

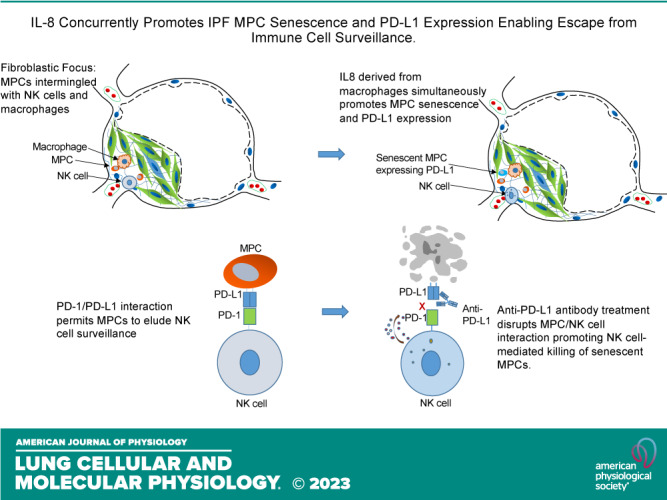
Keywords: idiopathic pulmonary fibrosis, mesenchymal progenitor cells, PD-L1, senescence
Abstract
Idiopathic pulmonary fibrosis (IPF) is a progressive fibrotic lung disease. We discovered fibrogenic mesenchymal progenitor cells (MPCs) in the lungs of IPF patients that display cell-autonomous fibrogenicity and drive fibrotic progression. In a study of the IPF MPC nuclear proteome, we identified DNA damage as one of the most altered functions in IPF MPCs. In prior work we found that IL-8 drives IPF MPC self-renewal. IL-8 can promote replicative stress and DNA damage and induce senescence through the CXCR2 receptor. We hypothesized that IL-8 promotes DNA damage-mediated senescence in IPF MPCs. We show that IL-8 induces DNA damage and promotes IPF MPC senescence. We discovered that IL-8 concurrently promotes senescence and upregulation of the programmed death ligand 1 (PD-L1) in a CXCR2-dependent manner. Disruption of programmed cell death protein-1 (PD-1)-PD-L1 interaction promotes natural killer (NK) cell killing of IPF MPCs in vitro and arrests IPF MPC-mediated experimental lung fibrosis in vivo. Immunohistochemical (IHC) analysis of IPF lung tissue identified PD-L1-expressing IPF MPCs codistributing with NK cells and β-galactosidase-positive cells. Our data indicate that IL-8 simultaneously promotes IPF MPC DNA damage-induced senescence and high PD-L1 expression, enabling IPF MPCs to elude immune cell-targeted removal. Disruption of PD-1-PD-L1 interaction may limit IPF MPC-mediated fibrotic progression.
NEW & NOTEWORTHY Here we show that IL-8 concurrently promotes senescence and upregulation of PD-L1 in IPF MPCs. IHC analysis identifies the presence of senescent IPF MPCs intermingled with NK cells in the fibroblastic focus, suggesting that senescent MPCs elude immune cell surveillance. We demonstrate that disruption of PD-1/PD-L1 interaction promotes NK cell killing of IPF MPCs and arrests IPF MPC-mediated experimental lung fibrosis. Disruption of PD-1/PD-L1 interaction may be one means to limit fibrotic progression.
INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a progressive fibrotic lung disease (1, 2). Although currently available antifibrotic pharmaceuticals modestly slow disease progression, they do not arrest the fibrotic process (3). To arrest fibrotic progression, its obligatory drivers need to be identified. We made several discoveries studying primary cells and extracellular matrix (ECM) from IPF patients that provide a path forward toward understanding mechanisms that drive the relentless progression of fibrosis following disease initiation (4–9). Fibrosis progression involves cell-autonomous, immune, and ECM-driven mechanisms, and, as in cancer, there is cooperation between autonomous cells and their microenvironment. Cell-autonomous fibrogenicity was established when we discovered fibrogenic mesenchymal progenitor cells (MPCs) in the lungs of IPF patients that are one source of IPF fibroblasts (4, 5, 9). IPF MPCs display a distinct transcriptome, cause nonresolving interstitial lung fibrosis in a humanized mouse xenograft model, and are found concentrated in a highly cellular region at the perimeter of the fibroblastic focus in IPF lung tissue codistributing with macrophages and other immune cells (5, 9, 10).
We have previously shown that IL-8 promotes IPF MPC self-renewal (9). Interestingly, in a recent proteomics study of IPF MPC nuclear proteins, ingenuity pathway analysis (IPA) of altered nuclear proteins identified DNA damage/repair as one of the most altered cell functions in IPF MPCs compared with control MPCs (11). Surprisingly, we have discovered that in response to IL-8 IPF MPCs display higher levels of the DNA damage marker histone H2AX (γH2AX) compared with control MPCs. Furthermore, studies have shown that IL-8 signaling through CXCR2 reinforces both replication stress-induced and oncogene-induced senescence (12). These studies suggested to us that chemokine signaling through CXCR2 may induce IPF MPC senescence in response to increased DNA damage.
Accumulating evidence indicates that cell senescence is an important driver of IPF fibrotic progression (13–18). Typically, senescent cells are targeted and rapidly removed by immune cell surveillance to limit tissue injury induced by secretion of harmful molecules by senescent cells. Programmed cell death protein-1 (PD-1) is an immune checkpoint receptor expressed on the cell surface of immune cells that plays an important role in immune regulation (19). PD-1 delivers a negative signal when it interacts with its ligands: programmed death-ligand 1 (PD-L1) and PD-L2 (20). In normal human tissue cells, the ligation of PD-1/PD-L1 promotes immune cell tolerance and escape from host immunity. When PD-L1 is expressed in cancer cells, it can engage the immune checkpoint PD-1/PD-L1 axis to escape antitumor immune responses to prevent the immune system from killing cancer cells (19–22). Here we show that IL-8 concurrently induces IPF MPC DNA damage-mediated senescence and increases PD-L1 expression via the CXCR2 receptor. We demonstrate that disruption of PD-1-PD-L1 interaction markedly promotes natural killer (NK) cell-mediated killing of IPF MPCs in vitro. Using an adoptive transfer modified model of sustained pulmonary fibrosis, we demonstrate that anti-PD-L1 antibody treatment of mice receiving IPF MPCs and NK cells markedly reduced lung fibrosis compared with mice receiving control antibody. Importantly, immunohistochemical (IHC) analysis of IPF lung tissue shows IPF MPCs expressing PD-L1 codistributing with CD56+ NK cells in the highly cellular region at the periphery of the fibroblastic focus.
METHODS
Study Approval
Deidentified patient samples were obtained by our tissue procurement service (Bionet) under a waiver of informed consent from the University of Minnesota Institutional Review Board. Animal protocols were approved by and conducted in accordance with the regulations of the University of Minnesota Institutional Animal Care and Use Committee.
Primary Mesenchymal Cell Lines
Six primary lung mesenchymal cell lines were established from patients fulfilling diagnostic criteria for IPF including a pathological diagnosis of usual interstitial pneumonia (23). Patient control subjects were selected to be similar in age to IPF patients with nonfibrotic lung disorders. On the basis of these criteria, we utilized six nonfibrotic primary control fibroblast lines from lung tissue uninvolved by the primary disease process: adenocarcinoma (n = 3) or chronic obstructive pulmonary disease (COPD) (n = 3). Cell lines were derived from lungs, characterized as mesenchymal cells, and cultivated as previously described (4).
Isolation of Mesenchymal Progenitor Cells
IPF mesenchymal progenitor cells were isolated from primary IPF mesenchymal cell cultures at passage 0 (initial isolate before subcultivation) through passage 4. To isolate progenitors, primary IPF mesenchymal cells were labeled with mouse anti-human SSEA4 antibody conjugated to Alexa Fluor 647 (Clone MC-813-70, catalog no. 560796; BD Biosciences, Franklin Lake, NJ) and mouse anti-human SSEA1 conjugated to PE (Clone MC480, catalog no. 560142; BD Biosciences) as we previously described (4). Cells were sorted on a FACS Aria Cell Sorter (BD Biosciences). Cells that were SSEA4+ and SSEA1− (relative to mouse IgG3 κ isotype control conjugated to Alexa Fluor 647 and mouse IgM κ isotype control conjugated to PE, respectively) (clone J606, catalog no. 560803 and clone G155-228, catalog no. 555584; BD Biosciences) were collected. To generate sufficient numbers of MPCs for the in vivo mouse studies, SSEA4+ cells were expanded by culture in DMEM + 10% FCS for 7 days before use. The resulting MPC cultures were reanalyzed for SSEA4 expression by FACS analysis and for colony formation in vitro. Ninety-seven percent of day 7 MPCs were SSEA4+ and formed colonies in methylcellulose, indicating retention of progenitor self-renewal properties.
NK92 Cells
Human immortalized NK92 cells were purchased from ATCC (Manassas, VA). NK92 cells were cultured in αMEM (Gibco catalog no. 12571) with 0.1 mM 2-mercaptoethanol, 500 U/mL recombinant IL-2, 12.5% horse serum, 12.5% fetal bovine serum, and 1,000 U/mL penicillin-streptomycin.
Cell Viability Assay
IPF and control MPCs were cocultured with NK92 cells in the presence of PD-L1 antibody (BE0285, B7-H1; Bioxcell) or isotype control antibody. The cells were then incubated with CellTiter blue according to the manufacturer’s instructions (CellTiter Blue Cell Viability Assay Kit; Promega, Madison, WI). Fluorescence was recorded at 520ex/600em with a Spectra Max CM3 fluorescent plate reader according to the manufacturer’s instructions (Molecular Devices, San Jose, CA).
Flow Cytometry
Cells were dissociated with 0.05% trypsin-EDTA, blocked with 1% BSA at room temperature for 20 min, and incubated with the appropriate primary antibody for 1 h. Cells were washed with PBS containing 0.5% FBS and incubated with Alexa Fluor 488 donkey anti-mouse IgG secondary antibody (Invitrogen) at room temperature for 30 min. Flow cytometry analyses were performed with a BD Aria II instrument (BD Biosciences).
Quantitative PCR
Analysis of PD-L1 and CXCR2 gene expression was conducted by quantitative PCR (Q-PCR) as previously described (5). Total RNA was extracted with the RNeasy mini kit (Qiagen), and cDNA was synthesized with a cDNA mini kit (Qiagen). Total RNA was isolated and reverse transcribed with a Taqman Reverse Transcriptase Reagent Kit (Roche) and primed with random hexamers. Primer sequences were selected with NCBI Primer-BLAST. Real-time PCR (Q-PCR) was performed with a LightCycler FastStart DNA MasterPLUS SYBR Green I Kit (Roche). Primer sequences were as follows: GAPDH forward: 5′-TGTTGCCATCAATGACCCCTT -3′; GAPDH reverse: 5′- CTCCACGACGTACTCAGCG-3′; PD-L1 forward: 5′-TTG GGA AAT GGA GGA TAA GA-3′; PD-L1 reverse: 5′-GGA TGT GCC AGA GGT AGT TCT-3′; CXCR2 forward: 5′-CACCGATGTCTACCTGCTGA-3′; CXCR2 reverse: 5′-CACAGGGTTGAGCCAAAA GT-3′.
Samples were quantified at the log-linear portion of the curve with LightCycler analysis software and compared to an external calibration standard curve.
Self-Renewal Assay
Single-cell suspensions of SSEA4+ IPF MPCs were incorporated into methylcellulose gels (StemCell Technologies, Inc., Vancouver, BC, Canada) and maintained in MSC SFM CTS (Thermo Scientific/Gibco, Rochford IL) (37°C, 5% CO2; 1 wk). Enumeration of colonies was performed microscopically, and colony size was quantified by ImageJ (NIH, Bethesda, MD).
Plasmids/Constructs
For loss of function, PD-L1 or CXCR2 was knocked down with shRNA (pGIPZ-PD-L1 or pGIPZ-CXCR2 shRNA; IDT and UMN Genomics Center). Scrambled shRNA served as control.
Western Blotting and Immunoprecipitation
Western blotting was performed as previously described (24). For immunoprecipitation, nuclear fractions were isolated by NE-PER Nuclear and Cytoplasmic Extraction reagents. The samples were centrifuged at 120,000 g for 15 min at 4°C, and the lysates were precleared for 1 h at 4°C with protein A/G beads and immunoprecipitated for 2 h at 4°C with the appropriate primary antibody.
ELISA to Detect Secreted Proteins
IPF MPCs were seeded in tissue culture dishes containing serum-free DMEM plus IL-8 (5 ng/mL), vehicle control, or IPF conditioned media and incubated for 24 h. Cells were then washed and placed in serum-free DMEM and cultured for 48 h. The resulting cell culture medium was collected and analyzed by multiplex ELISA to detect secreted proteins (AAH-CYT-5 human cytokine array C5; EMD Raybiotech). For derivation of IPF MPC conditioned media, IPF MPCs were first cultured in DMEM + 5% serum for 24 h, followed by a wash step. The cells were then cultured in serum-free DMEM for 48 h, followed by collection of the conditioned media. Relative abundance of each protein was quantified by densitometry.
Immunohistochemistry
Immunohistochemistry was performed on 4-µm paraffin-embedded serial-sectioned IPF lung tissue and mounted on polylysine-coated slides. The sections were deparaffinized in xylene, rehydrated through a graded methanol series, quenched with 0.3% hydrogen peroxide in methanol, and immersed in a 98°C water bath for 30 min in citrate buffer (pH 6·0) for antigen retrieval. Sections were placed in 5% normal horse serum (Jackson Immunoresearch, West Grove, PA) to block nonspecific binding of secondary antibodies. A multiplex immunohistochemistry kit was used for antigen detection according to the manufacturer’s instructions (MULTIVIEW IHC Kit ADI-950-101-0001; Enzo Life Sciences, Farmingdale, NY). The tissue specimens were incubated overnight (18–20 h, 4°C) with the following primary antibodies: anti-human SSEA4 antibody (1:100) (Clone MC-813-70, catalog no.30402; BioLegend, San Diego, CA); CD56 antibody (1:400, MAB24081; R&D Systems, Minneapolis, MN), and PD-L1 antibody (1:600, EPR19759; Abcam, Boston, MA). Specimens were coverslipped with a Prolong Antifade Kit (Invitrogen/Molecular Probes) and stored overnight at room temperature without light before image analysis.
Mouse Xenograft Model of Fibrotic Progression
To assess the ability of PD-L1 to enable IPF MPCs to elude immune cell surveillance and drive fibrotic progression in vivo, we modified our IPF MPC-based mouse model of fibrotic progression. NOD/SCID/IL2rγ/B2M (NSG) male and female mice (Jackson Laboratories), average 10 wk of age, were administered intratracheal bleomycin (1.25 U/kg) to establish self-limited lung fibrosis. One week after administration of bleomycin the mice were irradiated (225 cGy) to deplete immune cells. Two weeks after bleomycin administration, IPF MPCs were suspended in PBS (106 cells/100 µL) and injected via the tail vein according to our published protocol (5). On day 3 and day 13 after IPF MPC administration, both NK cells (10 × 106 cells) and anti-PD-L1 antibody (2 mg/kg, BE0285, B7-H1; BioCell) or isotype control antibody were administered by peritoneal injection. Mice were euthanized 4 wk after adoptive transfer of human cells, and the lungs were harvested. Collagen content was quantified in left lung tissue by Sircol assay and served as the primary end point (5). As a secondary end point, semiquantitative analysis of lung collagen deposition by trichrome staining was performed. Three trichrome-stained images at low power (×5, scale bar = 500 µm) were randomly selected from each section. The blue regions (fibrotic stain) were defined and quantified with program ImageJ. Histological [hematoxylin and eosin (H&E) and trichrome staining] and immunohistochemical analysis were performed on paraffin-embedded and frozen right lung tissue. Cells positive for human procollagen (anti-human procollagen type I antibody; 1:500, catalog no. MAB1912; EMD Millipore, Burlington, MA) were identified as human. The presence of lung fibrotic lesions by histological analysis served as an additional secondary end point. IHC using the human procollagen antibody and PD-L1 antibody was performed to assess the distribution of PD-L1-expressing cells with human procollagen I-expressing cells.
Statistics
Comparisons among experiments were performed with the two-tailed Student’s t test. Experiments were independently replicated a minimum of three times. Data are expressed as means ± SE. P < 0.05 was considered significant.
RESULTS
IL-8 Induces IPF MPC DNA Damage
IPF MPCs display a distinct transcriptome, cause nonresolving interstitial lung fibrosis in a humanized mouse xenograft model, and are found concentrated in a highly cellular region on the perimeter of the fibroblastic focus in IPF lung tissue (4, 5, 10). However, the mechanism of cell-autonomous fibrogenicity has remained incompletely understood. To gain insight, in a recent study we employed quantitative mass spectrometry and interactomics to define protein alterations in the nuclear compartment of IPF MPCs. Ingenuity pathway analysis of the IPF and control MPC nuclear proteome identified DNA damage/repair as one of the most altered cell functions in IPF MPCs compared with control MPCs (11). Interestingly, during routine culture we found that several markers of DNA damage were increased in IPF MPCs compared with control, suggesting that IPF MPCs have sustained significant DNA damage (11). IPF MPCs codistribute with macrophages and other immune cells in a highly cellular region at the periphery of the IPF fibroblastic focus. We have previously shown that IPF macrophage release of IL-8 stimulates IPF MPC self-renewal (9). However, under chronic inflammatory conditions, cytokines such as IL-8 can also promote genomic instability by causing replicative stress and formation of double-stranded DNA breaks (25). To begin, we first examined for evidence that IPF MPCs have sustained DNA damage. Phosphorylation of the histone H2AX, termed γH2AX, is an initial step involved in the recruitment of DNA repair proteins to sites of double-stranded DNA breaks (26). In addition, activation of the p53-p21 pathway frequently occurs during DNA damage (27, 28). We found that compared with control MPCs IPF MPCs displayed higher levels of γH2AX and p21, indicating that IPF MPCs have sustained DNA damage (Fig. 1A). We next analyzed the effect of IL-8 on induction of DNA damage in IPF MPCs. We found that IL-8 increased γH2AX levels in IPF MPCs (Fig. 1B). Similarly, IL-8 increased p21 levels in IPF MPCs, indicating that IL-8 augments DNA damage in IPF MPCs (Fig. 1C). We also examined levels of the replication protein subunit 2 (RPA2), which is used as a marker of replication stress in IPF and control MPCs. Phosphorylated RPA2 levels were increased in IPF MPCs compared with control MPCs (Fig. 1A), and, importantly, we found that IL-8 increased the expression of phosphorylated RPA2, supporting the concept that IL-8 is promoting replication stress (Fig. 1D). We next examined the effect of IL-8 inhibition on induction of DNA damage. Pharmacological inhibition of IL-8 by reparixin, a specific inhibitor of IL-8, attenuated the ability of IL-8 to increase γH2AX levels and to promote DNA damage (Fig. 1E).
Figure 1.
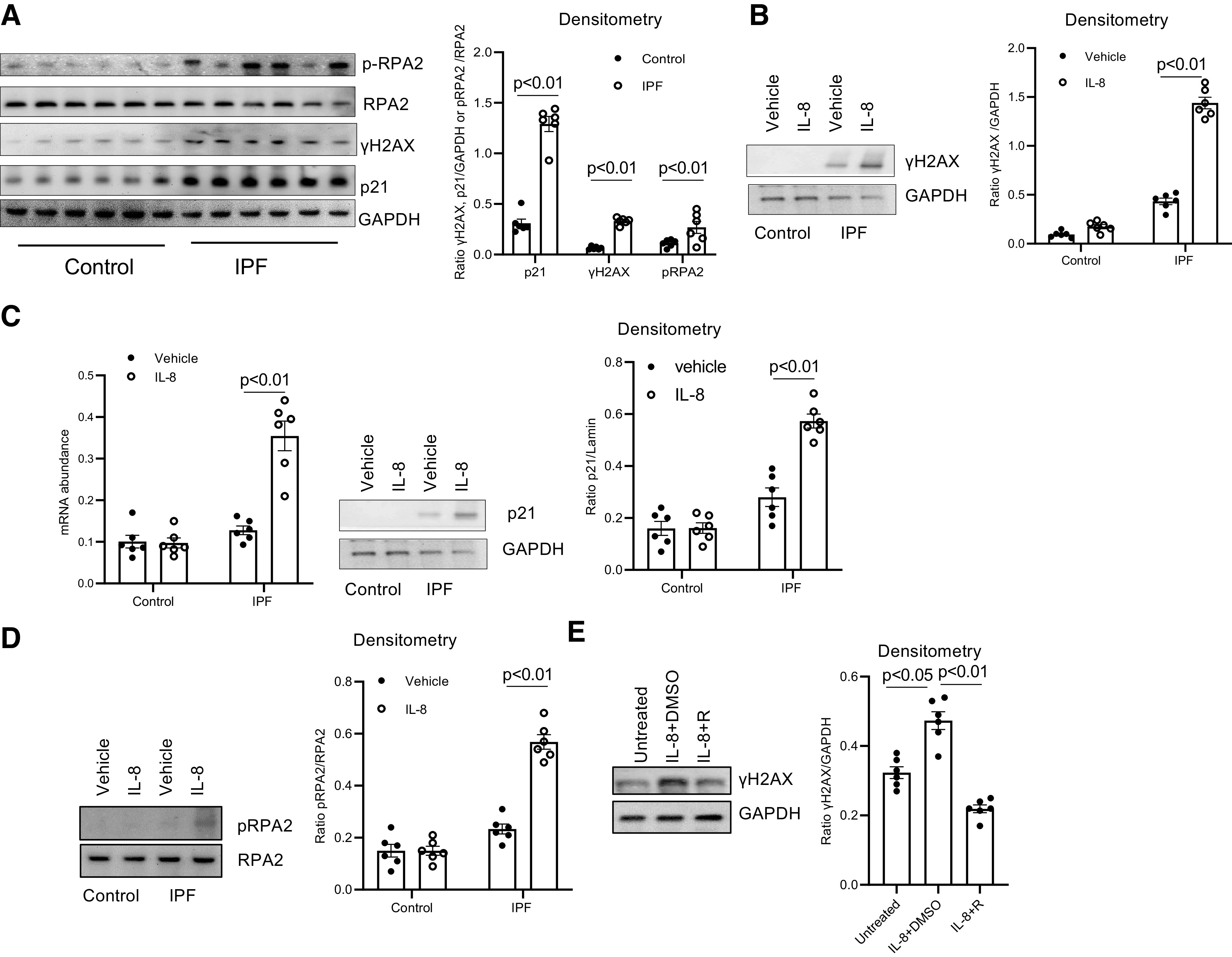
IL-8 induces idiopathic pulmonary fibrosis (IPF) mesenchymal progenitor cell (MPC) DNA damage. A: histone H2AX (γH2AX), p21, and phosphorylated (p) and total replication protein subunit 2 (RPA2) levels were quantified in IPF and control MPCs by Western blot analysis (left) (n = 6 IPF and control MPC cell lines each). GAPDH served as loading control. Densitometry values summarizing Western blot data are shown at right. B–D: IPF and control MPCs were treated with recombinant IL-8 (5 ng) or vehicle control (n = 6 IPF and control MPC cell lines each). B: a representative Western blot quantifying γH2AX levels in 1 IPF and 1 control MPC cell line (left). GAPDH served as loading control. Densitometry values summarizing Western blot data are shown at right. C: p21 levels were quantified by quantitative PCR (Q-PCR; left) and Western blot analysis (shown is a representative Western blot; center). Densitometry values summarizing Western blot data are shown at right. D: phosphorylated and total RPA2 levels were quantified by Western blot analysis (shown is a respresentative Western blot, left). Densitometry values summarizing the Western blot data are shown at right. E: IPF MPCs were treated with 5 ng of recombinant IL-8 in the presence of the IL-8 receptor inhibitor reparixin (IL-8 + R) or vehicle control (IL-8 + DMSO) (n = 6 IPF MPC cell lines). Cells not treated with IL-8 served as an additional control (Untreated). Shown is a representative Western blot quantifying γH2AX levels in 1 IPF MPC cell line (left). GAPDH served as loading control. Densitometry values summarizing the Western blot data are shown at right.
IL-8 Promotes IPF MPC Senescence via the CXCR2 Receptor
It has been established that DNA damage caused by replicative stress can provoke senescence (29–32). Because IPF MPCs display evidence of replicative stress and DNA damage, this suggested that these cells may be prone to undergoing senescence. Therefore, using β-galactosidase as a marker of senescence, we quantified β-galactosidase levels in IPF and control MPCs. IPF MPCs displayed higher levels of β-galactosidase compared with control (Fig. 2A). Since IL-8 treatment promoted IPF MPC replicative stress and DNA damage, we next examined whether IL-8 could induce IPF MPC senescence. In response to IL-8, 33% of IPF MPCs stained positive for β-galactosidase, whereas 15% of IPF MPCs treated with vehicle control were β-galactosidase positive (Fig. 2B). Since IL-8 promotes DNA damage and can induce IPF MPC senescence, we examined the effect of reparixin on the ability of IL-8 to induce senescence in IPF MPCs. Reparixin decreased the ability of IL-8 to promote induction of IPF MPC senescence (Fig. 2C). These data indicate that inhibition of IL-8 attenuates both DNA damage and senescence in IPF MPCs.
Figure 2.
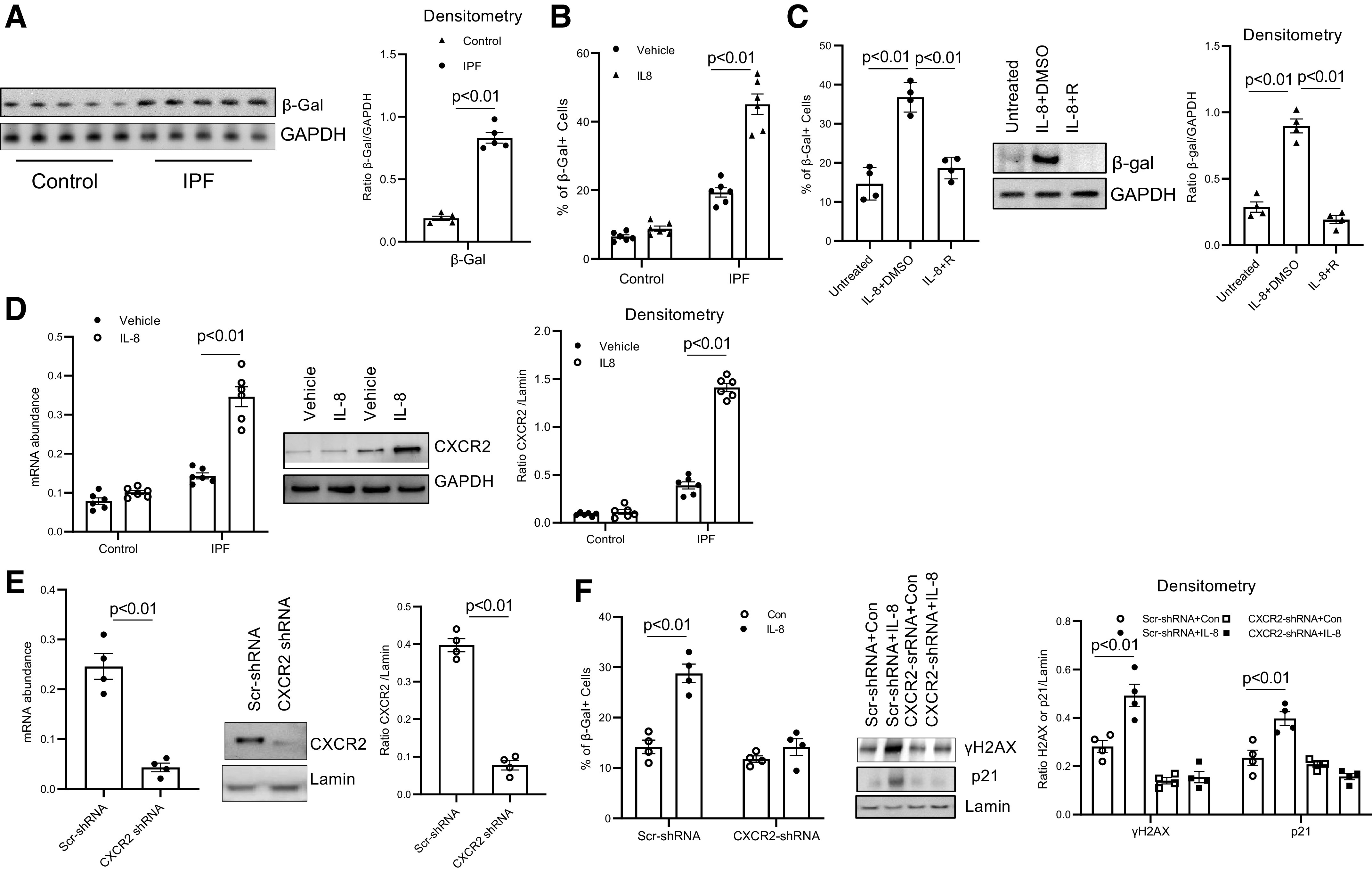
IL-8 promotes idiopathic pulmonary fibrosis (IPF) mesenchymal progenitor cell (MPC) senescence via the CXCR2 receptor. A: β-galactosidase (β-Gal) levels were quantified in IPF and control MPCs by Western blot analysis (left) (n = 5 IPF and control MPC cell lines each). GAPDH served as loading control. Densitometry values summarizing Western blot data are on right. B: IPF and control MPCs (n = 6 IPF and control MPC cell lines each) were treated with IL-8 (5 ng) or vehicle control; 72 h later the percentage of senescent cells was analyzed by β-Gal staining. C: IPF MPCs (n = 4 IPF MPC cell lines) were treated with IL-8 in the presence of the IL-8 receptor inhibitor reparixin (IL-8 + R) or vehicle control (IL-8 + DMSO). Cells not treated with IL-8 served as an additional control (Untreated). Senescence was quantified by β-Gal staining (left) and by quantification of β-Gal levels by Western blot analysis (shown is a respresentative Western blot; center). Densitometry values summarizing Western blot data are on right. D: IPF and control MPCs (n = 6 IPF and control MPC cell lines each) were treated with IL-8 or vehicle control. CXCR2 levels were quantified by quantitative PCR (Q-PCR; left) and Western blot analysis (shown is a respresentative Western blot; center). GAPDH = loading control. Densitometry values summarizing Western blot data are on right. E: IPF MPCs were transduced with CXCR2 (CXCR2-shRNA) or scrambled (Scr-shRNA) shRNA. CXCR2 levels were quantified by Q-PCR (left) and Western blot analysis (shown is a respresentative Western blot; center). Densitometry values summarizing Western blot data are on right. F: IPF MPCs transduced with CXCR2-shRNA or Scr-shRNA were treated with IL-8 or vehicle control (Con). Senescence was quantified by β-Gal staining (left), and histone H2AX (γH2AX) and p21 levels were quantified by Western blot analysis (center). Lamin = loading control. Shown is a respresentative Western blot. Densitometry values summarizing Western blot data are on right. The experiments in E and F were replicated with 4 IPF MPC cell lines.
Senescent cells can secrete CXCR2 ligands including IL-8 that coordinately induce CXCR2 expression, forming a self-amplifying loop whereby CXCR2-binding chemokines reinforce senescence (12). Therefore, we first examined the effect of IL-8 on CXCR2 expression in IPF MPCs. We found that IL-8 increased the expression of CXCR2 (Fig. 2D). We then examined whether IL-8 induced IPF MPC senescence and DNA damage via the CXCR2 receptor. CXCR2 expression was knocked down with CXCR2 shRNA. CXCR2 protein expression was decreased by 80% (Fig. 2E). Knockdown of CXCR2 inhibited IL-8-induced DNA damage and senescence of IPF MPCs (Fig. 2F). These data indicate that IL-8 increases CXCR2 expression and induces IPF MPC DNA damage and senescence in a CXCR2-dependent manner.
IL-8 Promotes IPF MPC Secretion of Cytokines
The secretion of chemokines including IL-8 by senescent cells is part of the senescence-associated secretory phenotype (SASP). To determine whether IL-8 stimulates IPF MPC secretion of cytokines in response to IL-8, we measured the release of chemokines in IPF MPCs treated with IL-8, using a chemokine/cytokine array assay. We found that IL-8 induced secretion of multiple cytokines consistent with induction of a SASP (Fig. 3A). Since some IPF MPCs treated with IL-8 become senescent, display the SASP, and have high expression of CXCR2, we next analyzed IPF lung tissue for IPF MPCs expressing CXCR2 cells (double-positive SSEA4 and CXCR2). We found SSEA4-positive cells expressing CXCR2 at the periphery of the fibroblastic focus (Fig. 3B). Importantly, senescent cells (β-galactosidase+) were detectable in the same location as SSEA4/CXCR2-positive cells (Fig. 3C), supporting the concept that IPF MPCs displaying CXCR2 and a SASP are present in active areas of fibrogenesis. To further examine for the presence of senescent MPCs at the periphery of the fibroblastic focus, we performed double staining for SSEA4 and p21. We found SSEA4+/p21+ MPCs at the periphery of the fibroblastic focus, providing evidence for senescent MPCs at regions of active fibrogenesis (Fig. 3D).
Figure 3.
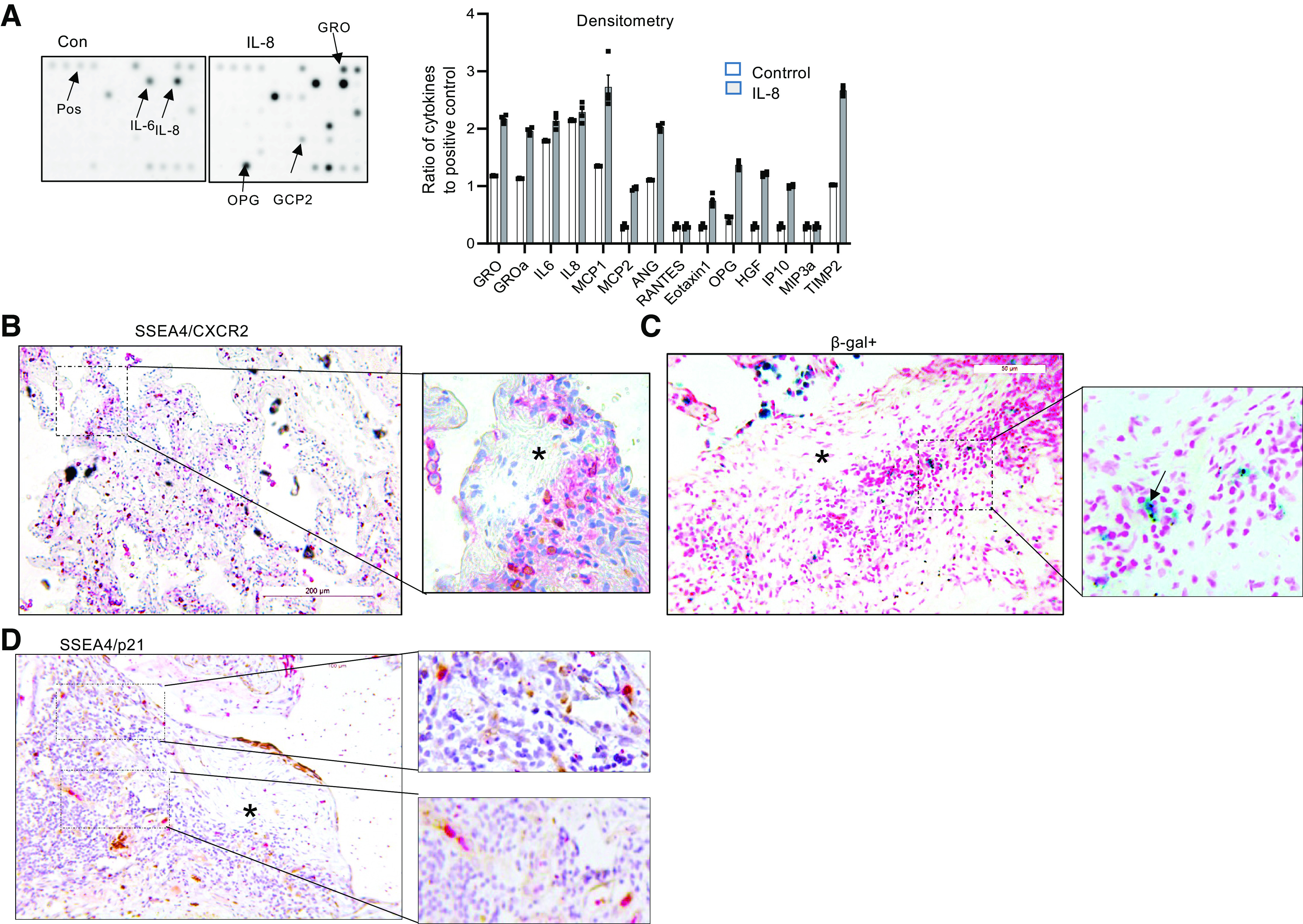
IL-8 promotes idiopathic pulmonary fibrosis (IPF) mesenchymal progenitor cell (MPC) secretion of cytokines. A: cytokine array assay was performed on conditioned media derived from IPF MPCs pretreated with IL-8 or vehicle control (Con); 4 IPF MPC cell lines were used. Left: representative cytokine array demonstrating increased cytokine levels in the conditioned medium derived from IPF MPCs treated with IL-8 compared with control. Right: densitometry values summarizing cytokine array data. Each data point represents the ratio of the cytokine to the positive control in the conditioned medium from 1 IPF cell line. B–D: immunohistochemical (IHC) staining of IPF lung tissue for SSEA4 (brownish-yellow stain) and CXCR2 (red stain) (B); β-galactosidase (β-gal; blue stain) (C); and SSEA4 (brownish-yellow stain) and p21 (red stain) (D). Asterisk denotes myofibroblast core of the fibroblastic focus. (n = 7 IPF patient specimens; 4 sections were imaged in each specimen).
PD-L1 Levels Are Elevated in IPF MPCs
The immune system plays a fundamental role in the elimination of senescent cells and the prevention of disease progression (19–22). This occurs when senescent cells are recognized by cytotoxic T cells and NK cells and are targeted for removal (33). However, in some disease states such as cancer pathological cells have acquired the ability to escape immune surveillance. In cancer, upregulation of the programmed death-ligand 1 (PD-L1) permits cancer cells to escape targeted removal by immune cells (19). An increasing body of evidence supports the concept that senescence is a driver of fibrosis in IPF (13). Although IL-8 promotes IPF MPC senescence and acquisition of a SASP in response to IL-8, the presence of senescent cells in regions of active fibrogenesis suggested to us that IPF MPCs might escape immune surveillance by upregulation of PD-L1. To examine this, we began by quantifying PD-L1 levels in IPF MPCs and control MPCs. IPF MPCs expressed higher levels of PD-L1 compared with control MPCs (Fig. 4A). We next analyzed the effect of IL-8 treatment on PD-L1 expression in IPF MPCs. IL-8 increased PD-L1 expression in a dose-dependent manner with a maximal response at ∼5 ng (Fig. 4B). We then examined the effect of IL-8 on PD-L1 expression in IPF and control MPCs. IL-8 markedly increased PD-L1 mRNA and protein expression in IPF MPCs compared with vehicle control (Fig. 4C). In contrast, compared with vehicle control. IL-8 had no significant effect on PD-L1 expression in control MPCs.
Figure 4.
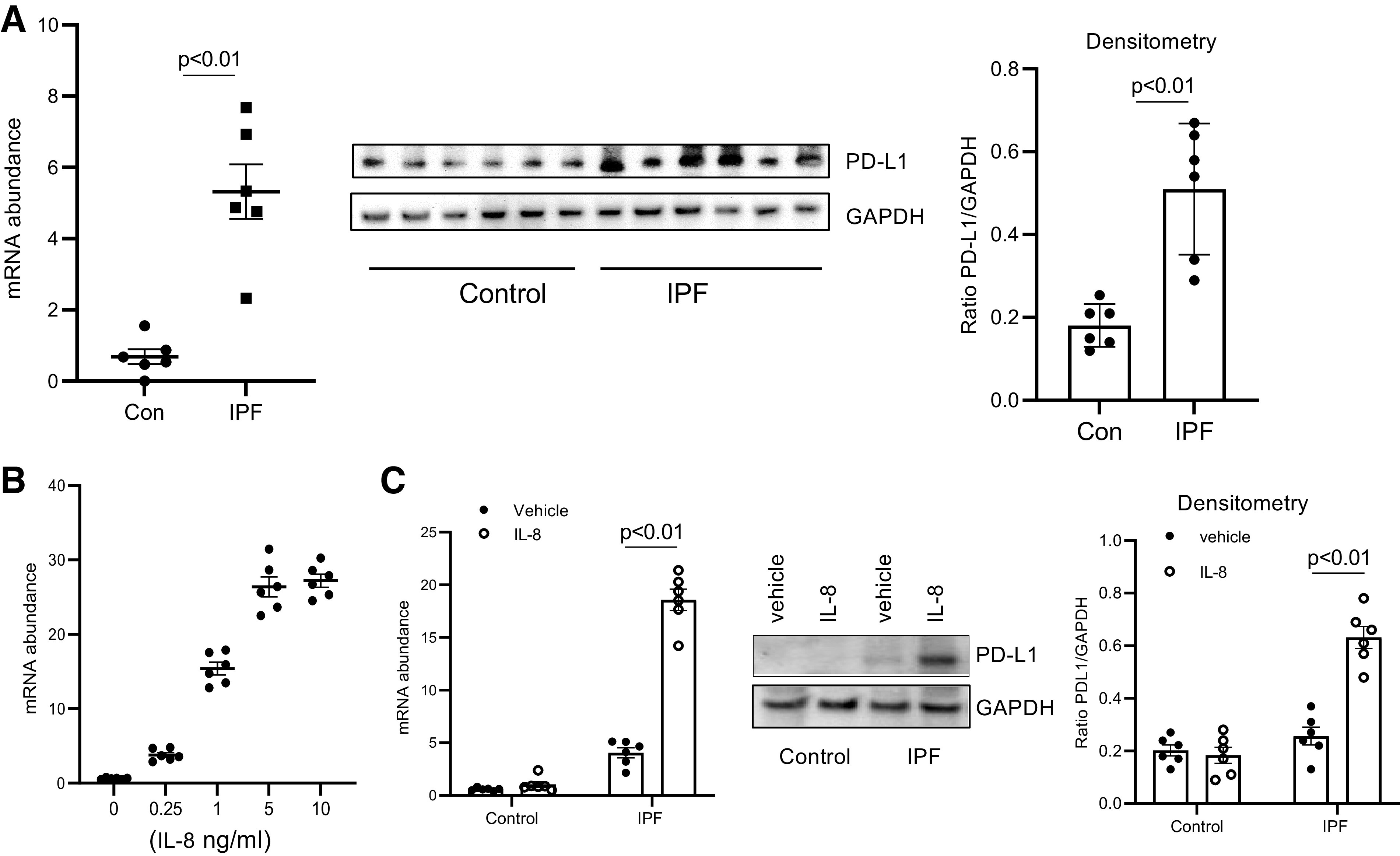
Programmed death ligand 1 (PD-L1) levels are elevated in idiopathic pulmonary fibrosis (IPF) mesenchymal progenitor cells (MPCs). A: PD-L1 expression was quantified in IPF and control (Con) MPCs (n = 6 IPF and control MPC cell lines each) by quantitative PCR (Q-PCR) (left) and Western blot analysis (center). GAPDH = loading control. Densitometry values summarizing Western blot data are on right. B: IPF MPCs were treated with various amounts of IL-8. PD-L1 levels were quantified by Q-PCR. C: IPF and control MPCs (n = 6 cell lines each) were treated with IL-8 (5 ng) or vehicle control. PD-L1 levels were quantified by Q-PCR (left) and Western blot analysis (shown is a respresentative Western blot quantifying PD-L1 levels in 1 IPF and 1 control MPC cell line; center). GAPDH = loading control. Densitometry values summarizing Western blot data are on right.
IL-8 Increases IPF MPC PD-L1 Expression via CXCR2
Since IL-8 promotes IPF MPC senescence via the CXCR2 receptor, we next analyzed the effect of knockdown of CXCR2 on IL-8 mediated induction of PD-L1 expression. Knockdown of CXCR2 prevented IL-8 from increasing PD-L1 expression in IPF MPCs (Fig. 5A). Taken together, these data indicate that IL-8 operating through CXCR2 concurrently increases both IPF MPC senescence and PD-L1 expression. We then explored the signaling pathway by which the IL-8/CXCR2 axis upregulates PD-L1. We found that antagonism of MAPK inhibited the ability of IL-8 to increase PD-L1 (Fig. 5B).
Figure 5.
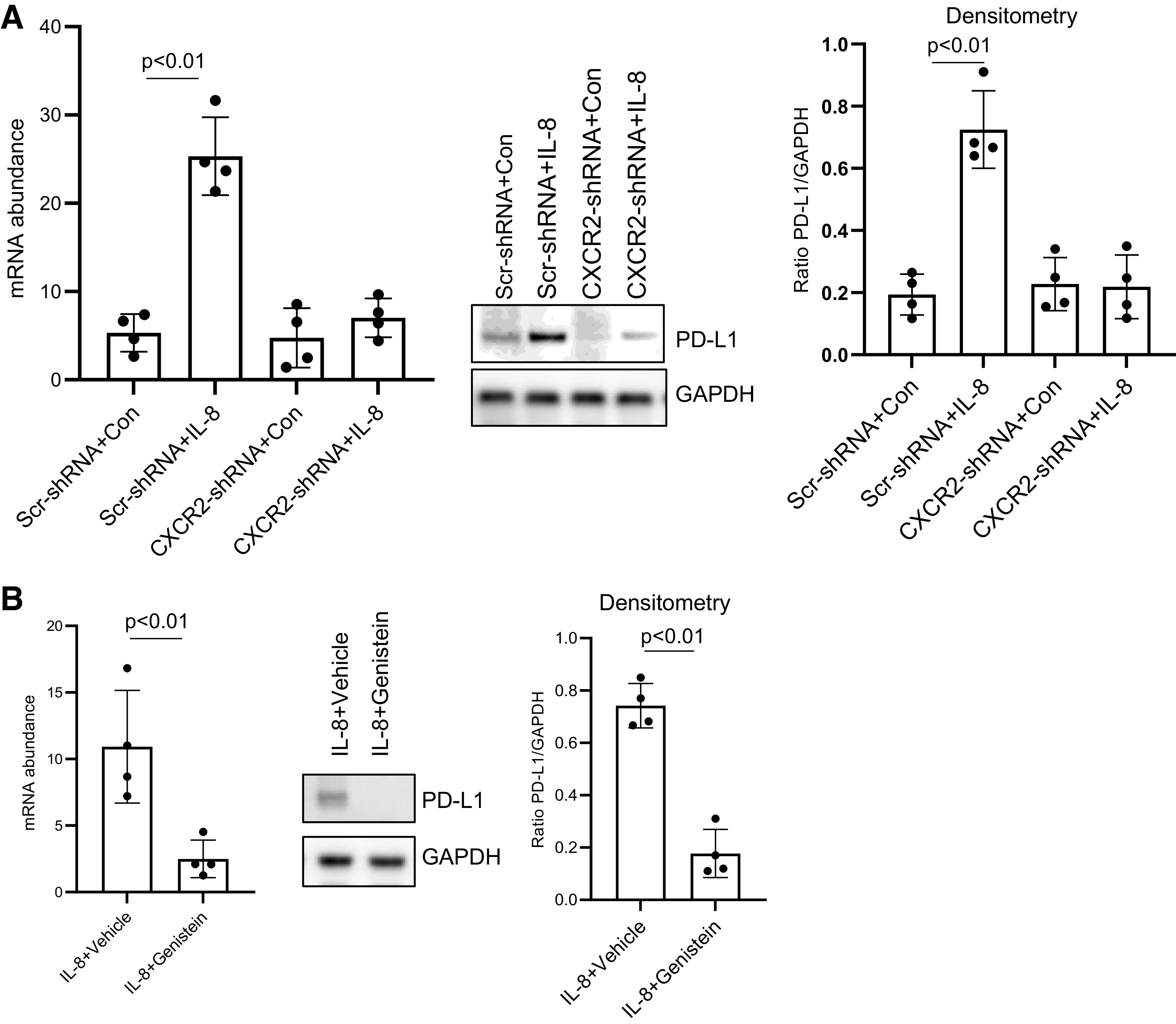
IL-8 increases idiopathic pulmonary fibrosis (IPF) mesenchymal progenitor cell (MPC) programmed death ligand 1 (PD-L1) expression via CXCR2. A: IPF MPCs transduced with CXCR2 (CXCR2-shRNA) or scrambled (Scr-shRNA) shRNA were treated with 5 ng of IL-8 or vehicle control (Con) and PD-L1 expression quantified by quantitative PCR (Q-PCR) (left) and Western blot analysis (shown is a respresentative Western blot; center). Densitometry values summarizing Western blot data are on right. B: IPF MPCs were treated with 5 ng of IL-8 and the MAPK inhibitor genistein or vehicle control. PD-L1 levels were quantified by Q-PCR (left) and Western blot analysis (shown is a respresentative Western blot; center). Densitometry values summarizing Western blot data are on right. Experiments in A and B were replicated with 4 IPF MPC cell lines.
PD-L1-Expressing IPF MPCs Codistribute with CD56+ NK Cells within the Highly Cellular Region at the Periphery of the Fibroblastic Focus
The IPF fibroblastic focus is a polarized structure containing a focus core and an active fibrotic front (5, 8, 9). The focus core consists of noncycling myofibroblasts actively synthesizing and organizing a type I collagen-rich matrix. The active fibrotic front is a highly cellular region at the perimeter of the focus core containing cycling fibrogenic MPCs. Our IHC studies demonstrate the presence of senescent cells within the highly cellular region at the periphery of the fibroblastic focus, and our in vitro data indicate that cytokines can induce IPF MPCs to become senescent and display the SASP. Senescent cells with a SASP can cause tissue damage and promote fibrosis and therefore are frequently targeted for removal by the immune system (29). Since IPF MPCs display elevated levels of PD-L1, we hypothesized that this may enable them to elude immune cell-mediated killing. We analyzed the distribution of NK cells in relation to PD-L1-expressing IPF MPCs in serial sections of IPF lung tissue by IHC using an antibody for CD56, a NK cell marker, SSEA4, that identifies MPCs and a PD-L1 antibody. We performed IHC double staining of human IPF lung tissue to examine the distribution of PD-L1-expressing MPCs (double positive for SSEA4 and PD-L1). PD-L1-expressing MPCs were found concentrated at the periphery of the fibroblastic focus (Fig. 6, top). We also performed IHC using a CD56 antibody that identifies NK cells. IHC analysis indicates the presence of numerous CD56-positive cells intermingled with SSEA4/PD-L1-positive IPF MPCs at the periphery of the fibroblastic focus (Fig. 6, bottom). The persistence of PD-L1-expressing IPF MPCs codistributing with NK cells in regions of active fibrogenesis suggests that these IPF MPCs are not being targeted for removal by the NK cells but instead are participating in driving fibrotic progression.
Figure 6.

Programmed death ligand 1 (PD-L1)-expressing idiopathic pulmonary fibrosis (IPF) mesenchymal progenitor cells (MPCs) codistribute with CD56+ natural killer (NK) cells within the highly cellular region at the periphery of the fibroblastic focus. Immunohistochemistry (IHC) was performed on serial sections of IPF lung tissue (n= 8 IPF patient specimens; 4 sections were imaged in each specimen). Top: hematoxylin and eosin (H&E; right) and trichrome (left) stain of a fibroblastic focus. Bottom: IHC staining for the NK cell marker CD56 (left) and IHC double staining for SSEA4 (brownish-yellow stain) and PD-L1 (red stain) (center and right). Asterisk denotes focus (myofibroblast) core.
Antagonism of PD-1-PD-L1 Interaction Sensitizes IPF MPCs to NK-Mediated Killing in Vitro
To examine whether high PD-L1 levels enable IPF MPCs to escape immune surveillance, we took two approaches: 1) knockdown of PD-L1 and 2) antagonism of PD-1-PD-L1 interaction by anti-PD-L1 blocking antibody. NK cells target senescent or damaged cells for removal (33). Therefore, we examined the ability of NK92 cells to eliminate IPF MPCs in which PD-L1 had been knocked down or PD-1-PD-L1 interaction had been disrupted. We first examined PD-1 levels in NK92 cells treated with IL-2, which activates NK92 cells and is required for their cytotoxicity (34). NK92 cells treated with IL-2 displayed increased levels of PD-1 compared with cells treated with vehicle control (Fig. 7A). We then knocked down PD-L1 in IPF MPCs with PD-L1 shRNA. PD-L1 mRNA expression was decreased by 92% and protein expression by 88% (Fig. 7B). We found that knockdown of PD-L1 amplified NK92 cell killing of IPF MPCs in vitro (Fig. 7C). We next cocultured IPF MPCs with NK92 cells and varying amounts of anti-PD-L1 antibody to antagonize PD-1-PD-L1 interaction. Anti-PD-L1 antibody induced NK92 cell-mediated killing in a dose-response manner, with 2 µg/mL inducing a maximal response (Fig. 7D, left). We also cocultured IPF MPCs with various ratios of NK92 cells in the presence of 2 µg/mL of anti-PD-L1 antibody. NK92 cells at a 5-to-1 ratio (NK92 cells to MPCs) promoted a maximal killing response (Fig. 7D, right). Therefore, analogous to knockdown of PD-L1, antagonism of PD-1-PD-L1 interaction with PD-L1 antibody markedly enhanced NK92 cell-mediated killing of IPF MPCs. Compared with control MPCs, IPF MPCs express higher levels of PD-L1 and display higher levels of DNA damage and senescence. Therefore, we next sought to determine whether there were differences in NK92 cell-mediated killing between IPF MPCs and control MPCs. Anti-PD-L1 antibody promoted NK92 cell-mediated killing of IPF MPCs but not control MPCs (Fig. 7E). IL-8 promotes PD-L1 expression in IPF MPCs. Therefore, we next examined the effect of IL-8 on NK92 cell-mediated killing of IPF MPCs. Compared with cells treated with vehicle control, NK92 cell-mediated killing was diminished in IL-8-treated IPF MPCs (Fig. 7F). We next examined the effect of anti-PD-L1 antibody on NK92 cell killing of IPF MPCs that were treated with IL-8. Inhibition of PD-1-PD-L1 interaction led to enhanced NK92 cell killing of IPF MPCs that had been treated with IL-8 (Fig. 7G). Together, these data support the concept that high PD-L1 levels enable IPF MPCs to escape immune cell surveillance and that disruption of PD-1-PD-L1 interaction promotes NK cell-mediated killing of IPF MPCs.
Figure 7.
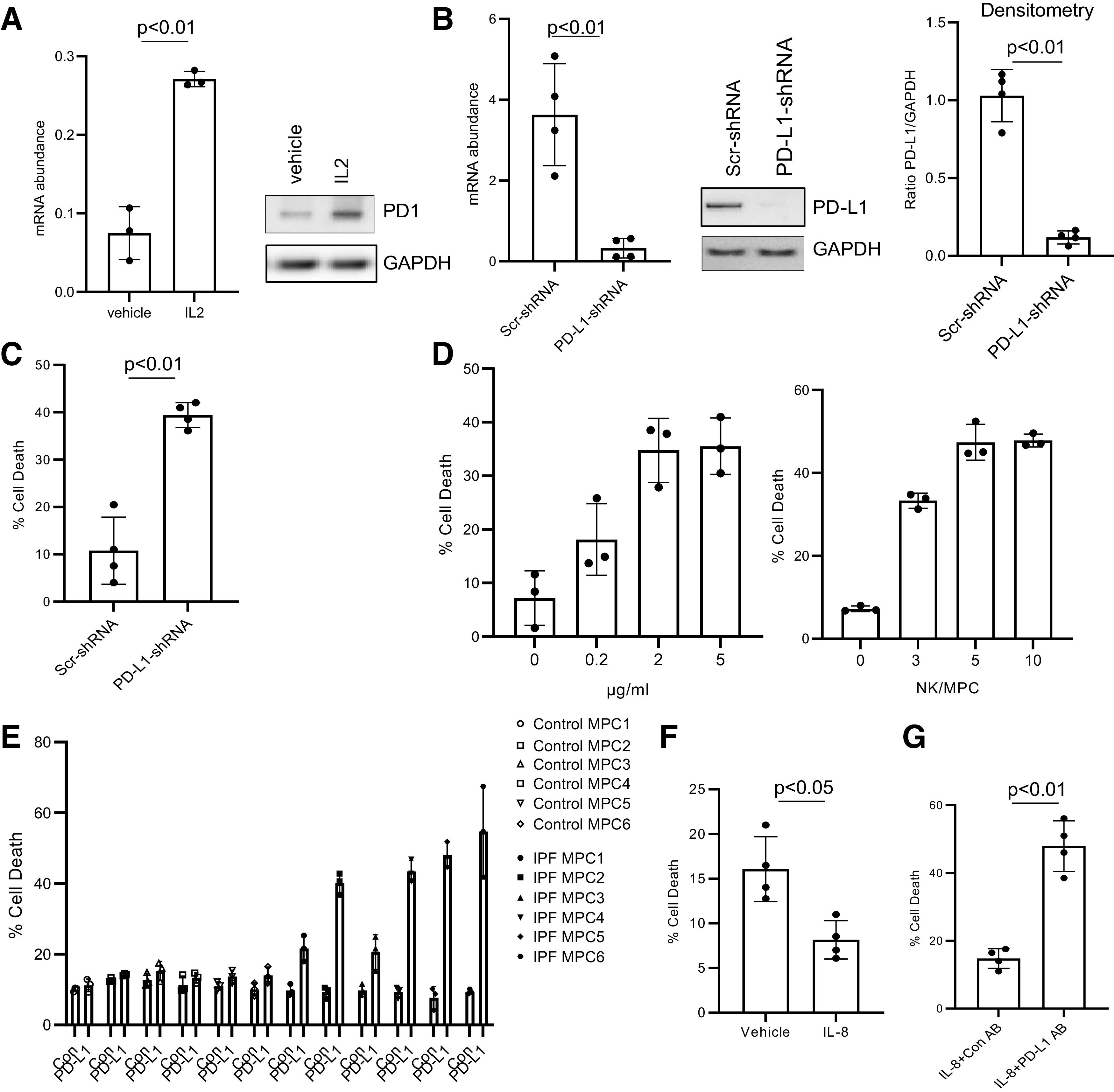
Programmed death ligand 1 (PD-L1) antagonism sensitizes idiopathic pulmonary fibrosis (IPF) mesenchymal progenitor cells (MPCs) to natural killer (NK) cell-mediated killing in vitro. A: programmed cell death protein-1 (PD-1) levels were quantified in NK92 cells treated with IL-2 or vehicle control by quantitative PCR (Q-PCR; left) and Western blot analysis (right). GAPDH served as loading control. The experiment was performed in triplicate. B: IPF MPCs were transduced with PD-L1 (PD-L1-shRNA) or scrambled (Scr-shRNA) shRNA. PD-L1 expression was quantified by Q-PCR (left) or Western blot analysis (shown is a respresentative Western blot; center). Densitometry values summarizing Western blot data are at right. C: transduced IPF MPCs were cocultured with NK92 cells in a 3-to-1 ratio (NK/MPC). Cell killing was quantified at 4 h. D, left: IPF MPCs were cocultured with NK92 cells in a 3-to-1 ratio (NK/MPC) in the presence of various concentrations of anti-PD-L1 antibody. Right: IPF MPCs were cocultured with various ratios of NK cells (0:1, 3:1, 5:1, 10:1; NK/MPC) in the presence of PD-L1 antibody (2 µg/mL). Cell killing was quantified at 4 h. The experiment was performed in triplicate. E: IPF or control MPCs (n = 6 cell lines each) were cocultured with NK92 cells in a 3-to-1 ratio (NK/MPC) in the presence of PD-L1 (2 µg/mL) or isotype control (Con) antibody. Cell killing was quantified at 4 h. F: IPF MPCs were pretreated with IL-8 (5 ng) or vehicle control and cocultured with NK cells. Cell killing was quantified at 4 h. G: IPF MPCs pretreated with IL-8 (5 ng) were cocultured with NK cells in the presence of PD-L1 or isotype control antibody (Con AB). Cell killing was quantified at 4 h. The experiment was replicated with 4 IPF MPC cell lines in B, C, F, and G.
Disruption of PD-1-PD-L1 Interaction Arrests IPF MPC-Mediated Experimental Lung Fibrosis in Vivo
We have developed a xenograft mouse model of sustained pulmonary fibrosis in which treatment of NSG mice with intratracheal bleomycin followed by adoptive transfer of IPF MPCs promotes durable interstitial fibrosis (5). To analyze the role of PD-1-PD-L1 interaction in enabling IPF MPCs to elude immune surveillance and promote sustained lung fibrosis, we modified this model (Fig. 8A). The NSG mice were first treated with low-dose intratracheal bleomycin to create self-limited lung injury. Seven days after administration of bleomycin, we irradiated the mice to eliminate all immune cells. One week after irradiation, we administered human IPF MPCs via tail vein injection. On day 3 and day 13 after IPF MPC administration, we delivered NK cells and anti-PD-L1 blocking antibody or isotype control antibody to the NSG mice by peritoneal injection. Irradiated mice treated with bleomycin only or IPF MPCs + anti-PD-L1 antibody or control antibody without NK cells served as additional controls. Four weeks after administration of IPF MPCs, the mice were euthanized and analyzed for lung fibrosis by Sircol assay and by immunohistochemical analysis. We found that mice receiving IPF MPCs and NK cells and treated with an anti-PD-L1 antibody have markedly reduced fibrosis compared with mice that received IPF MPCs and NK cells but were treated with control antibody. Lung collagen content as measured by Sircol assay was reduced by 67% in mice receiving anti-PD-L1 antibody (764.6 ± 35.4 µg/left lung in control groups vs. 252.8 ± 21.7 µg/left lung in anti-PD-L1 groups) (Fig. 8B). Semiquantitative analysis of lung collagen deposition by trichrome staining demonstrated a 68% decrease in lung collagen deposition in mice receiving anti-PD-L1 antibody (Fig. 8C). H&E and trichrome staining demonstrated more extensive fibrosis with corresponding collagen deposition in the lungs of mice treated with 1) IPF MPCs + control antibody (no NK cells) (Fig. 8, D and H); IPF MPCs + PD-L1 antibody (no NK cells) (Fig. 8, E and I); and IPF MPCs + NK cells + control antibody (Fig. 8, F and J) compared with mice receiving IPF MPCs + NK cells + anti-PD-L1 blocking antibody (Fig. 8, G and K). Consistent with this, IHC using an antibody recognizing human procollagen I showed heavy infiltration of human IPF MPCs expressing procollagen I colocalizing with regions of new collagen deposition as determined by trichrome staining in the lungs of mice receiving 1) IPF MPCs + control antibody (no NK cells) (Fig. 8, H and L); 2) IPF MPCs + PD-L1 antibody (no NK cells) (Fig. 8, I and M); and IPF MPCs + NK cells + control antibody (Fig. 8, J and N). In stark contrast, the lungs of mice treated with both NK cells and PD-L1 blocking antibody displayed few human cells expressing procollagen together with a paucity of new collagen deposition (Fig. 8, K and O). To further assess the number of human cells in mice receiving IPF MPCs and treated with PD-L1 or control antibody, we quantified human cell numbers in the lungs of the mice at the time of tissue harvest, using a real-time PCR method shown to be sensitive for the detection and quantification of human cells in mice (35). The lungs of mice receiving IPF MPCs and treated with NK cells and anti-PD-L1 antibody contained 52% lower levels of human DNA compared with mice that received IPF MPCs and were treated with NK cells and control antibody (Fig. 8P). Together, these data indicate that disruption of PD-1-PD-L1 interaction eliminates the ability of IPF MPCs to elude NK cell-mediated killing and abrogates the ability of IPF MPCs to promote sustained lung fibrosis in vivo.
Figure 8.

Disruption of programmed cell death protein-1 (PD-1)-programmed death ligand 1 (PD-L1) interaction arrests idiopathic pulmonary fibrosis (IPF) mesenchymal progenitor cell (MPC)-mediated experimental lung fibrosis in vivo. A: schematic of xenograft mouse model protocol. NOD/SCID/IL2rγ/B2M (NSG) mice were treated with intratracheal bleomycin (Bleo; 1.25 U/kg). One week later the mice were irradiated. Two weeks after bleomycin, IPF MPCs were administered via tail vein injection (106 cells/100 µL). At days 3 and 13 after IPF MPC administration, the mice received NK92 cells (10 × 106 cells) and either PD-L1 (2 mg/kg) or isotype control antibody (AB) by peritoneal injection. Mice receiving IPF MPCs and treated with PD-L1 or control antibody in the absence of NK cells served as controls. Mice treated with bleomycin and radiation only served to establish baseline collagen content in the absence of additional treatments. There were 10 mice/group. Tissue harvest occurred 4 wk after MPC administration. The experiment was replicated with IPF MPCs derived from 3 separate IPF patients. B: collagen content was quantified in left lungs by Sircol assay. C: semiquantitative analysis of lung collagen deposition by trichrome staining with ImageJ. D–O: representative hematoxylin and eosin (H&E) (D–G) and trichrome (H–K) stains assessing fibrosis and collagen deposition, respectively, and immunohistochemistry (IHC) using an antibody recognizing human procollagen to identify human cells and assess collagen synthesis (L–O). P: human IPF cell numbers were quantified by quantitative PCR (Q-PCR).
DISCUSSION
DNA damage caused by replicative stress can induce senescence (29–32, 36–38). In prior work we have found that IL-8 drives IPF MPC self-renewal (9). IL-8 can lead to genomic instability by promoting replicative stress and formation of double-stranded DNA breaks, and IL-8 signaling through the CXCR2 receptor can induce senescence (12). This prompted us to analyze IPF MPCs for markers of replicative stress and DNA damage. As markers of replicative stress and DNA damage we quantified γH2AX, phosphorylated (p)RPA2, and p21 levels in IPF and control MPCs. γH2AX, pRPA2, and p21 levels were increased in IPF MPCs compared with control, and IL-8 increased expression of these markers, supporting the concept that IPF MPCs have sustained replicative stress and associated DNA damage.
Work in cancer connects senescence with cancer progression (39–41). Cellular senescence has been linked to IPF fibrotic progression, and targeted removal of senescent cells may be a means to limit progressive fibrosis (13–18). Studies employing experimental models of liver fibrosis indicate that senescence of activated hepatic stellate cells, a precursor cell to myofibroblasts, has been associated with progression of liver fibrosis (42). Importantly, rapid clearance of senescent hepatic stellate cells by a functional innate immune response arrested experimental liver fibrosis (42). We have found that IPF MPCs have sustained significant DNA damage and can become senescent, a process magnified by IL-8. These data support the concept that IPF MPC senescence may drive fibrotic progression.
Senescent cells can secrete CXCR2 ligands, which coordinately induce CXCR2 expression, forming a self-amplifying loop whereby CXCR2-binding chemokines reinforce senescence (12). The secretion of chemokines by senescent cells is part of the SASP. Our data indicate that in response to IL-8 IPF MPCs exhibit high levels of CXCR2 and display increased secretion of chemokines consistent with acquisition of a SASP. IL-8 interaction with either CXCR1 or CXCR2 can regulate a variety of cellular functions and appears to be cell type specific (43). Although both CXCR1 and CXCR2 serve as receptors for IL-8 and are expressed in IPF MPCs, here we show that IL-8 interaction with CXCR2 can induce senescence in IPF MPCs.
Programmed cell death protein-1 (PD-1), an immune checkpoint receptor expressed on the cell surface of immune cells, plays an important role in immune regulation (19). In normal cells, the ligation of PD-1/PD-L1 promotes immune cell tolerance and escape from host immunity (19). When PD-L1 is expressed by cancer cells, it can engage the immune checkpoint PD-1/PD-L1 axis to escape an antitumor immune response (19–22). We found that IPF MPCs display high levels of PD-L1 and that IL-8 simultaneously induces IPF MPC senescence and upregulation of PD-L1. High levels of PD-L1 may enable senescent IPF MPCs to escape immune cell killing. In support of this, our in vitro data demonstrate that elevated PD-L1 levels lead to impaired clearance of IPF MPCs by NK cells. Disruption of PD-1-PD-L1 interaction markedly enhances the elimination of IPF MPCs by NK cells. Control MPCs, which do not display significant DNA damage or senescence, are not targeted for removal by NK cells, despite showing low levels of PD-L1. It has been determined that, in addition to alterations in PD-L1 expression, changes in HLA class I molecules are important in determining the outcome of immune cell surveillance, suggesting that control MPCs may display a repertoire of PD-L1 and HLA class I molecules that serve as a negative targeting signal for immune cells.
Our data provide a paradigm of IPF fibrotic progression, where senescent IPF MPCs at the periphery of the fibroblastic focus serve as a fibrotic niche. This paradigm is supported by our in vivo studies and by our IHC analysis. Using a modified version of our mouse xenograft model in which administration of intrinsically fibrogenic human IPF MPCs in immune-deficient mice pretreated with low dose of IT bleomycin creates sustained interstitial lung fibrosis (5), we demonstrate that mice receiving NK cells and anti-PD-L1 antibody had significantly reduced lung fibrosis with few human cells expressing human procollagen I. In contrast, the mice receiving NK cells and isotype control antibody had increased fibrosis, and numerous human cells expressing procollagen were present in fibrotic regions. Although it is not possible to demonstrate NK cell elimination of human IPF cells in the mouse lung, we used the number of human cells in the mouse lungs as a surrogate marker of NK cell clearance. The paucity of human IPF cells in the mice treated with both NK cells and anti-PD-L1 antibody provides evidence that human IPF MPCs had been eliminated by NK cells. These data support the concept that disruption of PD-1-PD-L1 interaction leads to NK-mediated killing of IPF MPCs and arrest of experimental pulmonary fibrosis.
Our IHC analysis of human IPF lung tissue identified PD-L1-expressing MPCs at the periphery of the fibroblastic focus colocalizing with immune cells including NK cells, T cells, and macrophages (9). The persistence of PD-L1-expressing IPF MPCs in regions of active fibrogenesis near immune cells further supports the concept that they elude immune cell surveillance and participate in fibrotic progression. The concept that high PD-L1 expression on mesenchymal cells is involved in fibrosis is supported by previous studies showing that PD-L1 expression was required for an invasive fibroblast phenotype and fibroblast-to-myofibroblast transdifferentiation (44, 45). Whether IPF MPCs elude immune surveillance solely via upregulation of PD-L1 or there is concomitant immune cell dysfunction is unclear. In this regard, several studies implicate NK cell dysfunction with IPF progression (35, 46).
Our data indicate that IL-8 promotes IPF MPC DNA damage-induced senescence and high PD-L1 expression, enabling IPF MPCs to elude immune cell elimination. Disruption of the PD-1/PD-L1 axis led to NK killing of IPF MPCs in vitro and the attenuation of experimental lung fibrosis in vivo. Disruption of PD-1-PD-L1 interaction may be a means to limit IPF MPC-mediated fibrotic progression.
DATA AVAILABILITY
Data will be made available upon reasonable request.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute (NHLBI) Grants R01 HL125227 (to C. A. Henke) and R01 HL125236 (to P. B. Bitterman) and by funds provided by the O’Brien family.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.Y. and C.A.H. conceived and designed research; L.Y., H.X., A.G., K.S., and E.R. performed experiments; L.Y., H.X., A.G., K.S., and E.R. analyzed data; L.Y., H.X., P.B.B., and C.A.H. interpreted results of experiments; L.Y., H.X., and A.G. prepared figures; L.Y. and C.A.H. drafted manuscript; L.Y., P.B.B., and C.A.H. edited and revised manuscript; L.Y. and C.A.H. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge the assistance of the Flow Cytometry Core Facility of the Masonic Cancer Center, a comprehensive cancer center designated by the National Cancer Institute (NCI), supported in part by NCI Grant P30 CA77598 and the University of Minnesota Imaging Center. We also acknowledge the Center for Mass Spectrometry and Proteomics.
REFERENCES
- 1. Noble PW. Idiopathic pulmonary fibrosis: natural history and prognosis. Clin Chest Med 27: S11–S16, 2006. doi: 10.1016/j.ccm.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 2. Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 174: 810–816, 2006. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 3. Raghu G, Selman M. Nintedanib and pirfenidone. New antifibrotic treatments indicated for idiopathic pulmonary fibrosis offer hopes and raises questions. Am J Respir Crit Care Med 191: 252–254, 2015. doi: 10.1164/rccm.201411-2044ED. [DOI] [PubMed] [Google Scholar]
- 4. Xia H, Bodempudi V, Benyumov A, Hergert P, Tank D, Herrera J, Braziunas J, Larsson O, Parker M, Rossi D, Smith K, Peterson M, Limper A, Jessurun J, Connett J, Ingbar D, Phan S, Bitterman PB, Henke CA. Identification of a cell-of-origin for fibroblasts comprising the fibrotic reticulum in idiopathic pulmonary fibrosis. Am J Pathol 184: 1369–1383, 2014. doi: 10.1016/j.ajpath.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xia H, Gilbertsen A, Herrera J, Racila E, Smith K, Peterson M, Griffin T, Benyumov A, Yang L, Bitterman PB, Henke CA. Calcium-binding protein S100A4 confers mesenchymal progenitor cell fibrogenicity in idiopathic pulmonary fibrosis. J Clin Invest 127: 2586–2597, 2017. doi: 10.1172/JCI90832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Parker MW, Rossi D, Peterson M, Smith K, Sikström K, White ES, Connett JE, Henke CA, Larsson O, Bitterman PB. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest 124: 1622–1635, 2014. doi: 10.1172/JCI71386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Herrera J, Beisang DJ, Peterson M, Forster C, Gilbertsen A, Benyumov A, Smith K, Korenczuk CE, Barocas VH, Guenther K, Hite R, Zhang L, Henke CA, Bitterman PB. Dicer1 deficiency in the IPF fibroblastic focus promotes fibrosis by suppressing microRNA biogenesis. Am J Respir Crit Care Med 198: 486–496, 2018. doi: 10.1164/rccm.201709-1823OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Herrera J, Forster C, Pengo T, Montero A, Swift J, Schwartz MA, Henke CA, Bitterman PB. Registration of the extracellular matrix components constituting the fibroblastic focus in idiopathic pulmonary fibrosis. JCI Insight 4: e125185, 2019. doi: 10.1172/jci.insight.125185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang L, Herrera J, Gilbertsen AJ, Xia H, Smith K, Benyumov A, Bitterman PB, Henke CA. IL-8 mediates idiopathic pulmonary fibrosis mesenchymal progenitor cell fibrogenicity. Am J Physiol Lung Cell Mol Physiol 314: L127–L136, 2018. doi: 10.1152/ajplung.00200.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beisang DJ, Smith K, Yang L, Benyumov A, Gilbertsen A, Herrera J, Lock E, Racila E, Forster C, Sandri BJ, Henke CA, Bitterman PB. Single cell sequencing reveals that lung mesenchymal progenitor cells in IPF exhibit pathological features early in their differentiation trajectory. Sci Rep 10: 11162, 2020. doi: 10.1038/s41598-020-66630-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang L, Gilbertsen A, Smith K, Xia H, Higgins L, Guerrero C, Henke CA. Proteomic and interactomic analysis of the IPF MPC proteome identifies abnormalities in key nodal proteins that underlie their fibrogenic phenotype. Proteomics 22: e2200018, 2022. doi: 10.1002/pmic.202200018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d’Adda di Fagagna F, Bernard D, Hernando E, Gil J. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133: 1006–1018, 2008. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 13. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, Pirtskhalava T, Prakash YS, Tchkonia T, Robbins PD, Aubry MC, Passos JF, Kirkland JL, Tschumperlin DJ, Kita H, LeBrasseur NK. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8: 14532, 2017. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Waters DW, Blokland KE, Pathinayake PS, Burgess JK, Mutsaers SE, Prele CM, Schuliga M, Grainge CL, Knight DA. Fibroblast senescence in the pathology of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 315: L162–L172, 2018. doi: 10.1152/ajplung.00037.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yao C, Guan X, Carraro G, Parimon T, Liu X, Huang G, Mulay A, Soukiasian HJ, David G, Weigt SS, Belperio JA, Chen P, Jiang D, Noble PW, Stripp BR. Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis. Am J Respir Crit Care Med 203: 707–717, 2021. doi: 10.1164/rccm.202004-1274OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Monkley S, Overed-Sayer C, Parfrey H, Rassl D, Crowther D, Escudero-Ibarz L, Davis N, Carruthers A, Berks R, Coetzee M, Kolosionek E, Karlsson M, Griffin LR, Clausen M, Belfield G, Hogaboam CM, Murray LA. Sensitization of the UPR by loss of PPP1R15A promotes fibrosis and senescence in IPF. Sci Rep 11: 21584, 2021. doi: 10.1038/s41598-021-00769-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, Chu SG, Raby BA, DeIuliis G, Januszyk M, Duan Q, Arnett HA, Siddiqui A, Washko GR, Homer R, Yan X, Rosas IO, Kaminski N. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv 6: eaba1983, 2020. doi: 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Álvarez D, Cárdenes N, Sellarés J, Bueno M, Corey C, Hanumanthu VS, Peng Y, D’Cunha H, Sembrat J, Nouraie M, Shanker S, Caufield C, Shiva S, Armanios M, Mora AL, Rojas M. IPF lung fibroblasts have a senescent phenotype. Am J Physiol Lung Cell Mol Physiol 313: L1164–L1173, 2017. doi: 10.1152/ajplung.00220.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Salmaninejad A, Valilou SF, Shabgah AG, Aslani S, Alimardani M, Pasdar A, Sahebkar A. PD-1/PD-L1 pathway: Basic biology and role in cancer immunotherapy. J Cell Physiol 234: 16824–16837, 2019. doi: 10.1002/jcp.28358. [DOI] [PubMed] [Google Scholar]
- 20. Shen X, Zhang L, Li J, Li Y, Wang Y, Xu ZX. Recent findings in the regulation of programmed death ligand 1 expression. Front Immunol 10: 1337, 2019. doi: 10.3389/fimmu.2019.01337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol 18: 153–167, 2018. doi: 10.1038/nri.2017.108. [DOI] [PubMed] [Google Scholar]
- 22. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity 48: 434–452, 2018. doi: 10.1016/j.immuni.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am. J. Respir. Crit. Care Med 161: 646–664, 2020. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- 24. Xia H, Khalil W, Kahm J, Jessurun J, Kleidon J, Henke CA. Pathologic caveolin-1 regulation of PTEN in idiopathic pulmonary fibrosis. Am J Pathol 176: 2626–2637, 2010. doi: 10.2353/ajpath.2010.091117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aivaliotis IL, Pateras IS, Papaioannou M, Glytsou C, Kontzoglou K, Johnson EO, Zoumpourlis V. How do cytokines trigger genomic instability. J Biomed Biotechnol 2012: 536761, 2012. doi: 10.1155/2012/536761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mah LJ, El-Osta A, Karagiannis TC. γH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia 24: 679–686, 2010. doi: 10.1038/leu.2010.6. [DOI] [PubMed] [Google Scholar]
- 27. Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282: 1497–1501, 1998. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 28. Macleod KF, Sherry N, Hannon G, Beach D, Tokino T, Kinzler K, Vogelstein B, Jacks T. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev 9: 935–944, 1995. doi: 10.1101/gad.9.8.935. [DOI] [PubMed] [Google Scholar]
- 29. Collado M, Blasco MA, Serrano KM. Cellular senescence in cancer and aging. Cell 130: 223–233, 2007. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 30. Minieri V, Saviozzi S, Gambarotta G, Lo Iacono M, Accomasso L, Cibrario Rocchietti E, Gallina C, Turinetto V, Giachino C. Persistent DNA damage-induced premature senescence alters the functional features of human bone marrow mesenchymal stem cells. J Cell Mol Med 19: 734–743, 2015. doi: 10.1111/jcmm.12387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sperka T, Wang J, Rudolph KL. DNA damage checkpoints in stem cells, ageing and cancer. Nat Rev Mol Cell Biol 13: 579–590, 2012. doi: 10.1038/nrm3420. [DOI] [PubMed] [Google Scholar]
- 32. Erol A. Deciphering the intricate regulatory mechanisms for the cellular choice between cell repair, apoptosis or senescence in response to damaging signals. Cell Signal 23: 1076–1081, 2011. doi: 10.1016/j.cellsig.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 33. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, Azimi CS, Scheer AK, Randolph HE, Thompson TW, Zhang L, Iannello A, Mathur N, Jardine KE, Kirn GA, Bell JC, McBurney MW, Raulet DH, Ardolino M. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest 128: 4654–4668, 2018. doi: 10.1172/JCI99317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 8: 652–658, 1994. [PubMed] [Google Scholar]
- 35. Huang Y, Oldham JM, Ma SF, Unterman A, Liao SY, Barros AJ, Bonham CA, Kim JS, Vij R, Adegunsoye A, Strek ME, Molyneaux PL, Maher TM, Herazo-Maya JD, Kaminski N, Moore BB, Martinez FJ, Noth I. Blood transcriptomics predicts progression of pulmonary fibrosis and associated natural killer cells. Am J Respir Crit Care Med 204: 197–208, 2021. doi: 10.1164/rccm.202008-3093OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res 62: 1876–1883, 2002. [PubMed] [Google Scholar]
- 37. Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, Takaoka M, Nakagawa H, Tort F, Fugger K, Johansson F, Sehested M, Andersen CL, Dyrskjot L, Ørntoft T, Lukas J, Kittas C, Helleday T, Halazonetis TD, Bartek J, Gorgoulis VG. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444: 633–637, 2006. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 38. Mirzayans R, Andrais B, Hansen G, Murray D. Role of p16(INK4A) in replicative senescence and DNA damage-induced premature senescence in p53-deficient human cells. Biochem Res Int 2012: 951574, 2012. doi: 10.1155/2012/951574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mavrogonatou E, Pratsinis H, Kletsas D. The role of senescence in cancer development. Semin Cancer Biol 62: 182–191, 2020. doi: 10.1016/j.semcancer.2019.06.018. [DOI] [PubMed] [Google Scholar]
- 40. Lecot P, Alimirah F, Desprez PY, Campisi J, Wiley C. Context-dependent effects of cellular senescence in cancer development. Br J Cancer 114: 1180–1184, 2016. doi: 10.1038/bjc.2016.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hernandez C, Huebener P, Schwabe RF. Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene 35: 5931–5941, 2016. doi: 10.1038/onc.2016.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell 134: 657–667, 2008. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bi H, Zhang Y, Wang S, Fang W, He W, Yin L, Xue Y, Cheng Z, Yang M, Shen J. Interleukin-8 promotes cell migration via CXCR1 and CXCR2 in liver cancer. Oncol Lett 18: 4176–4184, 2019. doi: 10.3892/ol.2019.10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Geng Y, Liu X, Liang J, Habiel DM, Kulur V, Coelho AL, Deng N, Xie T, Wang Y, Liu N, Huang G, Kurkciyan A, Liu Z, Tang J, Hogaboam CM, Jiang D, Noble PW. PD-L1 on invasive fibroblasts drives fibrosis in a humanized model of idiopathic pulmonary fibrosis. JCI Insight 4: e125326, 2019. doi: 10.1172/jci.insight.125326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guo X, Sunil C, Adeyanju O, Parker A, Huang S, Ikebe M, Tucker TA, Idell S, Qian G. PD-L1 mediates lung fibroblast to myofibroblast transition through Smad3 and β-catenin signaling pathways. Sci Rep 12: 3053, 2022. doi: 10.1038/s41598-022-07044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aquino-Galvez A, Pérez-Rodríguez M, Camarena A, Falfan-Valencia R, Ruiz V, Montaño M, Barrera L, Sada-Ovalle I, Ramirez R, Granados J, Pardo A, Selman M. MICA polymorphisms and decreased expression of the MICA receptor NKG2D contribute to idiopathic pulmonary fibrosis susceptibility. Hum Genet 125: 639–648, 2009. doi: 10.1007/s00439-009-0666-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available upon reasonable request.