

Abstract
Mesenchymal stem/stromal cells (MSCs) are multipotent stem cells that can be isolated from bone marrow, adipose tissue, the umbilical cord, dental pulp, etc. These cells have unique properties that give them excellent therapeutic potential, including immunoregulation, immunomodulation, and tissue regeneration functions. MSC-based products are considered advanced therapy medicinal products (ATMPs) under European regulations (1394/2007); thus, they must be manufactured under good manufacturing practices and via effective manufacturing methods. The former can be achieved via a proper laboratory design and compliance with manufacturing protocols, whereas the latter requires an approach that ensures that the quality of the products is consistent regardless of the manufacturing procedure. To meet these daunting requirements, this study proposes an exchangeable approach that combines optimized and equivalent manufacturing processes under the Quality by Design (QbD) principle, allowing investigators to convert from small laboratory-scale to large-scale manufacturing of MSC-based products for clinical applications without altering the quality and quantity of the cell-based products.
Keywords: Mesenchymal stem/stromal cells, Cellular manufacturing process, Quantum cell expansion system, HYPERflask, Quality by design
Graphical abstract
1. Introduction
Mesenchymal stem/stromal cells (MSCs) are multipotent stem cells that were first isolated from bone marrow aspirate [1] and later found in other tissues, including adipose tissue [2], the umbilical cord (UC) [3], umbilical cord blood [4], the placenta membrane [5], dental pulp [6], etc. Among these sources, bone marrow, adipose tissue, and the UC are the three most widely used sources of MSCs for therapeutic applications [7]. MSC-based therapy has evolved rapidly both scientifically and clinically in recent decades, with approximately 1400 clinical trials being conducted worldwide [8]. The popularity of this therapy originates from the unique nature of MSCs themselves, which have low immunogenicity due to a lack of HLA-DR expression, supporting their use as “allogeneic” or “off-the-shelf” products for therapeutic treatments [9]. In addition, MSCs have the ability to participate in immunoregulatory functions [10], immunosuppression and modulation of the host inflammatory response via cell-to-cell contacts and paracrine effects mediated by a wide range of secreted growth factors [11], cytokines and chemokines [12]. As many studies of MSCs have supported their safety and potential therapeutic effects in certain diseases, they have been considered advanced therapeutic medicinal products in Europe since 2006 [[13], [14], [15]]. Although the first administration of stem cells could be traced back to the late 1950s, MSC-based therapies for diverse medical indications have begun to be popular recently due to the rapid development of cultivation methods and standardized xeno-free and serum-free conditions [16]. To meet the high demand for MSCs in the clinical setting, the manufacturing process plays a critical role in ensuring that high-quality MSCs are generated and provided in time for patients in need. The manufacture of MSCs for clinical applications typically comprises several steps, such as the determination of MSC sources, the acquisition of the raw materials, isolation, expansion, harvest, purification, formulation, fill and finish, cryopreservation, thawing and quality control. Although the principle underlying the MSC manufacturing process is similar to that of other pharmaceutical products, the variability in cell sources, proliferation rates, and nonstandardized culture conditions introduce complexity and challenges to the manufacture of MSC-based products.
To address these challenges of the MSC manufacturing process, we propose a combination of standardized xeno- and serum-free culture conditions, cell line screening and selection, the advantages of the Quality by Design (QbD) approach, and a standardized manufacturing process using either manual planar culture or an automated system [17]. Since their initial discovery, MSCs have been cultured with media containing fetal bovine serum (FBS), which is of animal origin, has undefined components and exhibits batch-to-batch variations [18]. The multitude of components found in serum greatly affect the proliferation and potency of cultured cells, especially MSCs, resulting in variations in their therapeutic potential [19]. Moreover, serum also has the risk of high endotoxin levels, hemoglobin effects, infectious agents, and supply concerns related to the availability of GMP-grade serum for the cell manufacturing process [20]. Thus, the use of xeno-free and serum-free culture conditions has emerged as an alternative approach for MSC expansion for downstream clinical applications. The recent establishment of standardized xeno-free and serum-free culture conditions that allow the use of a single platform to support the expansion of MSCs derived from bone marrow, adipose tissue, and the UC provides a powerful tool to directly compare the similarities and differences of these sources [16]. In addition, xeno- and serum-free culture conditions allow more consistent performance, better reproducibility, and stable large-scale production of cell-based products. As culture conditions could be standardized and stabilized using xeno- and serum-free culture conditions, for a consistent cellular manufacturing process, a stable cell line is needed to provide a good master and working cell bank. Umbilical cord tissue is easy to obtain using a noninvasive method, there is no ethical barrier, and the acquisition procedure does not introduce any potential risk or harm to newborns and mothers. We proved that UC-MSCs cultured under standardized xeno- and serum-free culture conditions had great proliferation ability and could be expanded in a large-scale process [16]. These advantages provide us with a unique opportunity to establish stable cell lines via genetic screening and biological function selection processes. Therefore, it has been suggested that when manufacturing such complex and variable products as cell-based therapeutic agents, it is of the utmost importance to acknowledge that any changes and variations in the expansion process, regardless of how minor, might directly affect the quality of the final products, which is a critical point for a cell-based therapy in relation to the safety and efficacy of the treatment.
An increasing number of stem cell-based therapies are being developed and used, together with an increasing number of regulatory approvals being issued worldwide, resulting in a pressing need for large-scale production procedures and industrial translation [21,22]. Thus, an efficient, cost-effective manufacturing process and compliance with regulations are important for the success of stem cell-based clinical trials. Currently, cellular manufacturing processes are manual and semiautomated and are mostly performed under laboratory-scale and planar culture conditions [21]. They are time consuming, labor intensive, and often involve open processes, which limit their applications in large-scale expansion at the industrial scale [23]. As a result, they are prone to human error and largely experience-dependent, and they can result in batch-to-batch variability, lack of reproducibility, increased production costs, and an increased risk of culture contamination. To address the current challenges of manual cell culture, the introduction of multilayered cell culture flasks to enhance the scale-up process from single-layered T-flasks provides an alternative approach for the cellular manufacturing industry [24]. Recent advances in culture vessel design allow researchers to not only upscale the process but also reduce medium consumption, make the process less labor intensive and increase cell numbers. In terms of automated platforms, bioreactor systems are the most widely used methods for culturing cells to harvest proteins, as they are scalable, amenable to the integration of environmental control and real-time monitoring of the cell culture, and capable of an automated response to changes in culture conditions to maintain a high protein yield [25]. However, these systems are designed and optimized to obtain the highest yield of the protein of interest but do not prioritize the quality of the cells themselves. Thus, the use of bioreactors needs to be carefully considered to allow cells of the best quality to be produced and harvested. However, there has been no direct comparison of the quality of MSCs produced via either multilayered flasks or automated culture platforms to date.
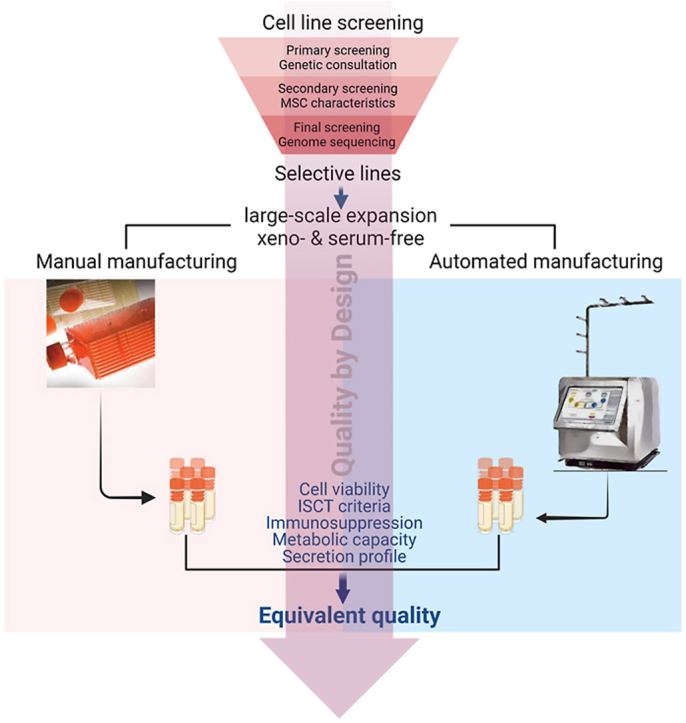
A direct comparison between a manual manufacturing process and its automated counterpart allows researchers and investigators to transform from laboratory-based manufacturing to the industrial scale without extensive investigations to optimize the process. In our opinion, a successful optimization process that demonstrates that the equivalent quality of cell products generated via the manual manufacturing process and the automated culture platform would become a driving force for the cell-based therapy process (Fig. 1). One might use the manual process to generate enough cells for small-scale applications of targeted cell-based therapy, such as phase I and II clinical trials or the early stage of the commercialization process, where a small number of patients are included and the target doses are used at a lower pace. At a larger scale with a high demand for high-quality cells, one might switch from a manual process to an automated culture process, allowing a higher number of cells to be produced with equivalent quality and characteristics similar to those of the cells produced via the manual process (Fig. 1A). According to the guidelines CPMP/ICH/5721/03 ICH topic Q5E “Comparability of Biotechnological/Biological products” and EMA/CAT/499821/2019 Committee for Advanced Therapies (CAT), the comparability is a key concept in the development, manufacture and evaluation of the biological product, in which, any changes before and after manufacturing process or any other relevant factors should not alter the quality of the product. Thus, achieving a consistent efficacy of cell-based therapy heavily depends on the consistent quality of the cell products, which is achieved via a reliable manufacturing process. In addition, the QbD principle was implemented to ensure that the quality of MSC-based products generated using either manual manufacturing (multilayered flasks) or an automated cell culture platform under xeno-free and serum-free conditions was maintained throughout the procedure [17]. The QbD is defined as a scientific, risk-based approach for cellular manufacturing design that is based on product and process attributes that contribute to product quality. The success of the QbD principle relies on a thorough understanding of the cellular biology and culture process underlying a product and its manufacture. It usually starts with a detailed description of the quality characteristics of the desired product, in our case, MSCs, followed by a detailed investigation of the attributes that directly influence product quality throughout the manufacturing process and the development of a design space that considers the effects of these attributes on quality to identify potential solutions to minimize their detrimental effects (Fig. 1B). A comprehensive review on the QbD principle can be found in a previous publication [17,26]. Thus, an experimental design in accordance with QbD principle plays a critical role in manufacturing process and involves the following steps: (1) define the objective/quality target product profile (QTPP), (2) identify critical process parameters (CPPs), (3) develop a design space, (4) define the control strategy, (5) conduct experiments, (6) analyze the results, (7) develop a final control strategy, and (8) monitor and review. Here, we describe the process of screening and selecting stable MSCs, followed by identification the QTPP of the MSC products according to the QbD principle. Based on the QTPP, the experiment design was conducted to direct comparisons of MSCs produced via a manual manufacturing process and their counterparts produced via an automated platform to identify the CPPs and critical quality attributes (CQAs) of the two processes (Fig. 1). The results of our study provide a standardized platform supporting the manufacturing process of allogeneic MSC-based therapy under manual and automated culture conditions.
Fig. 1.

Manufacturing process of an allogeneic MSC-based product under the Quality by Design principle for clinical applications. (A) Manufacturing process of an allogeneic MSC-based product under manual and automated culture conditions. MSC-based products can be derived from bone marrow, adipose tissue, or umbilical cord tissue, followed by culture under xeno- and serum-free conditions to establish a Token cell bank, master cell bank, and working cell bank for the manufacturing process. Conventional manufacturing processes (manual vs. automated) have both advantages and limitations, but there is a need for an interchangeable system that allows investors to easily switch from one to another depending on the scale of downstream applications of the product. (B) Cell-based product with the Quality by Design (QbD) principle and Quality Target Product Profile (QTPP). In the context of MSC-based products, a QTPP describes the desired characteristics of the product, including MSC morphology, cell viability/purity, MSC markers, karyotype, MSC differentiation, immunosuppression, and metabolic function. The QTPP was pre-defined to cover the identification of MSCs, the potency, the purity, and the safety requirements. The manufacturing process of an allogeneic MSC-based product under the QbD principle involves a systematic approach that ensures the comparability, consistency, and reproducibility of the manufacturing process. This approach involves identifying critical quality attributes (CQAs), determining the impact of process parameters on the CQAs, and optimizing the process parameters within a design space. The resulting product is evaluated against the pre-defined QTPP to ensure that it meets the desired characteristics for clinical applications. The QbD strategy presented in this study provides a robust framework for the development and manufacture of MSC-based products that adhere to the highest standards of quality and safety, making them suitable for regulatory approval and clinical use.
2. Results
2.1. Establishment of clinical-grade UC-MSC lines for the cellular manufacturing process
In the primary screening process, 22 out of 30 parents participated in genetic consulting and answered 97 questions related to inherited diseases, infant medical history, parental medical background, and lifestyle and behavior. The results indicated that two families had scores higher than 32, which is our established threshold for cut-off; thus, the two UC-MSC lines derived from these two families were excluded from the secondary screening (Supplementary Fig. 1A). The results of cellular characterization showed that all UC-MSC lines expressed normal MSC markers (Supplementary Figs. 1B–C) and had population doubling times from 18 to 24 h. However, five UC-MSC lines had CFU-F numbers lower than 200 CFU-F/1000 cells (Supplementary Fig. 1D), three UC-MSC lines were unable to differentiate into either osteocytes or chondrocytes (Supplementary Table 3), and one UC-MSC line did not undergo the differentiation assay. These lines and one UC-MSC line derived from the same donor as another line were excluded. The 20 remaining UC-MSC lines were subjected to final screening by genome sequencing of 6700 genes using the TRUESIGHT ONE EXPANDED kit. The genomic sequencing strategy and quality results are shown in Supplementary Table 4. Our data showed that three out of 20 UC-MSC lines had pathogenic and dominant mutations at G6PD and TTR, which led us to exclude these lines from the pool. Although the remaining 17 UC-MSC lines carry genetic mutations, these mutations are mostly recessive with normal phenotypes (Table 1). Taken together, the combination of the primary, secondary and final screening processes indicated that 17 out of 30 UC-MSC lines met the requirement for production of the master cell bank at passage 2 (P2) and the working cell bank at P4. Three out of 17 lines were then randomly selected for the cellular manufacturing process.
Table 1.
Genome sequencing of 20 cell lines.
| Cell line | Mutation gene | Position | Variations | Location | Homozygous/heterozygous | Variant classification | Traits | Disease |
|---|---|---|---|---|---|---|---|---|
| UC-1 | TGM1 | 14:24,261,783 | c.420A > G p.(Ile140Met) |
Missense | Heterozygous | Likely pathogenic | Recessive | Congenital Ichthyosis |
| ADSL | 22:40,358,950 | c.569G > A p.(Arg190Gln) |
Missense | Heterozygous | Likely pathogenic | Recessive | Adenylosuccinase deficiency | |
| MYO15A | 17:18,154,745 | c.8215delG p.(Ala2739ProfsTer15) |
Frameshift | Heterozygous | Not reported on ClinVar (**) | Recessive | Deafness 3 | |
| UC-2 | SLC22A5 | 5:132,385,435 | c.832C > T p.(Arg278Ter) |
Stop-gained | Heterozygous | Pathogenic | Recessive | Systemic primary carnitine deficiency |
| POLG | 15:89,319,065 | c.3139C > T p.(Arg1047Trp) |
Missense | Heterozygous | Likely Pathogenic | Recessive | Alpers disease | |
| CCDC88C | 14:91,321,256 | c.1391G > A p.(Arg464His) |
Missense | Heterozygous | Not reported on ClinVar (**) | Dominant | Spinocerebellar ataxia | |
| UC-3 | AGPAT2 | 9:136,674,750 | c.646A > T p.(Lys216Ter) | Stop-gained | Heterozygous | Likely pathogenic | Recessive | Lipodystrophy type 1 |
| UROC1 | 3:126,508,419:126,508,418 | c.405_408dupGGCT p.(Gln137GlyfsTer57) |
Frameshift | Heterozygous | Not reported on ClinVar (*) | Recessive | Urocanase deficiency | |
| UC-4 | MMACHC | 1:45,508,870: 45,508,882 |
c.507_519delAGAGGTGCCAGAT p.(Glu170CysfsTer36) |
Frameshift | Heterozygous | Pathogenic | Recessive | Methylmalonic Aciduria and homocystinuria, type cblC |
| USH2A | 1:215,671,222 | c.13883delC p.(Pro4628LeufsTer6) |
Frameshift | Heterozygous | Not reported on ClinVar (*) | Recessive | Usher syndrome 2A/Retinitis pigmentosa | |
| UC-5 | HPS4 | 22:26,458,578 | c.1714-1G > T | Splice acceptor | Heterozygous | Not reported on ClinVar (*) | Recessive | Hermansky-Pudlak syndrome 4 |
| PAH | 12:102,855,326 | c.517G > T p.(Gln172His) | Missense | Heterozygous | Not reported on ClinVar (**) | Recessive | Phenylketonuria | |
| G6PD | X:154,533,122 | c.961G > A p.(Val321Met) |
Missense | Heterozygous | Likely pathogenic | Dominant | G6PD deficiency | |
| ITGA3 | 17:50,081,328 | c.2839C > T p.(Arg947Ter) |
Stop_gained | Heterozygous | Not reported on ClinVar (**) | Recessive | Congenital interstitial lung disease, nephrotic syndrome, epidermolysis bullosa | |
| UC-6 | ATP7B | 13:51,950,132 | c.2605G > A p.(Gly869Arg) |
Missense | Heterozygous | Pathogenic | Recessive | Wilson disease |
| TTR | 18:31,592,974 | c.148G > A p.(Val50Met) |
Missense | Heterozygous | Pathogenic | Dominant | Familial amyloidosis with polyneuropathy type 1 | |
| SLC34A2 | 4:25,662,843 | c.250+1G > A | Splice donor | Heterozygous | Not reported on ClinVar (*) | Recessive | Pulmonary alveolar microlithiasis | |
| TPM3 | 1:154,159,025 | c.688C > T p.Arg230Ter |
Missense | Heterozygous | Not reported on ClinVar (*) | Dominant/Recessive | Myopathy | |
| ALDH5A1 | 6:24,502,592:24,502,593 | c.424_425delAT p.(Ile142HisfsTer3) |
Frameshift | Heterozygous | Not reported on ClinVar (*) | Recessive | Succinic semialdehyde dehydrogenase deficiency | |
| UC-8 | GCNT2 | 6:10,528,925 | c.14G > A p.Trp5Ter |
Stop-gained | Heterozygous | Not reported on ClinVar (**) | Recessive | Cataract |
| DTNA | 18:34,838,756 | c.1184delG p.(Ser395ThrfsTer2) |
Frameshift | Heterozygous | Not reported on ClinVar (**) | Dominant | Left ventricular noncompaction 1 | |
| UC-9 | RYR1 | 19:38,443,612 | c.325C > T p.(Arg109Trp) |
Missense | Heterozygous | Likely pathogenic | Recessive | Congenital myopathy |
| AMPD1 | 1:114,677,465 | c.1373G > A p.(Arg458His) | Missense | Heterozygous | Likely pathogenic | Recessive | Myopathy due to myoadenylate deaminase deficiency | |
| USH2A | 1:215,648,564 | c.14546G > A p.(Trp4849Ter) | Stop_gained | Heterozygous | Not reported on ClinVar (*) | Recessive | Usher syndrome 2A/Retinitis pigmentosa | |
| UC-12 | PAH | 12:102,855,326 | c.517G > T p.(Gln172His) | Missense | Heterozygous | Not reported on ClinVar (*) | Recessive | Phenylketonuria |
| UC-16 | COL12A1 | 6:75,181,213 | c.1892-2A > G | Splice acceptor | Heterozygous | Not reported on ClinVar (*) | Dominant/Recessive | Bethlem myopathy 2 /Ullrich congenital muscular dystrophy 2 |
| ANO5 | 11:22,272,913:22,272,912 | c.2159_2160dupTA p.(Ala721Ter) |
Frameshift | Heterozygous | Not reported on ClinVar (**) | Dominant/Recessive | Gnathodiaphyseal dysplasia/Muscular dystrophy | |
| UC-18 | SLCO1B1 | 12:21,222,355 | c.1738C > T p.(Arg580Ter) |
Stop gained | Heterozygous | Pathogenic | Multiple recessive gene | Digenic hyperbilirubinemia |
| PSEN2 | 1:226,885,652 | c.472delG p.(Val158CysfsTer15) |
Frameshift | Heterozygous | Not reported on ClinVar (**) | Dominant | Alzheimer/Dilated Cardiomyopathy | |
| UC-20 | CPOX | 3:98,585,631 | c.982C > T p.(Arg328Cys) | Missense | Heterozygous | Likely pathogenic | Dominant | Coproporphyria |
| SLC22A5 | 5:132,392,565 | c.1472C > G p.(Ser491Cys) |
Missense | Heterozygous | Pathogenic | Recessive | Systemic primary carnitine deficiency | |
| MVK | 12:109,596,554 | c.1168C > T p.(Gln390Ter) | Stop-gained | Heterozygous | Not reported on ClinVar (**) | Dominant/Recessive | Porokeratosis/Mevalonic aciduria/Hyper-IgD syndrome | |
| GJB2 | 13:20,189,473 | c.109G > A p.(Val37Ile) |
Missense | Heterozygous | Pathogenic | Recessive | Digenic deafness | |
| G6PD | X:154,533,122 | c.961G > A p.(Val321Met) |
Missense | Heterozygous | Pathogenic | Dominant | G6PD deficiency | |
| IGSF3 | 1:116,600,246 | c.1784G > A p.(Trp595Ter) |
Stop-gained | Heterozygous | Pathogenic | Recessive | Lacrimal duct defect | |
| PDE6A | 5:149,933,932 | c.715C > T p.(Gln239Ter) |
Stop-gained | Heterozygous | Not reported on ClinVar (*) | – | Retinitis pigmentosa | |
| UC-21 | ABCA4 | 1:94,008,252 | c.5881G > A p.(Gly1961Arg) |
Missense | Heterozygous | Pathogenic | Recessive | Stargardt disease |
| FANCD2 | 3:10,081,465 | c.3224+1G > T | Spice donor | Heterozygous | Not reported on ClinVar (*) | Recessive | Fanconi anemia | |
| UC-22 | ATP7B | 13:51,949,772 | c.2755C > T p.(Arg919Trp) |
Missense | Heterozygous | Likely pathogenic | Recessive | Wilson syndrome |
| ITGA7 | 12:55,694,633 | c.2259delC p.(Met754Ter) |
Frameshift | Heterozygous | Not reported on ClinVar (**) | Recessive | Congenital muscular dystrophy | |
| UC-23 | CHAT | 10:49,619,786:49,619,785 | c.451_466dupCGACACTTGGTGTCTG p.(Glu156AlafsTer6) |
Frameshift | Heterozygous | Not reported on ClinVar (**) | Recessive | Congenital myasthenic syndrome |
| UC-25 | PIGT | 20:45,416,673 | c.344G > A p.(Trp115Ter) |
Stop-gained | Heterozygous | Not reported on ClinVar (**) | Dominant /Recessive |
Paroxysmal nocturnal hemoglobinuria/Multiple congenital anomalies-hypotonia-seizures syndrome |
| LMF1 | 16:854,610:854,609 | c.1610_1626dupGGAAGAGGATCGGAGCC p.(Tyr543GlyfsTer16) |
Frameshift | Heterozygous | Not reported on ClinVar (**) | Recessive | Lipase deficiency | |
| GJB2 | 13:20,189,347 | c.235delC p.(Leu79CysfsTer3) |
Frameshift | Heterozygous | Pathogenic | Recessive | Digenic deafness | |
| UC-27 | CLPB | 11:72,372,923 | c.736+2T > C | Splice donor | Heterozygous | Not reported on ClinVar (*) | Recessive | 3-methylglutaconic aciduria type 7, cataracts, neurologic involvement and neutropenia |
| UC-28 | ACE | 17:63,494,382 | c.3292C > T p.(Gln1098Ter) | Stop-gained | Heterozygous | Not reported on ClinVar (*) | Recessive | Renal tubular dysgenesis |
| EIF2B5 | 3:184,138,239 | c.758delC p.(Ser253PhefsTer25) |
Frameshift | Heterozygous | Not reported on ClinVar (*) | Recessive | Leukoencephalopathy/Ovarioleukodystrophy | |
| TRMT1 | 17:18,154,745 | c.394_395delAG p.(Ser132Ter) |
Frameshift | Heterozygous | Not reported on ClinVar (*) | Recessive | Intellectual development disorder | |
| UC-29 | EVC2 | 4:5,622,562 | c.2476C > T p.(Arg826Ter) |
Stop-gained | Heterozygous | Pathogenic | Recessive | Ellis-van Creveld syndrome |
| COG4 | 16:70,514,334 | c.544+1G > T | Splice donor | Heterozygous | Not reported on ClinVar (*) | Dominant/Recessive | Saul-Wilson syndrome/Congenital disorders of glycosylation | |
| OFD1 | X:13,751,358 | c.1045G > T p.(Glu349Ter) | Stop-gained | Heterozygous | Not reported on ClinVar (*) | X-linked dominant/recessive | Orofaciodigital syndrome/Joubert syndrome, Simpson-Golabi-Behmel syndrome, retinitis pigmentosa | |
| GIGYF2 | 2:232,844,096:232,844,095 | c.3002_3003insGC p.(Gln1002HisfsTer70) |
Frameshift | Heterozygous | Not reported on ClinVar (**) | – | Parkinson disease | |
| OTOA | 16:21,736,318 | c.2359G > T p.(Glu787Ter) |
Stop-gained | Heterozygous | Not reported on ClinVar (**) | Recessive | Deafness | |
| UC-30 | WNT10B | 12:48,968,025 | c.632G > A p.(Arg211Gln) |
Missense | Heterozygous | Likely pathogenic | Dominant | Selective tooth agenesis |
| GJB2 | 13:20,189,473 | c.3292C > T p.(Gln1098Ter) | Missense | Heterozygous | Pathogenic | Recessive | Digenic deafness |
(*): Not reported on ClinVar, pathogenic.
(**): Not reported on ClinVar, likely pathogenic.
2.2. Optimization of the automated cell culture platform and manual manufacturing process for large-scale expansion of UC-MSCs under xeno-free and serum-free conditions
As our UC-MSC lines cultured under xeno-free and serum-free culture conditions required CellStart coating for long-term expansion using single-layered T flasks, we first investigated whether this coating is required when culturing UC-MSCs using HYPERFlask. Two different concentrations of CellStart were used, 1:300 and 1:500 dilutions, and compared to the uncoated flask. The results showed that there were no significant differences in terms of cell yield, days in culture, cell viability, MSC markers, or karyotype between coated and noncoated flasks (Supplementary Table 5). These results confirm that coating is not required for MSC culture using HYPERflasks. Although there were fluctuations in cell yield between batches, these differences resulted from the HYPERFlask handling skills, which were later optimized to allow an equal distribution of cells across the 10 layers of the flask. At a seeding density of 3500 cells/cm2, the maximum yield of UC-MSCs peaked at 110 × 106 cells/flask after 4 days of culture.
Next, we investigated the coating reagents for the Quantum bioreactor using either 2 mL CellStart or vitronectin at different concentrations (2.5, 4.5, or 10 mg). Coating the bioreactor with 2 mL CellStart yielded the lowest number of harvested viable cells (53 × 106 cells) after 9 days in culture. While coating the bioreactor with 2.5 mg vitronectin generated 380 × 106 cells from 17 × 106 input cells after 8 days in culture, the use of 4.5 mg vitronectin with 17 × 106 fresh input cells resulted in 270 × 106 cells and 380 × 106 cells after 3 days and 6 days in culture, respectively. However, we noticed a reduction in CD105 marker expression in the two batches, with values of 90.8% and 85.6%, respectively. Further increasing the concentration of vitronectin to 10 mg enhanced the numbers of harvested viable cells to (773 ± 140) × 106 cells from (26 ± 2) × 106 fresh-thawed cells (n = 3, mean ± SEM), with normal MSC marker expression (Supplementary Table 6). Taken together, these results suggested that 10 mg vitronectin is the most suitable coating reagent for a quantum bioreactor that supports the attachment and expansion of UC-MSCs under xeno-free and serum-free conditions.
2.3. Comparative analysis of UC-MSCs produced from Quantum Expansion system and the HYPERFlask platform
2.3.1. Biological analysis
To explore the differences in terms of cell production quality between the Quantum Cell Expansion system and the HYPERFlask platform, the biological characteristics of UC-MSCs generated from these two systems (called QUANT-MSCs and HYPER-MSCs) were first explored using the minimal guidelines of ISCT for MSC identification [27]. As three UC-MSC lines (passage 4) were used as the starting cell sources for both platforms, these cells (called P4-MSCs) were used as controls for a direct comparison between the two systems. Although there was a difference in cell yield between the two systems, the harvesting method was optimized, with equivalent and high cell viability (96.05 ± 1.4% for QUANT-MSCs and 98.25 ± 0.4% for HYPER-MSCs) (Fig. 2A). Analysis of MSC surface markers, including positive markers (CD73, CD90, and CD105) and negative markers (CD11, CD19b, CD34, CD45, and HLR-DR), indicated no significant differences between P4-MSCs (99.79 ± 0.08% CD73, 99.04 ± 0.71% CD90, 98.27 ± 0.93% CD105, and 0.19 ± 0.04% negative markers), QUANT-MSCs (99.86 ± 0.04% CD73, 99.83 ± 0.04% CD90, 98.91 ± 0.34% CD105, and 0.35 ± 0.09% negative maker), and HYPER-MSCs (98.78 ± 0.52% CD73, 98.83 ± 0.33% CD90, 96.73 ± 0.38% CD105, and 0.14 ± 0.07% negative makers) (Fig. 2B). Both QUANT-MSCs and HYPER-MSCs were able to differentiate into osteocytes (Fig. 2C), adipocytes (Fig. 2D), and chondrocytes (Fig. 2E). Taken together, the results showed that QUANT-MSCs and HYPER-MSCs are equivalent in terms of MSC phenotype according to ISCT guidelines.
Fig. 2.

Characterization of UC-MSCs manufactured with the Quantum Cell Expansion system and HYPERflasks. (A) Cell viability for QUANT-MSCs (96.05 ± 1.4%) and HYPER-MSCs (98.25 ± 0.4%) after optimized harvesting. (B) Flow cytometry analysis of MSC surface markers (positive: CD73, CD90, and CD105; negative: CD11, CD19b, CD34, CD45, and HLR-DR) for P4-MSCs, QUANT-MSCs, and HYPER-MSCs showing no significant differences. (C–E) Trilineage differentiation potential of QUANT-MSCs and HYPER-MSCs into (C) osteocytes, (D) adipocytes, and (E) chondrocytes.
2.3.2. Immunoregulatory analysis
To reveal the immunoregulatory function of QUANT-MSCs and HYPER-MSCs, we investigated their ability to suppress the proliferation of activated T cells in vitro. The results showed that the percentages of proliferation of both CD4+ T cells (Fig. 3A) and CD8+ T cells (Fig. 3B) in the presence of UC-MSCs at ratios of PBMC:UC-MSC = 10:1 and 20:1 were markedly lower than those of T cells cultured in the presence of PBMCs only. In the absence of UC-MSCs, the percentage of CD4+ T cell proliferation was 32.17 ± 0.85%. However, in the presence of MSC at PBMC: MSC = 10: 1 and 20:1, percentages of CD4+ T cell proliferation were 9.4 ± 1.4% and 15.4 ± 3.9% (P4-MSCs) 8.3 ± 3% and 13.6 ± 5.4% (HYPER-MSCs), and 11.5 ± 2.9% and 18.9 ± 2.9% (QUANT-MSCs), respectively (Fig. 3A). Similarly, while the percentage of CD8+ T cell proliferation without MSCs was 29.3 ± 0.8%, the percentages of the cell proliferation was reduced when co-cultured with MSC at both ratios, PBMC: MSC = 10: 1 and 20:1 (7.8 ± 2.3% and 13.9 ± 4.8% for P4-MSC, 7.2 ± 1.5% and 12.7 ± 5.04% for HYPER-MSCs, and 7.1 ± 1.2% and 14.7 ± 2.6% for QUANT-MSCs, respectively) (Fig. 3B). These data indicated that both QUANT-MSCs and HYPER-MSCs significantly suppressed the proliferation of both CD4+ T cells (Fig. 3A) and CD8+ T cells (Fig. 3B). Importantly, there was no significant difference in the percentages of proliferation of both CD4+ T cells and CD8+ T cells when using UC-MSCs from the two systems and P4-MSCs, suggesting that manufacturing hUC-MSCs using both HYPERFlask and Quantum did not alter the immunoregulatory effects of the cells.
Fig. 3.

Immunomodulatory effects and metabolic activities of UC-MSCs manufactured with the Quantum Cell Expansion system and HYPERflasks. (A) Proliferation of CD4+ T cells in the presence of P4-MSCs, QUANT-MSCs, and HYPER/MSCs. Percentage of CD4+ T cell proliferation in the presence of P4-MSCs, HYPER-MSCs, and QUANT-MSCs at PBMC:MSC ratios of 10:1 and 20:1 compared to the control (PBMCs only). (B) Proliferation of CD8+ T cells in the presence of P4-MSCs, QUANT-MSCs, and HYPER/MSCs. Percentage of CD8+ T cell proliferation under the same conditions as in (A). Results demonstrate that both QUANT-MSCs and HYPER-MSCs significantly suppress the proliferation of CD4+ (A) and CD8+ (B) T cells. No significant differences were observed in the suppression of T cell proliferation among P4-MSCs, HYPER-MSCs, and QUANT-MSCs, indicating that both HYPERFlask and Quantum manufacturing methods did not alter the immunoregulatory effects of hUC-MSCs. (C) Glycolysis capacity of P4-MSCs, QUANT-MSCs, and HYPER/MSCs. QUANT-MSCs show reduced glycolysis and glycolytic capacity compared to P4-MSCs and HYPER-MSCs, with no significant differences in basal glycolytic rate or glycolytic reserve among the groups. (D) Mitochondrial respiratory capacity of P4-MSCs, QUANT-MSCs, and HYPER/MSCs including basal respiration rate, proton leakage, maximal respiration, and ATP production, are similar among P4-MSCs, QUANT-MSCs, and HYPER-MSCs. QUANT-MSCs exhibit a higher spare respiratory capacity than input cells (P4-MSCs) and HYPER-MSCs.
2.3.3. Metabolic activities
In terms of glycolysis, QUANT-MSCs exhibited a significant reduction in glycolysis and glycolytic capacity (1.2 ± 0.06 × 10−2 and 1.8 ± 0.07 × 10−2, respectively) compared to P4-MSCs (1.7 ± 0.04 × 10−2 and 2.3 ± 0.07 × 10−2, respectively) and HYPER-MSCs (1.6 ± 0.06 × 10−2 and 2.4 ± 0.07 × 10−2, respectively) (Fig. 3C and D). No significant differences in the basal glycolytic rate or glycolytic reserve in UC-MSCs were found among P4-MSCs, QUANT-MSCs and HYPER-MSCs. Analysis of mitochondrial activities showed that both QUANT-MSCs and HYPER-MSCs had a basal respiration rate, proton leakage, maximal respiration, and ATP production (1.8 ± 0.07 × 10−2, 0.3 ± 0.04 × 10−2, 3.0 ± 0.08 × 10−2, and 1.5 ± 0.05 × 10−2, respectively, for QUANT-MSCs and (2.0 ± 0.06 × 10−2, 0.4 ± 0.05 × 10−2, 2.8 ± 0.11 × 10−2, and 1.6 ± 0.06 × 10−2, respectively, for HYPER-MSCs) similar to those of the input cell sources (2.0 ± 0.07 × 10−2, 0.4 ± 0.05 × 10−2, 2.9 ± 0.10 × 10−2, and 1.6 ± 0.06 × 10−2, respectively. However, QUANT-MSCs exhibited a higher spare respiratory capacity (1.2 ± 0.08 × 10−2) than input cells (0.9 ± 0.05 × 10−2) and HYPER-MSCs (0.8 ± 0.08 × 10−2) (Fig. 3C and D).
2.3.4. Growth factor and cytokine secretion profile
To elucidate the growth factor and cytokine secretion profile of QUANT-MSCs and HYPER-MSCs, the levels of the 45 cytokines, chemokines, and growth factors were measured in both cell culture media and the UC-MSC-derived EVs of the three selected cell lines and their input P4-MSCs. In terms of secretion into the culture media, QUANT-MSCs, HYPER-MSCs and P4-MSCs did not induce cytokines related to Th9, Th17, Th22, or T regulatory cell responses, including IL-9, IL-10, IL-17A, IL-21, IL-22, IL-23, and IL-27, or inflammatory cytokines, including TNF-alpha, IL-1 alpha, IL-1RA, IL-7, IL-15, IL-31, and TNF-beta (Fig. 4A). HYPER-MSCs tended to secrete higher levels of IL-6 than QUANT-MSCs (Fig. 4B) (1327.4 ± 1074.7 pg/106 cells vs 648.4 ± 354.1 pg/106 cells), while QUANT-MSCs released higher levels of IL-8 than HYPER-MSCs (Fig. 4C) (1091.7 ± 81.4 pg/106 cells vs 750.3 ± 34.1 pg/106 cells). Both QUANT-MSCs and HYPER-MSCs released similar level of GRO-alpha (745.95 ± 169.3 pg/106 cells vs 757.1 ± 204.6 pg/106 cells, respectively), and significantly higher than that of P4-MSCs (229.1 ± 9.6 pg/106 cells) (Fig. 4D). The levels of the chemokines MCP-1 (Fig. 4E) and SDF-1α (Fig. 4G) were higher in HYPER-MSCs than in QUANT-MSCs (MCP-1: 11062.0 ± 3129.2 pg/106 cells vs 6351.1 ± 1861.6 pg/106 cells; SDF-1α: 2844.1 ± 1647.2 pg/106 cells vs 453.2 ± 453.2 pg/106 cells). However, MIP-1β (Fig. 4F) and PLGF-1 (Fig. 4H) were secreted by QUANT-MSCs (0.4 ± 33.9 pg/106 cells and 328.3 ± 94.6 pg/106 cells, respectively) but not HYPER-MSCs. LIF levels were higher in HYPER-MSCs (Fig. 4I) (759.9 ± 219.4 pg/106 cells vs 354.1 ± 48.4 pg/106 cells), but HGF levels were higher in QUANT-MSCs (Fig. 4J) (4623.9 ± 2560.7 pg/106 cells vs 2959.4 ± 355.1 pg/106 cells). Collectively, our data revealed that both methods generated UC-MSCs with the ability to produce high levels of cytokines, chemokines and growth factors. The UC-MSCs obtained via each method showed slightly different secretion profiles, and culturing UC-MSCs in the Quantum system tended to result in the release of secretion factors with higher levels of growth factors, chemokines and Th1/Th2 cytokines than those observed for cells cultured in HYPERflasks.
Fig. 4.

Supernatant cytokine secretion profiles of UC-MSCs expanded in the Quantum Cell Expansion system and HYPERflasks. Levels of 45 cytokines, chemokines, and growth factors were measured in the cell culture media for QUANT-MSCs, HYPER-MSCs, and P4-MSCs. (A) A heatmap of supernatant cytokines, chemokines, and growth factors released by UC-MSCs expanded in the Quantum Cell Expansion system and HYPERflasks. No induction of cytokines related to Th9, Th17, Th22, or T regulatory cell responses or inflammatory cytokines in any of the cell groups. (B) Higher levels of IL-6 secretion in HYPER-MSCs compared to QUANT-MSCs. (C) Higher levels of IL-8 secretion in QUANT-MSCs compared to HYPER-MSCs. (D) Similar GRO-alpha secretion levels in QUANT-MSCs and HYPER-MSCs, both significantly higher than in P4-MSCs. (E) Higher MCP-1 and (G) SDF-1α levels in HYPER-MSCs compared to QUANT-MSCs. (F) MIP-1β and (H) PLGF-1 secretion detected in QUANT-MSCs but not HYPER-MSCs. (I) Higher LIF levels in HYPER-MSCs compared to QUANT-MSCs. (J) Higher HGF levels in QUANT-MSCs compared to HYPER-MSCs. Overall, both QUANT-MSCs and HYPER-MSCs produce high levels of cytokines, chemokines, and growth factors with slightly different secretion profiles. QUANT-MSCs tend to release higher levels of growth factors, chemokines, and Th1/Th2 cytokines than HYPER-MSCs.
2.3.5. EV characterization
To characterize EVs from QUANT-MSCs (EV/QUANT-MSCs), HYPER-MSCs (EV/HYPER-MSCs) and P4-MSCs (EV/P4-MSCs), EV morphology was observed by TEM, and total protein was measured by a bicinchoninic acid assay (Fig. 5). EVs comprise a heterogeneous population of vesicles, including exosomes and microvesicles with different sizes and shapes (Fig. 5A, red arrow). EV/QUANT-MSCs and EV/HYPER-MSCs were found to be similar in morphology (Fig. 5A). EV/QUANT-MSCs and EV/HYPER-MSCs presented higher levels of total protein than EV/P4-MSCs (Fig. 5B), suggesting that QUANT-MSCs and HYPER-MSCs could release more EVs than P4-MSCs.
Fig. 5.

Characterization of EVs derived from QUANT-MSCs, HYPER-MSCs, and P4-MSCs. (A) TEM observation of EV morphology, showing a heterogeneous population of vesicles, including exosomes and microvesicles with different sizes and shapes (red arrow). EV/QUANT-MSCs and EV/HYPER-MSCs exhibit similar morphology. (B) Higher total protein levels in EV/QUANT-MSCs and EV/HYPER-MSCs compared to EV/P4-MSCs, indicating greater EV release by QUANT-MSCs and HYPER-MSCs. (C) Luminex assay (heat map) results showing no production of Th9, Th17, Th22, T regulatory cell-related cytokines, or inflammatory cytokines in EV/QUANT-MSCs, EV/HYPER-MSCs, or EV/P4-MSCs. (D) Significantly higher IL-8 levels in EV/QUANT-MSCs compared to EV/HYPER-MSCs. (E) Higher TNF-α levels in EV/HYPER-MSCs than in EV/QUANT-MSCs. (F, H, I) Higher levels of Th1/Th2-related cytokines (IL-8), chemokines (MCP-1), and growth factors (FGF-2 and HGF) in EV/QUANT-MSCs and EV/HYPER-MSCs compared to EV/P4-MSCs. (G) Higher RANTES levels in EV/QUANT-MSCs compared to EV/HYPER-MSCs, although not statistically significant. Overall, EV/QUANT-MSCs produce higher levels of growth factors, chemokines, and Th1/Th2 cytokines than EV/HYPER-MSCs. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Next, EV-derived growth factors, chemokines, and cytokines were examined by a Luminex assay. Similar to EV/P4-MSCs, EV/QUANT-MSCs and EV/HYPER-MSCs did not produce cytokines related to Th9, Th17, Th22, and T regulatory responses, including IL-9, IL-10, IL-17A, IL-21, IL-22, and IL-27, or inflammatory cytokines, including IFN-alpha, IL-1 alpha, IL-1RA, IL-7, IL-15, IL-31, and TNF-beta (Fig. 5C). The levels of Th1/Th2-related cytokines (IL-8), chemokines (MCP-1) and growth factors (FGF-2 and HGF) were found to be higher in EV/QUANT-MSCs (2.9 ± 1.7 pg/106 cells IL-8, 15.1 ± 8.8 pg/106 cells MCP-1, 63.48 ± 19.59 pg/106 cells FGF-2, and 37.5 ± 16.8 pg/106 cells HGF) and EV/HYPER-MSCs (0.31 ± 0.31 pg/106 cells IL-8, 5.5 ± 2.3 pg/106 cells MCP-1, 30.96 ± 11.17 pg/106 cells FGF-2, and 14.4 ± 1.5 pg/106 cells HGF) than in EV/P4-MSCs (0 pg/106 cells IL-8, 3.3 ± 1.98 pg/106 cells MCP-1, 7.3 ± 3.2 pg/106 cells FGF-2, and 2.2 ± 1.1 pg/106 cells HGF) (Fig. 5D, F, H, I). Regarding Th1/Th2 cytokines, while EV/QUANT-MSCs showed significantly higher levels of IL-8 than EV/HYPER-MSCs (Fig. 5D), EV/HYPER-MSCs had higher levels of TNF-α than EV/QUANT-MSCs (TNF-α: 3.8 ± 2.2 pg/106 cells vs 0.99 ± 1.7 pg/106 cells, respectively) (Fig. 5E). EV/QUANT-MSCs showed higher levels of growth factors (FGF-2 and HGF), chemokines (MCP-1) and Th1/Th2 cytokines (IL-8) than those of EV/HYPER-MSCs (Fig. 5D, F, H, I). A higher level of RANTES was found in EV/QUANT-MSCs (2.8 ± 2.5 pg/106 cells) than in EV/HYPER-MSCs (0.24 ± 0.15 pg/106 cells), but there was no statistically significant difference between the EVs (Fig. 5G). Taken together, these results confirmed that EV/QUANT-MSCs are able to produce higher levels of growth factors, chemokines and Th1/Th2 cytokines than EV/HYPER-MSCs.
3. Discussion
The establishment of a high-quality master cell bank plays a crucial role in the cellular manufacturing process for downstream clinical applications. In our study, 30 UC-MSC lines that were obtained in a previous study and cultured under xeno-free and serum-free culture conditions were subjected to a screening process to prepare a Token Cell stock (premaster stock) at P1, Master Cell Bank (P2, P3) and Working Cell Bank (P4). Following the International Stem Cell Bank Initiative (ISCBI) guidelines, our selected UC-MSCs were cultured under standardized xeno- and serum-free conditions, and all results met the minimal criteria for MSC identification established by the ISCT [28]. In most studies, the genetic stability of MSCs has been evaluated using diploid Geimsa-banded karyotype techniques to confirm the normal karyotype, which represents the genetic stability during culture [29,30]. All 30 UC-MSC lines used in this study have previously been proven to have a normal and stable karyotype after 7 passages [16]. However, it was reported that the accumulation of genetic changes can occur, which would not necessarily alter the higher-order characteristics of chromosomes or be detected by the G-banding technique [30]. Thus, to ensure high quality of the MSC bank, UC-MSCs that met our screening criteria were subjected to whole-genome sequencing, which is a high-resolution genetic analysis technique. Our results presented a genome-wide screen of UC-MSCs to identify cell lines that are suitable for downstream clinical application with minimal risks of carrying mutations related to cancer, metabolic disorder, cardiovascular disease, etc. In addition, our results also provided a starting data set for the identification of genetic variations in culture-expanded UC-MSCs, which further enhances the reliability of the cell sources during the manufacturing process. It was reported that the emergence of multiple low-frequency single nucleotide changes (SNCs) in primary MSCs does occur. However, early passages of MSCs in vitro did not show a dramatically increased mutation frequency, as human culture-expanded MSCs maintain a relatively stable genomic composition [31].
To establish a robust, reliable and reproducible manufacturing process, it is important that the principle of QbD be applied [32]. For any cell-based products generated under the QbD principle, the most important step is defining the quality target product profile, which describes in detail the properties of the desired end products, including production capacity, cellular identity, potency, and purity [33]. The definition and establishment of the QTPP are also the starting points of the QbD process. Thus, in our study, once the master and working cell banks were established, we created our QTPP for cell products generated via either manual or automated manufacturing processes. Typically, MSC-based products must meet all minimal criteria established by the ISCT to ensure the identification of MSCs throughout the manufacturing process. In fact, the minimal criteria are necessary but are not sufficient to ensure the high quality of MSC-based products. Thus, potency tests are introduced to provide a solid background on the functions of MSCs for their downstream clinical applications. In our study, potency tests, including investigations of immunosuppressive activity and MSC secretion profiles post-manufacture, were performed. As MSCs are known to deliver their therapeutic effects via effectors such as growth factors, cytokines and extracellular vesicles (EVs), it is important to evaluate the components and concentrations of secreted factors from MSCs post-manufacture to assess their potency prior to clinical applications. Identifying the QTPP and the CQAs are critical steps to ensure UC-MSC safety and efficacy and to provide background data for measurements and quality control to ensure robust manufacturing processes.
One of the major challenges of clinical trials is the conversion for laboratory-scale manufacturing to larger-scale for large trials (Phase III) or commercialization of products, which is also one of the critical process parameters (CPPs) [34]. This issue arises from the fact that the methods that are used at the laboratory scale to support small-scale trials might not be able to provide enough cells for larger trials, where billions of cells with equivalent quality and quantity are needed. Several studies have tried to adapt the manufacturing process from 2D culture to 3D platforms with automated culture systems without assessing the potency of MSC-based products [35]. In addition, several CQAs and CPPs of the QbD change significantly when methods are switched from small-scale to large-scale manufacturing, resulting in alteration of the therapeutic potential of the final product. Thus, the results of this study provide a solution to address these challenges. We proposed a system that allows easy switching from a small scale (i.e., using the HYPERflask system) to an adapted scale-out platform (i.e., Quantum Automated Cell Expansion System) to provide acceptable and equivalent MSC products under xeno-free and serum-free culture conditions without any changes in CQAs or QTPP under the QbD principle.
This study provides the first data set that directly compares the biological properties and potency of UC-MSCs generated via either manual (HYPERflasks) or automated manufacturing platforms (the Quantum Cell Expansion System). In terms of MSC characteristics, both QUANT-MSCs and HYPER-MSCs showed similar morphology, MSC markers, differentiation ability, and normal karyotypes. Our previous study demonstrated that providing a coating substrate allowed MSCs to proliferate and maintain their characteristics during prolonged culture on T-flasks under xeno- and serum-free conditions. However, in this study, our results supported the elimination of the coating substrate for the HYPERflasks, which in turn reduced the manufacturing cost per cell product without changing the cell quality. These results were not surprising, as MSCs were propagated under either serum-free or serum-containing conditions and did not require a coating substrate when they were maintained in the HYPERflasks due to the unique treated culture surface [36]. For HYPERflasks, the maximum number of harvested cells was approximately 1 × 108 cells/flask, equivalent to a yield of approximately 25 T225 flasks, whereas the maximum number of harvested cells obtained from the Quantum system was 9 × 108 cells/run using our optimized protocol. The number of harvested cells was higher than that previously reported [[37], [38], [39]]. As cell quality and potency are tightly regulated by metabolic activities, it is important to understand how these cells behave under the culture conditions of HYPERflasks and the Quantum system. Our metabolic analysis revealed that QUANT-MSCs generated an amount of ATP equivalent to that generated by P4-MSCs and HYPER-MSCs while maintaining a lower glycolysis rate and glycolytic capacity. These data suggest that MSCs cultured on dynamic porous scaffolds (in the Quantum bioreactor with automated real-time feeding) showed higher glycolytic activity than cells cultured under the static 2D culture conditions found in HYPERflasks. As glycolysis is a critical energy production pathway in MSCs, recent studies demonstrated that increases in the glycolytic activity of MSCs improved their therapeutic effects in several disease conditions [40], upregulating their proliferation ability, enhancing their survival after transplantation, and increasing the secretion of growth factors and cytokines involved in tissue regeneration [41,42]. Interestingly, QUANT-MSCs also show higher respiratory spare capacity, supporting the notion that these cells confer better energy production via either glycolysis or mitochondrial respiration. This finding is consistent with the previous suggestion that mitochondrial activities play important roles in signal transmission in MSCs, especially for cell signals related to reactive oxygen species (ROS) [43], calcium homeostasis, and membrane potential. In fact, we speculated that the lower glycolysis rate and glycolytic capacity together with the higher respiratory spare capacity found in QUANT-MSCs illustrate energy compensation between glycolysis and mitochondrial respiration to maintain low levels of ROS, supporting previous findings [44,45].
MSCs usually exhibit therapeutic potential via a wide range of signaling molecules, including cytokines and growth factors. Our study showed that among 19 interleukins (ILs), only IL-6 and IL-8 were highly secreted from manufactured MSCs. The level of IL-6 was similar among all tested cell types, whereas QUANT-MSCs secreted the highest level of IL-8, followed by HYPER-MSCs and P4-MSCs. The secretion of IL-6 and IL-8 together with GRO-α and MCP-1 supports the ability of UC-MSCs from either the Quantum system or HYPERflasks to suppress PBMC proliferation in vitro [46]. Our findings are consistent with previous studies suggesting that MSCs from bone marrow, adipose tissue, and the umbilical cord secrete high levels of IL-6, IL-8, GRO-α, and MCP-1, and these chemokines can be detected in the supernatants of MSC-conditioned medium under xeno- and serum-free culture conditions [47,48]. Interestingly, we detected a high level of MIP-1β, a specific chemokine involved in the anti-inflammatory response of MSCs, produced by HYPER-MSCs in their resting state [49]. Manufactured MSCs also showed high levels of growth factors, including PLGF-1, LIF and HGF, reflecting their high quality and potency after manufacture. It is now accepted that MSCs exert their therapeutic potential by communicating with other cells through a wide range of EVs. Our data showed a significant improvement in EV production from both HYPER-MSCs and QUANT-MSCs, with approximately 10 μg EV/1 million cells. The number of EVs produced via our optimized manufacturing platform could potentially meet the needs of clinical trials, such as trials of treatments for bronchopulmonary dysplasia (requiring 200 pmol EV/kg patient body weight, NCT03857841), dry eye (10 μg EV/drop, NCT04213248), or ischemic stroke (200 μg EV/patient, NCT03384433). A detectable number of exosomes was also found in conditioned medium from both the Quantum and Hyperflask culture platforms in our study, with the size of exosomes ranging from 30 to 150 nm. Detailed analysis of EV components, mostly cytokines, chemokines and growth factors, notably IL-8, TNF-α, MCP-1, RANTES, FGF-2, and HGF, showed a diverse range of EVs from both QUANT-MSCs and HYPER-MSCs. These factors are produced at high levels by manufactured MSCs.
In our study, we aimed to directly compare two manufacturing process of MSCs, including an automated cell culture system (Quantum Expansion System, Terumo) and a manual process using multilayer HYPERflask. By optimizing for both manufacturing processes, the results demonstrated the feasibility of generating high-quality MSC products with equivalent QTPPs. Our predefined QTPPs encompassed the CQAs such as MSC morphology, cell viability, MSC marker expression, karyotype, cytokine profile, exosome production and metabolic function. Despite differences in yield, the two manufacturing processes produced MSC products with comparable QTPPs. Our findings not only align with the principles set forth in QbD development, but also highlight the optimization process required in the CPMP/ICH/5721/03 ICH Topic Q5E “Comparability of Biotechnological/Biological Products” and EMA/CAT/499821/2019 Committee for Advanced Therapies (CAT) guidelines [50]. The results of our study illustrated the value of implementing a systematic approach for MSC-based product development, ensuring comparability, consistency, and reproducibility of the manufacturing process regardless of the chosen approach – automated culture system or multiplayer flask. By using the data provided from this study, researchers or manufacturers would save time and effort in conducting experimental design for MSC-based product, as our system employs xeno- and serum-free culture conditions with all materials being GMP-graded and commercially available. This optimized processes ensures that manufacturing platforms can be utilized interchangeably without compromising the quality, safety, or efficacy of the MSC-based products.
4. Conclusions
Overall, our study demonstrated that MSCs generated from the automated Quantum Expansion system and the manual HYPERflask approach under xeno- and serum-free conditions exhibited equivalent biological phenotypes, metabolic profiles, immunosuppression abilities, and secretion profiles (supernatant and extracellular vesicles). By adhering to the principles of the CPMP/ICH/5721/03 ICH Topic Q 5 E “Comparability of Biotechnological/Biological Products” and EMA/CAT/499821/2019 Committee for Advanced Therapies (CAT) guidelines, as well as incorporating the QbD strategy, our study contributes to the ongoing efforts to standardize and improve MSC manufacturing processes in line with regulatory expectations. Furthermore, our research offers a valuable resource for other researchers or manufacturers looking to streamline the development of MSC-based products using either automated culture systems or multilayer flask approaches. Although the regulatory path for MSC-based products as advanced cell therapies has been proposed and accepted, certain challenges remain. One such problem might be the problem of MSC heterogeneity, as it is difficult to obtain a monoclonal MSC population. In addition, as our protocols generate MSCs without priming or stimulating factors, it is difficult to anticipate the potential therapeutic potential of these cells in their downstream applications.
5. Material and methods
5.1. Cell culture
All cell culture was conducted at 37 °C and 5% CO2 in a humidified atmosphere. All culture reagents were purchased from Thermo Fisher and Miltenyi Biotec unless otherwise stated. All 30 UC-MSC lines were established under xeno- and serum-free culture conditions in a previous study. A single vial of each cell line at passage 0 (P0) was thawed and cultured to establish a master cell bank (at P1 and P2) and working cell bank (at P3).
5.2. Primary and secondary screening of UC-MSCs
Prior to initiating the cellular manufacturing process, we conducted screening of 30 established UC-MSC lines, which were previously fully characterized under xeno-free and serum-free conditions [16]. To start the screening process, all parents who participated in the previous project and donated their child’s umbilical cord to establish the UC-MSC lines were invited to Vinmec International Hospital (Hanoi, Vietnam) to undergo genetic counseling using a prepared questionnaire containing 97 questions related to the infant’s medical history, general health, family genetic background, clinical history of cancer, cardiovascular disease, diabetes, metabolic disorders, etc., and lifestyle and behavior (Supplementary Table 1). The scoring system was applied to select 20 lines that had a score less than 32 to enter the secondary screening. The secondary screening focused on the biological characterization of the UC-MSC lines following the International Society for Cell and Gene Therapy (ISCT) guidelines. Several criteria were set to evaluate the quality of the UC-MSCs, including population doubling time, karyotyping, number of colony-forming units (CFUs), MSC markers as ISCT guidelines, and differentiation ability (Supplementary Table 2). After two rounds of screening, the selected UC-MSC lines were subjected to whole-genome sequencing to exclude UC-MSC lines that carry genetic mutations related to cardiovascular disease, cancer, metabolic disorders, etc.
5.3. Genome sequencing of UC-MSCs
Once the culture of selected UC-MSCs at P2 reached 80% confluence, they were harvested for further expansion, and two million UC-MSCs were collected for DNA extraction using a QIAamp DNA Mini Kit (Qiagen). The DNA concentration and quality were measured with a Nanodrop and Quibit dsDNA BR Assay kits (Invitrogen, US). The TruSight One Expanded Sequencing Panel, which covers the coding regions of 6794 genes and 16.6 Mb of genomic content, was used for DNA library preparation, and then the size and quality of the DNA library were calculated by Quibit and Tape Station (Agilent, US). The NextSeq 550 High Output Kit (v2.5) was used for DNA sequencing. BWA-MEM and DRAGEN-GATK software were used for alignment and variant calling. Variants were classified according to ACMG and AMP guidelines by TAPE, ANNOVAR, and Variant Interpreter (Illumina, US). Three cell lines that met all screening criteria were randomly selected for the manufacturing process.
5.4. Manual manufacturing process of UC-MSCs using Corning® CellBIND® surface HYPERFlasks®
The selected UC-MSC lines at P4 were thawed in prewarm StemMACS™ MSC expansion medium (Miltenyi Biotec) and centrifuged at 500×g for 5 min at room temperature. The cells were then resuspended in StemMACS medium and reseeded into a T75 Nunc™ EasYFlask™ Cell Culture Flask at a density of 3200 cells/cm2. Once the culture reached 80% confluence, the cells were incubated with TrypLE Select for 4 min at 37 °C and diluted with StemMACS medium, followed by centrifugation at 400×g for 5 min. The cells were counted and seeded into Corning® CellBIND® Surface HYPERFlask® cell culture vessels at a density of 3400 cells/cm2. When the culture reached 80% confluence, the cells were harvested for further analysis and cryopreserved in CryoStor CS10 cell freezing medium (Stemcell Technologies, Canada), followed by storage in liquid nitrogen for further use.
5.5. Automated manufacturing process of UC-MSCs using the Quantum® Cell Expansion System
To expand UC-MSCs under xeno- and serum-free culture conditions using the Quantum® Cell Expansion System (Terumo BCT, USA), the cell expansion set was installed according to the manufacturer’s protocol and primed with PBS. The standard operation protocol followed a previous study with modifications of a particular section [51]. The bioreactor was then coated with vitronectin (A14700, Thermo Fisher Scientific, 2.5–10 mg) in PBS overnight. On the following day, the bioreactor was washed with PBS to remove vitronectin residue followed by inoculation of 25 × 106 UC-MSCs at P4. The cells were cultured automatically under continuous flow of StemMACS medium at a low feeding rate with a premixed gas supply (20% O2, 5% CO2, and 75% N2). The feeding rate was changed daily to meet the nutrient requirements of the cultured cells based on measurement of the glucose consumption rate using a glucose meter (Arkrey, Japan) and lactate production levels using a lactate measurement device (Nova Biomedical, USA). The cells were harvested after 7 days in culture using 200 mL TrypLE Select, followed by neutralization using 400 mL PBS supplemented with human albumin (0.1%, vol/vol). The harvested cells were then subjected to other analyses, cryopreserved in CryoStor CS10 cell freezing medium (Stem Cell Technologies) in cryogenic vials at 20 × 106 cells/vial using a CryoMed™ Controlled-Rate Freezer (Thermo Fisher Scientific) and stored in liquid nitrogen for further use.
5.6. Comparative analysis of UC-MSCs generated via the manual and automated manufacturing platforms
5.6.1. Biological characterization
UC-MSCs harvested from either Corning® CellBIND® Surface HYPERFlask® cell culture vessels or the Quantum® Cell Expansion System were subjected to quality control tests, including tests for cell yield, cell viability (Trypan Blue), MSC markers and differentiation. The identification of MSCs included evaluations of the expression of positive MSC markers (CD73, CD90, and CD105) and negative markers (CD11b, CD19, CD34, CD45 and HLA-DR) using the Human MSC Analysis Kit (Becton Dickinson, USA). The cells were analyzed using a FACSLyric flow cytometer (Becton Dickinson, USA), followed by data analysis using FlowJo software. To assess differentiation ability, the harvested UC-MSCs were tested using a StemPro Osteogenesis, Adipogenesis, and Chondrogenesis Differentiation kit following the manufacturer’s instructions. Once differentiation was completed, the cells were fixed with 4% paraformaldehyde and stained with Alizarin Red S, Oil Red O, and Alcian Blue to detect osteogenic, adipogenic, and chondrogenic lineages, respectively.
5.6.2. Metabolic activities
The cellular mitochondrial stress and glycolytic stress assays were performed using an Agilent Seahorse XFe96 Analyzer (Agilent Technologies) as previously described [52]. Briefly, 1 × 104 UC-MSCs in 100 μL of StemMACS medium were seeded into each well of Seahorse XF96 cell culture microplates (Agilent Technologies) coated with CTS™ CellStart™ (1:300 in PBS) and were then incubated overnight at 37 °C in 5% CO2. The XFe96 Extracellular Flux sensor cartridges (Agilent Technologies) were hydrated with distilled water for 24 h, followed by incubation with Agilent Seahorse XF Calibrant (pH 7.4) (Agilent Technology) for 1 h at 37 °C in a non-CO2 incubator before the assay. On the day of the experiment, the StemMACS medium from the microplates was removed, and the wells were rinsed with prewarm XF assay medium containing 10 mM glucose (Agilent Technologies, #103577-100), 2 mM glutamine (Agilent Technologies, #103579-100), and 1 mM pyruvate (Agilent Technologies, #103578-100). The wells were then filled with 180 μL of the XF assay medium, and the cell culture plates were incubated for 1 h at 37 °C without CO2.
To measure the glycolytic function of hUC-MSCs, glucose (10 mM), oligomycin (1 μM), and 2-deoxy-glucose (2-DG; 50 mM) from the Seahorse XF Cell Glycolysis Stress Test Kit (Agilent Technologies, #103020-100) were sequentially injected to establish parameters including basal acidification, glycolysis rate, glycolytic capacity, and glycolytic reserve. To measure the mitochondrial function of hUC-MSCs, oligomycin (1 μM), fluoro-carbonyl cyanide phenylhydrazone (FCCP; 2.0 μM) and rotenone (Rot; 0.5 μM) together with antimycin A (AA; 0.5 μM) from the Seahorse XF Cell Mito Stress test kit (Agilent Technologies, #103015-100) were sequentially injected to establish parameters, including basal respiration, proton leakage, maximal respiration, spare respiratory capacity, and ATP production. The cells were fixed with 4% paraformaldehyde (PFA) and stained with DAPI staining solution (1:5000, Abcam, #ab228549) after the assay. The ImageXpress® Micro Confocal High-content Imaging System (Molecular Devices) was used to count the cells in each well via DAPI staining for normalization.
5.6.3. Immunoregulation assay
Peripheral blood mononuclear cells (PBMCs) were isolated by the Ficoll gradient centrifugation method as previously described. The cells were then stored in CryoStor CS10 cell freezing medium at −80 °C until use. One day before coculture with MSCs, the cells were plated in RPMI medium supplemented with 10% FBS and 1% PS overnight. On the day of coculture, PBMCs were harvested and stained with carboxyfluorescein succinimidyl ester (CFSE) according to the manufacturer’s instructions. Cryopreserved hUC-MSCs were thawed and plated in StemMACS medium for 3 days at 37 °C and 5% CO2. The hUC-MSCs were then harvested and seeded in 96-well flat-bottom plates containing StemMACS medium at serial dilutions overnight for cell attachment.
For the T-cell proliferation assay, hUC-MSCs were washed with PBS and cocultured with 5 × 104 PBMCs/well in 200 μl RPMI in the presence of anti-CD2, anti-CD3, and anti-CD28 (αCD3/CD28) T-cell activation beads (Miltenyi Biotec) according to the manufacturer’s instructions. After 5 days, all cells were harvested. To measure T-cell proliferation, all cells were stained with anti-CD3-V450, anti-CD4-APC-Vio770, and CD8-PE-Vio777 (Miltenyi Biotec) for 30 min at 4 °C. The cells were then stained with Cytox Red to discriminate a viable cell population and subjected to flow cytometry. All stained cells were acquired on a FACSLyric flow cytometer (BD). Fluorescence compensation was optimized using cells individually labeled with each fluorochrome-conjugated antibody. Data were obtained from the live population based on cell size- and morphology-based gating, which was used to eliminate cell debris and dead cells. The data were analyzed using FlowJo software (Tree Star).
5.6.4. Growth factor and cytokine secretion profile
The concentrations of 45 cytokines, chemokines, and growth factors were measured in the supernatant from MSC cultures and EV lysates using Cytokine/Chemokine/Growth Factor 45-Plex Human ProcartaPlex™ Panel 1 (EPX450-12171-901) in accordance with the manufacturer’s recommendations (MAN0019825) with a Luminex 200 instrument (Luminex Corporation, Austin, Texas, USA) and analyzed using ProcartaPlexAnalyst software; the evaluated cytokines included the following: Th1/Th2 cytokines: granulocyte-macrophage colony-stimulating growth factor (GM-CSF), interferon - gamma (IFN-γ), interleukin-1 beta (IL-1β), IL-2, IL-4, IL-5, IL-6, IL-8, IL-12p70, IL-13, IL-18, and tumor necrosis factor-alpha (TNF-α); Th9/Th17/Th22/Treg cytokines: IL-9, IL-10, IL-17A (Cytotoxic T-lymphocyte-associated antigen-8 – CTLA-8), IL-21, IL-22, IL-23, and IL-27; inflammatory cytokines: IFN-α, IL-1α, interleukin-1 receptor antagonist (IL-1RA), IL-7, IL-15, IL-31, and TNF-β; chemokines: Eotaxin (C–C motif chemokine ligand 11 – CCL11), Growth-related Oncogene alpha (GRO-α or C-X-C motif ligand 1 – CXCL1), Interferon-gamma inducible protein 10 (IP-10 or CXCL10), Monocyte chemoattractant protein-1 (MCP-1 or CCL2), Macrophage Inflammatory Protein 1 alpha (MIP-1α or CCL3), MIP-1β (CCL4), RANTES (CCL5), and Stromal cell-derived factor 1 alpha (SDF-1α); and growth factors: brain-derived neurotrophic factor (BDNF), epidermal growth factor (EGF), fibroblast growth factor 2 (FGF-2), hepatocyte growth factor (HGF), nerve growth factor-beta (NGF-β), platelet-derived growth factor BB (PDGF-BB), placenta growth factor-1 (PIGF-1), stem cell factor (SCF), vascular endothelial growth factor-D (VEGF-D), vascular endothelial growth factor-A (VEGF-A) and leukemia inhibitory factor (LIF). After measurement, the values were normalized to the total number of harvested cells. Values less than the lowest limit of quantification (LLOQ) were assigned a value of 0.0 pg/mL. All samples were analyzed in duplicate according to the manufacturer’s instructions.
5.6.5. Isolation and characterization of extracellular vesicles
After harvesting UC-MSCs via either the manual manufacturing process or the automated culture platform, the cell culture supernatants were collected for extracellular vesicle (EV) collection using the ultracentrifugation method as previously described [53]. Briefly, the supernatants were centrifuged at 300×g for 10 min at 4 °C to remove the cell pellet. The collected supernatants were then centrifuged at 2000×g for 20 min at 4 °C to remove dead cells and large EVs and then at 10,000×g for 30 min at 4 °C to remove cell debris. The supernatants were then centrifuged at 100,000×g for 70 min at 4 °C to collect pellets containing EVs and contaminating proteins. The pellets were washed with 10 mL PBS and subjected to an additional centrifugation at 100,000×g for 70 min at 4 °C to remove contaminating protein. The remaining EVs were resuspended in 100 μl PBS and stored at −80 °C for further use. To visualize and measure the EV size, TEM images of EV samples were obtained using a JEOL 1100 Transmission Electron Microscope (TEM, JEOL Ltd., Tokyo, Japan) at 80 kV. For visualization, EV samples were dropped and dried on a formvar- and carbon-coated copper grid (Ted Pella, Inc.).
5.6.6. Statistical analysis
Data are presented as the means ± SEM. Significant differences among multiple groups were assessed using one-way ANOVA followed by Bonferroni’s correction (ANOVA/Bonferroni). Pvalues < 0.05 were considered to be statistically significant. All analyses were performed using GraphPad PRISM software (GraphPad).
Author contribution statement
Duc M. Hoang: Conceived and designed the experiments; Performed the experiments; Analyzed and interpreted the data; Contributed reagents, materials, analysis tools or data; Wrote the paper.
Quyen T. Nguyen, Trang T.K. Phan, Anh T.L. Ngo, Phuong T. Pham: Performed the experiments; Analyzed and interpreted the data; Contributed reagents, materials, analysis tools or data; Wrote the paper.
Trung Q. Bach: Performed the experiments; Contributed reagents, materials, analysis tools or data; Wrote the paper.
Phuong T.T. Le, Hoa T.P. Bui: Performed the experiments.
Liem Nguyen Thanh: Conceived and designed the experiments; Contributed reagents, materials, analysis tools or data; Wrote the paper.
Funding statement
Dr Duc M. Hoang, Liem T. Nguyen were supported by Tập đoàn Vingroup - Công ty CP {PRO.19.47}.
Data availability statement
Data will be made available on request.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper
Acknowledgments
The author would like to thank the Vingroup Scientific Research and Clinical Application Fund (Grant number: PRO19.47) for supporting this work. We would like to thank Dr. Layka Abbasi, Dr. Sharon Shouhui, and the Support team from Terumo BCT and Phuong Dong Company for their knowledge and support in preparation and optimization of Quantum Expansion system. All figures were created with Biorender.com.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.heliyon.2023.e15946.
Contributor Information
Duc M. Hoang, Email: v.duchm3@vinmec.com.
Liem Nguyen Thanh, Email: v.liemnt@vinmec.com.
Appendix A. Supplementary data
The following are the Supplementary data to this article.
Primary screening of UC-MSC lines. (A) Thirty parents of 30 UC-MSC lines were participated in genetic consultation. Lines with scores higher than 32 were eliminated from further screening. (B) The expression of MSC markers of thirty UC-MSC lines was measured by flow cytometry analysis. (C) Population doubling time of the thirty UC-MSC lines was investigated. PDT of the cell lines was ranged from 18 to 24. (D) Colony forming unit of the thirty UC-MSC lines was calculated. Lines with CFU-F number lower than 200 was eliminated.
{kind=link}
References
- 1.Friedenstein A.J., Chailakhyan R.K., Gerasimov U.V. Bone marrow osteogenic stem cells: in vitro cultivation and transplantation in diffusion chambers. Cell Tissue Kinet. 1987;20(3):263–272. doi: 10.1111/j.1365-2184.1987.tb01309.x. [DOI] [PubMed] [Google Scholar]
- 2.Han S., et al. Adipose-derived stromal vascular fraction cells: update on clinical utility and efficacy. Crit. Rev. Eukaryot. Gene Expr. 2015;25(2):145–152. doi: 10.1615/critreveukaryotgeneexpr.2015013057. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y., et al. Effects of storage solutions on the viability of human umbilical cord mesenchymal stem cells for transplantation. Cell Transplant. 2013;22(6):1075–1086. doi: 10.3727/096368912X657602. [DOI] [PubMed] [Google Scholar]
- 4.Chen K., et al. Human umbilical cord mesenchymal stem cells hUC-MSCs exert immunosuppressive activities through a PGE2-dependent mechanism. Clin. Immunol. 2010;135(3):448–458. doi: 10.1016/j.clim.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 5.Torre P., Flores A.I. Current status and future prospects of perinatal stem cells. Genes (Basel) 2020;12(1) doi: 10.3390/genes12010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alkhalil M., Smajilagic A., Redzic A. Human dental pulp mesenchymal stem cells isolation and osteoblast differentiation. Med. Glas. (Zenica) 2015;12(1):27–32. [PubMed] [Google Scholar]
- 7.Zhou T., et al. Challenges and advances in clinical applications of mesenchymal stromal cells. J. Hematol. Oncol. 2021;14(1):24. doi: 10.1186/s13045-021-01037-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoang D.M., et al. Stem cell-based therapy for human diseases. Signal Transduct. Targeted Ther. 2022;7(1):272. doi: 10.1038/s41392-022-01134-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grau-Vorster M., et al. HLA-DR expression in clinical-grade bone marrow-derived multipotent mesenchymal stromal cells: a two-site study. Stem Cell Res. Ther. 2019;10(1):164. doi: 10.1186/s13287-019-1279-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song Y., et al. Human mesenchymal stem cells derived from umbilical cord and bone marrow exert immunomodulatory effects in different mechanisms. World J. Stem Cell. 2020;12(9):1032–1049. doi: 10.4252/wjsc.v12.i9.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kyurkchiev D., et al. Secretion of immunoregulatory cytokines by mesenchymal stem cells. World J. Stem Cell. 2014;6(5):552–570. doi: 10.4252/wjsc.v6.i5.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han Y., et al. The secretion profile of mesenchymal stem cells and potential applications in treating human diseases. Signal Transduct. Targeted Ther. 2022;7(1):92. doi: 10.1038/s41392-022-00932-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mellman I., Coukos G., Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ancans J. Cell therapy medicinal product regulatory framework in Europe and its application for MSC-based therapy development. Front. Immunol. 2012;3:253. doi: 10.3389/fimmu.2012.00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin J.Q., Zhu J., Ankrum J.A. Manufacturing of primed mesenchymal stromal cells for therapy. Nat. Biomed. Eng. 2019;3(2):90–104. doi: 10.1038/s41551-018-0325-8. [DOI] [PubMed] [Google Scholar]
- 16.Hoang V.T., et al. Standardized xeno- and serum-free culture platform enables large-scale expansion of high-quality mesenchymal stem/stromal cells from perinatal and adult tissue sources. Cytotherapy. 2021;23(1):88–99. doi: 10.1016/j.jcyt.2020.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Lipsitz Y.Y., Timmins N.E., Zandstra P.W. Quality cell therapy manufacturing by design. Nat. Biotechnol. 2016;34(4):393–400. doi: 10.1038/nbt.3525. [DOI] [PubMed] [Google Scholar]
- 18.Jung S., et al. Ex vivo expansion of human mesenchymal stem cells in defined serum-free media. Stem Cell. Int. 2012;2012 doi: 10.1155/2012/123030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Popov A., et al. Impact of serum source on human mesenchymal stem cell osteogenic differentiation in culture. Int. J. Mol. Sci. 2019;20(20) doi: 10.3390/ijms20205051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kirsch M., et al. Comparative analysis of mesenchymal stem cell cultivation in fetal calf serum, human serum, and platelet lysate in 2D and 3D systems. Front. Bioeng. Biotechnol. 2020;8 doi: 10.3389/fbioe.2020.598389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jossen V., et al. Manufacturing human mesenchymal stem cells at clinical scale: process and regulatory challenges. Appl. Microbiol. Biotechnol. 2018;102(9):3981–3994. doi: 10.1007/s00253-018-8912-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kouroupis D., Correa D. Increased mesenchymal stem cell functionalization in three-dimensional manufacturing settings for enhanced therapeutic applications. Front. Bioeng. Biotechnol. 2021;9 doi: 10.3389/fbioe.2021.621748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hassan M., et al. Large-scale expansion of human mesenchymal stem cells. Stem Cell. Int. 2020;2020 doi: 10.1155/2020/9529465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bellani C.F., et al. Scale-up Technologies for the manufacture of adherent cells. Front. Nutr. 2020;7 doi: 10.3389/fnut.2020.575146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phelps J., et al. Bioprocessing of mesenchymal stem cells and their derivatives: toward cell-free therapeutics. Stem Cell. Int. 2018;2018 doi: 10.1155/2018/9415367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maillot C., et al. Quality by design to define critical process parameters for mesenchymal stem cell expansion. Biotechnol. Adv. 2021;50 doi: 10.1016/j.biotechadv.2021.107765. [DOI] [PubMed] [Google Scholar]
- 27.Dominici M., et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 28.Crook J.M., Hei D., Stacey G. The international stem cell banking initiative (ISCBI): raising standards to bank on. In Vitro Cell. Dev. Biol. Anim. 2010;46(3–4):169–172. doi: 10.1007/s11626-010-9301-7. [DOI] [PubMed] [Google Scholar]
- 29.Borgonovo T., et al. Genetic evaluation of mesenchymal stem cells by G-banded karyotyping in a Cell Technology Center. Rev. Bras. Hematol. Hemoter. 2014;36(3):202–207. doi: 10.1016/j.bjhh.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stultz B.G., et al. Chromosomal stability of mesenchymal stromal cells during in vitro culture. Cytotherapy. 2016;18(3):336–343. doi: 10.1016/j.jcyt.2015.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cai J., et al. Whole-genome sequencing identifies genetic variances in culture-expanded human mesenchymal stem cells. Stem Cell Rep. 2014;3(2):227–233. doi: 10.1016/j.stemcr.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lipsitz Y.Y., et al. Achieving efficient manufacturing and quality assurance through synthetic cell therapy design. Cell Stem Cell. 2017;20(1):13–17. doi: 10.1016/j.stem.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 33.Guadix J.A., et al. Principal criteria for evaluating the quality, safety and efficacy of hMSC-based products in clinical practice: current approaches and challenges. Pharmaceutics. 2019;11(11) doi: 10.3390/pharmaceutics11110552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bieback K., Kinzebach S., Karagianni M. Translating research into clinical scale manufacturing of mesenchymal stromal cells. Stem Cell. Int. 2011;2010 doi: 10.4061/2010/193519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lechanteur C., et al. MSC manufacturing for academic clinical trials: from a clinical-grade to a full GMP-compliant process. Cells. 2021;10(6) doi: 10.3390/cells10061320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clark K.C., et al. Canine and equine mesenchymal stem cells grown in serum free media have altered immunophenotype. Stem Cell Rev. Rep. 2016;12(2):245–256. doi: 10.1007/s12015-015-9638-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanley P.J., et al. Efficient manufacturing of therapeutic mesenchymal stromal cells with the use of the Quantum Cell Expansion System. Cytotherapy. 2014;16(8):1048–1058. doi: 10.1016/j.jcyt.2014.01.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Russell A.L., Lefavor R.C., Zubair A.C. Characterization and cost-benefit analysis of automated bioreactor-expanded mesenchymal stem cells for clinical applications. Transfusion. 2018;58(10):2374–2382. doi: 10.1111/trf.14805. [DOI] [PubMed] [Google Scholar]
- 39.Mizukami A., et al. A fully-closed and automated hollow fiber bioreactor for clinical-grade manufacturing of human mesenchymal stem/stromal cells. Stem Cell Rev. Rep. 2018;14(1):141–143. doi: 10.1007/s12015-017-9787-4. [DOI] [PubMed] [Google Scholar]
- 40.Moya A., et al. Quiescence preconditioned human multipotent stromal cells adopt a metabolic profile favorable for enhanced survival under ischemia. Stem Cell. 2017;35(1):181–196. doi: 10.1002/stem.2493. [DOI] [PubMed] [Google Scholar]
- 41.Zhang W., et al. Cathepsin K deficiency promotes alveolar bone regeneration by promoting jaw bone marrow mesenchymal stem cells proliferation and differentiation via glycolysis pathway. Cell Prolif. 2021;54(7) doi: 10.1111/cpr.13058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen L.C., Wang H.W., Huang C.C. Modulation of inherent niches in 3D multicellular MSC spheroids reconfigures metabolism and enhances therapeutic potential. Cells. 2021;10(10) doi: 10.3390/cells10102747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu C., et al. Regulation of the mitochondrial reactive oxygen species: strategies to control mesenchymal stem cell fates ex vivo and in vivo. J. Cell Mol. Med. 2018;22(11):5196–5207. doi: 10.1111/jcmm.13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burnham A.J., Foppiani E.M., Horwitz E.M. Key metabolic pathways in MSC-mediated immunomodulation: implications for the prophylaxis and treatment of graft versus host disease. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.609277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lo T., et al. Glucose reduction prevents replicative senescence and increases mitochondrial respiration in human mesenchymal stem cells. Cell Transplant. 2011;20(6):813–825. doi: 10.3727/096368910X539100. [DOI] [PubMed] [Google Scholar]
- 46.Chinnadurai R., et al. Potency analysis of mesenchymal stromal cells using a combinatorial assay matrix approach. Cell Rep. 2018;22(9):2504–2517. doi: 10.1016/j.celrep.2018.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ivanova-Todorova E., et al. Adipose tissue-derived mesenchymal stem cells are more potent suppressors of dendritic cells differentiation compared to bone marrow-derived mesenchymal stem cells. Immunol. Lett. 2009;126(1–2):37–42. doi: 10.1016/j.imlet.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 48.Ivanova-Todorova E., et al. Conditioned medium from adipose tissue-derived mesenchymal stem cells induces CD4+FOXP3+ cells and increases IL-10 secretion. J. Biomed. Biotechnol. 2012;2012 doi: 10.1155/2012/295167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dorner B.G., et al. MIP-1alpha, MIP-1beta, RANTES, and ATAC/lymphotactin function together with IFN-gamma as type 1 cytokines. Proc. Natl. Acad. Sci. U. S. A. 2002;99(9):6181–6186. doi: 10.1073/pnas.092141999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salazar-Fontana L.I. A regulatory risk-based approach to ATMP/CGT development: integrating scientific challenges with current regulatory expectations. Front. Med. (Lausanne) 2022;9 doi: 10.3389/fmed.2022.855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frank N.D., et al. Evaluation of reagents used to coat the hollow-fiber bioreactor membrane of the Quantum(R) Cell Expansion System for the culture of human mesenchymal stem cells. Mater Sci. Eng. C Mater. Biol. Appl. 2019;96:77–85. doi: 10.1016/j.msec.2018.10.081. [DOI] [PubMed] [Google Scholar]
- 52.Ngo A.T.L., et al. Clinically relevant preservation conditions for mesenchymal stem/stromal cells derived from perinatal and adult tissue sources. J. Cell Mol. Med. 2021;25(22):10747–10760. doi: 10.1111/jcmm.17016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Purushothaman A. Springer; 2019. Exosomes from Cell Culture-Conditioned Medium: Isolation by Ultracentrifugation and Characterization; pp. 233–244. (The Extracellular Matrix). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primary screening of UC-MSC lines. (A) Thirty parents of 30 UC-MSC lines were participated in genetic consultation. Lines with scores higher than 32 were eliminated from further screening. (B) The expression of MSC markers of thirty UC-MSC lines was measured by flow cytometry analysis. (C) Population doubling time of the thirty UC-MSC lines was investigated. PDT of the cell lines was ranged from 18 to 24. (D) Colony forming unit of the thirty UC-MSC lines was calculated. Lines with CFU-F number lower than 200 was eliminated.
Data Availability Statement
Data will be made available on request.