

One sentence summary:
In the Phase 3 SPRINT trial, pre-symptomatic gene therapy demonstrated impressive clinical outcomes in infants with a genetic diagnosis of SMA; long-term safety follow-up of these patients must now be a key priority.
Onasemnogene abeparvovec (OA) is an in vivo viral-mediated gene therapy – one of only two such therapies in clinical use – which is approved for one-time intravenous administration to infants under 2 years of age with early-onset spinal muscular atrophy (SMA). Despite the small number of in vivo gene therapies in clinical use, more than a hundred are in clinical trials (www.clinicaltrials.gov). The greatest benefit from these rapidly emerging treatments is likely to come when initiated prior to disease onset, particularly when disease progression claims non-replicating cells such as neurons — as is the case for SMA. In paired papers in this issue of Nature Medicine, Strauss et al. report the two-year outcomes of a Phase 3 trial (SPR1NT) in which infants with SMA were treated with a single intravenous dose of the gene therapy onasemnogene abeparvovec (OA) prior to overt disease manifestation.1,2
SMA is an early onset motor neuron disease caused by recessive loss-of-function mutations of the survival motor neuron 1 (SMN1) gene and retention of a paralogous alternatively spliced SMN2 gene, with consequent insufficient expression of SMN protein. SMA-associated genotypes vary in copy number of the SMN2 gene. Infants with just 2 copies of the gene experience profound muscle weakness of the trunk and limb muscles with failure to achieve any motor milestones including head control, rolling, or sitting; while infants with 3 copies of SMN2 generally achieve the ability to sit and sometimes to stand. Three treatments that increase SMN levels are now approved for treating infants with SMA, having shown some benefit in patients with symptomatic disease: the splice-switching antisense oligonucleotide nusinersen, the small molecule risdiplam, and OA. OA consists of a SMN cDNA transgene whose expression is driven by a human cytomegalovirus enhancer and β actin promoter, packaged within an adeno-associated virus 9 (AAV9) capsid. Following a single intravenous administration of OA, persistence of a functional SMN transgene offers the potential for lifelong SMN expression in motor neurons (Fig.1).
Figure 1. Onasemnogene abeparvovec (OA).
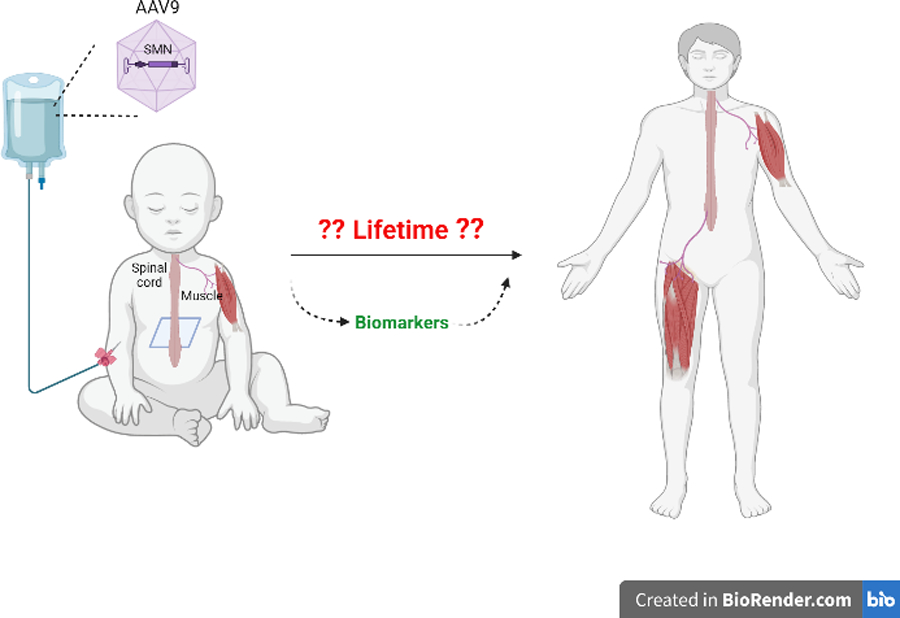
an AAV9 SMN gene replacement therapy, is administered as a one-time intravenous injection. Transduction of motor neurons likely results in improved motor neuron development, function, and survival with the potential for near normal attainment of motor milestones. Whether OA remains effective and safe throughout life requires further assessment.
Strauss et al. treated infants with genetically confirmed, presymptomatic SMA — including 14 infants with 2 copies of SMN2 at a median age of 21 days,1 and 15 infants with 3 copies at a median age of 32 days.2 Remarkably, by 18 months of follow up, 79% of those with 2 copies achieved independent standing and 64% achieved independent walking, compared to only 3–5% of postsymptomatically-treated genotype-matched infants treated at a median age of ~4 months.3,4 For infants with 3 copies of the gene (also treated pre-symptomatically), 93% walked independently in the 24 month post-treatment observation period. It is worth noting that these markedly enhanced outcomes with presymptomatic treatment are strikingly similar to those observed in trials of pre-symptomatic infants treated with the other two SMN-enhancing therapies, nusinersen5 and risdiplam.6 This suggests that the optimal levels of SMN induction can be achieved with any of the 3 therapies when given very early, despite their distinct molecular mechanisms and biodistributions. With the recent approval of risdiplam for neonatal use (and nusinersen already approved in this setting), families now have the choice of starting one of three distinct SMN enhancing treatments at the time of newborn diagnosis.
These impressive results have encouraged increasing newborn screening for SMA internationally. But even with such early diagnosis, optimal outcomes may be difficult to achieve. In the Strauss et al. studies, 14 of 44 newborns screened were excluded due to evident symptoms. Furthermore, although all infants enrolled in the study were “presymptomatic” by clinical neurologic examination at the time of dosing, those infants with 2 copies of SMN2 fared less well than those with 3 copies. Scored on the Bayley gross motor scale, one-third of infants with 2 copies scored below the normal range at study end, suggesting that pre-treatment pathology emerges in the form of mild motor impairment by this age. All those with 3 copies scored within the normal range. These observations, mirrored in the presymptomatic trials of other therapies, emphasise that the timing of treatment initiation and the magnitude of neurodegeneration at that time are key drivers of therapeutic response — possibly even more so than the choice of specific treatment. Evidence from autopsy studies7 and analyses of serum neurofilament levels (a marker of active neurodegeneration)5 indicate that disease-associated pathology in infants with 2-copies of SMN2 begins in utero with rapid degeneration occurring neonatally, even before overt symptoms. In this group it is truly a race against time, and we strongly encourage initiation of any one of the therapeutics as soon after birth as possible. Biomarkers that can predict functional outcomes within the range manifest for each of the SMN2 copy number-defined genotype groups are urgently needed. Serum neurofilaments have shown promise as safety and efficacy biomarkers in interventional studies of SMA5 and other neurodegenerative diseases; these were not measured in the SPR1NT study, but warrant investigation in this context.
Characteristics of early childhood suggest particular promise for gene transfer therapies8 because the nervous system is still developing and highly plastic at this age, the immune system is relatively tolerant, and broad transgene biodistribution may be enhanced by small body size and an immature blood-brain barrier. OA offers the potential advantage of a single dose as compared to other SMA treatments. But a major potential disadvantage of OA is that once administered, treatment cannot be withdrawn. Clinical trials and early post-marketing surveillance have identified short-term toxicities including acute liver toxicity and rare thrombotic microangiopathy. But these post-market, short-term assessments are ill-suited for identifying potential long-term toxicities that might arise.9,10 Although AAV gene therapies generally do not integrate in the genome if this occurs at low frequency there would be associated concern over insertional oncogenesis. There are also ongoing concerns about potential toxicity from SMN overexpression, epigenetic silencing of recombinant AAV encapsulated genomes,11 or slow progressive loss of OA transgenes by other means. With this in mind, the follow up of just 2 years for a putative lifelong treatment is relatively short.
Although challenging, long-term studies will be essential for both assessment of safety and to identify ways to improve efficacy.9 The establishment of an infrastructure to obtain autopsy tissues from individuals treated with SMN-enhancing therapies would be particularly valuable and informative. To date, autopsies have been reported for only 2 patients treated with OA,12 9 treated with nusinersen,13 and none treated with risdiplam.
The reversal of a relentless degenerative disorder of infancy with a one-time treatment is an extraordinary accomplishment; future work must build on this foundation with vigilant follow-up and biomarker studies.
Footnotes
Author conflict of interests: CJS has been a consultant to Novartis, Ionis Pharmaceuticals, Biogen, PTC Therapeutics, Roche, Genentech, Cytokinetics, Sarepta, Nura Bio, Atalanta, Shift, Argenx, Biomarin, Scholar Rock, GenEdit, Epirium, and Capsigen. CJS has received research grant from Ionis Pharmaceuticals and currently receives grant support from Roche and Biogen. CJS is a coholder of 2 pending patent applications (BIOL0274USA and BIOL0293WO) with Ionis Pharmaceuticals on antisense oligonucleotides targeting SMN-AS1. CJS receives royalties from Elsevier for the book Spinal Muscular Atrophy: Disease Mechanisms and Therapy (editors, CJ Sumner, S Paushkin, CP Ko; Elsevier, 2017). TOC has been a consultant to Avexis/Novartis, Biogen, Catalyst, Cytokinetics, Erydel, Genentech, Ionis, and Scholar Rock. He is/has been a site principal or co–principal investigator for the Biogen EMBRACE, NURTURE, and DEVOTE clinical trials, the Avexis/Novartis STR1VE and STRONG clinical trials, and individual clinical trials with Catabasis, Catalyst, Cytokinetics, Santherra, and Sarepta.
References
- 1).Strauss KA, Farrar MA, Muntoni F, Saito K, Mendell JR, Servais L, McMillan HJ, Finkel RS, Swoboda KJ, Kwon JM, Zaidman CM, Chiriboga CA, Iannaccone ST, Krueger JM, Parsons JA, Shieh PB, Kavanagh S, Tauscher-Wisniewski S, McGill BE, Macek TA; on behalf of the SPR1NT Study Group. The Phase III SPR1NT trial: Onasemnogene abeparvovec for presymptomatic infants with two copies of SMN2 at risk for spinal muscular atrophy type 1. Nat Med, In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Strauss KA, Farrar MA, Muntoni F, Saito K, Mendell JR, Servais L, McMillan HJ, Finkel RS, Swoboda KJ, Kwon JM, Zaidman CM, Chiriboga CA, Iannaccone ST, Krueger JM, Parsons JA, Shieh PB, Kavanagh S, Tauscher-Wisniewski S, McGill BE, Macek TA; on behalf of the SPR1NT Study Group. The Phase III SPR1NT trial: Onasemnogene abeparvovec for presymptomatic infants with three copies of SMN2 at risk for spinal muscular atrophy type 1. Nat Med, In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Day JW, et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol 20, 284–293 (2021). [DOI] [PubMed] [Google Scholar]
- 4).Mercuri E, et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile onset spinal muscular atrophy type 1 (STR1VE-EU): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol 20, 832–841 (2021). [DOI] [PubMed] [Google Scholar]
- 5).De Vivo DC, et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: interim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul. Disord 29, 842–856 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Finkel RS, et al. RAINBOWFISH: a study of risdiplam in newborns with presymptomatic spinal muscular atrophy (SMA). Neurology 96 (15 Supplement), 4281 (2021). [Google Scholar]
- 7).Kong L, et al. Impaired prenatal motor axon development necessitates early therapeutic intervention in severe SMA. Sci. Transl. Med 13, eabb6871 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Uchitel J et al. Viral-Mediated Gene Replacement Therapy in the Developing Central Nervous System: Current Status and Future Directions. Pediatric Neurol 110, 5–9 (2020). [DOI] [PubMed] [Google Scholar]
- 9).Crawford TO and Sumner CJ. Assuring long-term safety of highly effective gene-modulating therapeutics for rare diseases. J. Clin. Invest 2021;131:e152817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Long Term Follow-up After Administration of Human Gene Therapy Products (FDA, 2020). [Google Scholar]
- 11).Das A et al. Epigenetic silencing of recombinant adeno-associated virus genomes by NP220 and he HUSH complex. J. Virol 96, e0203921 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Thomsen G, et al. Biodistribution of onasemnogene abeparvovec DNA, mRNA and SMN protein in human tissue. Nat. Med 27, 1701–1711 (2021). [DOI] [PubMed] [Google Scholar]
- 13).Ramos DM et al. Age-dependent SMN expression in disease-relevant tissue and implications for SMA treatment. J. Clin. Invest 129, 4817–4831 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]