL also rescued parental B16 cells from cell death following treatment with TBK1i plus TNFα/IFNγ in a clonogenic assay (Extended Data Fig. 8f–h). Transcriptional upregulation of MLKL was observed in response to IFNγ +/− TNFα (Extended Fig. 8i), consistent with previous reports40. Upregulation of MLKL expression was also more pronounced in Tbk1-null B16 cells following TNFα/IFNγ (Extended Fig. 8j). In addition to pRIPK1 and cleavage of caspase 8 and caspase 3, increased phosphorylated (Ser358) and total MLKL was also observed in Tbk1-null B16 cells following TNFα/IFNγ treatment (Extended Data Fig. 8k), which was reversed with RIPK1 +/− caspase inhibition.
Consistent with a primary role for TBK1 in regulating necroptosis following TNFα/IFNγ treatment, Tbk1-null B16 cells did not exhibit baseline or induced differences in apoptotic priming, compared to control sgRNA B16. BCL2-homology domain (BH3) profiling41 demonstrated that cytochrome c release following mitochondrial exposure to pro-apoptotic BH3-only peptides (e.g. BIM BH3, PUMA BH3) was similar in control and Tbk1-null B16 cells (Extended Data Fig. 9a). Following TNFα/IFNγ treatment, the impact of TBK1 loss on apoptotic priming was also modest with the most dramatic shifts driven by the differential response to TNFα/IFNγ treatment in Tbk1-null B16 cells (Extended Data Fig. 9b). Consistent with these findings, control and Tbk1-null B16 cells exhibited identical sensitivity to the apoptosis-inducing pan-kinase inhibitor staurosporine (STS) in 2D and 3D culture (Extended Data Fig. 9c–d). In summary, loss of TBK1 did not fundamentally alter apoptotic priming or sensitivity to cytotoxic agents, whereas melanoma cells lacking TBK1 were more sensitive to RIPK- and caspase-dependent cell death following TNFα/IFNγ challenge.
STING is dispensable for necroptosis
TBK1 plays a central role in coordinating the innate immune response in response to cytosolic nucleic acids (e.g. cGAS-STING-TBK1-IRF3-IFN type I pathway)3, and enhanced sensitivity to TNFα has been shown to drive cGAS-STING-dependent interferon response and impact cell viability42. To evaluate the role of the STING-TBK1-IRF3 axis, we generated B16 cells in which STING (Tmem173) and Irf3 were deleted +/− Tbk1 deletion (Extended Data Fig. 9e–f). B16 cells lacking Tmem173 and Irf3 did not exhibit enhanced sensitivity to combined TNFα/IFNγ challenge (Extended Data Fig. 9g), and co-deletion of Tmem173 or Irf3 with Tbk1 did not alter sensitivity to TNFα/IFNγ (Fig. 5c). Lastly, treatment of melanoma PDOTS with a STING agonist (ADU-S10043,44) had no impact on PDOTS viability, in contrast to TNFα/IFNγ +/− TBK1i (Extended Data Fig. 9h). To confirm activity of the STING agonist in PDOTS, we performed multiplexed analysis of secreted cytokines and observed upregulation of several inflammatory cytokines and chemokines (e.g., CXCL10) following treatment with ADU-S100 (Extended Data Fig. 9i). Together with the observation of aberrant RIPK1 activation in cells lacking TBK1, these findings indicate the TNFα/IFNγ-driven death of Tbk1-null cells occurs independently of cytosolic nucleic acid sensing pathways (i.e., STING-TBK1-IRF3 axis).
Requirement for intact IFNγ sensing
To uncover genes/pathways required for sensitivity of Tbk1-null cells to TNFα/IFNγ, we performed a whole genome pooled in vitro CRISPR screen using both control sgRNA and Tbk1 sgRNA B16 cells. Single-guide RNAs targeting genes involved in IFNγ sensing (Ifngr1, Ifgnr2, Jak1, Jak2, and Stat1) were enriched in control and Tbk1-null cells (Fig. 5d, Extended Data Fig. 10a), consistent with previous in vivo and in vitro screens 4,29. In contrast, sgRNAs targeting key components of the TNFR and necroptosis signaling pathways (e.g. Ripk1, Ripk3, Birc2, Birc3, Casp8) were not significantly enriched (or depleted) in either control sgRNA or Tbk1-null B16 cells (Extended Data Fig. 10b), possibly reflecting the greater complexity of cell death signaling downstream of the TNF receptor. Surprisingly, no differences were observed in activation of IFN sensing pathways (e.g. JAK1/JAK2/STAT1), NF-кB (p65), or IRF3 between control and Tbk1-null B16 cells (Extended Data Fig. 10c). Pre-treatment with ruxolitinib (JAK1/2 inhibitor) completely rescued Tbk1-null B16 cells and TBK1i-treated parental B16 cells from TNFα/IFNγ-mediated cell death (Fig. 5e, Extended Data Fig. 10d). In addition to completely blocking STAT1 phosphorylation (Y701) in both control and Tbk1-null B16 cells, ruxolitinib pre-treatment abolished RIPK1 phosphorylation (S166/T169), caspase 8 cleavage, and caspase 3 cleavage in Tbk1-null B16 cells (Fig. 5f, Extended Data Fig. 10e). GR analysis confirmed that ruxolitinib restored viability of Tbk1-null B16 by converting the cytotoxic response to a cytostatic response, mirroring the response characteristics of parental and control sgRNA B16 cells (Fig. 5g, Extended Data Fig. 10f). Lastly, we observed that JAK1/2i rescued melanoma PDOTS treated with anti-PD-1 plus TBK1i (Fig. 5h). These data confirm that enhanced sensitivity to TNFα/IFNγ in B16 cells lacking TBK1 requires IFN sensing and provide a novel link between IFNγ-induced JAK-STAT signaling and TNFα-mediated RIPK1 activation (Fig. 5i).
Discussion
Here, we have shown that TBK1 is an immune evasion gene and that targeting TBK1 can enhance response to PD-1 blockade by sensitizing tumour cells to effector cytokine-induced cell death. Using syngeneic murine tumour models and novel, patient-derived ex vivo models, we have demonstrated that targeting TBK1 sensitizes tumours to immune challenge. In contrast to other recently characterized immune evasion genes4,8,45, tumour-specific loss of TBK1 did not result in dramatic remodeling of the immune compartment. Rather, TBK1 loss sensitized tumour cells to immune cell-derived effector cytokines (TNFα and IFNγ), a finding confirmed in an independent, whole genome, in vitro CRISPR screen and subsequent validation studies. Whereas a key role for TNFα signaling has been demonstrated in immunotherapy-resistant melanoma cell lines devoid of IFNγ signaling11, our findings demonstrate critical interplay between TNFα and IFNγ signaling that can be therapeutically exploited to sensitize tumour cells to immune attack.
Despite multiple loss-of-function CRISPR screens (in vivo and in vitro) identifying TBK1 as a potential immune evasion gene4,9–11, the finding that TBK1 inhibition can enhance the response to cancer immunotherapy is surprising. Intact TBK1 signaling is required for response to STING agonists, innate immune stimulatory molecules that mimic response to cytosolic DNA, which have been shown to limit tumour growth alone or in combination with cancer immunotherapy46–48. However, TBK1 has an emerging role in regulating death receptor signaling distinct from its role in innate immune response and viral sensing35,36,49. Here, we demonstrate that loss of TBK1 leads to RIPK- and caspase-dependent cell death following challenge with TNFα and IFNγ and confirm that STING and IRF3 are dispensable for this tumour-intrinsic cell death phenotype.
While loss of TBK1 signaling did not impact tumour growth in immune-deficient mice or in isolated cancer cell lines, moderate anti-tumour activity was observed following pharmacologic inhibition of TBK1 in models containing tumour cells and autologous immune cells, suggesting a tumour-extrinsic effect of TBK1 inhibition. Consistent with these observations, TBK1i treatment increased the proportion of early exhausted/effector CD8 T cells and M1 macrophages in the tumour immune microenvironment, and enhanced expression/production of effector cytokines in vitro using isolated CD8 T cells and macrophages. Thus, TBK1i treatment not only lowered the cytotoxic threshold of tumour cells to TNFα and IFNγ, but also promoted elaboration of TNFα and IFNγ from tumour-infiltrating immune cells. While marked systemic upregulation of TNFα and IFNγ can promote tissue damage50, observations from patients with inherited TBK1 deficiency suggests that loss of TBK1 signaling is associated with a milder TNF-driven autoinflammatory syndrome, but not sepsis or increased incidence/severity of viral illnesses49. Importantly, treatment of mice with TBK1i +/− anti-PD-1 did not result in weight loss or other signs of systemic toxicity. Future studies will be required to further deconvolute the roles of TBK1 in distinct immune cell populations and determine the therapeutic potential of disrupting TBK1 signaling in melanoma patients resistant to immunotherapy.
Two central challenges in the field of cancer immunotherapy are (1) the need for pre-clinical models that translate to human immunity and (2) strategies to effectively and efficiently assess cancer immunotherapy combinations51. With over 1,000 cancer immunotherapy combination trials under evaluation5, novel approaches are needed to deprioritize ineffective treatment strategies and to better understand mechanisms of response and resistance to novel therapeutic strategies. Murine models are amenable to in vivo, ex vivo, and in vitro manipulation and iterative experimentation, but lack the heterogeneity observed in human cancer. Patient-derived models are inherently heterogeneous and more complex but offer greater clinical relevance and enable evaluation of the distribution of treatment response across multiple patients using clinically relevant biospecimens. Our results not only support further evaluation and development of TBK1-directed therapeutic strategies, but also provide a framework to evaluate potential immune evasion targets across multiple model systems using a combination of genetic and pharmacologic tools.
Online Content
Methods, along with any additional Extended and Supplemental Data display items and Source Data, are available in the online version of the paper; references unique to these sections appear only in the online paper.
METHODS
Generation of CRISPR-edited tumour cell lines.
For in vivo and in vitro validation experiments, confirmatory epistasis experiments, Tbk1 was deleted in B16 cells using transient transfection of a Cas9-sgRNA plasmid (pX459, Addgene) with Lipofectamine 3000 (Thermo Fisher Scientific, L3000015) followed by puromycin selection. For epistasis experiments, Cas9 was expressed using the pLX311 backbone, transient transfection was used to introduce the first guide(s), and the final epistasis guides were expressed using the pXPR_BRD024 lentiviral expression system. Cell lines were tested every 3–6 months for mycoplasma contamination.
Animal treatment and tumour challenges.
The designs of animal studies and procedures were approved by the Broad Institute, Massachusetts General Hospital, and Charles River Laboratories IACUC committees. Ethical compliance with IACUC protocols and institute standards was maintained. Specific pathogen-free facilities at the Broad Institute were used for the storage and care of all mice. Murine pathogen testing and mycoplasma testing was performed prior to tumour inoculations. Wild-type female C57BL/6J mice (7 weeks old) were obtained from Jackson Laboratories. A colony of NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were bred on site at the Broad Institute. Mice were age-matched to be 6–12 weeks old at the time of tumour inoculation. For tumour challenges, 2.0 × 106 B16 tumour cells were resuspended in Hanks balanced salt solution (Gibco), mixed 1:1 by volume with Matrigel (Corning) and subcutaneously injected into the right flank on day 0. Each tumour injected contained only a single sgRNA targeting each indicated gene or control sgRNA. Vaccination on days 1 and 4 with 1.0 × 106 previously irradiated GM-CSF-secreting B16 (GVAX) cells (kindly provided by G. Dranoff) was performed where indicated. For validation experiments, mice were treated with 200 μg of rat monoclonal anti-PD1 antibodies (#BP0273, BioXCell, clone: 29F.1A12) via intraperitoneal injection on days 6, 9 and 12. Beginning on day 6 after challenge, tumour volumes (TV) were estimated using longest dimension (length) and the longest perpendicular dimension (width), using the formula (L × W2)/2. Tumour volumes were assessed every 3–4 days until either the survival endpoint was reached, or no palpable tumour remained. Pre-specified endpoints for tumour size were adhered to as defined by IACUC protocols, including 2.0 cm in maximum dimension for validation studies and 2.5 cm in maximum dimension for screens with daily monitoring. CO2 inhalation was used to euthanize mice. Statistical methods were not used to predetermine sample size. At least five mice were included in each group for all experiments. Animals were randomized before treatment. No blinding was performed.
TBK1i in vivo studies were performed by Charles River Laboratories. For TBK1i in vivo treatment studies, wild-type female C57BL/6J mice (7-8 weeks old) were obtained from Charles River Laboratories. 1.0 × 106 B16-ova cells (kindly provided by Dr. Debattama Sen, MGH) were resuspended in sterile Ca- and Mg-free PBS (Gibco), mixed 1:1 by volume with Matrigel (Corning) and subcutaneously injected into the flank on day 0. Mice were randomized into four groups of 10 using the stratified method via Study Log program based on tumour size. Randomization and treatment initiated on Day 1; mean TV at the start of dosing of 110.05 mm3. Vehicle (0.5% hydroxypropyl methylcellulose K100LV / 0.4% Tween 80 / 99.1% 0.05N hydrochloric acid) or TBK1i (Compound 1, Gilead Sciences; 40 mg/kg)14 was administered by oral gavage daily (Days 1-18) and isotype control IgG (#BE0089, clone 2A3, BioXCell) or anti–PD-1 (#BP0273, BioXCell, clone: 29F.1A12) (10 mg/kg) was administered three times weekly for a total of six doses. Investigators were not blinded to treatment groups. Combination studies using MC38 and MB49 were performed by vivoPharm (Hummelstown, PA, USA). MB49 cells (used for in vivo studies only) were licensed from Dr. K Esuvaranathan (University of Singapore) by vivoPharm in collaboration with Gilead Sciences. All procedures used in the performance of these studies were carried out in accordance with vivoPharm’s Standard Operating Procedures, with particular reference to US_SOPvP_EF0314 “General Procedures for Efficacy Studies.” Vehicle, TBK1i (40 mg/kg) was administered by oral gavage daily for 26 days and isotype control or a reverse chimera anti-PD-L1 cloned from literature reports and placed into a mouse IgG1 framework (10mg/kg)52 was administered every 5 days for a total of six doses. Investigators were not blinded to treatment groups.
For MDOTS studies, mice were euthanized 8-14 days after inoculation and tumours were harvested. B16 and B16-ova MDOTS were prepared from tumours using wild-type female C57BL/6J mice (6 weeks old, Jackson Labs). D4M.3A (Braf/Pten) MDOTS were generated using wild-type male C57BL/6J mice (6 weeks old, Jackson Labs). CT26 MDOTS were prepared using wild-type female BALB/c mice (6-8 weeks old, Jackson Labs).
Isolation and Culture of Primary Murine T Cells.
Spleens harvested from C57BL/6J mice were mechanically dissociated, filtered through a 70-μm filter, and incubated in 1 mL ACK lysing buffer per spleen for 1 minute. Cells were quenched in 10X the lysis buffer volume with a PBS + 2% FBS + 5 mM EDTA solution. T cells were isolated with the mouse CD8a+ T Cell Isolation Kit (Miltenyi Biotec) as per the manufacturer’s instructions. T cells were cultured on a plate coated with Purified NA/LE Hamster Anti-Mouse CD3e antibody and in T/NK cell media supplemented with 1 μg/mL Purified NA/LE Hamster Anti-Mouse CD28 antibody, 100 U/mL rhIL-2, and either 1 μM TBK1i (Compound 1) or an equal volume of DMSO. After 24 hours incubation, T cells were spun out of their media to remove the CD3e and CD28 antibodies and transferred to Ultra-Low Attachment plates. Cells were cultured in T/NK media supplemented with rhIL-2 and either 1 μM TBK1i (Compound 1) or an equal volume of DMSO for an additional 96 hours with a 50% media volume refresh every 48 hours. On the 6th day post isolation, T cells were pooled by TBK1i treatment status, replated at a final concentration of 2x106 cells/mL, and stimulated with ionomycin (0.5 ug/mL, Millipore Sigma #I0634) and PMA (5 ng/mL, Millipore Sigma #P8139). After two hours of stimulation, Brefeldin A (1X, Invitrogen/eBioscience) was added to the culture media. Cells were incubated for an additional two hours before collection for analysis by flow cytometry.
Analysis of Primary Murine T Cells by Flow Cytometry.
Cells were stained with conjugated fluorescent monoclonal antibodies against CD69 (#104527, clone H1.2F3, BioLegend) and CD25 (#102024, clone PC61, BioLegend). After washing, cells were fixed and permeabilized with the BD Cytofix/Cytoperm Fixation/Permeabilization kit (BD Biosciences) as per the manufacturer’s instructions. Cells were stained with conjugated fluorescent monoclonal antibodies against IFNγ (#505807, clone XMG1.2, BioLegend), TNFα (#506303, clone MP6-XT22, BioLegend) and IL-2 (#503821, JES6-5H4, BioLegend). All samples were acquired on a Beckman Coulter Cytoflex LX flow cytometry system using single-color compensation controls to set gate margins and analyzed with FlowJo software (FlowJo LLC).
Isolation, culture, and stimulation of bone marrow-derived macrophages (BMDMs).
Murine BMDMs were generated by flushing bone marrow from the bones of the hind legs (Day 0) and differentiated to macrophages by culturing for 7 days in twelve-well plates (2 x 106 cells/well) in DMEM with 10% FCS plus M-CSF (20 ng/mL, vendor details) with media exchange and fresh M-CSF added on Day 4 and Day 6. On Day 8, media was exchanged with fresh M-CSF with TBK1i (1 μM) or DMSO (0.1%) for 24 hours. On Day 9, lipopolysaccharide (LPS, 20 ng/mL, Sigma-Aldrich #L4391) and IFNγ (20ng/mL, PeproTech #315-05) or vehicle control (PBS) were added. After 2 hours, media was aspirated from dishes and cells were collected in RNA Later for subsequent qRT-PCR analysis.
Analysis of tumour-infiltrating immune cells by flow cytometry.
Tbk1-null (sgRNA-1) or control sgRNA-1 B16 tumour cells (2 × 106) were implanted in Matrigel into abdominal subcutaneous tissue of C57BL/6 female mice. On day 13, tumours were dissected from the surrounding fascia, mechanically minced, and dissociated with the mouse Tumor Dissociation Kit (Miltenyi Biotec) per manufacturer’s instructions. After filtering through a 70-μm filter, live cells were isolated using a gradient with Lympholyte-M separation media (Fisher Scientific) per manufacturer’s instructions. Tumour-infiltrating lymphocytes were enriched by CD45+ MACS positive selection (Miltenyi Biotec). Cells were then stained with conjugated fluorescent monoclonal antibodies against CD45 (#103139, clone 30-F11, BioLegend), F4/80 (#157306, clone QA17A29, BioLegend), CD8α (#100749, clone 53-6.7, BioLegend), CD4 (100538, clone RM4-5, BioLegend), NK1.1 (#404-5941-82, clone PK136, Invitrogen), and TCRβ (#109220, clone H57-597, BioLegend). After washing, cells were fixed and permeabilized with the FoxP3/Transcription Factor Staining Buffer Set (eBiosciences) per manufacturer’s instructions. Cells were stained with conjugated fluorescent monoclonal antibodies against FoxP3 (#12-5773-82, clone FJK-16s, Invitrogen) and Granzyme B (#515403, clone GB11, BioLegend). All samples were acquired using a Beckman Coulter Cytoflex instrument and analyzed with FlowJo software (FlowJo, LLC).
Analysis of tumour-infiltrating immune cells by single cell RNA-seq.
For TBK1i +/− anti-PD-1 studies, subcutaneous B16-ova tumours were implanted into C57BL/6J mice and treated with control IgG or anti-PD-1 therapy in the presence of either Vehicle or TBK1i as described above. Tumours were dissected on day 14 post-inoculation and disassociated using the Miltenyi Mouse Tumor Dissociation Kit and gentleMACS Octo-Dissociator (Miltenyi) using the m-TKD-1 program. After filtering through a 70-μm filter, live cells were isolated using a density gradient with Lympholyte-M separation media (Cedarlane Labs) per the manufacturer’s specifications. CD45+ tumour-infiltrating immune cells were enriched by positive selection with MicroBeads (Miltenyi) and magnetic separator (Miltenyi). Four representative samples from each of the Control (Vehicle/IgG-treated), αPD-1 (Vehicle/anti-PD-1-treated), TBK1i (TBK1i/IgG-treated), and αPD-1 + TBK1i groups were selected and droplet-based isolation of single cells was performed with the Chromium Controller (10X Genomics) according to the manufacturer’s specifications. Subsequent generation of 3′ sequencing libraries was performed per manufacturer’s instructions (10X Genomics). Libraries were prepared utilizing Chromium Next GEM Single Cell Reagent Kits 3′v2 chemistry (10X Genomics). Characterization of the sequencing library was performed with TapeStation (Agilent) and Qubit (ThermoFisher) instruments.
Pooled equimolar 3’ 10X output libraries were sequenced using two Illumina SP flow cells and two paired-end 150 bp cycle kits. Downstream preprocessing steps were performed using cellranger version 5.0.1. Individual replicate quality was evaluated based on the number of cells recovered, mean reads per cell, and median genes per cell. Before preprocessing, filtering, or sample exclusion, 106,949 cells were recovered across all conditions. Early QC metrics determined that a single sample from the Control (Vehicle/IgG-treated) arm should be excluded based on a low capture rate of CD45+ cells. Additional cell and gene filtering was performed using Scanpy version 1.7.253. Cells with greater than 10 percent mitochondrial gene content were excluded. Cells with more than 2,500 genes were excluded as suspected doublets, while cells that had less than 500 genes were excluded due to poor gene capture. In addition, genes that were not recovered in any cell were also excluded from the downstream analysis. Downstream analysis revealed unequal capture of contaminating B16OVA tumour cells (Ptprc−, Mlana+, Mitf+, Dcn+) across treatment conditions so suspected tumour cells were also excluded. Gene counts were library size normalized to 100,000 and log transformed with pseudocount of 1. Principal component analysis and nearest neighbor graphs were calculated in order to visualize on a Uniform Manifold Approximation and Projection (UMAP) plot. Harmony batch correction was then used to correct PCA embeddings for technical batch effects between experiments 54. Cells were then grouped into 26 distinct clusters using the leiden algorithm. Clusters driven by a high doublet score or markers of low cell viability, like long noncoding RNAs, were excluded. After this additional filtering, 53,637 immune (Ptprc+) cells were left for downstream analysis. Cells were re-clustered and classified based on the built-in scanpy function one-vs-rest differential expression and immune-related gene signatures. To gain more granularity between the myeloid cell subtypes and T and NK cell subtypes, sub-clustering was performed on cells in specific clusters with specific marker gene expression profiles (clusters of cells expressing Itgam, Itgax, and Itgae transcripts or on clusters of cells expressing Cd8a, Cd4, and Ncr1 transcripts, respectively). New PCA embeddings, nearest neighborhood graphs, and harmony batch corrections were calculated for this subgroup on a set of 10,000 highly variable genes. Differentially expressed genes between treatment conditions were calculated using a logistic regression model55. Ranked lists of differential genes were created using signed p-values calculated by the logistic regression model and passed to GSEA Prerank to search for enriched gene sets by treatment56.
For tumour-specific TBK1 CRISPR studies, Tbk1-null (sgRNA-2) or control sgRNA-1 B16 tumour cells (2 × 106) were implanted in Matrigel into the stomach of C57BL/6 female mice. On day 13, tumours were dissected from the surrounding fascia, mechanically minced, and dissociated with the mouse Tumor Dissociation Kit (Miltenyi Biotec) as per manufacturer’s instructions. After filtering through a 70-μm filter, live cells were isolated using a gradient with Lympholyte-M separation media (Fisher Scientific) as per manufacturer’s instructions. Tumour-infiltrating immune cells were enriched by CD45+ MACS positive selection (Miltenyi Biotec). Four representative samples each of Tbk1-null (sgRNA-1) or control sgRNA-1 samples were selected, counted, and loaded onto the Chromium Controller (10X Genomics). Subsequent generation of 3’ sequencing libraries was performed as per manufacturer’s instructions (10X Genomics). Characterization of the sequencing library was performed with TapeStation (Agilent) and Qubit (ThermoFisher) instruments. Pooled equimolar 3’ 10X libraries were sequenced with an Illumina NextSeq 500 instrument using paired-end 50bp reads. Sample demultiplexing, barcode processing, and alignment was performed using the Cell Ranger analysis pipeline (v3.0). Downstream analysis was performed using Scanpy (version 1.4.5post3). For each cell, two quality control metrics were calculated: (1) the total number of genes detected and (2) the proportion of UMIs contributed by mitochondrially encoded transcripts. Cells in which fewer than 200 or greater than 2500 genes were detected, or in which mitochondrially encoded transcripts constituted more than 10% of the total library, were excluded from downstream analysis. Tumour replicates were concatenated, and batch effect correction was performed using ComBat, implemented in Scanpy. The resulting expression matrix consisted of 34,223 cells by 31,053 genes. PCA dimensionality reduction was applied, and the first 50 principal components were used for UMAP projection into two-dimensional space. The Leiden algorithm was used to perform unsupervised clustering, and clusters were labeled via expression of canonical marker genes. For differential expression, between cell normalization was calculated using scran (version 1.14.6), and the expression matrix was subset to genes expressed in at least .1 percent of cells, a total of 19,780 genes. Differential expression was then performed via logistic regression.
Patient samples.
Tumour samples are collected and analyzed according to Dana-Farber/Harvard Cancer Center IRB-approved protocols. A cohort of patients (Supplementary Table 1) treated at Massachusetts General Hospital and Dana-Farber Cancer Institute was assembled for PDOTS profiling. These studies were conducted according to the Declaration of Helsinki and approved by the DF/HCC IRB. Response to treatment was determined radiographically, as previously described 14.
Organotypic tumour spheroid preparation and microfluidic culture.
Murine- and patient-derived organotypic tumour spheroids (MDOTS/PDOTS) were prepared and cultured, as previously described 14. Briefly, fresh tumour specimens received in media (DMEM or RPMI) on ice were minced in a standard 10cm dish using sterile forceps and scalpel. Minced tumours were resuspended in high-glucose DMEM (RPMI for CT26) supplemented with 10% FBS + 1% pen-strep and 100 U/mL type IV collagenase, and 15 mM HEPES (Life Technologies, Carlsbad, CA). After 15-30 min, equal volumes of media were added to minced tumour specimens. Cell suspensions were then pelleted and resuspended in fresh media and passed over 100 mm and 40 mm filters sequentially to obtain S1 (>100 μm), S2 (40-100 μm), and S3 (<40 μm) spheroid fractions, which are subsequently transferred to ultra-low attachment tissue culture plates. The S2 fraction was pelleted and resuspended in type I rat tail collagen (Corning) at a concentration of 2.5 mg/mL following the addition of 10× PBS with phenol red with pH adjusted using NaOH. PANPEHA Whatman paper (Sigma-Aldrich) was used to confirm pH 7.0–7.5. The spheroid-collagen mixture was injected into the center gel region of the 3D microfluidic culture device (10 μL per device). After incubation for 30 minutes at 37°C in sterile humidity chambers, collagen hydrogels containing MDOTS/PDOTS were hydrated with media with or without indicated treatments; untreated control, αPD-1 (250 μg/mL pembrolizumab), TBK1i (1 μM), or combined αPD-1+TBK1i. Control human IgG4 (Invivogen, anti-β-gal-hIgG4 (bgal-mab114) 100 μg/mL was used for indicated experiments
Viability assessment of MDOTS/PDOTS.
Dual label fluorescence live/dead staining was using acridine orange/propidium iodide (AO/PI) Staining Solution (Nexcelom, CS2-0106) or Hoechst/ propidium iodide (Ho/PI) staining solution (Nexcelom, CSK-V0005), as previously described14,18. Following incubation with acridine orange/propidium iodide AO/PI (20 min, room temp, protected from light) or Ho/PI (45 min, 37°C, 5% CO2), images were obtained. Image capture and analysis are performed using a Nikon Eclipse NiE fluorescence microscope equipped with Z-stack (Prior), motorized stage (ProScan) and ZYLA5.5 sCMOS Camera (Andor) and NIS-Elements AR software package. Live and dead cell quantitation was performed by measuring total cell area of each dye. Percent change and L2FC data were generated using raw fluorescence data (live) for given treatments relative to control conditions.
In vitro CRISPR screen.
B16 cells (control and Tbk1-null) expressing Cas9 (pLoxp311 cas9) were transduced with a library of lentiviral vectors encoding 78,637 sgRNAs targeting 19,674 genes in the mouse genome (Brie pXPR003, CP0044)57 at >100X coverage/sgRNA. Cells were selected and passaged in vitro for eight days to allow sufficient time for gene editing. Subsequently, cells were stimulated with murine TNFα and IFNγ (10ng/mL, each) or vehicle control (PBS) for 12 days. Subsequently, cell pellets were lysed in ATL buffer (QIAGEN) with proteinase K (QIAGEN) before genomic DNA extraction (QIAGEN Blood Maxi kit). DNA (120ug per sample condition) was PCR amplified and sequenced using Illumina HiSeq. Significantly depleted or enriched sgRNAs were identified using the STARS algorithm, as previously described4,58. Gene essentiality analysis was conducted by calculating the log fold changes between sgRNAs at day 12 and day 0 for both the TBK1 knockout condition and the control condition.
In vitro cytokine stimulations and growth inhibition assays.
Parental and CRISPR-edited B16.F10 tumour cells were plated in DMEM + 10% FBS containing the indicated combinations of cytokines. For cell growth and viability assays, 8,000 cells were plated in 96-well plates and viable cells were enumerated after 24 or 48h using Cell Titer-Glo (Promega, G7570) after treatment mouse IFNγ (40 ng/mL, R&D Systems), mouse TNFα (160 ng/mL, R&D Systems). For inhibitor studies, B16 cells were pre-treated for 2 hours with indicated doses of TBK1i (Compound 1, Gilead Sciences)14, MRT67307 (MedChemExpress, HY-13018), GSK8612 (MedChemExpress, HY-111941), TBK1 PROTAC 3i (Bio-techchne/TOCRIS, 7259), Nec-1s (MedChemExpress, HY-15760), HS-1371 (MedchemExpress, HY-114349), GW806742X (MedchemExpress, HY-112292A), Z-DEVD-fmk (R&D Systems, 2163/1), zVAD-FMK (R&D Systems, 2166/1), Q-VD-OPh (MedChemExpress, HY-12305), zIETD-FMK (R&D Systems, FMK007), staurosporine (APExBIO, A8192), birinapant (APExBIO, A4219), ADU-S100 (MedchemExpress, CT-ADUS100), and ruxolitinib (MedChemExpress, HY-50856). All compounds were dissolved in DMSO (0.1% final concentration) except ADU-S100 which was dissolved in sterile water. Plates were read on a Cytation 5 plate reader and analysis was performed using Prism9 (GraphPad Software). All conditions were tested in triplicate. The values represent the average of three replicates and a representative experiment from at least two independent experiments.
Normalized growth rate inhibition (GR) measurements.
Cell lines, maintained in their recommended growth conditions were seeded depending on cell line and growth rate, in 384-well CellCarrier plates (Perkin Elmer, Waltham, MA) using a Multidrop Combi Reagent Dispenser (Thermo Fisher Scientific, Waltham, MA) and allowed to adhere to for 24 hours prior to drug treatment. B16 cells – plated at 750 cells/well for 24 and 48 h time points; A375 (ATCC) and BRAFi/MEKi-resistant A375 cells (provided by Dr. Gao Zhang)59 were plated at 1000 cells/well for 24, 48 and 72h time points. Cells were treated with a half-log dilution series of TNFα (R&D Systems, Minneapolis, MN) 0.005 - 500 ng/mL and IFNγ (R&D Systems, Minneapolis, MN) 0.001-125 ng/mL in combination by HP D300e Digital Dispenser (HP, Palo Alto, CA). Cytokines were prepared in PBS containing 0.05% Tween-20 (necessary for dispensing aqueous solutions), which was diluted to a final concentration of < .0008% Tween-20 after addition to the wells. In conditions that included TBK1i, cells were pretreated for two hours with the TBK1i (Compound 1, Gilead Pharmaceuticals, Foster City, CA14) at various concentrations prior to the addition of cytokines, all dispensed by the HP D300e Digital Dispenser. Cells were stained and fixed for analysis at the time of drug delivery and after 24, 48 and 72 hours of treatment. Cells were stained with LIVE/DEAD Far Red Dead Cell Stain (LDR, 1:5000) (Thermo Fisher Scientific, Waltham, MA) and 1 μg/ml Hoechst 33342 (Sigma Aldrich, St. Louis, MO). Cells were then fixed with 4% formaldehyde (Sigma Aldrich, St. Louis, MO) for 30 minutes. Fixed cells were imaged with a 10x objective using an ImageXpress confocal microscope (Molecular Devices, San Jose, CA). MetaXpress software was used to segment nuclei based on their Hoechst signal, and the LDR intensity within each nuclear mask was extracted and used to classify cells as live or dead. Live cell counts were normalized to DMSO-treated controls on the same plates to yield normalized growth rate inhibition (GR) values as described previously24. Experiments were performed in technical duplicate or triplicate unless otherwise indicated. GR values shown in heatmaps depict the mean across three biological replicates.
Clonogenic assay.
Five hundred B16.F10 cells were plated onto 6-well type plates, and then cultured in the presence of TBK1i (Cmpd1: 0, 0.25, or 1.0 μM), Nec-1s (10μM), Q-VD-OPh (20 μM), HS-1371 (2 μM, or MLKL inhibitor GW806742X (2 μM), in the presence of TNFα (10ng/mL), IFNγ (10ng/mL), or combined TNFα + IFNγ for 9 days. Media was changed every 3 days with the indicated drugs. After 9 days of culture, cells were fixed with 4% paraformaldehyde, and then stained with 0.5% crystal violet solution [20% methanol+0.5% crystal violet]. After washing excessive dye, crystal violet was extracted by using 10% acetic acid for 20 min incubation with shaking, diluted in water as necessary, and images captured using the LiCOR Odyssey (fluorescence at 590 nm) converted to gray scale with color inversion for final visualization.
Patient-derived organoid (PDO) generation, culture, and viability assessment.
Tumour specimens were minced and dissociated in S-MEM media (Gibco) supplemented with Liberase (1:20, Sigma) on a heater-shaker (37°C, 45 min), followed by resuspending and pelleting dissociated cells twice in DMEM/F12 supplemented with 10% FBS, 1% Pen/Strep and 1% glutamate. Organoids were generated and cultured as previously described60. Briefly, dissociated cells were seeded on a 24-well plate in Matrigel and cultured in basal organoid growth media (30% DMEM/F12 supplemented with 20% FBS, 50% WNT3A conditioned media, 20% R-spondin conditioned media, 1x B27, 1x N-2, 10mM nicotinamide (Sigma, N0636), 1.25mM N-acetyl-L-cysteine (Sigma, A9165), 100 mg/ml Primocin (InvivoGen, ant-pm-2), 0.5mM A83-01 (Tocris, 2939), 10nM Gastrin (Sigma, G9145), 4nM R-spondin (R&D Systems, 4645-RS-100), 4nM Noggin (R&D Systems, 6057-NG-100), 5nM fibroblast growth factor (R&D Systems, 345-FG-250), 5ng/ml epidermal growth factor (R&D Systems, 236-EG-200), 3μM p38i SB202190 (Sigma, S7067), 10mM Rho-kinase inhibitor Y-27632 (Sigma, Y0503). Patient-derived organoids serially passaged at confluence by mechanical disruption of cooled Matrigel domes (1hr, 4°C) using Corning Cell Recovery Solution (Corning). Organoids were dissociated enzymatically with Tryple E (Gibco) at 37°C for 5 minutes followed by brief (1-2 sec) mechanical disruption using a 20-gauge needle. Dissociated organoids were pelleted by centrifugation (200 x g, 5 minutes) in culture medium (DMEM/F12 + 10% FBS + 1 % Pen/Strep) prior to resuspension in Matrigel for plating or expansion. After Matrigel polymerization (37°C, 15 min), basal growth media with Rho-Kinase inhibitor was added. For patient-derived organoid viability assays, organoids were seeded at a concentration of 1 × 104 cells/well in a 96-well plate coated with Matrigel. The cells were incubated for 24 hours before addition of TNFα (10 ng/mL) + IFNγ (10 ng/mL), TBK1i (1 μM), or combination TNFα/IFNγ + TBK1i in Basal Growth Medium in 2% Matrigel in triplicate. Organoids were treated for 12 days in total with inhibitors/cytokines refreshed every 4 days. On Day 12, viability assessment was performed using the 3D CellTiter-Glo (Promega) kit according to manufacturer’s protocol by adding 80 μL of CellTiter-Glo 3D Reagent to each well. The plate was incubated at room temperature for 30-minutes before recording the luminescence via the plate reader.
BH3 Profiling.
B16 cells (Tbk1 sgRNA 1&2 and Control sgRNA 1&2) were isolated, centrifuged at 200g for 5 minutes and subjected to flow cytometry based BH3 profiling, as previously described61. Briefly, cells were treated with BIM (peptide sequence Ac-MRPEIWIAQELRRIGDEFNA-NH2) or PUMA (peptide sequence Ac-EQWAREIGAQLRRMADDLNA-NH2) BH3 peptides (Vivitide) for 60 minutes at 28°C in MEB (10 mM HEPES (Sigma Aldrich) pH 7.5, 150 mM mannitol (Sigma Aldrich), 50 mM KCl (Sigma Aldrich), 0.02 mM EGTA (Sigma Aldrich), 0.02 mM EDTA (Sigma Aldrich), 0.1% BSA (Jackson ImmunoResearch), 5 mM succinate (Sigma Aldrich)) with 0.001% digitonin (Sigma Aldrich). Alamethicin (Sigma Aldrich) at 0.25 μM and DMSO at 1% were used as positive and negative controls, respectively. After peptide exposure, cells were fixed in 2% paraformaldehyde for 15 minutes which was then neutralized by addition of N2 buffer (1.7 M Tris base, 1.25 M glycine, pH 9.1). Cells were stained overnight with DAPI (1:1000, Abcam) and anti-Cytochrome c-AlexaFluor647 (1:2000, clone 6H2.B4, Biolegend) in a saponin-based buffer (final concentration 0.1% saponin (Sigma Aldrich), 1% BSA) and then analyzed by flow cytometry. Cytochrome c release in response to treatment was measured on an Attune NxT flow cytometer (Thermo Fisher Scientific) from the DAPI+ population. Greater cytochrome c negative percentage indicates a greater response to peptide treatment. To evaluate the effect of TNFα + IFNγ on apoptotic priming, BH3 profiling was performed as above at the indicated time points following treatment with TNFα (160ng/mL) and IFNγ (40ng/mL).
Annexin V/Propidium Iodide viability assay.
Cells were plated at 104 cells per well in 100 μl culture media on flat-bottom 96-well plates (Nest Scientific) and treated with TNFα (160 ng/mL) and IFNγ (40 ng/mL) alone or in combination and co-treated with JAK1/2i (0.5 or 1 μM ruxolitinib) or TBK1i (0.25 or 1 μM). All combination treatments were performed with simultaneous administration. After 12, 18, 24 or 48 hours under standard tissue culture conditions, media was collected and moved to a fresh 96-well V-bottom plate (Greiner Bio-One). 25 μL 0.0025% Trypsin (Gibco) was added to each well on the original plate, allowed to incubate for 5 minutes, then trypsinized cells were added back to the media on the V-bottom plate and stained with viability markers AxV and PI using the following protocol. A staining solution was prepared with 10x Annexin binding buffer (0.1 M Hepes (pH 7.4), 1.4 M NaCl, and 25 mM CaCl2 solution, sterile filtered) and AxV/PI. AlexaFluor488-conjugated AxV (a gift from Prof. Anthony Letai, Dana-Farber Cancer Institute) and PI (Abcam) was added to solution at a dilution of 1:500. The staining solution was then added to the cells in solution at 1:10 dilution and the cells were allowed to stain for 20 minutes on ice in the dark. AxV/PI positivity was then measured on an Attune NxT flow cytometer equipped with an autosampler (Thermo Fisher).
Western blotting.
Whole-cell lysates were prepared in RIPA Lysis Buffer (Millipore Sigma, #20–188). Protein concentration was measured with a BCA protein assay kit (Pierce). Samples clarified by centrifugation and boiled at 95°C in 4x fluorescent compatible Sample Buffer (Invitrogen). Protein lysates (30–50 mg) were loaded onto 4–12% Bolt Bis-Tris Plus gels (Life Technologies) in MES buffer (Life Technologies). Protein was transferred to a PVDF membrane using iBLOT2 dry transferring system (Invitrogen). Membranes were blocked in Tris-buffered saline plus 0.1% Tween 20 (TBS-T) containing FL fluorescence blocking buffer (Thermo Scientific) for 1 h at room temperature followed by overnight incubation with primary antibody at 4 °C. After washing, membranes were incubated with Blocking Buffer, and IRDye 800CW or 680RD conjugated secondary antibodies. Membranes were then visualized using the Odyssey CLx scanner (LI-COR), then analysed using ImageJ and Adobe Photoshop software. All data shown are representative of three independent experiments.
RNA isolation and Quantitative RT-PCR.
For BMDM qRT-PCR studies, cells were seeded in 12-well plates and cultured for 7 days. On day 8 TBK1i was added and on day 9, LPS and IFNγ were added. After 2 hours LPS/IFNγ stimulation, total RNA was extracted from the cells with RNeasy plus kit (QIAGEN, cat#:74034) according to the manufacturer’s instructions. The mRNAs were quantified by using Power SYBR Green qPCR kit (Applied Biosystems, Cat#:4389986) with the company’s protocol on a Thermo (Applied Biosystems) QStudio 6 FLX real-Time PCR System-105969. The primers were used as following: Tnf, forward 5′-CCCTCACACTCAGATCATCTTCT −3′ and reverse 5′-GCTACG ACGTGGGCTACA G-3′; IL1a, forward 5′-CGAAGACTACAGTTCTGCCATT-3′; and reverse 5′GACGTTTCAGAGGTTCTCAGAG-3′; 18s, forward 5′-GCA ATT ATT CCC CAT GAA CG-3′ and reverse 5′-GGC CTC ACT AAA CCA TCC AA-3′. Levels of mRNAs of interest were normalized to 18s using the formula 2Ct (18s)–Ct(mRNA X). The resulting normalized ratio was presented in the figures. For B16 qRT-PCR studies, cells were seeded in 60mm dishes (2x106/dish) and cultured for 24h at 37°C. The cells were treated with TNFα (160 ng/mL) and IFNγ (40 ng/mL) or PBS for 19h. Total RNA was extracted from the cells with RNAeasy micro kit (QIAGEN, cat#:74004) according to the manufacturer’s instructions. The mRNAs were quantified using Blaze Taq one-step SYBR Green RT-qPCR kit (GeneCopoeia, Cat#:QP070) with the company’s protocol on a ROCHE Lightcycler-96. The primers were used as following: Mlkl, forward 5′-TGAGGGAACTGCTGGATAGA-3′ and reverse 5′-CCGAATGGTGTAGCCTGTATAA-3′; Ripk3, forward 5′-GCACTCCTCAGATTCCACATAC-3′; reverse 5′-GTGTCTTCCATCTCCCTGATTC-3′; β-actin, forward 5′-GAG GTA TCC TGA CCC TGA AGT A-3′ and reverse 5′-CAC ACG CAG CTC ATT GTA GA-3′. Levels of mRNAs of interest were normalized to β-actin using the formula 2Ct (β-actin)–Ct(mRNA X). The resulting normalized ratio was presented in the figures.
Antibodies.
For western blotting, primary antibodies against TBK1 (#ab40676, Abcam), IKKε (#3416T, Cell Signaling), p-RIPK S166/T169 (#31122S, Cell Signaling), RIPK1 (#3493S, Cell Signaling), cleaved caspase 8 (#9429S, Cell Signaling), cleaved caspase 3 (#9661T Cell Signaling), cleaved PARP (#6544, Cell Signaling), c-FLIP (#56343S, Cell Signaling), p-STAT1 Y701 (#9167S, Cell Signaling), STAT1 (#14994S, Cell Signaling), STING (#13647S, Cell Signaling), p-IRF3 (#29047S, Cell Signaling), IRF3 (#4302S, Cell Signaling) p-JAK1 (#74129T, Cell Signaling), JAK1 (#3344T, Cell Signaling), p-JAK2 (#8082T, Cell Signaling), JAK2 (#3230T, Cell Signaling), p-p65 (#3033T, Cell Signaling), p65 (#8242T, Cell Signaling), p-MLKL S345 (#37333, Cell Signaling), and MLKL (#37705, Cell Signaling). Primary antibodies were used at 1:1000 dilution in LI-COR Blocking Buffer. IRDye secondary antibodies against rabbit IgG, mouse IgG or goat IgG were purchased from LI-COR Biosciences (Invitrogen) and used at 1:10,000. β-actin-680 (#MA5-15739-D680, Invitrogen) was used as a loading control. Flow cytometry antibodies are listed above.
Plasma collection and OLINK plasma proteomics assay.
Metastatic melanoma patients at MGH provided written informed consent for the collection of blood samples (DF/HCC IRB approved Protocol 11-181). Whole blood was collected in BD Vacutainer CPT tubes (BD362753) before (n=179; 93 responders, 86 non-responders) and during treatment with immune checkpoint blockade after 6 weeks (n=173; 93 responders, 80 non-responders) and 6 months (n=151; 88 responders, 63 non-responders). Plasma (3 mL) was isolated after centrifuging CPT tubes containing whole blood 25-30 min at room temperature and was subsequently stored at −80 °C. Olink Proximity Extension Assay (PEA) for high-multiplex analysis of proteins was performed as previously described62. Briefly, oligonucleotide-labeled monoclonal or polyclonal antibodies (PEA probes) are used to bind target proteins in a pair-wise manner, facilitating hybridization when the oligonucleotides are in close proximity, followed by an extension step that generates a unique sequence used for digital identification of the analyte using next-generation sequencing (NGS). The full library (Olink ® Explore 1536) consists of 1472 proteins and 48 control assays, divided into four 384-plex panels. Four overlapping assays of IL-6, IL-8 (CXCL8), and TNF are included for quality control (QC) purposes. In the immune reaction, 2.8 mL of sample is mixed with PEA probes and incubated overnight at 4°C. NPX is Olink’s relative protein quantification unit on a log2 scale and values are calculated from the number of matched counts on the NovaSeq run. Data generation of NPX consists of normalization to the extension control (known standard), log2-transformation, and level adjustment using the plate control (plasma sample).
Secreted Cytokine Profiling.
Multiplexed analysis of secreted cytokines was performed using the MILLIPLEX MAP Human Cytokine/Chemokine Magnetic Bead Panel (Cat# HCYTMAG-60K-PX30). Conditioned media samples (25 μL) from PDOTS were assayed neat. Concentration levels (pg/mL) of each protein were derived from 5-parameter curve fitting models. Fold changes relative to control samples were calculated and plotted as log2FC (L2FC). Lower and upper limits of quantitation (LLOQ/ULOQ) were imputed from standard curves for cytokines above or below detection.
CRISPR sgRNA sequences.
Target sequences for CRISPR interference were designed using the sgRNA designer (http://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design).
Control sgRNA 1 ATTGTTCGACCGTCTACGGG
Control sgRNA 2 ACGTGTAAGGCGAACGCCTT
Tbk1 sgRNA 1 CGGGAACAACTCAATACCGT
Tbk1 sgRNA 2 GACCGTCCACAAGAAGACGG
Tmem173 (STING) sgRNA 1 GAAGGCCAAACATCCACTG
Irf3 sgRNA 1 GCATGGAAACCCCGAAACCG
Sanger sequencing.
B16 clones were harvested and DNA was extracted using 50 uL QuickExtract DNA Extraction Solution (Lucigen). PCR was performed on 1 μL of the extracted DNA solution using Herculase II Fusion DNA Polymerase (Agilent Technologies) according to the manufacturer’s protocol for targets < 1 kb. PCR primers were designed to target the region of the Tbk1 gene flanking the expected CRISPR/Cas9 cut site. Tbk1 sgRNA 1 cut site forward primer CCTTCTGACGTCCCTCACAG. Tbk1 sgRNA 1 cut site reverse primer ACTGGTGAAAGTTATGATGGA
PCR products were purified using QIAquick PCR Purification Kit (Qiagen). PCR bands were visualized using the E-Gel Power Snap Electrophoresis System (Invitrogen). Sanger sequencing was performed at the MGH CCIB DNA Core using an ABI 3730XL DNA Analyzer (Thermo Fisher). Sanger sequencing results were analyzed using Synthego Inference of CRISPR Edits (ICE)63 and Tracking of Indels by DEcomposition (TIDE)64 software tools to determine insertions and deletions at the CRISPR cut site. Next-generation sequencing (NGS) was subsequently performed to confirm Sanger sequencing results.
Source data.
Single-cell RNA-seq data for CD45+ cells (SMART-Seq2) from melanoma patients treated with immune checkpoint blockade was previously described23 [accession number GEO: GSE120575 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120575)]. In each sample, the fraction of cells with a non-zero expression of either IFNG, TNF or both was calculated. The Wilcoxon rank-sum test was then used to determine the significance level of the difference between responding and non-responding patients. The Broad Single Cell Viewer (https://singlecell.broadinstitute.org/single_cell) was used for evaluation of TBK1 and IKBKE expression in CD45+ immune cells from human melanoma patients. Bulk RNA sequencing data from B16 cells treated with TNFα, IFNγ, or TNFα/IFNγ compared to untreated control was previously described [accession number GEO: GSE99299 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE99299)].
Statistical Methods, Data Analysis, and Software.
Statistical tests employed with the number of replicates and independent experiments are listed in the text and figure legends. All graphs with error bars report mean ± s.e.m. values except where indicated. t-tests were two-tailed in all cases. GraphPad/Prism (v9.0) was used for basic statistical analysis and plotting (www.graphpad.com). The R language and programming environment (www.r-project.org) was used for the remainder of the statistical analysis. Multiple hypothesis testing correction was applied where multiple hypotheses were tested and is indicated by the use of FDR. Data analysis software used included GraphPad/Prism (v9.0), Microsoft Excel(v15), FlowJo (v10), NIS Elements (v5.11), Cell Ranger (v3.0), Scanpy (v1.4.5post3), scran (v1.14.6), and MetaXpress (v6.5.3.427). Schematics generated using BioRender (biorender.com) using a purchased academic license.
Data availability.
The datasets generated and analyzed in this study are included in the manuscript. In vivo scRNA-seq data have been deposited in Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) under the accession codes GSE217160 (in vivo TBK1i study) and GSE217274 (in vivo TBK1 CRISPR-Cas9 study) and are available upon request. Descriptions on the analyses are provided in the Methods and Reporting Summary section.
Extended Data
Extended Data Fig. 1 |. Supporting evidence that loss of TBK1 sensitizes tumours to cancer immunotherapy.
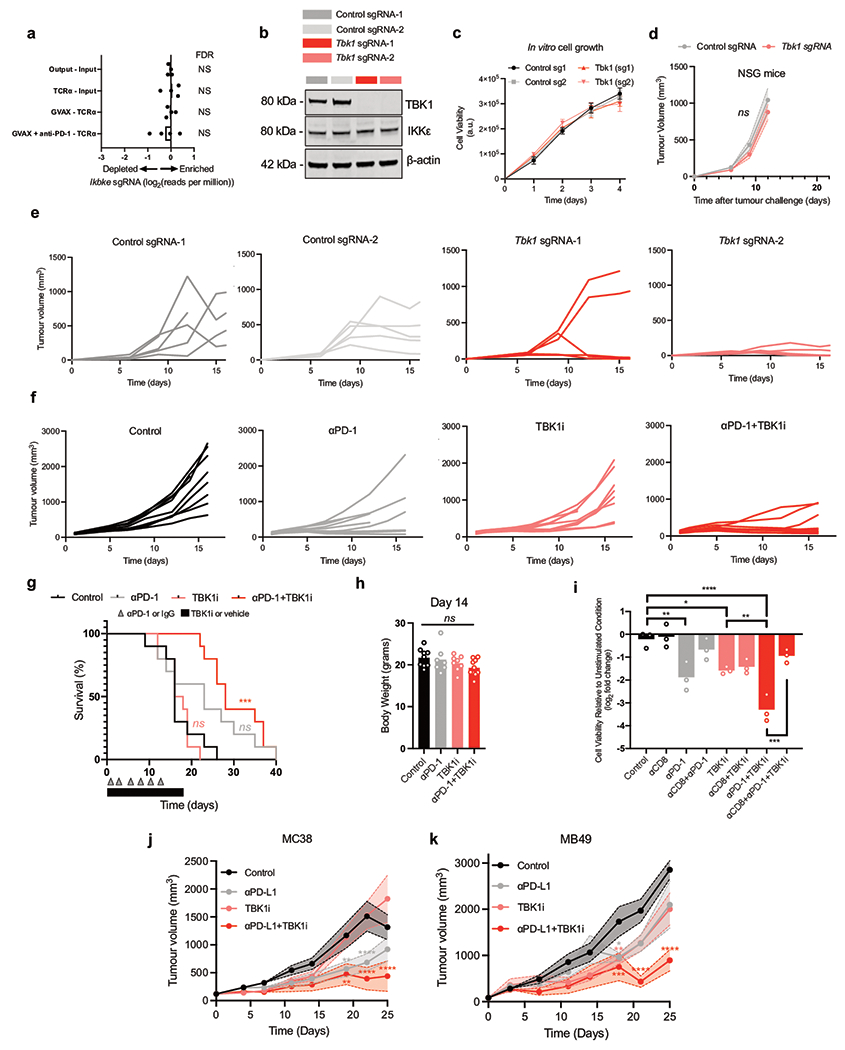
a, Relative depletion/enrichment of Ikbke sgRNAs from a pool of sgRNAs targeting 2,368 genes expressed by Cas9-expressing B16 melanoma cells (n = 4 independent guides targeting each gene; false discovery rate (FDR) was calculated using the STARS algorithm v1.3, as previously described 6,7). b, TBK1 and β-actin protein levels in control and Tbk1-null B16 cells. Results are representative of three independent experiments. c, Proliferation of Tbk1-null and control B16 tumour cells following at 1-4 days of in vitro culture (n = 9 per condition from three independent experiments). d, Tumour volume of control (grey), Tbk1-null (light red) B16 tumours in NSG mice (n=5 mice per group). Mean tumour volumes (solid circles) are shown +/− s.e.m. (shaded region). 2-way ANOVA with Sidak’s multiple comparisons test. e, Spider plots for tumour volume analysis for control sgRNA-1 (black), sgRNA-2 (grey), Tbk1 sgRNA-1 (pink), and Tbk1 sgRNA-2 (red) B16 tumours in anti-PD-1-treated wild-type C57BL/6 mice (see Fig. 1c). f-g, Spider plots for tumour volume analysis (f) and survival (g) for control (black), αPD-1 (grey), TBK1i (pink), and αPD-1+TBK1i (red) B16 tumours in C57BL/6 mice (see Fig. 1d). For survival analysis (g), pairwise testing was performed using the log-rank (Mantel-Cox) test for survival (g); n=10 mice per treatment group, ***P < 0.001; ns, not significant, compared to control group. h, body weight of mice bearing B16-ova tumours on Day 14 of indicated treatment. Means (bars) and individual values (open circles) are shown (n = 10 mice per group, 1-way ANOVA with Tukey’s multiple comparisons test; ns, not significant). i, Viability assessment of CT26 MDOTS with indicated treatments. Means (bars) and individual values (open circles) are shown (n = 3, biological replicates, one-way ANOVA with Tukey’s multiple comparisons test; *P < 0.05; **P < 0.01; ***P < 0.001; **** P < 0.0001). j-k, Tumour volume analyses of mice bearing MC38 (j) and MB49 (k) tumours treated with TBK1i, αPD-L1, or combination compared to control (IgG + vehicle); n=10 mice per treatment group. Mean tumour volumes (solid circles) are shown +/− s.e.m. (shaded region). 2-way ANOVA with Tukey’s multiple comparisons test ***P < 0.001; compared to control group.
Extended Data Figure 2 |. Supporting data that TBK1 inhibition enhances sensitivity to PD-1 blockade using PDOTS.
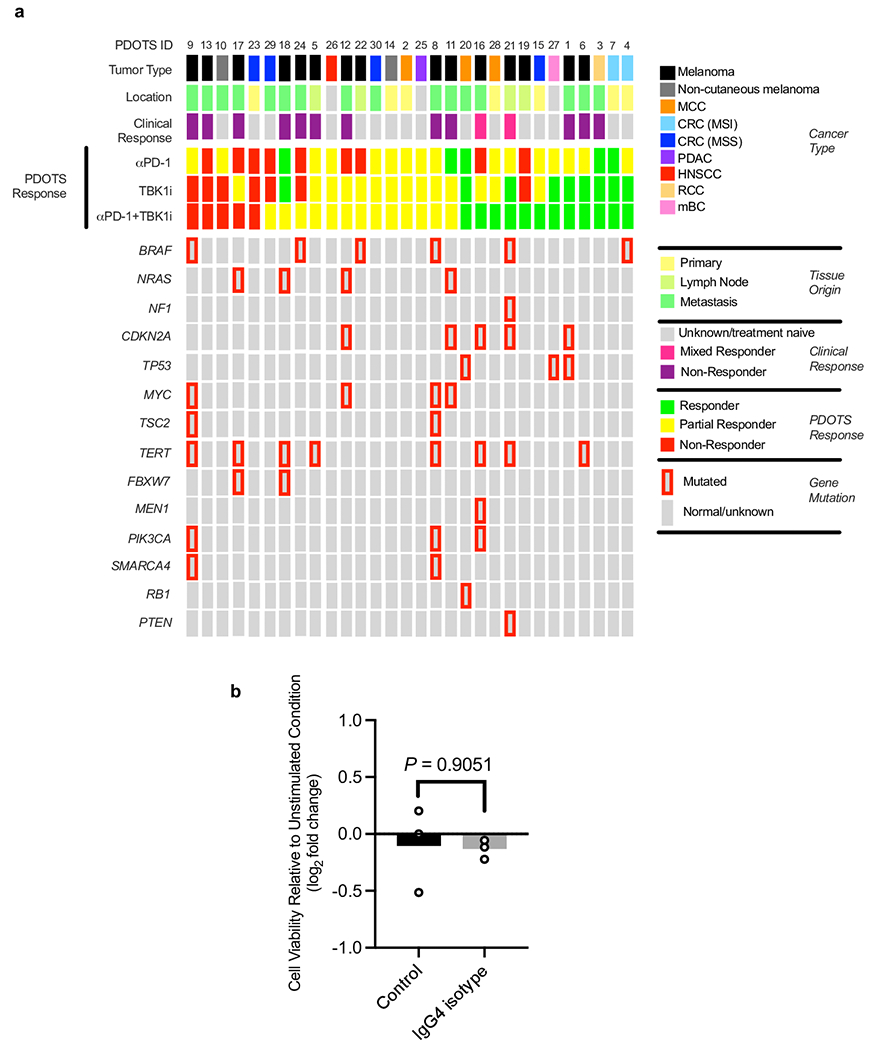
a, Tumour type, tissue source (location), clinical response data, PDOTS response data, and associated tumour mutation profile for specimens used for PDOTS profiling (samples ordered by ex vivo PDOTS response to combined αPD-1+TBK1i). PDOTS response parameters defined as follows: responder (reduction >30% compared to control), partial responder (<30% reduction and <20% growth compared to control), and non-responder (>20% growth compared to control). Red border around grey rectangle indicates presence of alteration in indicated gene. b, effect of IgG4 control monoclonal Ab on viability of PDOTS from a patient with melanoma. Means (bars) and individual values (open circles) are shown (n = 3, biological replicates, 2-sided unpaired t-test).
Extended Data Figure 3 |. Effect of TBK1 inhibition on the tumour immune microenvironment.
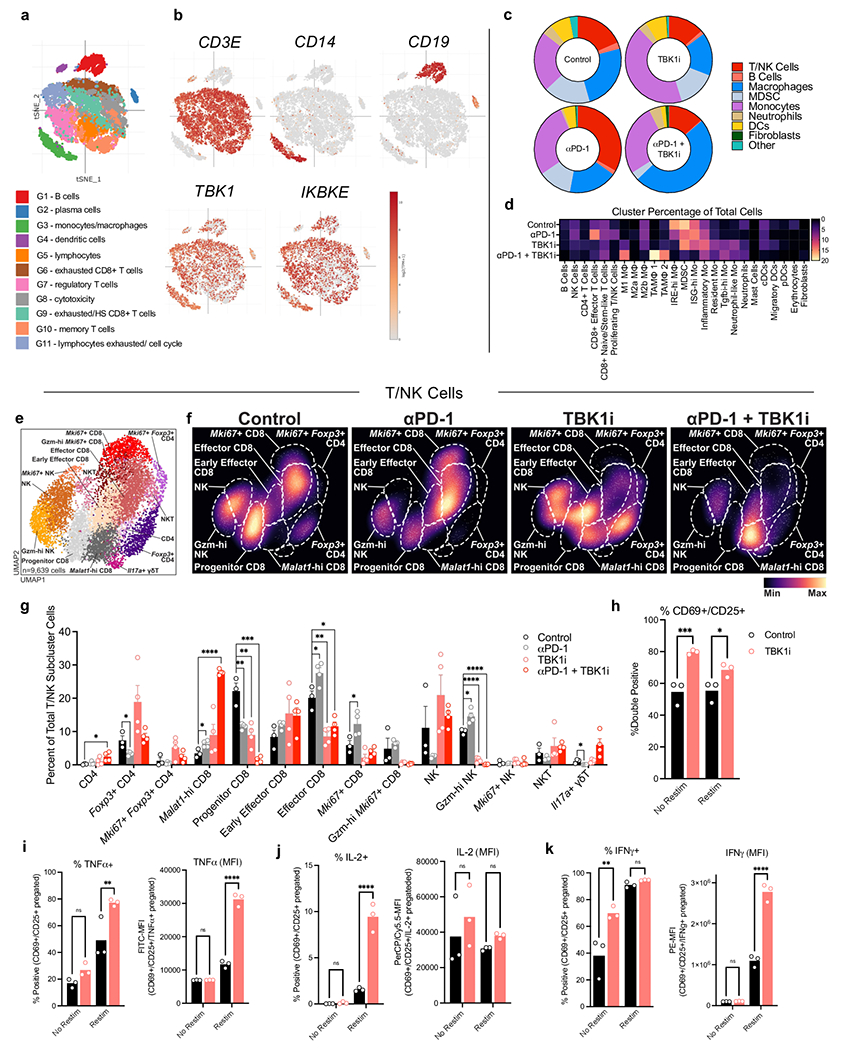
a-b, tSNE plot of 11 clusters of CD45+ cells (a) from patients with metastatic melanoma responsive (R) or non-responsive (NR) to immune checkpoint blockade (ref. Sade-Feldman et al. 2018), and t-SNE plots of RNA-sequenced single cells with colouring of CD3E (T cells), CD14 (myeloid cells), and CD19 (B cells) TBK1 and IKBKE expression (b). c-d, broad cluster proportions (c) and percent cells per cluster across indicated treatment groups (d). e-f, UMAP (c) and density (d) plots of reclustered lymphoid (T/NK) cells. g, cluster proportions of lymphoid (T/NK) cells. Means (bars) and individual values (circles) are shown +/− s.e.m (error bars). Multiple unpaired t-test, *P < 0.05; **P < 0.01; ***P < 0.001; **** P < 0.0001; ns, not significant. h, percentage of activated (CD69+CD25+) murine CD8+ splenocytes pre-treated with TBK1i (1 μM) or DMSO (0.1%) with/without restimulation; n=3 biologically independent samples, 2-way ANOVA, Sidak’s multiple comparisons test; *P < 0.05; ***P < 0.001. i-k, intracellular cytokine staining for TNFα (i), IL-2 (j), and IFNγ (k) of murine CD3+CD8+ splenocytes pre-treated with TBK1i (1 μM) or DMSO (0.1%) with/without restimulation with data shown as % CD69+CD25+ cells and MFI); n=3 biologically independent samples, 2-way ANOVA, Sidak’s multiple comparisons test; **P < 0.01; **** P < 0.0001; ns, not significant.
Extended Data Figure 4|. Effect of Tbk1 deletion on the tumour immune microenvironment.
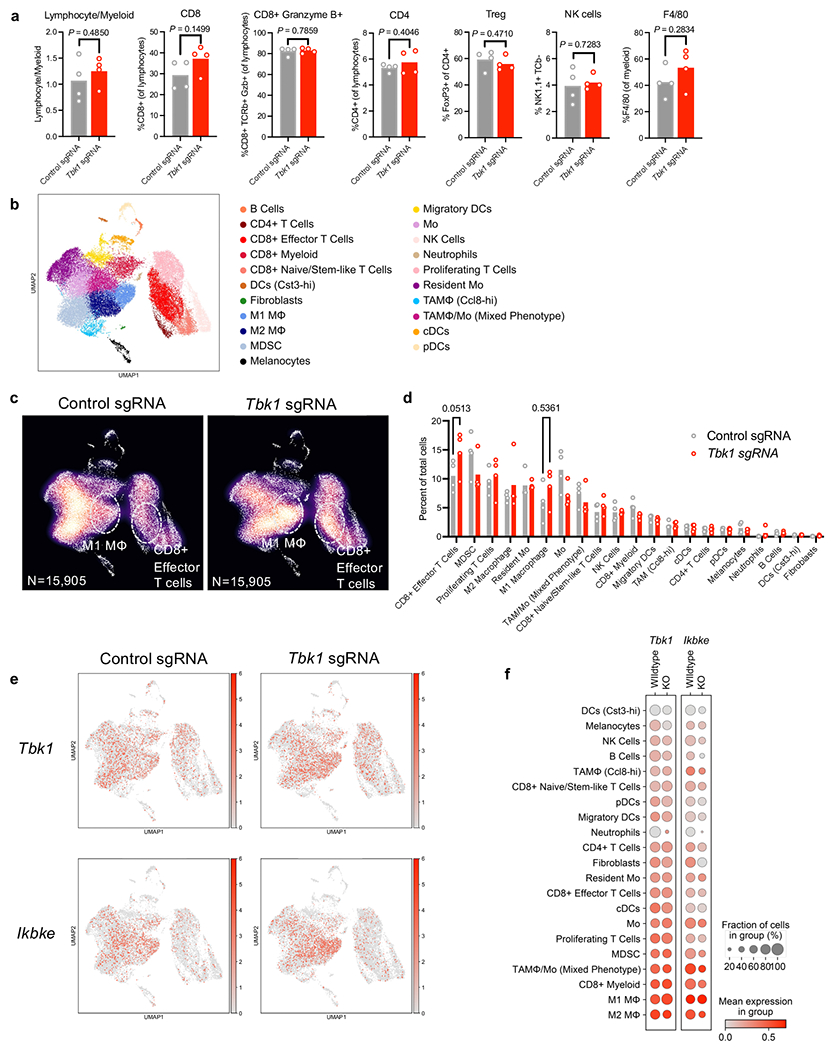
a, Flow cytometry of immune populations from control and Tbk1-null B16 tumours treated with anti-PD-1 (n=4 per group). Means (bars) and individual values (open circles) are shown (n = 4 biologically independent samples, 2-sided unpaired t-test). b-c, UMAP (b) and density (c) plots of 31,810 RNA-sequenced single cells from control and Tbk1-null B16 tumours following anti-PD-1 treatment (DC, dendritic cells; Tregs, regulatory T cells; MDSC, myeloid-derived suppressor cell; NK, natural killer cells; M1, M1 macrophages; M2, M2 macrophages). d, percent of cells in each lineage-defined cluster. Means (bars) and individual values (open circles) are shown (n = 4 biologically independent samples, 2-way ANOVA, Sidak’s multiple comparisons test; P values shown for M1 macrophages and CD8 T cells that did not reach statistical significance). e, UMAP plot of RNA-sequenced single cells with colouring of Tbk1 and Ikbke expression with cell types referenced (b). f, bubble plot indicating Tbk1 and Ikbke expression across UMAP-defined cell clusters.
Extended Data Figure 5|. TNFα and IFNγ expression in B16 melanoma tumours.
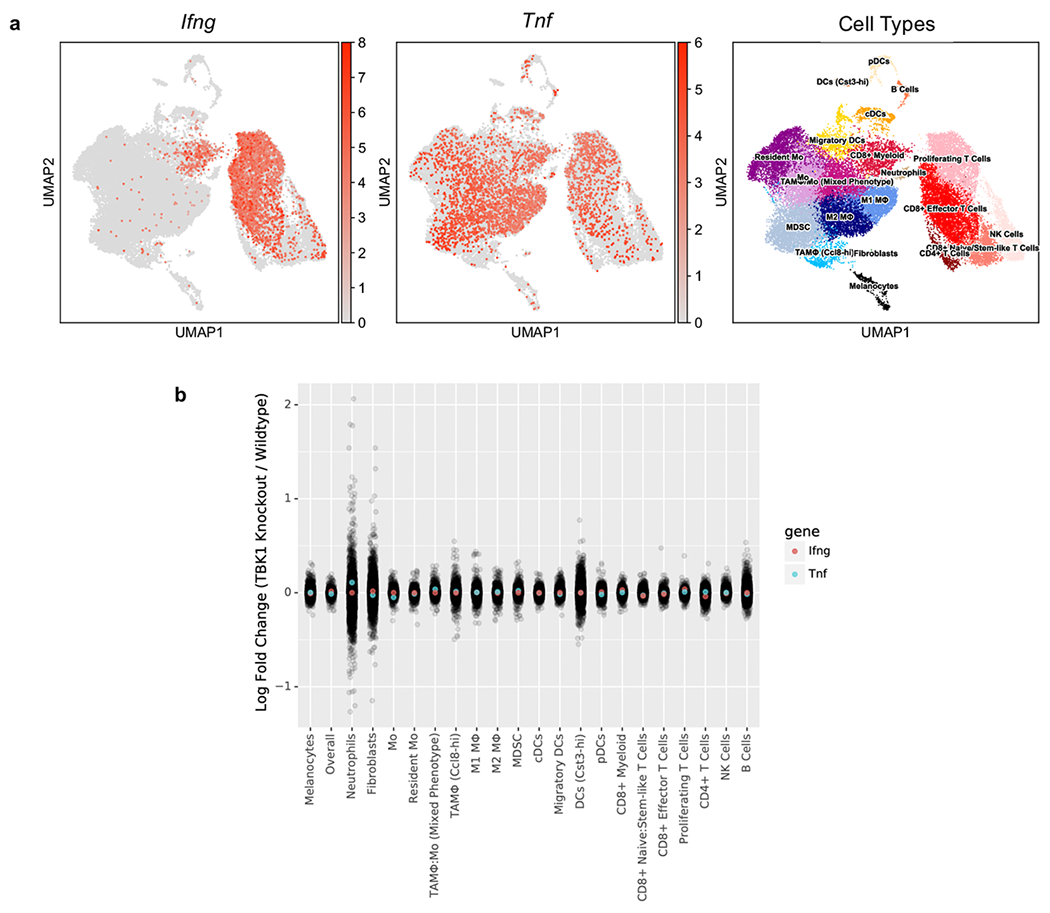
a, UMAP plot of RNA-sequenced single cells with colouring of Ifng and Tnfa expression with cell types referenced (right). b, log-fold change of Ifng (light red) and Tnfa (light blue) expression across lineage-defined cell clusters (Tbk1-null/control).
Extended Data Figure 6|. Supporting data that loss/inhibition of TBK1 sensitizes tumour cells to TNFα/IFNγ.
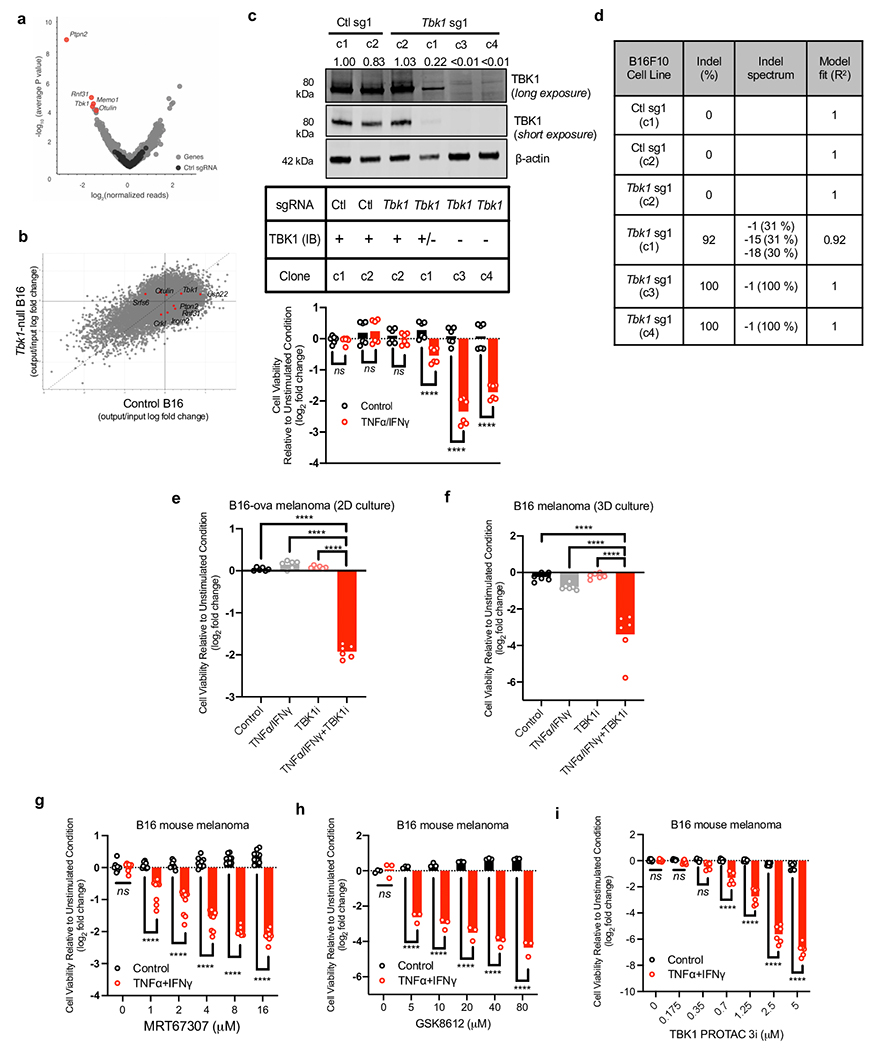
a, volcano plot depicting relative sgRNAs gene depletion/enrichment. Top 5 depleted sgRNAs indicated. b, scatter plot of gene essentiality from in vitro CRISPR screen (control and Tbk1-null B16 cells). c, TBK1 expression and cell viability (control vs. TNFα/IFNγ;) for single cell clones derived from polyclonal control and Tbk1-null B16 cells. Western blot is representative of three independent experiments. Means (bars) and individual values (open circles) are shown (n=6 across two independent experiments, 2-way ANOVA, Sidak’s multiple comparisons test; **** P < 0.0001; ns, not significant). d, TBK1 indel spectrum from control sgRNA and Tbk1 sgRNA B16 single cell clones. e, Viability assessment (Cell Titer Glo) of B16-ova cells in standard 2D culture after 24 hours treatment with TNFα (160 ng/mL) + IFNγ (40 ng/mL) compared to unstimulated cells (n=6, 2 independent experiments, 1-way ANOVA, Holm-Sidak’s multiple comparisons test). f, Viability assessment (Hoechst/propidium iodide) of B16 tumour spheroids (lacking immune cells) in 3D microfluidic culture after 96 hours treatment with TNFα (10 ng/mL) + IFNγ (10 ng/mL) compared to unstimulated cells (n=6, 2 independent experiments, 1-way ANOVA, Holm-Sidak’s multiple comparisons test). g, Cell viability assessment of B16 cells after 24 hours treatment with TNFα (200 ng/mL) + IFNγ (40 ng/mL) compared to unstimulated cells treated with increasing concentrations of MRT67307 (n=9, 3 independent experiments 2-way ANOVA, Sidak’s multiple comparisons test). h, Cell viability assessment of B16 cells in standard 2D culture after 24 hours treatment with TNFα (200 ng/mL) + IFNγ (40 ng/mL) compared to unstimulated cells treated with increasing concentrations of GSK8612 (n=3, 1 independent experiment, 2-way ANOVA, Sidak’s multiple comparisons test). i, Cell viability assessment of B16 cells in standard 2D culture after 24 hours treatment with TNFα (200 ng/mL) + IFNγ (40 ng/mL) with increasing concentrations of TBK1 PROTAC 3i (n=6, 2 independent experiments 2-way ANOVA, Sidak’s multiple comparisons test). **** P < 0.0001; ns, not significant.
Extended Data Figure 7|. Supporting data that TBK1 inhibition lowers the cytotoxicity threshold to TNFα/IFNγ.
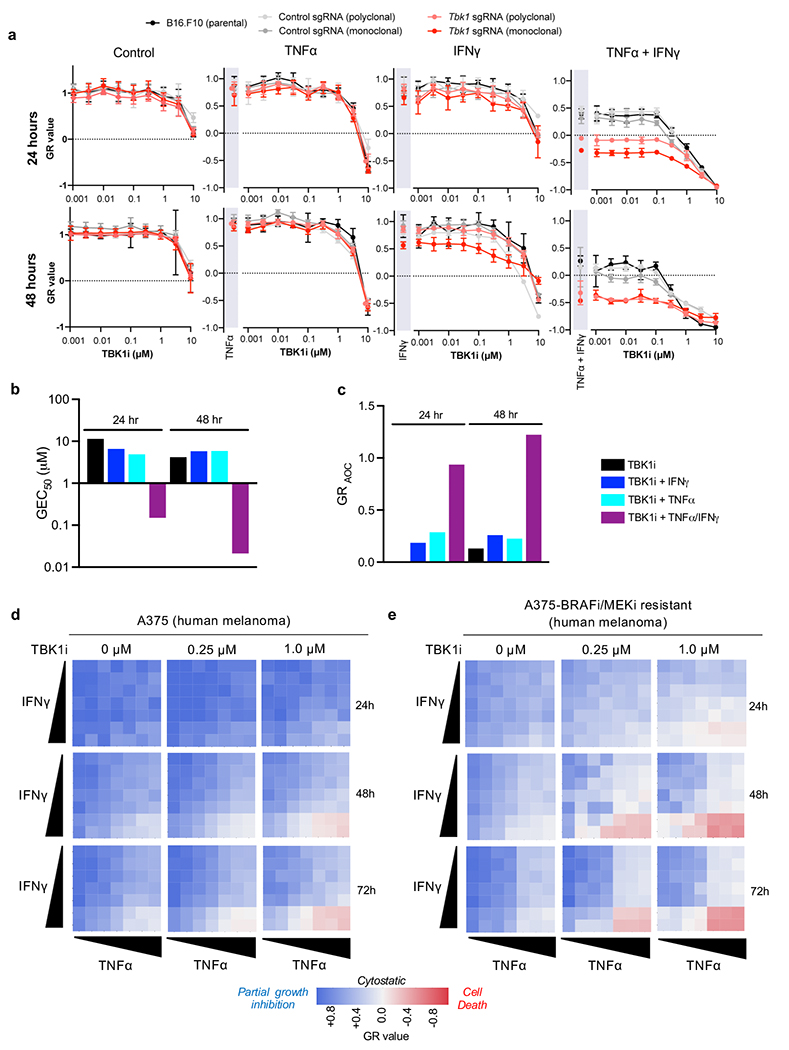
a, GR values for 9-point inhibitor titration of TBK1i in parental, control sgRNA (polyclonal and monoclonal), and Tbk1 sgRNA (polyclonal and monoclonal) B16 cells (2 independent experiments; representative data from single experiment with 6 technical replicates per condition). Means (solid circles) are shown +/− s.e.m (error bars). b-c, evaluation of TBK1i potency (b; half-maximal effect, GEC50) and overall efficacy (c; area over the GR curve, GRAOC) d-e, Heatmap of GR values for parental (d) and BRAF/MEK inhibitor resistant (e) A375 human melanoma cells treated with increasing concentrations of TNFα and IFNγ for 24, 24, and 72 hours with 0, 0.25, and 1.0 μM TBK1i (n=3).
Extended Date Figure 8|. Supporting data that Tbk1-null cells undergo RIPK- and caspase-dependent cell death.
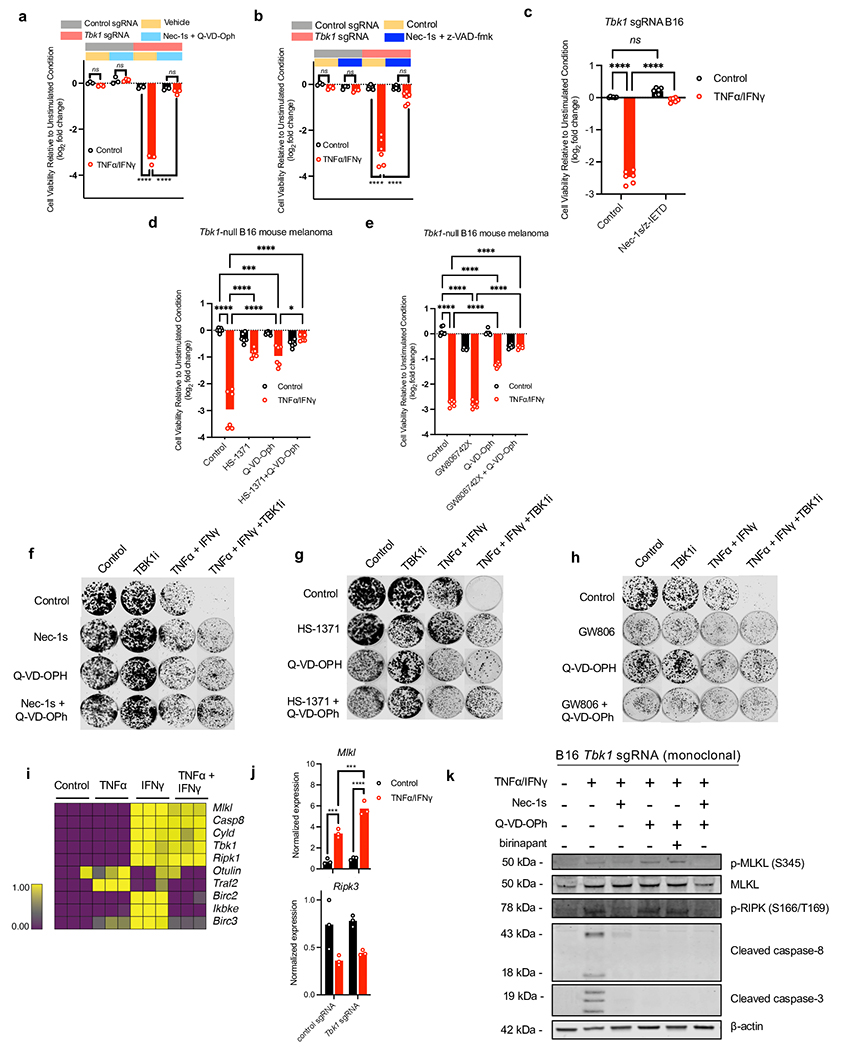
a-b, Cell viability assessment (Cell Titer Glo) in control and Tbk1-null B16 cells pre-treated with RIPK1 inhibitor (Nec-1s, 10 μM) and the pan-caspase inhibitor Q-VD-OPh (10 μM) +/− TNFα/IFNγ (n=3, 1 independent experiment: 2-way ANOVA, Dunnett’s multiple comparisons test). b, cell viability assessment (Cell Titer Glo) in control and Tbk1-null B16 cells pre-treated with RIPK1 inhibitor (Nec-1s, 10 μM) and the pan-caspase inhibitor z-VAD-fmk (20 μM) +/− TNFα/IFNγ (n=3-6, 1-2 independent experiments: 2-way ANOVA, Dunnett’s multiple comparisons test). c, cell viability assessment in Tbk1-null B16 cells pre-treated with RIPK1 inhibitor (Nec-1s, 10 μM) and the caspase 8 inhibitor z-IETD-fmk (2.5 μM) +/− TNFα/IFNγ (n=6, 2 independent experiments; 2-way ANOVA, Dunnett’s multiple comparisons test). d, cell viability assessment in Tbk1-null B16 cells pre-treated with RIPK3 inhibitor (HS-1371, 2 μM) and the pan-caspase inhibitor Q-VD-OPh (20 μM) +/− TNFα/IFNγ (n=6, 2 independent experiments: 2-way ANOVA, Dunnett’s multiple comparisons test). e, cell viability assessment in Tbk1-null B16 cells pre-treated with MLKL inhibitor (GW806742X, 5 μM) and the pan-caspase inhibitor Q-VD-OPh (20 μM) +/− TNFα/IFNγ (n=6, 2 independent experiments: 2-way ANOVA, Dunnett’s multiple comparisons test). f-h, Clonogenic assay of B16 cells treated with TNFα (10 ng/mL), IFNγ (10 ng/mL), or TNFα + IFNγ with control (0.1% DMSO), Q-VD-OPh (20 μM) with/without the RIPK1 inhibitor Nec-1s (10 μM, f), RIPK3 inhibitor HS-1371 (2 μM, g), and MLKL inhibitor GW806742X (2 μM, h) (representative images shown; n=3). i, normalized expression of selected genes in B16 cells treated with TNFα (10 ng/mL), IFNγ (100 ng/mL), or both, compared to control cells (source data for bulk RNA-seq – Manguso et al. 2017). j, normalized expression of Mlkl and Ripk3 in control and Tbk1-null B16 cells with/without TNFα/IFNγ treatment (18 hours) determined by qRT-PCR (n=3; 2-way ANOVA, Sidak’s multiple comparison test). *P < 0.05; **P < 0.01; ***P < 0.001; **** P < 0.0001; ns, not significant. k, Western blot of indicated proteins in Tbk1-null B16 cell lysates following 2-hour pre-treatment with vehicle control (0.1%DMSO), Q-VD-OPh (20μM), Nec-1s (10 μM), or Q-VD-OPh plus Nec-1s, or Q-VD-OPh plus birinapant (1 μM) followed by 10 hour treatment with TNFα (160 ng/mL) and IFNγ (40 ng/mL) or unstimulated (PBS) control. Data are representative of three independent experiments.
Extended Date Figure 9|. Supporting data regarding TNFα/IFNγ-induced cell death signaling in control and Tbk1-null cells.
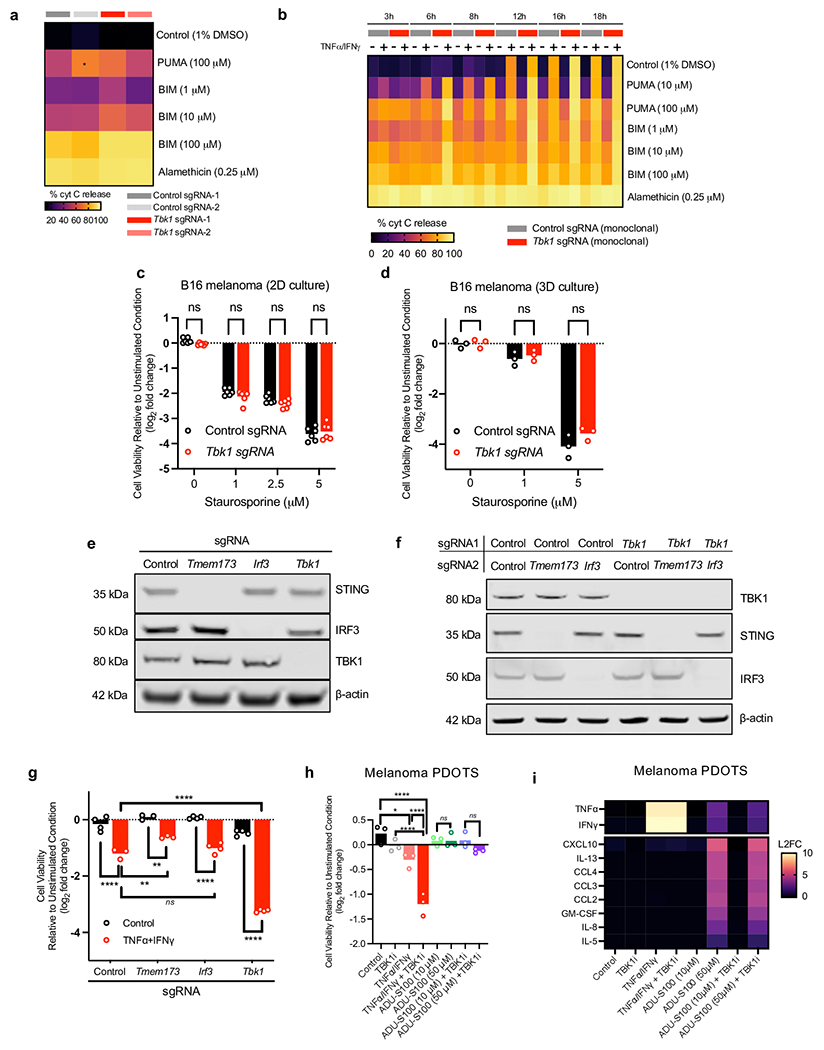
a, heatmap of % cytochrome C (cyt C) release for in vitro BH3 profiling of unstimulated control (sg1 and sg2) and Tbk1-null (sg1 and sg2) B16 cells. Mean values shown; n=3 biologically independent samples; 2-way ANOVA, Dunnett’s multiple comparisons test. b, heatmap of % cytochrome C (cyt C) release for in vitro BH3 profiling of control sgRNA and Tbk1 sgRNA B16 cells. Mean values shown; n=3 biologically independent samples; 2-way ANOVA, Tukey’s multiple comparisons test. No statistically significant differences observed between control sgRNA and Tbk1 sgRNA B16 cells at any time point. c, Viability assessment (Cell Titer Glo) of B16 cells in standard 2D culture after 24 hours treatment with indicated concentrations of staurosporine (STS) in control and Tbk1-null B16 cells. Means (bars) and individual values (open circles) are shown (n=6, 2 independent experiments, 2-way ANOVA, Sidak’s multiple comparisons test). d, Viability assessment (Hoechst/propidium iodide) of B16 tumour spheroids (lacking immune cells) in 3D microfluidic culture after 48 hours treatment indicated concentrations of staurosporine (STS) compared to unstimulated cells Means (bars) and individual values (open circles) are shown (n=6, 2 independent experiments, 1-way ANOVA, Holm-Sidak’s multiple comparisons test). e, Western blot for STING, IRF3, TBK1, and β-actin in B16 cells with single CRISPR cell lines with single-guide RNAs targeting Tmem173, Irf3, and Tbk1 compared to control sgRNA. Data are representative of three independent experiments. f, Western blot for STING, IRF3, TBK1, and β-actin in double CRISPR B16 cells with indicated sgRNA pairs. Data are representative of three independent experiments. g, Viability assessment (Cell Titer Glo) of indicated sgRNA B16 cells after 48 hours treatment with TNFα (160 ng/mL) + IFNγ (40 ng/mL) compared to unstimulated cells. Means (bars) and individual values (open circles) are shown (n = 4 biological replicates, 2-way ANOVA, Sidak’s multiple comparisons test, **P < 0.01; **** P < 0.0001; ns, not significant). h, PDOTS viability assessment from patients (n=2) with cutaneous melanoma with indicated treatments. Means (bars) and individual values (open circles) are shown (n = 6 biological replicates, 2 independent specimens; one-way ANOVA with Dunn’s multiple comparisons test, **P < 0.01; **** P < 0.0001; ns, not significant). i, heatmap of secreted cytokine profiles (L2FC) of conditioned media from PDOTS in response to indicated treatments (n=2). Mean values shown. **P < 0.01; **** P < 0.0001; ns, not significant.
Extended Date Figure 10|. Supporting data that IFNγ sensing is essential for effector cytokine-induced death in TBK1-null cells.
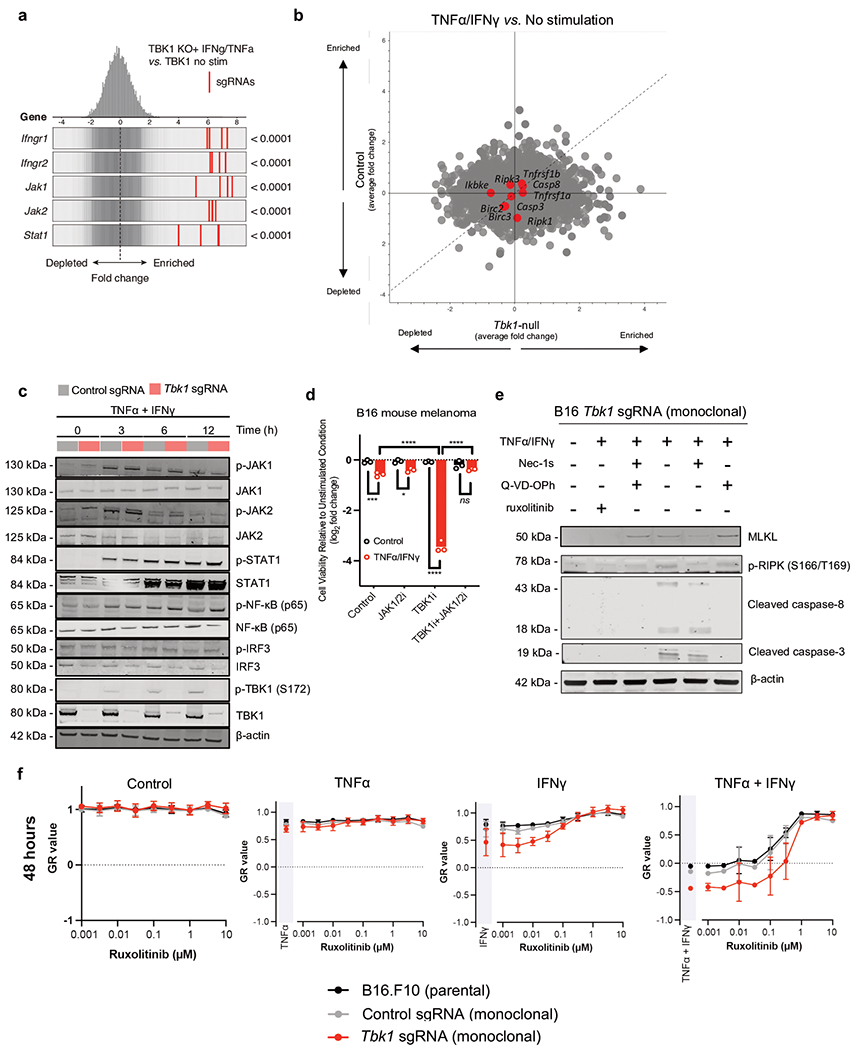
a, Frequency histograms of enrichment (z-score) for all sgRNAs per target in a Tbk1-null B16 cells +/− in vitro stimulation with TNFα (10ng/mL) and IFNγ (10ng/mL). b, scatter plot depicting relative depletion of sgRNAs targeting 19,674 genes in a Cas9+ B16 control and Tbk1 sgRNA cell line +/− in vitro stimulation with TNFα (10ng/mL) and IFNγ (10ng/mL). c, Western blot of control sgRNA and Tbk1-null B16 cells treated with TNFα (160 ng/mL) and IFNγ (40 ng/mL) for the indicates times. Data are representative of three independent experiments. d, cell viability assessment in parental B16 cells pre-treated with TBK1i (1μM) +/− JAK 1/2 inhibitor (ruxolitinib, 0.5 μM) +/− TNFα/IFNγ for 48 hours compared to unstimulated controls. Means (bars) and individual values (open circles) are shown (n=3, 1 independent experiment; 2-way ANOVA, Dunnett’s multiple comparisons test; *P < 0.05; ***P < 0.001; **** P < 0.0001; ns, not significant). e, Western blot of indicated proteins in Tbk1-null B16 cell lysates following 2-hour pre-treatment with vehicle control (0.1%DMSO), ruxolitinib (1 μM), Q-VD-OPh (20μM), Nec-1s (10 μM), or Q-VD-OPh plus Nec-1s followed by 10-hour treatment with TNFα (160 ng/mL) and IFNγ (40 ng/mL) or unstimulated (PBS) control. Data are representative of three independent experiments. f, GR values for 9-point inhibitor titration of ruxolitinib (JAK1/2i) in parental, control sgRNA (monoclonal), and Tbk1 sgRNA (monoclonal) B16 cells (2 independent experiments; representative data from single experiment with 6 technical replicates per condition). Means (solid circles) are shown +/− s.e.m (error bars).
Supplementary Material
Acknowledgements
This work was supported by NIH K08CA226391 (R.W.J.), P01CA24023 (S.I.P.), K99CA259511 (K.P.), T32CA207021 (J.H.C.), 5R01AR072304 (D.E.F.), 5P01CA163222 (D.E.F.), 5R01AR043369 (D.E.F.), and 5R01CA222871 (D.E.F.). Additional support provided by Melanoma Research Alliance Young Investigator Award (https://doi.org/10.48050/pc.gr.86371, R.W.J.), Karin Grunebaum Cancer Research Foundation Faculty Research Fellowship (R.W.J.), Termeer Early Career Fellowship in Systems Pharmacology (R.W.J.), and a generous gift from Steven B. And Joan W. Belkin. K.P. gratefully acknowledges support from the German Research Foundation (DFG), Stand Up to Cancer Peggy Prescott Early Career Scientist Award PA-6146, Stand Up to Cancer Phillip A. Sharp Award SU2C-AACR-PS-32. D.J. gratefully acknowledges the Susan Eid Tumor Heterogeneity Initiative and D.E.F. gratefully acknowledges grant support from the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation. The funding bodies had no role in the design of the study, and collection, analysis, and interpretation of the data, or in writing the manuscript. The authors thank all members of the Manguso and Jenkins laboratories at MGH, HMS, and the Broad Institute. Graphics in Fig. 2a and 5i were created with Biorender.com using a paid license.
Competing Interests
R.W.J. is a member of the advisory board for and has a financial interest in Xsphera Biosciences Inc., a company focused on using ex vivo profiling technology to deliver functional, precision immune-oncology solutions for patients, providers, and drug development companies. A.M. is a consultant for Third Rock Ventures, LLC., Asher Biotherapeutics and Abata Therapeutics. A.M. holds equity in Asher Biotherapeutics and Abata Therapeutics. S.I.P. has received consultancy payments from Abbvie, Astrazeneca/MedImmune, Cue Biopharma, Fusion Pharmaceuticals, MSD/Merck, Newlink Genetics, Oncolys Biopharma, Replimmune, Scopus Biopharma, and Sensei Biopharma. She has received grants/research support from Abbvie, Astrazeneca/MedImmune, Cue Biopharma, Merck, and Tesaro. S.J.K. has served a consultant/advisory role for Eli Lilly, Merck, BMS, Astellas, Daiichi-Sankyo, Pieris, and Natera. S.J.K. owns stock in Turning Point Therapeutics, Inc. D.J. received consulting fees from Novartis, Genentech, Syros, Eisai, Vibliome, Mapkure, and Relay Therapeutics. D.J. conducted contracted research with Novartis, Genentech, Syros, Pfizer, Eisai, Takeda, Pfizer, Ribon Therapeutics, Infinity, InventisBio, and Arvinas. D.J. has ownership interest in Relay Therapeutics and PIC Therapeutics. D.M.M. has received honoraria for participating on advisory boards for Checkpoint Therapeutics, EMD Serono, Castle Biosciences, Pfizer, Merck, Regeneron, and Sanofi Genzyme. D.M.M. owns stock for Checkpoint Therapeutics. D.E.F. has a financial interest in Soltego, a company developing salt inducible kinase inhibitors for topical skin-darkening treatments that might be used for a broad set of human applications. R.T.M. consults for Bristol Myers Squibb. M.S.F receives research funding from Bristol-Meyers Squib.
References
- 1.Jenkins RW, Barbie DA & Flaherty KT Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 118, 9–16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma P, Hu-Lieskovan S, Wargo JA & Ribas A Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168, 707–723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou R, Zhang Q & Xu P TBK1, a central kinase in innate immune sensing of nucleic acids and beyond. Acta Biochim. Biophys. Sin (2020) doi: 10.1093/abbs/gmaa051. [DOI] [PubMed] [Google Scholar]
- 4.Manguso RT et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang J, Shalabi A & Hubbard-Lucey VM Comprehensive analysis of the clinical immuno-oncology landscape. Annals of Oncology vol. 29 84–91 Preprint at 10.1093/annonc/mdx755 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Gogas H et al. Cobimetinib plus atezolizumab in BRAF(V600) wild-type melanoma: primary results from the randomized phase III IMspire170 study. Ann. Oncol 32, 384–394 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Long GV et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 20, 1083–1097 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Ishizuka JJ et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565, 43–48 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lawson KA et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature (2020) doi: 10.1038/s41586-020-2746-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan D et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vredevoogd DW et al. Augmenting Immunotherapy Impact by Lowering Tumor TNF Cytotoxicity Threshold. Cell 178, 585–599 e15 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Zhao C & Zhao W TANK-binding kinase 1 as a novel therapeutic target for viral diseases. Expert Opin. Ther. Targets 23, 437–446 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Kwon J & Bakhoum SF The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 10, 26–39 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jenkins RW et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov. 8, 196–215 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu L et al. TBKBP1 and TBK1 form a growth factor signalling axis mediating immunosuppression and tumourigenesis. Nat. Cell Biol 21, 1604–1614 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hasan M & Yan N Therapeutic potential of targeting TBK1 in autoimmune diseases and interferonopathies. Pharmacol. Res 111, 336–342 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lo JA et al. Epitope spreading toward wild-type melanocyte-lineage antigens rescues suboptimal immune checkpoint blockade responses. Sci. Transl. Med 13, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aref AR et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip 18, 3129–3143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Voabil P et al. An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancer. Nat. Med 27, 1250–1261 (2021). [DOI] [PubMed] [Google Scholar]
- 20.Sade-Feldman M et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 176, 404 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu J et al. Regulation of T-cell activation and migration by the kinase TBK1 during neuroinflammation. Nat. Commun 6, 6074 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee MSJ et al. B cell-intrinsic TBK1 is essential for germinal center formation during infection and vaccination in mice. J. Exp. Med 219, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin J et al. The kinase TBK1 controls IgA class switching by negatively regulating noncanonical NF-kappaB signaling. Nat. Immunol 13, 1101–1109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao Y et al. The kinase TBK1 functions in dendritic cells to regulate T cell homeostasis, autoimmunity, and antitumor immunity. J. Exp. Med 214, 1493–1507 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao T et al. Myeloid cell TBK1 restricts inflammatory responses. Proc. Natl. Acad. Sci. U. S. A 119, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagan RS, Torres-Castillo J & Doerschuk CM Myeloid TBK1 Signaling Contributes to the Immune Response to Influenza. American Journal of Respiratory Cell and Molecular Biology vol. 60 335–345 Preprint at 10.1165/rcmb.2018-0122oc (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barth RJ, Mule JJ, Spiess PJ & Rosenberg SA Interferon gamma and tumor necrosis factor have a role in tumor regressions mediated by murine CD8+ tumor-infiltrating lymphocytes. J. Exp. Med 173, 647–658 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benci JL et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 167, 1540–1554 e12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kearney CJ et al. Tumor immune evasion arises through loss of TNF sensitivity. Sci Immunol 3, (2018). [DOI] [PubMed] [Google Scholar]
- 30.Kearney CJ et al. PD-L1 and IAPs co-operate to protect tumors from cytotoxic lymphocyte-derived TNF. Cell Death Differ. 24, 1705–1716 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hafner M, Niepel M, Chung M & Sorger PK Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat. Methods 13, 521–527 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clark K et al. Novel cross-talk within the IKK family controls innate immunity. Biochem. J 434, 93–104 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Thomson DW et al. Discovery of GSK8612, a Highly Selective and Potent TBK1 Inhibitor. ACS Med. Chem. Lett 10, 780–785 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crew AP et al. Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. J. Med. Chem 61, 583–598 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Lafont E et al. TBK1 and IKKepsilon prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol 20, 1389–1399 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu D et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell 174, 1477–1491 e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinlich R, Oberst A, Beere HM & Green DR Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol 18, 127–136 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Park H-H et al. HS-1371, a novel kinase inhibitor of RIP3-mediated necroptosis. Exp. Mol. Med 50, 1–15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hildebrand JM et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. U. S. A 111, 15072–15077 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knuth A-K et al. Interferons Transcriptionally Up-Regulate MLKL Expression in Cancer Cells. Neoplasia 21, 74–81 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montero J et al. Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell 160, 977–989 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heijink AM et al. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat. Commun 10, 100 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sivick KE et al. Magnitude of Therapeutic STING Activation Determines CD8 T Cell-Mediated Anti-tumor Immunity. Cell Reports vol. 29 785–789 Preprint at 10.1016/j.celrep.2019.09.089 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Knelson EH et al. Activation of tumor-cell STING primes NK-cell therapy. Cancer Immunol Res (2022) doi: 10.1158/2326-6066.CIR-22-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Griffin GK et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature (2021) doi: 10.1038/s41586-021-03520-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu Y et al. STING: a master regulator in the cancer-immunity cycle. Mol. Cancer 18, 152 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang H et al. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc. Natl. Acad. Sci. U. S. A 114, 1637–1642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woo SR et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taft J et al. Human TBK1 deficiency leads to autoinflammation driven by TNF-induced cell death. Cell 184, 4447–4463 e20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karki R et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 184, 149–168.e17 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hegde PS & Chen DS Top 10 Challenges in Cancer Immunotherapy. Immunity 52, 17–35 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Deng R et al. Preclinical pharmacokinetics, pharmacodynamics, tissue distribution, and tumor penetration of anti-PD-L1 monoclonal antibody, an immune checkpoint inhibitor. MAbs 8, 593–603 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolf FA, Angerer P & Theis FJ SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Korsunsky I et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ntranos V, Yi L, Melsted P & Pachter L A discriminative learning approach to differential expression analysis for single-cell RNA-seq. Nat. Methods 16, 163–166 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Doench JG et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol 32, 1262–1267 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lu H et al. PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature 550, 133–136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van de Wetering M et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fraser C, Ryan J & Sarosiek K BH3 Profiling: A Functional Assay to Measure Apoptotic Priming and Dependencies. Methods Mol. Biol 1877, 61–76 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Filbin MR et al. Longitudinal proteomic analysis of severe COVID-19 reveals survival-associated signatures, tissue-specific cell death, and cell-cell interactions. Cell Rep Med 2, 100287 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Conant D et al. Inference of CRISPR Edits from Sanger Trace Data. CRISPR J 5, 123–130 (2022). [DOI] [PubMed] [Google Scholar]
- 64.Brinkman EK, Chen T, Amendola M & van Steensel B Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analyzed in this study are included in the manuscript. In vivo scRNA-seq data have been deposited in Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) under the accession codes GSE217160 (in vivo TBK1i study) and GSE217274 (in vivo TBK1 CRISPR-Cas9 study) and are available upon request. Descriptions on the analyses are provided in the Methods and Reporting Summary section.