

Abstract
Peanut-induced allergy is an IgE-mediated type I hypersensitivity reaction that manifests symptoms ranging from local edema to life-threatening anaphylaxis. While there are treatments for symptoms in allergic patients resulting from allergen exposure, there are few preventive therapies other than strict dietary avoidance or oral immunotherapy, neither of which are successful in all patients. We have previously designed a covalent heterobivalent inhibitor (cHBI) that binds in an allergen-specific manner as a preventive for allergic reactions. Building on previous in vitro testing, in this report we developed a humanized mouse model to test cHBI efficacy in vivo. Humanized mice (NSG-SCF/GM-CSF/IL-3) developed mature functional human mast cells in multiple tissues and displayed robust anaphylactic reactions when passively sensitized with patient-derived IgE monoclonal antibodies specific for peanut Arachis hypogaea 2 (Ara h 2) allergen. The allergic response in humanized mice is IgE dose-dependent and is mediated by human mast cells indicated by elevated serum tryptase and increased human mast cell degranulation. Using the validated humanized mouse model, we showed that cHBI prevented allergic reactions for more than two weeks when administered before allergen exposure. cHBI also prevented fatal anaphylaxis and attenuated allergic reactions when administered shortly after the onset of symptoms. cHBI impaired mast cell degranulation in vivo in an allergen-specific manner. Importantly, cHBI rescued the mice from lethal anaphylactic responses during oral Ara h 2 allergen-induced anaphylaxis. Together, these findings suggest that cHBI has the potential to be an effective preventative for peanut-specific allergic responses in patients.
One Sentence Summary:
Blockade of peanut-reactive epitopes prevents systemic anaphylaxis.
INTRODUCTION
Peanut allergy is a prominent IgE-mediated type I hypersensitivity affecting 25% of children with food allergy and 1.8% of adults in the United States (1, 2). Peanut-induced allergic reactions are systemic leading to severe reactions including tissue edema, shock, and death. IgE-dependent allergic reactions are initiated with the binding of the peanut allergen to the allergen-specific IgE bound to the high-affinity IgE epsilon receptor (FcεRI) on the surface of mast cells and basophils, eliciting their degranulation and the release of inflammatory mediators including histamine, proteases, prostaglandins, and cytokines/chemokines (3–5). To date, there is no specific therapy to prevent peanut allergic reactions. The current standard of care for sensitized individuals is strict peanut avoidance with accidental exposure treated immediately with epinephrine injection and anti-mast cell mediators such as antihistamines (3, 5–7).
The LEAP (Learning Early about Peanut Allergy) and LEAP-On studies demonstrated that peanut allergy can be minimized by early introduction of peanut into the diet (8, 9). Oral immunotherapy (OIT) can limit the severity of allergic reactions in already sensitized individuals, although it does not have sustained efficacy in all patients. Moreover, OIT requires a maintenance dose of allergen and reducing or eliminating dietary peanut increases the possibility of developing severe allergic reactions including anaphylaxis (10–13). Other therapies that prevent allergic reactions by targeting IgE antibodies and blocking allergic responses have been considered for peanut allergy treatment (14). Omalizumab, a monoclonal antibody, represents a clinically approved anti-IgE therapy for asthma patients. Although omalizumab has shown therapeutic efficacy in allergic asthma patients, it has not been approved for peanut allergies. Clinical trials with peanut allergic patients showed that the clinical benefit of omalizumab could only be achieved after multiple doses and long-periods of administration, or in combination with peanut OIT (15–17). Other anti-IgE molecules have been developed and introduced in human clinical trials that similarly disrupt IgE-receptor interactions (18–20). Yet, none of these anti-IgE molecules target allergen specific IgE, nor do they rapidly prevent peanut allergic reactions. Moreover, these approaches minimize any beneficial effects of IgE.
While peanut-allergic patients develop antibodies with broad specificities, there is evidence that restricted epitopes are required for the anaphylactic response (21), Evidence also suggests the existence of public specificities among patients (22). This suggested that epitope-targeted therapies might be a potential approach for treatment. We reported the design of a peanut allergen-specific inhibitor that we termed covalent heterobivalent inhibitor (cHBI) (23). cHBI selectively binds to allergen specific antibodies. Once bound, cHBI forms an irreversible covalent bond, thus preventing allergen-induced allergic reactions. Using two cHBI inhibitors targeting the IgE that recognize immunodominant epitopes of the peanut allergens Ara h 2 and Ara h 6 (24, 25), we demonstrated that cHBIs effectively inhibited basophil activation ex vivo and in vitro mast cell degranulation using sera from an array of peanut allergic patients (23), supporting both the epitope-targeting approach and the concept of public epitopes among patients. Here, we evaluated cHBI efficacy using an in vivo model that simulates anaphylaxis in patients. We demonstrated that pre-treatment with a single dose of cHBI prevented IgE-mediated allergic responses and anaphylaxis to Ara h 2 peanut allergen for at least two weeks, and that it can prevent fatal anaphylaxis if administered shortly after allergen exposure. These data support cHBI as a therapeutic option for patients with peanut food allergy.
RESULTS
Defining cHBI-targeted IgE monoclonal antibodies
To test the ability of cHBI to inhibit anaphylaxis in vivo, we wanted to identify peanut-specific epitopes that matched patient sensitivities and bound the cHBI defined in our previous studies (23) (Fig. 1, A and B). We utilized a method to produce monoclonal IgE from allergic patient B cells (26) to generate six mAbs (5D7, 15A7, 16A8, 38B7 13D9, 8F3) that recognized Ara h 2. To evaluate the epitope specificity of the Ara h 2-specific IgE mAbs in inducing mast cell degranulation, we primed RBL-SX38 cells with each of the mAbs and identified that cHBI inhibition worked with one of the six mAbs, clone 38B7. We then used two cocktails, one cocktail containing all six mAb clones, and another of the same mAbs excluding clone 38B7 to prime RBL-SX38 cells for 24 hours before challenging the cells using Ara h 2 allergen at various concentrations. There was a significant difference in degranulation of cells primed with six specific IgEs compared to cells primed without 38B7 (Fig. 1C). This result suggested a major contribution of 38B7 to Ara h 2-triggered degranulation. We observed >90 % inhibition of cell degranulation by cHBI using cells primed with all six specific IgE mAbs (Fig. 1D) but no inhibition when cells were primed with the five mAbs excluding 38B7 across a broad range of allergen dose (Fig. 1E).
Fig. 1: In vitro screening of human specific IgE mAbs for peanut allergen Ara h 2 epitope specificity.
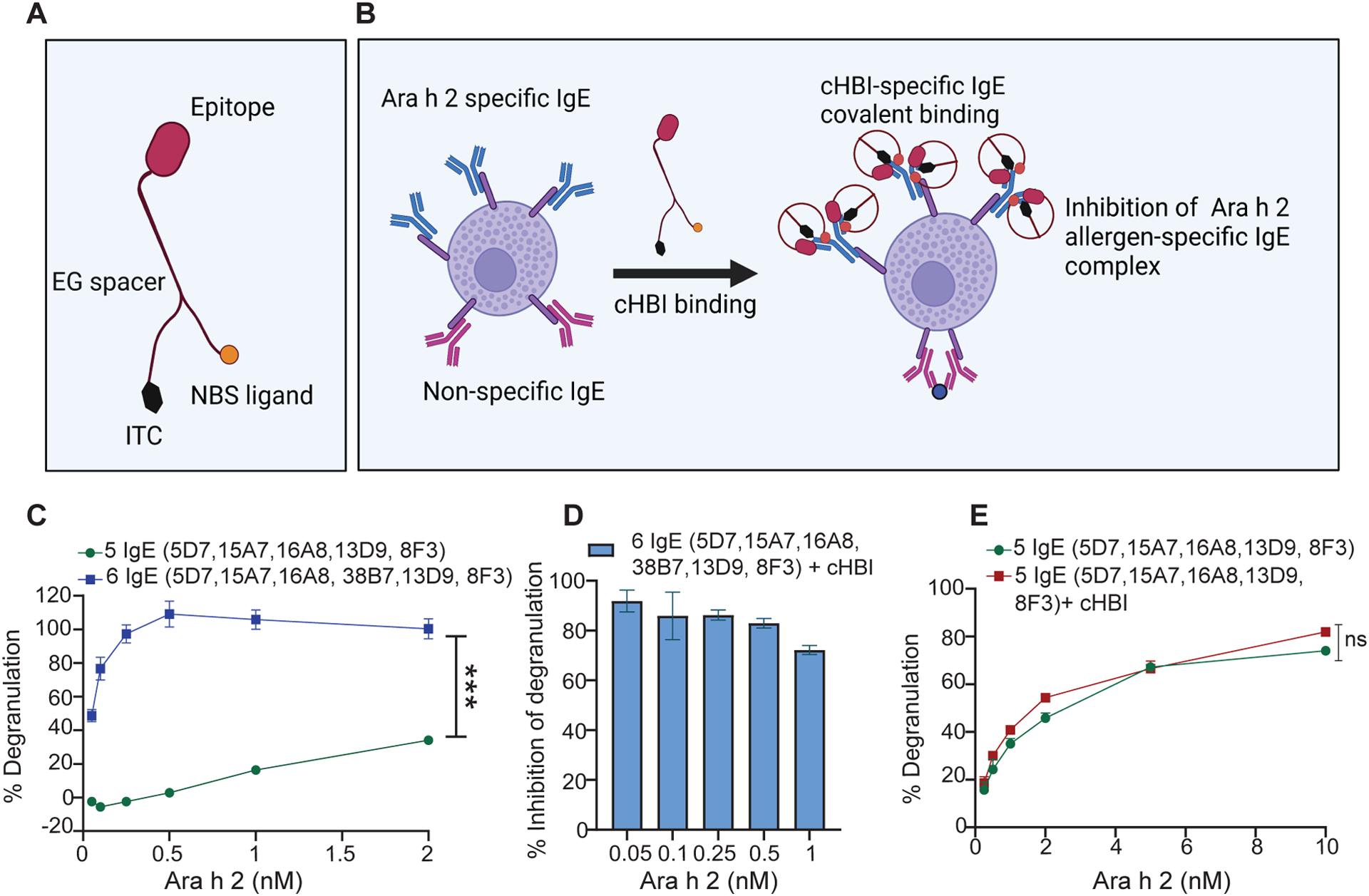
(A) Illustration of cHBI structure shows the antigen binding site (peptide epitope), nucleotide binding site (NBS), and isothiocyanate moiety (ITC). (B) Schematic illustration of cHBI mechanism of action. cHBI binds to Ara h 2 specific IgE on mast cells forming an irreversible covalent bond and preventing the binding of Ara h 2 allergen to specific IgE inhibiting the allergic reactions. (C) Line graph of percent RBL-SX38 cell degranulation. RBL-SX38 cells primed with two cocktails of IgE mAbs targeting different epitopes of Ara h 2 allergen. One cocktail with all six IgE mAbs, and one cocktail with 5 IgE mAbs without clone 38B7 mAb. The cells were challenged with Ara h 2 at (0.05, 0.10, 0.25, 0.5, 1.0, 2.0 μM) 24 hours post IgE priming. (D) Percent inhibition of RBL-SX38 cell degranulation by cHBI when RBL-SX38 cells primed with a cocktail of six IgE mAbs and challenged with Ara h 2 at (0.05, 0.10, 0.25, 0.5, 1.0 μM). (E) Line graph of percent RBL-SX38 cell degranulation. RBL-SX38 cells primed with a cocktail of 5 specific IgE mAbs without clone 38B7 mAb in the presence or absence of cHBI and then challenged with Ara h 2 at (0.05, 0.10, 0.25, 0.5, 1.0, 2.0, 5.0, 10.0 μM). Data represent mean ±SEM, mean in each graph represents three technical replicates. Data are representative of two independent experiments. One way ANOVA with t-test was used to compare means in (C), and (E). ***P <0.001. ns indicates no statistical difference. Illustrations in (A) and (B) created with BioRender.com.
HuNSGS mice develop mature functional human mast cells that mediate passive systemic anaphylaxis (PSA) to Ara h 2 allergen in an IgE-dependent manner
After identifying the dominant epitope that contributes to RBL-SX38 cell degranulation, we then tested the Ara h 2 specific IgE mAbs in a humanized mouse model as a robust and predictive preclinical model of anaphylaxis (27–30). To establish the model, we used the non-obese diabetic-severe combined immunodeficient γc-deficient mice expressing transgenes for human SCF, GM-CSF, IL-3 (NSG-SGM3 or NSGS mice) that support enhanced differentiation of human myeloid lineages (31). Sub-lethally irradiated NSGS mice were injected with hematopoietic CD34+ cells from the cord blood of human donors. The levels of human cell chimerism in the humanized NSGS mice (hereafter termed huNSGS mice) were determined by flow cytometry at 10–12 weeks post CD34+ transfer. As expected, huNSGS demonstrated development of a significant number of human leukocytes (CD45+ cells) in bone marrow, spleen, lung, and peritoneal cavity (Fig. 2, A and B) and in the stomach, intestine, and skin (fig. S1A). We used CD117 and FcεR1α as markers to identify human mast cells (32, 33). A substantial percentage of mature human mast cells among total human CD45+ cells was detected in huNSGS mouse tissues, including spleen, lung, peritoneal cavity (Fig. 2, C and D) intestine, and to a lesser extent in stomach and the skin (fig. S1B). Additionally, the majority of cells detected in the peripheral blood of huNSGS mice were human leukocytes (CD45+ cells) (Fig. 2, E and F). Thus, huNSGS mice develop mature, human mast cells in multiple tissues.
Fig. 2: HuNSGS mice develop human cells in tissues and peripheral blood.
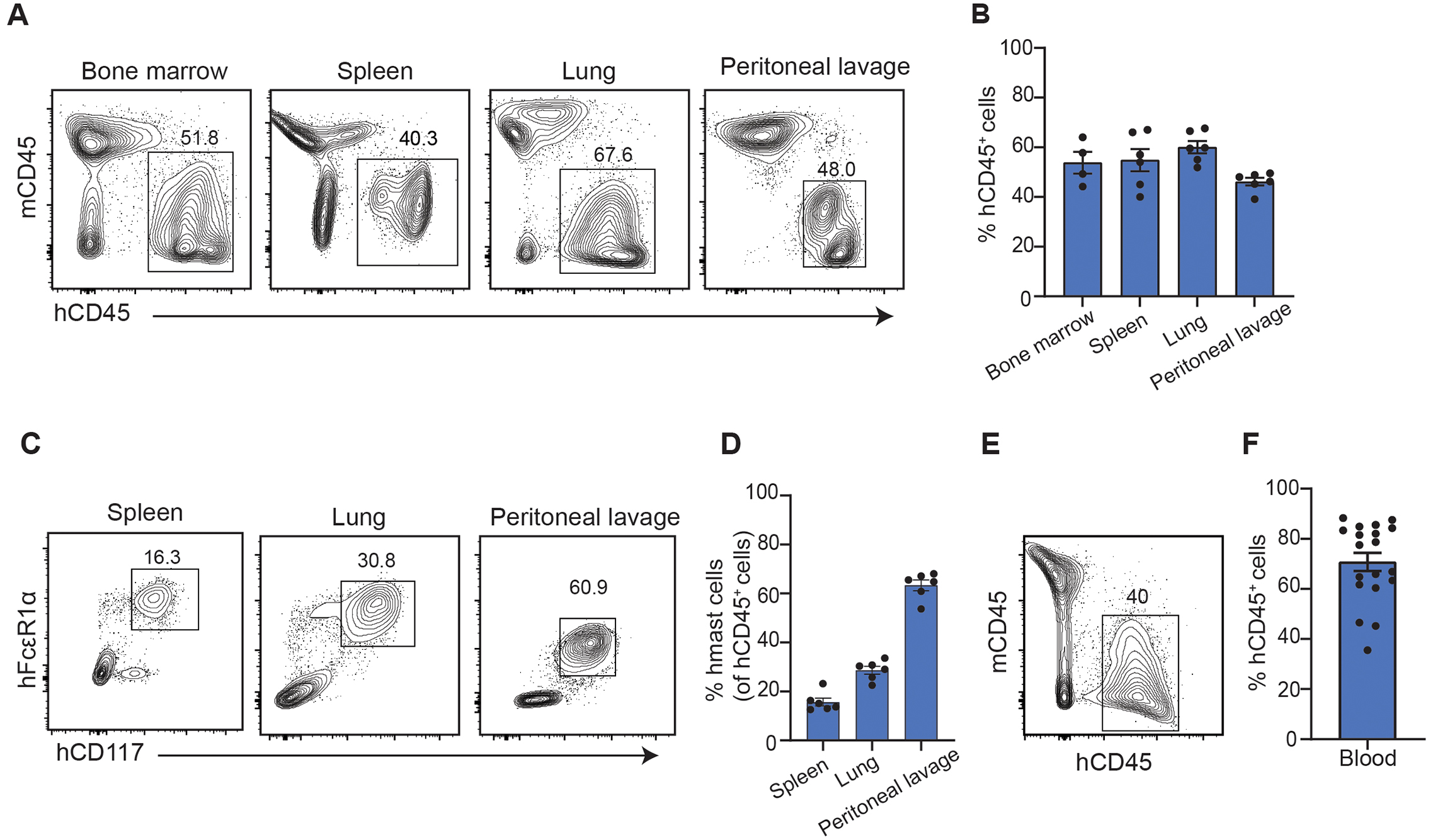
Flow cytometry analysis of human cells in huNSGS mice 10–12 weeks post engraftment (A) Representative flow plots of human (h) and murine (m) CD45+ cells in bone marrow, spleen, lung, and peritoneal lavage. (B) Scatter plot of the frequency of human CD45+ cells in bone marrow, spleen, lung, and peritoneal lavage of the NSGS humanized mice (n = 4–6). (C) Representative flow plots of human mast cells (hCD117+ hFcεR1α+) gated on hCD45+ cells in bone marrow, spleen, lung, and peritoneal lavage. (D) Scatter plot of the frequency of human mast cells of hCD45+ cells in bone marrow, spleen, lung, and peritoneal lavage of the huNSGS mice (n= 4–6). Flow cytometry analysis of human cells in whole blood from huNSGS mice 10 weeks post engraftment. Representative contour plot (E) and scatter plot (F) of the frequency of hCD45+ cells in the peripheral blood (n = 19). Results are representative of six independent experiments.
We next examined if the human mast cells were competent to undergo degranulation and promote anaphylaxis when challenged with Ara h 2. To induce PSA, we sensitized the mice with 100 μg of the cocktail of six Ara h 2-specific IgE mAbs (5D7, 15A7, 16A8, 38B7 13D9, 8F3) to better represent a patient polyclonal response. After 24 hours, mice were challenged with various Ara h 2 doses (Fig. 3A). HuNSGS mice challenged with a high doses of Ara h 2 developed fatal anaphylactic shock within 5–10 minutes of the challenge (Fig. 3B). Mice challenged with low Ara h 2 doses had less severe reactions but exhibited a significant decline in core body temperature (Fig. 3C). To demonstrate that human mast cells in huNSGS mice were functional, we determined whether these cells could undergo degranulation and mediate anaphylactic reactions by measuring serum tryptase levels. Tryptase, a serine protease released from mast cells, but not basophils, serves as a marker for mast cell activation (34). Post Ara h 2 challenge, huNSGS mouse serum had significant tryptase levels that increased in an allergen dose-dependent manner (Fig. 3D). Additionally, the mast cell degranulation markers CD107a and CD63 were significantly increased on mast cells after the Ara h 2 challenge in huNSGS mice (Fig. 3, E to H). Importantly, the anaphylactic response was entirely dependent on engrafted human cells as non-engrafted NSGS control mice exhibited no change of core body temperature when sensitized with human IgE mAbs and challenged with Ara h 2 (fig. S1C). Moreover, human IgE was detected only on the surface of human mast cells (hCD45+ hCD117+ hFcεR1α+) but not mouse mast cells (mCD45+ mCD117+ mFcεR1α+) after intravenously sensitizing huNSGS mice with a cocktail of six Ara h 2-specific IgE mAbs (Fig S1D and E). Human tryptase levels were only significantly increased in the supernatant of sorted human mast cells and not in the supernatant of sorted mouse mast cells after stimulation for 1 hour in vitro with Ara h 2 (Fig S1F).These data support what has been reported previously that human IgE cannot bind to mouse IgE receptor and cannot activate murine mast cells (35–37).
Fig. 3: HuNSGS mouse model predicts mast-cell mediated PSA to Ara h 2 allergen.
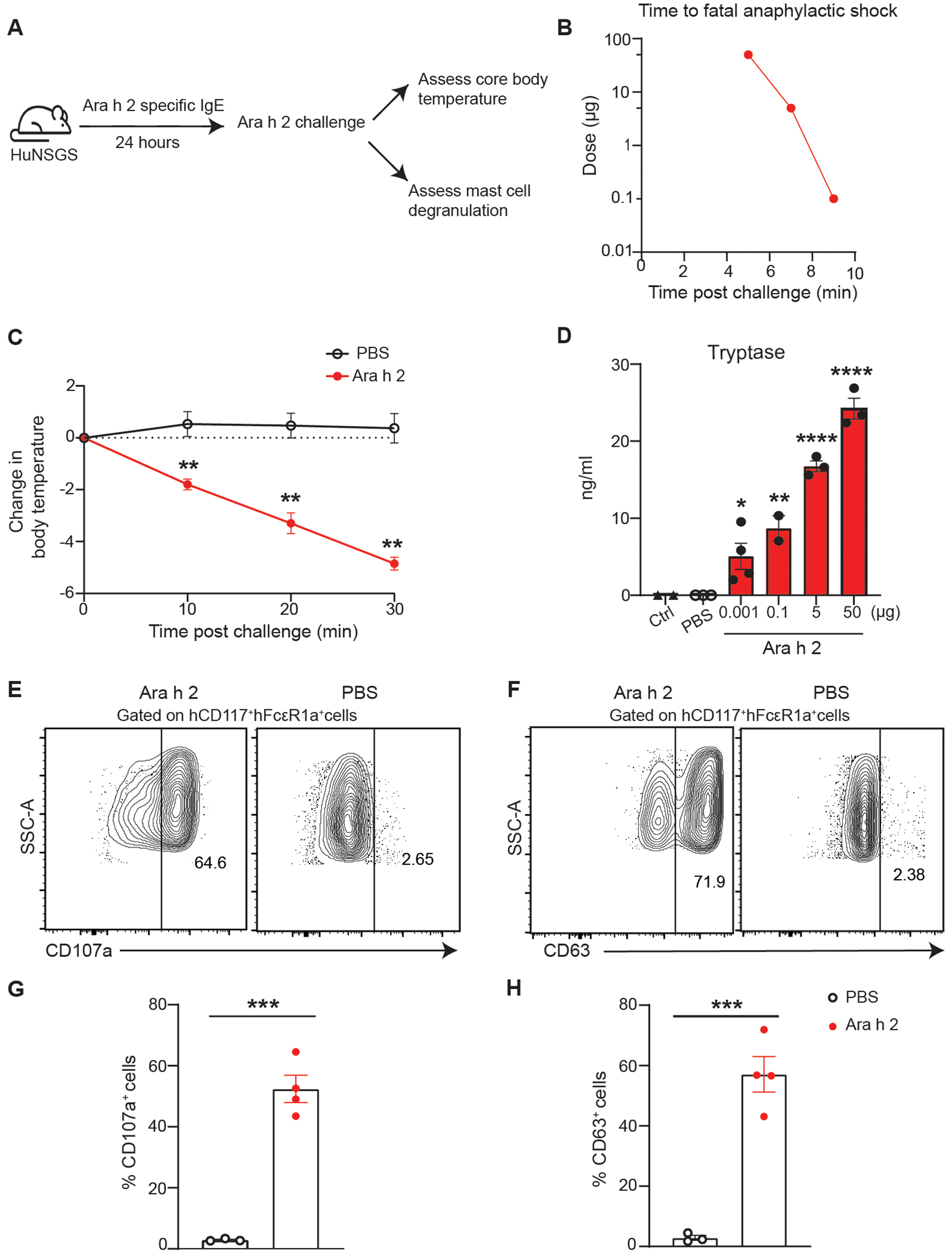
(A) Schematic illustration of PSA in huNSGS mice. Mice were sensitized with a cocktail of six Ara h 2 specific IgE mAbs (100 μg) and challenged intravenously 24 hours later with Ara h 2 at (50, 5,1, or 0.001 μg). (B) Time to fatal anaphylactic shock following challenge is Ara h 2 allergen-dose dependent (n = 3) (C) Change of core body temperature measured post Ara h 2 (1 ng) challenge. Data represent mean ±SEM, (n = 3–4). (D) Scatter plot of tryptase serum levels in huNSGS mice challenged with the indicated doses of Ara h 2 (n = 3–4). (E-H) Analysis of mast cell degranulation markers in huNSGS mice challenged with (1ng) of Ara h 2. (E,F) Representative contour plot of surface expression levels of splenic mast cells degranulation markers CD107a (E), and CD63 (F). (G,H) Scatter plot of the percentage of CD107a (G) and CD63 (H) positive cells (n = 3–4). One way ANOVA followed by unpaired t-test was used to compare means in (C), and (D); an unpaired Mann Whitney test was used to compare means in (G), and (H). ****P <0.0001, ***P <0.001, **P <0.01, *P <0.05. Ctrl indicates non-engrafted NSGS control mice.
Our results indicated that at a high dose of IgE, huNSGS mice developed severe anaphylactic reactions in an Ara h 2 dose-dependent manner. To investigate if anaphylaxis in huNSGS mice is IgE-dependent, we sensitized huNSGS mice with various doses of IgE mAbs ranging from 0–10 μg. Mice were then challenged 24 hours post sensitization with 0.5 ng Ara h 2 allergen. We found that only the mice that received at least 1 μg IgE mAbs had a substantial decline in body temperature and fatal response (fig. S1G). Although all mice had a similar percentage of mature human mast cells, the elevated serum tryptase levels was dependent on IgE dose (fig. S1, H and I). These results suggest that huNSGS mice develop mature and functional human mast cells that mediate anaphylaxis to Ara h 2 in IgE-dependent manner.
cHBI inhibits specific IgE-mediated anaphylaxis and mast cell degranulation in huNSGS mice
To determine the ability of cHBI to prevent human Ara h 2-specific IgE-mediated anaphylaxis in huNSGS mice, we sensitized the mice with a cocktail of six Ara h 2 specific IgE mAbs. The following day we intravenously injected the mice with 20 nmol of cHBI or PBS and challenged the mice 1 hour later with 1 ng of Ara h 2 (Fig. 4A). HuNSGS mice treated with cHBI exhibited no significant change in core body temperature or clinical symptoms compared with PBS-treated huNSGS mice (Fig. 4, B and D). Additionally, huNSGS or non-engrafted NSGS mice treated with cHBI and challenged with Ara h 2 in the absence of hIgE sensitization had no change in core body temperature (fig. S1, J and K). Tryptase serum levels were significantly lower in mice treated with cHBI (Fig. 4E), and the expression of CD107a and CD63 on human mast cells were significantly lower compared with PBS-treated mice (Fig. 4, F and G). Collectively, these data demonstrated that cHBI effectively inhibits IgE-dependent anaphylaxis and decreases human mast cell activation in the huNSGS mouse model.
Fig. 4: Inhibition of IgE-mediated PSA to Ara h 2 allergen by cHBI.
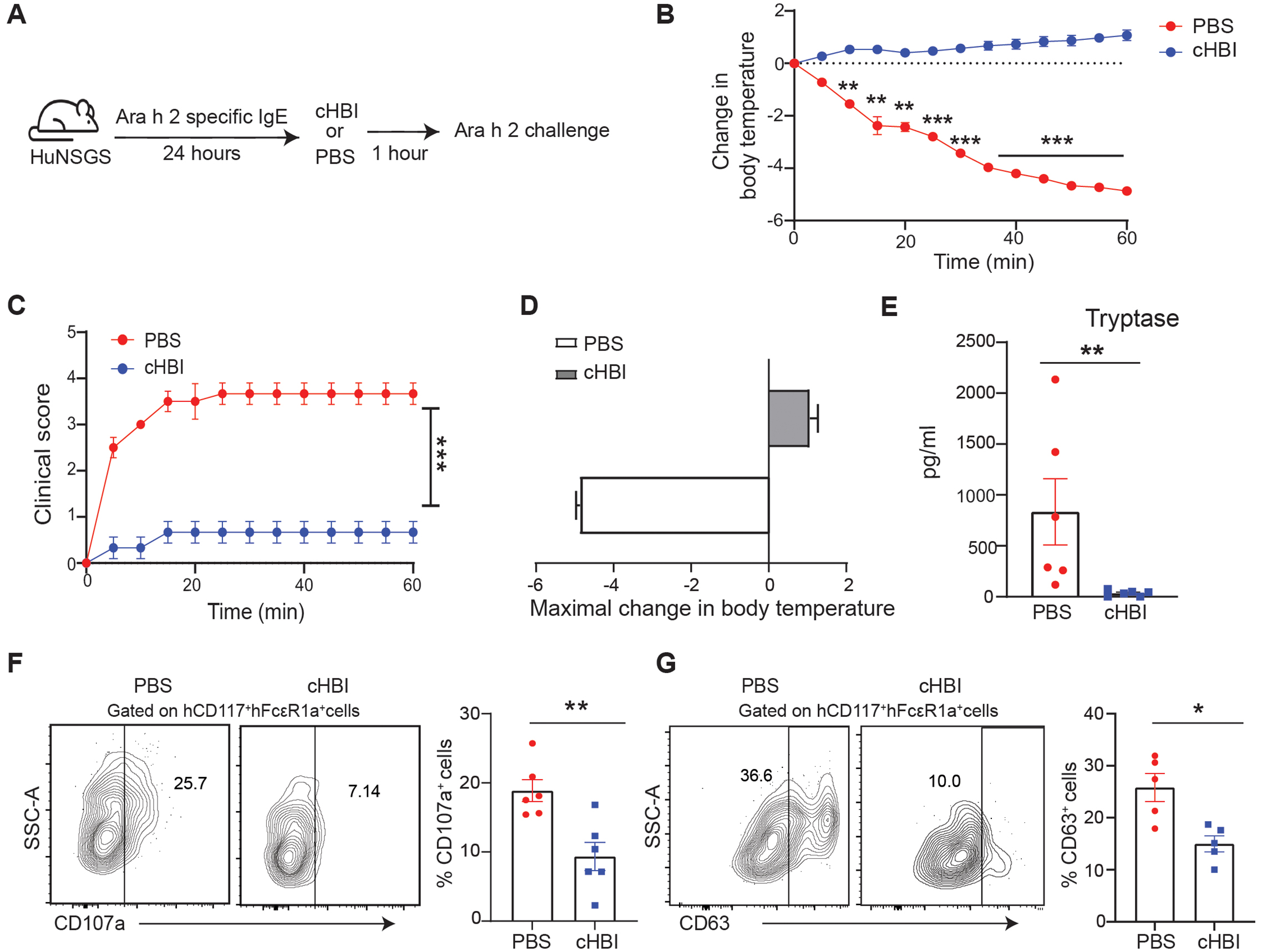
(A) Schematic illustration of PSA in huNSGS mice. Mice were sensitized with a cocktail of six Ara h 2 specific IgE mAbs (1 μg) and then were treated with cHBI (20 nmol) or PBS 1 hour prior to Ara h 2 allergen (1 ng) intravenous challenge. (B) Change in core body temperature, and (C) observable clinical scores in PBS-treated mice and cHBI-treated mice post Ara h 2 allergen challenge. Data represent mean ±SEM (n = 4). (D) Maximal change in body temperature measured every 5 minutes post Ara h 2 challenge in the PBS-treated mice and cHBI-treated mice. Data represent mean ±SEM, (n = 4). Data are representative of two independent experiments. (E) Scatter plot of serum tryptase levels in huNSGS mice (n = 6) data are pooled from two independent experiments. (F-G) Representative flow plot (left) of surface expression levels of splenic mast cell degranulation markers CD107a (F), and CD63 (G) scatter plots (right) (n = 6). Data are pooled from two independent experiments. One way ANOVA with Tukey post hoc test was used to compare means in (B) and (C). An unpaired Mann Whitney test was used to compare means in (E), (F), and (G). ***P <0.001, **P <0.01, *P <0.05.
A single cHBI injection protects huNSGS mice from anaphylactic responses for extended times
The effective inhibition of anaphylactic reactions in huNSGS by cHBI at 1 hour prompted us to examine cHBI efficacy for an extended time. HuNSGS mice were treated either with cHBI or PBS 24 hours after Ara h 2-specific IgE-sensitization. Mice were then challenged with Ara h 2 at four time-points 24 hours, 72 hours, 7 days, and 14 days after cHBI treatment. cHBI-treated mice had no significant change in core body temperature when challenged with 1 ng of Ara h 2 at 24 hours, 72 hours, or 7 days post cHBI treatment when compared with PBS-treated mice (Fig. 5, A–C and E). In contrast to cHBI-treated mice, PBS-treated mice had significant decrease in core body temperature, severe clinical symptoms and lethal responses at the three time points tested (Fig. 5, A–C, E, and data not shown). Challenging the huNSGS mice with 10 ng of Ara h 2 on day 14 provoked potent anaphylactic reactions and a significant decline in core body temperature in PBS-treated mice compared to cHBI-treated mice, demonstrating that cHBI is effective in preventing IgE-mediated anaphylaxis to Ara h 2 allergen for at least 14 days (Fig. 5, D and E). Moreover, cHBI effectively inhibited mast cell activation demonstrated by significantly lower serum tryptase levels in cHBI-treated mice compared with PBS-treated mice at all time points tested (Fig. 5F).
Fig. 5: PSA inhibition by cHBI is effective for 14 days.
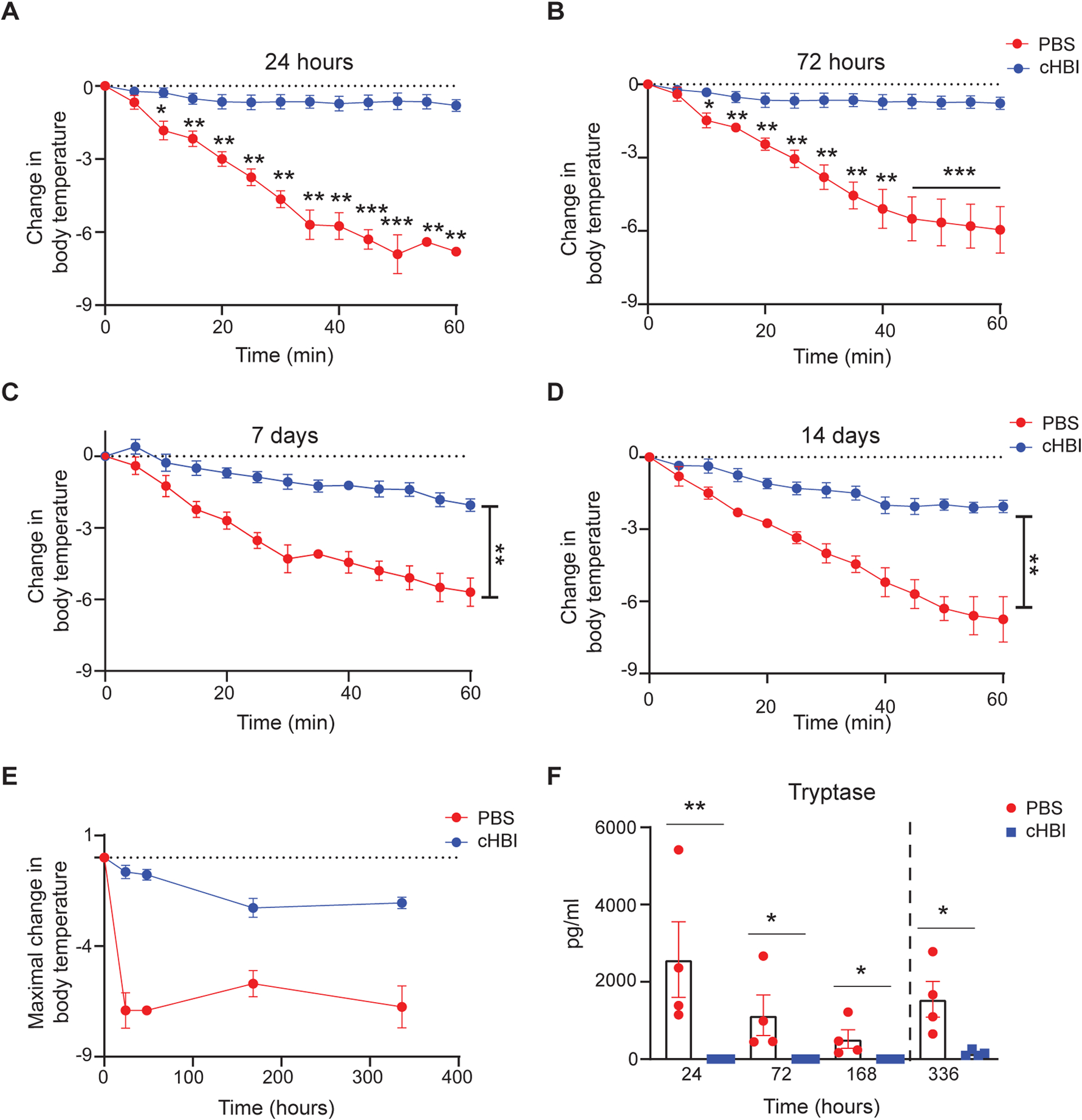
Mice were treated with either cHBI (20 nmol) or PBS post sensitization with a cocktail of six Ara h 2 specific IgE (1 μg) and then challenged intravenously with Ara h 2 after 24 hours, 72 hours, 7 days or 14 days. At 24, 72, 168 hour time points the Ara h 2 dose was 1ng; on day 14 the dose was 10 ng. (A-D) Change in core body temperature at the indicated time points post cHBI treatment. (E) Maximal change in body temperature post Ara h 2 challenge in the PBS-treated mice and cHBI-treated mice. Data represent mean ±SEM, (n = 4). Data are representative of two independent experiments. (F) Scatter plot of serum tryptase levels from mice at the indicated time points (n = 4). Data are representative of two independent experiments. One way ANOVA with Tukey post hoc test was used to compare means in (A-D). An unpaired Mann Whitney test was used to compare means in (F). ***P <0.001, **P <0.01, *P <0.05. Dashed line in F indicates different allergen dose.
To further define the duration of cHBI efficacy, we sensitized the huNSGS mice as described above and treated the mice with cHBI or PBS 24 hours post sensitization. HuNSGS mice were challenged intravenously with Ara h 2 at 4 or 6 weeks post cHBI treatment. While protection ranged from 60–70% and was similar at weeks 1 and 2 after cHBI injection, protection was decreased to less than 50% by 4 weeks and minimal protection was observed at week 6 (fig. S2A). This set of experiments also highlights one of the limitations of the model; IgE is not replenished in vivo and mice have diminished sensitivity to allergen over time which we interpret as loss of IgE bound to FceRI. Thus, past the one-week time point, mice required increasing doses of Ara h 2 to generate anaphylaxis (fig S2B). Overall, our data demonstrate that cHBI is protective for an extended period of time.
cHBI activity is allergen-specific
To investigate if the cHBI inhibition is allergen-specific, we compared cHBI inhibition of allergic reactions mediated by Ara h 2 allergen and a tree nut allergen, Anacardium occidentale 3 (Ana o 3) of cashew. HuNSGS mice were sensitized with a cocktail of six Ara h 2 specific IgE mAbs at 1 μg and with three IgE mAbs specific to cashew Ana o 3 at 1 μg. HuNSGS mice were then challenged with either 10 ng of Ara h 2 or Ana o 3 allergens 24 hours post cHBI or PBS treatment (Fig. 6A). Challenge with Ara h 2 provoked robust allergic reactions indicated by a significant drop of body temperature in PBS-treated mice compared with cHBI-treated mice which had minimal change in body temperature (Fig. 6B). In contrast to Ara h 2, Ana o 3 challenge provoked similar severe allergic reactions in both PBS and cHBI-treated mice (Fig. 6C). cHBI-treated mice challenged with Ara h 2 exhibited minimal drop in body temperature (−1.9 °C) compared to control mice (Fig. 6D). In contrast, cHBI-treated mice challenged with Ana o 3 exhibited a substantial drop of body temperature (−6.2 °C), comparable to the maximal drop of body temperature in PBS-treated mice (−5.4 °C, and −6.7 °C) challenged with Ara h 2 or Ana o 3, respectively (Fig. 6D). cHBI inhibited mast cell activation to Ara h 2 challenge indicated by the significant difference in serum tryptase levels (Fig. 6E), while PBS and cHBI treated mice challenged with Ana o 3 exhibited similar serum tryptase concentration (Fig. 6F). We further assessed cHBI specificity by comparing Ara h 2 allergen to a non-food allergen, house dust mite (HDM), dermatophagoides pteronyssinus 2 (Der p 2) allergen. HuNSGS mice were sensitized with a cocktail of six Ara h 2 specific IgE mAbs and two IgE mAbs specific to Der p 2. HuNSGS mice were then challenged with Ara h 2 or HDM extract 24 hours post cHBI treatment (fig. S2A). While cHBI attenuated the drop in core body temperature when challenged with Ara h 2 (fig. S2B), it had no effect on the temperature drop induced by Der p2.
Fig. 6: Inhibition of the allergic reactions by cHBI is allergen-specific.
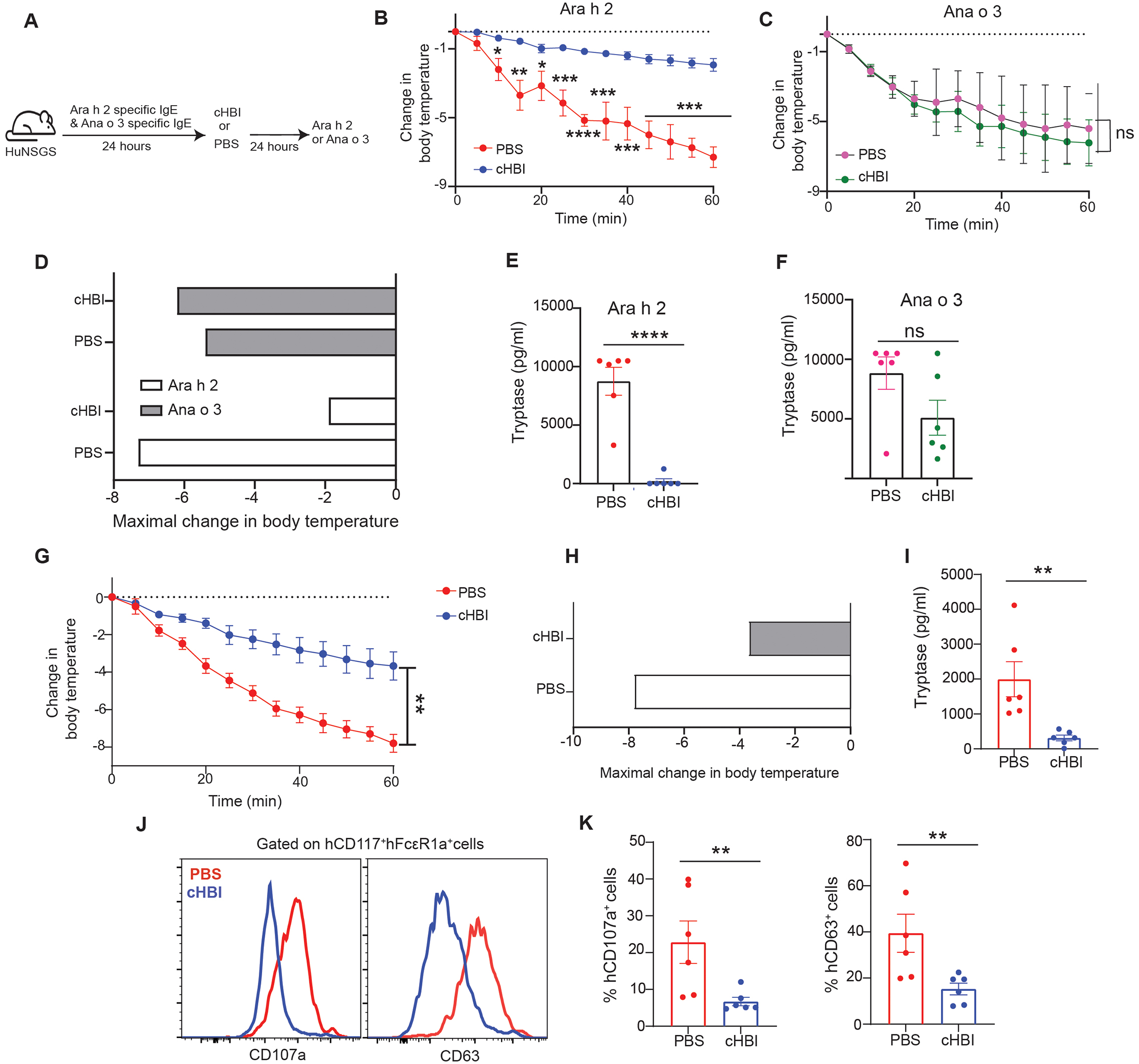
Schematic illustration of PSA where huNSGS mice were dual sensitized with (1 μg) of a cocktail of six Ara h 2 specific IgE and (1 μg) of 3 Ana o 3 specific IgE mAbs then treated with cHBI or PBS 24 hours before Ara h 2 (10 ng) or Ana o 3 (10 ng) intravenous challenge (B) Change in core body temperature in PBS-treated mice and cHBI-treated mice post the Ara h 2 challenge. (C) Change in core body temperature in PBS-treated mice and cHBI treated mice post Ana o 3 challenge. Data represent mean ±SEM (n = 6). (D) Maximal change in body temperature post Ara h 2 and Ana o 3 challenges in the PBS-treated mice and cHBI-treated mice. Scatter plots of serum tryptase levels in PBS-treated mice and cHBI-treated mice post Ara h 2 challenge (E) and Ana o 3 challenge (F) (n=6). (G-K) HuNSGS mice were sensitized with (1 μg) of a cocktail of six Ara h 2 specific IgE and treated after 24 hours with (20 nmol) cHBI or PBS. Mice were challenged with peanut extract (1ng) 24 hours post cHBI treatment. (G) Change in core body temperature in PBS-treated mice and cHBI treated mice post peanut extract challenge. Data represent mean ±SEM, (n = 6) (H) Maximal change in body temperature post peanut challenge in PBS-treated mice and cHBI-treated mice. (I) Scatter plot of serum tryptase levels in PBS- and cHBI-treated mice (n = 6). (J) Representative flow plot showing surface expression levels of degranulation markers CD107a, and CD63 on peritoneal mast cells. (K) Scatter plots of the percentage of CD107a, and CD63 positive cells (n = 6). (G-K) Data are pooled from two independent experiments. One way ANOVA with Tukey post hoc test was used to compare means in (B), (C), and (G). One way ANOVA followed by unpaired t-test was used to compare means in (E), (F), (I), and (K). ****P <0.0001, ***P <0.001, **P <0.01, *P <0.05. ns indicates no statistical difference.
We then tested the ability of cHBI to attenuate anaphylactic reactions when Ara h 2 was present with other allergens in crude peanut-extract. HuNSGS mice were sensitized as above and treated with cHBI or PBS as a control before peanut-extract intravenous challenge. Consistent with the protection observed with isolated Ara h 2 allergen, cHBI protected the mice from developing severe anaphylactic reactions demonstrated with significantly less decline in core body temperature when compared to PBS-treated mice (Fig. 6G and H). cHBI also effectively prevented mast cell activation to peanut challenge (Fig. 6I–K). Thus, cHBI demonstrates allergen-specificity in blocking Ara h 2 allergen-induced anaphylaxis and the ability to block Ara h 2-dependent anaphylaxis as part of crude peanut extract.
cHBI is effective as a treatment following the onset of anaphylaxis
Having demonstrated that cHBI inhibits anaphylaxis when administered as a preventive therapy, we also wanted to test whether it had efficacy after the onset of anaphylaxis. We administered cHBI two minutes after mice were challenged with Ara h 2, a time point when a patient would begin to feel symptoms of an allergic reaction. We observed that treatment with cHBI post allergen challenge attenuated the loss of core body temperature compared with mice treated with PBS (Fig. 7A). Treatment with cHBI post-allergen challenge also limited mast cell activation as demonstrated with significantly lower tryptase serum levels (Fig. 7B) and substantial lower levels of CD63 (Fig. 7C and D). These data suggest that administration of cHBI at the onset of allergic symptoms could decrease the severity of the allergic reaction.
Fig. 7: cHBI attenuates symptoms when administered after the onset of anaphylaxis.
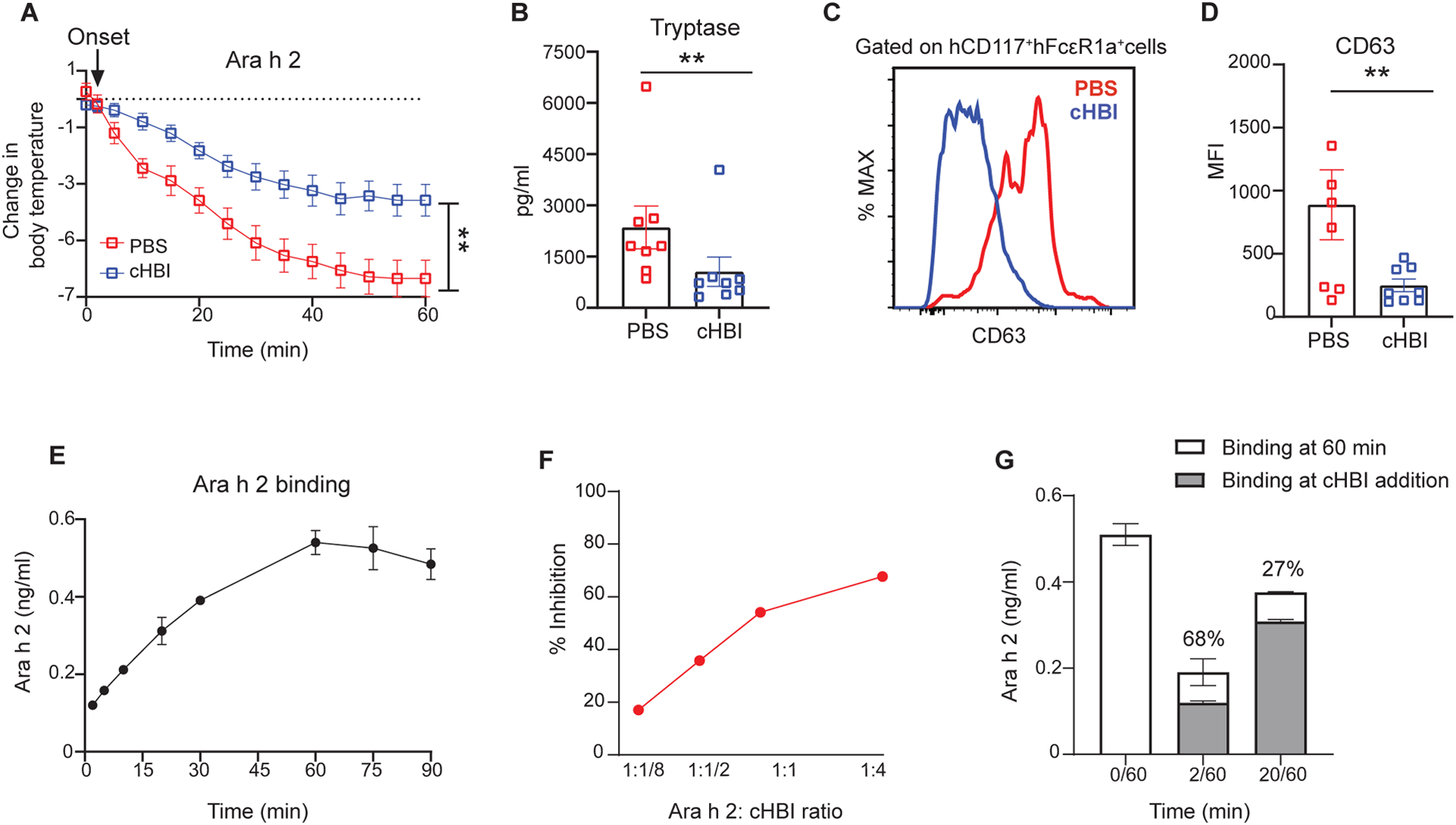
(A) HuNSGS mice were sensitized with a cocktail of six Ara h 2 specific IgE (1 μg), after 24 hours mice were challenged intravenously with Ara h 2 (1 ng). cHBI (20 nmol) or PBS was administrated two minutes post allergen challenge after the initial drop in core body temperature. (A) Change in core body temperature in PBS-treated mice and cHBI-treated mice. Data represent mean ±SEM (n=8). Data are pooled from two independent experiments. (B) Scatter plot of serum tryptase levels in PBS-treated mice and cHBI-treated mice. (C) Representative flow plot showing surface expression levels of degranulation marker CD63 on peritoneal mast cells. (D) Scatter plot of the median fluorescence intensity (MFI) of CD63 positive cells (n = 8). Data are pooled from two independent experiments. Arrow in (A) indicates the time of cHBI onset of anaphylaxis administration. (E) Time-dependent binding of Ara h 2 using ELISA with 38B7 as the capture IgE and 16A8 as the detection antibody. (F) Percent inhibition of Ara h 2 bound in the ELISA described in (E) when cHBI is added two minutes after Ara h 2. (G) Attenuation of Ara h 2 binding by cHBI assessed at 60 minutes when cHBI is added at 2 minutes (2/60) or 20 minutes (20/60). Gray bars represent Ara h 2 concentration at time of cHBI addition. Stacked white bars represent concentration of Ara h 2 after 60 minutes. Data represent mean ±SEM of technical replicates, and are representative of three independent experiments. One way ANOVA with Tukey post hoc test was used to compare means in (A). One way ANOVA followed by unpaired t-test was used to compare means in (B), and (D). **P <0.01.
These results raised the question of whether cHBI only prevented Ara h 2 binding, or whether cHBI displaced bound Ara h 2. To define these interactions, we developed an ELISA for Ara h 2 concentration using the cHBI-binding 38B7 as the capture antibody. Assessing the concentration of Ara h 2 over time, we found increasing binding over the first sixty minutes at which time binding was maximal and equivalent to the concentration added to the assay (Fig. 7E). At the time point of maximal Ara h 2 binding, we observed concentration dependent inhibition by cHBI when added two minutes after Ara h 2, and inhibition reached a peak of about 70% (Fig. 7F).
With this assay established we then tested whether cHBI attenuated binding of Ara h 2 or displaced Ara h 2 that was already bound to IgE. The cHBI was added at two minutes after Ara h 2, parallel with the in vivo experiment, or 20 minutes after Ara h 2 and the concentration of bound Ara h 2 was assessed after one hour. cHBI added at two minutes blocked nearly 70% Ara h 2 binding, as noted above, but the amount of Ara h 2 bound at 60 minutes in the presence of cHBI for 58 minutes still increased slightly over Ara h 2 bound at two minutes (Fig. 7G). The increased binding after cHBI addition suggested that the inhibitor was interfering with binding but not displacing bound Ara h 2. Similar results were seen when cHBI was added at 20 minutes with attenuation of Ara h 2 binding at 60 minutes, and overall inhibition less than 30% (Fig. 7G). Together, these data demonstrate that Ara h 2 binding increases over time, and that cHBI attenuates Ara h 2 binding but does not displace Ara h 2 already bound to IgE. It further suggests that cHBI added early following allergen challenge can inhibit enough Ara h 2 binding to block fatal anaphylaxis.
cHBI protects huNSGS mice from anaphylactic response to oral Ara h 2 challenge
While allergen injection is a widely used model of systemic anaphylaxis, we wanted to test the efficacy of cHBI as a preventive for peanut exposure through the gastrointestinal tract. We chose a dose of Ara h 2 that in a w/w ratio, based on 1 peanut being about 250 mg of protein (38) and Ara h 2 being about 7% of the total peanut protein (39) was equivalent to a human eating about 4 peanuts. This is likely more than an accidental exposure but less than a complete patient food challenge where the tolerated dose is cumulatively escalated to about 30 peanuts (40). HuNSGS sensitized with Ara h 2-specific IgE were gavaged with 25 μg Ara h 2 twenty-four hours after cHBI treatment. Remarkably, PBS-treated huNSGS mice exhibited anaphylactic reactions with a significant decline in core body temperature (maximal drop in temperature −8.0°C) (Fig. 8, A and C) and fatal anaphylactic responses observed in 6 mice during the 60 minute challenge (Fig. 8B). In contrast, cHBI treated mice had a minor change in core body temperature (maximal drop in temperature ~ −2°C) (Fig. 8, A and C). Importantly, cHBI prevented the huNSGS mice from developing a fatal anaphylactic response (Fig. 8B). We further confirmed that the host murine mast cells had no contribution to the anaphylactic response observed in huNSGS mice, showing that non-engrafted NSGS control mice sensitized with human specific IgE and challenged with oral Ara h 2 exhibited no change in core body temperature (fig. S1L).
Fig. 8: cHBI inhibited anaphylaxis and mast cell activation to oral Ara h 2 allergen challenge.
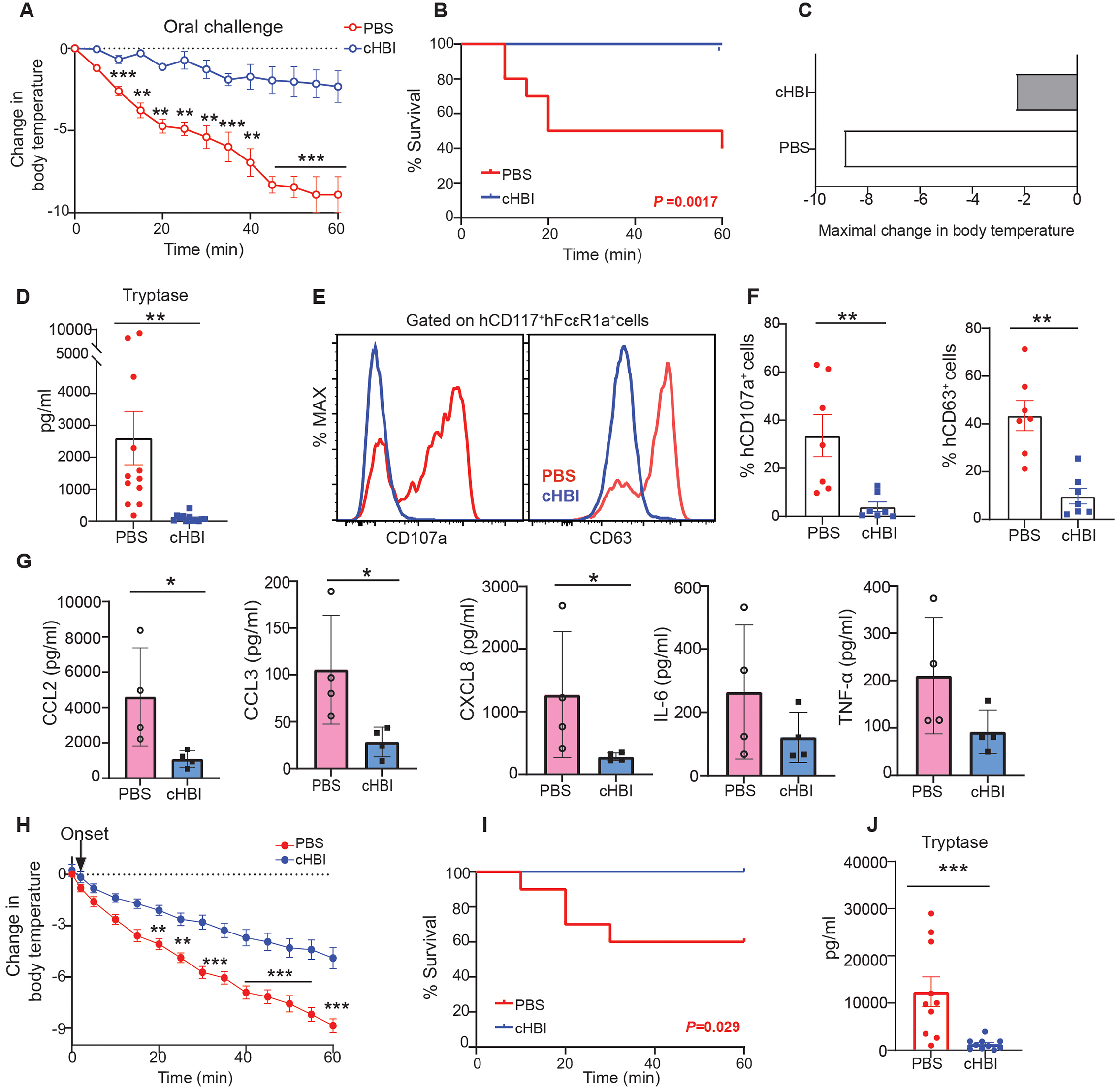
(A) Change in core body temperature in PBS-treated mice and cHBI (20 nmol) treated mice post Ara h 2 allergen (25 μg) oral challenge (B) Kaplan-Meyer survival plot for PBS- and cHBI-treated mice post oral Ara h 2 allergen challenge. Data represent mean ±SEM pooled from two independent experiments (n = 12). (C) Maximal change in body temperature post oral Ara h 2 allergen challenge in PBS-treated mice and cHBI-treated mice. Data represent mean ±SEM, (n = 12) (D) Scatter plot of serum tryptase levels in PBS- and cHBI-treated mice (n = 12). Data are pooled from two independent experiments. (E) Representative flow plot showing surface expression levels of degranulation markers CD107a, and CD63 on peritoneal mast cells. (F) Scatter plots of the percentage of CD107a, and CD63 positive cells (n = 6–7). Data are representative of two independent experiments. (G) Chemokine (CCL2, CCL3, and CXCL8) and cytokine (IL-6, and TNF-α) serum concentrations post Ara h 2 allergen oral challenge in PBS-treated mice and cHBI-treated mice (n = 4). Data are representative of two independent experiments. (H) Change in core body temperature in mice treated with PBS or cHBI two minutes after oral allergen challenge. Arrow indicated time of cHBI administration. (I) Kaplan-Meyer survival plot for mice treated with PBS or cHBI two minutes after oral allergen challenge. (J) Scatter plot of serum tryptase levels in mice treated with PBS or cHBI two minutes after oral allergen challenge. Data represent mean ±SEM. Data are pooled from two independent experiments (n=10). One way ANOVA with Tukey post hoc test was used to compare means in (A), and (H). A log-rank (Mantel-Cox) test was used for statistical analysis in (B), and (I). An unpaired Mann Whitney U test was used to compare means in (D), (F), (G), and (J). ***P <0.001, **P <0.01, *P <0.05.
We next assessed mast cell activation in huNSGS in response to oral Ara h 2 challenge. PBS-treated huNSGS mice had significantly elevated serum tryptase levels post challenge (Fig. 8D). Additionally, PBS-treated huNSGS mice had significantly increased expression of CD107a and CD63 surface expression on human mast cells (Fig. 8, E and F). In contrast, cHBI-treated mice had much lower levels of serum tryptase and low CD107a and CD63 expression on human mast cells (Fig. 8, D to F).
In addition to mast cell tryptase being used as a biomarker for anaphylaxis in the clinic, other mediators have been identified as potential markers for human anaphylaxis including the chemokine CCL2 (41). We measured the levels of CCL2, CCL3, CXCL8, IL-6, and TNF-α in the serum of huNSGS mice taken one hour after oral Ara h 2 challenge. We detected significantly elevated CCL2, CCL3, and CXCL8 levels in PBS-treated mice when compared with cHBI-treated mice (Fig. 8G). While IL-6 and TNFα were not significantly increased, this time point was likely too early to detect maximal concentrations of these cytokines in serum (42–44).
Finally, we tested whether cHBI administered after the onset of allergic symptoms following oral gavage was effective. HuNSGS mice sensitized with IgE and challenged with Ara h 2 as above were injected with cHBI two minutes after oral peanut challenge. We observed that cHBI significantly attenuated the allergic reactions when administered after the first temperature drop (Fig. 8H). Importantly, cHBI protected the mice from fatal anaphylactic responses (Fig. 8I). Treatment with cHBI post-oral challenge attenuated mast cell activation with significantly lower tryptase serum levels in cHBI treated mice compared with PBS-treated mice (Fig. 8J). Collectively, our results demonstrate that cHBI effectively protects the mice from anaphylactic responses and prevents mast cell activation following oral Ara h 2 challenge.
DISCUSSION
In this study, we developed a novel peanut-specific humanized mouse model that mimics key features of human anaphylaxis to peanut allergy. We passively sensitized humanized mice with a cocktail of human IgE mAbs targeting different epitopes of the clinically relevant Ara h 2 peanut allergen and showed that the humanized mice develop severe anaphylaxis when challenged with Ara h 2 allergen. To our knowledge, this establishes the use of human IgE mAbs specific to peanut Ara h 2 allergen generated from peanut-allergic patients for sensitization in a humanized mouse model. Other reports demonstrated that humanized mice develop hypersensitivity reactions and anaphylaxis using human chimeric IgE specific to the hapten 4-hydroxy-3 nitrophenacetyl acetyl (NP) for sensitization and NP-BSA for allergen-induced anaphylaxis (27, 28). However, models that use monoclonal IgE and polyvalent allergens have the potential for a biased assessment of allergen responses. The use of mAbs to multiple epitopes of an allergen in our model increases the physiological relevance to patients.
Anaphylaxis to Ara h 2 allergen in huNSGS mice correlates with the degranulation of mature human mast cells distributed across various tissues. Mast cells in humanized mice are phenotypically similar to mast cells in human, and functionally underwent degranulation in an IgE-dependent manner (32, 45). In clinic, serum mast cell tryptase concentrations are considered a diagnostic measurement for anaphylaxis (34, 46). Released tryptase can be detected in human serum within minutes after the first symptoms of anaphylaxis and can be detected for 24 to 48 hours (34, 46, 47). To parallel diagnostic methods of human anaphylaxis in the clinic, we used tryptase serum concentration as an indicator of anaphylaxis and mast cell activation in huNSGS mice. Our results demonstrate that in huNSGS mice, tryptase release is associated with the severity of the anaphylactic response and dependent on IgE and Ara h 2 doses. Overall, our humanized mouse model is advantageous over other mouse models as we demonstrate sensitivity and specificity for Ara h 2 using allergic patient-generated IgE mAbs. Additionally, the model can be utilized to investigate the mechanisms of mast cell-mediated allergic reactions to peanut allergens. Most importantly, it can be used as a screening model to examine therapeutic treatment specificity and efficacy for peanut allergies.
To date, there is no allergen-specific preventive treatment for peanut or food allergies, aside from dietary avoidance or OIT (6, 10). Although anti-IgE therapy can attenuate allergic reactions, anti-IgE treatments lack specificity, targeting and neutralizing all IgE antibodies. Nonspecific neutralization of all IgE can have undesirable effects. Parasite-specific IgE antibodies protect from parasite infections and mediate Th2 response during helminth-mediated infection (48, 49). Patients with IgE deficiency have an increased high risk of cancer susceptibility (50–52). Additionally, IgE antibodies have a critical role in regulating mast cell homeostasis and have immunomodulatory functions, which impact the ability of mast cells to respond to other stimuli (53, 54). Therefore, developing food allergen specific therapy targeting specific IgE is a putative approach to avoid the negative effect of neutralizing all IgE antibodies as it efficiently prevents anaphylaxis to food allergen exposure.
We previously described the development of cHBI, a peanut-specific IgE inhibitor, that binds irreversibly to the allergen binding site on specific IgE antibody preventing allergen binding thus inhibiting peanut allergen mediating allergic reactions. We demonstrated that using two cHBI molecules targeting the specific IgE that recognize immunodominant epitopes in Ara h 2 and Ara h 6 peanut allergens effectively prevented mast degranulation and basophil activation to crude peanut in vitro and ex-vivo (23, 55). Remarkably, in the huNSGS mouse model cHBI demonstrates a complete protection of anaphylaxis to peanut Ara h 2 allergen. An interesting finding in our study is that despite sensitizing the huNSGS mice with a cocktail of IgE mAbs targeting different epitopes of Ara h 2, cHBI effectively prevented allergic reactions in huNSGS mice to Ara h 2 indicating the ability of cHBI to prevent anaphylaxis in this model despite IgE epitope heterogeneity. As previously reported, high affinity IgE antibodies are cross-reactive and can bind to distinct major peanut allergens with differing affinity (22); cross-inhibition with allergens and mutant IgE abrogated patient IgE binding to the two peanut allergens Ara h 2 and Ara h 3 (21, 22). Results in the huNSGS model are in line with our previous report showing the ability of cHBI to prevent mast cell degranulation in vitro suggesting that the inhibition of one weak affinity epitope in the presence of high affinity epitopes can be effective in attenuating IgE-mediated responses (56, 57). The results demonstrating a major contribution of the 38B7-recognized epitope to anaphylaxis further highlight the importance of antibodies that are immunodominant in function even when present at concentration similar to other IgE. It further suggests that immunodominance is not simply a matter of abundance, but rather that there is functional dominance of the epitope. This finding fits with the ability of cHBI targeting one epitope on Ara h 2 and one on Ara h 6 to broadly impair degranulation responses to a panel of patient sera (14). Further experiments will yield new insights into allergen-IgE interactions.
There is still the question of when cHBI would be administered. We demonstrated that cHBI administered shortly after the onset of allergic symptoms attenuated the severity of the allergic reaction. However, cHBI appears to be more efficacious as a preventive measure. Our data show that a single dose of cHBI was sufficient to sustain complete protection from anaphylaxis and prevent lethal anaphylactic response in huNSGS mice for at least two weeks. Importantly, the cHBI molecule does not cause significant toxicity in a mouse model (23), and this is being further tested in non-human primates. Based on our results in the humanized mouse model, we propose that a bi-monthly dose of cHBI would efficiently prevent allergic reactions in patients sensitized to Ara h 2.
During anaphylaxis, several mediators are released from mast cells including histamine, tryptase, and cytokines (5). Preventing the release of these mediators has critical therapeutic implication on reducing the severity of anaphylactic reactions after allergen exposure. Here, we show that cHBI effectively prevents the activation of mast cells in systemic and oral challenges and attenuates the release of mast cell mediators. In addition, to decreased serum tryptase levels in huNSGS mice following Ara h 2 allergen challenge, we show that cHBI decreases CCL2 serum levels in the huNSGS mice. CCL2 is a chemokine recently identified as a mast cell-derived biomarker of human anaphylaxis that positively correlates with tryptase levels in patients’ blood during anaphylaxis (41, 58). The ability of cHBI to block the release of mast cell mediators in huNSGS mice suggest similar protective effects in peanut allergic patients after accidental peanut exposure.
In summary, we show that cHBI acts as a peanut allergen inhibitor and efficiently prevents anaphylaxis to peanut allergen in the humanized mouse model. If the effective protection by a single dose of cHBI observed in this study translates in human subjects, cHBI could be a potential preventative therapy for peanut allergies. Notably, the described cHBI design approach can be applied to other IgE-dependent food and drug allergies, as well as any other pathologic conditions that involve immunoglobulins, such as autoimmune diseases.
MATERIALS AND METHODS
Study design
This study was designed to investigate the efficacy of an allergen-specific inhibitor termed cHBI in preventing anaphylaxis to peanut Ara h 2 allergen in an in vivo model relevant to human clinical anaphylaxis. We developed a humanized mouse model of passive systemic anaphylaxis to peanut allergen using a cocktail of six human IgE mAbs specific to Ara h 2 generated from peanut allergic patients for sensitization and Ara h 2 allergen for challenge. We demonstrated that the model is predictive of anaphylaxis in an IgE and allergen-dose dependent manner. We then assessed cHBI efficacy in systemic and oral challenges and demonstrated that cHBI is effective and specific in preventing peanut Ara h 2 induced anaphylaxis. Data are representative or pooled from at least two independent experiments. Animal numbers and statistical analyses are detailed in the figure legends or materials and methods section. Randomization of the humanized mice was implemented in all experiments.
Humanized mouse model
All experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Indiana University. The triple transgenic (NOD. Cg-Prkdc scid Il2rg tm1wjl Tg (CMV-IL-3,CSF2,KITLG)1Eav./MloySzJ) were purchased from the Jackson Laboratory (Stock number 013062) and bred in house by the IUSCCC In Vivo Therapeutics Core. Mice were maintained under pathogen free conditions in ventilated cages and were given ad libitum irradiated Teklad Uniprim Medicated Diet (TD 06596, Harlan Laboratories, Indianapolis, IN) and autoclaved, acidified water. Prior to transplant, cryopreserved human cord blood CD34+ cells (Stem Cell Technologies or Lonza) were thawed and cultured for 4 hours at (5 × 105) cells per mL in StemSpan serum free media (StemCell Technologies, Vancouver Canada) supplemented with stem-cell factor (SCF) at (100ng/mL) (PeproTech, Rocky Hill, NJ). Cells were then counted in trypan blue and were 95–98% viable. Adult female NSGS mice 6–8 weeks were sublethally irradiated (100cGy) and injected intravenously 4 hours later with (2 ×104) human CD34+ cells.
Isolation of mononuclear cells from peripheral tissues
The reconstitution levels of human cells in huNSGS mice were analyzed 10–12 weeks after human CD34+ cell transplant in peripheral tissues and whole blood. Briefly, various tissues were harvested from huNSGS mice. Lung, stomach, and skin were minced, and digested in DMEM containing 10% FBS, 0.05% collagenase IV (Gibco), and 0.01% DNase I (Roche) at 37 °C for 30 minutes with orbital shaking at 300 rpm. Spleens were dissociated using gentleMACS C tubes (Miltenyi Biotech). Intestines were digested in DMEM containing 0.1% Collagenase I, 0.25%, Collagenase IV, and 7.5 ug/mL DNase I. The digested tissues were washed and mashed in 100μm filter in DMEM media. Peritoneal cells were recovered from huNSGS mice by flushing the peritoneal cavity with 5 ml of HBSS containing 0.5% EDTA. Bone marrow cell were obtained by flushing the bone with HBSS containing 0.5% EDTA. Red blood cells in all samples were lysed using (Ammonium-Chloride-Potassium) ACK lysis buffer (Gibco).
Flow cytometry
Mononuclear cells isolated from bone marrow, spleen, lung, peritoneal, stomach, intestine, and skin and RBC-lysed cells were incubated with Live/Dead flexible dye eFlour 780 (eBioscience) and Fcγ block (BD 1:50). Cells were washed, and surface stained with fluorochrome-conjugated monoclonal antibodies specific for human CD45 (clone: H130), CD117 (clone: 104D2), FcεR1α (clone: AER-37), CD107a (clone: H4A3), and CD63 (clone: H5C6), IgE (clone: MHE-18), and antibodies specific for mouse CD45 (clone: 30-F11), CD117 (clone: 2B8), FcεR1α (clone: MAR-1), and IgE (clone:R35–72) all from Biolegend or BD bioscience. All antibodies were used at 1 to 100 dilution. Stained cells were washed and fixed with 2% of paraformaldehyde. Cells were analyzed by LSR Fortessa (BD Bioscience), and data were analyzed with FlowJo 10.7.2 (Tree Star).
Human hybridoma generation and IgE mAb production
IgE secreting human hybridomas were generated using methodology described in detail (26). Peanut, cashew, and dust mite-specific IgE mAbs were created from peanut, cashew, and/or dust mite allergic research subjects recruited from within the Vanderbilt University Medical Center. Diagnosis was based on clinical history, skin prick testing, and testing serum for the presence and quantity of IgE antibody to peanut or dust mite. Cryopreserved peripheral blood mononuclear cells were grown in culture for 7 days prior to screening for IgE isotype secretion using an ELISA. A non-secreting myeloma cell was used to immortalize IgE secreting B-cells by electrical cytofusion. Hybridomas secreting IgE antibody were selected using HAT medium and cloned biologically by indexed single cell flow cytometric sorting. Screening for peanut and dust mite-specificity did not occur until IgE monoclonal antibodies were generated, thus no allergen-specific bias was introduced during the B cell screening or hybridoma generation process. Each hybridoma was expanded, IgE mAb expressed in a liter of serum free medium and purified by omalizumab immunoaffinity chromatography. IgE specificity was determined using allergen extract and recombinant proteins in binding assays.
Human IgE specific mAbs
The clones of the six Ara h 2 specific IgE mAbs were: 5D7, 15A7, 16A8, 38B7 13D9, 8F3. The clones for Ana o 3 specific IgE mAbs were: 2F5, 4A7, and 14C12.The clones for Der p 2 specific IgE mAbs were: 2G1 and 2F10. All IgE mAbs were generated as described (26).
cHBI generation
cHBI was synthesized using the methods previously described (23). Briefly, the epitope was synthesized as a linear peptide using Fmoc-amino acids activated with a 3.6-fold excess of HBTU with 20-fold DIEA for 5 min before addition. The Fmoc was deprotected and a HBTU-activated CF was added. Then the IvDdE group was deprotected with 2% hydrazine in DMF and epitope synthesis was performed. Following epitope synthesis, a Fmoc-EG8-OH spacer was added, followed by conjugation of Fmoc-Lys(IvDdE)-OH. To the Fmoc-protected amine, IBA-BOC was added. Following IBA-Boc addition, the IvDdE group of lysine was deprotected using 2% hydrazine in DMF in the same fashion. Fmoc-EG4-OH spacer was added to the e-amine of this lysine. ITC domain was added before cleavage from resin. Briefly, resins with deprotected primary amines were washed in anhydrous DMF three times. A 10-fold excess CS2 with a 20-fold excess of DIEA was added in DMF and allowed to react for 30 min. Resin was then drained and washed once with anhydrous DMF. One milliliter of DMF with a 20-fold excess of DIEA was added to resin and cooled to ~0°C in a −20 °C freezer. Then, a 2-fold excess of Boc2O and 0.2-fold of DMAP was added to the vessel and allowed to react for 20 min at −20 °C. The vessel was removed, allowed to warm to room temperature for 30 min, and then washed with DMF, DCM, and diethyl ether and allowed to dry in a vacuum chamber.
Degranulation assay
Degranulation assays were performed using nanoallergens as described previously in detail (23). RBL-SX38 cells were plated into 96-well plate for 24 hours, then primed with a cocktail of six Ara h 2 specific IgE mAbs or five Ara h 2 specific IgE mAbs each at (0.1 μg/mL) for 24 hours. After incubation, cells were washed and challenged with natural Ara h 2 allergen (Indoor Biotechnologies) at various amounts (0.05, 0.1,0.25, 0.5, 1.0, 2.0, 5.0, and 10.0 nM). Following allergen challenge, the supernatant was incubated with pNAG (poly-N-acetyl-D-glucosamine) solution for 45 min. The reaction was halted with a stopping solution and the degranulation was determined by reading absorbance at 405 nm with background subtraction. In the cHBI inhibition experiment, cells post IgE mAbs priming were incubated with cHBI at (1 μM) for 24 hours and challenged with Ara h 2 allergen at the indicated amounts. Percent degranulation was determined with Origin 7 software using methods described previously in detail (23, 59).
Passive systemic anaphylaxis
HuNSGS mice (10 to 12 weeks post-engraftment) were sensitized intravenously with doses of a cocktail of six Ara h 2 allergen specific IgE mAbs (as stated in the Fig. legends). Mice were challenged intravenously with Ara h 2 allergen at the indicated times and doses in the corresponding figure legend. Anaphylaxis was determined by measuring core body temperature using a rectal probe every 10 or 5 minutes for up to 60 minutes and assessing the survival rate. Visual clinical symptoms were scored as following 0 (no symptoms), 1 (stretching), 2 (edema around the eyes), 3 (labored breathing), 4 (unresponsive), 5 (death). All mice were euthanized at the end of the 60 minutes challenge with CO2 and tissues were collected immediately after euthanasia.
Anaphylaxis inhibition with cHBI
Mice were sensitized intravenously with a cocktail of six Ara h 2 specific IgE mAbs (1 μg), 24 hours post-sensitization mice were intravenously injected with cHBI (20 nmol) or PBS. Mice were then challenged intravenously with (1 ng) Ara h 2 allergen at the indicated time points. In some experiments mice were challenged with peanut extract at (1ng; Stallergenes Greer). In one experiment, cHBI was administered two minutes after allergen challenge. In the specificity experiment, mice were sensitized intravenously with (1 μg) of a cocktail of six peanut Ara h 2 allergen specific IgE mAbs, (1 μg) of three cashew Ana o 3 allergen specific IgE mAbs, and (1 μg) of two IgE mAbs specific for HDM Der p 2. 24 hours later post IgE sensitization mice were treated with cHBI (20 nmol) or PBS and then challenged after 24 hours with either Ara h 2 (10 ng) and Ano o 3 (10 ng; Indoor Biotechnologies) or Ara h 2 (1 ng) and HDM (10 ng; Stallergenes Greer). In all experiments, mice in the PBS group or cHBI group were chosen randomly regardless of human cell constitution levels.
Oral Ara h 2 allergen challenge
Mice were sensitized intravenously with a cocktail of six Ara h 2 specific IgE mAbs (1 μg), and 24 hours later, were injected intravenously with cHBI (20 nmol) or PBS. Ara h 2 challenge was performed by gavage feeding with (25 μg) of Ara h 2 allergen. In experiments where indicated, cHBI (20 nmol) was administered two minutes after allergen challenge. Anaphylaxis was assessed by core body temperature change every 5 minutes for a total of 60 minutes and survival rate.
ELISA and multiplex analysis
Blood samples were collected immediately after fatal anaphylactic shock during the challenge or at the end of the 60 minutes challenge. Serum was prepared by blood clotting at room temperature for 30 minutes. Human tryptase was quantified in the huNSGS mice serum using human tryptase alpha/beta1 ELISA kit from BIOMATIK following manufacture instructions. Human cytokine and chemokine (IFN-γ, IL-6, CCL2, CCL3, CXCL8, and TNFα) levels in the serum were analyzed using Bio-Plex 200 system and a custom multiplex assay (Milliplex MAP, Millipore).
In vitro mast cell stimulation
HuNSGS mice were sensitized intravenously with (1 μg) of six Ara h 2 specific IgE mAbs. After 24 hours, mice were euthanized, and peritoneal cells were recovered from huNSGS as described above. Human and mouse cells were enriched by flow sorting for live+ CD45+CD117+ cells. Sorted human and mouse cells were plated overnight at (1 × 10^5/well) into 96-well plate. On the following day, cells were stimulated with Ara h 2 at the indicated concentrations for 1 hour. Supernatants from the two cultures were analyzed for human tryptase by ELISA as described above.
Ara h 2 binding ELISA
Capturing IgE mAb clone 38B7 was coated into 96-well plate overnight at (1 μg/well). On the following day, Ara h 2 was added to the wells at (0.5 ng/ml) either alone or with cHBI added to the wells at the times indicated. cHBI was added at the indicated molar ratios relative to Ara h 2. Detection was performed with biotinylated detection IgE mAb clone 16A8 and streptavidin-horseradish peroxidase (HRP). A standard curve was constructed with Ara h 2 at 2-fold serial dilution in the range (2–0.015 ng/mL). The concentration of Ara h 2 was measured at the indicated times.
Statistics
All data was analyzed using JMP-14 or GraphPad Prism 9.1 (Statistical software). Statistical-significant was determined by unpaired two-tailed t test, Mann-Whitney U test, one-way analysis of variance (ANOVA) with Tukey post hoc test, and log-rank (Mantel-Cox) test. The statistical test used for each figure is stated in the corresponding figure legend. Data are presented as mean ± standard error of the mean (SEM). All dots on the plots represent biological replicates.
Supplementary Material
Acknowledgements:
The authors thank Amina Abdul Qayum for help in setting up the model and Barbara Bailey (IVT Core) for her expertise with preparation of CD34+ for transplantation studies. We thank Christopher Moreland for assistance in designing the competitive binding ELISA. We thank Dr. Alexander Dent for comments on the manuscript.
Funding:
Research in this report was supported by NIH R01 grants AI088884 (B.B., S.A.S and M.H.K.), AI129241 (M.H.K.), and AI155668 (S.A.S.). Portions of the research were supported by the Dr. Ralph and Marian Falk Medical Research Trust, Bank of America, Private Bank. Support provided by the Herman B Wells Center was in part from the Riley Children’s Foundation. This research was funded by the IU Simon Comprehensive Cancer Center Support Grant (P30CA082709) and by the IUSM NIDDK Cooperative Centers of Excellence in Hematology (CCEH) U54 DK106846.
Footnotes
Competing Interests: B.B. and M.H.K. are owners of IP to commercialize cHBI and similar molecules.
Data and materials availability:
All data associated with this paper are present in the paper or in the Supplementary Materials. Human IgE are available from S.A.S. under material transfer agreement.
References:
- 1.Gupta RS, Springston EE, Warrier MR, Smith B, Kumar R, Pongracic J, Holl JL, The prevalence, severity, and distribution of childhood food allergy in the united states. Pediatrics 128, e9–17 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Gupta RS, Warren CM, Smith BM, Jiang J, Blumenstock JA, Davis MM, Schleimer RP, Nadeau KC, Prevalence and severity of food allergies among us adults. JAMA network open 2, e185630 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burks AW, Peanut allergy. The Lancet 371, 1538–1546 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Anvari S, Miller J, Yeh CY, Davis CM, IgE-mediated food allergy. Clinical reviews in allergy & immunology 57, 244–260 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Reber LL, Hernandez JD, Galli SJ, The pathophysiology of anaphylaxis. The Journal of allergy and clinical immunology 140, 335–348 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Capucilli P, Wang KY, Spergel JM, Food reactions during avoidance: Focus on peanut. Annals of allergy, asthma & immunology : official publication of the American College of Allergy, Asthma, & Immunology 124, 459–465 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Cardona V, Ansotegui IJ, Ebisawa M, El-Gamal Y, Fernandez Rivas M, Fineman S, Geller M, Gonzalez-Estrada A, Greenberger PA, Sanchez Borges M, Senna G, Sheikh A, Tanno LK, Thong BY, Turner PJ, Worm M, World allergy organization anaphylaxis guidance 2020. The World Allergy Organization journal 13, 100472 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Du Toit G, Roberts G, Sayre PH, Bahnson HT, Radulovic S, Santos AF, Brough HA, Phippard D, Basting M, Feeney M, Turcanu V, Sever ML, Gomez Lorenzo M, Plaut M, Lack G, Randomized trial of peanut consumption in infants at risk for peanut allergy. The New England journal of medicine 372, 803–813 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du Toit G, Sayre PH, Roberts G, Sever ML, Lawson K, Bahnson HT, Brough HA, Santos AF, Harris KM, Radulovic S, Basting M, Turcanu V, Plaut M, Lack G, Effect of avoidance on peanut allergy after early peanut consumption. The New England journal of medicine 374, 1435–1443 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Patrawala M, Shih J, Lee G, Vickery B, Peanut oral immunotherapy: A current perspective. Current allergy and asthma reports 20, 14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chinthrajah RS, Purington N, Andorf S, Long A, O’Laughlin KL, Lyu SC, Manohar M, Boyd SD, Tibshirani R, Maecker H, Plaut M, Mukai K, Tsai M, Desai M, Galli SJ, Nadeau KC, Sustained outcomes in oral immunotherapy for peanut allergy (poised study): A large, randomised, double-blind, placebo-controlled, phase 2 study. Lancet (London, England) 394, 1437–1449 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones SM, Kim EH, Nadeau KC, Nowak-Wegrzyn A, Wood RA, Sampson HA, Scurlock AM, Chinthrajah S, Wang J, Pesek RD, Sindher SB, Kulis M, Johnson J, Spain K, Babineau DC, Chin H, Laurienzo-Panza J, Yan R, Larson D, Qin T, Whitehouse D, Sever ML, Sanda S, Plaut M, Wheatley LM, Burks AW, Efficacy and safety of oral immunotherapy in children aged 1–3 years with peanut allergy (the immune tolerance network impact trial): A randomised placebo-controlled study. Lancet (London, England) 399, 359–371 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vickery BP, Vereda A, Casale TB, Beyer K, du Toit G, Hourihane JO, Jones SM, Shreffler WG, Marcantonio A, Zawadzki R, Sher L, Carr WW, Fineman S, Greos L, Rachid R, Ibáñez MD, Tilles S, Assa’ad AH, Nilsson C, Rupp N, Welch MJ, Sussman G, Chinthrajah S, Blumchen K, Sher E, Spergel JM, Leickly FE, Zielen S, Wang J, Sanders GM, Wood RA, Cheema A, Bindslev-Jensen C, Leonard S, Kachru R, Johnston DT, Hampel FC Jr., Kim EH, Anagnostou A, Pongracic JA, Ben-Shoshan M, Sharma HP, Stillerman A, Windom HH, Yang WH, Muraro A, Zubeldia JM, Sharma V, Dorsey MJ, Chong HJ, Ohayon J, Bird JA, Carr TF, Siri D, Fernández-Rivas M, Jeong DK, Fleischer DM, Lieberman JA, Dubois AEJ, Tsoumani M, Ciaccio CE, Portnoy JM, Mansfield LE, Fritz SB, Lanser BJ, Matz J, Oude Elberink HNG, Varshney P, Dilly SG, Adelman DC, Burks AW, Ar101 oral immunotherapy for peanut allergy. The New England journal of medicine 379, 1991–2001 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Gould HJ, Sutton BJ, IgE in allergy and asthma today. Nature reviews. Immunology 8, 205–217 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Lieberman JA, Chehade M, Use of omalizumab in the treatment of food allergy and anaphylaxis. Current allergy and asthma reports 13, 78–84 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Savage JH, Courneya JP, Sterba PM, Macglashan DW, Saini SS, Wood RA, Kinetics of mast cell, basophil, and oral food challenge responses in omalizumab-treated adults with peanut allergy. The Journal of allergy and clinical immunology 130, 1123–1129.e1122 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brandström J, Vetander M, Sundqvist AC, Lilja G, Johansson SGO, Melén E, Sverremark-Ekström E, Nopp A, Nilsson C, Individually dosed omalizumab facilitates peanut oral immunotherapy in peanut allergic adolescents. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology 49, 1328–1341 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Gasser P, Eggel A, Targeting IgE in allergic disease. Current opinion in immunology 54, 86–92 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Gasser P, Tarchevskaya SS, Guntern P, Brigger D, Ruppli R, Zbären N, Kleinboelting S, Heusser C, Jardetzky TS, Eggel A, The mechanistic and functional profile of the therapeutic anti-IgE antibody ligelizumab differs from omalizumab. Nature communications 11, 165 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pennington LF, Gasser P, Brigger D, Guntern P, Eggel A, Jardetzky TS, Structure-guided design of ultrapotent disruptive IgE inhibitors to rapidly terminate acute allergic reactions. Journal of Allergy and Clinical Immunology 148, 1049–1060 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gould HJ, Ramadani F, Peanut allergen-specific antibodies go public. Science (New York, N.Y.) 362, 1247–1248 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Croote D, Darmanis S, Nadeau KC, Quake SR, High-affinity allergen-specific human antibodies cloned from single IgE b cell transcriptomes. Science (New York, N.Y.) 362, 1306–1309 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Deak PE, Kim B, Abdul Qayum A, Shin J, Vitalpur G, Kloepfer KM, Turner MJ, Smith N, Shreffler WG, Kiziltepe T, Kaplan MH, Bilgicer B, Designer covalent heterobivalent inhibitors prevent IgE-dependent responses to peanut allergen. Proceedings of the National Academy of Sciences of the United States of America 116, 8966–8974 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hemmings O, Du Toit G, Radulovic S, Lack G, Santos AF, Ara h 2 is the dominant peanut allergen despite similarities with ara h 6. The Journal of allergy and clinical immunology 146, 621–630.e625 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kukkonen AK, Pelkonen AS, Mäkinen-Kiljunen S, Voutilainen H, Mäkelä MJ, Ara h 2 and ara 6tyythe best predictors of severe peanut allergy: A double-blind placebo-controlled study. Allergy 70, 1239–1245 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Wurth MA, Hadadianpour A, Horvath DJ, Daniel J, Bogdan O, Goleniewska K, Pomés A, Hamilton RG, Peebles RS Jr., Smith SA, Human IgE mabs define variability in commercial aspergillus extract allergen composition. JCI insight 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bryce PJ, Falahati R, Kenney LL, Leung J, Bebbington C, Tomasevic N, Krier RA, Hsu CL, Shultz LD, Greiner DL, Brehm MA, Humanized mouse model of mast cell-mediated passive cutaneous anaphylaxis and passive systemic anaphylaxis. The Journal of allergy and clinical immunology 138, 769–779 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burton OT, Stranks AJ, Tamayo JM, Koleoglou KJ, Schwartz LB, Oettgen HC, A humanized mouse model of anaphylactic peanut allergy. The Journal of allergy and clinical immunology 139, 314–322.e319 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pagovich OE, Wang B, Chiuchiolo MJ, Kaminsky SM, Sondhi D, Jose CL, Price CC, Brooks SF, Mezey JG, Crystal RG, Anti-hige gene therapy of peanut-induced anaphylaxis in a humanized murine model of peanut allergy. The Journal of allergy and clinical immunology 138, 1652–1662.e1657 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Dispenza MC, Krier-Burris RA, Chhiba KD, Undem BJ, Robida PA, Bochner BS, Bruton’s tyrosine kinase inhibition effectively protects against human IgE-mediated anaphylaxis. The Journal of clinical investigation 130, 4759–4770 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takagi S, Saito Y, Hijikata A, Tanaka S, Watanabe T, Hasegawa T, Mochizuki S, Kunisawa J, Kiyono H, Koseki H, Ohara O, Saito T, Taniguchi S, Shultz LD, Ishikawa F, Membrane-bound human scf/kl promotes in vivo human hematopoietic engraftment and myeloid differentiation. Blood 119, 2768–2777 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bischoff SC, Role of mast cells in allergic and non-allergic immune responses: Comparison of human and murine data. Nature reviews. Immunology 7, 93–104 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Varricchi G, Raap U, Rivellese F, Marone G, Gibbs BF, Human mast cells and basophils-how are they similar how are they different? Immunological reviews 282, 8–34 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Schwartz LB, Metcalfe DD, Miller JS, Earl H, Sullivan T, Tryptase levels as an indicator of mast-cell activation in systemic anaphylaxis and mastocytosis. The New England journal of medicine 316, 1622–1626 (1987). [DOI] [PubMed] [Google Scholar]
- 35.Dombrowicz D, Brini AT, Flamand V, Hicks E, Snouwaert JN, Kinet JP, Koller BH, Anaphylaxis mediated through a humanized high affinity IgE receptor. Journal of immunology (Baltimore, Md. : 1950) 157, 1645–1651 (1996). [PubMed] [Google Scholar]
- 36.Fung-Leung WP, De Sousa-Hitzler J, Ishaque A, Zhou L, Pang J, Ngo K, Panakos JA, Chourmouzis E, Liu FT, Lau CY, Transgenic mice expressing the human high-affinity immunoglobulin (ig) e receptor alpha chain respond to human IgE in mast cell degranulation and in allergic reactions. The Journal of experimental medicine 183, 49–56 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kraft S, Kinet JP, New developments in fcepsilonri regulation, function and inhibition. Nature reviews. Immunology 7, 365–378 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Keet CA, Johnson K, Savage JH, Hamilton RG, Wood RA, Evaluation of ara h2 IgE thresholds in the diagnosis of peanut allergy in a clinical population. The journal of allergy and clinical immunology. In practice 1, 101–103 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koppelman SJ, Vlooswijk RA, Knippels LM, Hessing M, Knol EF, van Reijsen FC, Bruijnzeel-Koomen CA, Quantification of major peanut allergens ara h 1 and ara h 2 in the peanut varieties runner, spanish, virginia, and valencia, bred in different parts of the world. Allergy 56, 132–137 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Bird JA, Leonard S, Groetch M, Assa’ad A, Cianferoni A, Clark A, Crain M, Fausnight T, Fleischer D, Green T, Greenhawt M, Herbert L, Lanser BJ, Mikhail I, Mustafa S, Noone S, Parrish C, Varshney P, Vlieg-Boerstra B, Young MC, Sicherer S, Nowak-Wegrzyn A, Conducting an oral food challenge: An update to the 2009 adverse reactions to foods committee work group report. The journal of allergy and clinical immunology. In practice 8, 75–90.e17 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Korosec P, Turner PJ, Silar M, Kopac P, Kosnik M, Gibbs BF, Shamji MH, Custovic A, Rijavec M, Basophils, high-affinity IgE receptors, and ccl2 in human anaphylaxis. The Journal of allergy and clinical immunology 140, 750–758.e715 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mican JA, Arora N, Burd PR, Metcalfe DD, Passive cutaneous anaphylaxis in mouse skin is associated with local accumulation of interleukin-6 mrna and immunoreactive interleukin-6 protein. The Journal of allergy and clinical immunology 90, 815–824 (1992). [DOI] [PubMed] [Google Scholar]
- 43.Francis A, Bosio E, Stone SF, Fatovich DM, Arendts G, MacDonald SPJ, Burrows S, Brown SGA, Markers involved in innate immunity and neutrophil activation are elevated during acute human anaphylaxis: Validation of a microarray study. Journal of innate immunity 11, 63–73 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Urrutia A, Duffy D, Rouilly V, Posseme C, Djebali R, Illanes G, Libri V, Albaud B, Gentien D, Piasecka B, Hasan M, Fontes M, Quintana-Murci L, Albert ML, Standardized whole-blood transcriptional profiling enables the deconvolution of complex induced immune responses. Cell Rep 16, 2777–2791 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Moon TC, St Laurent CD, Morris KE, Marcet C, Yoshimura T, Sekar Y, Befus AD, Advances in mast cell biology: New understanding of heterogeneity and function. Mucosal immunology 3, 111–128 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Brown SG, Stone SF, Fatovich DM, Burrows SA, Holdgate A, Celenza A, Coulson A, Hartnett L, Nagree Y, Cotterell C, Isbister GK, Anaphylaxis: Clinical patterns, mediator release, and severity. The Journal of allergy and clinical immunology 132, 1141–1149.e1145 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Schwartz LB, Yunginger JW, Miller J, Bokhari R, Dull D, Time course of appearance and disappearance of human mast cell tryptase in the circulation after anaphylaxis. The Journal of clinical investigation 83, 1551–1555 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gurish MF, Bryce PJ, Tao H, Kisselgof AB, Thornton EM, Miller HR, Friend DS, Oettgen HC, IgE enhances parasite clearance and regulates mast cell responses in mice infected with trichinella spiralis. Journal of immunology (Baltimore, Md. : 1950) 172, 1139–1145 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Kelly BT, Grayson MH, Immunoglobulin e, what is it good for? Annals of allergy, asthma & immunology : official publication of the American College of Allergy, Asthma, & Immunology 116, 183–187 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCraw AJ, Chauhan J, Bax HJ, Stavraka C, Osborn G, Grandits M, López-Abente J, Josephs DH, Spicer J, Wagner GK, Karagiannis SN, Chenoweth A, Crescioli S, Insights from IgE immune surveillance in allergy and cancer for anti-tumour IgE treatments. Cancers 13, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferastraoaru D, Jordakieva G, Jensen-Jarolim E, The other side of the coin: IgE deficiency, a susceptibility factor for malignancy occurrence. The World Allergy Organization journal 14, 100505 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pellizzari G, Bax HJ, Josephs DH, Gotovina J, Jensen-Jarolim E, Spicer JF, Karagiannis SN, Harnessing therapeutic IgE antibodies to re-educate macrophages against cancer. Trends in molecular medicine 26, 615–626 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Mathias CB, Freyschmidt EJ, Caplan B, Jones T, Poddighe D, Xing W, Harrison KL, Gurish MF, Oettgen HC, IgE influences the number and function of mature mast cells, but not progenitor recruitment in allergic pulmonary inflammation. Journal of immunology (Baltimore, Md. : 1950) 182, 2416–2424 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oettgen HC, Fifty years later: Emerging functions of IgE antibodies in host defense, immune regulation, and allergic diseases. The Journal of allergy and clinical immunology 137, 1631–1645 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deak PE, Vrabel MR, Kiziltepe T, Bilgicer B, Determination of crucial immunogenic epitopes in major peanut allergy protein, ara h2, via novel nanoallergen platform. Scientific reports 7, 3981 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Handlogten MW, Serezani AP, Sinn AL, Pollok KE, Kaplan MH, Bilgicer B, A heterobivalent ligand inhibits mast cell degranulation via selective inhibition of allergen-IgE interactions in vivo. Journal of immunology (Baltimore, Md. : 1950) 192, 2035–2041 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Handlogten MW, Kiziltepe T, Serezani AP, Kaplan MH, Bilgicer B, Inhibition of weak-affinity epitope-IgE interactions prevents mast cell degranulation. Nature chemical biology 9, 789–795 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vantur R, Rihar M, Koren A, Rijavec M, Kopac P, Bidovec-Stojkovic U, Erzen R, Korosec P, Chemokines during anaphylaxis: The importance of ccl2 and ccl2-dependent chemotactic activity for basophils. Clinical and translational allergy 10, 63 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Handlogten MW, Deak PE, Bilgicer B, Two-allergen model reveals complex relationship between IgE crosslinking and degranulation. Chemistry & biology 21, 1445–1451 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this paper are present in the paper or in the Supplementary Materials. Human IgE are available from S.A.S. under material transfer agreement.