

Abstract

The Notch pathway is remarkably simple without the interventions of secondary messengers. It possesses a unique receptor–ligand interaction that imparts signaling upon cleavage of the receptor followed by the nuclear localization of its cleaved intracellular domain. It is found that the transcriptional regulator of the Notch pathway lies at the intersection of multiple signaling pathways that enhance the aggressiveness of cancer. The preclinical and clinical evidence supports the pro-oncogenic function of Notch signaling in various tumor subtypes. Owing to its oncogenic role, the Notch signaling pathway assists in enhanced tumorigenesis by facilitating angiogenesis, drug resistance, epithelial to mesenchymal transition, etc., which is also attributed to the poor outcome in patients. Therefore, it is extremely vital to discover a suitable inhibitor to downregulate the signal-transducing ability of Notch. The Notch inhibitory agents, such as receptor decoys, protease (ADAM and γ-secretase) inhibitors, and monoclonal/bispecific antibodies, are being investigated as candidate therapeutic agents. Studies conducted by our group exemplify the promising results in ablating tumorigenic aggressiveness by inhibiting the constituents of the Notch pathway. This review deals with the detailed mechanism of the Notch pathways and their implications in various malignancies. It also bestows us with the recent therapeutic advances concerning Notch signaling in the context of monotherapy and combination therapy.
Keywords: Notch ligands, Notch receptors, Notch pathway, cancer therapeutics, γ-secretase inhibitor, ADAM inhibitor, γ-secretase modulators
Decades of research reveal that every component of the signaling pathways is intertwined in an array of complex networks. Disruption in any of the components of a pathway as a consequence of oncogenic mutations, abnormal expression of signaling components, or inactivation of tumor suppressors results in the hyperactivation of the signaling pathway and elimination of the negative regulators of the pathway, respectively, therefore bestowing tumor cells with abnormal characteristics that enhance their survival in a complex environment. Additionally, aberrant mutations in the conserved developmental pathways are responsible for the modulation of proliferation, differentiation, and viability of the undifferentiated pluripotent stem cells and tumor initiating cells, which further leads to the enhanced aggressiveness of numerous malignancies. The most prevalent conserved developmental pathways in cancer are the Wnt, Hedgehog, and Notch signaling pathways that are generally responsible for the development and differentiation of embryonic and adult tissues.1−3 These pathways are known to function by converting the extracellular stimulus into a specific intracellular response by the underlying molecular circuits that govern diverse cellular processes.
The Notch signaling pathway is conserved throughout evolution, in both vertebrates and invertebrates. It was first studied in Drosophila neurogenesis, which occurs due to the phenomenon of lateral inhibition, i.e., an inhibitory cell–cell interaction.4 The Notch gene encodes a type I transmembrane protein, which responds to the regulatory and instructive signals upon interacting with its ligand, thereby facilitating cell specification and differentiation during embryogenesis. The Notch pathway signaling is pleiotropic; i.e., it not only affects cell specifications across a wide variety of species (both unicellular and multicellular) but also varies among various cell types and at subsequent stages of cell lineage progression.5 The current review deals with the biological roles of Notch signaling in human malignancies and the therapeutic efficacy that could be acquired upon blocking this pathway.
The Notch Pathway
Structural Organization of the Notch Receptors
The Drosophila Notch is a type 1 integral membrane protein (with a molecular weight of 300 kDa) that traverses the cellular membrane. It comprises an intracellular cytoplasmic domain and a ligand-binding domain present extracellularly, which is required for transducing signals upon activation of the Notch pathway.6 The Notch in Drosophila is homologous to that of the lin-12 gene in Caenorhabditis elegans. Early studies in C. elegans revealed the lin-12 and glp-1 genes encode the two LIN-12/Notch proteins, the ligands for which belong to the DSL protein family, an annotation defining the ligands Delta, Serrate (from Drosophila), and LAG-2 (C. elegans).7,8
In vertebrates, four Notch receptor genes are present (Notch1, Notch2, Notch3, and Notch4) that are synthesized individually from independent mRNA as single protein precursors (Figure 1A). All receptors possess an extracellular domain comprising several EGFR (epidermal growth factor receptor)-like repeats that assist in the binding of ligands. The different isoforms of the Notch receptors exhibit different numbers of EGFR repeats, such as Notch1 and Notch2 in vertebrates contain 36 tandem EGFR repeats, possessing striking similarity with the Drosophila Notch, while Notch3 comprises 34 and Notch4 comprises 29 tandemly arranged EGFR repeats.9 Moreover, the orderly arrangement of the EGFR is evolutionarily conserved and mutations in any one or more of these repeats produce different developmental phenotypes.10
Figure 1.

(A) Schematic representation of mammalian Notch receptor proteins and (B) the Notch ligands.
The downstream of the EGFR domain contains three LNR (LIN-12/Notch related region) repeats, which are composed of a cysteine-rich region and a hydrophobic region, altogether comprising the Notch extracellular domain (NECD). This motif negatively regulates receptor activation, thereby preventing ligand-independent signaling. The LNR region is followed by the transmembrane domain, consisting of a pair of conserved cysteine residues between themselves, known as HD (heterodimerization domain) that is thought to facilitate receptor dimerization.11
The cytoplasmic domain of the receptors, commonly known as the Notch intracellular domain (NICD), comprises multiple conserved regions: the RBPJ associated molecule (RAM) motif that lies between the TMD and ANK repeats, which interacts with the DNA binding protein CSL, a mammalian homologue of Suppressor of Hairless [Su(H)] in Drosophila.12 It also comprises six copies of the 33 residue CDC10-ankyrin (ANK) like repeats, which are involved in the protein–protein interactions of cytoskeleton and transcription factors.13 Moreover, the RAM domain has a high-affinity binding site for CSL, while the ankyrins form weak contacts with CSL and are vital in recruiting MAML.14 Additionally, the ANK repeats are spanned by two nuclear localization signaling (NLS) domains and a C-terminal PEST domain [containing the amino acids proline (P), glutamic acid (E), serine (S), and threonine (T)], which facilitates rapid proteolytic degradation of the Notch receptors. Additionally, a transcription activation domain (TAD) is also present in the Notch1 and Notch2 receptors, which enhances the recruitment and stabilization of the coregulators and/or coactivators at the site of the target promoters13,14 Moreover, the varying numbers of amino acids present within the ANK and PEST domains among different Notch proteins are responsible for the inconsistent size of the cytoplasmic domains.15
The Notch Ligands
The canonical Notch signaling is assisted by the DSL (Delta, Serrate, Lag2) ligands. Besides the receptors, Notch ligands are also type 1 transmembrane proteins possessing multiple signature EGF repeats in their extracellular domains. The domain of DSL shares a conserved flanking N-terminal region and two EGF moieties that are essential for receptor ligation and activation of Notch signaling.16 Interestingly, the cells expressing the Notch ligands (known as the signal-sending cells) possess an E3 ubiquitin ligase (such as Neur and MIB) that assists in the ubiquitination of the ligand’s intracellular domain. This phenomenon further promotes epsin-mediated endocytosis, which ultimately results in ligand activation.17
In vertebrates, the Notch ligands, as illustrated in Figure 1B, share structural homology with the Delta and Serrate ligands of Drosophila. They are categorized as either Serrate (e.g., Jagged1 and Jagged2) ligands or Delta-like (e.g., DLL1, DLL3, and DLL4) ligands. Interestingly, the Serrate ligands have more numbers of EGF repeats (16 in Jagged1 and Jagged2, 8 in DLL1 and DLL4, and 6 in DLL3), a von Willebrand factor type-C (vWF-c) region, and a supplementary cysteine-rich (CR) domain, in comparison to the Delta-like ligands. The cytoplasmic domains of Serrate ligands do not share significant amino acid identities. However, DLL1 and DLL4 have similar structures with approximately 60% similarity in their protein sequences. Additionally, the intracellular regions of Jagged1, DLL1 and DLL4, have a C-terminal domain that contains a PDZ (PSD-95/Dlg/ZO-1) [post synaptic density protein (PSD95) binding motif, Dlg-1 (Drosophila disk large tumor suppressor), and zonula occludens-1 protein (ZO-1)] binding motif, which facilitates the intracellular protein–protein associations. The PDZ motif also facilitates the anchoring of membrane proteins to the cytoskeleton.18 Moreover, the DLL1 Jagged domains also comprise the DOS (Delta and OSM-11) motifs, which resemble the EGF-like repeats that enhance the affinity of ligands to receptors.19
The Notch Receptor Processing
The activity of the receptor is modulated by the O-fucosyltransferases, furin-like convertases, and Fringe glycosyltransferases. Moreover, other modes of modulations include (i) modification of receptors or ligands, (ii) coexpression of receptors with ligands, (iii) mode of ligand presentation, and (iv) cell surface area in contact.20,21
The comparative magnitude of the receptor–ligand interactions is regulated by post-transcriptional changes of the Notch receptors. Initially, the extracellular EGF repeats are modified upon adjoining O-fucose facilitated by the enzyme O-fucosyltransferase1 (O-fut1) in the endoplasmic reticulum. This assists in the receptor glycosylation by Fringe glycosyltransferases (e.g., Manic, Radical, and Lunatic Fringe in mammals).22 The augmentation of N-acetylglucosamine (GlcNAc) sugar to the O-fucose moiety mediated by Fringe proteins further regulates the responsiveness of the receptors to its ligands. However, Fringe mediated post-transcriptional changes are quantitative; i.e., the expression level of the Notch downstream genes can be modulated. The expression of Fringe accords with either of the Delta or Serrate ligand domains (such as Fringe+/Jagged+ and Fringe+/DLL+) that possess a varying effect on tissue domain boundaries and tissue organization.23 Besides Fringe, glycosyltransferase Rumi (Poglut1) and two enzymes of the human glycosyltransferase 8 families also glycosylate the Notch receptors.24
Additionally, proteins such as Numb, α-adaptin, and E3 ubiquitin ligase (such as Deltex and Nedd4) are also responsible for the regulation of steady-state levels of Notch receptors on the cellular surface.25
Activation of the Notch Pathway
The activation of the Notch receptors involves a series of proteolytic events (Figure 2). Initially, a furin-like convertase mediates the S1 cleavage of the receptors in the trans-Golgi network, thereby generating a heterodimeric molecule, which is essential for the functionalization of the receptors on the cellular surface.26 The LNR domain regulates the relationship between the polypeptide products (formed after cleavage by the furin convertase), thereby downregulating receptor activity.
Figure 2.

Schematic of the Notch signaling pathway. (1) The Notch receptor processing takes place in the ER, by the enzyme O-fut (O-fucosyltransferase) that adds fucose to the Notch precursor. (2) In the trans-Golgi network, furin-like convertase cleaves full-length Notch (S1 cleavage) generating a heterodimer molecule, which is then glycosylated by Fringe proteins (3). (4) The heterodimer Notch receptors are then trafficked to the cell membrane. (5) Activation of Notch receptor takes place when a Delta/Serrate family of ligands binds to the extracellular receptor of Notch. (6) Upon ligand binding, S2 cleavage occurs by the ADAM protease (also known as TACE). (7) The ligand bound to the Notch extracellular domain (NECD) gets internalized by the signal sending cell and (8) gets degraded inside the lysosomes. (9) S3 cleavage occurs by the γ-secretase enzyme, which results in the release of the Notch intracellular domain (NICD) that gets internalized into the nucleus (10) and (11) binds to the DNA binding domain, CSL, thereby replacing the corepressors with the coactivators and facilitating the transcription of Notch targeted genes. (12) In the absence of NICD, the DNA binding domain, CSL, is bound to the corepressor complex, which inhibits the transcription of Notch targeted genes.
However, the initiation of Notch signaling takes place by the secondary proteolytic cleavage (S2 cleavage), which is facilitated upon ligand binding on the receptors in trans fashion (i.e., it binds at the surface of the adjacent cells), triggering a conformational change in the receptor.27 The structural investigation of the receptors reveals that, upon ligand binding, the receptors are extended to reveal the NRR (negative regulatory region), exposing the cleavage site.28 This enables cleavage by the ADAM protease, also known as TACE (TNF-α converting enzyme), within the juxtamembrane region.27 Altogether, these phenomena result in the elimination of the Notch extracellular domain (NECD), which is then recycled via trans-endocytosis into the ligand-expressing cells.
The residual membrane-bounded section of the receptor, termed the notch extracellular truncation (NEXT) domain, a substrate for the intramembrane proteolysis, undergoes “S3 cleavage” by the presenilin enzyme associated with the γ-secretase complex. This ultimately leads to the generation of the Notch intracellular domain (NICD). Of the entire Notch protein, the NICD solely has an intrinsic signal-transducing ability, whereas the extracellular domain regulates the activity of the NICD. It is suggested that the γ-secretase dependent cleavage is modulated with respect to the cleavage efficacy and location of the cleavage site present in the receptor, which is due to the different isoforms of presenilin (PSEN1 and PSEN2). This results in the Notch receptor endocytosis requiring monoubiquitination at the lysine 1749, followed by deubiquitination by elF 3f, which enables the cleavage by γ-secretase. Moreover, Deltex (an E3 ubiquitin ligase), serves as the linking protein between Notch and Elf3 in early endosomes. Deltex serves as both a positive and negative regulator of Notch and helps in the modulation and internalization of the Notch receptors.
Furthermore, the NICD thus generated traverses to the nucleus where it binds to the DNA binding protein, CSL {also known as longevity-assurance gene-1 (LAG-1) in C. elegans; Suppressor of Hairless [Su(H)] in flies, and C promoter-binding factor (CBF1) or recombination signal binding protein for immunoglobulin κ J region (RBPJ-κ) or κ-binding factor 2 (KBF2) in mammals}. The interaction of NICD and CSL results in the recruitment of the adaptor protein, Mastermind-like (MAML), which further recruits EP300 and components of the downstream transcription machinery. This further aids in the transcription of a number of helix–loop–helix transcription factors, such as the Hairy and Enhancer of Split1 (HES1), HES1-related (HESR1), HERP, and HEY. Additionally, the downstream Notch target genes also include the p21/WAF1 promoter, c-Myc, cyclin A, cyclin D1, a ubiquitin ligase SKP2 (that promotes the degradation of the CDK inhibitor, p27/KIP1), and members of the NF-κB pathway, along with the insulin-like growth factor and its receptor (IGF-1 and IGF-1R). Altogether, these transcription factors regulate numerous cellular processes, including proliferation, self-renewal, differentiation, and apoptosis, thus defining the canonical pathway of Notch signaling.29
However, in the absence of Notch signaling, CSL interacts with numerous proteins to form a corepressor complex (such as CIR, corepressor interacting with RBPJ; CtIP/CtBP, carboxy-terminal-binding protein, HDAC-I, histone deacetylase I; SHARP, SMRT/HDAC-associated repressor protein; and SMRT, silencing mediator of retinoid and thyroid hormone receptors), which is transcriptionally inactive. Upon the recruitment of NICD, the corepressor complex is dissociated and several transcriptional activators (including HATs, histone acetyltransferases; SKIP, skeletal muscle and kidney enriched inositol phosphatase; MAML, Mastermind-like proteins, PCAF, p300/CPB associated factor; and GCN5, general control nonderepressible 5) are recruited to the CSL protein, thereby activating the transcription of the downstream signaling genes.29 An overview of the Notch signaling pathway and its possible therapeutic targets are provided in Figure 3.
Figure 3.

(A) Detailed overview of the Notch signaling pathway and its possible therapeutic targets. (B) The function domains of the Notch receptor and their corresponding functions. (Adapted from ref (75). CC BY 4.0.)
The NICD
The S3 cleavage of the receptors mediated by the γ-secretase enzyme is heterogeneous regarding the cleavage site. The NICD fragments comprise either an N-terminal serine/leucine (Ser/Leu) or N-terminal valine (Val) fragment, whereby the former exhibits a shorter half-life as compared to the Val-NICD fragments, altogether affecting the duration of Notch signaling.30 The NICD is subjected to a wide range of post-translational modifications (Figure 4), such as acetylation hydroxylation, phosphorylation, and ubiquitylation, which are important for its proper activity, half-life, and stability, reviewed by Belle et al.31
Figure 4.
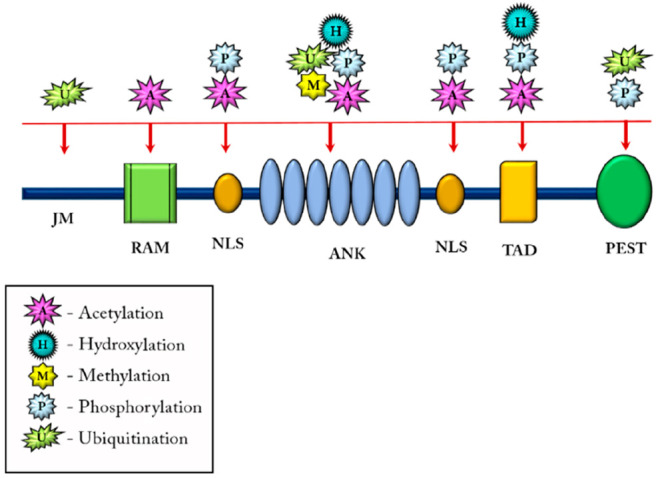
Post-translation modifications in the NICD. The NICD followed by its cleavage undergoes post-translational modifications like acetylation (responsible for finely tuning the half-life of Notch), hydroxylation (contributes to signaling diversity of NICD), methylation (increases stability and half-life of NICD), phosphorylation (regulates activity and turnover of NICD), and ubiquitination (activates Notch signaling, regulates the half-life of Notch).
Generally, the NICD is short-lived with a half-life of approximately 4 h. One molecule of NICD generates one signaling unit. Moreover, the efficacy of ligand–receptor interaction is directly proportional to the quantity of NICD present in the nucleus. Upon activation of transcriptional regulators, kinases (such as cyclin dependent kinase 8, CDK8) phosphorylate the NICD within its PEST domain, which is then targeted for proteasome-mediated degradation by Sel10 (also called Fbw7), an E3 ubiquitin ligase, thus limiting the half-life of the NICD that resets the cell for the next pulse of signaling.
Among the different components of the Notch signaling pathway, the cleaved intracellular domain has the inherent ability to transduce signals in both healthy and tumor cells. In tumor cells, the pleiotropic effects of NICD upon its interactions with the signaling components of the other pathways result in the enhanced tumorigenesis and dissemination of the cancer cells, as depicted in Figure 5.
Figure 5.

NICD (Notch intracellular domain) regulates the expression of various downstream genes that induces proliferation, aids in the EMT (epithelial to mesenchymal transition), and inhibits apoptosis, thereby promoting tumorigenesis.
The Noncanonical Notch Ligands
The noncanonical Notch ligands are structurally diverse and include the membrane-tethered ligands such as Delta-like noncanonical Notch ligands 1 and 2 (Dlk-1 and Dlk-2) that inhibit adipogenesis and function as cis inhibition of Notch signaling. Additionally, Delta/Notch-like EGF-related receptor (DNER), a Delta-like essential membrane protein, comprises tandem EGF repeats on its extracellular domain and lacks a DSL domain. DNER binds to Notch in a trans fashion and activates CSL transcription. Moreover, a DSL ligand-like protein known as Jagged and Delta protein (Jedi), is devoid of both transactivating activities and cis-inhibitory activity. F3/contactin1 and NB3/contactin6 encode GPI-linked neural cell adhesion molecules, which activate Notch to induce oligodendrocyte differentiation. The secreted non-DSL protein also includes Sca (involved in Notch-dependent patterning of the eye and sensory bristles in Drosophila), CCN3 (required for proper development of the vertebrate heart and skeleton, and its expression is associated with both positive and negative regulatory roles in cancer), MAGP1 and MAGP2 (present in vertebrates, assist in γ-secretase dependent NICD generation and result in autocrine Notch activation; MAGP-2 is the sole noncanonical ligand that mediates nonenzymatic dissociation of Notch).32,33
The lack of ligands invokes the Notch to participate in variable cellular processes. For example, Notch regulates the stability of β-catenin, a major component of the Wnt signaling pathway.34 Unbound Notch receptors are either recycled or targeted for lysosomal degradation, under normal physiological conditions. The pathway can also get activated without ligands in some pathological conditions, e.g., mutations in the NRR domain of the Notch receptors, overexpression of Notch on exposure to calcium chelators, etc. Furthermore, Notch signaling is constitutively activated due to the stabilization of the NICD by hypoxia-inducible factor-1 (HIF-1) in hypoxic conditions. Additionally, ion chelators such as EDTA and EGTA also result in the dissociation of Notch ligands, thereby mimicking the activation of Notch similar to the canonical pathway.35
The Noncanonical Notch Pathway
The noncanonical Notch pathway, also known as RBPJ/CSL-independent Notch signaling, is activated either upon binding of noncanonical ligands to the Notch receptors or in the absence of ligands.36 The Notch activation might be either γ-secretase dependent or independent, whereby the latter exerts its function in a membrane-bound form. The noncanonical pathway is also independent of CSL and instead is activated by interacting with PI3K/mTOR/Wnt/AKT/NF-κB/HIFα pathways, which might act with the NICD or other regulatory genes, either in the cytoplasm or in the nucleus to exert its function in various biological processes including cancer. Moreover, Notch can also be activated through R-Ras to promote cell adhesion. Alternatively, it may interact with IKKα in the NF-κB pathway or LEF1 in the Wnt pathway; Notch can also exert its function through its intracellular domain (NICD), which further activates multiple pathways.37
Implications of Notch in Cancer
Notch as an Oncogene
Notch signaling has a substantial contribution toward liver development, as well as in liver tumorigenesis. Notch facilitates the progression of hepatocellular carcinoma (HCC), an aggressive histological subtype among the primary liver cancers, by regulating the tumor microenvironment, along with proliferation, angiogenesis, invasion, and metastasis. Overexpression of Notch1, Notch3, and Notch4 is correlated with poor prognosis and is detected in more than 70% of HCC tissues.38
There are activating mutations of Notch1 receptors in T-cell acute lymphoblastic leukemia (T-ALL) and aggressive neoplasm of immature T-cells, with suppressed p53 functions.39 A high level of Jagged1 expression is observed in AML (acute myeloid leukemia).40 Inactivating mutations in Notch1 also promote squamous cell carcinoma.41 Constitutively active Notch1 promotes melanoma cell growth, which is mediated by β-catenin. In some cases of melanoma, Notch1 also facilitates the activation of the MAPK and PI3K–AKT pathways, thereby inducing the proliferation and dissemination of melanoma cells.42 In non-small cell lung carcinoma (NSCLC), Notch1 acts as an oncogene by providing critical survival signals under hypoxia. Also, there exists a negative correlation between the Notch and NUMB protease; for example, concomitant downregulation of NUMB is found in about 35% of NSCLC.29 In some cases, Notch3 is highly overexpressed, resulting in more aggressive forms of NSCLC and thymic cancer. Notch possesses exclusive oncogenic characteristics in the hematopoietic system, thereby promoting hematological malignancies.43
Moreover, a truncated and activated form of Notch4 has been found in many breast cancers (BCs) with hyperproliferation and enhanced invasiveness.44 Notch3 results in proliferation of Erb2-negative BC cell lines. In most of the BC cells, NUMB (a negative regulator of Notch) is lost due to ubiquitination and proteasomal degradation. Hyperactivation of Jagged1 and/or Notch1 along with Ras/MAPK pathways correlates with poor prognosis in patients with breast cancer.45 Constitutively active Notch 1 cross talks with the Erk pathway and mediates the transformation of immortalized BC cells. Notch also interacts with the HER2 (human epidermal growth factor receptor) signaling pathway resulting in more aggressive malignancies.
Similarly, the DLL4 ligand promotes the pro-inflammatory activation of macrophages in vitro.46 Notch signaling increases the level of vascular endothelial growth factor receptors (VEGFR1, VEGFR3) which in turn increases the DLL4, thereby promoting angiogenesis. Hypoxia-inducible factor (HIF) and Jagged1 potentiate the overexpression of Notch, leading to the epithelial to mesenchymal transition.47,48 Jagged2 and DLL4 dependent Notch signaling contributes to tumor initiation in pancreatic cancer.49 Altered Notch signaling, with Jagged1 upregulation and overexpression of Notch target transcription factor (HES1 and HEY2), induces pathogenesis of osteosarcoma.50
Notch has established cross talk with numerous signaling pathways that aid in the aggressiveness of the tumors.51 It confers angiogenesis, drug resistance, and invasive characteristics to the growing tumors and also assists in metastasis by facilitating the EMT, along with the generation of CSCs,52,53 therefore contributing to the poor prognosis in patients. Table 1 summarizes the role of the Notch pathway genes in various malignancies.
Table 1. Role of the Notch Genes in Cancer.
| Notch related genes | implications in cancer |
|---|---|
| Delta-like ligands | DLL1 promotes cell proliferation and angiogenesis in cancer. |
| DLL3 promotes migration and invasion by upregulating Snail. | |
| DLL4 promotes growth, proliferation, invasion, and angiogenesis. | |
| Serrate ligands | Jagged1 overexpression is implicated with the pathogenesis of various cancers and correlates with a poor clinical prognosis. |
| Jagged2 is involved in the regulation of migration and invasion of cancer cells. | |
| Notch receptors | Transmembrane receptors are involved in cell proliferation, maintenance, invasion, and chemoresistance. The different receptors are involved in different cancer subtypes. For example, Notch1 is implicated in the pathogenesis of numerous cancers such as breast cancer, lung cancer, and lymphomas, while constitutively active Notch2 gives rise to hepatic tumors. |
| ADAM protease | ADAM protease facilitates S2-mediated cleavage of the Notch receptors and other type I transmembrane proteases such as TNF-α and EGFR. Overexpression is associated with numerous malignancies. |
| γ-secretase enzyme | γ-Secretase enzyme facilitates S3-mediated cleavage of the Notch receptors, thereby generating NICD. |
| NICD domain | NICD domain activates the Notch pathway by dissociating the corepressors with the coactivators. It also interacts with numerous signaling pathways to facilitate the aggressiveness of a malignancy. |
| HES | Besides facilitating cellular proliferation and differentiation, HES contributes to multidrug resistance, along with metastasis. It also maintains the quiescent cells or cancer stem cells in a nondividing state. |
| HEY | HEY proteins have an effect on cancer proliferation and survival in some malignancies, while in some cancers it has a tumor suppressive role. |
| c-Myc | It triggers the activation of genes involved in protein biosynthesis, cancer metabolism, transcription factors, and the cell cycle. It also inhibits the expression of some tumor suppressor genes, thereby serving as a master regulator of cellular metabolism. |
| NF-κB | NF-κB mediates tumor cell proliferation, survival and angiogenesis |
The Notch receptors and its associated ligands, along with the downstream transcription factors, have potential oncogenic roles in a wide variety of malignancies.54 Thus, therapeutic interventions of this pathway hold promising results that might result in the inhibition of the critical hallmark of cancer cells, besides ablating cancer.
Notch as a Tumor Suppressor Gene
Besides having oncogenic roles in various malignancies, Notch ligands and/or receptors possess tumor suppressive properties in some cancers such as prostate,55 lung,56 brain,57 cervical,58 liver,59,60 skin cancers,61 etc.
It has been observed that different Notch ligands and receptors have different roles in a particular tumor. In some rare cases of hematological malignancies, Notch signaling has been found to suppress proliferation and induce apoptosis in B-cell lymphoma,62 Hodgkin lymphoma,63 and multiple myeloma, following upregulation of p21WAF/Cip expression.64 Contradictorily, Notch1 is highly expressed in some B-cell and T-cell derived tumors of Hodgkin lymphoma and anaplastic large-cell lymphoma.65 Aberrant expression of Notch1 is also associated with abnormal proliferation and survival of various lymphomas,66 altogether depicting the dual role of the Notch pathway in a particular tumor subtype; the underlying reason for this is still undetermined.
Furthermore, aberrant Notch1 induces the expression of p21WAF/Cip, thereby leading to growth arrest and differentiation of keratinocytes.67 Jagged1, DLL1, and DLL4 ligand mediated Notch activation is also tumor suppressive in nature; it inhibits the self-renewal capacity and growth of AML cells.68 Expression of intracellular Notch receptors (NICD1–NICD4), and Jagged ligands induced the transcription of HES1, which mediated inhibition of growth and induced apoptosis in B-cells.69 Notch is also observed to inactivate the Rb pathway, resulting in the inhibition of hepatocellular carcinoma.
Causal Mutations of Notch in Cancer
The role of the Notch pathway in human development and disease was recognized only when mutations in the Notch1 gene were associated with a form of T-ALL. Mutations in the Notch signaling pathway genes generally cause defective developmental phenotypes, which affect the heart, kidney, liver, skeleton, eye, and vasculature. The Notch-associated disorders are not restricted to cancer; rather they also include the autosomal dominant Alagille syndrome, autosomal recessive spondylocostal dysostosis, Hajdu–Cheney syndrome, cardiac diseases (such as CADASIL, an acronym for cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy), etc., which have been extensively reviewed by Penton et al.70
Analysis of a cohort of malignant cell lines divulges that genetic alterations (such as mutations, deletions, amplifications, and gain-of-function) in any Notch receptor are mutually exclusive to changes in other receptors. The case for the Notch ligands such as Jagged1 and Jagged2 is similar. Gain-of-function mutations are characteristics of hematopoietic cancers, whereby only one particular cell type is affected, such as T-ALL and B-CLL. However, in solid tumors, alteration in the signaling is bidirectional; i.e., the oncogenic or tumor suppressor function of Notch is context dependent, such as stage, cancer entity tissue of origin, tumor microenvironment, or the genetic background. For example, mutations in all four Notch receptors preferentially act either as a driver mutation that promotes tumor survival and progression or to also promote the progression and dissemination of malignancy at later stages. Similarly, genetically acquired gain-of-function mutations of Notch activity facilitate initiation and progression of gliomas,71 osteosarcomas,72 or SCLC (small cell lung cancer). Additionally, gain-of-function mutations in Notch also aid in EMT progression and invasive phenotypes of NSCLCs.73 Furthermore, they also promote the drug-resistant phenotype in lung cancers.74
Additionally, the various driver mutations that lead to malignant phenotypes are not restricted to a particular tumor subtype; rather it is implicated in the pathogenesis of various malignancies. For example, in breast cancer, a meta-analysis of approximately 4000 cases showed that gain-of-function mutations of the Notch pathway genes are associated with increased disease occurrence. Altered Notch1 signaling is related to ER+/PR+/HER2± breast cancers, while it is more prevalent in HER2– than in HER2+ tumors. The Jagged1/Notch signaling axis is either frequently deregulated or amplified in BCs. Activating Notch1 mutations or rearrangements in EGF and NRR regions have been observed in TNBC basal-like phenotypes. Moreover, nonsense mutations in the PEST domains of Notch1, Notch2, and Notch3 receptors have been observed in TNBC, which results in an elevated half-life of the NICD and leads to overexpression of the Notch downstream genes.75
Additionally, gain-of-function mutations in Notch1 and Notch2 implicate the pathogenesis of T-ALL and B-cell lymphomas, respectively. Also, in both cases, the mutations lead to enhanced activity and prolonged half-life of the NICDs. Moreover, activated Notch1 deregulates the Notch signaling in the majority of human T-ALL, whereas mutations leading to partial or complete deletion of the PEST domain or a single amino acid substitution in the C-terminus of the Notch2 proteins lead to the pathogenesis of a subset of B-cell lymphomas.76 Furthermore, in leukemic cells, FBW7 mutations activate the Notch pathway and provide resistance to γ-secretase inhibitors.77 The aberrant Notch pathway is involved in the pathogenesis of liver fibrosis. However, sustained Notch signaling in chronic liver diseases drives tumor formation without acquiring specific genome mutations. Additionally, it has also been observed that mutations in Notch2 facilitate the development of liver metastasis.78
Furthermore, the loss-of-function mutations in Notch are equivalently predominant in epithelial cancer types or carcinomas, such as squamous skin carcinoma and basal-cell carcinoma,79 along with pancreatic cancer,80 liver cancer,81 etc. Moreover, loss-of-function mutations in Notch also reduce the expression of regulatory genes such as FBXW7, which itself acts as a potent tumor suppressor, thereby resulting in cancer progression and dissemination.82 Some of the common mutations in the Notch receptors are depicted in Figure 6, as obtained from the cBioPortal database (https://www.cbioportal.org/).
Figure 6.

Mutational profile among the gene sequence of the Notch receptors. From the cBioPortal database (https://www.cbioportal.org/).
Targeting the Notch in Cancer
There are numerous reasons to target the Notch pathway due to its association with the aggressiveness of the malignancies. The targeting efficacy can be brought about by keeping in mind the three most important facts about Notch signaling. First, the half-life of the Notch intracellular domain (NICD) is very short (might be longer in transformed cells); thus, intermittent inhibition might be successful. Second, the downstream effects of Notch activation are dose dependent, i.e., “signal intensity” can be modulated very precisely by cellular regulatory mechanisms. Complete inhibition of the pathway might be not necessary since the signaling cascade triggered by Notch ligand interaction is devoid of an enzymatic amplification step. Third, the effects of Notch signaling are context dependent; i.e., it can act either as a tumor suppressor gene or as an oncogene, depending on the cancer type.83
Among the various underlying molecular components that aid in the initiation and functionalization of the Notch pathway, the potent molecular components having a supreme effect in the signaling pathway of Notch are described below.
The γ-Secretase Enzyme
γ-Secretase is a multisubunit intramembrane cleaving protease (i-CLiPs) having a molecular weight of approximately 230 kDa.84 It is composed of a tetrameric complex of proteins: nicastrin (NCT), anterior pharynx defective 1 (APH-1), presenilin enhancer 2 (PEN2), and presenilin (PSEN) and several associated proteins (such as TMP21, CD147, and Rer1p). In the human genome, there are two presenilin genes (PSEN1 and PSEN2) and two isoforms of the APH-1 gene (APH-1A and APH-1B), produced as a result of alternate splicing. Thus, the γ-secretase complex possesses six different conformations, each exhibiting distinct activities.85
Nicastrin is a type 1 integral membrane glycoprotein that facilitates complex maturation and stabilization.86 PEN2 belongs to the zinc metalloprotease site2 family, which is required for the endoproteolytic processing of the inactive presenilin into the catalytically active heterodimer, while APH-1 might be the substrate receptor for the protease complex. Additionally, APH-1A regulates Notch during embryogenesis, while APH-1B produces longer amyloid-β (Aβ) fragments.87 Further, presenilin is a multipass transmembrane aspartyl protease, having nine TMDs, and acts as the catalytic core of the γ-secretase complex.88 The active catalytic site lies between their sixth and seventh TMDs, with two aspartates (D257 and D385) of presenilin conferring the aspartate protease activity to the γ-secretase complex. It catalyzes the cleavage of various transmembrane proteins within the lipid bilayer, in a process termed “regulated intramembrane proteolysis”. Altogether, presenilin helps in regulating these domains to transduce signals to the nucleus.89
γ-Secretase is a highly regulated allosteric enzyme that possesses two conformations at equilibrium, which is distorted whenever a GSM (γ-secretase modulator) or a GSI (γ-secretase inhibitor) binds to it, thereby regulating its protease activity. Its principal role in the Notch pathway is the cleavage of the Notch receptor at its transmembrane domain near the cytoplasm and generating the cleaved intracellular domain (NICD), which in turn activates the Notch signaling and assists in the pathogenesis of cancer. Therefore, γ-secretase is validated as a significant therapeutic target in various cancers with elevated Notch signaling.
ADAM Protease
The ADAM acronym, “a disintegrin and metalloprotease”, is a transmembrane protein that mediates cell-surface proteolysis and modulates cell–cell and cell–matrix interactions. To date, 23 ADAMs have been identified in the human genome, out of which ADAM10 and ADAM17 facilitate the proteolytic processing of the Notch receptors via S2-mediated cleavage, which is potentiated upon ligand binding to the Notch receptor. The ADAMs contribute to the S2 cleavage in a tissue-specific manner. For example, ADAM10 is required in ligand-activated Notch signaling, while ADAM17 is required in ligand-independent Notch signaling.90,91
Research evidence suggests that ADAM cleaves the DSL ligands at the transmembrane region, which releases the extracellular part of the ligands as soluble proteins. These soluble proteins (DLL1, DLL4, and Jagged1) block the Notch activation, which in turn results in the inhibition of the target gene’s expression.92 The incompetence of the soluble ligands to expose the ADAM cleavage site is the reason behind their incapability to activate the Notch pathway.28 The ADAM protease plays an important role in eliciting the Notch signaling pathway, which in turn can be therapeutically regulated upon its inactivation.
NUMB
NUMB (found in Drosophila and vertebrates) and NUMB-like (in vertebrates) are endocytic adaptor proteins and act as suppressors of Notch.93 During asymmetric cell division in embryogenesis, the activity of Notch is antagonized by the cell fate determinant, NUMB, via endocytosis and a proteasomal dependent mechanism.94 NUMB interacts with E3 ubiquitin ligase Itch/AIP4 to promote intracellular trafficking and subsequent degradation of the Notch1 receptor.95 It also interacts with the AP-2 adaptor complex and the endocytic proteins α-adaptin and Eps15, in clathrin-coated pits and endosomes, to disrupt the endocytosis of Notch ligands and receptors. NUMB inhibits the Notch pathway by binding to the NICD and preventing its nuclear localization, which in turn impedes the NICD-dependent transcription initiation of the Notch downstream genes.
There exists a negative correlation between these two proteins, whereby cells expressing higher NUMB have low Notch expression and cells expressing high Notch have low NUMB expression. NUMB-mediated signaling control is lost in most cancers.96 Loss of NUMB by proteasomal degradation and ubiquitination is observed in approximately 50% of mammary carcinomas.
NUMB-mediated inhibition of Notch is used as a therapy against prostate cancer. According to a study conducted by Wang et al., NUMB binds to Par3 and E-cadherin, the interaction of which is regulated by tyrosine phosphorylation, which is induced by HGF/Src-mediated knockdown of NUMB. This in turn leads to apical translocation of E-cadherin, mislocation of Par3, and F-actin polymerization, which induced migration and proliferation.97 NUMB acts as a brake for a complete EMT by inhibiting Notch signaling.98,99 The reduced NUMB expression is often associated with the EMT and metastasis in TNBC.100 Therefore, NUMB-mediated Notch inhibition might possess a potential therapeutic approach.
Therapeutic Realms Concerning the Notch Pathway
There are numerous ways in which the Notch pathway can be regulated and/or inhibited. The crux of the therapeutic realms is illustrated in Figure 7. However, some of the mechanisms by which this pathway can efficiently be targeted are described below.
Figure 7.

Illustration depicting the possible strategies for Notch inhibition.
Modification of Receptor–Ligand Interaction
Blocking the ubiquitination and trans-endocytosis of the ligands and inhibiting the ability of Fringe and O-fut to glycosylate the Notch receptor would hinder the ligand–receptor interaction from initiating signal transduction. Additionally, the soluble Notch peptides (decoys) mimicking the ligand disrupt the receptor–ligand interaction by competitive inhibition. Research suggests that decoys of DSL ligands such as Delta101 and Jagged102 act as antagonists of Notch and fail to induce and/or activate the Notch signaling pathway.
Targeting Notch by Oligonucleotide Methods
Antisense oligonucleotides (ASOs) are short stretches of synthetically and/or chemically synthesized DNA or RNA that target gene of interest by penetrating the cell membranes. They possess high sequence specificity and, therefore, show fewer off-target effects as compared to other therapeutic approaches, as reviewed by Lundin et al.103
Notch1 phosphorothioate antisense oligonucleotides reduce the expression of HES1 in the H-82 cell line.104 An antisense Jagged oligomer, synthesized by Zimrin et al., demonstrated FGF2-induced collagen in invasion and tube formation by endothelial cells.105 The expression of Notch1 antisense oligonucleotide (NAS) in Notch antisense transgenic mice exhibited lower levels of Notch and showed several defects in learning and memory.106
Using Monoclonal Antibodies (mAbs), Bispecific Antibody (bs-Ab), Antibody–Drug Conjugate (ADC), and CAR-T Therapy
Antibodies that specifically inhibit or reduce the proteolytic cleavage of Notch are divided into two classes: (i) ligand competitive antibodies, which bind within the 1–13 region of the ligand-binding domain of the EGF repeat on the receptors and exhibit strong dependency on the specific ligand-expressing cell line; (ii) allosteric antibodies or NRR-binding antibodies, which are targeted to the extracellular negative regulatory region (NRR) on the Notch receptors and have a meager dependency on the type of ligand used for transactivation.107
The NRR antibodies are reported to bind and thereafter inhibit the ligand-independent activation of Notch1 receptors harboring T-cell acute lymphoblastic leukemia (T-ALL).108 The two potent antagonist mAbs (namely 256A-4 and 256A-8) have shown to inhibit the activity of Notch3 by competing with DSL ligands (including Jagged1, Jagged2, DLL1, and DLL4).109 Neutralizing DLL4 with DLL4 selective antibodies (e.g., anti-DLL4 antibody and soluble DLL4-Fc fusion protein) diminished the development of tumors in multiple tumor models, along with reduction of vascularization; DLL4 is necessary for vascularization.110
Apart from monoclonal antibodies, bispecific antibody, antibody–drug conjugate, and CAR-T therapy have also been proven to be effective in abrogating Notch signaling,113 listed in Table 2. While monoclonal antibodies are synthesized against a particular epitope having monoclonal affinity, the bispecific antibodies can simultaneously bind to two antigens or to the two epitopes of the same antigen. For example, the DLL-mediated Notch pathway activation plays an essential role in cancer by facilitating the maintenance of cancer stemness and induction of tumorigenesis. Despite numerous efforts in discovering a potent inhibitor against this particular pathway, the overall clinical outcomes are insufficient to overcome the clinical barriers. Therefore, to overcome the slender therapeutic window of DLL/Notch inhibitors, combination therapy with angiogenic inhibitors as well as the use of bispecific antibodies against the DLL/Notch and VEGF/VEGFR signaling axes are currently being employed to obtain an effective therapeutic outcome.114
Table 2. Notch Inhibition Using Different Forms of Antibodies and Antigens, under Clinical Trials.
| trade name | in combination | target | cancer subtypes | clinical trial | reference |
|---|---|---|---|---|---|
| mAbs | |||||
| OMP-59R5, Tarextumab | Nab-P and Gemcitabine | Notch2/Notch3 | stage IV, pancreatic cancer | phase Ib/II | NCT01647828 |
| OMP21M18 | FOLFIRI | DLL4 | advanced colorectal cancer | phase Ib | NCT01189942 |
| Carboplatin | DLL4 | advanced nonsquamous NSCLC | phase Ib | NCT01189968 | |
| Gemcitabine and Decizumab | DLL4 | advanced or metastatic pancreatic cancer | phase Ib | NCT01189929 | |
| bs-Ab | |||||
| Navicixizumab (OMP-305B83) | – | DLL4/VEGF | advanced solid tumors | phase I | NCT02298387 |
| AMG-757 | – | DLL3/CD3 | small cell lung cancer | phase I | NCT03319940, Owen et al.111 |
| ADC | |||||
| Rova T (Rovalpituzumab Tesirine) | – | DLL3 | SCLC | phase I/II | NCT01901653 |
| – | DLL3 | SCLC | phase II | NCT02674568 | |
| – | DLL3 | SCLC | phase III | NCT03033511 | |
| – | DLL3 | SCLC | phase II | NCT02674568 | |
| Topotecan | DLL3 | SCLC | phase III | NCT03061812 | |
| Nivolumab, Ipilimumab | DLL3 | SCLC | phase I/II | NCT03026166 | |
| Dexamethasone | DLL3 | SCLC | phase III | NCT03033511 | |
| PF-06650808 | – | Notch3 | breast cancer and other neoplasm | phase I | NCT02129205, Rosen et al.112 |
| Belantamab mafodotin (GSK2857916) | – | B-cell maturation antigen (BCMA) | relapsed or refractory multiple myeloma | phase II | NCT04126200 |
| CAR-T | |||||
| AMG-119 | – | DLL3 | SCLC | phase I | NCT03392064, Owen et al.111 |
Additionally, the antibody–drug conjugate (ADC), on the other hand, is an association of small molecular therapeutic drugs with the antibody, having a permanent or liable linker that is synthesized to target the specific antigens expressed by the cancer cells. CAR-T is an adoptive cellular therapy in which the T-cells of a patient are genetically modified ex vivo to express a chimeric antigen receptor (CAR). This in turn targets a particular receptor and readdresses cytotoxic T-cells to the specific receptor-positive cells, thereby resulting in cell death.115
Using GSIs (γ-Secretase Inhibitors)
The first class of inhibitors to reach clinical trials were the γ-secretase inhibitors (GSIs). They were initially developed as Aβ inhibitors that were proposed to halt the progression of Alzheimer’s disease (AD). Generally, the GSIs possess high affinity toward the catalytic core of the γ-secretase protein complex, presenilin. This results in the inhibition of the Notch pathway, or the inhibition of APPs (amyloid protein precursors) in AD.116 The list of GSIs (Figure 8) used to date is given in Table 3.
Figure 8.

Two-dimensional structures of γ-secretase inhibitors of different classes: (i) azepine (transition state analogues), e.g., LY411575 (A), and azepine (nontransition state analogues), e.g., DAPT (B); (ii) sulfonamides (nontransition state analogues), e.g., MK0752 (C); and (iii) peptide isoesterase (transition state analogues), e.g., GSI-1 (D). Two-dimensional structures of ADAM inhibitors, such as ZLDI-8 (E), INCB3619 (F), and Marimastat (G). Two-dimensional structures of γ-secretase modulators such as BPN15606 (H), Indomethacin (I), GSM-1 (J), Tarenflurbil (K), E2012 (L), CHF5074 (M), SGSM36 (N), FRM36143 (O), and BPN15606 (P).
Table 3. Notch Inhibition by GSI (in Monotherapy or Combination Therapy), under Clinical Trials.
| type and category of GSI | drug | in combination with another drug | target | clinical trials | reference |
|---|---|---|---|---|---|
| azepine (transition state analogues) | RO4929097 (RG4733) | a | pancreatic cancer | phase II | NCT01232829 |
| RO4929097 (RG4733) | a | advanced or metastatic breast cancer or recurrent TNBC | phase II | NCT01151449 | |
| RO4929097 (RG4733) | a | renal cell carcinoma | phase II | NCT01141569 | |
| RO4929097 (RG4733) | a | colorectal cancer | phase II | NCT01116687 | |
| RO4929097 (RG4733) | a | NSCLC | phase II | NCT01193868 | |
| RO4929097 (RG4733) | a | recurrent or metastatic ovarian cancer, fallopian tube cancer, or peritoneal cancer | phase II | NCT01175343 | |
| RO4929097 (RG4733) | a | melanoma | Huynh et al.117 | ||
| RO4929097 (RG4733) | a | metastatic colorectal cancer | phase II | Schell et al.118 | |
| RO4929097 (RG4733) | Vismodegib | advanced or metastatic sarcoma | phase I/II | NCT01154452 | |
| RO4929097 (RG4733) | Letrozole | ER+/PR+ stage I or II breast cancer | phase Ib | NCT01208441 | |
| azepine (nontransition state analogues) | Nirogacestat, PF-03084014 | a | fibromatosis | phase II | NCT01981551 |
| Nirogacestat, PF-03084014 | a | desmoid tumor/aggressive fibromatosis | phase III | NCT03785964 | |
| sulfonamide (nontransition state analogues) | MK-0752 | a | early stage ER+ breast cancer | presurgical | NCT00756717 |
| MK-0752 | Gemcitabine | stage III and IV pancreatic cancer | phase I/II | NCT01098344 | |
| MK-0752 | Docetaxel | locally advanced or metastatic breast cancer | phase I/II | NCT00645333 | |
| MK-0752 | Tamoxifen + Letrozole | early stage ER+ breast cancer | phase IV | NCT00756717 |
In monotherapy.
Generally, GSIs are categorized into three classes: (i) azepines, (ii) peptide isosteres, and (iii) sulfonamides. However, azepines and sulfonamides are prevalently used in combination therapy with chemotherapeutic drugs or inhibitors. Furthermore, GSIs are classified into two types depending on the structure and binding site. The first type is small molecule nontransition state inhibitors, which bind to a site other than the active site of the γ-secretase. It is probably at the interface of the γ-secretase complex dimer. The second type is aspartyl protease transition state analogues, which bind competitively to the catalytic cores of presenilins. They mimic the transition state of substrate cleavage by γ-secretase.
Using GSMs (γ-Secretase Modulators)
Unlike the GSIs, GSMs (Figure 8) are a group of small molecules that allosterically modulate the activity of the γ-secretase enzyme, by inducing a conformational change, thereby rendering it inactive or nonfunctional in nature. Some examples of GSMs are indomethacin, CHF5074, and Tarenflurbil, an NSAID (nonsteroidal anti-inflammatory drug). GSM-1 is a potent second-generation acidic GSM, and E2012 is a nonacidic GSM. Additionally, 2-aminothiazole γ-secretase modulators (SGSM-36), FRM36143, and BPN15606 decrease Aβ42 levels in Alzheimer’s disease.119,120
Using ADAM Inhibitors
While GSIs have been widely used in the inhibition of the Notch pathway, some α-secretase inhibitors (ASIs) have been developed that specifically block the activity of the ADAM (also known as TACE) enzyme (Figure 6). A major theoretical advantage of these ASIs might be their nonrequirement to enter the cell for eliciting their action.121
The TACE inhibitors are divided into various subtypes, such as (i) succinate-based inhibitors, (ii) macrocyclic inhibitors, (iii) sulfonamide inhibitors, (iv) γ-lactam inhibitors, (v) β-benzamido inhibitors, (vi) benzothiadiazine inhibitors, and (vii) nonhydroxamate inhibitors.122 Besides the Notch pathway, TACE is also responsible for the activation of numerous other pathways (TNFα, EGF, etc.) in cancer.
Research evidence suggests that inhibition of the ADAM proteases can regulate the Notch signaling pathway, thereby inhibiting the aggressive properties of various cancer subtypes. For example, Marimastat, an ADAM inhibitor, has been shown to decrease the expression of Notch1 and HES1 in renal cell carcinoma.123 Another potent ADAM inhibitor, INCB3619, inhibits the Notch pathway and reduces glioblastoma stem cells,124 and it enhances Gefitinib sensitivity in NSCLC.125 ZLDI-8, an ADAM17 inhibitor, decreases downstream proteins Survivin, CIAP1, and CIAP2 and also reverses the EMT by inhibiting the Notch pathway in NSCLC.126 It also enhances the effects of Sorafenib, Etoposide, and Paclitaxel on HCC cells.127
Moreover, an in silico study conducted by our group demonstrates the various binding affinities of the different classes of molecules and their functional derivatives (obtained from PubChem) against the ADAM protease.128 Through molecular docking studies, it was ascertained that the derivatives of the γ-lactam hydroxamate IK862 exhibit superior binding properties, followed by ZLDI8, a competitive and irreversible lymphoid tyrosine phosphatase inhibitor, in comparison to the derivatives of the other class of molecules. However, the in vitro assessment of TMI-1, a thiomorpholine sulfonamide hydroxamate bearing novel propargylic P1 group, endowed us with the efficacy of the compound in inhibiting the EMT, along with incorporating differentiated epithelial phenotype (CD44+/CD24+). It also induced apoptosis and decreased the colony-forming ability and wound-healing properties of the TNBC cells.129
Other Modes of Notch Inhibition
Abrogation of the Notch pathway has also been achieved using RNAi (ribonucleic acid interference) and shRNA (short-hairpin RNA). RNAi-mediated silencing of the Notch receptor DLL4 in ovarian tumor cells inhibited cell growth and angiogenesis. Inhibition of DLL4 and Jagged2 by shRNA has been shown to decrease angiogenesis and CSC130 and has therapeutic potential against retinoblastoma.131 Knockdown of CSL by shRNA also inhibits Notch effectively and inhibits tumor formation and overcomes chemoresistance in colorectal cancer.132 Knockdown of Notch2 mediated by shRNA induces apoptosis in human glioma;133 it also inhibits migration and proliferation of retinal pigment epithelial cells.134 Bevacizumab, an inhibitor of the vascular endothelial growth factor-A (VEGF), blocks angiogenesis, while Bevacizumab along with si-RNA mediated DLL4 silencing results in more potent inhibition of tumor growth.
Therapies using recombinant proteins, naturally occurring compounds, and metabolites have also been proven to be successful in blocking the activity of this pathway. It is reported that gliotoxin (a fungal secondary metabolite) is a compelling Notch2 transactivation inhibitor. It is observed that in chronic lymphocytic leukemia (CLL) cells, gliotoxin induces apoptosis.135 Additionally, it inhibited melanoma in a xenograft mouse model.136
Further, RITA (RBPJ interacting and tubulin associated), is a conserved 36 kDa protein that interacts with the tubulin in the cytoplasm. It is known to shuttle rapidly between cytoplasm and nucleus mediating nuclear export of RBPJ/CSL, thus acting as a negative regulator of the Notch pathway that downregulates Notch-mediated transcription of target genes.137 It is also known to upregulate p53 and downregulate cyclin E levels, thereby suppressing tumor growth and inducing apoptosis in HCC cells.138
Lidamycin (LDM) is an antibiotic having a discrete antitumor effect in various types of malignancies such as liver, breast, lungs, and colon. In hepatocellular carcinomas, Lidamycin inhibits the Notch pathway, thereby decreasing the expression of CD133, along with the mRNA of HES1, HEY1, and Notch1 genes.139
Resveratrol (RES), a naturally occurring phytoalexin, produced by plants in response to stress, injury, and fungal infection, has chemopreventive properties. It inhibits the survival signaling pathways such as Notch and PI3k/Akt and their downstream genes.140 Sulforaphane, a compound present in broccoli, in combination therapy with various chemotherapeutic drugs has been shown to inhibit Notch1, along with ALDH1 and C-rel expressing cancer stem cells.141
Conclusion and Future Prospectives
In order to explicate the molecular mechanisms facilitating cancer progression and dissemination, it is essential to identify the cellular factors that contribute to the aggressiveness of a malignancy, via the aberrant activation of the developmental pathways. The intriguing probability of Notch having either an oncogenic or a tumor suppressive effect in a particular tumor is highly controversial. The Notch pathway potentiates various hallmarks of cancer such as angiogenesis, drug resistance, metastasis, invasion, and maintenance of CSCs, by possessing established cross talk with several downstream pathways and transcription factors. A recent study conducted by our group led us to the conclusion that inhibiting the Notch pathway in TNBC cells results in diminishing the aggressive characteristics of the TNBC cells.129 The study elucidates the comparative analysis of the dual-targeted drug Lomitapide along with the γ-secretase inhibitor LY411575 and the ADAM inhibitor TMI-1. It was observed that, besides significant generation of ROS and inhibition of sphere formation by LY411575, TMI-1 exhibited no significant impact on the TNBC cells. Also, the IC50 for LY411575 was found to be much lower in comparison to that for TMI-1. However, TMI-1 showed better efficacy in colony formation assay and wound healing assay in TNBC cells. Immunoflow cytometry assay for the detection of CD24/CD44 stemness markers revealed a differentiated epithelial phenotype, following treatment with TMI-1 and LY411575 in both TNBC cell lines. However, the effect of LY411575 on the Notch downstream proteins such as HES1 and NICD showed significant downregulation in comparison to TMI-1 treated cells. Moreover, there was a significant reduction in the formation of autophagosomes and autolysosomes after treatment with LY411575, followed by TMI-1, suggesting their role in inhibiting autophagy via inhibition of the Notch pathway. Altogether, this study suggests the plausible role of the Notch pathway in supporting tumorigenesis, the inhibition of which results in the inhibition and/or downregulation of the aggressive characteristics of the TNBC cells.
Research evidence demonstrates that, besides the various positive aspects of targeting the Notch pathway, there still exhibit some limitations which are needed to be overcome in order to obtain an effective therapeutic outcome. For example, Notch receptor/ligand-specific targeting agents induce less adverse effects in comparison to the γ-secretase inhibitors. However, appropriate scheduling, reduced dosage, and amalgamating the therapeutic module with glucocorticoids might alleviate the adversative impact of GSIs. Monoclonal antibodies have minimal toxicity with restricted biodistribution and prolonged half-lives, which renders them unsuitable for precision therapy. On the other hand, solvable decoys of the extracellular domains of receptor/ligands block their subsequent interactions. However, the efficacy of these small molecular inhibitors regarding their bioavailability and pharmacodynamic properties needs to be further evaluated. Additionally, the benefits of pan-Notch inhibitors, which possess the capability of targeting all the Notch receptors rather than a single receptor, might hold promising results which need to be examined further. However, long-term therapeutic success with monotherapy targeting the Notch pathway is rarely achieved in controlling tumorigenesis. Besides, only some patients show aberrantly high Notch expressions. Thus, targeting the Notch pathway solely might not exhibit a significant impact on the clinical outcome. Hence, the discovery of a smarter drug to target specific drivers of tumor cell survival, progression, dissemination, and therapeutic resistance is equally important. Additionally, the identification of a potent Notch inhibitor exhibiting synergistic interactions in combination with conventional and systemic treatments is required, which might lead to faster clinical implementation, thereby benefiting patients.
Acknowledgments
The support of the Department of Biosciences and Bioengineering, Department of Biotechnology, Government of India (BT/PR44695/NER/95/1880/2021 and BT/PR41449/NER/95/1687/837 2020), Central Instruments Facility (CIF) and Centre for Nanotechnology (ICMR Centre for Excellence, Grant 5/3/8/20/2019-ITR, INUP, MeitY Grant 5(1)/2021-NANO) at IIT Guwahati is acknowledged. We also acknowledge the School of Health Science and Technology, Param-Ishan, and the BSL facilities of IIT Guwahati.
Glossary
Abbreviations
- AD
Alzheimer’s disease
- ADAM
a disintegrin and metalloprotease
- ADC
antibody–drug conjugate
- AIP4
aryl hydrocarbon receptor-interacting protein 4
- AKT
Ak strain transforming
- ALDH
aldehyde dehydrogenase
- AML
acute myeloid leukemia
- ANK
ankyrin
- APH
anterior pharynx-defective
- APP
amyloid protein precursor
- ASI
α-secretase inhibitor
- Aβ
amyloid-β
- BC
breast cancer
- bs-Ab
bispecific antibody
- CAR
chimeric antigen receptor
- CART
chimeric antigen receptor therapy
- CBF1
C promoter-binding factor
- CCN3
cellular communication network factor 3
- CDC10
cell division cycle
- CDK8
cyclin dependent kinase 8
- CIAP
cellular inhibitor of apoptosis proteins
- CIR
corepressor interacting with RBPJ 1
- CLL
chronic lymphocytic leukemia
- CR
cysteine-rich
- CSCs
cancer stem cells
- CSL
CBF1, Suppressor of Hairless, Lag-1
- CtBP
carboxy-terminal-binding protein 1
- Dlg-1
Drosophila disk large tumor suppressor
- Dlk
human Delta-like
- DLL
Delta Like Canonical Notch Ligand 1
- DNER
Delta/Notch-like EGF-related receptor
- DOS
Delta and OSM-11 motif
- DSL
Delta, Serrate, and LAG-2
- E(spl)
Enhancer of Split1
- EDTA
ethylenediaminetetraacetic acid
- EGFR
epidermal growth factor receptor
- EGTA
ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- EMT
epithelial to mesenchymal transition
- EPS15
epidermal growth factor receptor pathway substrate 15
- FGF
fibroblast growth factor
- GCN5
general control non-derepressible 5
- GlcNAc
O-linked N-acetylglucosaminyltransferase
- Glp-1
glucagon-like peptide 1
- GSI
γ-secretase inhibitor
- GSM
γ-secretase modulator
- HAT
histone acetyltransferase
- HCC
hepatocellular carcinoma
- HD
heterodimerization domain
- HDAC-I
histone deacetylase I
- HER2
human epidermal growth factor receptor 2
- HES
Hairy and Enhancer of Split1
- HEY2
Hairy/Enhancer-of-split related with YRPW motif protein 2
- HGF
hepatocyte growth factor
- HIF-1
hypoxia-inducible factor-1
- i-CLiPs
intramembrane cleaving protease
- IGF
insulin-like growth factor 1
- IKK
inhibitor of nuclear factor-κB (IκB) kinase
- Itch
Itchy E3 ubiquitin protein ligase
- KBF2
κ-binding factor 2
- LAG-1
Longevity-Assurance Gene-1
- LC3
microtubule-associated protein 1A/1B-light chain 3
- LDM
Lidamycin
- LEF
lymphoid enhancer factor
- LNR
LIN-12/Notch related region
- mAbs
monoclonal antibodies
- MAGP
microfibril-associated glycoproteins
- MAML
Mastermind-like
- MAPK
mitogen-activated protein kinase
- mRNA
messenger ribonucleic acid
- mTOR
mammalian target of rapamycin
- NAS
Notch1 antisense oligonucleotide
- NCT
nicastrin
- NICD
Notch intracellular domain
- NECD
Notch extracellular domain
- NEXT
Notch extracellular truncation
- NF-κB
nuclear factor κB
- NLS
nuclear localization signaling
- NRR
negative regulatory region
- NSCLC
non-small cell lung cancer
- NSAID
nonsteroidal anti-inflammatory drug
- O-fut
O-fucosyltransferase1
- OSM
Oncostatin M
- PCAF
P300/CPB associated factor
- PDZ
PSD-95/Dlg/ZO-1
- PEN
presenilin enhancer
- PEST
proline (P), glutamic acid (E), serine (S), and threonine (T)
- PI3K
phosphoinositide 3-kinase
- Poglut1
protein O-glucosyltransferase 1
- PSD95
post synaptic density protein (PSD95) binding motif
- PSEN
presenilin
- RAM
RBPJ associated molecule
- RBPJ
recombination signal binding protein for immunoglobulin κ J region
- RES
Resveratrol
- RITA
RBPJ interacting and tubulin associated
- RNAi
ribonucleic acid interference
- ROS
reactive oxygen species
- SKIP
skeletal muscle and kidney-enriched inositol phosphatase
- SHARP
SMRT/HDAC-associated repressor protein
- shRNA
short-hairpin RNA
- SKP2
S-phase kinase associated protein 2)
- SMRT
silencing mediator of retinoid and thyroid hormone receptors
- Su(H)
Suppressor of Hairless
- TACE
TNF-α converting enzyme
- TAD
transcription activation domain
- T-ALL
T-cell acute lymphoblastic leukemia
- TMD
transmembrane domain
- TMP
tumor-associated membrane protein
- TNBC
triple negative breast cancer
- TNFα
tumor necrosis factor-α
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
- vWFC
von Willebrand factor type-C
- ZO-1
zonula occludens-1
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Pharmacology & Translational Science virtual special issue “New Drug Modalities in Medicinal Chemistry, Pharmacology, and Translational Science”.
References
- Briscoe J.; Thérond P. P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. 10.1038/nrm3598. [DOI] [PubMed] [Google Scholar]
- Siebel C.; Lendahl U. Notch signaling in development, tissue homeostasis, and disease. Physiological reviews. 2017, 97, 1235–1294. 10.1152/physrev.00005.2017. [DOI] [PubMed] [Google Scholar]
- Steinhart Z.; Angers S. Wnt signaling in development and tissue homeostasis. Development 2018, 145 (11), dev146589. 10.1242/dev.146589. [DOI] [PubMed] [Google Scholar]
- Gridley T. Notch signaling in vertebrate development and disease. Molecular and Cellular Neurosciences 1997, 9, 103–108. 10.1006/mcne.1997.0610. [DOI] [PubMed] [Google Scholar]
- Demarest R. M.; Ratti F.; Capobianco A. J. It’s T-ALL about Notch. Oncogene 2008, 27, 5082–5091. 10.1038/onc.2008.222. [DOI] [PubMed] [Google Scholar]
- Weinmaster G. The ins and outs of Notch signaling. Molecular and Cellular Neurosciences. 1997, 9, 91–102. 10.1006/mcne.1997.0612. [DOI] [PubMed] [Google Scholar]
- Greenwald I. LIN-12/Notch signaling in C. elegans. WormBook. 2005, 1–16. 10.1895/wormbook.1.10.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald I. LIN-12/Notch signaling: Lessons from worms and flies. Genes & development. 1998, 12, 1751–1762. 10.1101/gad.12.12.1751. [DOI] [PubMed] [Google Scholar]
- Kelley M. R.; Kidd S.; Deutsch W. A.; Young M. W. Mutations altering the structure of epidermal growth factor-like coding sequences at the Drosophila Notch locus. Cell. 1987, 51 (4), 539–548. 10.1016/0092-8674(87)90123-1. [DOI] [PubMed] [Google Scholar]
- Hartley D. A.; Xu T. A.; Artavanis-Tsakonas S. The embryonic expression of the Notch locus of Drosophila melanogaster and the implications of point mutations in the extracellular EGF-like domain of the predicted protein. EMBO J. 1987, 6, 3407–3417. 10.1002/j.1460-2075.1987.tb02664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald I.; Seydoux G. Analysis of gain-of-function mutations of the lin-12 gene of Caenorhabditis elegans. Nature. 1990, 346 (6280), 197–199. 10.1038/346197a0. [DOI] [PubMed] [Google Scholar]
- Honjo T. The shortest path from the surface to the nucleus: RBP-Jκ/Su(H) transcription factor. Genes to Cells. 1996, 1 (1), 1–9. 10.1046/j.1365-2443.1996.10010.x. [DOI] [PubMed] [Google Scholar]
- Kovall R. A.; Blacklow S. C. Mechanistic insights into notch receptor signaling from structural and biochemical studies. Current Topics in Developmental Biology. 2010, 92, 31–71. 10.1016/S0070-2153(10)92002-4. [DOI] [PubMed] [Google Scholar]
- Gerhardt D. M.; Pajcini K. V.; D’altri T.; Tu L.; Jain R.; Xu L.; Chen M. J.; Rentschler S.; Shestova O.; Wertheim G. B.; Tobias J. W.; Kluk M.; Wood A. W.; Aster J. C.; Gimotty P. A.; Epstein J. A.; Speck N.; Bigas A.; Pear W. S. The Notch1 transcriptional activation domain is required for development and reveals a novel role for Notch1 signaling in fetal hematopoietic stem cells. Genes & development. 2014, 28 (6), 576–593. 10.1101/gad.227496.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald I. Structure/function studies of lin-12/Notch proteins. Current opinion in genetics & development. 1994, 4 (4), 556–562. 10.1016/0959-437X(94)90072-B. [DOI] [PubMed] [Google Scholar]
- Fiúza U. M.; Arias A. M. Cell and molecular biology of Notch. Journal of Endocrinology 2007, 194 (3), 459–474. 10.1677/JOE-07-0242. [DOI] [PubMed] [Google Scholar]
- Matsuno K. Notch signaling. Development Growth Differentiation. 2020, 62 (3), 3. 10.1111/dgd.12642. [DOI] [PubMed] [Google Scholar]
- Olsauskas-kuprys R.; Zlobin A.; Osipo C. Gamma secretase inhibitors of Notch signaling. OncoTargets and Therapy 2013, 6, 943–955. 10.2147/OTT.S33766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q.; Li J.; Zheng J.; Wei A. The carcinogenic role of the notch signaling pathway in the development of hepatocellular carcinoma. Journal of Cancer. 2019, 10 (6), 1570–1579. 10.7150/jca.26847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöqvist M.; Andersson E. R. Do as I say, Not(ch) as I do: Lateral control of cell fate. Developmental biology. 2019, 447 (1), 58–70. 10.1016/j.ydbio.2017.09.032. [DOI] [PubMed] [Google Scholar]
- Sato M.; Yasugi T.; Minami Y.; Miura T.; Nagayama M. Notch-mediated lateral inhibition regulates proneural wave propagation when combined with EGF-mediated reaction diffusion. PNAS. 2016, 113 (35), 5153–5162. 10.1073/pnas.1602739113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hussaini H.; Subramanyam D.; Reedijk M.; Sridhar S. S. Notch signaling pathway as a therapeutic target in breast cancer. Molecular cancer therapeutics. 2011, 10 (1), 9–15. 10.1158/1535-7163.MCT-10-0677. [DOI] [PubMed] [Google Scholar]
- Andersson E. R.; Sandberg R.; Lendahl U. Notch signaling: Simplicity in design, versatility in function. Development. 2011, 138 (17), 3593–3612. 10.1242/dev.063610. [DOI] [PubMed] [Google Scholar]
- Pandey A.; Niknejad N.; Jafar-Nejad H. Multifaceted regulation of Notch signaling by glycosylation. Glycobiology. 2020, 31 (1), 8–28. 10.1093/glycob/cwaa049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R. Notch signaling. Cold Spring Harbor perspectives in biology. 2012, 4 (10), a011213. 10.1101/cshperspect.a011213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logeat F.; Bessia C.; Brou C.; LeBail O.; Jarriault S.; Seidah N. G.; Israël A. The Notch1 receptor is cleaved constitutively by a furin-like convertase. PNAS. 1998, 95 (14), 8108–8112. 10.1073/pnas.95.14.8108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza B.; Miyamoto A.; Weinmaster G. The many facets of Notch ligands. Oncogene. 2008, 27 (38), 5148–5167. 10.1038/onc.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habets R. A.; Groot A. J.; Yahyanejad S.; Tiyanont K.; Blacklow S. C.; Vooijs M. Human NOTCH2 Is Resistant to Ligand-independent Activation by Metalloprotease Adam17. Journal of biological chemistry. 2015, 290 (23), 14705–14716. 10.1074/jbc.M115.643676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzo P.; Bocchetta M. Notch signaling in lung cancer. Expert review of anticancer therapy. 2011, 11 (4), 533–540. 10.1586/era.10.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagami S.; Okochi M.; Yanagida K.; Ikuta A.; Fukumori A.; Matsumoto N.; Ishizuka-Katsura Y.; Nakayama T.; Itoh N.; Jiang J.; Nishitomi K.; Kamino K.; Morihara T.; Hashimoto R.; Tanaka T.; Kudo T.; Chiba S.; Takeda M. Regulation of Notch Signaling by Dynamic Changes in the Precision of S3 Cleavage of Notch-1. Molecular and cellular biology. 2008, 28 (1), 165–176. 10.1128/MCB.00863-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belle V. A.; McDermott N.; Meunier A.; Marignol L. NUMB inhibition of NOTCH signalling as a therapeutic target in prostate cancer Victoria. Nature Reviews Urology. 2014, 11 (9), 499–507. 10.1038/nrurol.2014.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J. T.; Miyamoto A.; Weinmaster G. Notch Signaling - Constantly on the Move. Traffic 2007, 8 (8), 959–969. 10.1111/j.1600-0854.2007.00592.x. [DOI] [PubMed] [Google Scholar]
- Ayaz F.; Osborne B. A. Non-canonical Notch signaling in cancer and immunity. Frontiers in Oncology. 2014, 4, 1–6. 10.3389/fonc.2014.00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders P. G. T.; Munoz-Descalzo S. M.; Balayo T.; Wirtz-Peitz F.; Hayward P.; Arias A. M. Ligand-independent traffic of notch buffers activated armadillo in Drosophila. PLoS Biology 2009, 7 (8), e1000169. 10.1371/journal.pbio.1000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand M. D.; et al. Calcium Depletion Dissociates and Activates Heterodimeric Notch Receptors. Molecular and cellular biology. 2000, 20 (5), 1825–1835. 10.1128/MCB.20.5.1825-1835.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R.; Ilagan M. X. G. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell. 2009, 137 (2), 216–233. 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram M. V. The love-hate relationship between Ras and Notch. Genes & development. 2005, 19 (16), 1825–1839. 10.1101/gad.1330605. [DOI] [PubMed] [Google Scholar]
- Meurette O.; Stylianou S.; Rock R.; Collu G. M.; Gilmore A. P.; Brennan K. Notch activation induces Akt signaling via an autocrine loop to prevent apoptosis in breast epithelial cells. Cancer Res. 2009, 69 (12), 5015–5022. 10.1158/0008-5472.CAN-08-3478. [DOI] [PubMed] [Google Scholar]
- Roy M.; Pear W. S.; Aster J. C. The multifaceted role of Notch in cancer. Current Opinion in Genetics and Development. 2007, 17 (1), 52–59. 10.1016/j.gde.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Chiaramonte R.; Basile A.; Tassi E.; Calzavara E.; Cecchinato V.; Rossi V.; Biondi A.; Comi P. A wide role for NOTCH1 signaling in acute leukemia. Cancer Letters. 2005, 219 (1), 113–120. 10.1016/j.canlet.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Agrawal N.; Frederick M. J.; Pickering C. R.; Bettegowda C.; Chang K.; Li R. J.; Fakhry C.; Xie T. X.; Zhang J.; Wang J.; Zhang N.; El-Naggar A. K.; Jasser S. A.; Weinstein J. N.; Treviño L.; Drummond J. A.; Muzny D. M.; Wu Y.; Wood L. D.; Hruban R. H.; Westra W. H.; Koch W. M.; Califano J. A.; Gibbs R. A.; Sidransky D.; Vogelstein B.; Velculescu V. E.; Papadopoulos N.; Wheeler D. A.; Kinzler K. W.; Myers J. N. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011, 333 (6046), 1154–1157. 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Previs R. A.; Coleman R. L.; Harris A. L.; Sood A. K. Molecular pathways: Translational and therapeutic implications of the notch signaling pathway in cancer. Clinical cancer research. 2015, 21 (5), 955–961. 10.1158/1078-0432.CCR-14-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinakis A.; Lobry C.; Abdel-Wahab O.; Oh P.; Haeno H.; Buonamici S.; van De Walle I.; Cathelin S.; Trimarchi T.; Araldi E.; Liu C.; Ibrahim S.; Beran M.; Zavadil J.; Efstratiadis A.; Taghon T.; Michor F.; Levine R. L.; Aifantis I. A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature. 2011, 473 (7346), 230–233. 10.1038/nature09999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamatsu I.; Nagamatsu I.; Onishi H.; Matsushita S.; Kubo M.; Kai M.; Imaizumi A.; Nakano K.; Hattori M.; Oda Y.; Tanaka M.; Katano M.; et al. NOTCH4 Is a Potential Therapeutic Target for Triple-negative Breast Cancer. Anticancer Research 2014, 34 (1), 69–80. [PubMed] [Google Scholar]
- Mittal S.; Sharma A.; Balaji S. A.; Gowda M. C.; Dighe R. R.; Kumar R. V.; Rangarajan A. Coordinate hyperactivation of notch1 and Ras/MAPK pathways correlates with poor patient survival: Novel therapeutic strategy for aggressive breast cancers. Molecular cancer therapeutics. 2014, 13 (12), 3198–3209. 10.1158/1535-7163.MCT-14-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano T.; Fukuda D.; Koga J.; Aikawa M. Delta-Like Ligand 4-Notch Signaling in Macrophage Activation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2016, 36, 2038–2047. 10.1161/ATVBAHA.116.306926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Imanaka N.; Chen J.; Griffin J. D. Hypoxia potentiates Notch signaling in breast cancer leading to decreased E-cadherin expression and increased cell migration and invasion. British journal of cancer. 2010, 102 (2), 351–360. 10.1038/sj.bjc.6605486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Francesco E. M.; Maggiolini M.; Musti A. Crosstalk between Notch, HIF-1 α and GPER in Breast Cancer EMT. International Journal of Molecular Sciences 2018, 19 (7), 2011. 10.3390/ijms19072011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullendore M. E.; Koorstra J. B.; Li Y. M.; Offerhaus G. J.; Fan X.; Henderson C. M.; Matsui W.; Eberhart C. G.; Maitra A.; Feldmann G. Ligand-dependent Notch Signaling Is Involved inT umor Initiation and T umor Maintenance in Pancreatic Cancer. Clinical cancer research. 2009, 15 (7), 2291–2302. 10.1158/1078-0432.CCR-08-2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin F.; Bertin T.; Ma O.; Jiang M. M.; Wang L.; Sutton R. E.; Donehower L. A.; Lee B. Notch signaling contributes to the pathogenesis of human osteosarcomas. Human molecular genetics. 2009, 18 (8), 1464–1470. 10.1093/hmg/ddp057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borggrefe T.; Lauth M.; Zwijsen A.; Huylebroeck D.; Oswald F.; Giaimo B. D. The Notch intracellular domain integrates signals from Wnt, Hedgehog, TGF β/BMP and hypoxia pathways. BBA - Molecular Cell Research. 2016, 1863, 303–313. 10.1016/j.bbamcr.2015.11.020. [DOI] [PubMed] [Google Scholar]
- Sahlgren C.; Gustafsson M. V.; Jin S.; Poellinger L.; Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. PNAS. 2008, 105 (17), 6392–6397. 10.1073/pnas.0802047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa Iglesias V. S.; Giuranno L.; Dubois L. J.; Theys J.; Vooijs M. Drug Resistance in Non-Small Cell Lung Cancer: A Potential for NOTCH Targeting?. Frontiers in Oncology 2018, 8, 267. 10.3389/fonc.2018.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allenspach E. J.; Maillard I.; Aster J. C.; Pear W. S. Notch Signaling in Cancer Notch Signaling in Cancer. Cancer Biology & Therapy 2002, 1 (5), 466–476. 10.4161/cbt.1.5.159. [DOI] [PubMed] [Google Scholar]
- Shou J.; Ross S.; Koeppen H.; De Sauvage F. J.; Gao W. Q. Dynamics of Notch expression during murine prostate development and tumorigenesis. Cancer Res. 2001, 61 (19), 7291–7297. [PubMed] [Google Scholar]
- Sriuranpong V.; Borges M. W.; Ravi R. K.; Arnold D. R.; Nelkin B. D.; Baylin S. B.; Ball D. W. Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Res. 2001, 61 (7), 3200–3205. [PubMed] [Google Scholar]
- Fan X.; Mikolaenko I.; Elhassan I.; Ni X.; Wang Y.; Ball D.; Brat D. J.; Perry A.; Eberhart C. G. Notch1 and Notch2 have opposite effects on embryonal brain tumor growth. Cancer Res. 2004, 64 (21), 7787–7793. 10.1158/0008-5472.CAN-04-1446. [DOI] [PubMed] [Google Scholar]
- Sun L.; Liu M.; Sun G. C.; Yang X.; Qian Q.; Feng S.; Mackey L. V.; Coy D. H. Notch signaling activation in cervical cancer cells induces cell growth arrest with the involvement of the nuclear receptor NR4A2. Journal of Cancer. 2016, 7 (11), 1388–1395. 10.7150/jca.15274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobry C.; Oh P.; Aifantis I. Oncogenic and tumor suppressor functions of Notch in cancer: It’s NOTCH what you think. Journal of Experimental Medicine. 2011, 208 (10), 1931–1935. 10.1084/jem.20111855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viatour P.; Ehmer U.; Saddic L. A.; Dorrell C.; Andersen J. B.; Lin C.; Zmoos A. F.; Mazur P. K.; Schaffer B. E.; Ostermeier A.; Vogel H.; Sylvester K. G.; Thorgeirsson S. S.; Grompe M.; Sage J. Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. Journal of experimental medicine. 2011, 208 (10), 1963–1976. 10.1084/jem.20110198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyghobadi F.; Mehdipour M.; Nekoukar V.; Firouzi J.; Kheimeh A.; Lahrood F. N.; Zavared V. A.; Azimi M.; Mohammadi M.; Sodeifi N.; Ebrahimi M. Long-Term Inhibition of Notch in A-375 Melanoma Cells Enhances Tumor Growth Through the Enhancement of AXIN1, CSNK2A3, and CEBPA2 as Intermediate Genes in Wnt and Notch Pathways. Frontiers in Oncology 2020, 10, 531. 10.3389/fonc.2020.00531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimura T.; Goitsuka R.; Zhang Y.; Saito I.; Reth M.; Kitamura D. Cell cycle arrest and apoptosis induced by Notch1 in B cells. Journal of biological chemistry. 2000, 275 (47), 36523–36531. 10.1074/jbc.M006415200. [DOI] [PubMed] [Google Scholar]
- Romer S.; Saunders U.; Jäck H. M.; Jehn B. M. Notch1 enhances B-cell receptor-induced apoptosis in mature activated B cells without affecting cell cycle progression and surface IgM expression. Cell death and differentiation. 2003, 10 (7), 833–844. 10.1038/sj.cdd.4401253. [DOI] [PubMed] [Google Scholar]
- Nefedova Y.; Cheng P.; Alsina M.; Dalton W. S.; Gabrilovich D. I. Involvement of Notch-1 signaling in bone marrow stroma-mediated de novo drug resistance of myeloma and other malignant lymphoid cell lines. Blood. 2004, 103 (9), 3503–3510. 10.1182/blood-2003-07-2340. [DOI] [PubMed] [Google Scholar]
- Weniger M. A.; Küppers R. Molecular biology of Hodgkin lymphoma. Leukemia. 2021, 35 (4), 968–981. 10.1038/s41375-021-01204-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jundt F.; Anagnostopoulos I.; Förster R.; Mathas S.; Stein H.; Dörken B. Activated Notch1 signaling promotes tumor cell proliferation and survival in Hodgkin and anaplastic large cell lymphoma. Blood. 2002, 99 (9), 3398–3403. 10.1182/blood.V99.9.3398. [DOI] [PubMed] [Google Scholar]
- Rangarajan A.; Talora C.; Okuyama R.; Nicolas M.; Mammucari C.; Oh H.; Aster J. C.; Krishna S.; Metzger D.; Chambon P.; Miele L.; Aguet M.; Radtke F.; Dotto G. P. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO Journal. 2001, 20 (13), 3427–3436. 10.1093/emboj/20.13.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohda S.; Murata-Ohsawa M.; Sakano S.; Nara N. Notch ligands, Delta-1 and Delta-4 suppress the self-renewal capacity and long-term growth of two myeloblastic leukemia cell lines. International Journal of Oncology 2003, 22 (5), 1073–1079. [PubMed] [Google Scholar]
- Zweidler-McKay P. A.; He Y.; Xu L.; Rodriguez C. G.; Karnell F. G.; Carpenter A. C.; Aster J. C.; Allman D.; Pear W. S. Notch signaling is a potent inducer of growth arrest and apoptosis in a wide range of B-cell malignancies. Blood. 2005, 106 (12), 3898–3906. 10.1182/blood-2005-01-0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penton A. L.; Leonard L. D.; Spinner N. B. Notch signaling in human development and disease. Seminars in cell & developmental biology. 2012, 23 (4), 450–457. 10.1016/j.semcdb.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmigiani E.; Taylor V.; Giachino C. Oncogenic and Tumor-Suppressive Functions of NOTCH Signaling in Glioma. Cells. 2020, 9 (10), 2304. 10.3390/cells9102304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J.; Jiang M. M.; Jiang L.; Salvo J. S.; Zeng H. C.; Dawson B.; Bertin T. K.; Rao P. H.; Chen R.; Donehower L. A.; Gannon F.; Lee B. H. Notch activation as a driver of osteogenic sarcoma. Cancer cell. 2014, 26 (3), 390–401. 10.1016/j.ccr.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augert A.; Eastwood E.; Ibrahim A. H.; Wu N.; Grunblatt E.; Basom R.; Liggitt D.; Eaton K. D.; Martins R.; Poirier J. T.; Rudin C. M.; Milletti F.; Cheng W. Y.; Mack F.; MacPherson D. Targeting NOTCH activation in small cell lung cancer through LSD1 inhibition. Science Signaling 2019, 12 (567), eaau2922 10.1126/scisignal.aau2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M.; Zhang L.; He C. S.; Xu F.; Liu J. L.; Hu Z. H.; Zhao L. P.; Tian Y. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. Journal of Cellular Biochemistry 2012, 113 (5), 1501–1513. 10.1002/jcb.24019. [DOI] [PubMed] [Google Scholar]
- Mollen E. W. J.; Ient J.; Tjan-Heijnen V. C. G.; Boersma L. J.; Miele L.; Smidt M. L.; Vooijs M. A. G. G. Moving Breast Cancer Therapy up a Notch. Frontiers in Oncology. 2018, 8, 518. 10.3389/fonc.2018.00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. Y.; Kumano K.; Nakazaki K.; Sanada M.; Matsumoto A.; Yamamoto G.; Nannya Y.; Suzuki R.; Ota S.; Ota Y.; Izutsu K.; Sakata-Yanagimoto M.; Hangaishi A.; Yagita H.; Fukayama M.; Seto M.; Kurokawa M.; Ogawa S.; Chiba S. Gain-of-function mutations and copy number increases of Notch2 in diffuse large B-cell lymphoma. Cancer science. 2009, 100 (5), 920–926. 10.1111/j.1349-7006.2009.01130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil J.; Grim J.; Strack P.; Rao S.; Tibbitts D.; Winter C.; Hardwick J.; Welcker M.; Meijerink J. P.; Pieters R.; Draetta G.; Sears R.; Clurman B. E.; Look A. T. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. Journal of experimental medicine. 2007, 204 (8), 1813–1824. 10.1084/jem.20070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anjanappa M.; Hao Y.; Simpson E. R.; Bhat-Nakshatri P.; Nelson J. B.; Tersey S. A.; Mirmira R. G.; Cohen-Gadol A. A.; Saadatzadeh M. R.; Li L.; Fang F.; Nephew K. P.; Miller K. D.; Liu Y.; Nakshatri H. A system for detecting high impact-low frequency mutations in primary tumors and metastases. Oncogene. 2018, 37 (2), 185–196. 10.1038/onc.2017.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demehri S.; Turkoz A.; Kopan R. Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer cell. 2009, 16 (1), 55–66. 10.1016/j.ccr.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon L.; Avila J. L.; Demarest R. M.; Troutman S.; Allen M.; Ratti F.; Rustgi A. K.; Stanger B. Z.; Radtke F.; Adsay V.; Long F.; Capobianco A. J.; Kissil J. L. Notch1 functions as a tumor suppressor in a model of K-ras-induced pancreatic ductal adenocarcinoma. Cancer research. 2010, 70 (11), 4280–4286. 10.1158/0008-5472.CAN-09-4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luiken S.; Fraas A.; Bieg M.; Sugiyanto R.; Goeppert B.; Singer S.; Ploeger C.; Warsow G.; Marquardt J. U.; Sticht C.; De La Torre C.; Pusch S.; Mehrabi A.; Gretz N.; Schlesner M.; Eils R.; Schirmacher P.; Longerich T.; Roessler S. NOTCH target gene HES5 mediates oncogenic and tumor suppressive functions in hepatocarcinogenesis. Oncogene. 2020, 39 (15), 3128–3144. 10.1038/s41388-020-1198-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh C. H.; Bellon M.; Nicot C. FBXW7: a critical tumor suppressor of human cancers. Molecular Cancer. 2018, 17, 115. 10.1186/s12943-018-0857-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo P.; Osipo C.; Foreman K.; Golde T.; Osborne B.; Miele L. Rational targeting of Notch signaling in cancer. Oncogene. 2008, 27 (38), 5124–5131. 10.1038/onc.2008.226. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Li Y.; Xu H.; Zhang Y. The γ -secretase complex: from structure to function. Frontiers in cellular neuroscience. 2014, 8 (427), 1–10. 10.3389/fncel.2014.00427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J.; Shen Q. Targeting γ-secretase in breast cancer. Breast cancer. 2012, 4, 83–90. 10.2147/BCTT.S26437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elad N.; De Strooper B.; Lismont S.; Hagen W.; Veugelen S.; Arimon M.; Horré K.; Berezovska O.; Sachse C.; Chávez-Gutiérrez L. The dynamic conformational landscape of gamma-secretase. Journal of cell science. 2014, 128 (3), 589–598. 10.1242/jcs.164384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll C. M.; Li Y. M. Physiological and pathological roles of the γ-secretase complex. Brain research bulletin. 2016, 126, 199–206. 10.1016/j.brainresbull.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D.; Kopan R. Notch and Presenilin: regulated intramembrane proteolysis links development and degeneration. Annual review of neuroscience. 2003, 26, 565–597. 10.1146/annurev.neuro.26.041002.131334. [DOI] [PubMed] [Google Scholar]
- Golde T. E.; Koo E. H.; Felsenstein K. M.; Osborne B. A.; Miele L. γ -Secretase inhibitors and modulators. BBA - Biomembrane. 2013, 1828 (12), 2898–2907. 10.1016/j.bbamem.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann D.; de Strooper B.; Serneels L.; Craessaerts K.; Herreman A.; Annaert W.; Umans L.; Lübke T.; Lena Illert A.; von Figura K.; Saftig P. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Human Molecular Genetics. 2002, 11 (21), 2615–2624. 10.1093/hmg/11.21.2615. [DOI] [PubMed] [Google Scholar]
- Bozkulak E. C.; Weinmaster G. Selective Use of ADAM10 and ADAM17 in Activation of Notch1 Signaling. Molecular and cellular biology. 2009, 29 (21), 5679–5695. 10.1128/MCB.00406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifonova R.; Small D.; Kacer D.; Kovalenko D.; Kolev V.; Mandinova A.; Soldi R.; Liaw L.; Prudovsky I.; Maciag T. The non-transmembrane form of Delta1, but not of Jagged1, induces normal migratory behavior accompanied by fibroblast growth factor receptor 1-dependent transformation. Journal of biological chemistry. 2004, 279 (14), 13285–13289. 10.1074/jbc.C300564200. [DOI] [PubMed] [Google Scholar]
- Santolini E.; Puri C.; Salcini A. E.; Gagliani M. C.; Pelicci P. G.; Tacchetti C.; Di Fiore P. P. Numb is an endocytic protein. Journal of cell biology. 2000, 151 (6), 1345–1352. 10.1083/jcb.151.6.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pece S.; Serresi M.; Santolini E.; Capra M.; Hulleman E.; Galimberti V.; Zurrida S.; Maisonneuve P.; Viale G.; Di Fiore P. P. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. Journal of cell biology. 2004, 167 (2), 215–221. 10.1083/jcb.200406140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z.; Mu L.; Zheng Y.; Shen W.; Li J.; Xu L.; Zhong B.; Liu Y.; Zhou Y. (2020). NUMB enhances Notch signaling by repressing ubiquitination of NOTCH1 intracellular domain. Journal of molecular cell biology. 2020, 12 (5), 345–358. 10.1093/jmcb/mjz088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman G.; Liu L.; Sahlgren C.; Dahlqvist C.; Lendahl U. High levels of Notch signaling down-regulate Numb and Numblike. Journal of cell biology. 2006, 175 (4), 535–540. 10.1083/jcb.200602009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Li S. S. C. Numb: A new player in EMT. Cell adhesion & migration. 2010, 4 (2), 176–179. 10.4161/cam.4.2.10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocci F.; Jolly M. K.; Tripathi S. C.; Aguilar M.; Hanash S. M.; Levine H.; Onuchic J. N. Numb prevents a complete EMT by modulating Notch signalling. Journal of the Royal Society, Interface 2017, 14 (136), 20170512. 10.1098/rsif.2017.0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. Y.; Huang W. X.; Zhou X.; Chen J.; Li Z. Numb inhibits epithelial-mesenchymal transition via RBP-Jκ-dependent Notch1/PTEN/FAK signaling pathway in tongue cancer. BMC Cancer. 2019, 19 (1), 1–10. 10.1186/s12885-019-5605-5. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang J.; Shao X.; Sun H.; Liu K.; Ding Z.; Chen J.; Fang L.; Su W.; Hong Y.; Li H.; Li H. NUMB negatively regulates the epithelial-mesenchymal transition of triple-negative breast cancer by antagonizing Notch signaling. Oncotarget. 2016, 7 (38), 61036–61053. 10.18632/oncotarget.11062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnum-Finney B.; Wu L.; Yu M.; Brashem-Stein C.; Staats S.; Flowers D.; Griffin J. D.; Bernstein I. D. Immobilization of Notch ligand, Delta-1, is required for induction of Notch signaling. Journal of cell science. 2000, 113, 4313–4318. 10.1242/jcs.113.23.4313. [DOI] [PubMed] [Google Scholar]
- Small D.; Kovalenko D.; Kacer D.; Liaw L.; Landriscina M.; Di Serio C.; Prudovsky I.; Maciag T. Soluble Jagged 1 Represses the Function of its Transmembrane Form to Induce the Formation of the Src-dependent Chord-like Phenotype. Journal of biological chemistry. 2001, 276 (34), 32022–32030. 10.1074/jbc.M100933200. [DOI] [PubMed] [Google Scholar]
- Lundin K. E.; Gissberg O.; Smith C. I. E. Oligonucleotide Therapies: The Past and the Present. Human gene therapy. 2015, 26 (8), 475–485. 10.1089/hum.2015.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan L.; Aster J. C.; Sklar J.; Sunday M. E. Notch-1 regulates pulmonary neuroendocrine cell differentiation in cell lines and in transgenic mice. American Journal of Physiology-Lung Cellular and Molecular Physiology 2007, 292 (2), L500–L509. 10.1152/ajplung.00052.2006. [DOI] [PubMed] [Google Scholar]
- Zimrin A. B.; Pepper M. S.; McMahon G. A.; Nguyen F.; Montesano R.; Maciag T. An Antisense Oligonucleotide to the Notch Ligand Jagged Enhances Fibroblast Growth Factor-induced Angiogenesis in Vitro. Journal of Biological Chemistry 1996, 271 (51), 32499–32502. 10.1074/jbc.271.51.32499. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Chan S. L.; Miele L.; Yao P. J.; Mackes J.; Ingram D. K.; Mattson M. P.; Furukawa K. Involvement of Notch signaling in hippocampal synaptic plasticity. PNAS. 2004, 101 (25), 9458–9462. 10.1073/pnas.0308126101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Cain-Hom C.; Choy L.; Hagenbeek T. J.; de Leon G. P.; Chen Y.; Finkle D.; Venook R.; Wu X.; Ridgway J.; Schahin-Reed D.; Dow G. J.; Shelton A.; Stawicki S.; Watts R. J.; Zhang J.; Choy R.; Howard P.; Kadyk L.; Yan M.; Zha J.; Callahan C. A.; Hymowitz S. G.; Siebel C. W. Therapeutic antibody targeting of individual Notch receptors. Nature. 2010, 464 (7291), 1052–1057. 10.1038/nature08878. [DOI] [PubMed] [Google Scholar]
- Aste-Amézaga M.; Zhang N.; Lineberger J. E.; Arnold B. A.; Toner T. J.; Gu M.; Huang L.; Vitelli S.; Vo K. T.; Haytko P.; Zhao J. Z.; Baleydier F.; L’Heureux S.; Wang H.; Gordon W. R.; Thoryk E.; Andrawes M. B.; Tiyanont K.; Stegmaier K.; Roti G.; Ross K. N.; Franlin L. L.; Wang H.; Wang F.; Chastain M.; Bett A. J.; Audoly L. P.; Aster J. C.; Blacklow S. C.; Huber H. E. Characterization of notch1 antibodies that inhibit signaling of both normal and mutated notch1 receptors. PLoS One 2010, 5 (2), e9094. 10.1371/journal.pone.0009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K.; Li Y.; Wu W.; Gordon W. R.; Chang D. W.; Lu M.; Scoggin S.; Fu T.; Vien L.; Histen G.; Zheng J.; Martin-Hollister R.; Duensing T.; Singh S.; Blacklow S. C.; Yao Z.; Aster J. C.; Zhou B. B. Modulation of notch signaling by antibodies specific for the extracellular negative regulatory region of NOTCH3. Journal of biological chemistry. 2008, 283 (12), 8046–8054. 10.1074/jbc.M800170200. [DOI] [PubMed] [Google Scholar]
- Ridgway J.; Zhang G.; Wu Y.; Stawicki S.; Liang W. C.; Chanthery Y.; Kowalski J.; Watts R. J.; Callahan C.; Kasman I.; Singh M.; Chien M.; Tan C.; Hongo J. A.; de Sauvage F.; Plowman G.; Yan M. Inhibition of DLL4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006, 444 (7122), 1083–1087. 10.1038/nature05313. [DOI] [PubMed] [Google Scholar]
- Owen D. H.; Giffin M. J.; Bailis J. M.; Smit M. D.; Carbone D. P.; He K. DLL3: An emerging target in small cell lung cancer. Journal of hematology & oncology. 2019, 12 (1), 4–11. 10.1186/s13045-019-0745-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen L. S.; Wesolowski R.; Baffa R.; Liao K. H.; Hua S. Y.; Gibson B. L.; Pirie-Shepherd S.; Tolcher A. W. A phase I, dose-escalation study of PF-06650808, an anti-Notch3 antibody-drug conjugate, in patients with breast cancer and other advanced solid tumors. Investigational new drugs. 2020, 38 (1), 120–130. 10.1007/s10637-019-00754-y. [DOI] [PubMed] [Google Scholar]
- Katoh M.; Katoh M. Precision medicine for human cancers with Notch signaling dysregulation. International journal of molecular medicine. 2019, 45 (2), 279–297. 10.3892/ijmm.2019.4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You W. K.; Schuetz T. J.; Lee S. H. Targeting the DLL/Notch Signaling Pathway in Cancer: Challenges and Advances in Clinical Development. Molecular cancer therapeutics. 2023, 22 (1), 3–11. 10.1158/1535-7163.MCT-22-0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S.; Yi M.; Qin S.; Wu K. Next generation chimeric antigen receptor T cells: Safety strategies to overcome toxicity. Molecular cancer. 2019, 18 (1), 125. 10.1186/s12943-019-1057-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton C. C.; Zhu L.; Chau D.; Yang L.; Wang R.; Djaballah H.; Zheng H.; Li Y. M. Modulation of gamma-secretase specificity using small molecule allosteric inhibitors. PNAS. 2009, 106 (48), 20228–20233. 10.1073/pnas.0910757106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh C.; Poliseno L.; Segura M. F.; Medicherla R.; Haimovic A.; Menendez S.; Shang S.; Pavlick A.; Shao Y.; Darvishian F.; Boylan J. F.; Osman I.; Hernando E. The Novel Gamma Secretase Inhibitor RO4929097 Reduces the Tumor Initiating Potential of Melanoma. PloS one. 2011, 6 (9), e25264 10.1371/journal.pone.0025264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strosberg J. R.; Yeatman T.; Weber J.; Coppola D.; Schell M. J.; Han G.; Almhanna K.; Kim R.; Valone T.; Jump H.; Sullivan D. A phase II study of RO4929097 in metastatic colorectal cancer. European journal of cancer. 2012, 48 (7), 997–1003. 10.1016/j.ejca.2012.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S. L.; Rynearson K. D.; Duddy S. K.; Zhang C.; Nguyen P. D.; Becker A.; Vo U.; Masliah D.; Monte L.; Klee J. B.; Echmalian C. M.; Xia W.; Quinti L.; Johnson G.; Lin J. H.; Kim D. Y.; Mobley W. C.; Rissman R. A.; Tanzi R. E. Pharmacological and Toxicological Properties of the Potent Oral γ-Secretase Modulator BPN-15606. Journal of pharmacology and experimental therapeutics. 2017, 362 (1), 31–44. 10.1124/jpet.117.240861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raven F.; Ward J. F.; Zoltowska K. M.; Wan Y.; Bylykbashi E.; Miller S. J.; Shen X.; Choi S. H.; Rynearson K. D.; Berezovska O.; Wagner S. L.; Tanzi R. E.; Zhang C. Soluble Gamma-secretase Modulators Attenuate Alzheimer’s β-amyloid Pathology and Induce Conformational Changes in Presenilin 1. EBioMedicine. 2017, 24, 93–101. 10.1016/j.ebiom.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notch Signaling in Embryology and Cancer; Reichrath J., Reichrath S., Eds.; Advances in Experimental Medicine and Biology 727; Springer: 2012 10.1007/978-1-4614-0899-4. [DOI] [PubMed] [Google Scholar]
- Dasgupta S.; Murumkar P. R.; Giridhar R.; Yadav M. R. Current perspective of TACE inhibitors: A review. Bioorganic & medicinal chemistry. 2009, 17 (2), 444–459. 10.1016/j.bmc.2008.11.067. [DOI] [PubMed] [Google Scholar]
- Guo Z.; Jin X.; Jia H. Inhibition of ADAM-17 more effectively down-regulates the Notch pathway than that of γ-secretase in renal carcinoma. Journal of Experimental & Clinical Cancer Research. 2013, 32, 26. 10.1186/1756-9966-32-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd D. H.; Kefas B.; Seleverstov O.; Mykhaylyk O.; Dominguez C.; Comeau L.; Plank C.; Purow B. Alpha-secretase inhibition reduces human glioblastoma stem cell growth in vitro and in vivo by inhibiting Notch. Neuro-Oncology. 2012, 14 (10), 1215–1226. 10.1093/neuonc/nos157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B. B.; Peyton M.; He B.; Liu C.; Girard L.; Caudler E.; Lo Y.; Baribaud F.; Mikami I.; Reguart N.; Yang G.; Li Y.; Yao W.; Vaddi K.; Gazdar A. F.; Friedman S. M.; Jablons D. M.; Newton R. C.; Fridman J. S.; Minna J. D.; Scherle P. A. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer cell. 2006, 10 (1), 39–50. 10.1016/j.ccr.2006.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H. Y.; Zu Y. X.; Jiang X. W.; Sun X. T.; Liu T. Y.; Li R. L.; Wu Q.; Zhang Y. S.; Zhao Q. C. Novel ADAM-17 inhibitor ZLDI-8 inhibits the proliferation and metastasis of chemo-resistant non-small-cell lung cancer by reversing Notch and epithelial mesenchymal transition in vitro and in vivo. Pharmacological research. 2019, 148, 104406. 10.1016/j.phrs.2019.104406. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Li D.; Jiang Q.; Cao S.; Sun H.; Chai Y.; Li X.; Ren T.; Yang R.; Feng F.; Li B. A.; Zhao Q. Novel ADAM-17 inhibitor ZLDI-8 enhances the in vitro and in vivo chemotherapeutic effects of Sorafenib on hepatocellular carcinoma cells. Cell death & disease. 2018, 9 (7), 743. 10.1038/s41419-018-0804-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen P.; Kandasamy T.; Ghosh S. S. In-silico evidence of ADAM metalloproteinase pathology in cancer signaling networks. Journal of Biomolecular Structure & Dynamics. 2022, 40 (22), 11771–11786. 10.1080/07391102.2021.1964602. [DOI] [PubMed] [Google Scholar]
- Sen P.; Kandasamy T.; Ghosh S. S. Multi-targeting TACE/ADAM17 and gamma-secretase of notch signalling pathway in TNBC via drug repurposing approach using Lomitapide. Cellular Signalling. 2023, 102, 110529. 10.1016/j.cellsig.2022.110529. [DOI] [PubMed] [Google Scholar]
- Schadler K. L.; Zweidler-McKay P. A.; Guan H.; Kleinerman E. S. Delta-like ligand 4 plays a critical role in pericyte/vascular smooth muscle cell formation during vasculogenesis and tumor vessel expansion in Ewing’s sarcoma. Clinical cancer research. 2010, 16 (3), 848–856. 10.1158/1078-0432.CCR-09-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asnaghi L.; Tripathy A.; Yang Q.; Kaur H.; Hanaford A.; Yu W.; Eberhart C. G. Targeting Notch signaling as a novel therapy for retinoblastoma. Oncotarget. 2016, 7 (43), 70028–70044. 10.18632/oncotarget.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikandar S. S.; Pate K. T.; Anderson S.; Dizon D.; Edwards R. A.; Waterman M. L.; Lipkin S. M. NOTCH signaling is required for formation and self-renewal of tumor-initiating cells and for repression of secretory cell differentiation in colon cancer. Cancer research. 2010, 70 (4), 1469–1478. 10.1158/0008-5472.CAN-09-2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; He X.; Tian W.; Wang J. Short hairpin RNA targeting Notch2 inhibits U87 human glioma cell proliferation by inducing cell cycle arrest and apoptosis in vitro and in vivo. Molecular medicine reports. 2014, 10 (6), 2843–2850. 10.3892/mmr.2014.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Jin G.; Long C.; Zhou X.; Tang Y.; Huang S.; Kuang X.; Wu L.; Zhang Q.; Shen H. Blockage of notch signaling inhibits the migration and proliferation of retinal pigment epithelial cells. Scientific World Journal. 2013, 178708. 10.1155/2013/178708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubmann R.; Hilgarth M.; Schnabl S.; Ponath E.; Reiter M.; Demirtas D.; Sieghart W.; Valent P.; Zielinski C.; Jäger U.; Shehata M. Gliotoxin is a potent NOTCH2 transactivation inhibitor and efficiently induces apoptosis in chronic lymphocytic leukaemia (CLL) cells. British journal of haematology. 2013, 160 (5), 618–629. 10.1111/bjh.12183. [DOI] [PubMed] [Google Scholar]
- Hubmann R.; Sieghart W.; Schnabl S.; Araghi M.; Hilgarth M.; Reiter M.; Demirtas D.; Valent P.; Zielinski C.; Jäger U.; Shehata M. Gliotoxin targets nuclear NOTCH2 in human solid tumor derived cell lines in vitro and inhibits melanoma growth in xenograft mouse model. Frontiers in pharmacology. 2017, 8, 319. 10.3389/fphar.2017.00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker S. A.; Alvarado C.; von Wichert G.; Knippschild U.; Wiedenmann J.; Clauss K.; Nienhaus G. U.; Hameister H.; Baumann B.; Borggrefe T.; Knöchel W.; Oswald F. RITA, a novel modulator of Notch signalling, acts via nuclear export of RBP-J. EMBO journal. 2011, 30 (1), 43–56. 10.1038/emboj.2010.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Chen G.; Wang H.; Liu C. RITA inhibits growth of human hepatocellular carcinoma through induction of apoptosis. Oncology research. 2012, 20 (10), 437–445. 10.3727/096504013X13685487925059. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Sun W.; He R.; Zhang F.; Wang H.; Li P.; Shao R. G.; Xu X. Lidamycin decreases CD133 expression in hepatocellular carcinoma via the notch signaling pathway. Oncology letters. 2017, 14 (6), 7889–7895. 10.3892/ol.2017.7248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecchinato V.; Chiaramonte R.; Nizzardo M.; Cristofaro B.; Basile A.; Sherbet G. V.; Comi P. Resveratrol-induced apoptosis in human T-cell acute lymphoblastic leukaemia MOLT-4 cells. Biochemical pharmacology. 2007, 74 (11), 1568–1574. 10.1016/j.bcp.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Kallifatidis G.; Labsch S.; Rausch V.; Mattern J.; Gladkich J.; Moldenhauer G.; Büchler M. W.; Salnikov A. V.; Herr I. Sulforaphane increases drug-mediated cytotoxicity toward cancer stem-like cells of pancreas and prostate. Molecular therapy. 2011, 19 (1), 188–195. 10.1038/mt.2010.216. [DOI] [PMC free article] [PubMed] [Google Scholar]