

Abstract
Purpose:
We recently reported that the transcription factor NFATC4, in response to chemotherapy, drives cellular quiescence to increase OvCa chemoresistance. The goal of this work was to better understand the mechanisms of NFATC4 driven OvCa chemoresistance.
Experimental Design:
We used RNA-seq to identify NFATC4 mediated differential gene expression. CRISPR-Cas9 and FST neutralizing antibody were used to assess impact of loss of FST function on cell proliferation and chemoresistance. ELISA was used to quantify FST induction in patient samples and in vitro in response to chemotherapy.
Results:
We found that NFATC4 upregulates follistatin (FST) mRNA and protein expression predominantly in quiescent cells and FST is further upregulated following chemotherapy treatment. FST acts in at least a paracrine manner to induce a p-ATF2 dependent quiescent phenotype and chemoresistance in non-quiescent cells. Consistent with this, CRISPR KO of FST in OvCa cells or antibody mediated neutralization of FST sensitizes OvCa cells to chemotherapy treatment. Similarly, CRISPR KO of FST in tumors increased chemotherapy-mediated tumor eradication in an otherwise chemotherapy resistant tumor model. Suggesting a role for FST in chemoresistance in patients, FST protein in the abdominal fluid of OvCa patients significantly increases within 24 hours of chemotherapy exposure. FST levels decline to baseline levels in patients no longer receiving chemotherapy with no evidence of disease. Furthermore, elevated FST expression in patient tumors is correlated with poor progression free, post-progression free, and overall survival.
Conclusions:
FST is a novel therapeutic target to improve OvCa response to chemotherapy and potentially reduce recurrence rates.
Introduction
Ovarian cancer (OvCa) remains one of the deadliest cancers in terms of survival outcomes. This relates in large part to the fact that, while ~70% of patients obtain a complete clinical remission with a combination surgery and chemotherapy, 70% of these responders will have residual resistant cancer cells that drive relapse and ultimately the patient’s death from ovarian cancer. Consequently, identifying novel, therapeutically actionable drivers of chemoresistance is a critical need for ovarian cancer research.
The ability of cells to enter a reversible non-dividing state, termed quiescence, contributes to chemotherapy resistance (1–3). Quiescent cells have been shown to contribute therapeutic resistance in many settings and tumor types including breast, liver and pancreatic cancer, glioblastoma, leukemia and melanoma (4). This is at least in part due to chemotherapeutics primarily targeting rapidly dividing cells. However, quiescent cancer cells are enriched in the pool of cancer stem-like cells (CSC), and other mechanism of chemotherapy resistance are also possible. Quiescence is linked with tumor dormancy, another driver of disease recurrence (5, 6).
Quiescence plays a significant role in ovarian cancer and chemotherapy resistance (7–9). We recently demonstrated that the transcription factor NFATC4 (also known as NFAT3) is enriched in ovarian CSCs and, in response to chemotherapy, is translocated to the nucleus to induce transcription (3). Transcriptional activation of NFATC4 results in the induction of cellular quiescence and a chemoresistant state. However, the mechanisms of NFATC4 induced quiescence remain unclear. Here we identified 141 genes upregulated 24h, 48h and 96h after NFATC4 activation. We validate induction of mRNA expression of 6 genes, including follistatin (FST).
FST is a secreted factor known to play a critical role in ovarian biology (10). Canonically, FST functions by binding to and inactivating extracellular TGF-BMP family members to regulate their signaling (10). More recent work demonstrated that FST has a nuclear localization signal and can function in the nucleolus to (i) inhibit nucleolar RNA synthesis and (ii) restrict cell proliferation in response to glucose deprivation (11). The role of FST in ovarian cancer is poorly studied. Most FST associated studies evaluate a role for FST in fertility therapy as a potential risk factor for developing ovarian cancer (12). However, indicating a possible role as a regulator of cancer cell proliferation, FST expression in ovarian cancer is decreased with the loss of BRCA1 resulting in an increase in cellular proliferation (13). Suggesting a potential role in therapeutic resistance, Weinberg and colleagues found FST was upregulated in a mouse model of CCNE1 amplified ovarian cancer drives immunotherapy resistance (14). Interestingly, a recent GWAS study linked the FST locus to increased ovarian cancer risks in women of African ancestry (15).
We find FST is upregulated in quiescent OvCa cells and secreted following both NFATC4 induction, serum withdrawal (a known driver of quiescence) and chemotherapy exposure. FST subsequently drives a quiescent, chemotherapy resistant phenotype in otherwise non-quiescent cells in a p-ATF2 dependent manner. FST inhibition with neutralizing antibody or FST CRISPR KO resulted in increased response to chemotherapy in vitro, and FST-KO significantly increased chemotherapy response and tumor eradication in vivo. Finally, we found that FST levels in the peritoneal fluid taken from patients immediately after chemotherapy treatment increased significantly. Following completion of chemotherapy, peritoneal FST declined to baseline levels. Together, this data suggests a novel mechanism whereby inherently chemotherapy resistant quiescent cells, in response to chemotherapy, secrete FST to induce therapy resistance in neighboring cancer cells.
Methods And Materials
Cell Culture
SKOV3, CaOV3 and HEY1 lines were purchased from ATCC (2018). OVSAHO cells were gifted from Dr. Deborah Marsh from the University of Sydney. PT412, provided by Dr Geeta Mehta, was derived from an abdominal metastasis from a patient with platinum sensitive high grade serous ovarian cancer as previously described (16). Pt340 cell line was derived from an abdominal metastasis from a patient with mixed high grade serous and clear cell carcinoma (Sup Fig 1) with ARID1A mutation. Both cell lines were cultured in RPMI-10 and were used at passage ~12-14. SKOV3 cells were culture in McCoy’s Medium, CAOV3 were in DMEM, and all others were culture in RPMI-1640 media. All media contained 10%FBS 1%PennStrep. Cells were cultured at 37°C and 5% CO2. All cell lines were tested bimonthly for presence Mycoplasma.
Constructs
NFATc4 constructs were generated and validated as previously described (3). A constitutively nuclear NFATC4-YFP fusion (cNFATC4) with the phospho-regulatory domain deleted or an YFP-only control (Control-YFP) were cloned into a pGIPZ lentiviral vector and transduced into the OVSAHO HGSOC cell line. A second, phospho-specific mutant constitutively active NFATC4 (17) was also cloned into the doxycycline-inducible Tet-One expression system (Clontech) to create an inducible and constitutive NFATC4 (IcNFATC4) in the HEY1 and SKOV3 ovarian cancer cell lines. An inducible luciferase (ILuc) construct served as a control.
RNA-sequencing
HEY1 and SKOV3 cell lines expressing the IcNFATC4 or ILUC constructs were treated for 24, 48 and 96h with 100 ng/mL doxycycline and RNA extracted using the miRNeasy Mini Kit (Qiagen, 217004) according to the manufacturer’s guidelines. RNA quality was determined using Bioanalyzer chip and RNA Integrity Number (RIN) >7 was deemed acceptable. Novogene Bioinformatics Technology Co., Ltd., (Beijing, China) constructed 250-300 bp insert polyA selected cDNA library and ran the sequencing on the NextSeq platform (Illumina) according to 150 bp paired-ends protocol. 20 million reads were sequenced per sample. Pre-processing of the sequencing data was performed by Novogene. Briefly, the sequencing reads were checked for quality, adapters were trimmed, and the reads were aligned to the hg19 by Novogene. RNA seq counts summarized at gene level were used in downstream analyses. Approximately 14,000 genes with an average RNA seq counts > 10 were included in subsequent analyses. Differential gene expression was performed by the R/Bioconductor package DESeq2 (https://doi.org/10.1186/s13059-014-0550-8). Gene Ontology analysis was conducted using the R/Bioconductor package clusterProfiler (https://doi.org/10.1016/j.xinn.2021.100141).
Quantitative PCR
RNA was extracted using RNeasy Mini Kit (Qiagen) and cDNA was made using SuperScript™ III Reverse Transcription kit (Thermo Fisher). qPCR was performed using SYBR™ Green PCR Master Mix (Applied Biosystems) using standard cycling conditions. The primers used for this study are available in supplemental material (Sup Table 1).
Protein Phosphorylation Arrays
C-Series phosphorylation arrays were performed according to manufacturer recommendations (RayBiotech, Inc # AAH-TGFB-1-2). Briefly, PT412 cells were treated with or without 200 ng/mL FST for 6hr before protein was extracted quantified and normalized between samples. Protein was incubated on antibody array membranes, followed antibody TGFβ signaling detection cocktail amplification and detection per manufacturer’s instructions. Chemiluminescent readings were taken using a ChemiDoc MP imaging system (Bio-Rad Laboratories, Inc.) and densitometry data extracted using ImageJ software. Readings were normalized to the positive loading controls and membrane background signal subtracted.
CellTrace Violet
Ovarian cancer cell lines were labeled using CellTrace Violet (CTV) (Thermo Fisher Scientific, C34557) following manufactures protocol. Labeled cells were grown for 7-10 days before cells were FACS isolated for subsequent experiment; the top 5% of labeled cells were collected as CTV-bright (slowly/non-dividing) while the lowest 5% of labeled cells were considered CTV-dim (rapidly dividing).
Transwell Assay
Transwell assays used 6.5 mm membrane inserts with a 0.4μm pore size (Costar Cat#3413). CTV-bright or dim cells PT412 cells were seeded in RPMI-10 in the top/bottom chamber respectively and chamber location was reversed for replicate experiments (Sup Fig 4A) 24 hours after seeding, cells were treated with Taxol 11nM for 48 hours and then adherent cells were fixed with 4% formaldehyde, stained with crystal violet, and counted.
Cell counting
Cell counts were performed using the Moxi Z automated counting system (ORLFO Technologies) and the Cassettes Type S. In cases where cell numbers were below the accurate threshold for the Moxi Z, manual hemocytometer counts were performed with trypan blue.
Cell Cycle analysis
Cell cycle analysis was performed on OVSAHO and CaOV3 cells lines, and the primary patent sample PT340 as previously published (18). Briefly, cells were grown at low seeding densities in 0 or 200 ng/mL FST for 48 h. Cells were fixed in 70% ethanol, incubated at −20°C for 20 minutes, before being treated with 0.1 ug/mL RNAse A for 1h at 37° C and 1 ug/mL propidium iodide (PI) for 20 minutes and run on the Cytoflex flow cytometer (Beckman Coulter) and at least 10,000 events were recorded. Cell cycle peaks analyzed using FlowJo™ v10.6.2.
Annexin V/PI staining
For apoptosis detection via Annexin V staining, CaOV3 and OVSAHO cells were grown in 0 or 200 ng/mL FST for 72 hrs. Cells were stained with the Annexin-V FITC apoptosis kit (BD Biosciences) according to the manufacturer’s instructions and at least 10,000 events were analyzed on the Cytoflex flow cytometer (Beckman Coulter). The percentage of Annexin V+, PI+, Annexin V+/PI+, and Annexin V−/PI− cells was quantified.
Western Blotting
Western blotting was performed as described (19). Briefly, OVSAHO and HEY1 cells expressing the cNFATC4/YFP or IcNFATC4/ILUC constructs were grown to 80% confluence with or without Dox treatment for 72h. Protein was lysed in RIPA buffer containing Halt Protease Inhibitor Cocktail (Thermo Fisher), sonicated, and quantified using Pierce BCA Protein Assay Kit. 30 μg of protein was run on a 4–12% NuPAGE SDS gel (Thermo Fisher) and transferred to a PVDF membrane (Thermo Fisher). Membranes were incubated overnight with 1:1000 anti-FST (Abcam, ab64490), 1:1000 anti-NFATC4 (Santa Cruz, sc-271597) or 1:5000 anti-GAPDH (Proteintech cat# 60004-1-1g) antibodies in 5% skim milk. Membranes were washed in TBST, then incubated for 1h with 1:10,000 anti-mouse HRP or anti-rabbit HRP (Cell Signaling) and rewashed with TBST. Visualization was performed with ECL Plus Western Blotting Substrate (Pierce). Densitometry and quantification were subsequently performed with ImageJ.
GolgiPlug
GolgiPlug experiments were used to evaluate FST protein levels following NFATC4 overexpression as due to its tendency to be secrete and hence make difficult to interpret western data. HEY1 and OVSAHO cell lines expressing the cNFATC4/YFP or IcNFATC4/ILUC constructs were treated with or without 1:1000 BD GolgiPlug Protein Transport Inhibitor (BD Biosciences) for 4h. Protein was harvested, and Western Blotting performed as described above.
Fucci cell cycle reporters
HEY1 cells expressing the p27-mVenus and CDT1-mCherry FUCCI cell cycle reporter constructs (20) were treated with or without 200 ng/mL FST and flow cytometry analysis was performed.
FST ELISA
To investigate FST secretion from cell lines and patients a FST ELISA was performed. Ascites samples from HGSOC patients who had or had not received primary chemotherapy was collected. HEY1 cells expressing IcNFATC4/ILUC constructs were treated with Dox for 72h, and culture media was collected. Culture media was also collected from CaOV3 and OVSAHO cells treated with or without Taxol and Cisplatin for 72h. Patient ascites samples were diluted 1:2, while cell culture media was undiluted. FST (FST) Human ELISA Kit (Thermo Fisher, #EHFST), was used to quantify FST levels using the manufactures instructions and an overnight antibody incubation. The ELISA plate was then run-on Infinite M plex, Tecan LTD plate reader and FST concentration were calculated for each sample using a standard curve.
Patient samples
All patient samples were collected as part of a University of Pittsburgh IRB approved protocol. Ascites samples and IP samples from cohort 1 were collected as part of standard of care therapy under tissue collection protocol UPCI 07-058. Pre/post chemo treatment samples were collected as part of a clinical trial (NCT03734692). All patients had chemonaive or platinum sensitive HGSOC. Prior to chemotherapy treatment 50 ml of saline was injected via intraperitoneal port and after ~5 minutes, 30-50 ml of fluid was retrieved. For cohort 2, patients treated with IP cisplatin chemotherapy Day 1, peritoneal washes were taken (as above) on day 2 prior to intraperitoneal therapy with Rintatolimod (TLR3 agonist) and IV therapy with pembrolizumab D3 (21).
Generation of KO Cells through CRISPR
The AMAXA 4D system was used to nucleoporate SKOV3 cells with 30uM of sgRNA FST-53482920 (UCUUGUACAGGACCUGGCAG) or (GUUCGGUCUUGUACAGGACC), 3.22 mg/mL of Cas9, for mock transfection water was used instead of sgRNA (Lonza Kit cat#V4XP-3032). SKOV3 cells were nucleoporated with pre-settings following the manufacturer’s instruction. The transfected cells were cultured under the standard McCoy’s media 5 days to assess survival. FST knockout was confirmed using western blotting (Fig 6A). The SgRNA sequences were and FST-53482926 sgRNA was designed and purchased through Synthego.
Figure 6. FST promotes chemoresistance and reduces apoptosis in vivo.
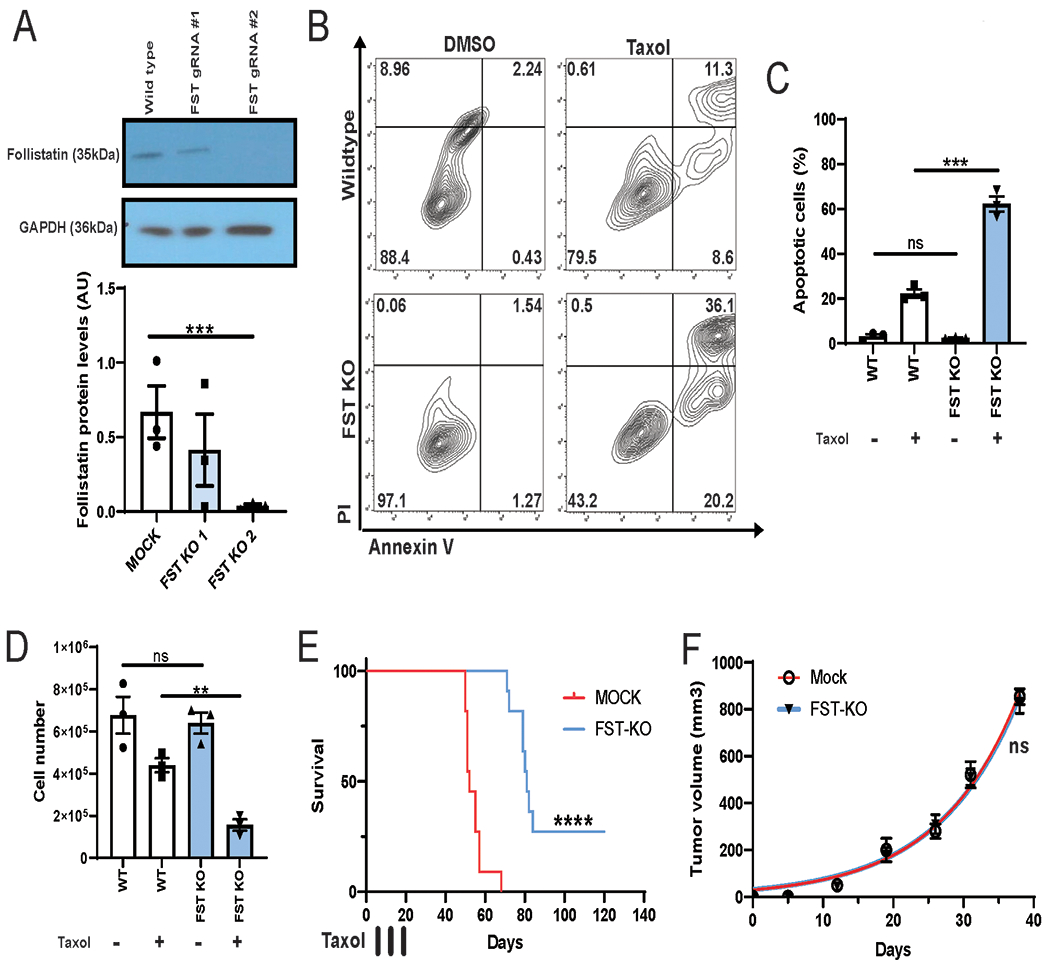
A. Western blotting for FST in SKOV3 cells expressing CRISPR-Cas9 and FST gRNA#1 or gRNA #2. B and C. AnnexinV/PI apoptosis assay and total apoptotic cell number for wild type (WT) and SKOV3-FST-KO cells treated with or without 4 nM taxol. D. Cell counts of SKOV3-FST-KO cells treated with or without 4 nM taxol. E. Survival plots of NSG mice (N=10) injected with 150,000 FST-KO SKOV3 or control cells into the intraperitoneal cavity. Mice received 3 doses of 10mg/kg doses of intraperitoneal Paclitaxel at days 7, 14 and 21. F. Tumor volumes of xenografted SKOV3 FST-KO or Mock cells over 38 days (N=3). **P<0.01, ***P<0.001, ****P<0.0001.
In vivo model
Six weeks old female NSG (NOD.Cg-Prkdcscid ) mice were acquired from The Jackson Laboratory (Bar Harbor, ME, USA) and allowed to acclimate one week in the animal facility before any intervention was initiated. All experimental procedures were conducted with the guidelines set by The Institute for Laboratory Animal Research (ILAR) of the National Academy of Sciences. For the FST and Ki67 IHC staining experiment, 500,000 OVSAHO cells were xenografted into mice and grown until reaching ∼1000 mm3 at which point mice were treated with DMSO or 30mg/kg Taxol. 72h after treatment mice were euthanized and where tumors were paraffin embedded and processed for IHC.
For the FST-KO in vivo experiments, 300,000 control or FST-KO SKOV3 cells were injected IP (n=10 animals/treatment group) in PBS. One week after tumor injection, animals were treated with Paclitaxel at a dose of 10mg/kg once a week for three weeks. Mouse weight was followed once a week. Criteria for euthanasia was as previously published (22), briefly: (i) Increase of 1cm of abdominal perimeter and/or (ii) changes in physiology and behavior (body weight, external ph or physical appearance) (iii) lower response to stimulation (inability to reach food and water, lethargy or decreased mental awareness, labored breathing or inability to remain upright).
Immunohistochemistry
Fresh tumors were harvested in linear growth phase (∼1000 mm3) and embedded in formalin. Then, they were processed and embedded in paraffin 5μm thickness, as in previously described protocols (23). Primary anti-rabbit Ki67 (1:500, Abcam #ab15580) and anti-rabbit FST (1:200, Abcam #ab64490) were incubated for overnight hours at 4°C. Subsequently, slides were incubated with a ready-to-use peroxidase-labeled anti-mouse HRP or anti-rabbit AP (Cell Signaling). Signal was visualized with Forangi Blue/HRP (to label Ki67 in blue) solution according to the manufacturer’s instructions and counterstained with Fast Red/AP (to label FST in red) Substrate Kit (Abcam, ab64254). Ki67 and FST immunohistochemistry (IHC) were performed on 6 to 8 independent sections of 3 independent OVSAHO cell–derived tumors (Paclitaxel or No Paclitaxel treatment, seven days prior to collection) derived tumors. Images were captured on an Olympus BX41 fluorescent microscope with a 12 MB digital camera at 16-bit depth/300 dpi. Total stain area/low power field (×100), as defined by pixel area (X:Y 1:1).
Statistical analysis and software
Statistically analysis was conducted using GraphPad Prism (8.0.2) and http://vassarstats.net/. All data was analyzed using two-tailed Student’s t-tests or one-way ANOVA. A minimum of 3 replicate experiments (n≥3) were used for statistical analysis. Data was plotted mean ± SEM. Biorender (Created with BioRender.com) was used to produce schematics.
Data Availability Statement
The data analyzed in this study were obtained from Gene Expression Omnibus (GEO) is GSE210439. Other data generated in this study are available upon request from the corresponding author.
Results
Identifying factors induced by NFATC4
We have previously shown that in NFATC4 transcriptional activity is activated in ovarian cancer cells in response to chemotherapy to drive a quiescent chemoresistant state (3). To identify NFATC4 transcription targets of, we performed RNA-sequencing on SKOV3 and HEY1 ovarian cell lines with an inducible constitutive NFATC4 expression construct (IcNFATC4) or luciferases control (ILUC) (3) 24h, 48h and 96h after induction (Sup Fig 2A). As expected, HEY and SKOV3 cells exhibit cell-type-specific gene expression profiles that are similar in ILUC (negative controls) across all time points (Fig 1A–B/Sup Fig 2B). Upon activation of NFACT4, while maintaining overall cell-type specific expression profiles, we saw significant changes in gene expression in both cell lines (Fig 1A–B/Sup Fig 2B–C). A total of 141 genes were found to be significantly differentially expressed at all time points in both cell lines upon NFATC4 induction (Fig 1C).
Figure 1. RNA-sequencing of SKOV3 and HEY1 cells overexpressing NFATC4.
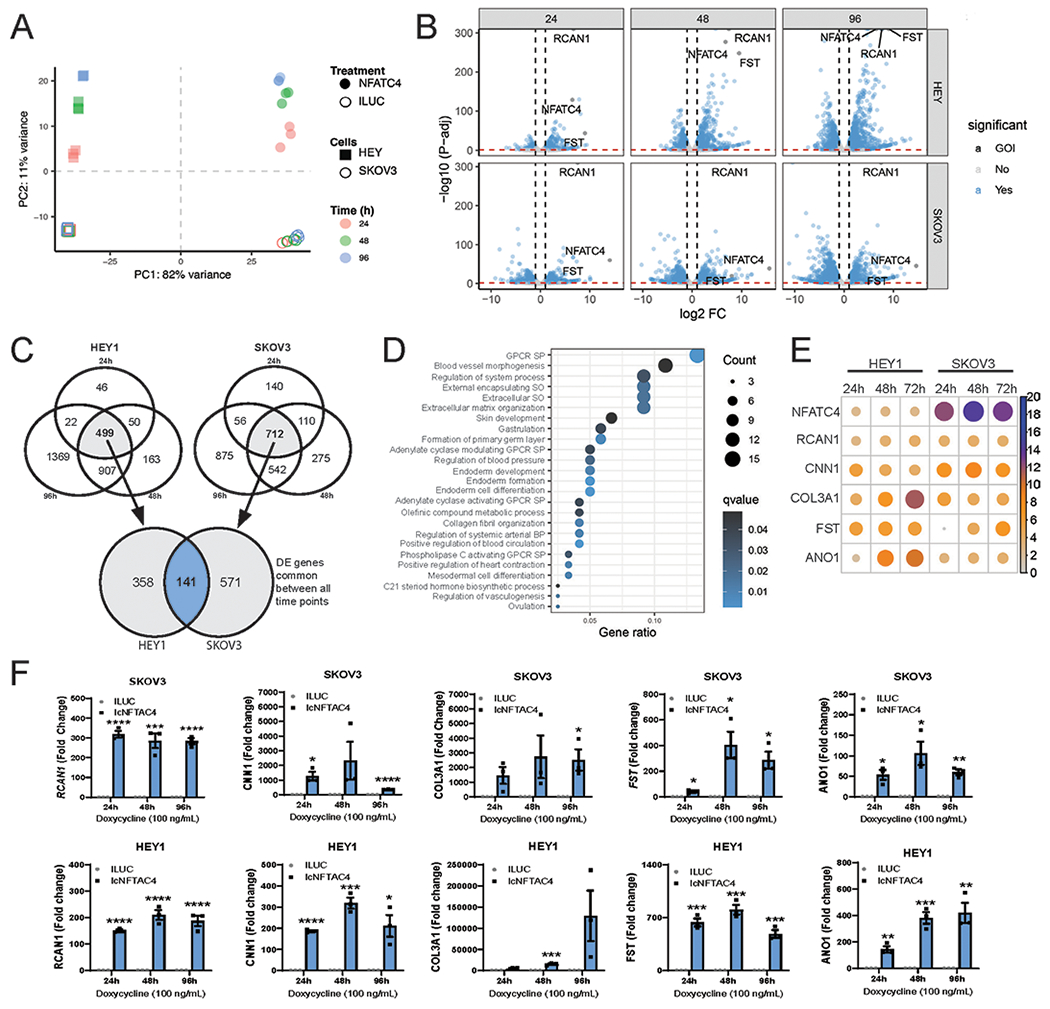
HEY1 and SKOV3 cells expressing NFATC4, or luciferase control were treated with 100 ng/mL doxycycline for 24h, 48h and 96h before RNA sequencing. A. Principal component analysis (PCA) of HEY1 and SKOV3 RNA-seq data sets. B. Volcano plots of differential gene expression between SKOV3 and HEY1 cell lines expressing NFATC4 compared to the Luciferases control. C. Venn Diagram demonstrating the number of common genes upregulated in each cell line at each time point compared with the luciferase control. D. Gene Ontology analysis of the top upregulated pathways following NFACT4 activation. G-protein couple receptor (GPCR) signaling pathway (SP) structural organization (SO) E. Bubble plot displaying the top 6 upregulated genes (log2 fold change) F. qPCR validation of the NFATC4 and the top five most upregulated mRNAa (RCAN1, CNN1, COL3A1, FST and ANO1) in the NFATC4 cells vs Luciferases control. **P<0.01, ***P<0.001.
Gene Ontology analysis identified 20 upregulated pathways (Fig 1D). Consistent with a role for NFATC4 expression in CSC and in regulating cell fate, changes in developmental and differentiation pathways were linked with NFATC4 expression (Fig 1D). The Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) analysis of the 141 genes (Sup Fig 2E) identified development, extracellular matrix, and secretion as the top pathways.
Evaluation of specific differentially expressed genes indicated that, as expected, NFATC4 was significantly upregulated gene in all 6 conditions (Fig 1E). RCAN1, a known NFATC4 target gene, was also highly upregulated. The top 6 most upregulated mRNA across all time points were NFATC4, RCAN1, CNN1, COL3A1, FST and ANO1 (Fig 1E). Consistent with our previous work (3), we also identified a decrease in MYC expression in both cell lines at 24h (Sup Fig 2D); however, MYC increased expression at 96h in SKOV3, suggesting alternate cell specific downstream signaling. qRT-PCR confirmed mRNA upregulation of RCAN1, CNN1, COL3A1, FST and ANO1 (Fig 1F); RCAN1, CNN1, ANO1 and FST were all expressed 24 hours after NFATC4 induction with expression peaked 48h post NFATC4 activation in both cell lines. COL3A1 mRNA was similarly induced in SKOV3 cells, but in HEY1 cells, induction was not seen until 48h, peaking at 96h.
NFATC4 increases FST protein levels and secretion
Given that FST plays a critical role in ovarian development and function (10) and has been found to be elevated in the serum of patients with ovarian cancer (12), we decided to further investigate the function of FST downstream of NFATC4. To confirm that an increase in FST mRNA expression translated to an increase in FST protein levels, we treated HEY1-IcNFATC4 or HEY1-ILUC control cells with doxycycline for 72h, in the presence or absence of GolgiPlug (to inhibit protein secretion), and then performed western blotting. While FST was not detectable in ILUC control cells, FST was clearly detectable in doxycycline induced, GolgiPlug treated NFATC4 cells (Fig 2A, p<0.05). To confirm this response in a high grade serous ovarian carcinoma (HGSOC) cell line, we transfected OVSAHO cell with a constitutively active cNFATC4 or control-YFP and treated with or without GolgiPlug. OVSAHO cells showed baseline FST protein expression in YFP transfected control cells, however expression of cNFATC4 resulted in a ~7-fold induction of FST compared to controls (Fig 2B).
Figure 2. NFATC4 overexpression, result in a significant increase in FST protein levels and secretion.
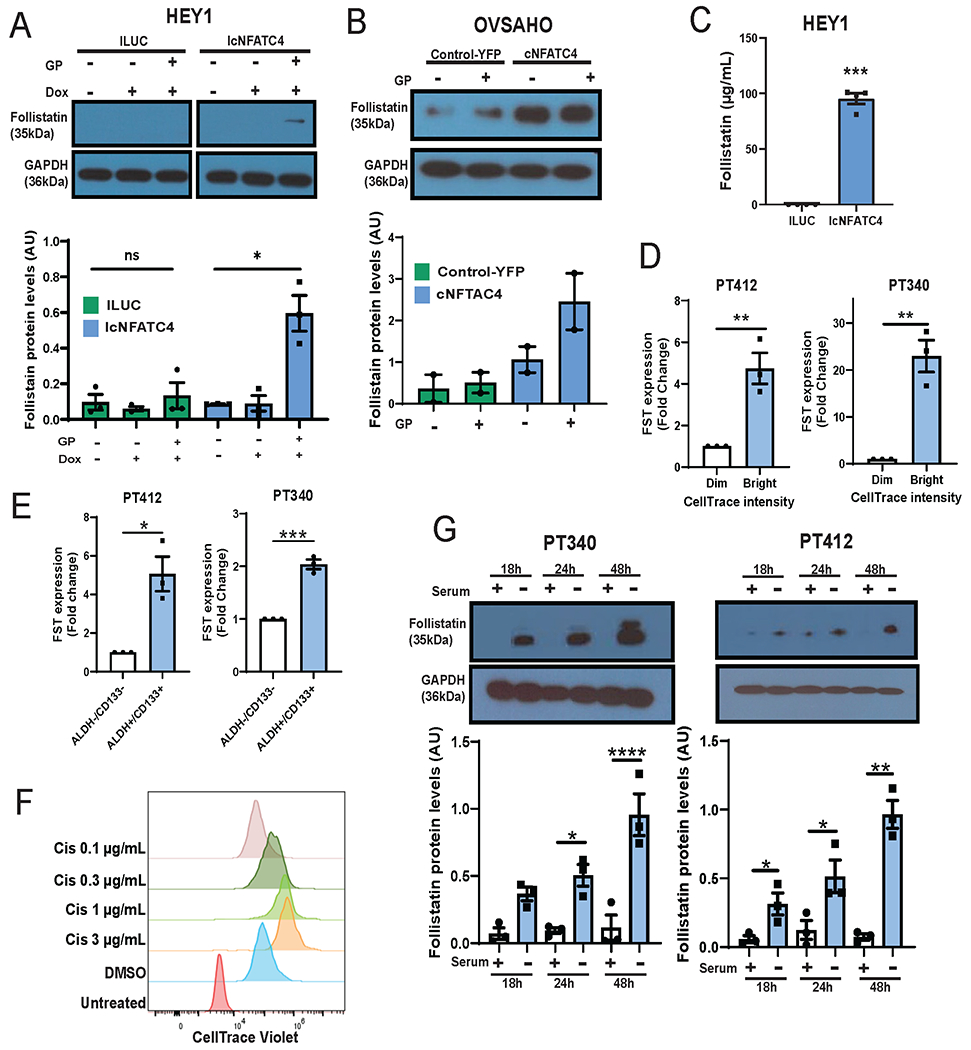
Immunoblotting for NFAT3 and FST protein levels in HEY1 (A) and OVSAHO (B) cells expressing an inducible or transient NFATC4 expression construct, respectively. Cells were treated with GolgiPlug (GP) for 4 h to retain secreted proteins. C. ELISA of FST section from HEY1 cells expressing ILUC or NFATC4 treated with Dox for 72h. D. FST mRNA expression in primary ovarian cancer cell lines (PT340, PT412) sorted based on CellTrace Violet retention. Bright=slowly diving, Dim=rapidly dividing. E. FST mRNA expression in PT340 and PT412 cells FACS sorted for CD133+/ALDH+ CSC populations and CD133−/ALDH− bulk cells. F. Flow cytometry histograms of CellTrace Violet retention in PT340 cell treated with various concentrations of cisplatin. G. FST protein levels in PT340 and PT412 cells grown in the presence or absence of serum for 18h, 24h or 48h. *P<0.05, **P<0.01, ***P<0.001 ****P<0.0001.
To confirm secretion of FST following NFATC4 activation, we once again treated HEY1 cells expressing IcNFATC4 or ILUC constructs with doxycycline for 72h and then performed a FST ELISA on the conditioned media. Conditioned media from NFATC4 expressing cell had 95 times higher FST levels than conditioned media from the ILUC control cells (Fig 2C, P<0.001).
FST is in enriched in quiescent cells and acts as a paracrine signaling factor to suppress cell proliferation
We previously demonstrated that NFATC4, whose expression drives FST expression, is enriched in slowly dividing CSC (3). To confirm FST is expressed in quiescent HGSOC, we performed vital dye labelling using CellTrace Violet (CTV) of two primary patient cell lines, PT340 and PT412. 10 days after labeling, we FACS isolated the slowly/non-dividing (CTV-bright) and rapidly dividing (CTV-dim) cells. Slowly dividing cells had 4.4- and 23-fold higher FST expression in PT412 (P<0.01) and PT340 (P<0.01) cells, respectively (Fig 2D). Consistent with our prior reports that primary human CD133+/ALDH+ HGSOC CSC are more slowly proliferating (24), FST expression is significant enriched in CD133+/ALDH+ ovarian CSCs, compared to the CD133−/ALDH− bulk cell population (PT412, P<0.05; PT340, P<0.001) (Fig 2E). Furthermore, cisplatin treatment resulted in a dose dependent enrichment of slowly/non-dividing (bright) quiescent cells, suggesting quiescent cells are chemoresistant (Fig 2F). As an independent means to evaluate the induction of quiescence on FST expression, we performed serum withdrawal studies; serum withdrawal is one of the first mechanism reported to induce quiescence (25). Induction of quiescence through serum withdrawal also resulted in a significant increase in FST protein levels (Fig 2G).
To determine whether FST secretion contributed to NFATC4 driven quiescence, we next treated HEY1, OVSAHO and CaOV3 OvCa cells with increasing concentrations of recombinant FST for 72h and evaluated total cell counts. FST treatment led to a dose dependent reduction in cell numbers, with a ~30% reduction total cell numbers in all cell lines at the highest FST dose (Fig 3A). Similarly, FST treatment of the two primary HGSOC patient samples also resulted in a significant decrease in cell number (PT340, P<0.001; PT412, P<0.0001) (Sup Fig 3A). Confirming the specificity of FST effect, FST driven reductions in cell growth were reversed with FST neutralizing antibody (Fig 3B).
Figure 3. FST decreases bulk cell proliferation.
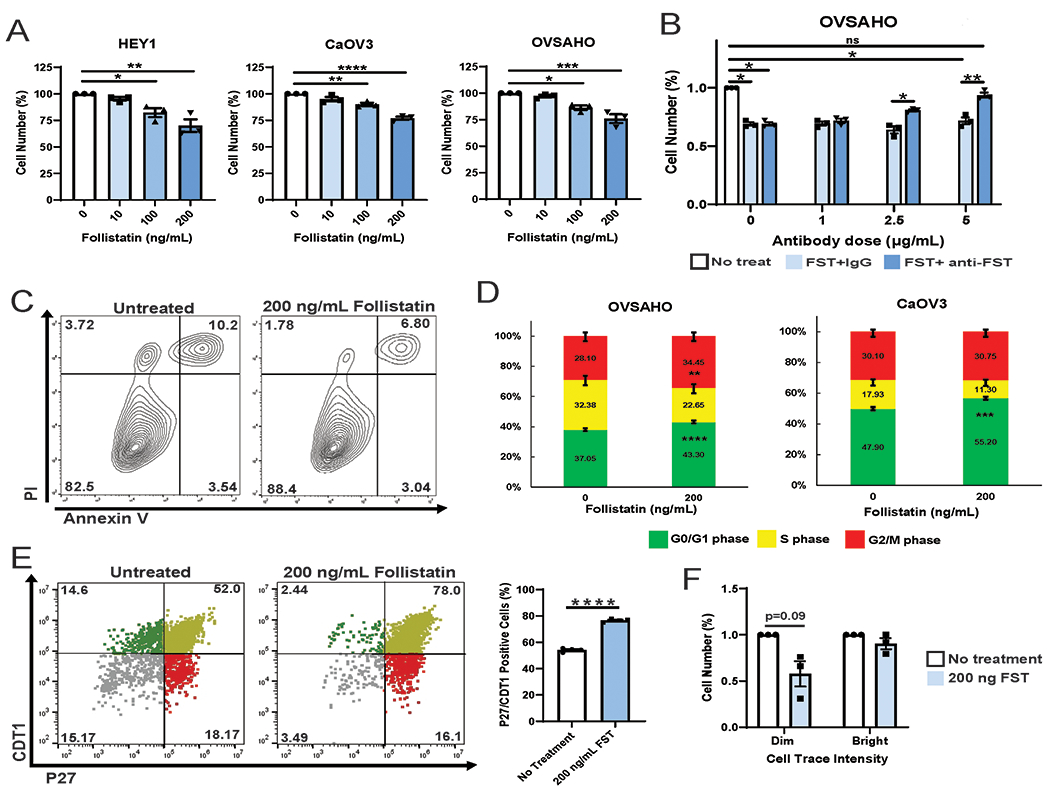
A. Cell counts of HEY1, CaOV3 and OVSAHO cell lines treated for 72h with 0 ng/mL 10 ng/mL, 100 ng/mL, or 200 ng/mL FST. B. Cell counts of OVSAHO cells treated with indicated concentrations of FST, IgG and anti-FST neutralizing antibody. C. Cell viability assay of CaOV3 and OVSAHO cells treated with vehicle or 200ng/mL FST for 72h. D. Cell cycle analysis of OVSAHO and CaOV3 cells treated with 0 ng/mL or 200 ng/mL FST for 48h. E. HEY1 cells expressing the p27-venus and CDT1-mCherry FUCCI cell cycle reporter constructs were treated with or without 200 ng/mL FST and flow cytometry analysis was performed and the percentage of p27 and CDT1 double positive cells was graphed. F. Average fold change in cell counts of PT340 and PT412 (pooled results) CellTrace Violet sorted Bright (slowly dividing), and Dim (rapidly dividing) cell populations treated with vehicle or 200 ng FST. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
To confirm the reduction in cell numbers is related to reduced proliferation and not related to increased cell death, we assessed the impact of FST on cell viability. CaOV3 and OVSAHO cells were treated with 200 ng/mL FST for 72h, and cell viability was assessed with Annexin V/PI staining. FST did not increase cell death in either cell line (Fig 3C). Consistent with reduced proliferation, cell cycle analysis on CaOV3 and OVSAHO treated with FST 48h demonstrated an increase in the percentage of cells in G0/G1 (P<0.001) (Fig 3D). Suggesting that FST may be increasing the number of cells in a quiescent state, FST treatment of HEY1 cells expressing the FUCCI cell cycle reporters (p27-mVenus/CDT1-mCherry) demonstrated an enrichment in the quiescent p27/CDT1 double positive population (P<0.001) (Fig 3E). To determine if reduce proliferation rates were a result of FST action on proliferating or quiescent cells, we treated isolated CTV-bright and CTV-dim cells with FST (Fig 3F). Only the rapidly dividing CTV-dim cells decreased cell proliferation, while the slowly dividing cells were unaffected. Together this data suggests FST contributes to NFATC4 driven quiescence, acting in a paracrine manner to reduce cell proliferation of rapidly dividing bulk ovarian cancer cells without affecting cell viability.
FST is upregulated and secreted in response to chemotherapy.
To investigate if FST could be induced in response to chemotherapy and drive chemotherapy resistance, we treated OVSAHO and CaOV3 cells with either cisplatin or Taxol and examined FST mRNA and protein expression. Treatment of cells with cisplatin or taxol resulted in a significant, 10-72-fold, increase in FST mRNA (Fig 4A, C). Suggesting that this is at least partially NFAT dependent, treatment with the NFAT selective peptide inhibitor VIVIT resulted is a significantly abrogated cisplatin mediated FST induction (Fig 4B, P<0.05). Cisplatin and Taxol treatment also resulted in a significant increase in FST protein levels as determined by both western blot (Fig 4D, P<0.05), and confirming secretion, ELISA which showed 4-50-fold increases in FST secretion (Fig 4E, CaOV3/OVSAHO cisplatin P<0.01, Fig 4F, CaOV3/OVSAHO Taxol P<0.05). Indicating quiescent cells are a major source of the FST, cisplatin treatment of slowly dividing CTV-bright cells vs. proliferating CTV-dim cells, demonstrated FST is secretion ~3-fold higher in the quiescent/bright cells (Fig 4G).
Figure 4. Chemotherapy increases FST expression and secretion preferentially in quiescent cancer cells.
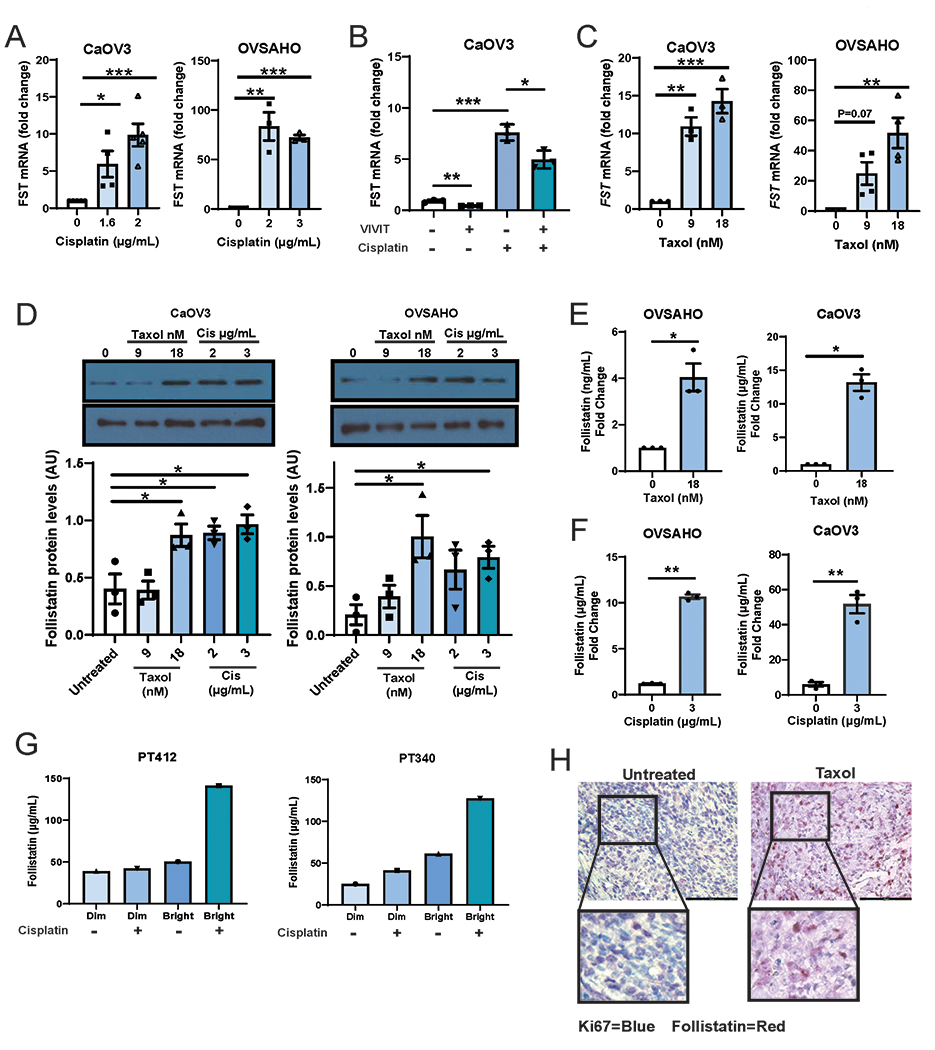
qPCR of FST expression in CaOV3 and OVSAHO cells treated with the indicated doses of (A) cisplatin or (C) Taxol. B. qPCR of FST mRNA expression in CaOV3 cells are treated with cisplatin (2μg/mL) without or with VIVIT (an NFAT inhibitor). C. Immunoblotting and densitometry of FST protein levels in CaOV3 and OVSAHO cells treated with various doses of cisplatin or Taxol. ELISA for FST protein in CaOV3 and OVSAHO cell culture media following treatment with (D) Taxol or (E) Cisplatin. F. FST ELISA of CellTrace Violet sorted Dim (rapidly dividing) and Bright (slowly diving) patient cell populations treated +/− cisplatin. G. IHC images of HGSOC mouse xenografts treated with or without taxol and stained with both Ki67 (Forangi Blue) and FST (Fast Red). *P<0.05, **P<0.01, ***P<0.001, ****P<0.001.
To confirm FST induction by chemotherapy in quiescent cells in vivo, we treated established OVSAHO xenografts with taxol chemotherapy and then resected tumors after therapy. Immunohistochemistry of the xenografts demonstrated untreated control tumors had high levels of Ki67 staining and low FST. In contrast, taxol treated xenografts demonstrated elevated levels of FST with a reduction in Ki67 staining (Fig 4H). Quantification indicates a >3-fold increase in FST+ cell bodies in taxol treated tumors (Sup Fig 3B). Furthermore, FST expressing cells were generally Ki67 negative/low. Together, this data shows that chemotherapy results in an increase in FST expression and secretion in quiescent cells in vitro and in vivo.
FST is increases OvCa chemotherapy resistance via ATF2 signaling
We next assessed if FST promoted chemotherapy resistance. CaOV3 and OVSAHO cells co-treated with Taxol or cisplatin and 200 ng/mL FST demonstrated a significant chemotherapy resistance in the presence of FST (Fig 5A). Cells treated with taxol in the presence of FST demonstrated a 1.5 (CaOV3, P<0.01) and 2.5 (OVSAHO, P<0.05) fold increase in cell viability compared to taxol alone. While cells treated with cisplatin in the presence of FST demonstrated 1.9 (CaOV3, P<0.01) and 2.2 (OVSAHO, P<0.01) fold increase in cell viability compared to cisplatin alone.
Figure 5. FST promotes chemoresistance and reduces apoptosis through the activation of ATF2.
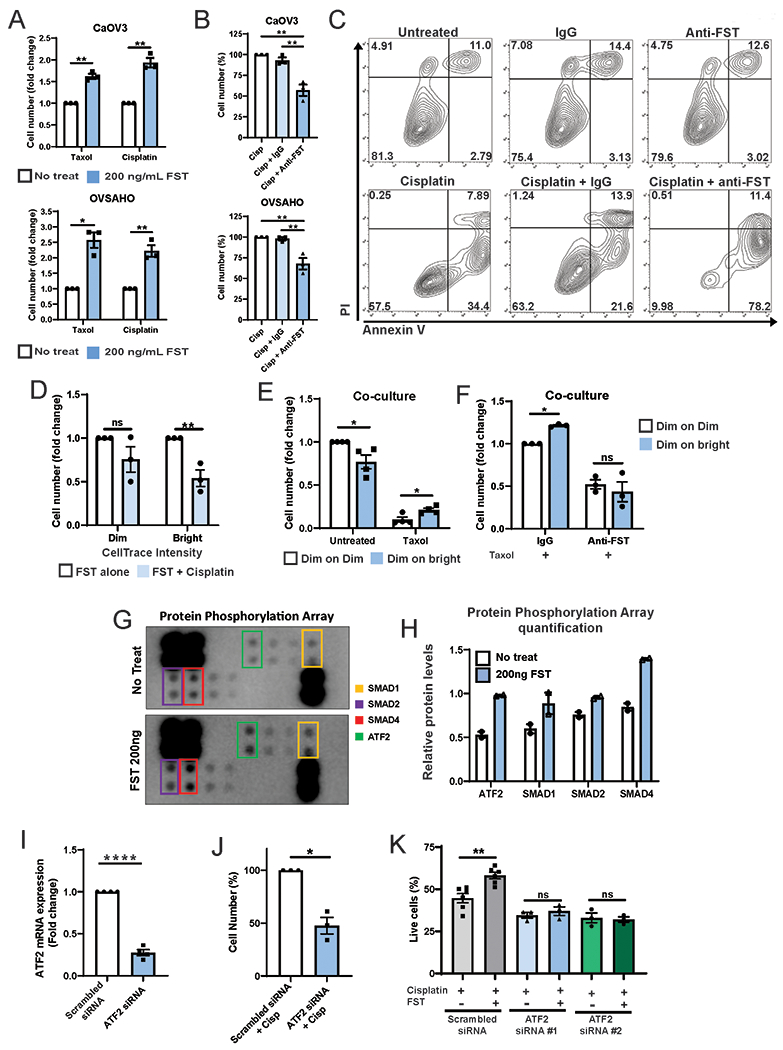
A. Cell counts of CaOV3 and OVSAHO cell lines co-treated with cisplatin without and with 200 ng/mL FST for 72h. B. Cell counts of CaOV3 and OVSAHO cell lines co-treated with cisplatin and 4ug/mL IgG or FST neutralizing antibody (FST-Nab). C. AnnexinV/PI apoptosis assay of OVSAHO cells treated with Cisplatin without and with IgG or FST-Nab. D. Fold change in cell number of CellTrace Violet sorted Dim (rapidly dividing) and Bright (slowly diving) cells treated with FST +/− Cisplatin. E Normalized CTV-dim cell number from transwell co-cultures (CTV-dim with CTV-dim (Dim:Dim) or CTV-dim with CTV-bright (Dim:Bright) Pt412 cells) (i) treated with or without taxol and (ii) with taxol and either IgG or FST-NAb. Data expressed as fold change in cell number. G and H. TGFβ Protein Phosphorylation Array and densitometry of cell lysates from PT412 cells treated with 200 ng/mL FST. I. ATF2 mRNA expression in siRNA knockdown cells vs scrambled control (pooled PT340 n=2, PT412 n=2). J. Cell counts of PT340 cells treated with cisplatin and ATF2 siRNA or scrambled siRNA control. K. Viable cell number of PT412 cells treated with scrambled siRNA or knocked down with 2 individual ATF2 siRNAs and treated with cisplatin +/− FST. *P<0.05, **P<0.01, ****P<0.0001, ns = not significant.
We next used FST neutralizing antibody (FST-NAb, R&D systems, Cat #AF669) to assess if neutralization of endogenous FST secreted in response to chemotherapy could enhance chemotherapy sensitivity. A titration assay indicated that 4 ug/mL of FST-NAb could neutralize >80% of the inhibitory effects of FST on cell growth (Sup Fig 3C). Using this dose, OVSAHO and CaOV3 cells were treated with cisplatin alone or in combination with an IgG control antibody or the anti-FST for 72h. Anti-FST significantly increased cell sensitivity to cisplatin in both CaOV3 and OVSAHO (P<0.01) (Fig 5B). AnnexinV/PI apoptosis analysis of cell viability confirmed an ~6-fold increased cisplatin induced apoptotic cell death in the presence of anti-FST (Fig 5C). To determine if FST induced chemoresistance was acting primarily on the quiescent or proliferating cells, we isolated fast growing CTV-dim and quiescent CTV-bright cells and treated them with FST +/− cisplatin. Dim cells were significantly more resistant to cisplatin in the presence FST compared with bright/quiescent cells (Fig 5D, P<0.01).
To demonstrate that quiescent cells could act in a paracrine manner to promote chemotherapy resistance, we tested chemotherapy response of CTV-dim and CTV-bright cells in co-culture using a transwell assay. We co-cultured (i) CTV-dim cells with CTV-bright cells or (ii) CTV-dim cells with CTV-dim cells (as a control), then treated with taxol (Sup Fig 4A). Compared to CTV-dim cells cultured with CTV-dim cells, CTV-dim cells cultured in the presence of CTV-bright cells were significantly more resistant to taxol (Fig 5Ei, p<0.05). To determine if this was driven by FST we repeated the experiment in the presence of FST-Nab or IgG control. As expected, IgG control treated CTV-dim cells were more resistant to taxol when co-cultured with CTV-bright (vs. CTV-dim) cells. (Fig 5Eii, p<0.05). However, FST-Nab treatment resulted in similar survival in both the CTV-dim:CTV-dim and CTV-dim:CTV-bright culture conditions (Fig 5Eii). This data indicates FST produced by quiescent cells can act in a paracrine manner to restrict the growth of nearby proliferating cells and increase their chemoresistance.
To determine what downstream signaling pathways may be regulating the FST driven chemoresistance, we conducted a TGFβ Protein Phosphorylation Array on PT412 cells treated with or without 200 ng of recombinant FST. FST treated cells had a significant enrichment in p-SMAD family members and p-ATF2 (Activating transcription factor-2) (Fig 5G–H). ATF2 has been reported to regulate chemotherapy and radiotherapy resistance in a range of malignancies, including breast, melanoma, and head and neck squamous cell carcinoma (26–28). Consequently, we knocked down ATF2 (Fig 5I, Sup Fig 4B) and assessed cell response to cisplatin. ATF2 knockdown cells we significantly more sensitive to cisplatin treatment (Fig 5J, p<0.05). Furthermore, when ATF2 siRNA knockdown cells were treated with FST and cisplatin, FST did not promote chemotherapy resistance when ATF2 was knocked down (Fig 5I, siRNA #1 P<0.05, siRNA #2, Sup Fig 4B). Consistent with previous finding, co-treatment of, siRNA scrambled cells with FST and cisplatin resulted in enhance chemotherapy resistance compared with cisplatin alone (Fig 5K, P<0.01 Sup Fig 4C P<0.05), however, the protective effects of FST on cisplatin treated cells was lost when ATF2 was knocked down (Fig 5K, ATF2 siRNA P=ns, Sup Fig 4C ATF2 siRNA P=ns).
Loss of FST activity increases OvCa chemotherapy resistance in vitro and in vivo
To confirm the impact of FST on chemotherapy response, we used CRISPR-Cas9 to delete FST with two independent sgRNA in platinum resistant SKOV3 cells. SKOV3 cells were chosen based on their platinum and taxane resistant status and ease of nucleofection. Western blotting for FST demonstrated reduced FST expression with sgRNA#1 and complete loss of expression with sgRNA #2 (Fig 6A, Sup Fig 5A, P<0.001). Given sgRNA#2 effectively eliminated FST expression, this cell pool was selected for all subsequent experiments. Compared to wild type cells, Taxol treatment of FST-KO cells resulted in a significant increase in apoptotic cells and decrease in viable cell number (Fig 6B–D). To assess whether FST KO makes tumors more sensitive to Taxol, we injected wild type or FST-KO SKOV3 cells into the intraperitoneal cavity of NSG mice. Tumor cells were allowed to engraft for three days and then mice were treated with Taxol IP (intraperitoneally) (Sup Fig 5B). Mice injected with FST-KO cells demonstrated a significant improvement in survival (Fig 6E, P<0.0001). By day 68, all mice injected with wild type cells had succumbed to metastatic disease (10/10), while 90% of the FST-KO tumor bearing mice were viable (9/10), the FST-KO group was monitored for a total of 120 days. At day 120 70% of FST KO mice had succumbed to metastatic disease (7/10), 30% of animals remained alive and showed no signs of disease. These animals were subsequently electively euthanized. Upon necropsy one animal had evidence of limited disease while the remaining 20% had no evidence of cancer. To confirm differences in mouse survival were due to FST induced chemoresistance rather than knockout of FST impacting basal cell growth, mice were xenografted with FST-KO or mock SKOV3 cells and tumor growth was recorded over 38 days. No significant difference in tumor growth was observed between mice xenografted with FST-KO or mock SKOV3 cells in the absences of Taxol (Figure 6F).
Together, this data demonstrates that tumor cells secrete FST in response to chemotherapy and that FST contributes to chemotherapy resistance. Furthermore, blocking this FST response sensitizes cells to chemotherapy both in vitro and in vivo.
FST is secreted in response to chemotherapy in ovarian cancer patients and correlates with poor survival outcomes
To investigate if FST is induced in response to chemotherapy in patients, we evaluated FST protein levels in ascites collected at the time of primary debulking surgery from patients with chemotherapy naive HGSOC (n=16) and compared to FST levels in ascites samples collected from patients with recurrent disease who had previously received chemotherapy (n=17, Fig 7A). We saw a positive trend of higher FST levels in treated patients; FST concentrations from ascites ranged from 4 to 320 ng/mL, the average FST concentration from ascites of patients of chemo naïve disease was 58 ng/mL, while the average level in patients with recurrent disease was 82 ng/mL (p=0.17). Unfortunately, due to limited clinical records, we were unable to control for the timing of ascites drainage relative to time of therapy (immediately after vs. weeks to months after).
Figure 7. FST is enriched in the ascites and peritoneum of HGSOC patient following treated with primary chemotherapy and correlates with worse patient outcomes.
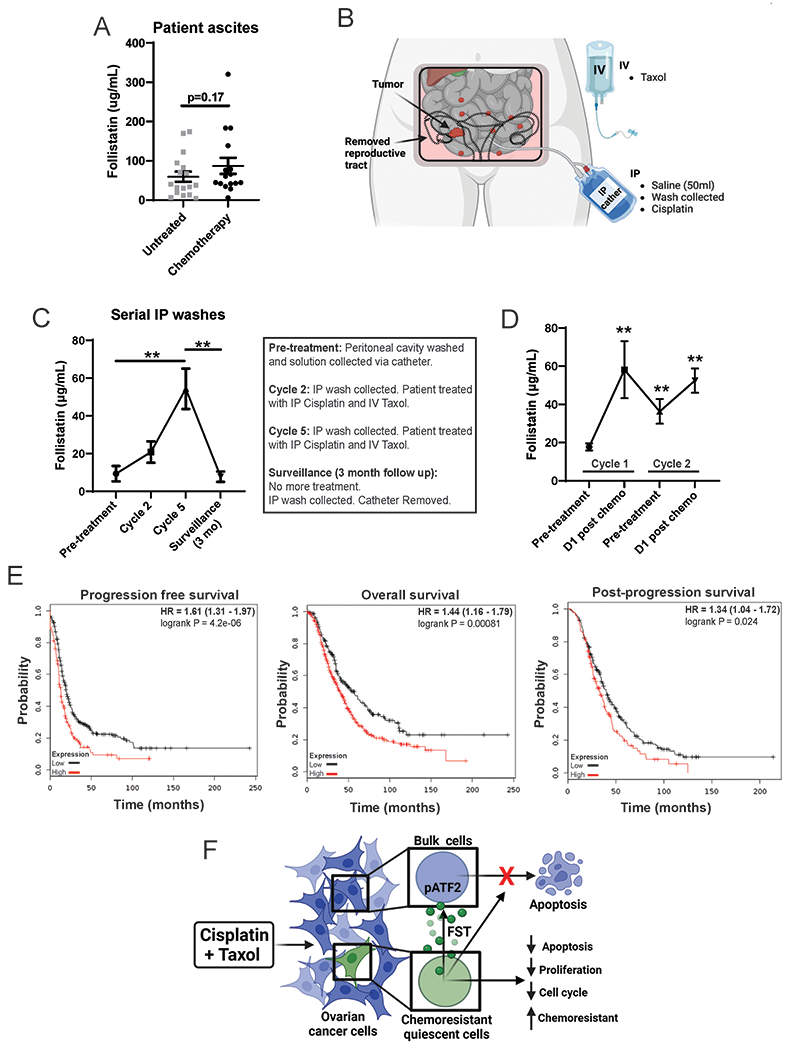
A. Ascites FST levels (quantified by ELISA) in HGSOC patients during primary debulking surgery (n=16), compared to ascites from patients who had received Carboplatin/Paclitaxel treatment regimens (n=17). B. Schematic of the experiment procedure for real time collection of serial IP washes from HGSOC patients (N=5), following IP cisplatin and IV Taxol treatments. C. FST levels in serial IP washes following cisplatin and Taxol treatment time course (Cycles 2 and 5). D. FST levels in serial IP washes following paired pre and post chem cisplatin and Taxol treatments (Cycles 1 and 2). E. Kaplan Meier survival curves for progression free survival (n=614), overall survival (n=655), and post-progression survival (n=382) of HGSOC patients with high vs low FST expression. F. Diagram of the ovarian cancer chemotherapy induces FST signaling pathway. Chemotherapy treatment of ovarian cancer cells normally results in apoptosis. However, quiescent cancer cells release FST in response to chemotherapy activating p-ATF2, which induces a chemoresistance state protecting the bulk cancer cells from apoptosis. *p<0.05, **p<0.01, ***p<0.001, ****p<0.001.
The lack of statistical significance in the ascites study above could be related to limited numbers or, given FST expression is induce by chemotherapy, due to lack to proximity to chemotherapy administration. To circumvent this limitation, we used samples from chemo-naïve patients with HGSOC (n=5) treated with intraperitoneal cisplatin chemotherapy. Peritoneal washings were collected from patients via an intraperitoneal catheter prior to treatment initiation, prior to cycle 2 and cycle 5 of chemotherapy, and 3 months after the completion of adjuvant therapy (Fig 7B). Initial patient FST concentrations before treatment averaged 9 μg/mL; however, by cycle two of treatment, FST concentration had increased to an average of 20 μg/mL and by cycle 5 it had increased to an average of 54 μg/mL (Fig 7C). When patients had completed chemotherapy and appeared to have no evidence of disease, peritoneal washings were obtained at the time of catheter removal. FST concentrations in peritoneal washings at this time dropped to 7 μg/mL, suggesting this increase in FST following treatment is an acute response. To confirm the temporal relationship of FST induction, we evaluated FST levels in peritoneal washes taken from patients prior to and the day after receiving IP cisplatin chemotherapy, as part of a chemo-immune therapy trial (21), at both cycle 1 and cycle 2. FST significantly increased following chemo cycle 1, before reducing post-chemo, then rising again following cycle 2 of chemotherapy (Fig 7D).
To further evaluate the clinical impact of FST, we interrogated the impact of FST expression on patient outcome using the ovarian cancer TCGA dataset. Supporting clinical importance, HGSOC and consistent with a role in chemotherapy resistance, HGSOC patients with high FST expression demonstrated significantly worse progression-free (p<0.0001, n=614), overall (p<0.001, n=655) and post-progression survival (p<0.05, n=382) (Fig 7E). Combined this data supports FST is induced in response to chemotherapy in patients and may be a driver of therapeutic resistance.
Discussion
In this study we have identified a novel mechanism of chemotherapy resistance whereby quiescent cells in response to chemotherapy increase FST secretion, activating p-ATF2 and reducing apoptosis, proliferation, and cell cycle, while increasing chemoresistance (Fig 7G). Targeting this chemoresistance mechanism by knocking out FST or inhibiting FST activity using an anti-FST antibody, results in an increase sensitivity to chemotherapy and a decrease in recurrence (Fig 7F).
FST and proliferation
FST is a 31-44 kDa glycosylated protein, expressed in many human tissues, in particular the ovary, ear, and larynx (29). The major function of FST is to bind and inhibit follicle stimulating hormone (FSH) and other members of the TGFβ superfamily of proteins, which include activins, inhibins, bone morphogenetic proteins (BMPs) and myostatin. Consequently, FST plays a key role in attenuating cellular response to these proliferation, differentiation, and apoptosis signals. Here we report that FST, in response to chemotherapy, is secreted primarily by quiescent cancer cells. FST then likely acts in at least a paracrine manner to induce chemotherapy resistance in bulk cells. Current studies cannot rule out an autocrine effect, however future studies with FST-KO cell lines can be used to further dissect autocrine vs. paracrine signaling.
Our data demonstrating FST inhibits cell cycle and proliferation is consistent with studies in ovarian sex cord-stromal tumors (30) and epithelial ovarian cancer cell lines (31). Although previous work has suggested FST inhibition of proliferation could result from attenuation of activin (31), our data in the a low activin cell line CaOV3 suggested other mechanisms may exist. Further, FST has been reported to promote proliferation in endothelial cells (32) and prostate cancer cell lines (33), thus suggesting tissue specific effects and alternative pathways.
FST as a stress response protein.
FST mRNA and protein expression increase in response to a number of cellular stresses including reactive oxygen species (34), ionizing radiation (35) ischemic reperfusion injury (36) and energy deprivation (11, 37). We similarly find serum withdrawal and chemotherapy exposure increase FST secretion from ovarian cancer cells. Combined, this suggest that FST may be part of a universal stress response mechanism. The regulation of FST following stress seems to be varied, with studies suggesting it is regulated both transcriptionally and post-translationally. In our data, the early expression of FST following NFATC4 induction suggests this is likely a direct NFATC4 effect, although more work is required to confirm this relationship.
FSTs and Cancer and Therapeutic Resistance
FST and the FST like protein family have been linked with carcinogenesis (29). Given FST regulates TGFβ signaling, and TGFβ is known dualistic role in cancer, it is not surprising that there are differing roles of FST in cancer. Suggesting a potentially tumor suppressive, FST is reported to reduce lung cancer metastasis in immune suppressed mice (38). However, it is possible that the reduction in metastasis could be related to the anti-proliferative effect we and others observe. FST is also reported to play a role in tumor angiogenesis, which would be consistent with a role of FST as a stress response gene, triggering angiogenesis to reverse adverse environmental conditions (39).
Our finding that FST promotes ovarian cancer chemotherapy resistance and reduces apoptosis, was corroborated in multiple cell lines, primary patient samples, and in a xenograft model. This is analogous to studies in breast and colorectal cancer indicating a role in therapeutic resistance role for the FST family member FSTL1 and FSTL3, respectively (40, 41). However, FST’s effects on apoptosis seem to be tissue specific, with studies in lung (34) and HeLa (11) cells demonstrating protection from apoptosis, while a study in endometrial stromal cells demonstrated the opposite (42).
This study suggests that FST induced chemotherapy resistance is mediated in part via ATF2 phosphorylation. Indeed expression of FST has been linked activation of the p38 MAPK/ERK pathways and subsequent phosphorylation ATF2 (43). ATF2 has been linked with chemotherapy resistance in non-small cell lung cancer (44), head and neck cancer (27) and laryngeal cancer (45). Similarly the, pATF2 also promotes radiation resistance in melanoma (28). The exact mechanism/s whereby ATF2 protects against apoptosis and promotes chemoresistance remain to be determined.
In addition to increases in pATF2 with FST exposure, we also observed modest increases in pSMAD1, 2, and 4. This is in line with a study linking pATF2 and activated SMAD2 and SMAD 1/5 driving quiescence and dormancy in breast cancer cells (46). As FST typically functions as a negative regulator of other TGF/BMP superfamily ligands, such as activins and inhibins, the increase in pSMADs at first appears counterintuitive. However, it is possible that FST could inhibit an inhibitor resulting in positive signaling. Alternatively, activation of SMADs may be a time dependent feedback response. Further studies will be necessary to determine the role of FST signaling.
FST was also recently linked with resistance to immune checkpoint inhibitor therapy in ovarian cancer (14). This study also identified IL33 and S100a4 as high-priority candidate genes significantly overexpressed in their therapy-resistant ovarian cancer model. This is intriguing, considering we also observe a significant increase in IL33 following NFATC4 induction. Together, this data links NFATC4 to both immunotherapy and chemotherapy resistance.
Lastly, our data demonstrating FST expression increase in HGSOC patients following primary chemotherapy and is correlated with poor progression free and overall survival, is consistent with an early study by Ren, et al, (2012) which reported FST to be elevated in ovarian cancer patients, where it correlated with poor prognosis (12).
Overall, this study has substantial implications for the treatment of ovarian cancer. Despite considerable progress with targeted therapy, chemotherapy remains the most effective means to treat ovarian cancer. The ability to improve response to chemotherapy offers the possibility to increase cure rates. Our results indicate that FST plays a significant role in driving chemotherapy resistance. As FST is a secreted factor, it represents an ideal target for antibody mediated neutralization. Our studies support the development of a human FST neutralizing antibody for translation into clinical trials.
Supplementary Material
Translational Relevance:
Quiescent cancer cells are resistant to chemotherapy and can drive disease recurrence and mortality. We find that innately quiescent ovarian cancer cells, in response to chemotherapy, secrete FST to induce a chemoresistant quiescent state in neighboring cells. Patient samples confirm a significant induction of follistatin within 24hrs of chemotherapy administration. Importantly, inhibition of FST sensitizes tumor cells to chemotherapy, and knockout of FST increases cancer cure rates in mice. Combined this work identifies FST as a clinical target to overcome chemotherapy resistance and potentially increase ovarian cancer cure rates.
Acknowledgements:
Funding for this work was provided by Ann and Sol Schreiber Mentored Investigator Award (599997) from the Ovarian Cancer Research Alliance (OCRA), DOD Award #W81XWH-15-1-0083 and NIH R01 award 1R01CA203810.
Footnotes
The authors have declared that no conflict of interest exists
References
- 1.Brown JA, Yonekubo Y, Hanson N, Sastre-Perona A, Basin A, Rytlewski JA, et al. TGF-β-induced quiescence mediates chemoresistance of tumor-propagating cells in squamous cell carcinoma. Cell stem cell. 2017;21(5):650–64. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen K, Zhang C, Ling S, Wei R, Wang J, and Xu X. The metabolic flexibility of quiescent CSC: Implications for chemotherapy resistance. Cell death & disease. 2021;12(9):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cole AJ, Iyengar M, Panesso-Gomez S, O’Hayer P, Chan D, Delgoffe GM, et al. NFATC4 promotes quiescence and chemotherapy resistance in ovarian cancer. JCI Insight. 2020;5(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cole AJ, Fayomi AP, Anyaeche VI, Bai S, and Buckanovich RJ. An evolving paradigm of cancer stem cell hierarchies: therapeutic implications. Theranostics. 2020;10(7):3083–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aguirre-Ghiso JA. Translating the Science of Cancer Dormancy to the Clinic. Cancer research. 2021;81(18):4673–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Risson E, Nobre AR, Maguer-Satta V, and Aguirre-Ghiso JA. The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nat Cancer. 2020;1(7):672–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shepherd TG, and Dick FA. Principles of dormancy evident in high-grade serous ovarian cancer. Cell Div. 2022;17(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao MQ, Choi YP, Kang S, Youn JH, and Cho NH. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene. 2010;29(18):2672–80. [DOI] [PubMed] [Google Scholar]
- 9.Bildik G, Liang X, Sutton MN, Bast RC, Jr., and Lu Z. DIRAS3: An Imprinted Tumor Suppressor Gene that Regulates RAS and PI3K-driven Cancer Growth, Motility, Autophagy, and Tumor Dormancy. Mol Cancer Ther. 2022;21(1):25–37. [DOI] [PubMed] [Google Scholar]
- 10.Lin SY, Morrison JR, Phillips DJ, and de Kretser DM. Regulation of ovarian function by the TGF-beta superfamily and follistatin. Reproduction. 2003;126(2):133–48. [DOI] [PubMed] [Google Scholar]
- 11.Gao X, Wei S, Lai K, Sheng J, Su J, Zhu J, et al. Nucleolar follistatin promotes cancer cell survival under glucose-deprived conditions through inhibiting cellular rRNA synthesis. The Journal of biological chemistry. 2010;285(47):36857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ren P, Chen FF, Liu HY, Cui XL, Sun Y, Guan JL, et al. High serum levels of follistatin in patients with ovarian cancer. J Int Med Res. 2012;40(3):877–86. [DOI] [PubMed] [Google Scholar]
- 13.Karve TM, Preet A, Sneed R, Salamanca C, Li X, Xu J, et al. BRCA1 regulates follistatin function in ovarian cancer and human ovarian surface epithelial cells. PloS one. 2012;7(6):e37697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iyer S, Zhang S, Yucel S, Horn H, Smith SG, Reinhardt F, et al. Genetically Defined Syngeneic Mouse Models of Ovarian Cancer as Tools for the Discovery of Combination Immunotherapy. Cancer Discov. 2021;11(2):384–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manichaikul A, Peres LC, Wang XQ, Barnard ME, Chyn D, Sheng X, et al. Identification of novel epithelial ovarian cancer loci in women of African ancestry. International journal of cancer. 2020;146(11):2987–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rashidi MRW, Mehta P, Bregenzer M, Raghavan S, Fleck EM, Horst EN, et al. Engineered 3D model of cancer stem cell enrichment and chemoresistance. Neoplasia (New York, NY). 2019;21(8):822–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, et al. L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature. 1999;401(6754):703–8. [DOI] [PubMed] [Google Scholar]
- 18.Iyengar M, O’Hayer P, Cole A, Sebastian T, Yang K, Coffman L, et al. CDK4/6 inhibition as maintenance and combination therapy for high grade serous ovarian cancer. Oncotarget. 2018;9(21):15658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chefetz I, Grimley E, Yang K, Hong L, Vinogradova EV, Suciu R, et al. A Pan-ALDH1A Inhibitor Induces Necroptosis in Ovarian Cancer Stem-like Cells. Cell Rep. 2019;26(11):3061–75 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oki T, Nishimura K, Kitaura J, Togami K, Maehara A, Izawa K, et al. A novel cell-cycle-indicator, mVenus-p27K−, identifies quiescent cells and visualizes G0–G1 transition. Scientific reports. 2014;4(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orr B, Mahdi H, Fang Y, Strange M, Uygun I, Rana M, et al. Phase I trial combining chemokine-targeting with loco-regional chemoimmunotherapy for recurrent, platinum-sensitive ovarian cancer shows induction of CXCR3 ligands and markers of type 1 immunity. Clinical Cancer Research. 2022;28(10):2038–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bai S, Ingram P, Chen Y-C, Deng N, Pearson A, Niknafs YS, et al. EGFL6 regulates the asymmetric division, maintenance, and metastasis of ALDH+ ovarian cancer cells. Cancer research. 2016;76(21):6396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silva IA, Bai S, McLean K, Yang K, Griffith K, Thomas D, et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer research. 2011;71(11):3991–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi YJ, Ingram PN, Yang K, Coffman L, Iyengar M, Bai S, et al. Identifying an ovarian cancer cell hierarchy regulated by bone morphogenetic protein 2. Proceedings of the National Academy of Sciences of the United States of America. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.White EZ, Pennant NM, Carter JR, Hawsawi O, Odero-Marah V, and Hinton CV. Serum deprivation initiates adaptation and survival to oxidative stress in prostate cancer cells. Scientific reports. 2020;10(1):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giannoudis A, Malki MI, Rudraraju B, Mohhamed H, Menon S, Liloglou T, et al. Activating transcription factor-2 (ATF2) is a key determinant of resistance to endocrine treatment in an in vitro model of breast cancer. Breast Cancer Research. 2020;22(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duffey D, Dolgilevich S, Razzouk S, Li L, Green R, and Gorti GK. Activating transcription factor‐2 in survival mechanisms in head and neck carcinoma cells. Head & neck. 2011;33(11):1586–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ze Ronai, Yang Y-m, Fuchs SY, Adler V, Sardana M, and Herlyn M. ATF2 confers radiation resistance to human melanoma cells. Oncogene. 1998;16(4):523–31. [DOI] [PubMed] [Google Scholar]
- 29.Shi L, Resaul J, Owen S, Ye L, and Jiang WG. Clinical and Therapeutic Implications of Follistatin in Solid Tumours. Cancer Genomics Proteomics. 2016;13(6):425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shikone T, Matzuk MM, Perlas E, Finegold MJ, Lewis KA, Vale W, et al. Characterization of gonadal sex cord-stromal tumor cell lines from inhibin-alpha and p53-deficient mice: the role of activin as an autocrine growth factor. Molecular endocrinology (Baltimore, Md). 1994;8(8):983–95. [DOI] [PubMed] [Google Scholar]
- 31.Di Simone N, Crowley WF Jr, Wang QF, Sluss PM, and Schneyer AL. Characterization of inhibin/activin subunit, follistatin, and activin type II receptors in human ovarian cancer cell lines: a potential role in autocrine growth regulation. Endocrinology. 1996;137(2):486–94. [DOI] [PubMed] [Google Scholar]
- 32.Kozian DH, Ziche M, and Augustin HG. The activin-binding protein follistatin regulates autocrine endothelial cell activity and induces angiogenesis. Lab Invest. 1997;76(2):267–76. [PubMed] [Google Scholar]
- 33.van Schaik RH, Wierikx CD, Timmerman MA, Oomen MH, van Weerden WM, van der Kwast TH, et al. Variations in activin receptor, inhibin/activin subunit and follistatin mRNAs in human prostate tumour tissues. Br J Cancer. 2000;82(1):112–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin C, Zhao X, Sun D, Zhang L, Fang W, Zhu T, et al. Transcriptional activation of follistatin by Nrf2 protects pulmonary epithelial cells against silica nanoparticle-induced oxidative stress. Scientific reports. 2016;6:21133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Forrester HB, Ivashkevich A, McKay MJ, Leong T, de Kretser DM, and Sprung CN. Follistatin is induced by ionizing radiation and potentially predictive of radiosensitivity in radiation-induced fibrosis patient derived fibroblasts. PloS one. 2013;8(10):e77119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y, Rothnie C, Spring D, Verrier E, Venardos K, Kaye D, et al. Regulation and actions of activin A and follistatin in myocardial ischaemia-reperfusion injury. Cytokine. 2014;69(2):255–62. [DOI] [PubMed] [Google Scholar]
- 37.Gao X, Dong H, Lin C, Sheng J, Zhang F, Su J, et al. Reduction of AUF1-mediated follistatin mRNA decay during glucose starvation protects cells from apoptosis. Nucleic acids research. 2014;42(16):10720–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiou J, Chang YC, Tsai HF, Lin YF, Huang MS, Yang CJ, et al. Follistatin-like Protein 1 Inhibits Lung Cancer Metastasis by Preventing Proteolytic Activation of Osteopontin. Cancer research. 2019;79(24):6113–25. [DOI] [PubMed] [Google Scholar]
- 39.Blank S, Deck C, Dreikhausen L, Weichert W, Giese N, Falk C, et al. Angiogenic and growth factors in gastric cancer. J Surg Res. 2015;194(2):420–9. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Tian M, Liu W, Wang D, Zhou Z, Pei Q, et al. Follistatin-Like 3 Enhances Invasion and Metastasis via beta-Catenin-Mediated EMT and Aerobic Glycolysis in Colorectal Cancer. Front Cell Dev Biol. 2021;9:660159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng S, Huang Y, Lou C, He Y, Zhang Y, and Zhang Q. FSTL1 enhances chemoresistance and maintains stemness in breast cancer cells via integrin beta3/Wnt signaling under miR-137 regulation. Cancer Biol Ther. 2019;20(3):328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coutinho LM, Vieira EL, Dela Cruz C, Casalechi M, Teixeira AL, Del Puerto HL, et al. Apoptosis modulation by activin A and follistatin in human endometrial stromal cells. Gynecol Endocrinol. 2016;32(2):161–5. [DOI] [PubMed] [Google Scholar]
- 43.Singh R, Braga M, Reddy ST, Lee S-J, Parveen M, Grijalva V, et al. Follistatin targets distinct pathways to promote brown adipocyte characteristics in brown and white adipose tissues. Endocrinology. 2017;158(5):1217–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lo Iacono M, Monica V, Vavalà T, Gisabella M, Saviozzi S, Bracco E, et al. ATF2 contributes to cisplatin resistance in non‐small cell lung cancer and celastrol induces cisplatin resensitization through inhibition of JNK/ATF2 pathway. International journal of cancer. 2015;136(11):2598–609. [DOI] [PubMed] [Google Scholar]
- 45.Tian L, Zhang J, Ren X, Liu X, Gao W, Zhang C, et al. Overexpression of miR-26b decreases the cisplatin-resistance in laryngeal cancer by targeting ATF2. Oncotarget. 2017;8(45):79023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nobre AR, Risson E, Singh DK, Di Martino JS, Cheung JF, Wang J, et al. Bone marrow NG2(+)/Nestin(+) mesenchymal stem cells drive DTC dormancy via TGFβ2. Nat Cancer. 2021;2(3):327–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data analyzed in this study were obtained from Gene Expression Omnibus (GEO) is GSE210439. Other data generated in this study are available upon request from the corresponding author.