

Abstract
Pancreatic cancer (PC) has a poor prognosis, and current therapeutic strategies are ineffective in advanced diseases. We and others have shown the aberrant expression of CXCR2 and its ligands in PC development and progression. Our objective for this study was to evaluate the therapeutic utility of CXCR2/1 targeting using an small molecule antagonist, SCH-479833, in different PC preclinical murine models (syngeneic or xenogeneic). Our results demonstrate that CXCR2/1 antagonist had both antitumor and anti-metastatic effects in PC. CXCR2/1 antagonist treatment inhibited tumor cell proliferation, migration, angiogenesis, and recruitment of neutrophils, while it increased apoptosis. Treatment with the antagonist enhanced fibrosis, tumor necrosis, and extramedullary hematopoiesis. Together, these findings suggest that selectively targeting CXCR2/1 with small molecule inhibitors is a promising therapeutic approach for inhibiting PC growth, angiogenesis, and metastasis.
Keywords: Pancreatic cancer, CXCR2 antagonist, tumor growth, angiogenesis, neutrophils, metastasis
INTRODUCTION
According to Cancer Statistics 2023, pancreatic cancer (PC) is the fourth leading cause of cancer-related death in both males and females in the United States [1]. Unfortunately, by the time PC gets diagnosed, almost 80–85% of the patients have already reached an advanced stage or have metastases, leaving minimal treatment options [2]. This indicates the need for novel and effective therapeutic measures and a better understanding of the molecular mechanisms implicated in disease progression.
The G-protein-coupled receptors (GPCR) CXCR1 and CXCR2 and their ligands can regulate the growth of the tumor, angiogenesis, and metastasis in various cancers [3, 4]. CXCR2/1 are known to orchestrate immune responses in multiple diseases, including cancer [5, 6]. Several studies have confirmed the presence of CXCR2/1 and their ligands in human pancreatic ductal adenocarcinoma (PDAC) tissues and cell lines [7–15]. In addition to tumor cells, CXCR 1 and 2 are expressed in endothelial cells neutrophils, and immunosuppressive granulocytic myeloid suppressor cells (GR-MDSC) [16–21]. Neutrophils are one of the innate immune cell types which originate from the myeloid precursor [22] and can play a pro-tumor role in cancer [23, 24]. The expression of CXCR2 has also been reported on PDAC fibroblasts [25] and is a known mediator of tumor progression, angiogenesis, and metastasis in PDAC [10, 26, 27]. Furthermore, our lab has demonstrated the critical role of this signaling axis in regulating KRAS(G12D)-induced autocrine growth of PDAC cells [28]. These findings suggest that CXCR2/1 can be a promising therapeutic target for PDAC.
Monoclonal antibodies and small molecule antagonists that block the CXCR2/1 axes have been undergoing clinical trials [4, 29, 30]. Earlier reports demonstrate that treatment with CXCR2/1 antagonists, SCH479833 and SCH527123, inhibited the in vitro cell proliferation and chemotaxis of malignant cells and inhibited tumor growth, angiogenesis, and metastasis in pre-clinical studies using human melanoma and colon cancer cells [31, 32]. Steele et al., [33] demonstrated that CXCR2 inhibition profoundly inhibits PDAC metastasis and augments immunotherapies. A recent report from Gulhati et al [34] demonstrated that targeting checkpoint in combination with CXCR2 antagonists have long-term impact in PDAC management. The utility of CXCR2 antagonists as anti-cancer agents in combination with immunotherapeutics is further supported by several published reports [12–15, 29, 33–42]. Our earlier report suggests a dynamic role of host CXCR2 axis in regulating PDAC immune suppression, tumor growth, and metastasis [43]. Host Cxcr2 loss led to increased extramedullary hematopoiesis and expansion of neutrophils and immature myeloid precursor cells in the spleen of tumor-bearing mice, leading to enhanced spontaneous and experimental metastasis [43]. However, the impact of pharmacological inhibition of CXCR2/1 in PDAC as monotherapy remains unclear.
The present study examined whether targeting the CXCR2/1 axis using an orally active small molecule antagonist SCH-479833 inhibits tumor growth, angiogenesis, neutrophil recruitment, fibrosis, necrosis, and metastasis in the preclinical murine models of PC. Our data demonstrate that selectively targeting CXCR2/1 with small molecule inhibitors is a promising therapeutic approach for inhibiting PC growth, angiogenesis, and metastasis.
MATERIALS AND METHODS
Cell lines and CXCR2/1 antagonists
Murine Cell line:
UN-KC-6141 cell line [44] (referred to as KC cells in this study) were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (HyClone®, Thermo Scientific, UT). We supplemented the medium with 5% Fetal Bovine Serum (FBS) (Atlanta Biologicals, GA), L-Glutamine (MediaTech, VA), Vitamin solution (MediaTech), and Gentamycin (Gibco, Life Technologies, NY).
Human Cell Lines:
CD18/HPAF are human PDAC cell lines maintained in DMEM supplemented with 5% FBS, L-Glutamine, Vitamin solution, and Gentamycin. HPNE & E6-E7-st with or without KRAS mutation are a model of immortalized human pancreatic duct-derived cell lines with or without exogenous KRASG12D expression. The model consisted of four cell lines hTERT-HPNE (referred to as HPNE), hTERT-HPNE-KRAS(G12D) (referred to as HPNE-KRAS), hTERT-HPNE-E6-E7-st (referred to as E6-E7-st), and hTERT-HPNE-E6-E7-st-KRAS(G12D) (referred to as E6-E7-st-KRAS). Generation and maintenance of hTERT-HPNE, E6-E7-st, and E6-E7-st-KRAS cells have been previously described by Campbell et al. [45]
All the cell lines were tested for mycoplasma using MycoAlert Plus Mycoplasma Detection kit (Lonza, Rockland, ME). We also had the cell lines authenticated by the Human DNA Identification Laboratory, UNMC, Omaha, NE, through short tandem repeat (STR) tests.
CXCR2/1 antagonists
SCH-479833 [46] was synthesized at Schering-Plough (Kenilworth, NJ) and formulated in hydroxypropyl-beta-cyclodextrin (HPβCD, Acros Organics, Morris Plains, NJ). The inhibition constant (Ki) of CXCR1 (17 nM) and CXCR2 (0.17 nM) for SCH-479833 were calculated from the IC50 value using the Cheng-Prusoff equation [47].
In vitro Cell proliferation assay
Tumor cells were seeded in 96-well plates at a low density (3000 cells/well). Following overnight adherence, cells were incubated with media alone or medium containing serum (5%) with SCH-479833 or media control for 72 h. Four different concentrations (5, 10, 25, and 50 ug/ml) of the antagonist were used. Cell proliferation was determined by MTT assay [48]. Growth inhibition was calculated as a percent , where A and B are the absorbances of untreated and treated cells, respectively.
Cell invasion assay
To investigate the effect of SCH-479833 on PC cell migration, cells (1×105 cells/well) in serum-free media were plated in the top chamber of Matrigel-coated polyethylene terephthalate membranes (six-well insert, 8 μm pore size; Becton Dickinson, Franklin Lakes, NJ). The bottom chamber contained 1.0 ml serum-free media with or without SCH-479833 (at different concentrations). The cells were incubated overnight at 37°C, and unmigrated cells were removed. Per the manufacturer’s instructions, cells that passed through the membrane pores were stained using the Hema 3 kit (Fisher Scientific Company L.L.C., Kalamazoo, MI). Cells were counted in ten random fields (200x) and expressed as the average number of cells per field of view. The data is represented as the average of three independent experiments.
Tumor growth and metastasis assay
All procedures performed in mice followed institutional guidelines approved by the University of Nebraska Medical Center (UNMC) Institution Animal Care and Use Committee.
Syngeneic Mouse Model:
C57BL/6 wild type (WT), 6–8 week old, immunocompetent mice were obtained from Charles River Laboratories (Wilmington, MA). Murine PC cells KC were inoculated (1×105/mouse in 50 μl HBSS) orthotopically into the pancreas of these mice. The mice were randomly divided into two groups a week following the inoculation. The control group (n = 5) was given intraperitoneal (IP) injections of 100 μl HPβCD ((2-Hydroxypropyl)-Beta-Cyclodextrin), while the treatment group (n = 5) was given CXCR2 antagonist (SCH-479833; 100 mg/kg body weight diluted in 100 μl HPβCD) for seven days a week. The mice were euthanized after 8 weeks; gross metastatic spread was evaluated, and a portion from each tumor was processed for histopathological and immunohistochemistry analysis.
Xenogeneic Mouse Model:
Athymic nude mice (6–8 weeks) were obtained from Charles River Laboratories (Wilmington, MA). Human PC cells CD18/HPAF and E6-E7-st-KRAS cells (1 × 106/mouse in 50 μl HBSS) were orthotopically inoculated into the pancreas of these mice. Seven days post-inoculation, the mice were randomly sorted into the HPβCD and SCH-479833 antagonist treatment groups, as mentioned in the syngeneic model. The mice were euthanized after four weeks and examined for tumor growth and metastasis. The primary tumors were resected and processed for histopathological and immunohistochemistry analysis.
Immunohistochemical analysis
To explore the expression of various tissue markers, we performed IHC staining. First, 4–5 μm thick, formalin-fixed, paraffin-embedded tissue sections were pre-warmed at 65 °C for 2 hours on the slide warmer and then deparaffinized with graded xylene. The slides were rehydrated through diluted concentrations of ethanol in water. Afterward, antigen retrieval was performed using sodium citrate buffer (pH = 6.0) and heating in the laboratory microwave for 10 minutes. Endogenous peroxidase was blocked by incubating with 3% hydrogen peroxide for 5 minutes. After blocking non-specific binding by incubating with serum, slides were probed with respective primary antibodies (Table 1) and incubated overnight at 4°C. The following day, slides were washed and incubated with appropriate secondary antibodies. The ABC Elite Kit and DAB substrate kit (3, 3 Diaminobenzidine) (Vector Laboratories, Burlingame, CA) were used to detect immunoreactivity per the manufacturer’s protocols. A positive IHC staining gave a brown color (cytoplasmic, nuclear, membrane, or all, depending on the proteins’ location). At the same time, the nuclei were counterstained blue with Meyer’s Hematoxylin (Thermo Scientific, Fremont, CA). Then the slides were dehydrated with ascending concentrations of ethanol and xylene. Finally, the slides were mounted with permanent media Permount (Fisher Scientific, Fair Lawn, NJ). Two independent observers evaluated IHC staining.
Table 1:
Antibodies used in this study
| No. | Primary Antibody | Marker For | Source | Catalog No. | Dilution |
|---|---|---|---|---|---|
| 1 | Anti-Ki-67 | Cell Proliferation | Santa Cruz, TX, USA | sc-23900 | 1:100 |
| 2 | Anti-MPO (Myeloperoxidase) |
Neutrophils | Abcam, MA, USA | ab9535 | 1:100 |
| 3 | Anti- CC3 (Cleaved Caspase 3) |
Apoptosis | Cell Signaling, MA, USA | asp175–5A1E-9664 | 1:200 |
| 4 | Anti- CD31 (Cluster of Differentiation 31) |
Blood Vessels | Abcam, MA, USA | ab28364 | 1:200 |
| 5. | F4/80 | Macrophages | Abcam | Ab6640 | 1:100 |
| 6 | Ly6 | Neutrophils | Thermo Scientific | MA1–40038 | 1:50 |
For quantitative evaluation, in each tissue section(n=3–5), positive cells were counted in five areas with significant staining at 200X magnification. The average was used for plotting the graph. Similarly, the number of positive endothelial cells was counted for blood vessels to evaluate microvessel density as described by Weidner et al. and Saad et al. [49, 50]. Single or clusters of positive endothelial cells were considered countable vessels, but the presence of blood cells or fibrin without any detectable endothelial cells was disregarded. All the above analyses were done, and the representative photomicrographs were captured with a Nikon Eclipse E800 microscope and its NIS-Elements BR 5.11.00 software (Nikon, Melville, NY).
Hematoxylin and Eosin Staining (H & E)
To demonstrate the general histological architecture of tissue, we did regressive hematoxylin and eosin (H&E) staining. Hematoxylin demonstrates nuclear detail, and eosin provides cytoplasmic and connective tissue details. The slides were stained in our Tissue Science Facility with an automated stainer Tissue-Tek® Prisma™ (Sakura, Torrance, CA). Briefly, 4–5 μm thick, formalin-fixed, paraffin-embedded tissue sections were stained with hematoxylin (CV select HemaMax, BBC Biochemical, McKinney, TX) for 5 minutes and with eosin (CV select Eosin Y, BBC Biochemical, McKinney, TX) for 90 seconds intermingled with pre-and post-staining xylene, alcohol, and water wash steps. The slides (n=3–5) were examined under Nikon Eclipse E800 light microscope after mounting with a permount mounting medium.
Masson Trichrome Staining (MT)
We used Masson trichrome to stain the tissue’s collagen, differentiating it from muscle fibers and cytoplasm. The 4–5 μm thick, formalin-fixed, paraffin-embedded tissue sections were stained with Masson Trichrome Stain Kit-Aniline Blue (BBC Biochemical, McKinney, TX) as per manufacture protocol. In brief, staining with Aniline Blue for 2 minutes and other reagents in the kit gives a blue color to the collagen, while the muscle fibers stain red and the cytoplasm stains pink. The slides (n=3–5) were mounted with a permount mounting medium and examined under the Nikon Eclipse E800 microscope. The amount of fibrosis (blue-stained collagen) was analyzed qualitatively & semi-quantitatively by the pathologist.
Pro- and anti-apoptotic gene expression analyais
Total RNA was isolated from homogenized tumor tissues using the standard Trizol (Invitrogen, Carlsbad, CA) protocol. Reverse Transcription was performed with 5 μg RNA using oligo (dT) (Fermentas, Hanover, MD, USA) and Superscript® II RT (Invitrogen) or iScript™ Reverse Transcription Supermix for RT-qPCR (BIO-RAD, Hercules, CA, USA). Quantitative real-time PCR reactions were performed using FastStart SYBR Green Master Mix (Roche; Indianapolis). Primer sets used for the study are; Bcl2, 5’-AATGTCCAGGTGGGTCAGAG-3’, and 5’-TCCTGCTGGATCTGCCTAGT-3’; Bax, 5’-TGCAGAGGATGATTGCTGAC-3’, and 5’-GGAGGAAGTCCAGTGTCCAG-3’; and Rpl13a, 5’-ACTCTGGAGGAGAAACGGAAGG-3’ and 5’-CAGGCATGAGGCAAACAGTC-3’. The Ct values of the target genes were normalized to mean Ct values of the endogenous control, ribosomal protein large 13 A (Rpl13a); . The ratio of mRNA expression of target genes versus Rpl13a was defined as 2(−∆Ct). Melting curve analysis was performed to check the specificity of the amplified product.
Statistical analysis
We performed the statistical analysis using GraphPad Prism 9.0.0 software (San Diego, CA). All the values are expressed as mean ± standard error of the mean (SEM). Differences between the groups were compared using an unpaired two-tailed t-test. In vivo analysis was performed using the Mann-Whitney U-test of significance. A value of p<0.05 was deemed significant.
RESULTS
SCH-479833 treatment decreased cell proliferation and invasion of PC cells in vitro
The overall growth of the tumor is determined by the interaction of tumor cells with the host microenvironment. In previous studies, it has been reported that chemokine signaling not only helps in the migration of endothelial cells and, thus, angiogenesis but may also help in the motility and invasion of malignant cells. Therefore, we investigated whether treating CXCR2/1 antagonists would affect tumor cell proliferation and motility. Our data demonstrated inhibition of cell proliferation with SCH-479833 treatment compared to the control (Fig. 1A and B). Cells treated with SCH-479833 also showed a significant inhibition (p<0.05) in the number of invading cells in the invasion assay (Fig. 1C and D).
Figure 1. SCH-479833 inhibits PC cell proliferation and invasiveness:
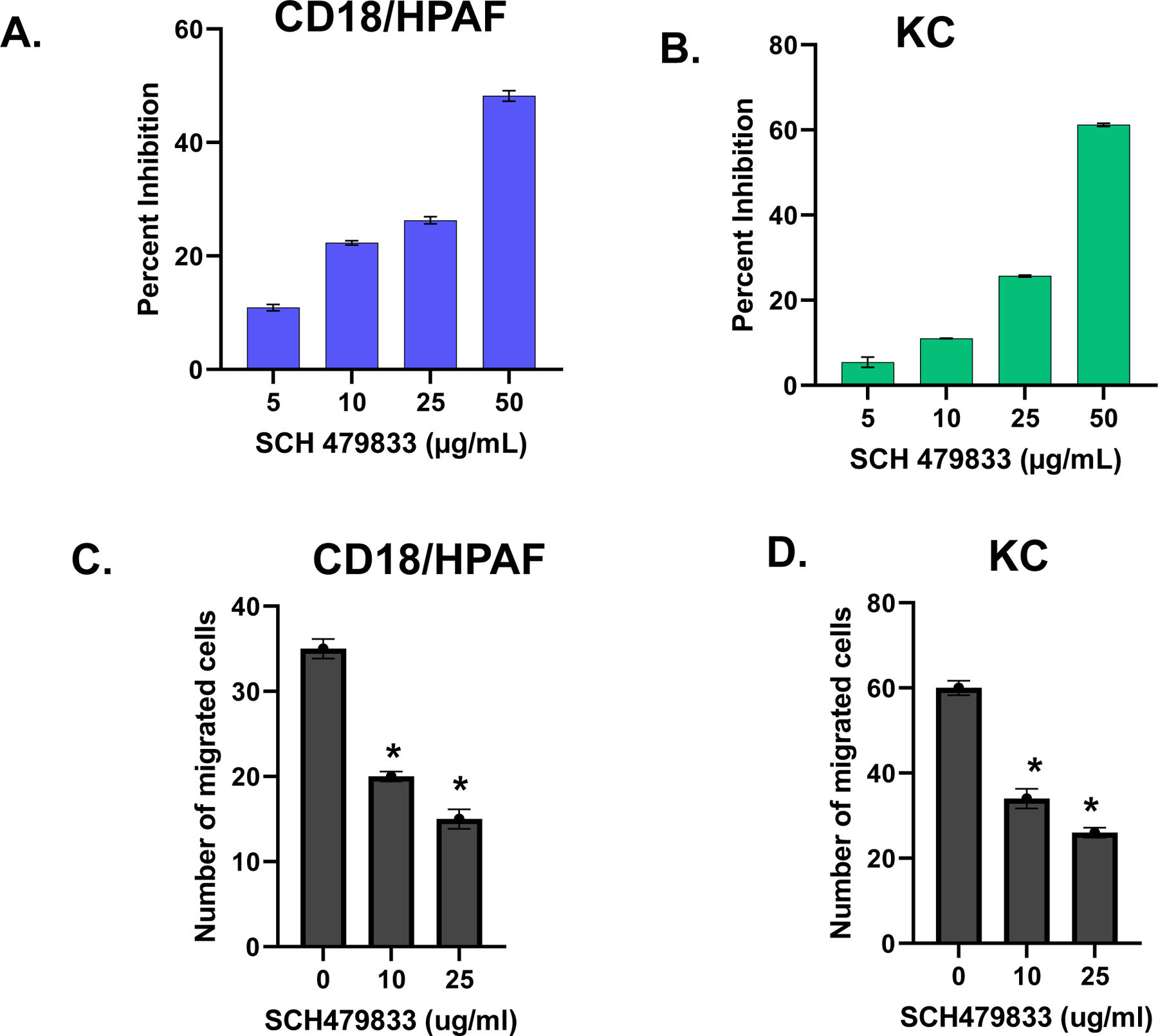
A and B. PC cells (3000 cells/well) in a 96-well plate were cultured in a medium with or without serum (5%), and SCH-479833 in different concentrations. Cellular proliferation was determined at 72h by MTT assay. The values are mean percent inhibition of proliferation ± SEM. C and D. Cells were plated on Matrigel-coated membranes for invasion assays and incubated overnight. Serum-free medium with SCH-479833 (different concentrations) or HPβCD was added to the lower chamber. The cells that did not migrate through the Matrigel and/or pores in the membrane were removed, and cells on the other side of the membrane were stained and photographed at 200x magnification. Cells were counted in ten random fields (200x) and expressed as the average number of cells per field of view. The values are the number of migrated cells ± SEM. This is representative of three experiments done in triplicate. *Significantly different from control (p<0.05).
CXCR2/1 antagonist inhibited tumor growth and metastasis.
Xenogenic models:
We examined the effect of the CXCR2/1 antagonists on mice bearing human PC cells CD18/HPAF. Animals were randomized into control and treatment groups (Fig. 2A). The control group received HPβCD, while the treatment group received the CXCR2/1 antagonist intraperitoneally. Mice were euthanized on day 28. We observed that the mice receiving the CXCR2/1 antagonist had low tumor weight (Fig. 2B) and reduced size (Fig. 2C) compared to control-treated mice. Also, the CXCR2/1 treated animals had a lower regional and distant metastasis incidence than controls (Fig. 1C & D).
Figure 2. CXCR2/1 antagonist to nude mice bearing CD18/HPAF-xenograft tumors inhibits tumor growth and metastasis.
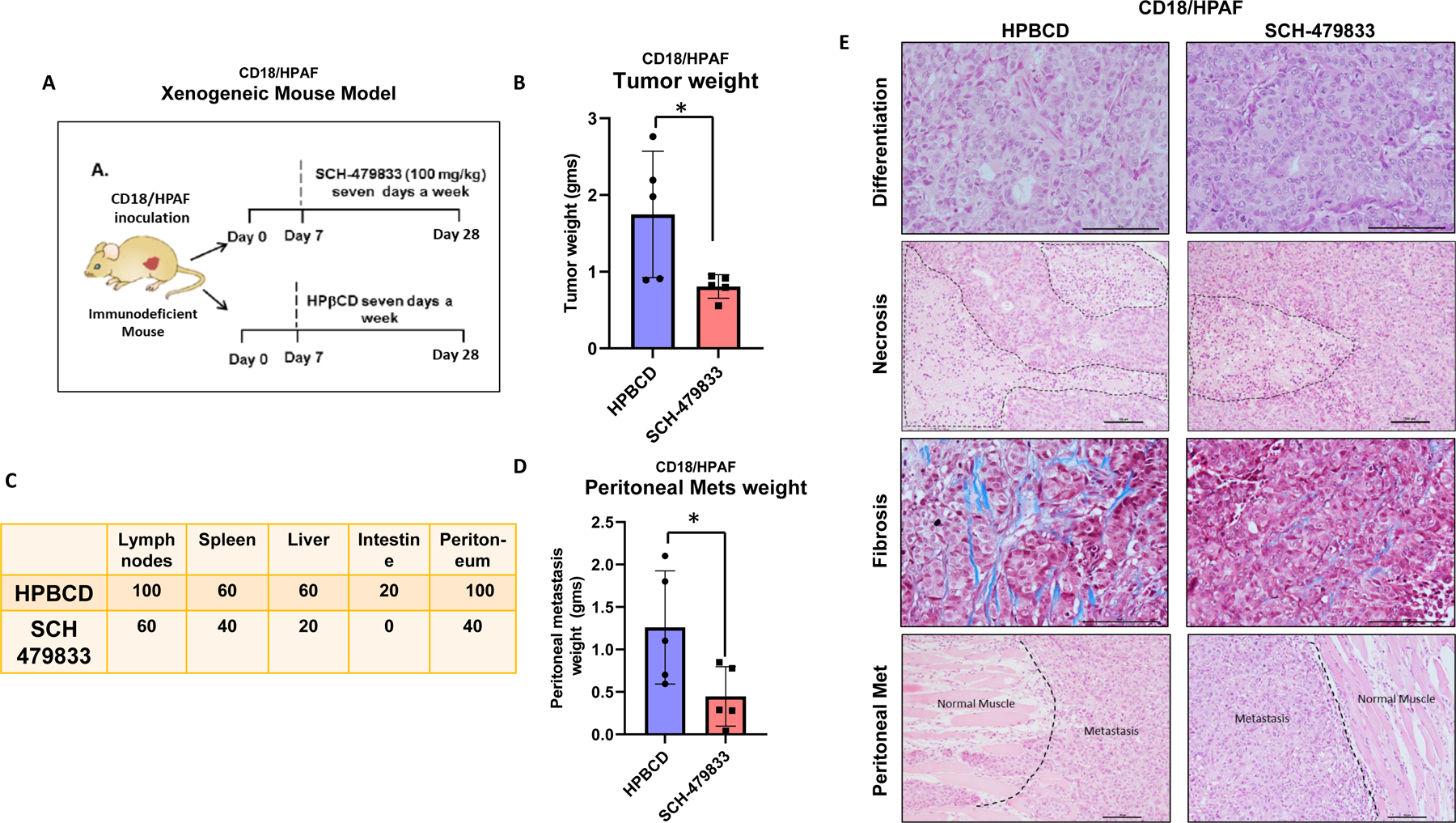
A. Experimental design for treating pancreatic cancer in CD18/HPAF-xenogeneic mouse model with HPβCD (control) and SCH-479833 (CXCR2 antagonist). B. The bar graph shows a difference in the weight of CD18/HPAF tumors derived from mice treated with HPβCD and SCH-479833. C. The incidence of various metastasis in two treatment groups. D. The graph showing difference in weight of peritoneal metastasis in the two treatment groups. E. Representative photomicrographs of H&E staining of tumors, showing similar morphology (moderately-to-poorly differentiated carcinoma) in both the groups; necrosis, fibrosis (Masson Trichrome staining), and peritoneal metastasis. The error bars in the graphs represent the SEM. Scale bar=100 μm. *Significantly different from control (p<0.05).
There was no difference in the histopathology of the tumors derived from the two different groups, as both have moderately-to-poorly differentiated morphology on H&E staining (Fig. 1E). There was decreased tumor necrosis in the CXCR2/1 antagonist group (Fig. 1E). We also investigated the ability of the CXCR2/1 antagonist to impact fibrosis in murine PC by staining with Masson’s Trichrome (MT). We observed decreased fibrosis in the tumors derived from mice treated with the CXCR2/1 antagonist (Fig. 1E).
In the xenogeneic mouse model bearing the human PC cell hTERT-HPNE-E6-E7-st-KRAS, animals were randomized into control and treatment groups (Fig. 3A). Mice were euthanized and examined for primary tumor growth and gross metastasis (day 28). The CXCR2/1 antagonist-treated mice had low tumor weights (Fig. 3B) compared to those receiving HPβCD. We observed no regional or distant metastasis in control or treated groups. We did not observe any noticeable difference in the histopathology of the tumors derived from the two different groups, as both have moderately-to-poorly differentiated morphology on H&E staining (Fig. 3C). We observed increased necrosis (Fig. 3C) and fibrosis (Fig. 3C) in the CXCR2/1 antagonist treatment group.
Figure 3. CXCR2/1 antagonist treatment to mice bearing E6-E7-st-KRAS xenograft tumors decreases tumor weight while increasing necrosis and fibrosis.
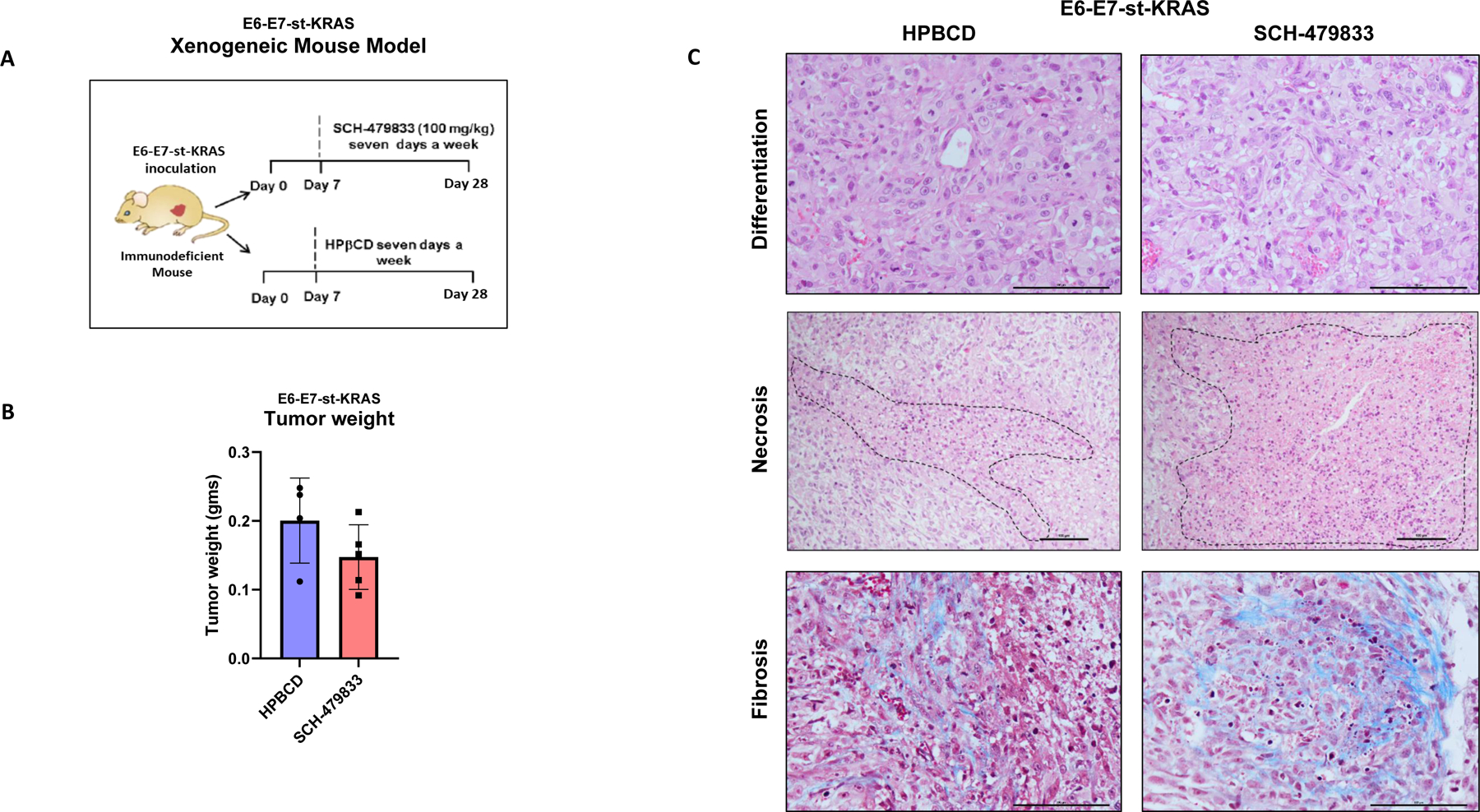
A. Experimental design for treating pancreatic cancer in E6-E7-st-KRAS-xenogeneic mouse model with HPβCD (control) and SCH-479833 (CXCR2 antagonist). B. The bar graph shows a difference in PC tumor weight of E6-E7-st-KRAS tumors derived from mice treated with HPβCD and SCH-479833. C. Representative photomicrographs of H&E staining of tumors showing similar morphology (Moderately to poorly differentiated carcinoma) in both the groups; the difference in necrosis (dotted area) between two groups (more necrosis with CXCR2 antagonist treatment); and fibrosis (Masson Trichrome staining) in different treatment groups. The error bars in the graphs represent the SEM. Scale bar=100 μm.
Syngeneic model:
We utilized the murine PC cell line, KC, to generate the syngeneic mouse model and examine the efficacy of the CXCR2/1 antagonist in immunocompetent animals. As per the schematic shown in Fig. 4A, the animals were randomized into control and treatment groups. The control group was given HPβCD, while the treatment group was given a CXCR2/1 antagonist intraperitoneally. The mice were euthanized at 8 weeks, and tumors were resected for analysis. We observed reduced primary tumor weight in CXCR2/1 treated group compared to controls (Fig. 4B). We observed enlarged spleen in all treated and control animals. There was no significant difference in the weight of the spleens in both groups (Fig. 4C). However, immunohistochemical analysis of the spleen of treated and untreated mice showed higher levels of Ly6G neutrophils in antagonist-treated animals as compared to untreated animals (Supplemental Fig. 1A).
Figure 4. CXCR2/1 antagonist inhibited syngenic KC tumor growth and metastasis:
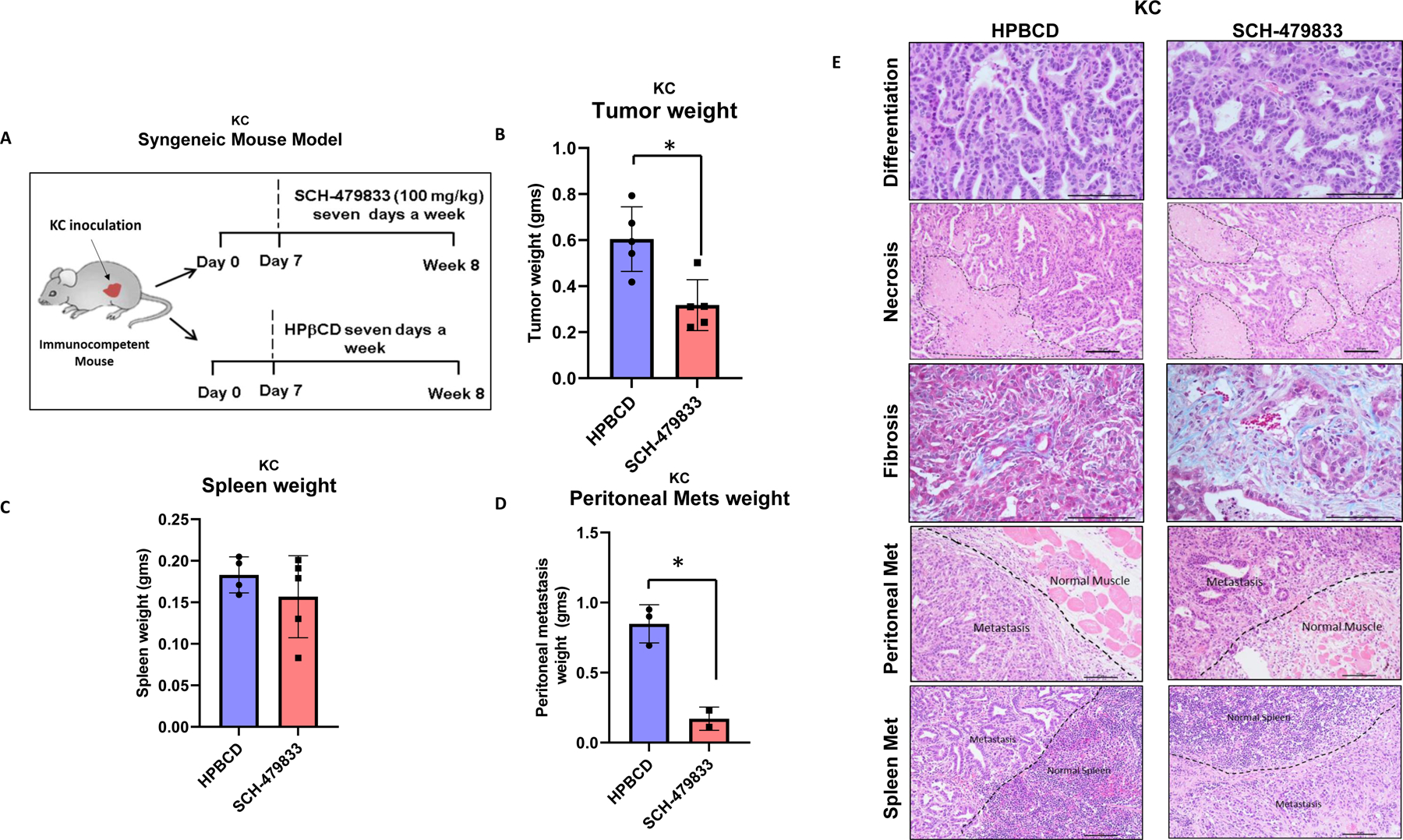
A. Experimental design for treating pancreatic cancer in KC-syngeneic mouse model with HPβCD (control) and SCH-479833 (CXCR2/1 antagonist). B. The bar graph shows a difference in the weight of KC tumors derived from mice treated with HPβCD and SCH-479833. C. The bar graph shows a difference in the weight of spleens from the mice in the two treatment groups. D. The bar graph shows a difference in the weight of peritoneal metastasis in the two treatment groups. E. Representative photomicrographs of H&E staining of tumors showing similar morphology (well-differentiated carcinoma) in both the groups; the difference in necrosis (dotted area) between two groups (more necrosis with CXCR2 antagonist treatment); increased fibrosis (Masson Trichrome staining) in CXCR2/1 antagonist treatment; H&E staining showing infiltration of muscles by malignant cells (metastasis) in the peritoneal region in both groups; and H&E staining shows infiltration of the spleen by malignant cells (metastasis) in both groups. The Error bars in the graphs represent the SEM. Scale bar=100 μm.
HPβCD and CXCR2/1 antagonist-treated mice had lymph node and distant metastasis, and the incidence of metastasis was lower in CXCR2/1 treated group as compared to the controls (Fig. 4D and E). Like the xenogeneic models, both groups had similar morphology. However, both groups have less aggressive well-differentiated tumors, as seen on H&E staining (Fig. 4E). Similarly, the necrosis and fibrosis were higher in the CXCR2/1 antagonist group than in the HPβCD group (Fig. 4E).
Decreased in-situ cell proliferation and increased apoptosis in SCH-479833 antagonist-treated animals
Next, we examined whether inhibition in tumor growth was due to reduced PC cell proliferation and/or survival by Ki-67 and CC3 immunostaining, respectively. Ki-67 is a nuclear protein and a biomarker used frequently to measure the proliferative activity of tumor tissues. At the same time, cleaved caspase 3 (CC3) is a standard method for detecting cellular apoptosis that results from caspase cascades. We observed that treating animals with the antagonist decreased tumor cell proliferation (Fig. 5 A and B) in both xenogeneic and syngeneic animal models. In contrast, the number of apoptotic cells was higher in the antagonist-treated group (Fig. 5C and D). In addition, we observed significantly higher expression levels of Bax and lower expression of Bcl2 in antagonist-treated tumors (Figure 5F).
Figure 5: SCH-479833 inhibits in-situ PC cell proliferation and survival.
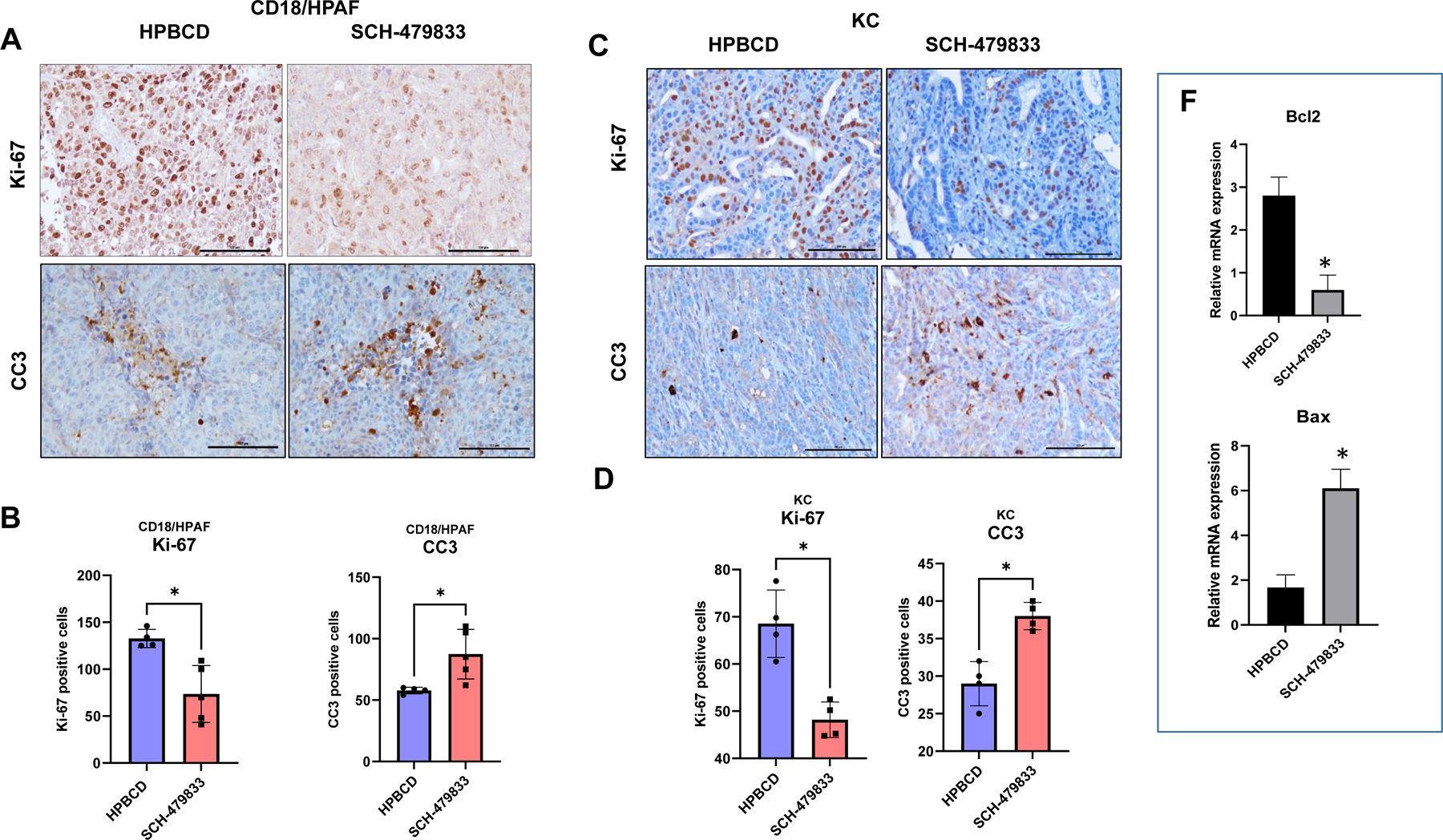
All figures show representative photomicrographs of IHC staining with different markers in CD18/HPAF (A and B) and KC (C and D) tumors derived from mice treated with HPβCD (control) and SCH-479833 (CXCR2/1 antagonist). A. Ki-67 and CC3 IHC shows a decrease in cell proliferation increased apoptosis after CXCR2 antagonist treatment in CD18/HPAF tumor-bearing mice. B. The bar graph shows the difference in the number of Ki-67 and CC3 positive cells in HPβCD and SCH-479833 treatment groups in CD18/HPAF tumor-bearing animals. C. Ki-67 and CC3 IHC show decreased cell proliferation and increased apoptosis after CXCR2 antagonist treatment in KC-tumor-bearing mice. D. The bar graph shows the difference in the number of Ki-67 and CC3 positive cells in HPβCD and SCH-479833 treatment groups in KC-tumor-bearing animals. The error bars in the graphs represent the SEM. Scale bar=100 μm. F. Relative mRNA expression of Bcl2 and Bax genes as determined by qRT-PCR.
Tumor angiogenesis and neutrophil recruitment were decreased in SCH-479833 antagonist-treated animals
The role of a CXCR2/1 is well-established in inflammation and angiogenesis [29]. Therefore, we examined whether inhibition of PC growth by SCH-479833 antagonist affected angiogenesis and inflammatory responses. We evaluated tumor angiogenesis by performing IHC using the blood vessel (angiogenesis) marker CD31. Microvessel density was lower in the tumors that received CXCR2/1 antagonist treatment than in HPβCD treatment (Fig. 6) in both syngenic and xenogenic animal models. Similarly, the number of MPO-positive neutrophils was significantly lower in the CXCR2/1 antagonist treatment group compared to controls (Fig. 6). We did not observe infiltration of tumor-associated macrophages in antagonist-treated and untreated murine KC tumors (Supplemental Fig. 2). Overall, treatment with the CXCR2/1 antagonist in xenogeneic and syngenic mouse models decreased angiogenesis and neutrophil infiltration in the pancreatic tumors.
Figure 6. SCH-479833 inhibits microvessel density and neutrophil recruitment in human and murine PC tumors.
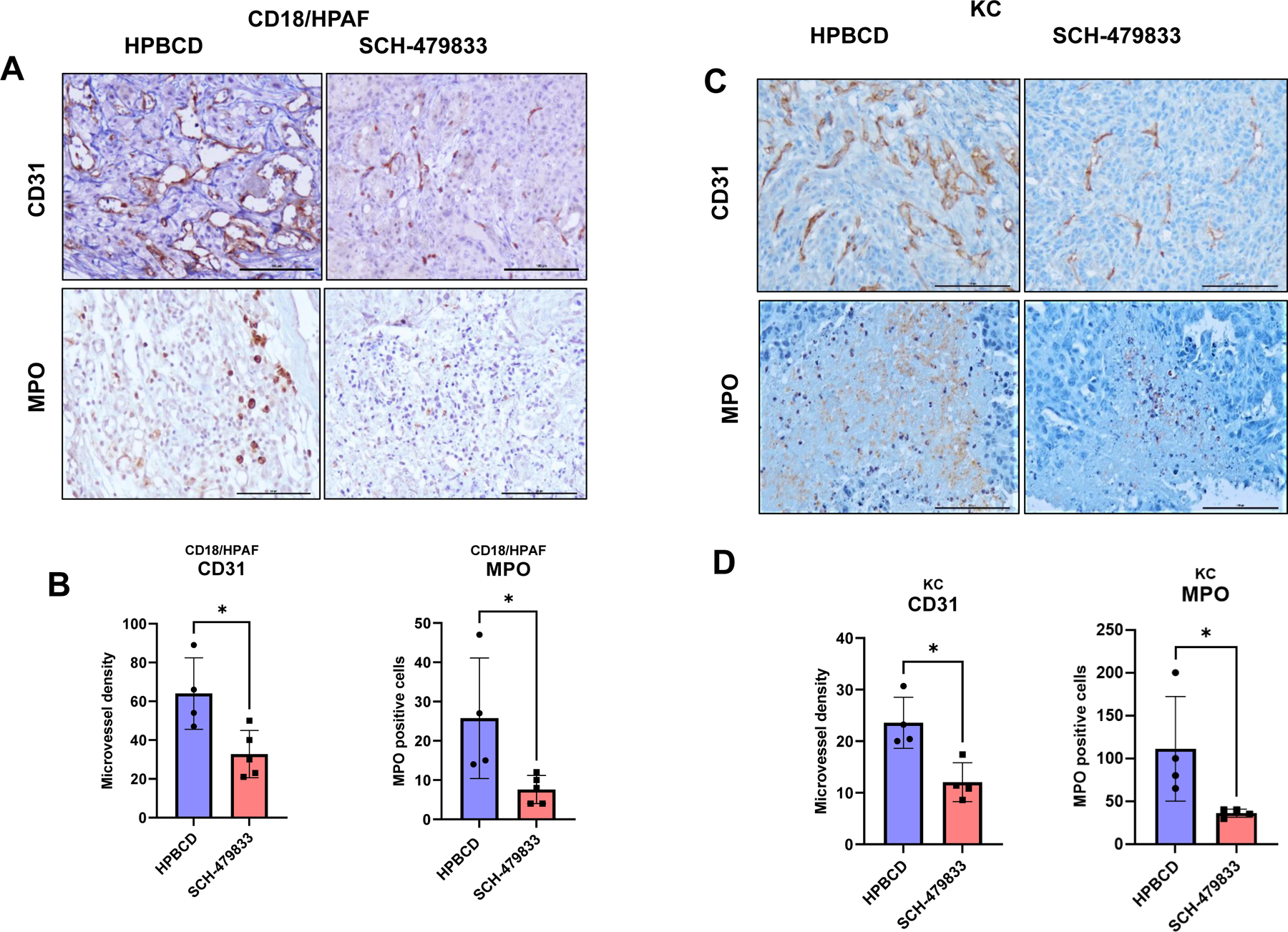
A. CD31 and MPO IHC show decreased cell proliferation and increased apoptosis after CXCR2 antagonist treatment in CD18/HPAF tumor-bearing mice. B. The bar graph shows the difference in the number of CD31 and MPO-positive cells in HPβCD and SCH-479833 treatment groups in CD18/HPAF-tumor-bearing animals. C. CD31 and MPO IHC show decreased cell proliferation and increased apoptosis after CXCR2 antagonist treatment in KC-tumor-bearing mice. D. The bar graph shows the difference in the number of CD31 and MPO-positive cells in HPβCD and SCH-479833 treatment groups. The error bars in the graphs represent the SEM. Scale bar=100 μm. The error bars in the graphs represent the SEM. Scale bar=100 μm.
DISCUSSION
In the present study, we demonstrated that intraperitoneal administration of the CXCR2/1 antagonist SCH-479833 in syngenic and xenogenic murine PC models inhibited the growth and incidence of metastasis. Our studies showed that the antitumor efficacy of SCH-479833 resulted from a decrease in in-situ cell proliferation, angiogenesis, neutrophil recruitment, and enhanced apoptosis. In addition, we observed reduced invasion and cell proliferation of human and murine PC cells upon treatment with the antagonists.
The use of small molecule inhibitors represents an attractive targeted therapeutic approach. Previously, we demonstrated that CXCR2 and its ligands play an important role in PC growth and metastasis [19, 29]. Therefore, targeting these receptors was a logical next step. CXCR2/1 antagonists have shown encouraging results for various cancer, including pancreatic cancer, melanoma, colon cancer, and esophageal and prostate cancer treatment [13, 14, 30, 31, 33, 34, 37, 46, 51–54]. Our in vivo studies revealed that mice treated with the CXCR2/1 antagonist reduced tumor volume and incidence of lymph node and distant metastasis in the treatment groups compared to the control group. These findings indicate the potential of small molecule CXCR2/1 antagonists as novel therapeutics for PC. Our previous observations support it, associating the CXCR2/1 axes with PC aggressiveness [13, 14, 29, 30, 33, 34, 37].
The overall growth of the tumor in vivo depends on several factors, including the tumor cell proliferation rate, survival, and vascularization of the tumor tissue, along with the invasive capacity of the tumor cells [55, 56]. While evaluating the effect of SCH-479833 antagonists on proliferation and apoptosis, we observed that tumors derived from antagonist-treated mice were associated with decreased cell proliferation and increased apoptosis, as detected by Ki-67 and CC3 immunostaining. The in vitro studies also supported the above finding, thus, demonstrating the antiproliferative effect of the CXCR2/1 antagonist. These results support our previous work that showed the critical role of CXCR2 and its ligands in modulating PC phenotypes associated with growth and metastasis [29, 43]. An earlier report by Steele et al. also supports that CXCR2 antagonists suppress metastasis in PDAC [33].
The CXCR2/1 axes are well-known in inflammation and angiogenesis. Our lab has already demonstrated that CXCR2 knockout reduced angiogenesis in pancreatic tumors [57]. In the present study, we observed that the treatment with CXCR2/1 antagonist also decreased tumor vascularity. This was similar to earlier observations from our laboratory and others in melanoma, breast, and colon cancer models [31, 32, 58, 59].
Neutrophils are known crusaders of inflammation and tumor progression. Our study demonstrated that treatment with the CXCR2/1 antagonist decreases neutrophil recruitment in the pancreatic tumor. These results affirmed our recently published CXCR2 knockout study [57]. White et al. showed that the CXCR2 antagonist inhibited the migration of neutrophils [60]. Moss et al. also concluded that CXCR2 antagonism might be a practical approach for modulating airway inflammation in patients with cystic fibrosis by reducing the recruitment of neutrophils at the site of inflammation [61]. As inflammation is critical in the progression of cancer, reducing it with the antagonist can also benefit pancreatic cancer. Tazzyman et al. had favorable results in decreasing neutrophils using CXCR2/1 antagonist in lung adenocarcinoma [62].
Many studies have revealed that CXCR2 expression in cancer cells can drive their proliferation, invasion, and migration [63, 64]. By inhibiting CXCR2, malignant cell proliferation, and motility can be controlled. Moreover, we discovered in our study that the CXCR2/1 antagonist inhibits PC cell proliferation and invasiveness. Steele et al. also demonstrated that tumor cell proliferation was reduced after treatment with CXCR2 antagonist in PDAC [33].
Fibrosis plays a vital role in pancreatic cancer. Studies have reported a cross-talk between tumor cells and cancer-associated fibroblasts (CAFs), aiding tumor progression, metastasis, and therapy resistance [48, 65, 66]. Our study shows that treatment with CXCR2/1 antagonist decreases fibrosis in CD18/HPAF xenogenic mouse model while increasing it in KC syngeneic models. Interestingly, fibrosis in these models was positively associated with necrosis. The precise cause for these differences remains unclear.
In conclusion, our data show that CXCR2/1 antagonists are effective against PC in preclinical models. The inhibitory effects of the antagonist resulted from decreased proliferation, survival, and invasion of PC cells. Furthermore, angiogenesis and recruitment of neutrophils were also inhibited, which is extremely important in tumor growth and progression. Based on these preclinical findings and published reports, CXCR2/1 represents a potential novel therapeutic agent for PC management.
Supplementary Material
Highlights.
This study evaluated the therapeutic utility of orally active small molecule antagonists targeting CXCR2/1, SCH-479833, using different PC preclinical murine models (syngeneic or xenogeneic).
We demonstrate that systemic administration of CXCR2/1 antagonist had both antitumor and anti-metastatic effects in PC. CXCR2/1 antagonist treatment inhibited tumor cell proliferation, angiogenesis, and recruitment of neutrophils, while it increased apoptosis.
Our data suggest that selectively targeting CXCR2/1 with small molecule inhibitors is a promising therapeutic approach for inhibiting PC growth, angiogenesis, and metastasis
Acknowledgments
This work was supported in part by grants R01CA228524, Cancer Center Support Grant (P30CA036727) from the National Cancer Institute, National Institutes of Health, Schering-Plough Research Institute (R.K.S.), and Samuel M., and Janel L. Cohen Professorship of Pathology and Microbiology Fund (R.K.S.). Caitlin Molczyk is supported by a T32CA009476 Eppley Institute Cancer Biology Training Grant from the National Cancer Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: The authors declare no potential conflicts of interest.
Declaration of Interest
The authors declare no competing interests.
REFERENCES
- [1].Siegel RL, Miller KD, Wagle NS, Jemal A, Cancer statistics, 2023, CA: A Cancer Journal for Clinicians, 73 (2023) 17–48. [DOI] [PubMed] [Google Scholar]
- [2].Vincent A, Herman J, Schulick R, Hruban RH, Goggins M, Pancreatic cancer, Lancet, 378 (2011) 607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Desurmont T, Skrypek N, Duhamel A, Jonckheere N, Millet G, Leteurtre E, Gosset P, Duchene B, Ramdane N, Hebbar M, Van Seuningen I, Pruvot F.o.R., Huet G, Truant S.p., Overexpression of chemokine receptor CXCR2 and ligand CXCL7 in liver metastases from colon cancer is correlated to shorter disease-free and overall survival, Cancer Science, 106 (2015) 262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Saxena S, Molczyk C, Purohit A, Ehrhorn E, Goel P, Prajapati DR, Atri P, Kaur S, Grandgenett PM, Hollingsworth MA, Batra SK, Singh RK, Differential expression profile of CXC-receptor-2 ligands as potential biomarkers in pancreatic ductal adenocarcinoma, American journal of cancer research, 12 (2022) 68–90. [PMC free article] [PubMed] [Google Scholar]
- [5].Chapman RW, Phillips JE, Hipkin RW, Curran AK, Lundell D, Fine JS, CXCR2 antagonists for the treatment of pulmonary disease, Pharmacology & therapeutics, 121 (2009) 55–68. [DOI] [PubMed] [Google Scholar]
- [6].Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris P.-á., Manova-Todorova K, Leversha M, Hogg N, Seshan V.-á., Norton L, Brogi E, Massagu+¬ J, A CXCL1 Paracrine Network Links Cancer Chemoresistance and Metastasis, Cell, 150 (2012) 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wang S, Wu Y, Hou Y, Guan X, Castelvetere MP, Oblak JJ, Banerjee S, Filtz TM, Sarkar FH, Chen X, Jena BP, Li C, CXCR2 macromolecular complex in pancreatic cancer: a potential therapeutic target in tumor growth, Translational oncology, 6 (2013) 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Frick VO, Rubie C, Wagner M, Graeber S, Grimm H, Kopp B, Rau BM, Schilling MK, Enhanced ENA-78 and IL-8 expression in patients with malignant pancreatic diseases, Pancreatology, 8 (2008) 488–497. [DOI] [PubMed] [Google Scholar]
- [9].Li A, King J, Moro A, Sugi MD, Dawson DW, Kaplan J, Li G, Lu X, Strieter RM, Burdick M, Go VL, Reber HA, Eibl G, Hines OJ, Overexpression of CXCL5 Is Associated With Poor Survival in Patients With Pancreatic Cancer, The American journal of pathology, 178 (2011) 1340–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Purohit A, Varney M, Rachagani S, Ouellette MM, Batra SK, Singh RK, CXCR2 signaling regulates KRAS (G12D) -induced autocrine growth of pancreatic cancer, Oncotarget, 9 (2016) 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Matsuo Y, Raimondo M, Woodward TA, Wallace MB, Gill KR, Tong Z, Burdick MD, Yang Z, Strieter RM, Hoffman RM, Guha S, CXC-chemokine/CXCR2 biological axis promotes angiogenesis in vitro and in vivo in pancreatic cancer, Int.J Cancer, 125 (2009) 1027–1037. [DOI] [PubMed] [Google Scholar]
- [12].Zhang M, Huang L, Ding G, Huang H, Cao G, Sun X, Lou N, Wei Q, Shen T, Xu X, Cao L, Yan Q, Interferon gamma inhibits CXCL8-CXCR2 axis mediated tumor-associated macrophages tumor trafficking and enhances anti-PD1 efficacy in pancreatic cancer, J Immunother Cancer, 8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sano M, Ijichi H, Takahashi R, Miyabayashi K, Fujiwara H, Yamada T, Kato H, Nakatsuka T, Tanaka Y, Tateishi K, Morishita Y, Moses HL, Isayama H, Koike K, Blocking CXCLs-CXCR2 axis in tumor-stromal interactions contributes to survival in a mouse model of pancreatic ductal adenocarcinoma through reduced cell invasion/migration and a shift of immune-inflammatory microenvironment, Oncogenesis, 8 (2019) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Morton JP, Sansom OJ, CXCR2 inhibition in pancreatic cancer: opportunities for immunotherapy?, Immunotherapy, 9 (2017) 9–12. [DOI] [PubMed] [Google Scholar]
- [15].Fu S, Chen X, Lin HJ, Lin J, Inhibition of interleukin 8/C‑X-C chemokine receptor 1,/2 signaling reduces malignant features in human pancreatic cancer cells, Int J Oncol, 53 (2018) 349–357. [DOI] [PubMed] [Google Scholar]
- [16].Alfaro C, Teijeira A, Oñate C, Pérez G, Sanmamed MF, Andueza MP, Alignani D, Labiano S, Azpilikueta A, Rodriguez-Paulete A, Garasa S, Fusco JP, Aznar A, Inogés S, De Pizzol M, Allegretti M, Medina-Echeverz J, Berraondo P, Perez-Gracia JL, Melero I, Tumor-Produced Interleukin-8 Attracts Human Myeloid-Derived Suppressor Cells and Elicits Extrusion of Neutrophil Extracellular Traps (NETs), American Association for Cancer Research, 22 (2016) 3924–3936. [DOI] [PubMed] [Google Scholar]
- [17].Li A, Dubey S, Varney ML, Dave BJ, Singh RK, IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis, J Immunol, 170 (2003) 3369–3376. [DOI] [PubMed] [Google Scholar]
- [18].Johnston RA, Mizgerd JP, Shore SA, CXCR2 is essential for maximal neutrophil recruitment and methacholine responsiveness after ozone exposure, Am.J.Physiol Lung Cell Mol.Physiol, 288 (2005) L61–L67. [DOI] [PubMed] [Google Scholar]
- [19].Li A, Varney ML, Valasek J, Godfrey M, Dave BJ, Singh RK, Autocrine role of interleukin-8 in induction of endothelial cell proliferation, survival, migration and MMP-2 production and angiogenesis, Angiogenesis, 8 (2005) 63–71. [DOI] [PubMed] [Google Scholar]
- [20].Salcedo R, Resau JH, Halverson D, Hudson EA, Dambach M, Powell D, Wasserman K, Oppenheim JJ, Differential expression and responsiveness of chemokine receptors (CXCR1–3) by human microvascular endothelial cells and umbilical vein endothelial cells, FASEB J, 14 (2000) 2055–2064. [DOI] [PubMed] [Google Scholar]
- [21].Addison CL, Daniel TO, Burdick MD, Liu H, Ehlert JE, Xue YY, Buechi L, Walz A, Richmond A, Strieter RM, The CXC chemokine receptor 2, CXCR2, is the putative receptor for ELR+ CXC chemokine-induced angiogenic activity, J Immunol, 165 (2000) 5269–5277. [DOI] [PubMed] [Google Scholar]
- [22].Kruger P, Saffarzadeh M, Weber AN, Rieber N, Radsak M, von Bernuth H, Benarafa C, Roos D, Skokowa J, Hartl D, Neutrophils: Between host defence, immune modulation, and tissue injury, PLoS pathogens, 11 (2015) e1004651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wu L, Awaji M, Saxena S, Varney ML, Sharma B, Singh RK, IL-17-CXC Chemokine Receptor 2 Axis Facilitates Breast Cancer Progression by Up-Regulating Neutrophil Recruitment, Am J Pathol, 190 (2020) 222–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wu L, Saxena S, Singh RK, Neutrophils in the Tumor Microenvironment, in: Birbrair A (Ed.) Tumor Microenvironment: Hematopoietic Cells – Part A, Springer International Publishing, Cham, 2020, pp. 1–20. [Google Scholar]
- [25].Ijichi H, Chytil A, Gorska AE, Aakre ME, Bierie B, Tada M, Mohri D, Miyabayashi K, Asaoka Y, Maeda S, Ikenoue T, Tateishi K, Wright CVE, Koike K, Omata M, Moses HL, Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma, The Journal of Clinical Investigation, 121 (2011) 4106–4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Matsuo Y, Ochi N, Sawai H, Yasuda A, Takahashi H, Funahashi H, Takeyama H, Tong Z, Guha S, CXCL8/IL-8 and CXCL12/SDF-1alpha co-operatively promote invasiveness and angiogenesis in pancreatic cancer, Int.J Cancer, 124 (2009) 853–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Matsuo Y, Campbell PM, Brekken RA, Sung B, Ouellette MM, Fleming JB, Aggarwal BB, Der CJ, Guha S, K-Ras promotes angiogenesis mediated by immortalized human pancreatic epithelial cells through mitogen-activated protein kinase signaling pathways, Mol Cancer Res, 7 (2009) 799–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Purohit A, Varney M, Rachagani S, Ouellette MM, Batra SK, Singh RK, CXCR2 signaling regulates KRAS(G(1)(2)D)-induced autocrine growth of pancreatic cancer, Oncotarget, 7 (2016) 7280–7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Saxena S, Singh RK, Chemokines orchestrate tumor cells and the microenvironment to achieve metastatic heterogeneity, Cancer Metastasis Rev, 40 (2021) 447–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Xie Y, Kuang W, Wang D, Yuan K, Yang P, Expanding role of CXCR2 and therapeutic potential of CXCR2 antagonists in inflammatory diseases and cancers, European Journal of Medicinal Chemistry, 250 (2023) 115175. [DOI] [PubMed] [Google Scholar]
- [31].Singh S, Sadanandam A, Nannuru KC, Varney ML, Mayer-Ezell R, Bond R, Singh RK, Small-molecule antagonists for CXCR2 and CXCR1 inhibit human melanoma growth by decreasing tumor cell proliferation, survival, and angiogenesis, Clin Cancer Res, 15 (2009) 2380–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Varney ML, Singh S, Li A, Mayer-Ezell R, Bond R, Singh RK, Small molecule antagonists for CXCR2 and CXCR1 inhibit human colon cancer liver metastases, Cancer Lett, 300 (2011) 180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, Foth M, Bryson S, McDaid K, Wilson Z, Eberlein C, Candido JB, Clarke M, Nixon C, Connelly J, Jamieson N, Carter CR, Balkwill F, Chang DK, Evans TRJ, Strathdee D, Biankin AV, Nibbs RJB, Barry ST, Sansom OJ, Morton JP, CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma, Cancer Cell, 29 (2016) 832–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gulhati P, Schalck A, Jiang S, Shang X, Wu CJ, Hou P, Ruiz SH, Soto LS, Parra E, Ying H, Han J, Dey P, Li J, Deng P, Sei E, Maeda DY, Zebala JA, Spring DJ, Kim M, Wang H, Maitra A, Moore D, Clise-Dwyer K, Wang YA, Navin NE, DePinho RA, Targeting T cell checkpoints 41BB and LAG3 and myeloid cell CXCR1/CXCR2 results in antitumor immunity and durable response in pancreatic cancer, Nat Cancer, 4 (2023) 62–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wu QY, Yang CK, Rong LJ, Li JC, Lei LM, Investigation of the association between C-X-C motif chemokine receptor subunits and tumor infiltration levels and prognosis in patients with early-stage pancreatic ductal adenocarcinoma, Oncology letters, 20 (2020) 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sun X, He X, Zhang Y, Hosaka K, Andersson P, Wu J, Wu J, Jing X, Du Q, Hui X, Ding B, Guo Z, Hong A, Liu X, Wang Y, Ji Q, Beyaert R, Yang Y, Li Q, Cao Y, Inflammatory cell-derived CXCL3 promotes pancreatic cancer metastasis through a novel myofibroblast-hijacked cancer escape mechanism, Gut, 71 (2022) 129–147. [DOI] [PubMed] [Google Scholar]
- [37].Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, Jacobs RC, Ye J, Patel AA, Gillanders WE, Fields RC, DeNardo DG, Hawkins WG, Goedegebuure P, Linehan DC, Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma, Gut, 67 (2018) 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lesina M, Wörmann SM, Morton J, Diakopoulos KN, Korneeva O, Wimmer M, Einwächter H, Sperveslage J, Demir IE, Kehl T, Saur D, Sipos B, Heikenwälder M, Steiner JM, Wang TC, Sansom OJ, Schmid RM, Algül H, RelA regulates CXCL1/CXCR2-dependent oncogene-induced senescence in murine Kras-driven pancreatic carcinogenesis, J Clin Invest, 126 (2016) 2919–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hoffman MT, Kemp SB, Salas-Escabillas DJ, Zhang Y, Steele NG, The S, Long D, Benitz S, Yan W, Margolskee RF, Bednar F, Pasca di Magliano M, Wen HJ, Crawford HC, The Gustatory Sensory G-Protein GNAT3 Suppresses Pancreatic Cancer Progression in Mice, Cell Mol Gastroenterol Hepatol, 11 (2021) 349–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chen Y, Kim J, Yang S, Wang H, Wu CJ, Sugimoto H, LeBleu VS, Kalluri R, Type I collagen deletion in αSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer, Cancer Cell, 39 (2021) 548–565.e546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Arnoletti JP, Reza J, Rosales A, Monreal A, Fanaian N, Whisner S, Srivastava M, Rivera-Otero J, Yu G, Phanstiel Iv O, Altomare DA, Tran Q, Litherland SA, Pancreatic Ductal Adenocarcinoma (PDAC) circulating tumor cells influence myeloid cell differentiation to support their survival and immunoresistance in portal vein circulation, PloS one, 17 (2022) e0265725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ando Y, Ohuchida K, Otsubo Y, Kibe S, Takesue S, Abe T, Iwamoto C, Shindo K, Moriyama T, Nakata K, Miyasaka Y, Ohtsuka T, Oda Y, Nakamura M, Necroptosis in pancreatic cancer promotes cancer cell migration and invasion by release of CXCL5, PloS one, 15 (2020) e0228015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Purohit A, Saxena S, Varney M, Prajapati DR, Kozel JA, Lazenby A, Singh RK, Host Cxcr2-Dependent Regulation of Pancreatic Cancer Growth, Angiogenesis, and Metastasis, The American Journal of Pathology, 191 (2021) 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Torres MP, Rachagani S, Souchek JJ, Mallya K, Johansson SL, Batra SK, Novel pancreatic cancer cell lines derived from genetically engineered mouse models of spontaneous pancreatic adenocarcinoma: applications in diagnosis and therapy, PloS one, 8 (2013) e80580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Campbell PM, Groehler AL, Lee KM, Ouellette MM, Khazak V, Der CJ, K-Ras promotes growth transformation and invasion of immortalized human pancreatic cells by Raf and phosphatidylinositol 3-kinase signaling, Cancer Res, 67 (2007) 2098–2106. [DOI] [PubMed] [Google Scholar]
- [46].Gonsiorek W, Fan X, Hesk D, Fossetta J, Qiu H, Jakway J, Billah M, Dwyer M, Chao J, Deno G, Taveras A, Lundell DJ, Hipkin RW, Pharmacological characterization of Sch527123, a potent allosteric CXCR1/CXCR2 antagonist, J Pharmacol Exp Ther, 322 (2007) 477–485. [DOI] [PubMed] [Google Scholar]
- [47].Xiong X, Liao X, Qiu S, Xu H, Zhang S, Wang S, Ai J, Yang L, CXCL8 in Tumor Biology and Its Implications for Clinical Translation, Frontiers in molecular biosciences, 9 (2022) 723846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Awaji M, Saxena S, Wu L, Prajapati DR, Purohit A, Varney ML, Kumar S, Rachagani S, Ly QP, Jain M, Batra SK, Singh RK, CXCR2 signaling promotes secretory cancer-associated fibroblasts in pancreatic ductal adenocarcinoma, Faseb j, 34 (2020) 9405–9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Weidner N, Semple JP, Welch WR, Folkman J, Tumor angiogenesis and metastasis--correlation in invasive breast carcinoma, N Engl J Med, 324 (1991) 1–8. [DOI] [PubMed] [Google Scholar]
- [50].Saad RS, Kordunsky L, Liu YL, Denning KL, Kandil HA, Silverman JF, Lymphatic microvessel density as prognostic marker in colorectal cancer, Mod Pathol, 19 (2006) 1317–1323. [DOI] [PubMed] [Google Scholar]
- [51].Wang B, Hendricks DT, Wamunyokoli F, Parker MI, A growth-related oncogene/CXC chemokine receptor 2 autocrine loop contributes to cellular proliferation in esophageal cancer, Cancer research, 66 (2006) 3071–3077. [DOI] [PubMed] [Google Scholar]
- [52].Wilson C, Wilson T, Johnston PG, Longley DB, Waugh DJ, Interleukin-8 signaling attenuates TRAIL- and chemotherapy-induced apoptosis through transcriptional regulation of c-FLIP in prostate cancer cells, Mol Cancer Ther, 7 (2008) 2649–2661. [DOI] [PubMed] [Google Scholar]
- [53].Dwyer MP, Yu Y, Chao J, Aki C, Chao J, Biju P, Girijavallabhan V, Rindgen D, Bond R, Mayer-Ezel R, Jakway J, Hipkin RW, Fossetta J, Gonsiorek W, Bian H, Fan X, Terminelli C, Fine J, Lundell D, Merritt JR, Rokosz LL, Kaiser B, Li G, Wang W, Stauffer T, Ozgur L, Baldwin J, Taveras AG, Discovery of 2-hydroxy-N,N-dimethyl-3-{2-[[(R)-1-(5-methylfuran-2-yl)propyl]amino]-3,4-dioxocyclobut-1-enylamino}benzamide (SCH 527123): a potent, orally bioavailable CXCR2/CXCR1 receptor antagonist, J Med Chem, 49 (2006) 7603–7606. [DOI] [PubMed] [Google Scholar]
- [54].Bertini R, Allegretti M, Bizzarri C, Moriconi A, Locati M, Zampella G, Cervellera MN, Di Cioccio V, Cesta MC, Galliera E, Martinez FO, Di Bitondo R, Troiani G, Sabbatini V, D’Anniballe G, Anacardio R, Cutrin JC, Cavalieri B, Mainiero F, Strippoli R, Villa P, Di Girolamo M, Martin F, Gentile M, Santoni A, Corda D, Poli G, Mantovani A, Ghezzi P, Colotta F, Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: prevention of reperfusion injury, Proc Natl Acad Sci U S A, 101 (2004) 11791–11796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].O’Hayre M, Salanga CL, Handel TM, Allen SJ, Chemokines and cancer: migration, intracellular signalling and intercellular communication in the microenvironment, Biochem J, 409 (2008) 635–649. [DOI] [PubMed] [Google Scholar]
- [56].Jain RK, Safabakhsh N, Sckell A, Chen Y, Jiang P, Benjamin L, Yuan F, Keshet E, Endothelial cell death, angiogenesis, and microvascular function after castration in an androgen-dependent tumor: role of vascular endothelial growth factor, Proc Natl Acad Sci U S A, 95 (1998) 10820–10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Purohit A, Saxena S, Varney M, Prajapati DR, Kozel JA, Lazenby A, Singh RK, Host Cxcr2-Dependent Regulation of Pancreatic Cancer Growth, Angiogenesis, and Metastasis, Am J Pathol, 191 (2021) 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].KC N, Singh S, S. R.K., Chemokines and Metastasis, in: Theicher B, Bagley RG (Eds.) Tumor Microenvironment: Cancer Drug Discovery and Development, Springer Sciences; 2010, pp. 601–632. [Google Scholar]
- [59].Ning Y, Labonte MJ, Zhang W, Bohanes PO, Gerger A, Yang D, Benhaim L, Paez D, Rosenberg DO, Nagulapalli Venkata KC, Louie SG, Petasis NA, Ladner RD, Lenz HJ, The CXCR2 antagonist, SCH-527123, shows antitumor activity and sensitizes cells to oxaliplatin in preclinical colon cancer models, Mol Cancer Ther, 11 (2012) 1353–1364. [DOI] [PubMed] [Google Scholar]
- [60].White JR, Lee JM, Young PR, Hertzberg RP, Jurewicz AJ, Chaikin MA, Widdowson K, Foley JJ, Martin LD, Griswold DE, Sarau HM, Identification of a potent, selective non-peptide CXCR2 antagonist that inhibits interleukin-8-induced neutrophil migration, J Biol Chem, 273 (1998) 10095–10098. [DOI] [PubMed] [Google Scholar]
- [61].Moss RB, Mistry SJ, Konstan MW, Pilewski JM, Kerem E, Tal-Singer R, Lazaar AL, Investigators CF, Safety and early treatment effects of the CXCR2 antagonist SB-656933 in patients with cystic fibrosis, J Cyst Fibros, 12 (2013) 241–248. [DOI] [PubMed] [Google Scholar]
- [62].Tazzyman S, Barry ST, Ashton S, Wood P, Blakey D, Lewis CE, Murdoch C, Inhibition of neutrophil infiltration into A549 lung tumors in vitro and in vivo using a CXCR2-specific antagonist is associated with reduced tumor growth, Int J Cancer, 129 (2011) 847–858. [DOI] [PubMed] [Google Scholar]
- [63].Matsuo Y, Ochi N, Sawai H, Yasuda A, Takahashi H, Funahashi H, Takeyama H, Tong Z, Guha S, CXCL8/IL-8 and CXCL12/SDF-1alpha co-operatively promote invasiveness and angiogenesis in pancreatic cancer, International journal of cancer. Journal international du cancer, 124 (2009) 853–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Matsuo Y, Raimondo M, Woodward TA, Wallace MB, Gill KR, Tong Z, Burdick MD, Yang Z, Strieter RM, Hoffman RM, Guha S, CXC-chemokine/CXCR2 biological axis promotes angiogenesis in vitro and in vivo in pancreatic cancer, International journal of cancer. Journal international du cancer, 125 (2009) 1027–1037. [DOI] [PubMed] [Google Scholar]
- [65].McCarthy JB, El-Ashry D, Turley EA, Hyaluronan, Cancer-Associated Fibroblasts and the Tumor Microenvironment in Malignant Progression, Front Cell Dev Biol, 6 (2018) 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Su S, Chen J, Yao H, Liu J, Yu S, Lao L, Wang M, Luo M, Xing Y, Chen F, Huang D, Zhao J, Yang L, Liao D, Su F, Li M, Liu Q, Song E, CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness, Cell, 172 (2018) 841–856 e816. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.