

Abstract
We describe a method for direct tRNA sequencing using the Oxford Nanopore MinION. The principal technical advance is custom adapters that facilitate end-to-end sequencing of individual transfer RNA (tRNA) molecules at subnanometer precision. A second advance is a nanopore sequencing pipeline optimized for tRNA. We tested this method using purified E. coli tRNAfMet, tRNALys, and tRNAPhe samples. 76–92% of individual aligned tRNA sequence reads were full length. As a proof of concept, we showed that nanopore sequencing detected all 43 expected isoacceptors in total E. coli MRE600 tRNA as well as isodecoders that further define that tRNA population. Alignment-based comparisons between the three purified tRNAs and their synthetic controls revealed systematic nucleotide miscalls that were diagnostic of known modifications. Systematic miscalls were also observed proximal to known modifications in total E. coli tRNA alignments, including a highly conserved pseudouridine in the T loop. This work highlights the potential of nanopore direct tRNA sequencing as well as improvements needed to implement tRNA sequencing for human healthcare applications.
Keywords: tRNA, modifications, nanopore, E. coli, pseudouridine, isodecoder, isoacceptor
Transfer RNA (tRNA) is the most numerous RNA species in living cells. It plays a central role in protein synthesis and has nontranslational regulatory functions.1 Individual tRNA strands adopt a cloverleaf secondary structure that typically includes four loops: the T loop, the variable loop, the anticodon loop, and the D loop. The tRNA cloverleaf structure further folds into an L shape important for binding and function in the ribosome.2 Mature tRNA molecules typically contain a terminal, single-stranded 3′NCCA end. Over 90 unique ribonucleotide modifications are documented among all tRNAs.3 Dysregulation of several of these tRNA modifications (‘tRNA modopathies’) has been implicated in human mitochondrial diseases, neurological disorders, and cancer.4
tRNA sequencing is typically performed using RNA-Seq.5,6 This method employs reverse transcription (RT) and then sequencing of synthesized cDNA products. Certain modified bases cause RT to stop or stall which in some methods is mitigated using demethylase treatments or thermostable group II intron reverse transcriptases.7 However, RNA-Seq cannot directly detect base modifications nor can it document multiple modifications on individual strands.
Nanopores coupled with machine learning have been used previously to capture and analyze single-folded tRNA molecules. Discrimination between five tRNA isotypes and between two valine tRNA isoacceptors was achieved using solid-state nanopores.8 Recently, tRNA was differentiated from other classes of small RNAs using the MspA pore.9
Nanopore RNA sequencing is a fundamentally different technique that reads nucleotides directly along linearized strands without RT or amplification steps,10 thus permitting detection of modified nucleotides as part of the sequencing process.11−14 Briefly, an applied voltage drives single-stranded RNA polyanions through a nanoscale pore in the 3′ to 5′ direction resulting in sequence-dependent partial blockades of the K+/Cl– ionic current. A processive helicase motor regulates translocation of each RNA strand at approximately 0.5 nm precision, on the millisecond time scale, so that individual current steps are detectable. This ionic current signature is base called using a proprietary Oxford Nanopore Technologies (ONT) Recurrent Neural Network.10
Anticipating the use of nanopore technology for tRNA sequencing, Smith et al.15 developed a strategy for unfolding and threading tRNA strands through single alpha-hemolysin (α-HL) pores. To accomplish this, double-stranded adapters were annealed to the tRNA NCCA 3′ overhangs and then ligated to the 3′ and 5′ termini. Using noncatalytic phi29 DNA polymerase (DNAP) as a brake,16 individual E. coli tRNAfMet and tRNALys molecules were classified based on ionic current duration and amplitude for three segments along each strand. However, in that 2015 study, RNA sequencing was not possible largely because the α-HL channel limiting aperture was too long to resolve nucleotides, likely confounded by the forward and backward slips of the phi29 DNAP-strand complex under load, as observed for DNA.17
In this study, we implement direct tRNA sequencing on the ONT MinION.18 As part of this strategy, we designed custom adapters to improve end-to-end base calling of full length tRNA strands. Analysis of gel-purified samples achieved 50,000+ individual, aligned, full length E. coli tRNA reads per experiment that could be categorized at the isoacceptor and isodecoder levels. Comparison between biological tRNA strand reads and analogous synthetic strand reads revealed base miscalls consistent with known nucleotide modifications.
Results and Discussion
Our early efforts to sequence E. coli tRNA on the ONT platform gave unsatisfactory results with approximately 28% of reads aligned to tRNA references, of which only 55% were full length. In those experiments, the double-stranded nanopore sequencing splint adapters were composed entirely of DNA except for six ribonucleotides on the 5′ adapter strand, similar to those used in our single channel experiments.15 In addition, those experiments were hindered by persistent channel blockages that necessitated multiple MinION restarts.
To address these issues and generate improved tRNA read quality, we developed the nanopore tRNA sequencing strategy diagrammed in Figure 1A: (i) tRNA molecules were ligated to double-stranded splint adapters (5′ adapter strand, red line; 3′ adapter strand, blue line) using RNA ligase 2. These adapters were optimized with an 18-nucleotide-long RNA segment that attached directly adjacent to the 5′ most tRNA nucleotide and a six-nucleotide RNA segment that attached directly adjacent to the 3′ most tRNA nucleotide. These added RNA nucleotides ensured that the ONT neural network base caller could read through and beyond the first and last tRNA nucleotides; (ii) the ligation product was run on an 8% denaturing PAGE gel, and the band corresponding to the ligated product (∼130 nt indicated by asterisks) was excised and purified; and (iii) this purified product was ligated to the ONT motor-associated sequencing adapter using T4 DNA ligase which yields the final sequence-ready product (iv).
Figure 1.

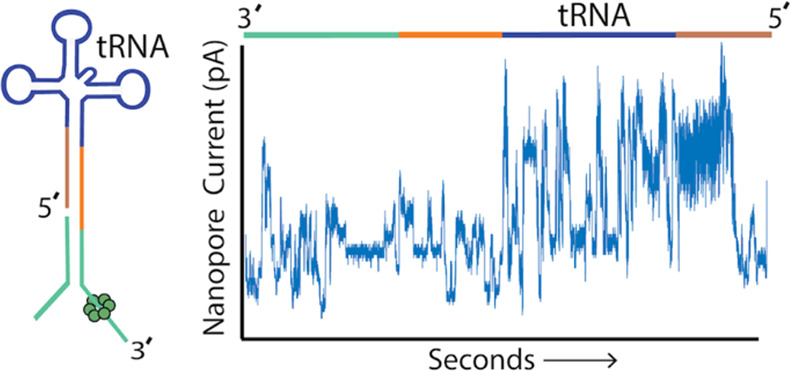
Overview of the tRNA sequencing strategy using synthetic canonical tRNAs. (A) tRNA adaptation for nanopore sequencing. From left to right: (i) The tRNA is ligated to a double-stranded splint adapter using RNA ligase 2. (ii) Gel purification of the ligation I product for synthetic tRNAs. The denaturing PAGE gel shows the first ligation of three synthetic tRNAs to the splint adapter. The lanes are as follows: 1, RNA ladder; 2, splint adapter; 3, synthetic tRNAfMet; 4, synthetic tRNAfMet ligation reaction; 5, synthetic tRNALys; 6, synthetic tRNALys ligation reaction; 7, synthetic tRNAPhe; 8, synthetic tRNAPhe ligation reaction; and 9, RNA ladder. The full length product (***) is excised and purified (see Figure S1 for biological tRNA). (iii) The purified product is ligated to the ONT sequencing adapters using T4 DNA Ligase. (iv) Adapted, nanopore sequencing-ready tRNA product. In the line drawing, the adapters and tRNA are not to scale. (B) An example ionic current trace of adapter-ligated synthetic tRNAfMet. Regions of the trace are indicated with colored bars corresponding to structures in (A): The 3′ strand of the ONT RMX adapter (teal); the 3′ strand of the splint adapter (blue); the tRNA (black); and the 5′ strand of the splint adapter (red). (C) Primary alignments of synthetic tRNAfMet to the reference sequence visualized using IGV. The reference sequence and its components are labeled as (i). The coverage at each position (coverage plot) is indicated by gray columns, where a maximally tall bar means every aligned read is covered at that position (ii). Beneath the coverage plot is a diagram of a randomly downsized sample of the aligned reads (iii). Gray rows denote continuous alignment and agreement with the reference nucleotide. Within each read, positions that do not match the reference (U(T) = red, A = green, C = blue, G = gold) are shown. White spaces bisected with a black bar within an aligned read indicate a deletion. Insertions are indicated in purple. The rows of aligned reads are presented as they were displayed on IGV.21
We first implemented this strategy using synthetic 5′-phosphorylated tRNAfMet composed of canonical nucleotides bearing no modifications. Figure 1B shows an ionic current trace associated with translocation of an adapted synthetic tRNAfMet strand through a CsgG nanopore.19 At 0 s, the open channel current is ∼ 245 pA (not shown). Upon strand capture, the current drops to approximately 60 pA. This is followed by discrete ionic current transitions corresponding to the 3′ adapter (teal and blue bars), synthetic tRNAfMet (black bar), and the 5′ splint (red bar).
Next, the nanopore ionic current data were base called using Guppy (v3.0.3). The resulting sequences were then aligned to a reference sequence using Burrows–Wheeler aligner, maximum exact match (BWA-MEM).20 This reference sequence contained the 18 ribonucleotides of the 5′ splint adapter strand, the E. coli tRNAfMet sequence, and the six ribonucleotides of the 3′ splint adapter strand. The resulting 83,956 aligned reads were visualized using integrated genome viewer (IGV) software21 (Figure 1C). Gray bars in the coverage plot (Figure 1C(i)) indicate positions where 80% or more of the quality weighted reads matched the expected nucleotide at that position.
Sequencing Purified Biological tRNAs and Corresponding Synthetic Canonical tRNAs
We applied this sequencing method to commercially available purified biological E. coli tRNAfMet, tRNALys, and tRNAPhe and their corresponding synthetic canonical controls. Because these tRNAs have an ACCA 3′ overhang, we used a splint adapter terminating with 5′-UrGrGrU-3′ (see Methods). Biological tRNA sequence reads and synthetic control sequence reads were aligned to their respective references using BWA-MEM and filtered for primary alignments (Figure 2). For the biological tRNAs, 76.4–92.1% of the aligned reads covered the entire tRNA (Table 1). The percentage of full length synthetic tRNA reads was lower (42.8–85.8%, Table 1). The reduced coverage seen at the 5′ end was most likely due to inefficient splint adapter ligation (Figure 2B(i), 2C(i)). This could arise from incomplete 5′-phosphorylation of the synthetic tRNA. Among 300 randomly selected unaligned reads for the three biological samples, a majority (172) had ionic traces recognizable as full-length adapted tRNA (i.e., broadly similar to the trace in Figure 1B). Alignment failure was likely due to: (i) suboptimal training of nanopore base callers for short RNA strands; (ii) nanopore RNA sequencing accuracy substantially below the 86% median; and (iii) parameter limitations of alignment software for short, error-prone, reads.
Figure 2.

Biological and canonical tRNA strand reads aligned against reference sequences. (A) tRNAfMet, (B) tRNAPhe, and (C) tRNALys. In each panel (i) is base coverage along the reference sequence at each position (coverage plot) and (ii) is a randomly selected subset of individual aligned nanopore reads. The total numbers of aligned reads are shown to the left of the coverage plots. The positions of expected modifications on biological tRNA3 are indicated above the coverage plots and are abbreviated: 4 = 4-thiouridine; D = Dihydrouridine; B = 2′-O-methylcytidine; 7 = 7-methylguanosine; T = 5-methyluridine; P = pseudouridine; X = 3-(3-amino-3-carboxypropyl)uridine; * = 2-methylthio-N6-isopentenyladenosine; S = 5-methyl-aminomethyl-2-thiouridine; and 6 = N6-threonylcarbamoyl-adenosine. Gray columns in the coverage plots indicate positions along the reference where 80% or more of the quality weighted reads are the expected canonical nucleotide. At positions where the value is under the 80% threshold, the proportion of each nucleotide call is shown in color where U(T) = red, A = green, C = blue, and G = gold. Similarly, the rows of individual aligned reads (A–C, ii) are gray at positions matching the reference and colored (using the previously mentioned convention) at positions with mismatches. The black horizontal bars in the aligned reads indicate a deletion, and purple bars indicate an insertion. The rows of aligned reads are presented as they were displayed on IGV.21
Table 1. Sequencing and Alignment Statistics for E. coli tRNAfMet, tRNALys, and tRNAPhea.
| synthetic |
biological |
|||||
|---|---|---|---|---|---|---|
| tRNAfMet | tRNALys | tRNAPhe | tRNAfMet | tRNALys | tRNAPhe | |
| no. of reads | 126,231 | 138,110 | 141,212 | 137,553 | 50,877 | 64,746 |
| no. of aligned reads | 83,956 | 103,834 | 72,870 | 67,396 | 22,087 | 36,731 |
| % of reads that aligned | 66.5% | 74.8% | 51.6% | 49.0% | 43.4% | 56.7% |
| no. of aligned reads that were full length | 72,034 | 72,683 | 31,188 | 51,490 | 20,342 | 33,682 |
| % of aligned reads that were full length | 85.8% | 70.0% | 42.8% | 76.4% | 92.1% | 91.7% |
Alignments were generated using references which contained adapter sequences and were filtered for MAPQ > 0. The “full length” categories refer to the number or percentage of aligned reads that span the full length of the tRNA reference sequence without the adapters.
Median alignment identities for the three biological tRNAs ranged from 75.3 to 78.9%, compared to 84 to 86.5% for their canonical controls (Table 2). The higher error rates for biological tRNA are consistent with prior nanopore studies that found base call errors proximal to modified ribonucleotides,11,13,14,22−24 which average about 10% of total tRNA nucleotides.25 Biological and synthetic tRNA primary alignments support this finding (Figure 2). Gray columns in these IGV coverage plots indicate positions where 80% or more of the reads matched reference sequences (Figure 2A(i),B(i),C(i)). When accuracy fell below this threshold, the proportion of reads for each nucleotide are indicated by red, blue, gold, and green columns (U(T), C, G and A bases, respectively). These tRNA base miscalls usually occurred at or near the positions of known modifications, which are denoted above the coverage plots (Figure 2 A–C(i)).
Table 2. Read alignment error profiles for E. coli tRNAfMet, tRNALys, and tRNAPhea.
| synthetic |
biological |
|||||
|---|---|---|---|---|---|---|
| tRNAfMet | tRNALys | tRNAPhe | tRNAfMet | tRNALys | tRNAPhe | |
| median alignment identity | 86.5% | 86.3% | 84.0% | 78.9% | 75.3% | 76.1% |
| median read coverage | 98.3% | 98.4% | 97.3% | 96.2% | 97.2% | 95.7% |
| median mismatches per aligned base | 5.7% | 5.8% | 7.7% | 12.9% | 15.5% | 15.2% |
| median deletions per read base | 4.1% | 4.1% | 4.4% | 4.0% | 4.3% | 4.1% |
| median insertions per read base | 1.4% | 1.5% | 1.8% | 2.9% | 1.7% | 3.2% |
Alignments were generated using references which did not contain adapter sequences and were filtered for MAPQ > 0. The first column to the left indicates the relevant error profile metrics. The following three columns contain the error profiles for synthetic E. coli tRNAfMet, tRNALys, and tRNAPhe. The last three columns contain the error profiles for biological E. coli tRNAfMet, tRNALys, and tRNAPhe.
Liquid chromatography tandem mass spectrometry (LC-MS/MS) was used to test for the presence of the expected modifications in one of the biological tRNA species (tRNAfMet). Comparison of the digested tRNA nucleoside products to commercially available standards verified the presence of 2′-O-methylcytidine, 7-methylguanosine, 5-methyluridine, and pseudouridine, but did not verify 4-thiouridine (Table S1, Figures S2–S7). The presence of dihydrouridine could not be tested using this strategy because a commercial standard is not available. However, its presence was supported based on expected parent and daughter ions (Figure S6).
Systematic Miscalls Identified by MarginCaller Occurred at or near Modified Nucleotides
The distribution of miscalls in the tRNA sequence alignments (Figure 2) suggested that some miscalls were caused by base modifications. As a quantitative test, we performed variant calling on control and biological tRNA alignments using marginCaller (Figure 3). Those miscalled positions that passed the marginCaller default posterior probability threshold of ≥ 30%26 were considered to be systematic miscalls.
Figure 3.

Systematic base miscalls in purified biological and canonical tRNAfMet, tRNALys, and tRNAPhe. The coverage plots (A–C) for biological and canonical synthetic tRNAs were generated from alignments using marginAlign. The number of aligned reads for each tRNA is shown under each coverage plot. Boxes surrounding base positions denote systematic miscalls (posterior probability of ≥30%). No systematic miscalls were identified in the synthetic canonical tRNAs. Colored bars are at positions where <80% of the quality weighted alignments match the reference. At these positions, the proportion of individual bases called are shown in color (U(T) = red, A = green, C = blue, and G = gold). The known modifications for the biological tRNAs3 are indicated above the coverage plots. Modified nucleotides are indicated above the reference sequence with abbreviations (4 = 4-thiouridine, D = dihydrouridine, B = 2′-O-methylcytidine, 7 = 7-methylguanosine, T = 5-methyluridine, P = pseudouridine, X = 3-(3-amino-3-carboxy-propyl)uridine, * = 2-methylthio-N6- isopentenyladenosine, S = 5-methylaminomethyl-2-thiouridine, and 6 = N6-threonylcarbamoyl-adenosine). The falloff in 5′ coverage of synthetic tRNALys (B, lower panel) and tRNAPhe (C, lower panel) is likely due to incomplete 5′ phosphorylation of these substrates.
This method identified systematic miscalls at three positions in biological tRNAfMet, six positions in biological tRNALys, and six positions in biological tRNAPhe (Figure 3, Table S2). None of these were associated with base variants among tRNA gene copies.27 All of these occurred within two nucleotides of a known modification. The highest posterior probability miscall in all tRNAs corresponded to pseudouridine in the T loop (position 55) (Table S2). This tRNA modification is the most conserved modification across phyla.25 In E. coli, all tRNAs have this modification at this position.3 The possible contribution of the neighboring 5-methyluridine (position 54) to the systematic miscall remains to be explored. No systematic miscalls were identified in the synthetic controls.
In biological tRNAs, not all known modification positions were detected at the default posterior probability threshold (Figure 3). Factors that could contribute to this include a modification’s chemical structure, neighboring nucleotide context, abundance at a given position, and the stringency used in defining a position as a systematic miscall. Miscall analysis is currently limited based on the constraints of the base caller and the quality of the alignments.4,28,29
Nanopore Detection of off-Target Biological tRNAs
We aligned nanopore reads for each of the purified biological tRNA samples against the total E. coli tRNA sequence reference (Table S3, Figure S8). Surprisingly, a fraction of the reads aligned to tRNAs other than the expected reference (4.6%, 7%, and 8.2% off-target reads for purified tRNAfMet, tRNALys, and tRNAPhe respectively, Table S3). This suggests that tRNA contaminants were carried over during purification of the commercial samples. The median alignment identities for these putative tRNA impurities to the alternate tRNA references ranged from 75.8% to 80.4%, consistent with gene-specific tRNA alignments summarized in Table 2. We conclude that nanopore direct tRNA sequencing could provide a fast and simple assay for tRNA purity in reference samples. This is consistent with prior work that detected 10 attomoles of an E. coli 16S rRNA against a background of human RNA.11 Confirmation of this strategy will require validation using LC-MS/MS.
Sequencing Total E. coli tRNA
As proof of concept, we used the nanopore method to sequence total tRNA from E. coli strain MRE600. This sample included tRNAs with four distinct 3′ NCCA overhangs (ACCA, UCCA, CCCA, and GCCA). For this reason, we used a combination of four double-stranded splint adapters for the first nanopore sequencing ligation. This ligation product was run on a denaturing PAGE gel (Figure 4), and full-length adapted tRNA strands were excised and extracted (Figure 4, lane 4). The purified ligation I products were then ligated to the ONT sequencing adapter, sequenced on the MinION, and base called. This generated 125,271 tRNA reads which were aligned to a reference set composed of the 43 E. coli tRNA isoacceptors (versions of tRNA that carry the same amino acid but have different anticodons) (Figure S8).
Figure 4.

PAGE gel of total tRNA ligated to splint adapters. Lane 1: The ssRNA ladder with sizes in nucleotides indicated to the left. Lane 2: E. coli MRE600 total tRNA. Lane 3: A 121 nt IVT human 5S rRNA used as a size marker. Lane 4: The products of the ligation reaction of total E. coli tRNA and the four types of splint adapters. The two bands under 50 nt are the 30 nt and 24 nt strands of the splint adapters that did not ligate to the tRNA. Successful ligation of the double-stranded splint will add 54 nt to the tRNA. As tRNA ranges from 75 to 93 nt, the expected ligation products are 129–140 nt. A block of gel encompassing fragments of approximately 110–180 nt, indicated on the gel as a black rectangle, was excised, purified, and carried forward for the library preparation.
Each of the 43 tRNA reference sequences was appended with the RNA portion of the splint adapters to maximize recovery of aligned reads. This resulted in 74,130 primary tRNA alignments of which 72,628 had a mapping quality (MAPQ) ≥ 1 (i.e., reads aligning uniquely to one of the sequences in the reference set).30 MAPQ ≥ 1 alignments were observed for all 43 isoacceptors (Table 3).
Table 3. Total E. coli tRNA Aligned Readsa.
| E. coli tRNA isoacceptor | no. of aligned reads | E. coli tRNA isoacceptor | no. of aligned reads |
|---|---|---|---|
| tRNA_Ala_VGC | 1861 | tRNA_Leu_BAA | 920 |
| tRNA_Ala_GGC | 1287 | tRNA_Leu_)AA | 775 |
| tRNA_Arg_ICG | 1455 | tRNA_Lys_SUU | 1729 |
| tRNA_Arg_CCG | 427 | tRNA_Met_MAU | 644 |
| tRNA_Arg_UCU | 382 | tRNA_Phe_GAA | 1289 |
| tRNA_Arg_CCU | 57 | tRNA_Pro_CGG | 1716 |
| tRNA_Arg_UCG | 18 | tRNA_Pro_GGG | 375 |
| tRNA_Asn_GUU | 4382 | tRNA_Pro_UGG | 1347 |
| tRNA_Asp_⊄UC | 5008 | tRNA_Sec_UCA | 291 |
| tRNA_Cys_GCA | 2682 | tRNA_Ser_UGA | 2738 |
| tRNA_Gln_UUG | 1061 | tRNA_Ser_CGA | 232 |
| tRNA_Gln_CUG | 2194 | tRNA_Ser_GCU | 1901 |
| tRNA_Glu_SUC | 6924 | tRNA_Ser_GGA | 1167 |
| tRNA_Gly_CCC | 822 | tRNA_Thr_GGU | 932 |
| tRNA_Gly {CC | 1136 | tRNA_Thr_CGU | 161 |
| tRNA_Gly_GCC | 8375 | tRNA_Thr_UGU | 814 |
| tRNA_His_GUG | 604 | tRNA_Trp_CCA | 1413 |
| tRNA_Ile_GAU | 5351 | tRNA_Tyr_QUA | 1259 |
| tRNA_Ile_CAU | 328 | tRNA_Val_VAC | 1708 |
| tRNA_Leu_CAG | 4049 | tRNA_Val_GAC | 792 |
| tRNA_Leu_GAG | 1006 | tRNA_Ini_CAU | 560 |
| tRNA_Leu_UAG | 456 | total | 72,628 |
Values are the number of nanopore reads aligned to each of the 43 tRNA isoacceptors with a MAPQ ≥ 1. The anticodon sequence is indicated for each tRNA isoacceptor. Noncanonical letters in the anticodons represent modified nucleotides.3 These results were from total biological tRNA sequencing using all four splint adapter versions. tRNAfMet (initiator tRNA) is listed as ‘tRNA_Ini’.
Among the 43 E. coli tRNA isoacceptors, 24 end in ACCA-3′, 13 end in GCCA-3′, 5 end in UCCA-3′, and one ends in CCCA-3′.3,31 We reasoned that separate tRNA capture reactions, each with a specific complimentary 4-mer overhang, could enrich for the corresponding tRNA isoacceptors. However, when we sequenced these four separate reactions on the MinION (Table S4), we observed only a modest enrichment for tRNA species with the targeted 3′ ends (2.3% to 33.9% enrichment, Figure S9). Surprisingly, reads for all 43 isoacceptor types were recovered from each experiment (Table S4).
When we compared the relative percentages of each isoacceptor in nanopore data to RNA fingerprinting data,32 we found a moderate positive correlation (R2 = 0.47, P < 0.0033) (Figure S10). These results were comparable to the correlation between RNA-seq isoacceptor abundances33 and the same RNA fingerprinting study32 (R2 = 0.5 and P < 0.0001). A high proportion of ACCA-terminating tRNAs were underrepresented in the nanopore data relative to the RNA fingerprinting study (blue circles below trendline in Figure S10). This could be caused by a limiting concentration of the ACCA specific adapter during sample preparation. Although ACCA terminated tRNAs comprise ∼ 60% of E. coli tRNA,32 we used an equimolar amount of each of the four NCCA adapters.
We note that tRNAHis in E. coli(3) and most other organisms34 has an extra 5′ G nucleotide that base pairs in the acceptor stem, resulting in a three nucleotide 3′ overhang rather than a typical four nucleotide overhang. This could account for the relatively low sequencing throughput for tRNAHis (Table 3). This problem could be resolved using a custom adapter that complements the CCA overhang rather than a CCCA overhang on the tRNA terminus.
Association of Nanopore Systematic Miscalls with Modifications Is Supported by Total tRNA Sequence Data
As an additional test of the association between nanopore base miscalls and base modifications, we examined sequence data for tRNAfMet, tRNALys, tRNAPhe, and tRNAAla1 acquired from total tRNA (see Materials and Methods). We chose these four tRNA because synthetic canonical control data were available for comparison. As anticipated, all systematic miscalls on the tRNA strands occurred at or adjacent to known modified positions3 (Table S5). Quantitative analysis with marginCaller supported two miscalls in tRNAAla1, three miscalls in tRNAfMet, nine miscalls in tRNALys, and seven miscalls in tRNAPhe. Each of these systematic miscalls was within three positions of a nucleotide that is known to be modified. In agreement with the purified tRNA sequence data (Table 2), the highest posterior probability miscalls were at position 55 which is a highly conserved pseudouridine in E. coli tRNA (Table S5).
Detection of Isodecoders within E. coli tRNA Isoacceptor Alignments
Among the total tRNA isoacceptor alignments, several contained consistent miscalls at positions where no modifications had been documented previously. This is illustrated for six positions in tRNAThrGGU (Figure 5i, vertical arrows). We reasoned that these miscalls might be associated with previously unidentified modifications; however, a more plausible explanation was commingling of isoacceptor subsets (‘isodecoders’)35 bearing additional nucleotide variants.
Figure 5.

Confirmation of two known isodecoders in nanopore tRNAThrGGU sequence alignments. The isoacceptor tRNAThrGGU has two isodecoder forms which have canonical sequence variations at positions 9 (G/A), 49 (C/G), 50 (C/G), 59 (C/A), 64 (G/C), and 65 (G/C).3 (i–iii) Black boxes on the IGV coverage plots surround the positions of these variations. Black arrows also point to variant positions for clarity. Above each black box, the reference nucleotide is enlarged, and the alternative nucleotide is above it, labeled and colored in accordance with IGV schema (A = green, G = gold, C = blue, and T(U) = red). (i) Alignments of total E. coli tRNA reads to the tRNAThrGGU isoacceptor. At positions that vary between tRNAThrGGU isodecoders, the colors representing the reference and alternative nucleotides are seen in the coverage plot. This can be interpreted as both isodecoder forms being present in the data. (ii) Alignments of total E. coli tRNA read to the tRNAThrGGU_A isodecoder. Gray in the coverage plot and the rows of aligned reads indicate agreement with the tRNAThrGGU_A reference. (iii) Alignments of total E. coli tRNA read to the tRNAThrGGU_B isodecoder. Gray in the coverage plot and the rows of aligned reads indicate agreement with the tRNAThrGGU_B reference. The known modifications D (dihydrouridine), E (N6-methyl-N6-threonylcarbomoyladenosine), 7 (7-methylguanosine), and P (pseudouridine) are denoted in black above the reference sequence.3 Adapter sequences were included in the alignments but are not shown.
To test this prediction, we generated an E. coli MRE600 isodecoder reference set using tRNAscan,31 a software program which predicts tRNA species based on genome sequence. This resulted in 15 predicted isodecoders: two each for isoacceptors tRNAArgUCU, tRNAIleCAU, tRNASerGGA, tRNAThrGGU, tRNATyrGUA, and tRNAValGAC and three predicted isodecoders for tRNALeuCAG.
A total of 701,706 E. coli tRNA reads from five MinION runs were aligned to a reference set composed of all isoacceptor sequences and the additional isodecoder sequences (Figure S8). These alignments were then compared to alignments against only the isoacceptor references. Figure 5i–iii shows tRNAThrGGU as an example. Alignments sorted to each of the two predicted tRNAThrGGU isodecoders (Figure 5ii,iii) resolved all six of the systematic miscalls observed among the isoacceptor alignments that were not proximal to known nucleotide modifications (Figure 5i).
Table 4 summarizes alignment statistics for 10 of the 15 predicted MRE600 E. coli tRNA isodecoders. More reads aligned uniquely (MAPQ ≥ 1) to 8 out of the 10 isodecoders relative to their corresponding isoacceptors. This was due to reads that were either previously unaligned or ambiguously aligned at the isoacceptor level, but could be aligned unambiguously at the isodecoder level. This was not true for the two tyrosine isodecoders. The most plausible explanation is ambiguous alignments (MAPQ = 0) between error-prone nanopore reads and tyrosine isodecoder sequences that differ by only two nucleotides. Importantly, no reads were lost when aligned at the isodecoder level. Five isodecoders (corresponding to tRNALeuCAG and tRNASerGGA) were not included in this analysis because they are differentiated solely by homopolymer lengths which cannot be accurately resolved using the ONT R9.4 chemistry.
Table 4. E. coli tRNA Nanopore Reads That Align to 10 E. coli Isodecoder Sequencesa.
| I | II | III | IV | V |
|---|---|---|---|---|
| E. coli MRE600 tRNA isoacceptor | no. of reads aligned to isoacceptor (MAPQ ≥ 1) | E. coli MRE600 tRNA isodecoder | no. of reads aligned to isodecoder (MAPQ ≥ 1) | no. of nucleotide differences between isodecoders |
| ArgUCU | 1623 | ArgTCT_A | 1619 | 17 |
| ArgTCT_B | 9 | |||
| IleCAU | 2094 | IleCAT_A | 1328 | 7 |
| IleCAT_B | 857 | |||
| ThrGGU | 6659 | ThrGGT_A | 2913 | 6 |
| ThrGGT_B | 4681 | |||
| TyrQUA | 11,518 | TyrGTA_A | 1933 | 2 |
| TyrGTA_B | 5904 | |||
| ValGAC | 5900 | ValGAC_A | 2911 | 6 |
| ValGAC_B | 3320 | |||
| Total | 27,794 | total | 25,475 |
Columns I and II are E. coli MRE600 isoacceptors and their counts in total E. coli tRNA reads. Column III lists MRE600 E. coli isodecoders predicted by tRNAScan.31 Each isodecoder species is appended with the letter A or B. We used ‘A’ or ‘B’ because there is no consistent nomenclature for E. coli isodecoders. Column IV contains read counts for each isodecoder when aligned against a reference composed of all isoacceptors and 10 isodecoders. Column V contains the number of nucleotide differences between isodecoders. The isodecoders for tRNALeuCAG and tRNASerGGA were not included because their canonical sequences differ by the length of homopolymer regions which cannot be accurately resolved using the ONT R9.4 chemistry.
Conclusions
In this study, we sequenced individual full-length tRNA strands using nanopore technology. All 43 E. coli isoacceptors and 10 out of 15 predicted isodecoders were documented, with a majority of individual reads mapping to a single reference (MAPQ ≥ 1). For each of these tRNA species, systematic base miscalls correlated with known modifications.
Looking ahead, Suzuki4 summarized the role of aberrant tRNA modifications in human disease.36−39 That review also noted that only 18 out of 200 human cytosolic tRNA species had documented modification profiles. It is reasonable to project that nanopore tRNA sequencing could add to these profiles; however, a number of technical advances will be required. Some of these are ONT platform dependent. Notably, the ONT canonical RNA base caller has remained at 86% median accuracy since 2019.11,23 We anticipate improvements in RNA base calling with the development of the ONT platform, as has been documented for the proprietary ONT DNA base caller over the past 5 years.40 Further, ONT has recently engineered a pore with two reading heads (R10.4 Chemistry) that could further improve accuracy and resolution of homopolymeric regions.41 This could be applied to resolve tRNA species that contain homopolymer sequences similar to E. coli tRNALeuCAG and tRNASerGGA.
Other tRNA-specific advances will come from the research community. In particular, to date, we and others have relied upon base miscalls to indirectly infer RNA modifications.13,14,22−24 A more principled method will employ algorithms trained on modification-dependent ionic current patterns, as has been done for 5-methylcytosine modifications in DNA40,42,43 and for m6A in mRNA.44 Verification of these tRNA modification signals will be essential45 using LC-MS/MS (the gold standard for chemical validation) and enzyme knockouts or knockdowns. The prospects for comprehensive direct tRNA nanopore sequencing are promising.
Methods
Materials
Enzymes
T4 RNA ligase 2 (10,000 units/mL), T4 DNA ligase (2,000,000 units/mL), T4 polynucleotide kinase (10,000 units/mL), and corresponding buffer solutions were purchased from New England Biolabs. Nuclease P1 (Sigma Aldrich), antarctic phosphatase (5,000 units/mL) (New England Biolabs), and phosphodiesterase 1 (Thermo Fisher Scientific) were used to prepare samples for liquid chromatography/mass spectrometry.
Biological tRNAs
Purified biological tRNAs included E. coli tRNA fMet (Subriden RNA, purity unknown), tRNA Lys(Sigma-Aldrich, amino acid acceptor activity ≥ 1000 pmol/A260 unit, 14–22 unit/mg solid) and tRNAPhe (Sigma-Aldrich, amino acid acceptor activity ≥ 900 pmol/A260 unit, ≥ 10 unit/mg solid)). E. coli total tRNA from strain MRE600 was obtained from Roche Pharmaceuticals.
Synthetic Canonical tRNA
Synthetic canonical tRNAs were ordered from IDT or Dharmacon. The 5′ phosphorylation was performed either during synthesis (for tRNAfMet) or following synthesis using T4 polynucleotide kinase (NEB) (for tRNALys and tRNAPhe) per manufacturer′s protocol. The sequences of these tRNAs are: E. coli synthetic tRNAfMet (initiator tRNA): 5′PCGCGGGGUGGAGCAGCCUGGUAGCUCGUCGGGCUCAUAACCCGAAGGUCGUCGGUUCAAAUCCGGCCCCCGCAACCA3′; E. coli synthetic tRNALys: 5′PGGGUCGUUAGCUCAGUUGGUAGAGCAGUUGACUUUUAAUCAAUUGGUCGCAGGUUCGAAUCCUGCACGACCCACCA3′; and E. coli tRNAPhe: 5′PGCCCGGAUAGCUCAGUCGGUAGAGCAGGGGAUUGAAAAUCCCCGUGUCCUUGGUUCGAUUCCGAGUCCGGGCACCA3′.
Splint Adapter Oligonucleotides
There were four distinct double-stranded splint adapters, each designed for one of the four different 3′ overhangs of E. coli tRNAs. Each was composed of two synthetic oligomers purchased from IDT. The oligonucleotide that ligates to the 3′ end of the tRNA was common to all splint adapters (Figure 1A(i), blue strand). It was composed of six ribonucleotides followed by 24 DNA nucleotides. The 30 nt sequence for this oligonucleotide was: 5′P-rGrGrCrUrUrCTTCTTGCTCTTAGGTAGTAGGTTC-3′.
The four different oligonucleotides that ligate to the 5′ end of the tRNA were identical except for the terminal base (Figure 1A(i), red strand). Each is composed of six DNA followed by 18 RNA nucleotides. The 24 nt sequences of these oligonucleotides are: 5′P-CCTAAGrArGrCrArArGrArArGrArArGrCrCrUrGrGrA-3′ (UCCA complement); 5′P-CCTAAGrArGrCrArArGrArArGrArArGrCrCrUrGrGrU-3′ (ACCA complement); 5′P-CCTAAGrArGrCrArArGrArArGrArArGrCrCrUrGrGrC-3′ (GCCA complement); and 5′P-CCTAAGrArGrCrArArGrArArGrArArGrCrCrUrGrGrG-3′ (CCCA complement).
Annealing Splint Adapters
Ten μM stocks of each double-stranded splint adapters were prepared in 10 mM Tris (pH 8.0), 50 mM NaCl, and 1 mM EDTA by adding 100 pmol of each strand in a total volume of 10 μL. The solution was heated to 75 °C for 15 s and slowly cooled to 25 °C to hybridize the adapter strands.
Library Preparation
tRNA libraries were prepared using the SQK-RNA002 (Oxford Nanopore Technologies) kit as described below.
First Ligation: tRNA to Splint Adapter
In the first ligation reaction, the splint adapter to tRNA molar ratio was 1:1.25. The tRNA sample is first heated to 95 °C for 2 min, allowed to cool for 2 min, then placed on ice for 2 min. The reaction was carried out at room temperature in a DNA LoBind tube for 45 min. Its constituents were 1× RNA ligase 2 buffer (NEB) supplemented with 5% PEG 8000, 2 mM ATP, 6.25 mM DTT, 6.25 mM MgCl2, and 0.5 units/μL T4 RNA ligase 2 (10,000 units/mL). For single tRNA isotype libraries (for tRNAfMet, tRNALys, or tRNAPhe), 16 pmol of splint adapter and 20 pmol of tRNA (∼500 ng), in a total reaction volume of 20 μL, were used. The tRNAs for these libraries have ACCA 3′ termini. The splint adapter used was the form with a UGGU overhang. Total tRNA reactions using all four splint adapters were performed using 32 pmol of adapter (8 pmol of each of the four adapters) and 40 pmol (∼1 μg) of total tRNA in a reaction volume of 40 μL. For total tRNA runs using only one of the four adapters, 16 pmol of adapter and 20 pmol (∼500 ng) of total tRNA were used in a reaction volume of 20 μL.
Gel Purification of Ligation I Product
Gel excision and purification was performed for all samples in this study. It is recommended for this procedure. Unligated and partially ligated tRNA carried forward to subsequent reactions may decrease the throughput and coverage. To perform this procedure without gel purification, see the Note Regarding Protocol without Gel Purification section.
PAGE Gel Separation and Excision of the tRNA/Splint Ligation Product
The ligation reaction was diluted to 1× with 2× RNA loading dye (NEB). Standards (low range ssRNA ladder, NEB) and 10 pmol of unligated tRNA sample were prepared in an equivalent buffer to the first ligation reaction (1× RNA ligase 2 buffer (NEB), 5% PEG 8000, 2 mM ATP, 6.25 mM DTT, 6.25 mM MgCl2) and diluted with an equal volume of 2× RNA loading dye.
The size standard, unligated tRNA, and ligation reaction samples were run on a denaturing 7 M urea/TBE PAGE gel (8%) in 1× TBE buffer for ∼1.5 h at 28 W. The gel was poststained in the dark, in a 1× TBE solution containing 2× (diluted 1 to 5000) SybrGoldTM (Life Technologies) for 20 min. The gel was transferred to a piece of saran wrap, and using UV shadowing, the gel region corresponding to the fully ligated product (∼130 nt for tRNAfMet, tRNALys, and tRNAPhe or from ∼ 120–170 nt for total tRNA) was excised.
Gel Purification of tRNA/Splint Ligation Product by Electroelution
The excised gel fragment was electroeluted in 1× TAE buffer using 3.5 kDa MWCO D-tube dialyzers (Novagen) according to the manufacturer’s protocol with minor modifications. For the ethanol precipitation step, the solution was precipitated overnight at −20 °C with 0.1× sodium acetate (pH 5.2), 1 μL of glycogen (20 mg/ml, RNA grade), and 2.5–3× ethanol. Following centrifugation at 4 °C for 30 min at 12,000g, the solution was removed. 200 μL of 80% ethanol was added to wash each pellet. After centrifugation at 4 °C for 15 min, the ethanol was removed, and the pellets were air-dried briefly. The pellets were resuspended and pooled using NF H2O. For single isotype tRNA libraries or total tRNA libraries where one of the four adapters was used, a total of 12 μL of nuclease-free (NF) H2O was used to resuspend the pellets. For total tRNA libraries where all four adapters were ligated, the resuspension volume was 24 μL of NF H2O. The concentration of the sample at this point may be quantified by nanodrop or the Qubit fluorometer RNA HS assay. The amount of material recovered and carried forward to the second ligation varied between ∼ 60 and 200 ng for single isotype tRNA libraries and total tRNA libraries using a single version of double stranded splint adapter. Approximately 500 ng was recovered in total tRNA libraries ligated to all four adapter versions. For this library, the amount of input tRNA (1 μg) was twice as much as other libraries (0.5 μg). On the order of 25% of the material by weight of the input tRNA is recovered following purification of the full length product, but this can vary substantially.
Note Regarding Protocol without Gel Purification
We recommend following the procedure in First Ligation: tRNA to Splint Adapter section with 3.2× fold greater concentration adapter than tRNA (i.e., 32 pmol adapter to 10 pmol tRNA). Then bring the remaining ligation reaction up to 40 μL with NF H2O, clean up with 1.8× RNAclean AMPure XP beads (Novagen), and elute in a final volume of 11 μL NF H2O. Next, follow the procedures starting in the Second Ligation: Splint-Ligated tRNA and RMX Adapter section.
Second Ligation: Splint-Ligated tRNA and RMX Adapter
For single isotype tRNA libraries or total tRNA libraries where one version of the splint adapters was used, the second ligation reaction was composed of 11 μL of the gel purified splint ligation product, 5 μL of 5× quick ligation reaction buffer (NEB: B6058S), 6 μL of the RMX adapter, and 3 μL of T4 DNA ligase (2,000,000 units/mL). RMX adapter is included in ONT’s RNA sequencing kits.
For total tRNA libraries previously ligated to all four types of double-stranded splint adapters, the second ligation reaction included 23 μL of the purified splint ligation products, 8 μL of 5× quick ligation reaction buffer (NEB: B6058S), 6 μL of the RMX adapter, and 3 μL of T4 DNA ligase (2,000,000 units/mL).
Ligation reactions were carried out at room temperature for 30 min. A 1.5× volume of Ampure RNAClean XP beads (Beckman-Coulter) was then added and mixed into the reaction by pipetting up and down. The tube was incubated for 15 min at room temperature with occasional light tapping and pelleted on the magnet, and the supernatant was removed. Two 150 μL washes with WSB (wash buffer in the ONT kit) were conducted, during which the pellet was vigorously resuspended by flicking and returned to the magnet to pellet, and the wash solution was removed. Following the second wash, the pellet was resuspended in 12.5 μL elution buffer (EB) and incubated for 20 min at room temperature with light tapping. Following pelleting of the beads on the magnet, the eluate was recovered to a fresh tube.
Flow Cell Quality Control, Priming the Flow Cell, and Loading the Sample on the MinION
The ONT SQK-RNA002 protocol was followed for minION flow cell (FLO-MIN-106) quality control, priming, and preparation of the sample in RNA running buffer and for loading the library onto the flow cell.
RNA Handling Practices
Care was taken to avoid introducing RNases into the samples or into stock solutions by wearing gloves at all times, using RNase-free filter tips and NF water. Pipettes, benches, and equipment were cleaned with RNase AWAY.
MinION Running Parameters
Sequencing runs were done with live base-calling off. The experiments were set for the standard 48 h period, but were typically run for <24 h due to a deterioration in functional channels over time seen using our samples. For sequencing runs where the nanopores in the flow cell became clogged (indicated by reduced functional pores on the MinKnow GUI), the experiment was restarted up to five times.
Bioinformatics
Total tRNA and Isodecoder Reference Curation
The reference used for the total tRNA experiments was designed to encompass all isoacceptor families (total tRNA reference, Figure S8).24 This included the grouping of tRNAs differing in their anticodons for all 20 amino acids, tRNASec and the initiator tRNA (tRNAfMet). Composed of 43 tRNAs, it was generated from 38 modomics sequences,3 4 sequences from gtRNAdb,27 and 1 additional MRE600 isoacceptor, tRNAArgUCG. tRNAArgUCG and isodecoder sequences were predicted for the MRE600 genome46 with tRNA-scan.31 The reference used to detect the presence of isodecoders in total tRNA reads, contained all 43 isoacceptors with 5 additional isodecoder sequences. References contained the ribonucleotide portions of the splint adapter for all analyses except for the error profile assessment, which contained only the tRNA sequences (Table 2). These references are shown in Figure S8.
Base Calling and Alignment
Base calling was done with Guppy v3.0.3 using the flipflop model. The resulting reads FASTQ file was then processed to convert all Us to Ts (further analysis software will not work without this step). Sequence alignment was accomplished using BWA-MEM v0.7.17-r1188 (parameter “-W 13 -k 6 -x ont2d”).20 The SAM files were filtered for primary alignments and for a mapping quality of ≥1 (removing nonspecific alignments) using Samtools v1.630 and visualized using the Broad Institute′s Integrated Genome Viewer (IGV) v2.4.14.21 The error model statistics were calculated using marginStats v0.126 (Table 2).
Alignment Statistics
We determined the quality of our alignments using the marginAlign subprogram maginStats.26 This program utilizes a metric called “alignment identity” which can be defined as the percent of each read (both full and partial length) that matched the reference. The equation for calculating the alignment identity is as follows:
We report median alignment identity over all aligned reads (Table 2). This is distinct from “full length aligned reads” (Table 1), which is the number or percentage of reads that cover the entire length of the tRNA reference sequence end-to-end.
Miscall Analysis
Three error models were generated with marginAlign v0.1 (EM training enabled - BWA-MEM; parameter “-W 13 -k 6 -xont2d”) from the synthetic canonical tRNAfMet, tRNALys, and tRNAPhe alignments. These error models (HMM file) were then used as input models to generate their corresponding biological tRNA alignments (under the same marginAlign settings, see usage on: https://github.com/mitenjain/marginAlign-tRNA). Alignments for the synthetic canonical tRNAs were also generated using their own error models, as a control.11 The alignment files were then filtered for primary alignments using Samtools v1.6.30 Systematic miscall identification was performed for the biological and synthetic alignments using marginCaller v0.1 (parameter “-alignmentModel= file.hmm - errorModel= file.hmm > output.vcf”) with their respective trained synthetic error models. We used a posterior probability threshold of ≥30%, which is the default for marginCaller.11
For tRNAAla1 miscall analysis, the same strategy was applied; however, the RNA-based error model was generated from canonical IVT data (Supporting Information and Methods).
Analysis of off-Target tRNAs
The procedure for identifying off-target tRNAs in commercially acquired tRNA samples (tRNAfMet, tRNALys and tRNAPhe) shown in Table S3 is diagrammed in Figure S11.
Liquid Chromatography Tandem Mass Spectrometry Confirmation of tRNAfMet Modifications
Liquid chromatography tandem mass spectrometry (LC-MS/MS) with selective ion monitoring was performed to determine if the expected RNA modifications were present in biological E. coli tRNAfMet. The samples were digested to ribonucleosides using a three enzyme protocol (similar to ref (47)). All water used was HPLC grade. 1–4 μg of tRNA was digested with 1 unit of nuclease P1 (Sigma Aldrich) in a 10 μL solution of 10 mM NH4OAc at 45 °C for 2 h. The solution was adjusted to 50 mM NH4HCO3 and 2.5 mM MgCl2 and treated with 0.004 units of phosphodiesterase 1 (Thermo Fisher Scientific) in a total volume of 20 μL at 37 °C for 3 h. 0.5 μL of 10× antarctic phosphatase buffer and 0.5 units of antarctic phosphatase (NEB) were added, and the solution was brought to 25 μL with water and incubated at 37 °C for 1 h. Mock digest solutions without substrate were used to prepare the standards to maintain uniform buffer conditions. Both samples and standards were brought up to 55 μL with 0.1% formic acid and purified on 3.5 MWCO/Nanosep 3K spin columns (Pall) for 10 min at 14,000 RCF. The flowthrough was retained for analysis. The amount of material for LC-MS/MS runs was 0.7–1.1 μg for digested tRNAfMet samples and 60 ng for each standard.
The standards included 5-methyluridine (TCI America), B-pseudouridine (TRC Canada), uridine (Sigma), 4-thiouridine (MP Biomedicals), 2′-O-methylcytidine (Alfa Aesar), and 7-methylguanosine (Sigma).
LC-MS/MS was done at the UC Santa Cruz Mass Spectrometry facility, with an LTQ-Orbitrap Velos Pro Mass Spectrometer (ThermoFisher) in positive ion mode. The column used was a Synergi 4 μm Fusion-RP 80Å C18 column (Phenomenex). Two solvents were used: 0.1% formic acid in H2O (A) and 0.1% formic acid in acetonitrile (B). The solvents gradients were: time (t) = 0–15 min: 100% A, t = 15–15.1 min 60% A, t = 15.1–20.1 min: 10% A, t = 20.1–30 min: 100% B. The flow rate of the chromatography was 200 μL/min.
The Xcalibur software (Thermo Fisher) was used to control the LC-MS/MS and for data analysis. Selective ion monitoring was performed and the following transitions3,47,48 were evidence of the presence of a modification: pseudouridine (245 > 209, 177, 155), 5-methyluridine (259 > 127), 4-thiouridine (261 > 129), 2′-O-methylcytidine (258 > 112), and 7-methylguanosine (298 > 166). For these modifications, we assessed whether the retention time for the samples was comparable to that of the standards. A commercially available dihydrouridine standard was not available, so we relied solely on published base peak (247) and daughter ion values (115) to confirm its presence.
Acknowledgments
A. Smith conceived and implemented the original tRNA ligation strategy that was modified in this study. K. Lieberman and H. Noller provided purified tRNAfMet, tRNALys, and tRNAPhe samples. Q. Zhang at the UCSC Mass Spectrometry Facility provided expert help and advice for detecting tRNA modifications using LC-MS/MS. S. Islam provided technical assistance. This work was supported by NIH grant HG010053 (M.A.) and Oxford Nanopore Technologies grant SC20130149 (M.A.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.1c06488.
Table S1: Verification of modifications in tRNAfMet with LC-MS/MS. Table S2: Systematic miscalls in purified biological tRNAfMet, tRNALys and tRNAPhe. Table S3: Off-target tRNA in biological tRNAfMet, tRNALys, and tRNAPhe sequencing experiments. Table S4: Total tRNA aligned read counts using all adapters and single NCCA complementing adapters. Table S5: Systematic miscalls in tRNAfMet, tRNALys, tRNAPhe, and tRNAAla1 from total E. coli tRNA. Figure S1: Biological tRNAfMet ligation reaction. Figure S2: LC-MS/MS analysis for pseudouridine in RNAfMet sample. Figure S3: LC-MS/MS analysis for 2′-O-methylcytidine in RNAfMet sample. Figure S4: LC-MS/MS analysis for 7-methylguanosine in RNAfMet sample. Figure S5: LC-MS/MS analysis for 5-methyluridine in RNAfMet sample. Figure S6: LC-MS/MS analysis for dihydrouridine in RNAfMet sample. Figure S7: LC-MS/MS analysis for 4-thiouracil in RNAfMet sample. Figure S8: tRNA reference sequences. Figure S9: Assessing enrichment of tRNAs using NCCA specific adapters. Figure S10: Nanopore vs RNA fingerprinting for individual tRNA abundance. Figure S11: Computational method for identification of off-target tRNAs in purified samples. Methods: IVT method for making unmodified tRNAAla1 (VGC) control (PDF)
Author Contributions
† These authors contributed equally.
A version of this manuscript was previously submitted as a preprint: Thomas, N.; Poodari, V.; Jain, M.; Olsen, H.; Akeson, M.; Abu-Shumays, R. Direct Nanopore Sequencing of Individual Full Length tRNA Strands. 2021, 2021.04.26.441285. bioRxiv. https://doi.org/10.1101/2021.04.26.441285 (accessed Apr 26, 2021).
The authors declare the following competing financial interest(s): M.A. holds shares in Oxford Nanopore Technologies (ONT). M.A. is a paid consultant to ONT. M.A. and M.J. received reimbursement for travel, accommodations, and conference fees to speak at events organized by ONT. M.A. is an inventor on 11 UC patents licensed to ONT (6,267,872, 6,465,193, 6,746,594, 6,936,433, 7,060,50, 8,500,982, 8,679,747, 9,481,908, 9,797,013, 10,059,988, and 10,081,835). M.A. received research funding from ONT.
Notes
Data availability statement: The sequencing data described in this manuscript can be publicly accessed at the European Nucleotide Archive https://www.ebi.ac.uk/ena. The study accession number is PRJEB47590.
Supplementary Material
References
- Raina M.; Ibba M. tRNAs as Regulators of Biological Processes. Front. Genet. 2014, 5, 1–14. 10.3389/fgene.2014.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Ferré-D’amaré A. R. The tRNA Elbow in Structure, Recognition and Evolution. Life 2016, 6, 3. 10.3390/life6010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccaletto P.; MacHnicka M. A.; Purta E.; Pitkowski P.; Baginski B.; Wirecki T. K.; De Crécy-Lagard V.; Ross R.; Limbach P. A.; Kotter A.; Helm M.; Bujnicki J. M. MODOMICS: A Database of RNA Modification Pathways. 2017 Update. Nucleic Acids Res. 2018, 46, D303–D307. 10.1093/nar/gkx1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T. The Expanding World of tRNA Modifications and Their Disease Relevance. Nat. Rev. Mol. Cell Biol. 2021, 22, 375–392. 10.1038/s41580-021-00342-0. [DOI] [PubMed] [Google Scholar]
- Zheng G.; Qin Y.; Clark W. C.; Dai Q.; Yi C.; He C.; Lambowitz A. M.; Pan T. Efficient and Quantitative High-Throughput tRNA Sequencing. Nat. Methods 2015, 12, 835–837. 10.1038/nmeth.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozen A. E; Quartley E.; Holmes A. D; Hrabeta-Robinson E.; Phizicky E. M; Lowe T. M ARM-Seq: AlkB-Facilitated RNA Methylation Sequencing Reveals a Complex Landscape of Modified tRNA Fragments. Nat. Methods 2015, 12, 879–884. 10.1038/nmeth.3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens A.; Rodschinka G.; Nedialkova D. D. High-Resolution Quantitative Profiling of tRNA Abundance and Modification Status in Eukaryotes by Mim-tRNAseq. Mol. Cell 2021, 81, 1802–1815. 10.1016/j.molcel.2021.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley R. Y.; Ashcroft B. A.; Farrell I.; Cooperman B. S.; Lindsay S. M.; Wanunu M. Electrophoretic Deformation of Individual tRNA Molecules Reveals Their Identity. Nano Lett. 2016, 16, 138–144. 10.1021/acs.nanolett.5b03331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Guan X.; Zhang S.; Liu Y.; Wang S.; Fan P.; Du X.; Yan S.; Zhang P.; Chen H. Y.; Li W.; Zhang D.; Huang S. Structural-Profiling of Low Molecular Weight RNAs by Nanopore Trapping/Translocation Using Mycobacterium Smegmatis Porin A. Nat. Commun. 2021, 12, 3368. 10.1038/s41467-021-23764-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garalde D. R.; Snell E. A.; Jachimowicz D.; Sipos B.; Lloyd J. H.; Bruce M.; Pantic N.; Admassu T.; James P.; Warland A.; Jordan M.; Ciccone J.; Serra S.; Keenan J.; Martin S.; McNeill L.; Wallace E. J.; Jayasinghe L.; Wright C.; Blasco J.; et al. Highly Parallel Direct RNA Sequencing on an Array of Nanopores. Nat. Methods 2018, 15, 201–206. 10.1038/nmeth.4577. [DOI] [PubMed] [Google Scholar]
- Smith A. M.; Jain M.; Mulroney L.; Garalde D. R.; Akeson M. Reading Canonical and Modified Nucleobases in 16S Ribosomal RNA Using Nanopore Native RNA Sequencing. PLoS One 2019, 14, e0216709 10.1371/journal.pone.0216709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz D. A.; Sathe S.; Einstein J. M.; Yeo G. W. Direct RNA Sequencing Enables M6A Detection in Endogenous Transcript Isoforms at Base-Specific Resolution. RNA 2020, 26, 19–28. 10.1261/rna.072785.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker M. T.; Knop K.; Sherwood A. V.; Schurch N. J.; Mackinnon K.; Gould P. D.; Hall A. J. W.; Barton G. J.; Simpson G. G. Nanopore Direct RNA Sequencing Maps the Complexity of Arabidopsis MRNA Processing and M6A Modification. eLife 2020, 9, e49658. 10.7554/eLife.49658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Begik O.; Lucas M. C.; Ramirez J. M.; Mason C. E.; Wiener D.; Schwartz S.; Mattick J. S.; Smith M. A.; Novoa E. M. Accurate Detection of M6A RNA Modifications in Native RNA Sequences. Nat. Commun. 2019, 10, 4079. 10.1038/s41467-019-11713-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. M.; Abu-Shumays R.; Akeson M.; Bernick D. L. Capture, Unfolding, and Detection of Individual tRNA Molecules Using a Nanopore Device. Front. Bioeng. Biotechnol. 2015, 3, 1–11. 10.3389/fbioe.2015.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman K. R.; Cherf G. M.; Doody M. J.; Olasagasti F.; Kolodji Y.; Akeson M. Processive Replication of Single DNA Molecules in a Nanopore Catalyzed by Phi29 DNA Polymerase. J. Am. Chem. Soc. 2010, 132, 17961–17972. 10.1021/ja1087612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherf G. M.; Lieberman K. R.; Rashid H.; Lam C. E.; Karplus K.; Akeson M. Automated Forward and Reverse Ratcheting of DNA in a Nanopore at 5-Å Precision. Nat. Biotechnol. 2012, 30, 344–348. 10.1038/nbt.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M.; Olsen H. E.; Paten B.; Akeson M. The Oxford Nanopore MinION: Delivery of Nanopore Sequencing to the Genomics Community. Genome Biol. 2016, 17, 239. 10.1186/s13059-016-1103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Verren S. E.; Van Gerven N.; Jonckheere W.; Hambley R.; Singh P.; Kilgour J.; Jordan M.; Wallace E. J.; Jayasinghe L.; Remaut H. A Dual-Constriction Biological Nanopore Resolves Homonucleotide Sequences with High Fidelity. Nat. Biotechnol. 2020, 38, 1415–1420. 10.1038/s41587-020-0570-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv (Genomics), May 26, 2013, https://arxiv.org/abs/1303.3997, 1303.3997, ver. 2.
- Robinson J. T.; Thorvaldsdóttir H.; Winckler W.; Guttman M.; Lander E. S.; Getz G.; Mesirov J. P. Integrative Genome Viewer. Nat. Biotechnol. 2011, 29, 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenjaroenpun P.; Wongsurawat T.; Wadley T. D.; Wassenaar T. M.; Liu J.; Dai Q.; Wanchai V.; Akel N. S.; Jamshidi-Parsian A.; Franco A. T.; Boysen G.; Jennings M. L.; Ussery D. W.; He C.; Nookaew I. Decoding the Epitranscriptional Landscape from Native RNA Sequences. Nucleic Acids Res. 2021, 49, e7 10.1093/nar/gkaa620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman R. E.; Tang A. D.; Tang P. S.; Jain M.; Tyson J. R.; Razaghi R.; Zuzarte P. C.; Gilpatrick T.; Payne A.; Quick J.; Sadowski N.; Holmes N.; de Jesus J. G.; Jones K. L.; Soulette C. M.; Snutch T. P.; Loman N.; Paten B.; Loose M.; Simpson J. T.; Olsen H. E.; Brooks A. N.; Akeson M.; Timp W. Nanopore Native RNA Sequencing of a Human Poly(A) Transcriptome. Nat. Methods 2019, 16, 1297–1305. 10.1038/s41592-019-0617-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begik O.; Lucas M. C.; Pryszcz L. P.; Ramirez J. M.; Medina R.; Milenkovic I.; Cruciani S.; Liu H.; Vieira H. G. S.; Sas-Chen A.; Mattick J. S.; Schwartz S.; Novoa E. M. Quantitative Profiling of Pseudouridylation Dynamics in Native RNAs with Nanopore Sequencing. Nat. Biotechnol. 2021, 39, 1–14. 10.1038/s41587-021-00915-6. [DOI] [PubMed] [Google Scholar]
- Machnicka M. A.; Olchowik A.; Grosjean H.; Bujnicki J. M. Distribution and Frequencies of Post-Transcriptional Modifications in tRNAs. RNA Biol. 2014, 11, 1619–1629. 10.4161/15476286.2014.992273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M.; Fiddes I. T.; Miga K. H.; Olsen H. E.; Paten B.; Akeson M. Improved Data Analysis for the MinION Nanopore Sequencer. Nat. Methods 2015, 12, 351–356. 10.1038/nmeth.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P. P.; Lowe T. M. GtRNAdb 2. 0: An Expanded Database of Transfer RNA Genes Identified in Complete and Draft Genomes. Nucleic Acids Res. 2016, 44, D184–D189. 10.1093/nar/gkv1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L.; Seki M. Recent Advances in the Detection of Base Modifications Using the Nanopore Sequencer. J. Hum. Genet. 2020, 65, 25–33. 10.1038/s10038-019-0679-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlan M.; Tanaka I.; Leonardi T.; de Pretis S.; Pelizzola M. Direct RNA Sequencing for the Study of Synthesis, Processing, and Degradation of Modified Transcripts. Front. Genet. 2020, 11, 1–12. 10.3389/fgene.2020.00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Handsaker B.; Wysoker A.; Fennell T.; Ruan J.; Homer N.; Marth G.; Abecasis G.; Durbin R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P. P.; Lowe T. M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol. Biol. 2019, 1962, 1–14. 10.1007/978-1-4939-9173-0_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H.; Nilsson L.; Kurland C. G. Co-Variation of tRNA Abundance and Codon Usage in Escherichia Coli at Different Growth Rates. J. Mol. Biol. 1996, 260, 649–663. 10.1006/jmbi.1996.0428. [DOI] [PubMed] [Google Scholar]
- Wei Y.; Silke J. R.; Xia X. An Improved Estimation of tRNA Expression to Better Elucidate the Coevolution between tRNA Abundance and Codon Usage in Bacteria. Sci. Rep. 2019, 9, 3184. 10.1038/s41598-019-39369-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann I. U.; Nakamura A.; O’Donoghue P.; Eiler D.; Söll D. tRNA His-Guanylyltransferase Establishes tRNA His Identity. Nucleic Acids Res. 2012, 40, 333–344. 10.1093/nar/gkr696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenbour J. M.; Pan T. Diversity of tRNA Genes in Eukaryotes. Nucleic Acids Res. 2006, 34, 6137–6146. 10.1093/nar/gkl725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres A. G.; Batlle E.; Ribas de Pouplana L. Role of tRNA Modifications in Human Diseases. Trends Mol. Med. 2014, 20, 306–314. 10.1016/j.molmed.2014.01.008. [DOI] [PubMed] [Google Scholar]
- Abbott J. A.; Francklyn C. S.; Robey-Bond S. M. Transfer RNA and Human Disease. Front. Genet. 2014, 5, 1–18. 10.3389/fgene.2014.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri I.; Kouzarides T. Role of RNA Modifications in Cancer. Nat. Rev. Cancer 2020, 20, 303–322. 10.1038/s41568-020-0253-2. [DOI] [PubMed] [Google Scholar]
- Macken W. L.; Vandrovcova J.; Hanna M. G.; Pitceathly R. D. S. Applying Genomic and Transcriptomic Advances to Mitochondrial Medicine. Nat. Rev. Neurol. 2021, 17, 215–230. 10.1038/s41582-021-00455-2. [DOI] [PubMed] [Google Scholar]
- Jain M.; Koren S.; Miga K. H.; Quick J.; Rand A. C.; Sasani T. A.; Tyson J. R.; Beggs A. D.; Dilthey A. T.; Fiddes I. T.; Malla S.; Marriott H.; Nieto T.; O’Grady J.; Olsen H. E.; Pedersen B. S.; Rhie A.; Richardson H.; Quinlan A. R.; Snutch T. P.; et al. Nanopore Sequencing and Assembly of a Human Genome with Ultra-Long Reads. Nat. Biotechnol. 2018, 36, 338–345. 10.1038/nbt.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson T.; Koskela S.; Yohannes D. A.; Partanen J.; Saavalainen P. Targeted RNA-Based Oxford Nanopore Sequencing for Typing 12 Classical HLA Genes. Front. Genet. 2021, 12, 273–283. 10.3389/fgene.2021.635601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand A. C.; Jain M.; Eizenga J. M.; Musselman-Brown A.; Olsen H. E.; Akeson M.; Paten B. Mapping DNA Methylation with High Throughput Nanopore Sequencing. Nat. Methods 2017, 14, 411–413. 10.1038/nmeth.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson J. T.; Workman R. E.; Zuzarte P. C.; David M.; Dursi L. J.; Timp W. Detecting DNA Cytosine Methylation Using Nanopore Sequencing. Nat. Methods 2017, 14, 407–410. 10.1038/nmeth.4184. [DOI] [PubMed] [Google Scholar]
- Pratanwanich P. N.; Yao F.; Chen Y.; Koh C. W. Q.; Wan Y. K.; Hendra C.; Poon P.; Goh Y. T.; Yap P. M. L.; Chooi J. Y.; Chng W. J.; Ng S. B.; Thiery A.; Goh W. S. S.; Göke J.. Identification of Differential RNA Modifications from Nanopore Direct RNA Sequencing with XPore. Nat. Biotechnol. 2021 10.1038/s41587-021-00949-w. [DOI] [PubMed] [Google Scholar]
- Schaefer M.; Kapoor U.; Jantsch M. F. Understanding RNA Modifications: The Promises and Technological Bottlenecks of the “Epitranscriptome.”. Open Biol. 2017, 7, 170077. 10.1098/rsob.170077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurylo C. M.; Alexander N.; Dass R. A.; Parks M. M.; Altman R. A.; Vincent C. T.; Mason C. E.; Blanchard S. C. Genome Sequence and Analysis of Escherichia Coli MRE600, a Colicinogenic, Nonmotile Strain That Lacks RNase i and the Type i Methyltransferase, EcoKI. Genome Biol. Evol. 2016, 8, 742–752. 10.1093/gbe/evw008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crain P. F. Preparation and Enzymatic Hydrolysis of DNA and RNA for Mass Spectrometry. Methods Enzymol. 1990, 193, 782–790. 10.1016/0076-6879(90)93450-Y. [DOI] [PubMed] [Google Scholar]
- Pomerantz S. C.; McCloskey J. A. Analysis of RNA Hydrolyzates by Liquid Chromatography-Mass Spectrometry. Methods Enzymol. 1990, 193, 796–824. 10.1016/0076-6879(90)93452-Q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.