
Figure 2. Hypothesized convergent/divergent mechanisms between (S)- and (R)-ketamine.
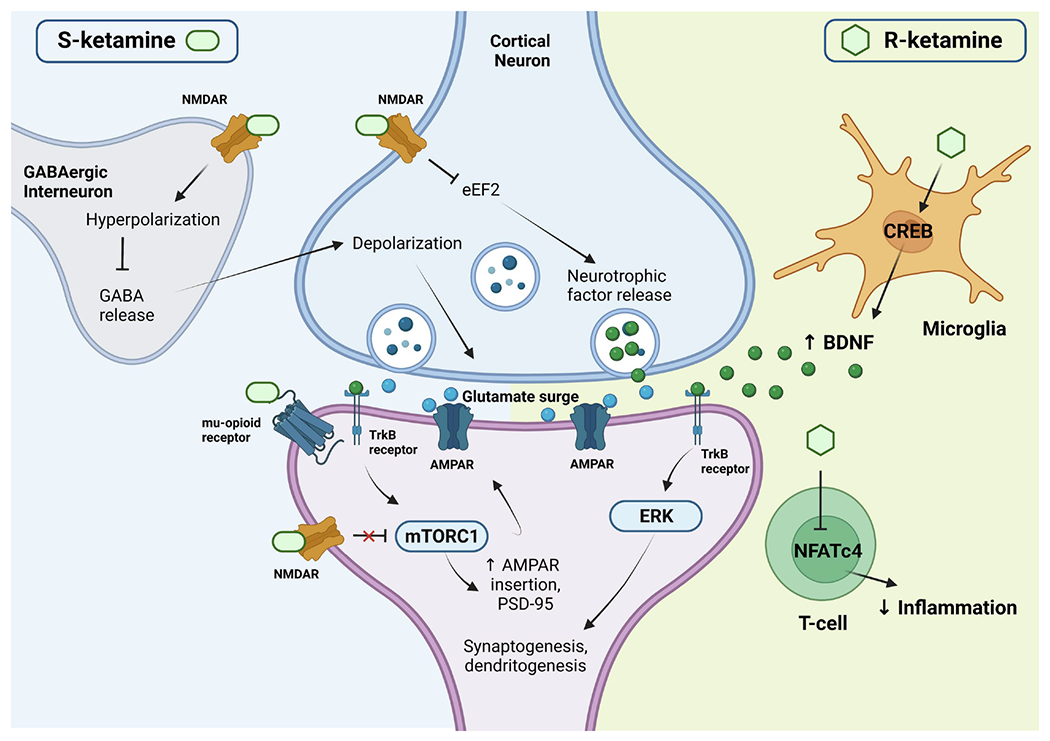
Both (S)- and (R)-ketamine increase the probability of glutamate release into the synaptic cleft, increasing AMPAR throughput and activating downstream cellular signaling mechanisms, as well as increasing synaptic protein translation of AMPAR subunits and PSD-95, contributing to further synaptogenesis and dendritogenesis. The actions of (S)-ketamine appear to be primarily facilitated through preferential binding to NMDARs expressed in GABAergic interneurons, leading to a depolarization of cortical excitatory neurons. This depolarization causes the observed glutamate release, as well as a release of neurotrophic factors such as BDNF, which binds to TrkB receptors. This, in turn, activates the mTORC1 signaling pathway, leading to the upregulation of synaptogenesis and dendritogenesis discussed earlier. In addition, (S)-ketamine binds to extrasynaptic NMDARs, disinhibiting mTORC1 signaling by deactivating eEFK2 (not pictured here). Binding to mu-opioid receptors may facilitate antidepressant effects but may also contribute to increased adverse events. In contrast, (R)-ketamine seems to primarily facilitate immune modulation by affecting microglial signaling and increasing BDNF release. This BDNF release binds to TrkB and activates the ERK signaling pathway, upregulating synaptogenesis and dendritogenesis. While displayed as distinct mechanisms here, please note that there may be overlap and other potential mechanisms not noted in this figure. Figure is approximate for illustrative purposes. Abbreviations: AMPAR: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; BDNF: brain-derived neurotrophic factor; CREB: cyclic adenosine monophosphate response element-binding protein; eEFK2: eukaryotic elongation factor-2 kinase; ERK: extracellular signal-related kinase; GABA: gamma aminobutyric acid; mTORC1: mechanistic target of rapamycin complex 1; NFATc4: nuclear factor of activated T cells 4; NMDAR: N-methyl-D-aspartate receptor; PSD-95: postsynaptic density protein 95; TrkB: tropomyosin receptor kinase B.