

Abstract
Neutrophils sense microbes and host inflammatory mediators, and traffic to sites of infection where they direct a broad armamentarium of antimicrobial products against pathogens. Neutrophils are also activated by damage-associated molecular patterns (DAMPs), which are products of cellular injury that stimulate the innate immune system through pathways that are similar to those activated by microbes. Neutrophils and platelets become activated by injury, and cluster and cross-signal to each other with the cumulative effect of driving antimicrobial defense and hemostasis. In addition, neutrophil extracellular traps are extracellular chromatin and granular constituents that are generated in response to microbial and damage motifs and are pro-thrombotic and injurious. Although neutrophils can worsen tissue injury, neutrophils may also have a role in facilitating wound repair following injury. A central theme of this review relates to how critical functions of neutrophils that evolved to respond to infection and damage modulate the tumor microenvironment (TME) in ways that can promote or limit tumor progression. Neutrophils are reprogrammed by the TME, and, in turn, can cross-signal to tumor cells and reshape the immune landscape of tumors. Importantly, promising new therapeutic strategies have been developed to target neutrophil recruitment and function to make cancer immunotherapy more effective.
Keywords: Neutrophils, myeloid-derived suppressor cells, T cells, platelets, endothelial cells, cancer
Introduction
The presence of white cells within tumors was observed in the 19th century by Rudolf Virchow and raised the notion that inflammation may play a role in cancer. It is now recognized that inflammation plays a critical role at multiple stages of cancer: initiation, growth, metastasis, and response to therapy. As examples of inflammation-induced cancer, chronic hepatitis B infection is a major cause of hepatocellular carcinoma, and inflammatory bowel disease increases the risk of colorectal cancer. Inflammation and oxidant stress increases genetic damage required for tumor initiation. Moreover, recruitment of inflammatory cells may contain or eradicate the tumor or be the “fuel that feeds the flames”1. As an example, nuclear factor kappa B (NF-kB), is a critical driver of inflammation-associated cancer and acts in a cell type-specific manner by activating survival genes within cancer cells and inflammation-promoting genes in cells in the tumor microenvironment (TME)2,3.
Once a tumor is established, it creates a stroma comprised of inflammatory cells, fibroblasts, vasculature, and extracellular matrix. The similarity between tumor stroma and non-healing wounds has been recognized for decades4. The TME is characterized by persistent cellular injury and the production of cytokines and chemokines that recruit mixed populations of inflammatory cells that form the immune landscape of the TME. In this context, tumor cells and stroma are altered by tissue-resident and recruited immune cells and vice versa. The landscape of tumor-associated myeloid cells and lymphoid cells is critical for prognosis and a target for therapy5–8.
Circumstantial evidence for a role of neutrophils in the TME was inferred by several studies. For instance, an elevated circulating neutrophil count and a high circulating neutrophil to lymphocyte ratio, are both associated with worse prognosis in multiple solid tumors9–19. In patients with newly diagnosed solid tumors, a significant expansion of circulating hematopoietic stem and progenitor cells was demonstrated to occur with a myeloid cell bias, which is characterized by increased granulocyte-monocyte progenitors skewed toward granulocytic differentiation20. These myeloid precursors were enriched in tumor tissues, and high levels of circulating granulocyte-monocyte progenitors were correlated with more advanced disease20. Gentles at al.21 analyzed expression signatures from ~18,000 human tumors with overall survival outcomes across 39 malignancies and identified intra-tumoral neutrophil signatures as the most significant adverse cancer-wide prognostic marker. Si et al.22 noted intra-tumoral hotspots for co-localization of neutrophils and T cells in primary head and neck tumors. In these areas, T cells had significantly reduced proliferation and granzyme B expression, suggesting that intra-tumoral neutrophils can suppress T cell activation.
Definite characterization of these neutrophils within the TME in human cancer has remained incomplete to date. Granulocytic myeloid-derived suppressor cells (PMN-MDSC) are commonly defined as immature populations of granulocytes resulting from disordered granulopoiesis that suppress T cells and are barriers to antitumor immunity23. However, the distinction between PMN-MDSC and normal circulating neutrophils that have acquired a suppressor function in the TME is also suggested by tumor histology, with important conceptual and therapeutic consequences. Results from our labs and others increasingly support the notion of normal neutrophils being recruited to and reprogrammed in the TME to acquire suppressor function and other features typically associated with PMN-MDSC.
Importantly, induction of suppressor function in neutrophils is not a universal feature of the TME, and different inflammatory or tumor-derived cues can prime neutrophil responses that enhance T-cell immunity24,25. In a chemically induced sarcoma model, Ponzetta et al.26 observed that neutrophils mediated resistance to primary carcinogenesis by stimulating type 1 responses by a subset of unconventional CD4− CD8− αβ T cells. Neutrophil infiltration was associated with better prognosis and higher interferon-γ expression in human undifferentiated pleomorphic sarcomas and in other selected tumors. Neutrophils are also capable of direct lysis of tumor cells27 and killing tumor cells by antibody-dependent cell-mediated cytotoxicity (ADCC)28,29. The potential for tumor-associated neutrophils to be friends or foes in cancer30 underscores the importance of context-dependent factors in the tumor microenvironment and neutrophil heterogeneity that modulate the interactions between neutrophils and cancer.
In addition to modulating T cell accumulation and function in the TME, neutrophils can influence the TME and have direct signaling effects on tumor cells. Neutrophils and platelets cross-signal to each other and cumulatively can cause endothelial cell injury, thrombosis, and angiogenesis, as well as direct signaling to tumor cells. Paraneoplastic thrombocytosis is associated with worse outcomes in multiple solid tumors31,32. Neutrophil extracellular traps (NETs), complement, and coagulation pathways can cross-signal to and amplify each other33, and, in the TME, create niches for metastatic seeding34,35. NET generation is a distinct injurious mode of neutrophil death that accelerates metastasis in several tumor-bearing mouse models. NETs can stimulate thrombosis, have direct signaling effects on tumor cells, and disable T cell-driven anti-tumor immunity in the TME.
To date, the predominance of the evidence points to neutrophils in the TME being more likely to be foes than friends regarding antitumor immunity. Promising targets for novel therapies that include disabling neutrophil trafficking to the TME and abrogating their suppressor function. In addition, neutrophils have plasticity, and there is potential to modify neutrophil function in ways that augment tumor cell attack.
Neutrophils respond to and are modified by injury and inflammatory cues in the TME
In the late 19th century, Metchnikoff made seminal observations on the injury-induced recruitment of phagocytic cells in starfish and posited that phagocytes digest microbes. After impaling transparent starfish larvae with thorns, he observed moving cells surrounding the foreign material, similar to what happens to a human finger with a splinter36. Starfish are echinoderms with an open circulatory system and contain circulating cells (coelomocytes) that perform diverse functions, such as clot formation, phagocytosis, encapsulation, and clearance of bacteria and other foreign materials37. Both invertebrates with primitive immune systems, such as starfish, and mammals with complex innate and adaptive immune systems must respond to injury as a condition for survival. Metchnikoff’s observations provide evolutionary context for innate immune responses to injury that we believe apply to the TME, a pathologic condition characterized by persistent injury.
Metchnikoff further made the distinction between macrophages and neutrophils (the latter referred to as microphages) based on morphology, and recognized their “protective role against injurious agents which are either formed by the organism itself or enter the organism from outside.”38 Today, it is well-recognized that non-infectious injury (e.g., from trauma or a caustic insult) results in the release of damage-associated molecular patterns (DAMPs) and pro-inflammatory cytokines, chemokines, and leukotrienes that rapidly recruit and activate neutrophils. Although DAMPs and microbial products can stimulate similar pattern recognition receptors, neutrophils can have distinct signaling responses to bacteria and DAMPs released by sterile injury39. From an adaptive standpoint, this rapid mobilization of neutrophils defends against pathogens that can be introduced following injury.
Neutrophils respond to a broad range of damage motifs through pattern recognition receptors rather than having a tailored response to specific insults. In this rule-of-thumb concept, damage and inflammatory cues in cancer aren’t unique to the TME, but simply reflect persistent injury, and neutrophils will respond to these cues in a similar fashion to other chronic injuries (e.g. from unresolved infection or a persisitent foreign body). While in the non-cancer setting, these neutrophil responses are adaptive in defending against infection and likely supporting wound repair, these same responses in the TME can either accelerate or limit tumor growth. Furthermore, direct tissue injury during surgical oncologic tumor resections could further exacerbate neutrophil responses in the TME, especially if the resection is incomplete. We believe that this wound-centric model in which neutrophilic responses in the TME are driven by generalized wound repair responses as opposed to tumor-specific responses provides a foundation for understanding common pathways driving neutrophil phenotypes and therapeutic approaches that modify these phenotypes.
The initial response to acute insults involves the rapid recruitment of neutrophils and platelets to the site of injury. Neutrophil recruitment to extravascular sites of injury is a hallmark of early innate immunity and is mediated by coordinated neutrophil and endothelial cell interactions. Once outside the vessel, neutrophils have coordinated chemotaxis and cluster formation, referred to as neutrophil swarms, in which, integrins, leukotriene B4 and other chemoattractants promote local neutrophil interactions resulting in a wound seal40. In addition, mitochondrial-derived DNA and formylated peptides (reflecting the bacterial origin of mitochondria) are released following cellular injury and recruit and activate neutrophils and can guide their localization to sites of injury41,42. Neutrophils recruited to sites of injury can exacerbate organ damage through the release of reactive oxidant species (ROS), granular proteases, and other injurious products. However, there is growing recognition for neutrophils promoting the resolution of inflammation and facilitating wound repair43. For example, neutrophils transport newly formed extracellular matrix to sites of traumatic injury via CD11b/CD18 binding to collagens, an effect predicted to heal wounds44.
Following the elimination of the acute insult, termination of the acute inflammatory response is necessary to avert excessive inflammatory injury. Macrophages have multiple roles in wound repair, including forming extravascular thrombus-like structures in response to injury that mimics the function of platelets and promotes scar formation45. Macrophages also help to clear neutrophilic inflammation through efferocytosis (removal of dying cells by phagocytosis). The next stage involves a more chronic process characterized by tissue remodeling and fibrosis.
In contrast to normal wound healing, pathological wound healing is characterized by a persistent and disorganized inflammatory response in which neutrophil recruitment and injury are ongoing and regenerative responses are ineffective46. Solid tumors are composed of malignant cells and tumor stroma that can support tumor growth by providing vascular supply, signaling cues, and a barrier to immunologic attack. In the 1980s, Dvorak noted the similarities between tumor stroma and wound healing4. The TME releases several cues that mimic other chronic wounds, including DAMPs and pro-inflammatory cytokines and chemokines that recruit and activate neutrophils. Indeed, data from our groups and others point to neutrophils being reprogrammed in the TME in ways that can impair anti-tumor immunity by T cells. While this suppressive function of neutrophils may be a generalized response to injury and serve to avoid excessive T cell responses that can impair wound healing (e.g., by the generation of tissue granulomata), this suppressive phenotype is expected to be a barrier to T cell activation and expansion in the TME required for durable antitumor immunity. There is precedent for similar suppressive responses in macrophages that are adaptive in supporting wound repair but can disable T cell responses in the TME. Although the concept of M1/M2 polarization in macrophages is an oversimplification, in general terms, M2 macrophages facilitate normal wound healing by suppressing inflammatory responses and producing pro-angiogenic factors47,48. Tumor-associated macrophages are commonly similarly M2-polarized, impede T cell-driven anti-tumor immunity, and are promising targets for new cancer therapies49.
Based on our work and that of others, we propose that the acquisition of T cell suppressor function in the TME is a predominant mode by which neutrophils impair anti-tumor immunity. There are multiple other modes in which neutrophils can promote tumor progression, including cross signaling with platelets, promoting thrombosis and angiogenesis, remodeling matrix, and direct signaling to tumor cells. However, neutrophils in other contexts can injure or kill tumor cells and enhance anti-tumor immunity. These results underscore the heterogeneity of neutrophil effector activities in the TME, which can reflect factors driving granulopoiesis of myeloid progenitor cells and phenotypic changes that occur within the TME.
The overall premise of this review is that whether tumor-associated neutrophils are friends or foes, their responses in the TME are governed by evolutionary conserved responses to injury and inflammatory cues that characterize wounds in general rather than unique tumor-derived factors. In this light, we evaluate the interactions of neutrophils with endothelial cells, platelets, and T cells in the TME. We argue that in most cases, these interactions favor tumor progression rather than control, and are targets for therapeutic modulation. Finally, we discuss the direct and indirect interactions of neutrophils with tumor cells. Although neutrophils can accelerate tumor progression through several mechanisms, they can also be modified therapeutically to attack tumor cells.
Challenges in defining neutrophil sub-populations and their effects on T cell function
In addressing the subject of neutrophil interactions with the TME and specifically their inhibitory effects on T cells, we need to review how neutrophil sub-populations are defined in the field. In the tumor immunology field, the definitions of neutrophils in circulation and in the TME are changing, the terminology is often confusing and contradictory, and different investigators refer to the same granulocyte populations with different terminology. MDSC are widely described as an immature subset of myeloid cells that are expanded as a result of disordered myelopoiesis and are defined by their ability to suppress T cell immunity. Disordered myelopoiesis is a central mechanism for MDSC expansion in cancer. Tumor-derived factors (e.g., G-CSF or GM-CSF) produced by tumor cells or the TME drive this pathologic myeloid cell expansion in the marrow that results in the release of MDSC systemically and subsequent accumulation in the TME. Human MDSC populations have been grouped into granulocytic (PMN-MDSC) and monocytic populations (M-MDSC), with the understanding that further heterogeneity may exist within each of these groups50. PMN-MDSC are typically defined as low density (co-sedimenting with peripheral blood mononuclear cells (PBMC) in density centrifugation) granulocytes that inhibit T cell responses51, and express surface markers have been attributed to PMN-MDSC52. PMN-MDSC can also drive tumor progression independently of their effect on adaptive immunity. They can stimulate tumor angiogenesis by releasing pro-angiogenic factors and promote the epithelial-to-mesenchymal (EMT) transition, which drives tumor progression and metastasis53. A meta-analysis of 40 studies showed that pre-treatment circulating PMN-MDSC and M-MDSC were associated with worse prognosis in patients with solid tumor malignancies54. However, we make the important caveat that a validated signature immunophenotype for human PMN-MDSC does not exist to date and that low density has become a widely used criterion to identify and quantify PMN-MDSC.
In consensus criteria for MDSC nomenclature, PMN-MDSC surface markers were defined based on CD11b+CD14−CD15+ or CD11b+CD14−CD66b+ expression50. However, these surface markers are also expressed on normal neutrophils. In tumor-bearing mice, depletion or inhibition of trafficking of granulocytes can enhance antitumor responses55–60. However, most of these approaches, such as anti-Ly6G61,62 or cytotoxic chemotherapy63, deplete both neutrophils and PMN-MDSC. These ideas about the origin, phenotype, and activity of PMN-MDSC are mostly derived from mouse studies, many of which have not been confirmed in human studies, suggesting the existence of fundamental differences between PMN-MDSC in these species64, complicating extrapolations from experimental mouse studies. Compounding the difficulty in interpreting experimental results, granulocytic cells within human tumors and tumor-bearing mice are typically defined as PMN-MDSC based on these non-specific markers that do not distinguish them from neutrophils. Whether these granulocytic cells are called neutrophils or PMN-MDSC usually reflects the background of the investigators rather than a clearly defined distinction between these populations.
These observations raise a central question: Should PMN-MDSC be considered a distinct granulocytic cell population with intrinsic T cell suppressor activity that results from disordered granulopoiesis? Alternatively, are PMN-MDSC simply a subset of neutrophils with suppressor function that can be acquired within the TME or systemically but not driven by disordered granulopoiesis? Given the diversity of neutrophil sub-populations, which can include differences in immunoregulatory functions, some investigators argue that PMN-MDSC should be called “immunosuppressive neutrophils”64. We agree with this point and would argue that if authors define PMN-MDSC based on disordered granulopoiesis, then they should provide proof in their experimental system that the specific PMN-MDSC population developed due to a dysregulated marrow and is distinguishable from normal neutrophils. Alternatively, authors may choose to define PMN-MDSC solely based on their T cell suppressor function and without regard to where and how the suppressor function was acquired; using this definition, PMN-MDSC are de facto a functional subset of neutrophils that are expanded in cancer and other diseases. Our research (described below) supports this latter concept – specifically that neutrophils can acquire an MDSC-like phenotype following activation by microbial motifs65 and products in the TME66,67. However, this notion does not negate that PMN-MDSC can also be the result of disordered granulopoiesis, but simply that there are diverse pathways that can result in an MDSC phenotype.
In contrast to the expansion of an immature population of MDSC, Engblom et al.68 showed a different mechanism for tumors inducing the expansion of cancer-promoting neutrophils. They showed that lung adenocarcinomas can remotely activate bone-resident osteocalcin-expressing osteoblasts. These osteoblasts caused an expansion of SiglecFhigh neutrophils that were rare in the healthy tissue but expanded ~70-fold in tumor-bearing lungs. SiglecFhigh neutrophils had a distinct transcriptional profile that mimicked profiles associated with PMN-MDSC. The SiglecFhigh neutrophil gene signature was associated with a worse outcome in patients with lung adenocarcinoma, and depletion of osteocalcin-expressing osteoblasts reduced lung tumor growth in mice. SiglecFhigh neutrophils are morphologically mature, long-lived, and are enriched in tumor, but not found in marrow or circulation69, which suggests that their tumor-promoting phenotype is at least in part acquired in the TME. In a non-cancer setting, SiglecF-expressing neutrophils were associated with collagen I production and renal fibrosis70, raising the possibility that this neutrophil subset might have a wound-healing function71.
Fridlender’s group proposed the definition of N1 and N2 tumor-associated neutrophils. In mice, N1 neutrophils can promote anti-tumor effector responses both by directly targeting tumor cells and by stimulating T cell immunity, while N2 neutrophils facilitate tumor progression by suppressing T cell responses and upregulating angiogenic factors such as VEGF and matrix metalloproteinase-9 (MMP-9)72–74. Sagiv et al.75 proposed three distinct neutrophil populations observed in tumor-bearing mice and in patients with cancer: mature high-density neutrophils, mature low-density neutrophils, and immature low-density neutrophils. The mature high-density neutrophils had an N1-like phenotype and the capacity to kill tumor cells. The low-density population included immature neutrophils (equated with PMN-MDSC) and mature neutrophils with T cell suppressive capacity. When treated with TGF-β, the mature high-density neutrophils polarized to become low-density neutrophils and acquired a suppressive phenotype, pointing to the plasticity of neutrophil subpopulations. The N1 and N2 tumor-associated neutrophil designation has been largely characterized in mouse models. Human neutrophils can be polarized ex vivo to acquire features associated with N1 and N2 neutrophils76, and the role of these neutrophil sub-populations in shaping the human TME is still unclear and an important area for research.
Neutrophils in the TME are heterogeneous and have plasticity in which their phenotypes are altered by various cues, such as DAMPs, cytokines, and chemokines64,77,78. There is greater recognition that specific phenotypes such as T cell suppressor function, low density, and surface expression of various markers can be induced by the TME. Low-density neutrophils (LDN) are observed in patients with cancer and other inflammatory conditions (e.g., sepsis, autoimmune conditions, and trauma)79–81. Riça et al.82 observed heterogeneity in circulating neutrophils in patients with acute trauma, including a distinct LDN population with activated CD11b/CD18 but reduced surface marker expression of nearly all other surface markers including receptors for formyl peptides, leukotrienes, chemokines and complement. LDNs are also present in normal individuals, are morphologically mature, and have increased ROS production and phagocytic capacity consistent with an activated phenotype83. Veglia et al.51 argued that low density is one of the defining characteristics of PMN-MDSC and that the term LDN creates an incorrect impression of the existence of multiple neutrophil populations with immunosuppressive function when in fact they are all PMN-MDSC. However, we would argue that LDN can be expanded under a number of pathologic conditions, and likely represent activated neutrophils. Indeed, normal human neutrophils stimulated ex vivo with formylated peptide become low density and suppress T cell activation through a ROS-dependent pathway84. LDNs may or may not have become pre-activated during their maturation in the bone marrow by tumor-derived factors released into the blood and hence show a different neutrophil density, activated phenotype, and differ functionally. As we ourselves have not observed a significant number of LDNs in treatment-naïve cancer patients85, not all cancer patients seem to have increased numbers of LDNs with T-cell suppressive MDSC activity. We should note that neutrophil isolation remains a delicate procedure and artifactual findings by pre-activation in the laboratory setting may easily occur.
The idea of (pre)activation or priming of circulating neutrophils may also explain previous observations of increased MDSC activity in LDNs. The presence of such LDNs with MDSC activity points to neutrophil activation as a mechanism for acquiring suppressive activity rather than suppressor function being an inherent characteristic of a specific neutrophil subset. In support of this notion, we observed that neutrophils activated by fMLF and other stimuli acquire a T cell suppressor phenotype through pathways requiring neutrophil-T cell contact, CD11b/CD18, neutrophil degranulation, and NADPH oxidase activation65 (Figure 1). Neutrophils can damage extracellular parasites86 and tumor cells87 by trogocytosis, a process involving the transfer of plasma membrane fragments between conjugated cells. We observed trogocytosis of T cells by activated neutrophils88 (Figure 1). During trogocytosis, neutrophils may cause the target cell to shrink to a level where it is no longer functional or viable. These results led us to consider that neutrophil effector functions may collectively impair the tumor-infiltrated T cells from their cytotoxic activity against tumor cells, thereby impairing durable antitumor immunity65.
Figure 1: Neutrophils exert PMN-MDSC function following activation.
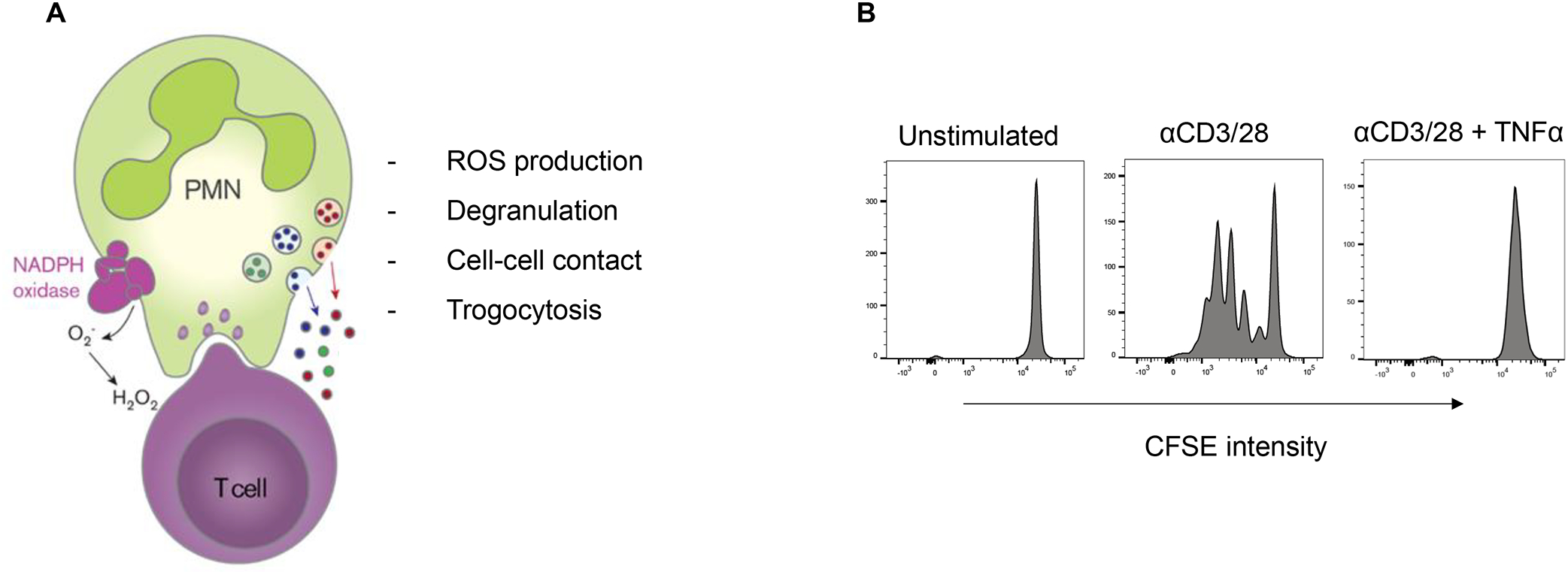
A) Activation of neutrophils by a number of stimuli (e.g., fMLF or TNF-α) can induce T cell suppressor function dependent on cell-cell contact by adhesion, ROS formation, degranulation and trogocytosis. B) The processes of adhesion, ROS production and degranulation have been proven essential for strongly suppressing T cells an in vitro model resulting in reduced T cell proliferation and irreparable T cell damage (right panel). Data are adapted from Aarts et al.65
Neutrophils are reprogrammed in the TME
When evaluating various surface markers or functional phenotypes (e.g., suppressor function or tumoricidal activity) of neutrophils in the TME, it is challenging to determine how and where these features were acquired. As described above, PMN-MDSC are traditionally defined by abnormal granulopoiesis and, therefore, the features that define PMN-MDSC should be present both in circulation and in the TME. By contrast, if the specific neutrophil populations are enriched or are almost exclusively found in the TME, then we would conclude that they have been re-programmed in the TME. The term “tumor-educated neutrophils” defines neutrophil features acquired by cues in the TME89.
A number of markers have been proposed to define PMN-MDSC, including lectin-type oxidized LDL receptor-1 (LOX-1)90, arginase I (a constituent of neutrophil tertiary granules91), CD14high (in mice)52, CD8492, and CD101neg (CD101 is a marker of neutrophil maturity93), and require validation. Condamine et al.90 identified LOX-1 (receptor of oxidized LDL) as a PMN-MDSC marker, but it is also inducible in normal neutrophils by endoplasmic reticulum (ER) stress. LOX-1+ granulocytes were enriched in tumors, had distinct transcriptome profiles, suppressed T cell immunity, and were associated with tumor niches with reduced T cell accumulation and activation22,52,90. Si et al.22 examined intratumoral interactions of neutrophils and T cells in head and neck tumors. Some of these regions contained colocalization of neutrophils and T cells in which neutrophils expressed LOX-1 and arginase I and T cell activation, assessed by the expression of Granzyme B and Ki67 (proliferation marker), was suppressed. This study raises the question of whether LOX-1 expression reflects an activated neutrophil in which LOX-1 has mobilized to the plasma membrane and whether LOX-1 is driving suppressor function as opposed to being simply a marker of suppressor neutrophils.
LOX-1 surface expression seemed to be promising as a PMN-MDSC marker, but our data indicates this surface marker is an excellent marker to assess specific granules in human neutrophils and its immediate upregulation upon neutrophil activation. As we and many others have already shown three decades ago94, rapid upregulation of surface markers is observed by the isolation of neutrophils per se or intended in vitro activation. Under these conditions, the expression of many surface markers is subject to upregulation from intracellular stores, which is also consistent with our previous cellular proteomics studies on neutrophils on the different protein content of azurophil, secondary, and tertiary granules during neutrophil developmental stages and their activation95. We observed that human neutrophils become LOX-1 positive upon neutrophil activation. In line with this, almost 90% of neutrophils stained by the neutrophil-specific CD66b marker in TME sections are also labeled by anti-LOX-1 monoclonal antibodies (data not shown), suggesting that this marker cannot be used to discriminate PMN-MDSC as a separate subset. However, caution should be taken with the interpretation of immunohistochemistry, knowing that it is impossible to distinguish between intracellular stores and surface staining in histology samples.
Consistent with these findings, we observed that healthy donor neutrophils exposed to ascites fluid supernatants from patients with newly diagnosed metastatic epithelial ovarian cancer acquire features of PMN-MDSC: low density, increased LOX-1 surface expression, and T-cell suppressor function67 (Figure 2). Ascites fluid exposure induces surface mobilization of CD11b and granule membrane constituents, including CD66b and the gp91phox/p22phox heterodimer of the NADPH oxidase67. The increased surface expression of these proteins is dependent on C5aR signaling, pointing to a role for complement in priming neutrophil effector functions. Surface CD10 expression has been linked to a suppressive population of mature neutrophils96. Neutrophil CD10 expression can be increased after C5a exposure97, and we observed that ascites fluid exposure induces CD10 surface expression in healthy donor neutrophils67. In addition, CyTOF analysis of neutrophils exposed to epithelial ovarian cancer ascites fluid supernatants showed that neutrophils could be divided into 15 phenotypically distinct clusters, whose relative populations change over 3 hours67. Linking these neutrophil sub-populations to T cell suppressor function will be pursued in future studies. We also observed that ascites fluid exposure results in normal neutrophils having an increased lifespan and nuclei acquiring a hypersegmented morphology similar to nuclear morphologic changes induced by Helicobacter pylori infection67,98. These results provide further evidence for neutrophils being modified in the TME by various cues (e.g., DAMPs, cytokines, chemokines) in ways that mimic features attributed to PMN-MDSC and in additional ways that may be relevant to tumor progression or control.
Figure 2. Neutrophils can be reprogrammed in the tumor microenvironment to acquire features of PMN-MDSC: low density, increased LOX-1 expression, and T-cell suppressor function.
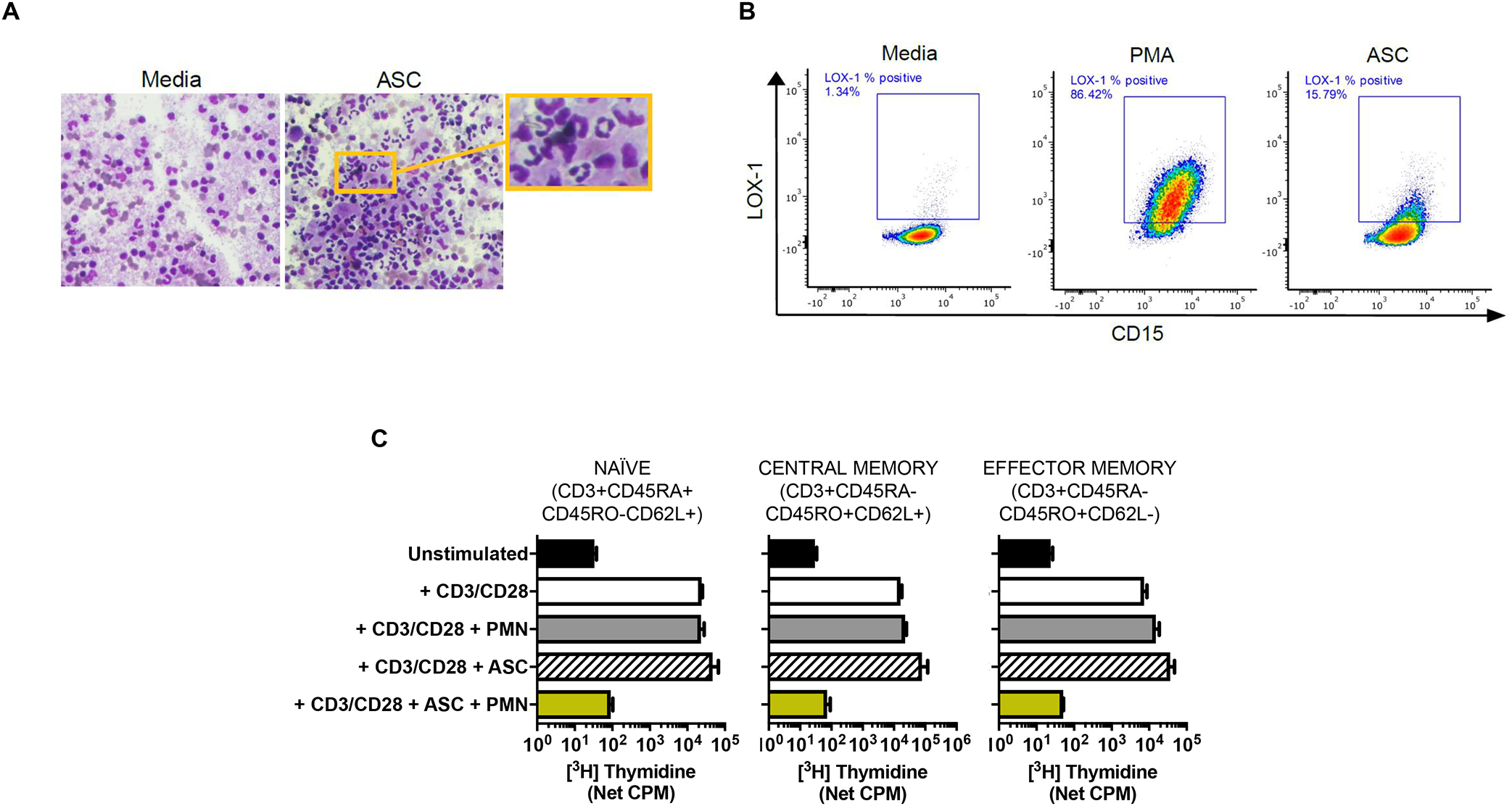
Ascites fluid supernatants (ASC) from patients with newly diagnosed metastatic ovarian cancer were used as a biologically important component of the TME. A) ASC exposure increased the proportion of low-density neutrophils (LDN). Representative Wright-Giemsa-stained slides show abundance of LDN in the PBMC monolayer following ASC exposure. Buffy coat cells from healthy donor blood were incubated in ASC or media for 1hr, then subjected to gradient centrifugation. The PBMC monolayer consisted of 34.5% vs. 10.5% LDN following exposure to ASC or media, respectively. Data representative of two independent experiments. B) ASC exposure increased LOX-1 expression on neutrophils. Healthy donor neutrophils (CD15+) were incubated in media, ASC or 20nM PMA (positive control) for 1.5 hours, then analyzed by flow cytometry. Representative gating shows effects of media, PMA, and ASC on LOX-1 expression. C) Healthy donor naïve, central memory, and effector memory T cells were purified, and incubated with autologous neutrophils (PMN), ASC, or ASC + PMN, and anti-CD3/CD28-stimulated T cell proliferation was assessed at 96h by thymidine incorporation. Data are from Singel et al.66 and Emmons et al.67.
While neutrophils were considered to be glycolytic with very limited mitochondrial metabolism, tumor-associated neutrophils can shift to mitochondrial fatty acid metabolism (likely an adaptation to limited glucose in the TME), generate increased NADPH oxidase-dependent ROS, and suppress T cell responses99. In mice, fatty acid transport protein 2 (FATP-2) is upregulated in PMN-MDSC and linked to their suppressive function, further supporting an association between metabolism and suppressor function100. FATP2-mediated suppressive activity involves the uptake of arachidonic acid and the synthesis of prostaglandin E2100. In addition, FATP2 inhibition enhanced the antitumor efficacy of checkpoint inhibitors100.
Extracellular nucleotides, particularly ATP and adenosine, are major biochemical constituents of the TME. Purinergic receptor signaling can promote tumor growth through a direct effect on tumor cells and by modulating immune cell function. The purinergic receptor, P2RX1, is an ATP-gated ion channel. Wang et al.101 identified a subset of P2RX1-negative neutrophils that are recruited to murine pancreatic cancer metastatic lesions and are characterized by increased mitochondrial metabolism and immunosuppressive activity. The ROS-inducible transcription factor, nuclear erythroid factor-2 (NRF-2), was upregulated in P2RX1-deficient neutrophils and contributed to immunosuppression and metabolic reprogramming101. Together, these experimental studies suggest that the TME can modify the phenotype of neutrophils, including metabolic reprogramming, and raise the notion of modulation of metabolic pathways in neutrophils as a therapeutic approach.
Additional questions relate to the stability of neutrophil sub-populations over time and within specific immunologic niches in the TME. As an example, mobilization of neutrophil granular membrane products to the plasma membrane will result in increased surface expression of these products, but this can be a dynamic process with surface membrane expression changing over time. Damage and inflammatory cues in the TME can also induce changes in transcription and protein synthesis in neutrophils resulting in phenotypic changes that might persist or be transient. We also expect that neutrophil-driven modulation of T cell immunity will be influenced by the proximity of neutrophils to T cells and local cues within the TME. In addition, since neutrophils are short-lived cells, the capacity for neutrophils to cause durable changes to the TME relies on ongoing neutrophil recruitment. Finally, the mode of neutrophil death – by apoptosis, NET generation, necrosis, or another pathway – is another example of neutrophil heterogeneity. We therefore expect neutrophil sub-populations within the TME to have plasticity in their phenotypes, which are influenced by multiple signaling cues and the time that neutrophils are exposed to these cues.
Neutrophils as drivers of cellular injury in the TME
Our own data in treatment-naïve patients with either breast cancer or head and neck squamous cell cancer65 did not show either the presence of circulating PMN-MDSC subsets by immunostaining of a set of surface markers or evidence for spontaneous PMN-MDSC activity. We observed that all circulating neutrophils can be stimulated to exert suppressive activity in T cell proliferation or cytokine release assays, but only following their activation65. We tested in the past various stimuli to activate neutrophils to obtain so-called MDSC activity of infectious or inflammatory origin, such as fMLF, LPS or TNF-α. However, we would argue that fMLF is a biologically relevant stimulus that mimics the immune activation of mitochondrial formylated proteins (which are bona fide DAMPs) that are released following cellular injury102. This also applies to the broader range of many endogenous TLR4 ligands other than the classical microbial LPS from Gram-negative bacteria.
Considering the TME as a local inflammatory or wound healing process, local endogenous activation of neutrophils within the TME is further supported by studies on the induction of PMN-MDSC activity by ascites of patients with metastatic epithelial ovarian cancer66,67,103. Using live cell imaging, we observed that upon coculture in OC ascites fluid supernatants, healthy donor neutrophils and T cells form stable interactions. Neutrophils were more likely to be elongated and flattened and caused stretching of T-cell membranes. Trogocytosis by neutrophils of T cell membranes was observed by confocal microscopy and quantified by flow cytometry. Trogocytosis was increased by ascites fluid exposure and decreased using anti-CD11b, suggesting that CD11b/CD18 drives trogocytosis by enabling neutrophil adherence to T cells67. RNAseq of T cells exposed to neutrophil suppressors showed induction in fatty acid biosynthesis and transport and cholesterol efflux pathways (unpublished observations), a finding consistent with the important role of lipid synthesis and transport in the repair response to plasma membrane injury104.
Although a likely obstacle to T cell immunity, neutrophil-driven trogocytosis can also have the benefit of injuring opsonized tumor cells. SIRPα is a transmembrane protein expressed on myeloid cells with a cytoplasmic region containing immunoreceptor tyrosine-based inhibitory motifs that facilitate binding of the tyrosine phosphatases SHP-1 and SHP-2 and serves as a myeloid-specific immune checkpoint pathway. Inhibition of SIRP-α or its ligand, CD47, is an exciting investigational approach to arm monocytes/macrophages and neutrophils to kill tumor cells105. In addition, Matlung et al.87 showed that neutrophils can kill tumor cells through antibody-mediated trogocytosis, an effect augmented by CD47-SIRPα blockade. These studies raise the potential for directing the injurious activity of activated neutrophils to tumor cells.
A reflection of PMN-MDSC activity in vivo
Upon infiltration into the TME human neutrophils may express FcγR type I (CD64), which can be induced locally or already prior to or during the process of infiltration itself. Expression can be induced by several growth factors and cytokines, including G-CSF, GM-CSF, IFN-γ, TNF-α and IL-1. A clear indication of the exact moment when or where the neutrophils start to express this IgG receptor remains obscure. Therefore, if possible, local staining of molecular markers that have been affected by ROS production (oxidized amino acid or nucleotide residues) may be of help to detect neutrophil activation within the TME. However, these molecular changes in tissue sections are not definite proof of neutrophil-derived ROS since the latter may also be generated by tumor cells themselves.
Other options to investigate local neutrophil activation are possible by staining of extracellular MPO or neutrophil elastase released by activated neutrophils from the azurophilic granules upon local activation. However, extracellular MPO or neutrophil elastase staining could indicate degranulation by viable activated neutrophils or local association with NETs from adjacent neutrophils. Both ROS and neutrophil elastase were shown to be important in both functional PMN-MDSC activity65 and NET generation106, which has been further substantiated by experiments with neutrophils from patients with well-characterized genetic defects. The presence of NETs within the TME strongly suggests local neutrophil activation and its subsequent death. Another option would be to stain neutrophils for activated CD11b/CD18, indicating integrin engagement by neutrophils in the TME, since we have recently identified enhanced surface expression of these markers in circulating neutrophils from patients with tissue injury.
Targeting Neutrophils to Enhance Anti-tumor Immunity
In recent years, treatment of multiple cancer types has been changed by the introduction of immunotherapies, mainly in the form of antibody-mediated inhibitors of immune checkpoints and adoptive T cell therapies. These therapies are effective by boosting anti-tumor T cell immune responses. In the wake of these immunotherapies, interest in the cellular composition of and more particular the role of neutrophils in the TME has grown over the years. The pathologist does not usually report on the presence of neutrophils within tumor since their relevance to clinical practice and treatment strategies is still largely lacking. However, tumor-associated neutrophils may exert strong suppression of T cell mediated anti-tumor responses and are targets for immunotherapy.
Neutrophils are recruited to the TME through multiple pathways that can include DAMPs (e.g. release of formylated peptides from mitochondria107), leukotrienes108, complement and cytokines and chemokines, such as ligands of CXCR255–57,59,109. In addition to recruitment, these mediators cumulatively alter the function of neutrophils in the TME. IL-17A is a pro-inflammatory cytokine secreted predominately by CD4+ Th17 cells, but also can be produced by other immune cells such as γδ T cells. IL-17A plays a major role in neutrophil-mediated host defense. IL-23 supports the expansion of IL-17A-producing Th17 cells and also regulates granulopoiesis110. While important for antimicrobial host defense, the IL-23/IL-17A axis is also associated with autoimmunity and is the basis for the development of FDA-approved monoclonal antibody drugs that target this pathway. The IL-23/IL-17A axis has also been shown to have important effects on the TME, raising the notion of applying these same drugs to cancer. IL-23 promoted tumor incidence and growth in mice111, which was associated with the expansion of IL-17A-producing lymphocytes that, in turn, stimulated myeloid growth factors and pro-inflammatory cytokines that drive neutrophilic inflammation. IL-23 produced by myeloid cells augmented the intratumoral IL-17 response and promoted tumor growth in murine colorectal cancer112. The authors proposed a model in which barrier erosion by the tumor enabled mucosal invasion by bowel flora that triggered IL-23/IL-17-dependent inflammation and acceleration of tumor growth112. In addition, enterotoxigenic Bacteroides fragilis, a human colonic bacterium, can activate Th17 responses that result in myeloid cell recruitment and colon cancer113,114. Coffelt et al.115 showed that IL-17 production by γδ T cells stimulated G-CSF-dependent expansion of neutrophils in a mouse model of spontaneous mammary tumor development. Tumor-induced neutrophils suppressed CTL responses, and depletion of IL-17 or G-CSF and absence of γδ T cells prevented neutrophil accumulation within tumors and abrogated the T cell suppressive phenotype of neutrophils.
Experiments in tumor-bearing mice provide strong premise for inhibiting neutrophil recruitment to the TME to enhance anti-PD-1 therapy. CXCR2 inhibition resulted in impaired neutrophil accumulation (referred to by the authors as neutrophils/MDSCs) in tumor and reduced metastasis57. Moreover, combined inhibition of CXCR2 and PD-1 significantly extended survival in murine pancreatic ductal cancer57. In another study complement C5a inhibition resulted in reduced accumulation of neutrophils (referred to by the authors as PMN-MDSC) in tumors and enhanced the efficacy of anti-PD-1 therapy116. These studies raise the potential for inhibiting neutrophil recruitment and activation as strategies to augment checkpoint inhibitor therapy, including in tumors that typically do not respond to existing immunotherapies.
Neutrophil extracellular traps, coagulation, and effects on T cell immunity
Our laboratory has made observations about neutrophil and complement interactions in the human TME that provide further rationale for inhibiting complement to augment T cell immunity. Though our work is mostly based on epithelial ovarian cancer (OC), we expect that the principles are broadly relevant to the TME in metastatic solid tumors. Ascites fluid is common in metastatic OC, and both the presence and volume of ascites at diagnosis are negative predictors of survival117–120. OC ascites can impair antitumor immunity through several mechanisms103,121–124, can facilitate tumor spread and chemotherapy resistance125, and is often associated with miliary dissemination and worse clinical outcomes118,120,126–129. Ascites fluid is a distinct part of the OC TME that contains specific TAL populations130 and immunosuppressive myeloid cells121,131 and exosomes123 that are obstacles to antitumor immunity. While OC is an immunogenic tumor and increased infiltration of CTL correlates with longer survival132,133, single-agent checkpoint inhibitors are largely ineffective in relapsed/refractory OC, with response rates of 8–15%134–136. These results suggest that abrogating suppressive pathways in the OC TME might make checkpoint inhibitors and other approaches to expand T cell immunity more effective.
Our results support a model in which products of cellular injury (DAMPs) and pro-inflammatory cytokines and chemokines in the TME recruit neutrophils and induce a T cell suppressor phenotype66,137,138. We consider this suppressor function to be a generalized response to injury rather than to tumor-specific factors, and to be mechanistically distinct from PMN-MDSC that are traditionally defined by suppressor function resulting from disordered granulopoiesis23. Neutrophils in the circulation and from OC ascites fluid are morphologically mature, and only ascites neutrophils suppress the ex vivo proliferation and activation of T cells131. Circulating neutrophils from OC patients are not intrinsically immunosuppressive but acquire a T cell suppressor phenotype following exposure to OC ascites fluid supernatants66. This same suppressor phenotype is also induced in normal donor neutrophils exposed to ASC. Neutrophil suppressors inhibit the proliferation of naïve, central memory and effector memory T cells and of tumor-associated lymphocytes (TALs) from patients with newly diagnosed OC. The induction of neutrophil suppressor function requires a number of neutrophil signaling pathways, including complement signaling and NADPH oxidase activity138. A similar complement-dependent neutrophil suppressor phenotype is induced by malignant effusions from patients with different metastatic cancers, demonstrating the generalizability of our findings. Neutrophil suppressors cause a profound immunoparalysis in T cells, characterized by inhibition of stimulated cytokine responses, NFAT translocation, glucose uptake, mitochondrial mass and depolarization, and mTOR activation138 (Figure 3). Results in tumor-bearing mice116,139–151, including in syngeneic OC152, support targeting the complement system to limit tumor progression and enhance antitumor immunity. In addition, C5a is a potent neutrophil chemoattractant, and C3a, C4a, and C5a are anaphylatoxins that cause vascular leakiness and extravascular fluid accumulation. Thus, C3 inhibition, by depletion anaphylatoxins, may result in reduced accumulation of malignant effusions.
Figure 3. Model of neutrophil-mediated T cell injury and immunoparalysis in the TME.
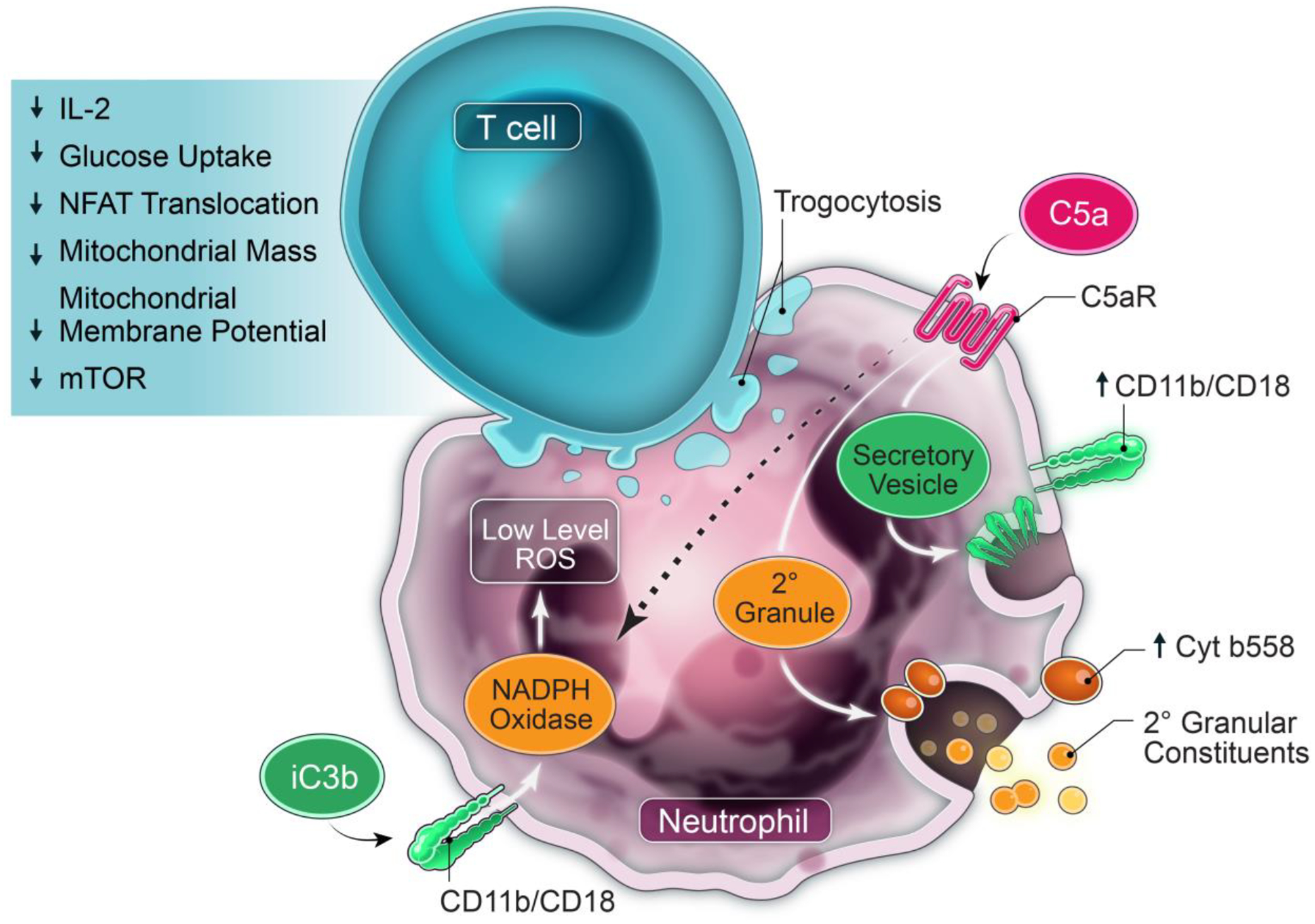
Circulating neutrophils are recruited to the TME where they acquire suppressor function directed at T cells. Malignant effusions have multiple products, including DAMPs, cytokines, chemokines, and activated complement that chemoattract and activate neutrophils and induce suppressor function. Multiple neutrophil effector functions including complement signaling, CD11b/CD18 (CR3; receptor for iC3b), NADPH oxidase, SNARE-mediated intracellular transport and exocytosis of granular and/or vesicular constituents, and phosphatidylserine drive this suppressor phenotype. A positive feedback loop through C5aR activation increases surface expression of CD11b/CD18 and cytochrome b558 (membrane-bound gp91phox/p22phox component of NADPH oxidase), predicted to amplify complement signaling and surface NADPH oxidase. Activated neutrophils also cause CD11b-dependent trogocytosis of T cell membranes, expected to cause membrane injury. These neutrophil-generated signaling and injury cues to T cells cumulatively result in non-responsiveness, characterized by suppression of stimulated cytokine responses, NFAT translocation, glucose uptake, mitochondrial mass and depolarization, and mTOR activation. Model is from Emmons et al.67.
Based on this scientific premise, we will evaluate APL-2 (pegcetacoplan), a peptide C3 inhibitor, in a phase 2 trial in patients with recurrent OC and persistent malignant effusions (NCT04919629). APL-2 was safe and superior to eculizumab in paroxysmal nocturnal hemoglobinuria (PNH)153, and is FDA-approved for this indication; its use in cancer is novel. Following a safety lead-in, patients will be randomized to the following cohorts: (i) bevacizumab (anti-VEGF); (ii) APL-2 + pembrolizumab (anti-PD1); and (iii) APL-2 + pembrolizumab + bevacizumab. This trial will enable us to gain safety and preliminary efficacy data on APL-2-based regimens in recurrent OC with malignant effusions and a detailed understanding of how these regimens modulate the immune landscape in the TME.
Neutrophils, Platelets and Endothelial Cells: Partnership in Tumor Spread
Neutrophils and platelets become activated as emergency responders to infection and injury, and are critical for defense against infection, controlling bleeding, and promoting wound repair. Neutrophil recruitment requires coordinated neutrophil and endothelial cell interactions, characterized by neutrophil rolling along endothelial cells mediated by lectin-glycan interactions, firm neutrophil adhesion along the vascular endothelium mediated principally by CD11b/CD18-ICAM-1 binding, and extravasation from the blood stream into affected tissues. Leukocyte adhesion deficiency 1 results from an inherited CD18 deficiency and is characterized by recurrent infections and lack of pus formation at infected sites, a reflection of the major role of CD11b/CD18 in neutrophil trafficking. Firm adhesion between platelets and activated endothelial cells is mediated by platelet αIIbβ3 (CD41/CD61) and contributes to thrombosis154. The interactions between neutrophils and platelets are orchestrated by both their surface molecules that enable physical interactions between these cells and secreted products. Activated platelets express P-selectin (CD62P), which binds to P-selectin glycoprotein ligand-1 (PSGL-1) on the surface of neutrophils. Platelet-leukocyte interactions are augmented via the interaction of the myeloid cell integrin CD11b/CD18 with platelet glycoprotein Ibα, junctional adhesion molecule A, or fibrinogen bound to the activated platelet integrin αIIbβ3155–157. Platelets release microparticles during inflammation, which are internalized by activated neutrophils and amplify inflammation in arthritis158,159. In addition, activated platelets release mitochondria into the extracellular environment, which, similar to bacteria, activate and interact with neutrophils in vivo, triggering neutrophil adhesion to the endothelial wall160. Although these coordinated neutrophil-platelet interactions are required to induce hemostasis and limit infection, excessive activation of these pathways, which can result from both severe infection and non-infectious injury, exacerbates organ injury. Consequently, inhibition of neutrophil-platelet interactions can be protective in experimental models of acute injury161–163.
Neutrophils and platelet activation can drive thrombosis, a required component of the vascular repair response, and modulate wound repair responses in the vascular space. Of central importance to the TME is the pro-angiogenic function of neutrophils and platelets. Tumor angiogenesis is stimulated by local factors in the TME, such as hypoxia and thrombosis, and is augmented by tumor cell-derived products that activate platelets. Antiangiogenic therapies that inhibit vascular endothelial growth factor (VEGF) signaling (e.g., bevacizumab and tyrosine kinase inhibitors directed against the VEGF receptor) have been used widely as first- and second-line treatments for cancer. Activated platelets release vascular endothelial growth factor (VEGF) and act as transporters of VEGF, and their contribution to tumor angiogenesis has been posited for more than 20 years164. Activated platelets can also induce tumor cell signaling that promotes proliferation and metastasis165–168. Platelet-derived TGF-β, a cytokine that drives collagen deposition required for wound healing, and direct platelet-tumor cell contacts synergistically activate the TGFβ/SMAD and NF-kB pathways in cancer cells, resulting in their transition to a mesenchymal-like phenotype and enhanced metastasis in mice168. Paraneoplastic thrombocytosis predicted worse outcomes in patients with a number of solid tumors31,32.
Neutrophil-derived pro-angiogenic factors include VEGF, Bv8, MMP-9, and S100A8/S100A9169. MMP-9 remodels the extracellular matrix and promotes angiogenesis indirectly by interacting with VEGF. Nozawa et al.170 showed that infiltrating neutrophils activated MMP-9-dependent angiogenesis during early stage pancreatic tumors in mice. MMP-9-expressing neutrophils accumulated within the tumor, and neutrophil depletion suppressed VEGF:VEGF-receptor association and inhibited angiogenic induction.
These results point to neutrophil-platelet interactions favoring tumor spread through a number of mechanisms, including angiogenesis and direct signaling to tumor cells. Labelle et al.34 showed that platelet-derived signals stimulated the recruitment of granulocytes to tumor cells to form early metastatic niches. In mice, granulocyte recruitment was dependent on CXCL5 and CXCL7 chemokines by platelets upon contact with tumor cells, and inhibition of the CXCL5/7 receptor CXCR2 or transient depletion of either platelets or granulocytes prevented the formation of early metastatic niches and significantly reduced metastasis34. In addition, platelet infiltration into tumors after anti-angiogenic therapy withdrawal was associated with enhanced tumor rebound in patients with epithelial ovarian cancer171.
These results support platelets being recruited to an activated by the TME, where they drive thrombosis and metastasis. In ascites fluid collected at primary surgery from patients with newly diagnosed metastatic epithelial ovarian cancer, we observed fibrin aggregates embedded with neutrophils and tumor cells (Figure 4). Since fibrin adheres to surfaces, these fibrin-neutrophil-tumor cell networks are likely to be important for seeding serosal surfaces, a characteristic feature of advanced ovarian cancer. Activated platelets can also interact with tumor cells and promote their proliferation via TGF-β-dependent signaling167. Guglietta et al.172 observed increased spontaneous intestinal tumorigenesis in APCmin mice associated with the accumulation of low-density neutrophils with a pro-tumorigenic N2 phenotype and increased capacity for NETs. They propose a model in which systemic LPS derived from bowel flora primes complement activation and neutrophil activation, NET generation, and coagulation, which, in turn, promote tumorigenesis172 (Neutrophil-driven cross-signaling to tumor cells is discussed below in Neutrophil Interactions with Tumor Cells).
Figure 4. Model of DAMPs and neutrophil-platelet responses in the ascites of patients with advanced epithelial ovarian cancer.
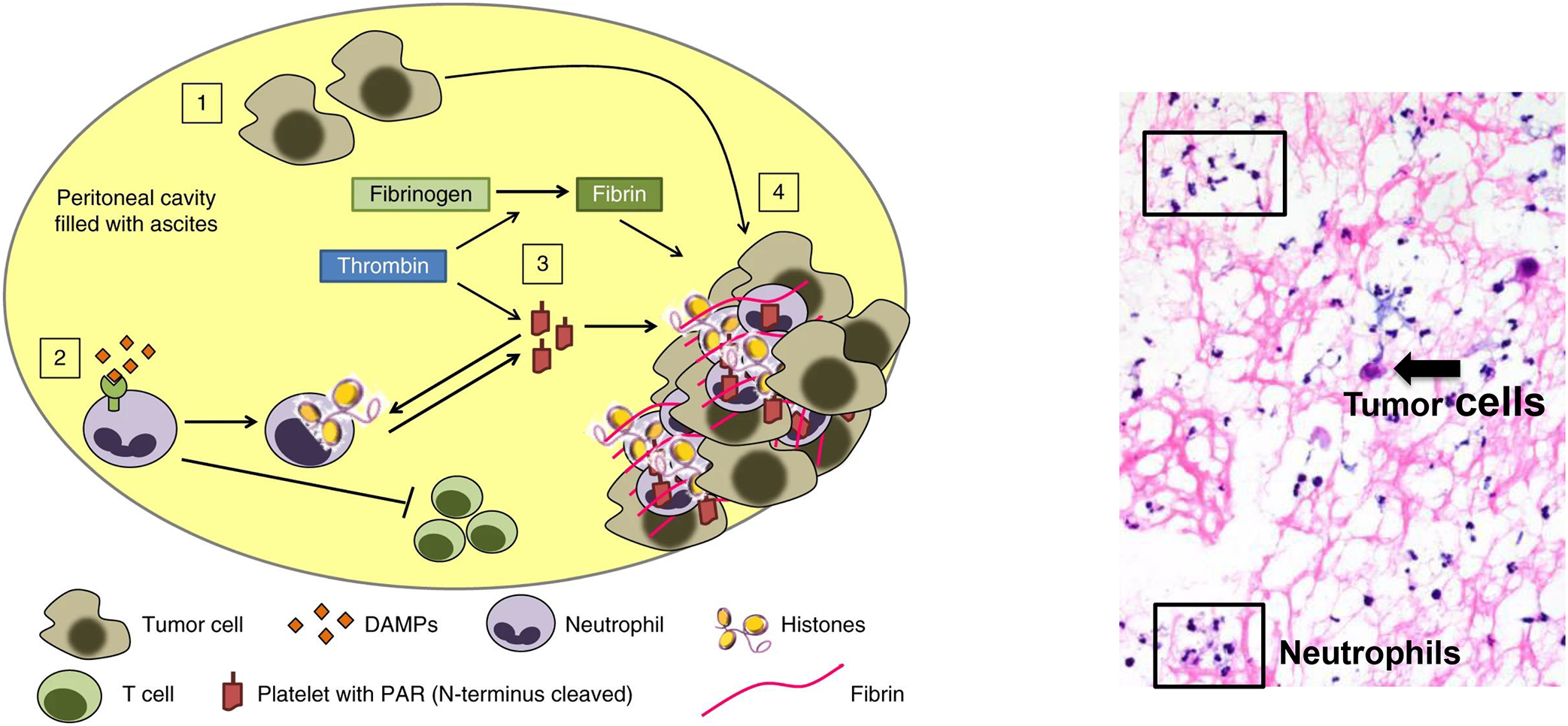
(1) A hallmark of advanced cancer is cellular necrosis, which releases DAMPs, and minor numbers of tumor cells into the ascites. (2) DAMPs and other inflammatory mediators recruit and activate neutrophils and induce NETs, and (3) activate platelets. (4) Platelet activation and aggregation with NETs and fibrin filaments are expected to trap tumor cells and enhance seeding to the serosa and local dissemination within the peritoneal cavity. Right panel: Floating aggregates in ascites fluid of patients with newly diagnosed epithelial ovarian cancer shows abundant neutrophils (boxes) and a sparse number of tumor cells (arrow) embedded in fibrin deposits (pink filaments). Model and imaging are from Singel et al.137.
Together, these results support a model for neutrophil-platelet interactions and their downstream effects on vasculature and tissue remodeling as being pro-tumorigenic and accelerating metastasis. This model does not rely on specific tumor-derived factors but posits these interactions as a generalized response to injury and has functional similarity to circulating cells in invertebrates that seal wounds and have phagocytic capacity. We proposed a model of OC progression that links injury, neutrophilic inflammation, and thrombosis137. This model is not expected to be unique to OC, but is based on our observations from patients with OC. This combination of factors in the TME enables tumor seeding of serosal surfaces where early metastasis in OC occurs. In addition to the direct adhesive effect of fibrin, activated platelets cross signal with neutrophils and release pro-proliferative and angiogenic factors that enhance tumor progression and metastasis. In addition, ascites fluid chemoattracts and induces a suppressor phenotype in neutrophils that is expected to be a barrier to tumor immunity. The capacity for the TME to stimulate NET generation supports the concept of neutrophils as sensors of damage and as drivers of inflammation, injury, and thrombosis in cancer.
Neutrophil-generated Reactive Oxygen Species
The phagocyte NADPH oxidase generates ROS in response to infection and other stimuli and is critical for antimicrobial defense. Chronic granulomatous disease (CGD), an inherited disorder of the phagocyte NADPH oxidase, is characterized by severe infections and by excessive inflammatory responses (e.g., inflammatory bowel disease and granulomatous cystitis)173. Following activation, NADPH oxidase converts molecular oxygen to superoxide anion and to downstream metabolites, including H2O2 and hydroxyl anion. In neutrophils, myeloperoxidase (MPO) converts H2O2 to HOCl174–176. In a series of studies by Clark and colleagues, neutrophil-mediated killing of tumor cells ex vivo depended on activation of the MPO-H2O2-halide system177–180. In addition, cationic proteins from human neutrophil granules, which can be activated by NADPH oxidase181, were cytotoxic against tumor cells182. Although ex vivo studies cannot model the complexity of the TME, these studies raise the notion that specific tumors might have different levels of sensitivity to neutrophil-generated ROS, which is likely driven in part by induction of ROS-scavenging pathways such as NRF-2 and other cytoprotective pathways183.
Zhong et al.184 showed a role for the phagocyte NADPH oxidase in promoting lung tumor seeding after intravenous administration of melanoma tumor cells. Mice deficient in NADPH oxidase (p47phox-deficient) had fewer lung metastatic lesions. Neutrophil depletion by anti-Ly6G increased lung metastasis in wildtype, but not in NADPH oxidase-deficient, mice. This is an unexpected finding since in most tumor-bearing models, anti-Ly6G either has no effect or reduces tumor progression and metastasis. NADPH oxidase-generated ROS can signal to tumor cells and to other immune cells in the TME. The neutrophil NADPH oxidase/MPO system can also inhibit T cell and natural killer (NK) cell function185–187. Aydin et al.188 showed that NADPH oxidase in phagocytes can inhibit the function of NK cells, thereby facilitating metastasis.
NADPH oxidase can cross-signal to T cells and limit T cell responses189–192; this dampening effect on T cell activation likely averts excessive granulomatous responses and autoimmunity. Neutrophil NADPH oxidase-generated ROS and downstream metabolites could oxidize residues on the T cell receptor (TCR) leading to impaired T cell activation193. To delineate the role of NADPH oxidase in neutrophil suppressor function, we evaluated neutrophils from patients with CGD. Normal donor neutrophils suppressed stimulated T cell proliferation following exposure to ovarian cancer ascites fluid, while similarly treated neutrophils from gp91phox-deficient CGD patients did not67. However, incubation of normal donor neutrophils in OC ascites fluid supernatants did not stimulate extracellular ROS production or result in increased peroxynitrite formation on T cells, and addition of superoxide dismutase and catalase to neutralize ROS did not abrogate neutrophil suppressor function67. These data suggest that suppression of T cell proliferation does not occur from a direct effect of neutrophil-generated ROS on the T cells, but likely involves indirect signaling pathways. Aarts et al.65 showed that neutrophils activated by N-Formylmethionyl-leucyl-phenylalanine (fMLF) and other stimuli acquire a suppressor phenotype through pathways that include NADPH oxidase. We speculate that phagocyte-derived ROS limit T cell activation as an adaptive response to avert excessive T cell responses following injury that may manifest as exuberant tissue granulomata that characterize CGD. In the TME, these same pathways might limit T cell expansion required for tumor control.
Neutrophil extracellular traps, coagulation, and effects on T cell immunity
In addition to pathogen killing via oxidant injury, NADPH oxidase can augment host defense by intracellular activation of granular proteases181 and generation of NETs194,195. NET production is a distinct response to microbial products and DAMPs, characterized by the breakdown of membranes and extracellular release of stretches of DNA, histones, granular proteases and other constituents195. NET generation can be stimulated by infections196,197 and by non-infectious insults that activate innate immune receptors198–201, and can be stimulated by NADPH oxidase--dependent and -independent pathways202. NETs limit pathogen spread and directly injure or kill extracellular microbes. While apoptosis of neutrophils results in non-inflammatory cell death, NETs release products such as proteases and histones that can cause tissue injury200,201,203,204. In the TME, these NET products that degrade and remodel the extracellular matrix may promote tumor cell migration and invasion.
During neutrophil activation myeloid-specific protein arginine deiminase-4 (PAD4) facilitates the ‘loosening’ of the densely packed DNA by histone citrullination, which can be visualized by staining of citrullinated histone 3 (CitH3). The presence of NETs within tumor may not only reflect the local activation of neutrophils within the TME but could also drive tumor progression, as was reported in several experimental mouse models59,205–210, and which still needs to be substantiated in the human cancer setting.
NET generation occurs at different stages of tumor development and dissemination. Several hypotheses need to be explored to understand how NETs interact with the tumor immune landscape. Broadly speaking, NETs have the potential to promote thrombosis and angiogenesis in the local TME and to predispose to large vessel thrombosis and embolic events commonly observed in patients with advanced cancer. NETs can also modulate anti-tumor immunity, remodel matrix, and directly signal to tumor cells.
NETs are pro-thrombogenic, likely due to the released extracellular stretches of chromatin211–216. and tissue factor217. In mice, depletion of NETs resulted in limiting thrombosis215,216. PAD4, an enzyme that catalyzes histone citrullination required for NET generation, mediated experimental deep venous thrombosis218. Activated platelets can also stimulate NET production219,220, supporting the notion of a positive feedback loop driving neutrophil activation and thrombosis. Thus, activated neutrophils and platelets can cross-signal in a positive feedback loop in the context of inflammation and injury.
Patients with cancer are at high risk of developing arterial and venous thromboembolism (VTE). Depending on the type of tumor, cancer therapy, and other risk factors, 1% to 25% of patients with cancer will develop thrombosis, and risk assessment models can identify patients at the highest risk for cancer-associated thrombosis who might benefit from thromboprophylaxis221. There is growing evidence for NETs playing a role in cancer-associated venous and arterial thrombosis214,222, and circulating citrullinated histone H3, a biomarker of NETs, predicted the risk of VTE in cancer patients223
As mentioned earlier, NETs can affect local tumor vasculature and cross-signal to stromal and tumor cells that facilitate tumor spread. NETs have been identified in a number of cancers including sarcoma,224, pancreatic cancer225, and ovarian cancer137. NETs, complement, and coagulation pathways can cross-signal and amplify each other’s effects in the TME33, creating niches for metastatic seeding34,35. Several studies point to NETs accelerating tumor progression in mice and inhibiting NETs as a therapeutic approach59,172,206,208,209. We observed NETs within resected human epithelial ovarian cancer, and ovarian cancer ascites fluid induced NET production by healthy donor neutrophils137. These results are consistent with various DAMPs expected to be abundant in the TME that ligate neutrophil receptors and induce NET generation198,199,225,226. In addition, tumor-secreted products can recruit neutrophils and induce NETs. Xiao et al.227 showed that tumor-secreted protease cathepsin C-promoted metastasis in mice by recruitment of neutrophils and stimulation of ROS and NETs, and inhibiting cathepsin C reduced metastasis. Cathepsin C, or dipeptidyl aminopeptidase I, is a lysosomal protease capable of removing dipeptides from the amino terminus of protein substrates including elastase, cathepsin G and granzymes. Nonetheless, patients with Papillon Lefevre syndrome, a disorder of CTSC function, do not have significant immunodeficiency, but their neutrophils do not form NETs228. These results show important species-dependent requirements for neutrophil-driven antimicrobial defense, and, by extension, their effects on the TME.
In addition to accelerating thrombosis, NETs are capable of disabling T cell immunity and signaling to tumor cells. Zhang et al.229 showed that IL-17-recruited neutrophils, stimulated NET production and excluded CTLs from tumors in murine pancreatic cancer, and inhibition of IL-17 enhanced the efficacy of checkpoint inhibitors. Teijeira et al.59 observed that CXCR1 and CXCR2 ligands produced by tumors were the major mediators of cancer-associated NET production. NETs coated tumor cells and impaired their contact with immune cells, resulting in diminished CD8+ T cell- and NK cell-directed cytotoxicity. A limitation of the experimental model was that tumor cells were pre-incubated with neutrophils undergoing NET formation, which is somewhat artificial since pre-NET-ed tumor cells will not exist in real life. In addition, PAD4 inhibitors enhanced the anti-tumor effect of immune checkpoint inhibitors. The extent that NETs drive MDSC activity either by a direct effect on NK or T cells or by shielding tumor cells from immune attack is an interesting subject to pursue both mechanistically and as a therapeutic target to abrogate neutrophil-driven immunosuppression.
Interestingly, there are settings in which NETs are potentially beneficial. Intrapleural instillation of methotrexate packaged tumor cell-derived microparticles to treat malignant pleural effusions resulted in recruitment of neutrophils to the pleural space and NET formation that was associated with tumor cell killing and pleural effusion resolution in tumor-bearing mice230. These studies reinforce a common theme of neutrophils having pro- or anti-tumor effects that can depend on specific tumors and the tumor immune landscape as well as interactions with specific therapeutics.
Neutrophil Interactions with Tumor Cells
Neutrophils are involved in regulating tumor progression at multiple stages from establishing primary tumors to preparing the pre-metastatic niche for seeding and influencing metastatic spread. Neutrophils can affect these stages via direct interaction with the tumor cells and by interactions with cells in the TME such as the stromal fibroblasts, platelets and immune cells. In this section, we discuss the major concepts regarding the more direct effects of neutrophils on tumor cells. We note that this is a simplification since signaling interactions between neutrophils and tumor cells and between neutrophils and other cells that constitute the TME occur concurrently, and cumulatively affect tumor growth and metastasis.
Neutrophils can be recruited early during tumor initiation and affect the growth of the primary tumor. In addition, the presence of neutrophilic inflammation can affect the likelihood of tumor establishment either following tumor injection or tumor initiation in genetically predisposed mice. For example, in a K-ras mutant mouse model with experimental COPD, the influx of neutrophils and the resultant increase in production of IL-6 were associated with an increased propensity for lung tumor development231. Neutrophil-dependent IL-6 production was also implicated in colorectal cancer incidence and progression in an inflammatory colitis-associated cancer model where infiltrating neutrophils activated by the commensal gut flora produce IL-1b and subsequently, IL-6 by macrophages and dendritic cells, promoting the proliferation of intestinal epithelial cells232. Similar observations were made by Katoh et al.233 where they describe a role for infiltration of CXCR2-expressing granulocytes in tumor establishment. Depletion of neutrophils in a Helicobacter hepaticus-driven mammary adenocarcinoma model in genetically susceptible C3–1-TAg mice abrogated tumorigenesis234. Depletion of infiltrating neutrophils abrogated Myc-inducible liver tumorigenesis in a zebrafish model235.
NETs are implicated in transforming dormant cancer cells into aggressive lung metastasis in an experimental lung inflammation model. Exposure to tobacco smoke or intranasal LPS stimulated metastasis in previously dormant tumor cells, an effect attributed to NETs. Neutrophil elastase and MMP-9, which are released following neutrophil degranulation and NET generation, remodeled tumor cell-associated laminin, resulting in the activation of integrin and ERK signaling in tumor cells that drove their metastatic potential207. However, Cui et al.236 show a tumoricidal role for neutrophil elastase. Human neutrophils release neutrophil elastase that kills many cancer cell types through a mechanism dependent on cleavage of the CD95 death domain in cancer cells236. Mouse neutrophils lacked this effect due to increased production of secretory leukocyte peptidase inhibitor (SLPI), which inactivates neutrophil elastase. Intratumoral administration of neutrophil elastase reduced tumor growth and induced a CD8+ T cell-mediated abscopal effect against distant metastatic sites236. Breast cancer cells have been observed to take up neutrophil elastase, generating an increase in neo-antigen presentation, which further allows for CTL-dependent tumor clearance237. These findings support a recurring theme of this review: neutrophils and their specific products can have different effects on tumor progression based on the signaling cues they deliver to tumor cells and the TME.
Epithelial-mesenchymal transition (EMT) is characterized by changes in gene expression and post-translational regulation that leads to the loss of these epithelial characteristics and the acquisition of mesenchymal characteristics. EMT in cancer cells is a reversible process associated with increased invasiveness, migratory and metastatic capacity, and chemotherapy resistance. Cancer stem cells (CSCs) are a fraction of undifferentiated cancer cells that exhibit stem cell-like features, drive tumor initiation and relapses, and are associated with resistance to therapy. Overexpression of several EMT transcription factors is associated with stemness in cancer cells and suggests a link between EMT and CSCs238. In addition, TGF-β can promote tumor invasion and metastasis by inducing EMT and stem cell-like features239.
Xia et al.240 showed that post-operative infections can stimulate NET production, an adaptive response to control infection. In gastric cancer, NETs promoted cell proliferation, invasion and EMT, an effect that was dependent on TGF-β signaling, and TGF-β blockade reduced metastatic spread in tumor-bearing mice. These findings raise the question of whether the ability of NETs to induce EMT is due to neutrophil-derived products, platelet cross-activation that releases TGF-β, or the concurrent activation of both neutrophils and platelets (see Neutrophils, Platelets and Endothelial Cells: Partnership in Tumor Spread).
Multiple studies have shown the potential for neutrophils/PMN-MDSC to induce stem cell-like features in tumor cells241 242 243. Prostaglandin E2 produced by myeloid cells/MDSCs increased the stem cell-like features and PD-L1 expression in murine epithelial ovarian cancer244. Neutrophil–tumor cell interactions may result in a positive feedback loop in a model of hepatocellular carcinoma resulting in the bolstering of stemness in cancer cells and increased recruitment of neutrophils to the TME. In this setting, tumor-associated neutrophils secrete BMP-2 and TGF-β, which induce miR-301b-3p expression in tumor cells, increasing stemness. These stem cell-like tumor cells secrete increased levels of CXCL5 that, in turn, recruit neutrophils to the TME245. Tumor-associated neutrophils in gastric cancer models secrete inflammatory factors such as IL-17, IL-23 and TNF-α, which can transform mesenchymal stem cells in the tumor stroma to cancer-associated fibroblasts by activation of PI3K/Akt pathways89. Cancer-associated fibroblasts can induce stemness in tumor cells by stimulating tumor production of TGF-β and CXCL6246.
Neutrophils can affect tumor cell invasiveness and metastasis through a direct effect on tumor cells and by modifying endothelial cell function and extracellular matrix. Studies in vitro showed that neutrophils can adhere to the tumor cells and facilitate migration across the endothelial layer via CD11b/CD18 and ICAM-1 interactions247–249. Spiegel et al.250 showed that tumor-associated neutrophils enhance metastasis by two mechanisms. Neutrophils inhibited NK cell function, which reduces tumor cell killing, and neutrophils facilitate extravasation of tumor cells through the secretion of IL1-β and matrix metalloproteinases250. As discussed earlier, there is precedent for the neutrophil NADPH oxidase dampening NK cell function188.
Circulating tumor cells (CTCs) are precursors of metastasis in several cancers and are occasionally found in the bloodstream in association with white blood cells. Neutrophils can adhere directly to tumor cells and influence their proliferation and metastatic potential. Neutrophils promoted liver metastasis via CD11b/CD18-mediated interactions with circulating tumor cells251 Intravital microscopy showed direct adhesion of the tumor cells on the cell surface of arrested neutrophils on the liver sinusoids, an initial step in hepatic seeding of tumor. This group subsequently showed in a model of concurrent sepsis and tumor challenge that NETs bound to circulating tumor cells and facilitated hepatic metastasis206. Wculek et al.252 demonstrated a role for neutrophil-derived leukotrienes in driving breast cancer metastasis to the lungs. Szczerba et al.253 characterized CTCs embedded within white blood cell clusters in patients with breast cancer and in tumor-bearing mice. CTCs associated with neutrophils, and neutrophil clustering induced transcriptome profiles in tumor cells characterized by cell cycle progression, leading to more efficient metastasis. Yang et al.35 showed that NETs in the liver or lungs enhanced metastasis by recruiting cancer cells to these sites. They further identified the transmembrane protein CCDC25 as a NET-DNA receptor on cancer cells that senses extracellular DNA and drives signaling responses in tumor cells that enhance motility. In patients, CCDC25 expression on primary cancer cells was associated with poor prognosis. NETs can also alter the metabolic programming of cancer cells characterized by increased mitochondrial biogenesis and ATP production to increase tumor growth254.
Activated neutrophils can reactivate dormant tumor cells. Cancer cell dormancy is defined as tumor cells in a reversible state of cell cycle arrest and can be associated with a senescence-like state induced by genes, chemotherapy, or radiation. Perego et al.255 observed that tumor recurrence in mice was inducible by stress-activated neutrophils through a mechanism dependent on S100A8/A9 and oxidized lipids. Albrengues et al.207 observed that the NET-associated proteases, neutrophil elastase and matrix metalloproteinase 9, cleaved laminin resulting in a proliferative signal to tumor cells and abrogation of dormancy.
Activated neutrophils and NETs can also create a “pre-metastatic niche” in which neutrophils enhance metastasis by preparing a distant site for metastatic seeding208,256–258. Chronic nicotine exposure in mice resulted in the recruitment of N2 neutrophils (defined based on CD206, CCL2, and ARG2 expression) to the lungs that facilitated mammary cancer metastasis to the lungs259. Consistent with these results, lung metastasis was more common in breast cancer patients who were current or ex-smokers compared to never smokers259. This pre-metastatic niche is likely mediated by a number of factors driven by activated neutrophils, including their direct interactions with tumor cells and modulation of local vasculature, thrombosis, tissue remodeling, and dampening of T cell responses260. The notion of neutrophils forming a pre-metastatic niche also applies to platelets261. As previously discussed, neutrophils and platelets cross-signal and form aggregates in response to infection and injury, and activated platelets rapidly degranulate, and therefore may not be detectable in the TME without specific imaging techniques or detection of platelet microparticles137,171.
Recent studies provide mechanistic knowledge about contexts in which neutrophils are friends rather than foes of both direct and T-cell mediated anti-tumor responses. The TME has distinct immunologic niches that can vary within the same patient’s tumor262. This concept applies to distinct neutrophil populations within different tumor niches whose function can be regulated by multiple cues that change as a function of tumor location (e.g., tumor-specific features, alterations in oxygen levels, DAMPs, local cytokines, chemokines, and cell populations). As an example of susceptibility to neutrophil cytotoxicity, tumor cell expression of TRPM2, an H2O2-dependent Ca2+ channel, was upregulated after EMT and rendered tumor cells more susceptible to neutrophil cytotoxicity263. Mahiddine et al.264 showed that tumor hypoxia regulated the recruitment and function of neutrophils and that relief of tumor hypoxia improved neutrophil-driven tumor control, an effect that was T-cell independent. These studies underscore the importance of niches within tumors that influence both tumor cell and immune cell function, including whether recruited neutrophils are pro- or anti-tumorigenic.
Future Directions for Research
One of the important challenges in the field of tumor-associated neutrophils relates to nomenclature. As we have discussed, the criteria to define PMN-MDSC are inconsistent, and different investigators use different terms for defining the same cells (e.g., neutrophil, PMN-MDSC, or the compromise of neutrophil/PMN-MDSC). In addition to the value of consistent and accurate nomenclature, these questions are important both from the standpoint of mechanisms driving neutrophil suppressor function and therapeutic approaches to reverse suppression. If the principal mode that neutrophils/PMN-MDSC acquire suppressive function is through disordered granulopoiesis, then therapies should be designed to correct this bone marrow disorder. If on the other hand, neutrophils acquire suppressor function based on signaling within the circulation or the TME, then therapies should be tailored to abrogating this suppressor function. Neutrophils can acquire suppressor function through multiple mechanisms, and we consider PMN-MDSC as a neutrophil subset. We also argue that there should be evidence of disordered granulopoiesis before concluding that this is the principal mode for neutrophils acquiring suppressor function.
Although neutrophil heterogeneity was proposed 40-years ago265, we now have the tools to dissect neutrophil heterogeneity at a single cell level in circulation and in inflamed tissue266. Mass cytometry and metabolic and functional studies (e.g., in causing T cell suppression) provide additional tools to characterize neutrophil subsets and how they are modified by the TME. Neutrophil phenotypes in circulation and in the TME should be compared and contrasted with those observed in other conditions, such as major trauma. An exciting area of research relates to distinct neutrophil sub-populations within different tumor regions, e.g., in close proximity to T cells, tumor cells, or perivascular, and the extent that this spatial localization affects neutrophil function.
Another important area of investigation relates to when neutrophils engage tumor cells and the TME. This engagement can occur during initial tumorigenesis, tumor growth at the primary site, within vasculature, at the pre-metastatic niche, microscopic metastasis that may enter a dormant phase, or uncontrolled metastatic disease with extensive tumor burden. The damage and inflammatory cues that recruit neutrophils differ based on these conditions, and these cues can direct neutrophil responses in ways that limit or worsen tumor progression and metastasis.
Mechanistic knowledge about neutrophil recruitment to the TME and how the TME alters neutrophil function has paved the way for new therapeutic approaches. In general, strategies that deplete neutrophils or disable their recruitment to the TME favor tumor control – but this is not a universal observation. For example, studies in tumor-bearing mice support the notion of CXCR2 inhibitors to limit tumor growth and enhance the effect of checkpoint inhibitors. This approach is attractive since CXCR2 antagonists are safe (but not effective) in clinical trials of asthma267,268, and are now being evaluated in patients with cancer. In most tumor-bearing models, NETs promote tumor progression through a number of mechanisms including thrombosis, entrapping and mobilizing tumor cells, and direct signaling to tumor cells. These results establish the rationale for a clinical trial of NET inhibition. Yet another approach would be drugs targeting the IL-23/IL-17 axis for immunotherapy for metastatic tumors. Our group and others are investigating complement inhibition as a means of limiting neutrophil recruitment to the TME and abrogating their suppressor function.
Given the plasticity of neutrophils, can we arm them to have enhanced tumor cell cytotoxicity or alter their effect on T cells in ways that promote anti-tumor immunity? One approach is to enhance neutrophil and monocyte/macrophage killing of tumor cells through blockade of SIRPα and other myeloid checkpoint receptors that limit phagocytic attack87,269. The combination of IgA antibodies against tumor antigens and CD47-SIRPα checkpoint inhibition might be particularly effective in neutrophil-driven tumor attack270. Such approaches could in principle shift the overall effect of tumor-infiltrating neutrophils from being a barrier to antitumor immunity to enhancing tumor control (Figure 5). Therapies directed against myeloid checkpoint pathways might be of particular value in patients with myeloid cell-rich tumors, which could be the basis for selecting such patients for clinical trials.
Figure 5. Potential to shift the balance between neutrophil-driven injury directed at T cells (foe) to tumor cells (friend) in the TME.
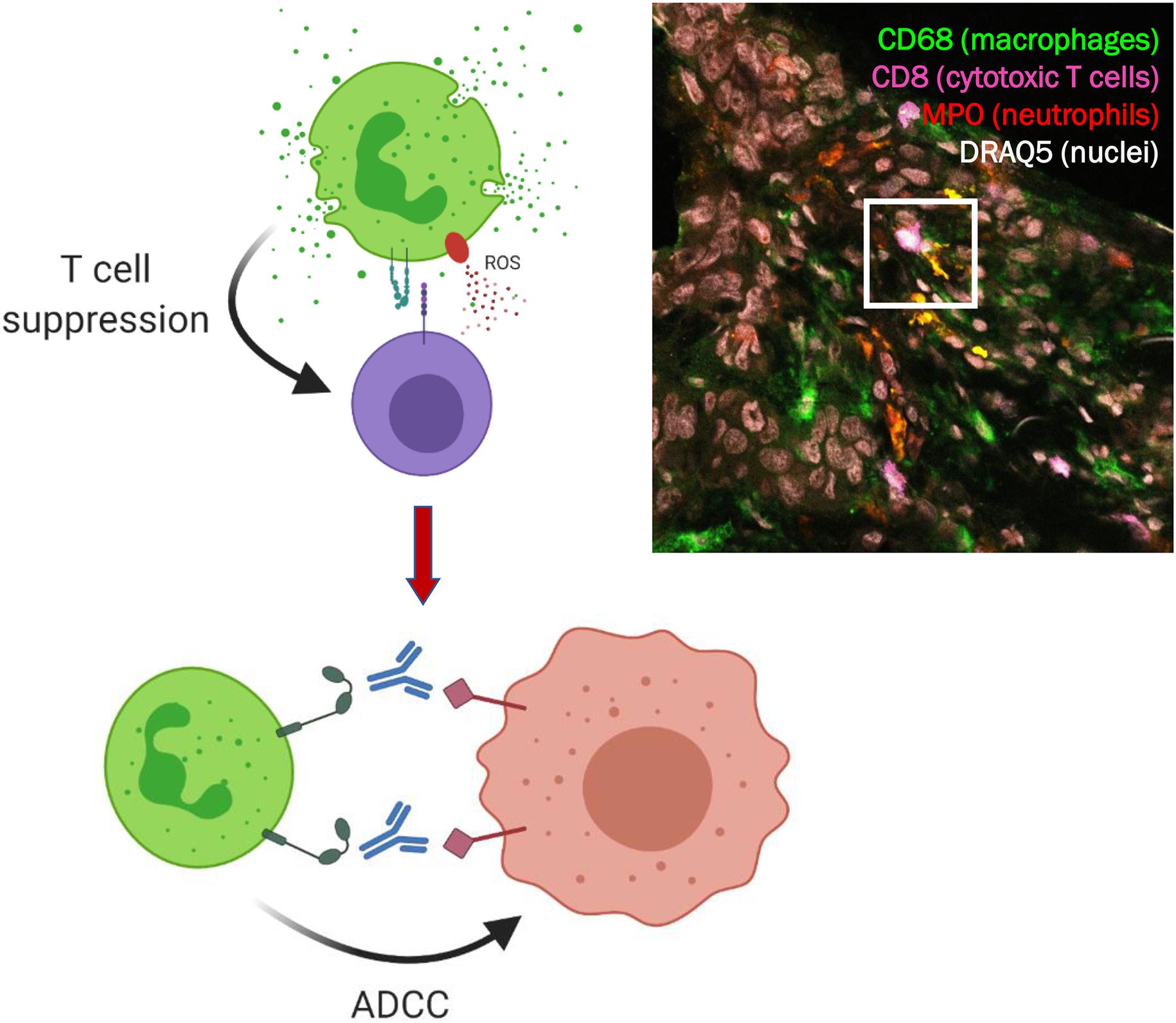
Model to change neutrophils (green cells) with PMN-MDSC activity against tumor-infiltrating T cells (purple) into PMN-ADCC activity against tumor cells (red) when aided by target recognition by specific anti-tumor antibodies. One approach to do so is the combination of myeloid checkpoint blockade (e.g., directed at SIRPα) and antibodies directed against tumor antigens. Right panel: Histology section of colon carcinoma. With 8-color Airyscan confocal microscopy the cellular composition of the TME can be dissected into areas with tumor-associated neutrophils (red), macrophages (green), and CD8+ T cells (magenta) with DRAQ5 as nuclear staining. Within the TME, contact between PMN and T cells is observed (quadrant), and is expected to inhibit T cell expansion and activation.
Acknowledgments:
This work was supported by R01CA188900 (BHS) and R01CA267690 (EZ and BHS).
References
- 1.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–545. [DOI] [PubMed] [Google Scholar]
- 2.Pikarsky E, Porat RM, Stein I, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431(7007):461–466. [DOI] [PubMed] [Google Scholar]
- 3.DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev. 2012;246(1):379–400. [DOI] [PubMed] [Google Scholar]
- 4.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315(26):1650–1659. [DOI] [PubMed] [Google Scholar]
- 5.Lavin Y, Kobayashi S, Leader A, et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell. 2017;169(4):750–765 e717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng C, Zheng L, Yoo JK, et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell. 2017;169(7):1342–1356 e1316. [DOI] [PubMed] [Google Scholar]
- 7.Angelova M, Mlecnik B, Vasaturo A, et al. Evolution of Metastases in Space and Time under Immune Selection. Cell. 2018;175(3):751–765 e716. [DOI] [PubMed] [Google Scholar]
- 8.Pages F, Mlecnik B, Marliot F, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018;391(10135):2128–2139. [DOI] [PubMed] [Google Scholar]
- 9.Thavaramara T, Phaloprakarn C, Tangjitgamol S, Manusirivithaya S. Role of neutrophil to lymphocyte ratio as a prognostic indicator for epithelial ovarian cancer. J Med Assoc Thai. 2011;94(7):871–877. [PubMed] [Google Scholar]
- 10.Azab B, Bhatt VR, Phookan J, et al. Usefulness of the neutrophil-to-lymphocyte ratio in predicting short- and long-term mortality in breast cancer patients. Annals of surgical oncology. 2012;19(1):217–224. [DOI] [PubMed] [Google Scholar]
- 11.Absenger G, Szkandera J, Pichler M, et al. A derived neutrophil to lymphocyte ratio predicts clinical outcome in stage II and III colon cancer patients. Br J Cancer. 2013;109(2):395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donskov F. Immunomonitoring and prognostic relevance of neutrophils in clinical trials. Semin Cancer Biol. 2013;23(3):200–207. [DOI] [PubMed] [Google Scholar]
- 13.Perez DR, Baser RE, Cavnar MJ, et al. Blood neutrophil-to-lymphocyte ratio is prognostic in gastrointestinal stromal tumor. Annals of surgical oncology. 2013;20(2):593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perisanidis C, Kornek G, Poschl PW, et al. High neutrophil-to-lymphocyte ratio is an independent marker of poor disease-specific survival in patients with oral cancer. Med Oncol. 2013;30(1):334. [DOI] [PubMed] [Google Scholar]
- 15.Stotz M, Gerger A, Eisner F, et al. Increased neutrophil-lymphocyte ratio is a poor prognostic factor in patients with primary operable and inoperable pancreatic cancer. Br J Cancer. 2013;109(2):416–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szkandera J, Absenger G, Liegl-Atzwanger B, et al. Elevated preoperative neutrophil/lymphocyte ratio is associated with poor prognosis in soft-tissue sarcoma patients. Br J Cancer. 2013;108(8):1677–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang QT, Zhou L, Zeng WJ, et al. Prognostic Significance of Neutrophil-to-Lymphocyte Ratio in Ovarian Cancer: A Systematic Review and Meta-Analysis of Observational Studies. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2017;41(6):2411–2418. [DOI] [PubMed] [Google Scholar]
- 18.Heng DY, Xie W, Regan MM, et al. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol. 2009;27(34):5794–5799. [DOI] [PubMed] [Google Scholar]
- 19.Mitchell KG, Diao L, Karpinets T, et al. Neutrophil expansion defines an immunoinhibitory peripheral and intratumoral inflammatory milieu in resected non-small cell lung cancer: a descriptive analysis of a prospectively immunoprofiled cohort. J Immunother Cancer. 2020;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu WC, Sun HW, Chen HT, et al. Circulating hematopoietic stem and progenitor cells are myeloid-biased in cancer patients. Proc Natl Acad Sci U S A. 2014;111(11):4221–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gentles AJ, Newman AM, Liu CL, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21(8):938–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Si Y, Merz SF, Jansen P, et al. Multidimensional imaging provides evidence for down-regulation of T cell effector function by MDSC in human cancer tissue. Sci Immunol. 2019;4(40). [DOI] [PubMed] [Google Scholar]
- 23.Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19(2):108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eruslanov EB, Bhojnagarwala PS, Quatromoni JG, et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J Clin Invest. 2014;124(12):5466–5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singhal S, Bhojnagarwala PS, O’Brien S, et al. Origin and Role of a Subset of Tumor-Associated Neutrophils with Antigen-Presenting Cell Features in Early-Stage Human Lung Cancer. Cancer Cell. 2016;30(1):120–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ponzetta A, Carriero R, Carnevale S, et al. Neutrophils Driving Unconventional T Cells Mediate Resistance against Murine Sarcomas and Selected Human Tumors. Cell. 2019;178(2):346–360 e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lichtenstein A, Kahle J. Anti-tumor effect of inflammatory neutrophils: characteristics of in vivo generation and in vitro tumor cell lysis. Int J Cancer. 1985;35(1):121–127. [DOI] [PubMed] [Google Scholar]
- 28.Otten MA, Rudolph E, Dechant M, et al. Immature neutrophils mediate tumor cell killing via IgA but not IgG Fc receptors. J Immunol. 2005;174(9):5472–5480. [DOI] [PubMed] [Google Scholar]
- 29.Stockmeyer B, Beyer T, Neuhuber W, et al. Polymorphonuclear granulocytes induce antibody-dependent apoptosis in human breast cancer cells. J Immunol. 2003;171(10):5124–5129. [DOI] [PubMed] [Google Scholar]
- 30.Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe? Carcinogenesis. 2012;33(5):949–955. [DOI] [PubMed] [Google Scholar]
- 31.Lin RJ, Afshar-Kharghan V, Schafer AI. Paraneoplastic thrombocytosis: the secrets of tumor self-promotion. Blood. 2014;124(2):184–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stone RL, Nick AM, McNeish IA, et al. Paraneoplastic thrombocytosis in ovarian cancer. The New England journal of medicine. 2012;366(7):610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Bont CM, Boelens WC, Pruijn GJM. NETosis, complement, and coagulation: a triangular relationship. Cellular & molecular immunology. 2019;16(1):19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Labelle M, Begum S, Hynes RO. Platelets guide the formation of early metastatic niches. Proc Natl Acad Sci U S A. 2014;111(30):E3053–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang L, Liu Q, Zhang X, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. 2020;583(7814):133–138. [DOI] [PubMed] [Google Scholar]
- 36.Gordon S Phagocytosis: The Legacy of Metchnikoff. Cell. 2016;166(5):1065–1068. [DOI] [PubMed] [Google Scholar]
- 37.Andrade C, Oliveira B, Guatelli S, et al. Characterization of Coelomic Fluid Cell Types in the Starfish Marthasterias glacialis Using a Flow Cytometry/Imaging Combined Approach. Frontiers in immunology. 2021;12:641664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaufmann SH. Immunology’s foundation: the 100-year anniversary of the Nobel Prize to Paul Ehrlich and Elie Metchnikoff. Nat Immunol. 2008;9(7):705–712. [DOI] [PubMed] [Google Scholar]
- 39.Deng Q, Harvie EA, Huttenlocher A. Distinct signalling mechanisms mediate neutrophil attraction to bacterial infection and tissue injury. Cell Microbiol. 2012;14(4):517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lammermann T, Afonso PV, Angermann BR, et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature. 2013;498(7454):371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McDonald B, Pittman K, Menezes GB, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330(6002):362–366. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peiseler M, Kubes P. More friend than foe: the emerging role of neutrophils in tissue repair. J Clin Invest. 2019;129(7):2629–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fischer A, Wannemacher J, Christ S, et al. Neutrophils direct preexisting matrix to initiate repair in damaged tissues. Nat Immunol. 2022;23(4):518–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zindel J, Peiseler M, Hossain M, et al. Primordial GATA6 macrophages function as extravascular platelets in sterile injury. Science. 2021;371(6533). [DOI] [PubMed] [Google Scholar]
- 46.Singel KL, Segal BH. Neutrophils in the tumor microenvironment: trying to heal the wound that cannot heal. Immunol Rev. 2016;273(1):329–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krzyszczyk P, Schloss R, Palmer A, Berthiaume F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front Physiol. 2018;9:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oishi Y, Manabe I. Macrophages in inflammation, repair and regeneration. Int Immunol. 2018;30(11):511–528. [DOI] [PubMed] [Google Scholar]
- 49.Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bronte V, Brandau S, Chen SH, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nature communications. 2016;7:12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Veglia F, Hashimoto A, Dweep H, et al. Analysis of classical neutrophils and polymorphonuclear myeloid-derived suppressor cells in cancer patients and tumor-bearing mice. J Exp Med. 2021;218(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kramer ED, Abrams SI. Granulocytic Myeloid-Derived Suppressor Cells as Negative Regulators of Anticancer Immunity. Frontiers in immunology. 2020;11:1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang PF, Song SY, Wang TJ, et al. Prognostic role of pretreatment circulating MDSCs in patients with solid malignancies: A meta-analysis of 40 studies. Oncoimmunology. 2018;7(10):e1494113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jamieson T, Clarke M, Steele CW, et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J Clin Invest. 2012;122(9):3127–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Highfill SL, Cui Y, Giles AJ, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. 2014;6(237):237ra267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Steele CW, Karim SA, Leach JD, et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2016;29(6):832–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu Z, Zou J, Li S, et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature. 2020;579(7798):284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teijeira A, Garasa S, Gato M, et al. CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps that Interfere with Immune Cytotoxicity. Immunity. 2020;52(5):856–871 e858. [DOI] [PubMed] [Google Scholar]
- 60.Kumar S, Wilkes DW, Samuel N, et al. DeltaNp63-driven recruitment of myeloid-derived suppressor cells promotes metastasis in triple-negative breast cancer. J Clin Invest. 2018;128(11):5095–5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83(1):64–70. [DOI] [PubMed] [Google Scholar]
- 62.Wang JX, Bair AM, King SL, et al. Ly6G ligation blocks recruitment of neutrophils via a beta2-integrin-dependent mechanism. Blood. 2012;120(7):1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vincent J, Mignot G, Chalmin F, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70(8):3052–3061. [DOI] [PubMed] [Google Scholar]
- 64.Quail DF, Amulic B, Aziz M, et al. Neutrophil phenotypes and functions in cancer: A consensus statement. J Exp Med. 2022;219(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aarts CEM, Hiemstra IH, Beguin EP, et al. Activated neutrophils exert myeloid-derived suppressor cell activity damaging T cells beyond repair. Blood Adv. 2019;3(22):3562–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Singel KL, Emmons TR, Khan ANH, et al. Mature neutrophils suppress T cell immunity in ovarian cancer microenvironment. JCI Insight. 2019;4(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Emmons TR, Giridharan T, Singel KL, et al. Mechanisms driving neutrophil-induced T-cell immunoparalysis in ovarian cancer. Cancer Immunol Res. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Engblom C, Pfirschke C, Zilionis R, et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecF(high) neutrophils. Science. 2017;358(6367). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pfirschke C, Engblom C, Gungabeesoon J, et al. Tumor-Promoting Ly-6G(+) SiglecF(high) Cells Are Mature and Long-Lived Neutrophils. Cell reports. 2020;32(12):108164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ryu S, Shin JW, Kwon S, et al. Siglec-F-expressing neutrophils are essential for creating a profibrotic microenvironment in renal fibrosis. J Clin Invest. 2022;132(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hu J, Yao B, Yang X, Ma F. The immunosuppressive effect of Siglecs on tendon-bone healing after ACL reconstruction. Med Hypotheses. 2015;84(1):38–39. [DOI] [PubMed] [Google Scholar]
- 72.Mishalian I, Bayuh R, Levy L, Zolotarov L, Michaeli J, Fridlender ZG. Tumor-associated neutrophils (TAN) develop pro-tumorigenic properties during tumor progression. Cancer immunology, immunotherapy : CII. 2013;62(11):1745–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tazawa H, Okada F, Kobayashi T, et al. Infiltration of neutrophils is required for acquisition of metastatic phenotype of benign murine fibrosarcoma cells: implication of inflammation-associated carcinogenesis and tumor progression. Am J Pathol. 2003;163(6):2221–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest. 2010;120(4):1151–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sagiv JY, Michaeli J, Assi S, et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell reports. 2015;10(4):562–573. [DOI] [PubMed] [Google Scholar]
- 76.Ohms M, Moller S, Laskay T. An Attempt to Polarize Human Neutrophils Toward N1 and N2 Phenotypes in vitro. Frontiers in immunology. 2020;11:532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hegde S, Leader AM, Merad M. MDSC: Markers, development, states, and unaddressed complexity. Immunity. 2021;54(5):875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Giese MA, Hind LE, Huttenlocher A. Neutrophil plasticity in the tumor microenvironment. Blood. 2019;133(20):2159–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sun R, Huang J, Yang Y, et al. Dysfunction of low-density neutrophils in peripheral circulation in patients with sepsis. Scientific reports. 2022;12(1):685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tay SH, Celhar T, Fairhurst AM. Low-Density Neutrophils in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2020;72(10):1587–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bryk JA, Popovic PJ, Zenati MS, Munera V, Pribis JP, Ochoa JB. Nature of myeloid cells expressing arginase 1 in peripheral blood after trauma. J Trauma. 2010;68(4):843–852. [DOI] [PubMed] [Google Scholar]
- 82.Riça IG, Joughin BA, Teke ME, et al. Neutrophil heterogeneity and emegergence of a distinct population of CD11b/CD18-activated low-density neutrophils after trauma. The journal of trauma and acute care surgery. 2022. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blanco-Camarillo C, Aleman OR, Rosales C. Low-Density Neutrophils in Healthy Individuals Display a Mature Primed Phenotype. Frontiers in immunology. 2021;12:672520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001;61(12):4756–4760. [PubMed] [Google Scholar]
- 85.Aarts CEM, Hiemstra IH, Tool ATJ, et al. Neutrophils as Suppressors of T Cell Proliferation: Does Age Matter? Frontiers in immunology. 2019;10:2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mercer F, Ng SH, Brown TM, Boatman G, Johnson PJ. Neutrophils kill the parasite Trichomonas vaginalis using trogocytosis. PLoS Biol. 2018;16(2):e2003885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Matlung HL, Babes L, Zhao XW, et al. Neutrophils Kill Antibody-Opsonized Cancer Cells by Trogoptosis. Cell Rep. 2018;23(13):3946–3959 e3946. [DOI] [PubMed] [Google Scholar]
- 88.Joly E, Hudrisier D. What is trogocytosis and what is its purpose? Nat Immunol. 2003;4(9):815. [DOI] [PubMed] [Google Scholar]
- 89.Zhang J, Ji C, Li W, et al. Tumor-Educated Neutrophils Activate Mesenchymal Stem Cells to Promote Gastric Cancer Growth and Metastasis. Front Cell Dev Biol. 2020;8:788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Condamine T, Dominguez GA, Youn JI, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. 2016;1(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jacobsen LC, Theilgaard-Monch K, Christensen EI, Borregaard N. Arginase 1 is expressed in myelocytes/metamyelocytes and localized in gelatinase granules of human neutrophils. Blood. 2007;109(7):3084–3087. [DOI] [PubMed] [Google Scholar]
- 92.Alshetaiwi H, Pervolarakis N, McIntyre LL, et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci Immunol. 2020;5(44). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Evrard M, Kwok IWH, Chong SZ, et al. Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity. 2018;48(2):364–379 e368. [DOI] [PubMed] [Google Scholar]
- 94.Kuijpers TW, Tool AT, van der Schoot CE, et al. Membrane surface antigen expression on neutrophils: a reappraisal of the use of surface markers for neutrophil activation. Blood. 1991;78(4):1105–1111. [PubMed] [Google Scholar]
- 95.Hoogendijk AJ, Pourfarzad F, Aarts CEM, et al. Dynamic Transcriptome-Proteome Correlation Networks Reveal Human Myeloid Differentiation and Neutrophil-Specific Programming. Cell reports. 2019;29(8):2505–2519 e2504. [DOI] [PubMed] [Google Scholar]
- 96.Marini O, Costa S, Bevilacqua D, et al. Mature CD10(+) and immature CD10(−) neutrophils present in G-CSF-treated donors display opposite effects on T cells. Blood. 2017;129(10):1343–1356. [DOI] [PubMed] [Google Scholar]
- 97.Werfel T, Sonntag G, Weber MH, Götze O. Rapid increases in the membrane expression of neutral endopeptidase (CD10), aminopeptidase N (CD13), tyrosine phosphatase (CD45), and Fc gamma-RIII (CD16) upon stimulation of human peripheral leukocytes with human C5a. The Journal of Immunology. 1991;147(11):3909–3914. [PubMed] [Google Scholar]
- 98.Whitmore LC, Weems MN, Allen LH. Cutting Edge: Helicobacter pylori Induces Nuclear Hypersegmentation and Subtype Differentiation of Human Neutrophils In Vitro. J Immunol. 2017;198(5):1793–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rice CM, Davies LC, Subleski JJ, et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nature communications. 2018;9(1):5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Veglia F, Tyurin VA, Blasi M, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. 2019;569(7754):73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang X, Hu LP, Qin WT, et al. Identification of a subset of immunosuppressive P2RX1-negative neutrophils in pancreatic cancer liver metastasis. Nature communications. 2021;12(1):174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pugin J How tissue injury alarms the immune system and causes a systemic inflammatory response syndrome. Ann Intensive Care. 2012;2(1):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Singel KL, Grzankowski KS, Khan A, et al. Mitochondrial DNA in the tumour microenvironment activates neutrophils and is associated with worse outcomes in patients with advanced epithelial ovarian cancer. Br J Cancer. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Horn A, Jaiswal JK. Structural and signaling role of lipids in plasma membrane repair. Curr Top Membr. 2019;84:67–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Advani R, Flinn I, Popplewell L, et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N Engl J Med. 2018;379(18):1711–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191(3):677–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Weiss E, Kretschmer D. Formyl-Peptide Receptors in Infection, Inflammation, and Cancer. Trends Immunol. 2018;39(10):815–829. [DOI] [PubMed] [Google Scholar]
- 108.Tian W, Jiang X, Kim D, Guan T, Nicolls MR, Rockson SG. Leukotrienes in Tumor-Associated Inflammation. Frontiers in pharmacology. 2020;11:1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang M, Huang L, Ding G, et al. Interferon gamma inhibits CXCL8-CXCR2 axis mediated tumor-associated macrophages tumor trafficking and enhances anti-PD1 efficacy in pancreatic cancer. J Immunother Cancer. 2020;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Smith E, Zarbock A, Stark MA, et al. IL-23 is required for neutrophil homeostasis in normal and neutrophilic mice. J Immunol. 2007;179(12):8274–8279. [DOI] [PubMed] [Google Scholar]
- 111.Langowski JL, Zhang X, Wu L, et al. IL-23 promotes tumour incidence and growth. Nature. 2006;442(7101):461–465. [DOI] [PubMed] [Google Scholar]
- 112.Grivennikov SI, Wang K, Mucida D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491(7423):254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Thiele Orberg E, Fan H, Tam AJ, et al. The myeloid immune signature of enterotoxigenic Bacteroides fragilis-induced murine colon tumorigenesis. Mucosal Immunol. 2017;10(2):421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wu S, Rhee KJ, Albesiano E, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15(9):1016–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Coffelt SB, Kersten K, Doornebal CW, et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522(7556):345–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ajona D, Ortiz-Espinosa S, Moreno H, et al. A Combined PD-1/C5a Blockade Synergistically Protects against Lung Cancer Growth and Metastasis. Cancer discovery. 2017;7(7):694–703. [DOI] [PubMed] [Google Scholar]
- 117.Puls LE, Duniho T, Hunter JE, Kryscio R, Blackhurst D, Gallion H. The prognostic implication of ascites in advanced-stage ovarian cancer. Gynecol Oncol. 1996;61(1):109–112. [DOI] [PubMed] [Google Scholar]
- 118.Huang H, Li YJ, Lan CY, et al. Clinical significance of ascites in epithelial ovarian cancer. Neoplasma. 2013;60(5):546–552. [DOI] [PubMed] [Google Scholar]
- 119.Szender JB, Emmons T, Belliotti S, et al. Impact of ascites volume on clinical outcomes in ovarian cancer: A cohort study. Gynecologic oncology. 2017;146(3):491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Eng KH, Morrell K, Starbuck K, et al. Prognostic value of miliary versus non-miliary sub-staging in advanced ovarian cancer. Gynecol Oncol. 2017;146(1):52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer research. 2011;71(24):7463–7470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Khan AN, Kolomeyevskaya N, Singel KL, et al. Targeting myeloid cells in the tumor microenvironment enhances vaccine efficacy in murine epithelial ovarian cancer. Oncotarget. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shenoy GN, Loyall J, Maguire O, et al. Exosomes Associated with Human Ovarian Tumors Harbor a Reversible Checkpoint of T-cell Responses. Cancer Immunol Res. 2018;6(2):236–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gong Y, Yang J, Wang Y, Xue L, Wang J. Metabolic factors contribute to T-cell inhibition in the ovarian cancer ascites. Int J Cancer. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ford CE, Werner B, Hacker NF, Warton K. The untapped potential of ascites in ovarian cancer research and treatment. Br J Cancer. 2020;123(1):9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Szender JB, Emmons T, Belliotti S, et al. Impact of ascites volume on clinical outcomes in ovarian cancer: A cohort study. Gynecol Oncol. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Heintz AP, Van Oosterom AT, Trimbos JB, Schaberg A, Van der Velde EA, Nooy M. The treatment of advanced ovarian carcinoma (I): clinical variables associated with prognosis. Gynecol Oncol. 1988;30(3):347–358. [DOI] [PubMed] [Google Scholar]
- 128.Kuhn W, Rutke S, Spathe K, et al. Neoadjuvant chemotherapy followed by tumor debulking prolongs survival for patients with poor prognosis in International Federation of Gynecology and Obstetrics Stage IIIC ovarian carcinoma. Cancer. 2001;92(10):2585–2591. [DOI] [PubMed] [Google Scholar]
- 129.Janco JM, Glaser G, Kim B, et al. Development of a prediction model for residual disease in newly diagnosed advanced ovarian cancer. Gynecol Oncol. 2015;138(1):70–77. [DOI] [PubMed] [Google Scholar]
- 130.Idorn M, Olsen M, Halldorsdottir HR, et al. Improved migration of tumor ascites lymphocytes to ovarian cancer microenvironment by CXCR2 transduction. Oncoimmunology. 2018;7(4):e1412029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Khan AN, Kolomeyevskaya N, Singel KL, et al. Targeting myeloid cells in the tumor microenvironment enhances vaccine efficacy in murine epithelial ovarian cancer. Oncotarget. 2015;6(13):11310–11326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang L, Conejo-Garcia JR, Katsaros D, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348(3):203–213. [DOI] [PubMed] [Google Scholar]
- 133.Sato E, Olson SH, Ahn J, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(51):18538–18543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hamanishi J, Mandai M, Ikeda T, et al. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J Clin Oncol. 2015;33(34):4015–4022. [DOI] [PubMed] [Google Scholar]
- 136.Matulonis UA, Shapira-Frommer R, Santin AD, et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: results from the phase II KEYNOTE-100 study. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2019;30(7):1080–1087. [DOI] [PubMed] [Google Scholar]
- 137.Singel KL, Grzankowski KS, Khan A, et al. Mitochondrial DNA in the tumour microenvironment activates neutrophils and is associated with worse outcomes in patients with advanced epithelial ovarian cancer. Br J Cancer. 2019;120(2):207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Emmons TR, Giridharan T, Singel KL, et al. Mechanisms Driving Neutrophil-Induced T-cell Immunoparalysis in Ovarian Cancer. Cancer Immunol Res. 2021;9(7):790–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Corrales L, Ajona D, Rafail S, et al. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol. 2012;189(9):4674–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cho MS, Vasquez HG, Rupaimoole R, et al. Autocrine effects of tumor-derived complement. Cell reports. 2014;6(6):1085–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gunn L, Ding C, Liu M, et al. Opposing roles for complement component C5a in tumor progression and the tumor microenvironment. J Immunol. 2012;189(6):2985–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kwak JW, Laskowski J, Li HY, et al. Complement Activation via a C3a Receptor Pathway Alters CD4(+) T Lymphocytes and Mediates Lung Cancer Progression. Cancer Res. 2018;78(1):143–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Medler TR, Murugan D, Horton W, et al. Complement C5a Fosters Squamous Carcinogenesis and Limits T Cell Response to Chemotherapy. Cancer Cell. 2018;34(4):561–578 e566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nabizadeh JA, Manthey HD, Steyn FJ, et al. The Complement C3a Receptor Contributes to Melanoma Tumorigenesis by Inhibiting Neutrophil and CD4+ T Cell Responses. J Immunol. 2016;196(11):4783–4792. [DOI] [PubMed] [Google Scholar]
- 145.Vadrevu SK, Chintala NK, Sharma SK, et al. Complement c5a receptor facilitates cancer metastasis by altering T-cell responses in the metastatic niche. Cancer Res. 2014;74(13):3454–3465. [DOI] [PubMed] [Google Scholar]
- 146.Markiewski MM, DeAngelis RA, Benencia F, et al. Modulation of the antitumor immune response by complement. Nat Immunol. 2008;9(11):1225–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Markiewski MM, Vadrevu SK, Sharma SK, et al. The Ribosomal Protein S19 Suppresses Antitumor Immune Responses via the Complement C5a Receptor 1. J Immunol. 2017;198(7):2989–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zha H, Wang X, Zhu Y, et al. Intracellular Activation of Complement C3 Leads to PD-L1 Antibody Treatment Resistance by Modulating Tumor-Associated Macrophages. Cancer Immunol Res. 2019;7(2):193–207. [DOI] [PubMed] [Google Scholar]
- 149.Roumenina LT, Daugan MV, Noe R, et al. Tumor Cells Hijack Macrophage-Produced Complement C1q to Promote Tumor Growth. Cancer Immunol Res. 2019;7(7):1091–1105. [DOI] [PubMed] [Google Scholar]
- 150.Wang Y, Sun SN, Liu Q, et al. Autocrine Complement Inhibits IL10-Dependent T-cell-Mediated Antitumor Immunity to Promote Tumor Progression. Cancer discovery. 2016;6(9):1022–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Magrini E, Di Marco S, Mapelli SN, et al. Complement activation promoted by the lectin pathway mediates C3aR-dependent sarcoma progression and immunosuppression. Nat Cancer. 2021;2(2):218–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nunez-Cruz S, Gimotty PA, Guerra MW, et al. Genetic and pharmacologic inhibition of complement impairs endothelial cell function and ablates ovarian cancer neovascularization. Neoplasia. 2012;14(11):994–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hillmen P, Szer J, Weitz I, et al. Pegcetacoplan versus Eculizumab in Paroxysmal Nocturnal Hemoglobinuria. N Engl J Med. 2021;384(11):1028–1037. [DOI] [PubMed] [Google Scholar]
- 154.Coenen DM, Mastenbroek TG, Cosemans J. Platelet interaction with activated endothelium: mechanistic insights from microfluidics. Blood. 2017;130(26):2819–2828. [DOI] [PubMed] [Google Scholar]
- 155.Simon DI, Chen Z, Xu H, et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med. 2000;192(2):193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Santoso S, Sachs UJ, Kroll H, et al. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J Exp Med. 2002;196(5):679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Weber C, Springer TA. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet-activating factor. J Clin Invest. 1997;100(8):2085–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Boilard E, Nigrovic PA, Larabee K, et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. 2010;327(5965):580–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Duchez AC, Boudreau LH, Naika GS, et al. Platelet microparticles are internalized in neutrophils via the concerted activity of 12-lipoxygenase and secreted phospholipase A2-IIA. Proc Natl Acad Sci U S A. 2015;112(27):E3564–3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Boudreau LH, Duchez AC, Cloutier N, et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood. 2014;124(14):2173–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zarbock A, Singbartl K, Ley K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Invest. 2006;116(12):3211–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Grommes J, Alard JE, Drechsler M, et al. Disruption of platelet-derived chemokine heteromers prevents neutrophil extravasation in acute lung injury. Am J Respir Crit Care Med. 2012;185(6):628–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sreeramkumar V, Adrover JM, Ballesteros I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. 2014;346(6214):1234–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pinedo HM, Verheul HM, D’Amato RJ, Folkman J. Involvement of platelets in tumour angiogenesis? Lancet. 1998;352(9142):1775–1777. [DOI] [PubMed] [Google Scholar]
- 165.Asghar S, Parvaiz F, Manzoor S. Multifaceted role of cancer educated platelets in survival of cancer cells. Thromb Res. 2019;177:42–50. [DOI] [PubMed] [Google Scholar]
- 166.Mai S, Inkielewicz-Stepniak I. Pancreatic Cancer and Platelets Crosstalk: A Potential Biomarker and Target. Front Cell Dev Biol. 2021;9:749689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Cho MS, Bottsford-Miller J, Vasquez HG, et al. Platelets increase the proliferation of ovarian cancer cells. Blood. 2012;120(24):4869–4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20(5):576–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ozel I, Duerig I, Domnich M, Lang S, Pylaeva E, Jablonska J. The Good, the Bad, and the Ugly: Neutrophils, Angiogenesis, and Cancer. Cancers. 2022;14(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006;103(33):12493–12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Haemmerle M, Bottsford-Miller J, Pradeep S, et al. FAK regulates platelet extravasation and tumor growth after antiangiogenic therapy withdrawal. J Clin Invest. 2016;126(5):1885–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Guglietta S, Chiavelli A, Zagato E, et al. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nature communications. 2016;7:11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Seger R, Roos D, Segal BH, Kuijpers TW. Chronic Granulomatous Disease: Genetics, Biology and Clinical Management. Open access textbook, NOVA Publishers. 2017. [Google Scholar]
- 174.Harrison JE, Schultz J. Studies on the chlorinating activity of myeloperoxidase. The Journal of biological chemistry. 1976;251(5):1371–1374. [PubMed] [Google Scholar]
- 175.Albrich JM, McCarthy CA, Hurst JK. Biological reactivity of hypochlorous acid: implications for microbicidal mechanisms of leukocyte myeloperoxidase. Proceedings of the National Academy of Sciences of the United States of America. 1981;78(1):210–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Weiss SJ, Klein R, Slivka A, Wei M. Chlorination of taurine by human neutrophils. Evidence for hypochlorous acid generation. The Journal of clinical investigation. 1982;70(3):598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Clark RA, Klebanoff SJ. Neutrophil-mediated tumor cell cytotoxicity: role of the peroxidase system. J Exp Med. 1975;141(6):1442–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Clark RA, Klebanoff SJ. Role of the myeloperoxidase-H2O2-halide system in concanavalin A-induced tumor cell killing by human neutrophils. J Immunol. 1979;122(6):2605–2610. [PubMed] [Google Scholar]
- 179.Clark RA, Klebanoff SJ, Einstein AB, Fefer A. Peroxidase-H2O2-halide system: Cytotoxic effect on mammalian tumor cells. Blood. 1975;45(2):161–170. [PubMed] [Google Scholar]
- 180.Clark RA, Szot S. The myeloperoxidase-hydrogen peroxide-halide system as effector of neutrophil-mediated tumor cell cytotoxicity. J Immunol. 1981;126(4):1295–1301. [PubMed] [Google Scholar]
- 181.Reeves EP, Lu H, Jacobs HL, et al. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002;416(6878):291–297. [DOI] [PubMed] [Google Scholar]
- 182.Clark RA, Olsson I, Klebanoff SJ. Cytotoxicity for tumor cells of cationic proteins from human neutrophil granules. J Cell Biol. 1976;70(3):719–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tao S, Wang S, Moghaddam SJ, et al. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014;74(24):7430–7441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zhong J, Li Q, Luo H, Holmdahl R. Neutrophil-derived reactive oxygen species promote tumor colonization. Commun Biol. 2021;4(1):865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.El-Hag A, Clark RA. Down-regulation of human natural killer activity against tumors by the neutrophil myeloperoxidase system and hydrogen peroxide. J Immunol. 1984;133(6):3291–3297. [PubMed] [Google Scholar]
- 186.el-Hag A, Clark RA. Immunosuppression by activated human neutrophils. Dependence on the myeloperoxidase system. J Immunol. 1987;139(7):2406–2413. [PubMed] [Google Scholar]
- 187.el-Hag A, Lipsky PE, Bennett M, Clark RA. Immunomodulation by neutrophil myeloperoxidase and hydrogen peroxide: differential susceptibility of human lymphocyte functions. J Immunol. 1986;136(9):3420–3426. [PubMed] [Google Scholar]
- 188.Aydin E, Johansson J, Nazir FH, Hellstrand K, Martner A. Role of NOX2-Derived Reactive Oxygen Species in NK Cell-Mediated Control of Murine Melanoma Metastasis. Cancer Immunol Res. 2017;5(9):804–811. [DOI] [PubMed] [Google Scholar]
- 189.Holmdahl R, Sareila O, Pizzolla A, et al. Hydrogen Peroxide As an Immunological Transmitter Regulating Autoreactive T Cells. Antioxid Redox Signal. 2012. [DOI] [PubMed] [Google Scholar]
- 190.Kraaij MD, Savage ND, van der Kooij SW, et al. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc Natl Acad Sci U S A. 2010;107(41):17686–17691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Gelderman KA, Hultqvist M, Pizzolla A, et al. Macrophages suppress T cell responses and arthritis development in mice by producing reactive oxygen species. J Clin Invest. 2007;117(10):3020–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Horvath R, Rozkova D, Lastovicka J, et al. Expansion of T helper type 17 lymphocytes in patients with chronic granulomatous disease. Clin Exp Immunol. 2011;166(1):26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Brito C, Naviliat M, Tiscornia AC, et al. Peroxynitrite inhibits T lymphocyte activation and proliferation by promoting impairment of tyrosine phosphorylation and peroxynitrite-driven apoptotic death. J Immunol. 1999;162(6):3356–3366. [PubMed] [Google Scholar]
- 194.Bianchi M, Hakkim A, Brinkmann V, et al. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. 2009;114(13):2619–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Fuchs TA, Abed U, Goosmann C, et al. Novel cell death program leads to neutrophil extracellular traps. The Journal of cell biology. 2007;176(2):231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. [DOI] [PubMed] [Google Scholar]
- 197.Rohm M, Grimm MJ, D’Auria AC, Almyroudis NG, Segal BH, Urban CF. NADPH oxidase promotes neutrophil extracellular trap formation in pulmonary aspergillosis. Infection and immunity. 2014;82(5):1766–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Itagaki K, Kaczmarek E, Lee YT, et al. Mitochondrial DNA released by trauma induces neutrophil extracellular traps. PLoS ONE. 2015;10(3):e0120549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Arai Y, Nishinaka Y, Arai T, et al. Uric acid induces NADPH oxidase-independent neutrophil extracellular trap formation. Biochem Biophys Res Commun. 2014;443(2):556–561. [DOI] [PubMed] [Google Scholar]
- 200.Caudrillier A, Kessenbrock K, Gilliss BM, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. The Journal of clinical investigation. 2012;122(7):2661–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Thomas GM, Carbo C, Curtis BR, et al. Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood. 2012;119(26):6335–6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci U S A. 2015;112(9):2817–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Narasaraju T, Yang E, Samy RP, et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol. 2011;179(1):199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Rossaint J, Herter JM, Van Aken H, et al. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap mediated sterile inflammation. Blood. 2014. [DOI] [PubMed] [Google Scholar]
- 205.Nie M, Yang L, Bi X, et al. Neutrophil Extracellular Traps Induced by IL8 Promote Diffuse Large B-cell Lymphoma Progression via the TLR9 Signaling. Clin Cancer Res. 2019;25(6):1867–1879. [DOI] [PubMed] [Google Scholar]
- 206.Cools-Lartigue J, Spicer J, McDonald B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Albrengues J, Shields MA, Ng D, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018;361(6409). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lee W, Ko SY, Mohamed MS, Kenny HA, Lengyel E, Naora H. Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J Exp Med. 2019;216(1):176–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Tohme S, Yazdani HO, Al-Khafaji AB, et al. Neutrophil Extracellular Traps Promote the Development and Progression of Liver Metastases after Surgical Stress. Cancer Res. 2016;76(6):1367–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Demers M, Wong SL, Martinod K, et al. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology. 2016;5(5):e1134073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Semeraro F, Ammollo CT, Morrissey JH, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011;118(7):1952–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Fuchs TA, Brill A, Duerschmied D, et al. Extracellular DNA traps promote thrombosis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(36):15880–15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014;123(18):2768–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Demers M, Krause DS, Schatzberg D, et al. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(32):13076–13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Brill A, Fuchs TA, Savchenko AS, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. 2012;10(1):136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.von Bruhl ML, Stark K, Steinhart A, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. The Journal of experimental medicine. 2012;209(4):819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Stakos DA, Kambas K, Konstantinidis T, et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. European heart journal. 2015;36(22):1405–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Martinod K, Demers M, Fuchs TA, et al. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(21):8674–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13(4):463–469. [DOI] [PubMed] [Google Scholar]
- 220.Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. 2015;126(2):242–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Elshoury A, Schaefer JK, Lim MY, Skalla DP, Streiff MB. Update on Guidelines for the Prevention of Cancer-Associated Thrombosis. J Natl Compr Canc Netw. 2022:1–8. [DOI] [PubMed] [Google Scholar]
- 222.Thalin C, Demers M, Blomgren B, et al. NETosis promotes cancer-associated arterial microthrombosis presenting as ischemic stroke with troponin elevation. Thromb Res. 2016;139:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Mauracher LM, Posch F, Martinod K, et al. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J Thromb Haemost. 2018;16(3):508–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Berger-Achituv S, Brinkmann V, Abed UA, et al. A proposed role for neutrophil extracellular traps in cancer immunoediting. Front Immunol. 2013;4:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Boone BA, Orlichenko L, Schapiro NE, et al. The receptor for advanced glycation end products (RAGE) enhances autophagy and neutrophil extracellular traps in pancreatic cancer. Cancer Gene Ther. 2015;22(6):326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Liu S, Feng M, Guan W. Mitochondrial DNA sensing by STING signaling participates in inflammation, cancer and beyond. Int J Cancer. 2016. [DOI] [PubMed] [Google Scholar]
- 227.Xiao Y, Cong M, Li J, et al. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell. 2021;39(3):423–437 e427. [DOI] [PubMed] [Google Scholar]
- 228.Sorensen OE, Clemmensen SN, Dahl SL, et al. Papillon-Lefevre syndrome patient reveals species-dependent requirements for neutrophil defenses. J Clin Invest. 2014;124(10):4539–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Zhang Y, Chandra V, Riquelme Sanchez E, et al. Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J Exp Med. 2020;217(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Xu P, Tang K, Ma J, et al. Chemotherapeutic Tumor Microparticles Elicit a Neutrophil Response Targeting Malignant Pleural Effusions. Cancer Immunol Res. 2020;8(9):1193–1205. [DOI] [PubMed] [Google Scholar]
- 231.Gong L, Cumpian AM, Caetano MS, et al. Promoting effect of neutrophils on lung tumorigenesis is mediated by CXCR2 and neutrophil elastase. Mol Cancer. 2013;12(1):154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Wang Y, Wang K, Han GC, et al. Neutrophil infiltration favors colitis-associated tumorigenesis by activating the interleukin-1 (IL-1)/IL-6 axis. Mucosal Immunol. 2014;7(5):1106–1115. [DOI] [PubMed] [Google Scholar]
- 233.Katoh H, Wang D, Daikoku T, Sun H, Dey SK, Dubois RN. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell. 2013;24(5):631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Lakritz JR, Poutahidis T, Mirabal S, et al. Gut bacteria require neutrophils to promote mammary tumorigenesis. Oncotarget. 2015;6(11):9387–9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Zhao Y, Huang X, Ding TW, Gong Z. Enhanced angiogenesis, hypoxia and neutrophil recruitment during Myc-induced liver tumorigenesis in zebrafish. Sci Rep. 2016;6:31952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Cui C, Chakraborty K, Tang XA, et al. Neutrophil elastase selectively kills cancer cells and attenuates tumorigenesis. Cell. 2021;184(12):3163–3177 e3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Mittendorf EA, Alatrash G, Qiao N, et al. Breast cancer cell uptake of the inflammatory mediator neutrophil elastase triggers an anticancer adaptive immune response. Cancer Res. 2012;72(13):3153–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Sato R, Semba T, Saya H, Arima Y. Concise Review: Stem Cells and Epithelial-Mesenchymal Transition in Cancer: Biological Implications and Therapeutic Targets. Stem Cells. 2016;34(8):1997–2007. [DOI] [PubMed] [Google Scholar]
- 239.Kim BN, Ahn DH, Kang N, et al. TGF-beta induced EMT and stemness characteristics are associated with epigenetic regulation in lung cancer. Scientific reports. 2020;10(1):10597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Xia X, Zhang Z, Zhu C, et al. Neutrophil extracellular traps promote metastasis in gastric cancer patients with postoperative abdominal infectious complications. Nature communications. 2022;13(1):1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Ai L, Mu S, Sun C, et al. Myeloid-derived suppressor cells endow stem-like qualities to multiple myeloma cells by inducing piRNA-823 expression and DNMT3B activation. Mol Cancer. 2019;18(1):88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Peng D, Tanikawa T, Li W, et al. Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res. 2016;76(11):3156–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kuroda H, Mabuchi S, Yokoi E, et al. Prostaglandin E2 produced by myeloid-derived suppressive cells induces cancer stem cells in uterine cervical cancer. Oncotarget. 2018;9(91):36317–36330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Komura N, Mabuchi S, Shimura K, et al. The role of myeloid-derived suppressor cells in increasing cancer stem-like cells and promoting PD-L1 expression in epithelial ovarian cancer. Cancer Immunol Immunother. 2020;69(12):2477–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Zhou SL, Yin D, Hu ZQ, et al. A Positive Feedback Loop Between Cancer Stem-Like Cells and Tumor-Associated Neutrophils Controls Hepatocellular Carcinoma Progression. Hepatology. 2019;70(4):1214–1230. [DOI] [PubMed] [Google Scholar]
- 246.Song M, He J, Pan QZ, et al. Cancer-Associated Fibroblast-Mediated Cellular Crosstalk Supports Hepatocellular Carcinoma Progression. Hepatology. 2021;73(5):1717–1735. [DOI] [PubMed] [Google Scholar]
- 247.Wu QD, Wang JH, Condron C, Bouchier-Hayes D, Redmond HP. Human neutrophils facilitate tumor cell transendothelial migration. Am J Physiol Cell Physiol. 2001;280(4):C814–822. [DOI] [PubMed] [Google Scholar]
- 248.Strell C, Lang K, Niggemann B, Zaenker KS, Entschladen F. Neutrophil granulocytes promote the migratory activity of MDA-MB-468 human breast carcinoma cells via ICAM-1. Exp Cell Res. 2010;316(1):138–148. [DOI] [PubMed] [Google Scholar]
- 249.Slattery MJ, Dong C. Neutrophils influence melanoma adhesion and migration under flow conditions. Int J Cancer. 2003;106(5):713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Spiegel A, Brooks MW, Houshyar S, et al. Neutrophils Suppress Intraluminal NK Cell-Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells. Cancer discovery. 2016;6(6):630–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Spicer JD, McDonald B, Cools-Lartigue JJ, et al. Neutrophils promote liver metastasis via Mac-1-mediated interactions with circulating tumor cells. Cancer Res. 2012;72(16):3919–3927. [DOI] [PubMed] [Google Scholar]
- 252.Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature. 2015;528(7582):413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Szczerba BM, Castro-Giner F, Vetter M, et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature. 2019;566(7745):553–557. [DOI] [PubMed] [Google Scholar]
- 254.Yazdani HO, Roy E, Comerci AJ, et al. Neutrophil Extracellular Traps Drive Mitochondrial Homeostasis in Tumors to Augment Growth. Cancer Res. 2019;79(21):5626–5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Perego M, Tyurin VA, Tyurina YY, et al. Reactivation of dormant tumor cells by modified lipids derived from stress-activated neutrophils. Sci Transl Med. 2020;12(572). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.van Kempen LC, Coussens LM. MMP9 potentiates pulmonary metastasis formation. Cancer Cell. 2002;2(4):251–252. [DOI] [PubMed] [Google Scholar]
- 257.Kaplan RN, Psaila B, Lyden D. Bone marrow cells in the ‘pre-metastatic niche’: within bone and beyond. Cancer Metastasis Rev. 2006;25(4):521–529. [DOI] [PubMed] [Google Scholar]
- 258.Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006;8(12):1369–1375. [DOI] [PubMed] [Google Scholar]
- 259.Tyagi A, Sharma S, Wu K, et al. Nicotine promotes breast cancer metastasis by stimulating N2 neutrophils and generating pre-metastatic niche in lung. Nature communications. 2021;12(1):474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Jablonska J, Lang S, Sionov RV, Granot Z. The regulation of pre-metastatic niche formation by neutrophils. Oncotarget. 2017;8(67):112132–112144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Gkolfinopoulos S, Jones RL, Constantinidou A. The Emerging Role of Platelets in the Formation of the Micrometastatic Niche: Current Evidence and Future Perspectives. Frontiers in oncology. 2020;10:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Marusyk A, Janiszewska M, Polyak K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell. 2020;37(4):471–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Yee PP, Wei Y, Kim SY, et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nature communications. 2020;11(1):5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Mahiddine K, Blaisdell A, Ma S, Crequer-Grandhomme A, Lowell CA, Erlebacher A. Relief of tumor hypoxia unleashes the tumoricidal potential of neutrophils. J Clin Invest. 2020;130(1):389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Gallin JI. Human neutrophil heterogeneity exists, but is it meaningful? Blood. 1984;63(5):977–983. [PubMed] [Google Scholar]
- 266.Grieshaber-Bouyer R, Radtke FA, Cunin P, et al. The neutrotime transcriptional signature defines a single continuum of neutrophils across biological compartments. Nature communications. 2021;12(1):2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Nair P, Gaga M, Zervas E, et al. Safety and efficacy of a CXCR2 antagonist in patients with severe asthma and sputum neutrophils: a randomized, placebo-controlled clinical trial. Clin Exp Allergy. 2012;42(7):1097–1103. [DOI] [PubMed] [Google Scholar]
- 268.O’Byrne PM, Metev H, Puu M, et al. Efficacy and safety of a CXCR2 antagonist, AZD5069, in patients with uncontrolled persistent asthma: a randomised, double-blind, placebo-controlled trial. The Lancet Respiratory medicine. 2016;4(10):797–806. [DOI] [PubMed] [Google Scholar]
- 269.Ring NG, Herndler-Brandstetter D, Weiskopf K, et al. Anti-SIRPalpha antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc Natl Acad Sci U S A. 2017;114(49):E10578–E10585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Treffers LW, Ten Broeke T, Rosner T, et al. IgA-Mediated Killing of Tumor Cells by Neutrophils Is Enhanced by CD47-SIRPalpha Checkpoint Inhibition. Cancer Immunol Res. 2020;8(1):120–130. [DOI] [PubMed] [Google Scholar]