

Abstract
Background:
Cleft palate is one of the most prevalent birth defects. Mice are useful for studying palate development because of their morphological and genetic similarities to humans. In mice, palate development occurs between embryonic days (E)11.5 to 15.5. Single cell transcriptional profiles of palate cell populations have been a valuable resource for the craniofacial research community, but we lack a single cell transcriptional profile for anterior palate at E13.5, at the transition from proliferation to shelf elevation.
Results:
A detailed single cell RNA sequencing analysis reveals heterogeneity in expression profiles of the cell populations of the E13.5 anterior palate. Hybridization chain reaction RNA fluorescent in situ hybridization (HCR RNA FISH) reveals epithelial populations segregate into layers. Mesenchymal populations spatially segregate into four domains. One of these mesenchymal populations expresses ligands and receptors distinct from the rest of the mesenchyme, suggesting that these cells have a unique function. RNAVelocity analysis shows two terminal cell states that contribute to either the proximal or distal palatal regions emerge from a single progenitor pool.
Conclusion:
This single cell resolution expression data and detailed analysis from E13.5 anterior palate provides a powerful resource for mechanistic insight into secondary palate morphogenesis for the craniofacial research community.
Introduction
Cleft palate is one of the most prevalent birth defects affecting 1 in every 1,700 babies resulting from a failure of fusion of the palatal shelves during embryonic development1. Perturbation of over three hundred genes leads to cleft palate2–4. Exposure to various pharmacological medications, nicotine, or alcohol increases incidence of cleft palate suggesting that palate development is especially susceptible to genetic and environmental insult5–11. Children with cleft palate have difficulty with feeding, speech, and hearing12. Surgical intervention is costly and places a large financial burden on affected families13. Palatal development is a highly conserved process among mammals14. Mice have traditionally been used as a model for studying palate closure. In mice, this process occurs between embryonic day (E)11.5 to E15.515–17. The secondary palate originates as paired outgrowths from either oral side of the developing maxillary prominences on E11.516,17. The palate undergoes a period of vertical outgrowth from E11.5 to E13.5, increasing greatly in size due to high rates of cellular proliferation18. At E14.0, the palatal shelves elevate above the tongue, grow horizontally at E14.5, adhere at E15.0, and fuse by E15.516. The precise molecular mechanisms controlling these transitions have not been completely characterized. Specifically, the transition from a period of high proliferation at E13.5 to a dynamic elevation above the tongue at E14.0 is particularly understudied.
Microarrays and bulk RNA sequencing can provide important information about gene expression allowing for novel pathway identification and hypothesis development. Previous work within the palate has utilized both methods to gain more information about the developmental landscape of palatogenesis. Affymetrix GeneChip microarrays have provided transcriptional profiles of whole palates at E13.5, E14.5, and E15.519. Laser capture microdissection combined with RNA-seq has allowed for more spatial compartmentalization. Researchers have used this technique to analyze differences in medial versus lateral and oral versus nasal compartments in E14.5 palatal shelves20. However, these techniques still do not delineate gene expression differences among cells within a population or compartment. With the advent of single cell RNA sequencing (scRNA seq), we can obtain a more accurate transcriptional profile of different populations of cells that would reveal any heterogeneity. As a result, we now have single cell developmental atlases of many key timepoints in craniofacial development including upper lip and primary palate fusion21, midfacial morphogenesis22, soft palate myogenesis23, tooth development24, mandibular development25,26, and cranial suture formation27–29. Although we now have bulk RNA sequencing of fused palatal shelves, scRNA-seq data from palatal precursor populations, posterior palate, and Shox2+ E13.5 palatal mesenchyme20–23,30, we do not have a complete atlas at a single cell resolution for the E13.5 anterior palate which would greatly increase our understanding of the complex gene regulatory processes at work.
Mesenchymal and epithelial heterogeneity has not been explored at this key timepoint prior to palatal elevation and horizontal outgrowth. Previous methods have not distinguished whether anterior palate marker genes such as Msx1 and Shox2 for example, which are used to delineate this population from the middle and posterior mesenchyme31, are co-expressed among the same cells or occupy distinct cell populations. Gene summaries for both genes use the same language: “Msx1/Shox2 is expressed in the mesenchyme of the anterior region of the developing palate, in both nasal and oral sides”32,33. Similarly, most genes described with anterior palate expression are not fully parsed apart. scRNA seq sequencing allows us to look at the expression of these genes at a higher resolution than previous technologies have allowed. scRNA seq data further allows for various computational pipelines to be employed giving insights into ligand-receptor interactions, predicted transcription factor activity, and pseudotime analyses.
Here, we show unexpected complexity and heterogeneity among the mesenchymal cells of the E13.5 anterior palate. Using scRNA seq, we cluster the anterior palatal mesenchyme into four populations based on gene expression profiles. Hybridization chain reaction (HCR) confirms these populations and reveals spatial segregation into four domains: oral, lingual, proximal, and central. We find that anterior palate genes Msx1 and Shox2 mark distinct and transcriptionally separable populations within the anterior palate. Msx1 spans the proximal and central mesenchyme while Shox2 is restricted to the distal oral and lingual mesenchyme. Using cell chat, we identify a mesenchymal subpopulation (Mes_3/proximal) with distinct signaling profiles from the remaining mesenchyme. RNAVelocity analysis shows the emergence of two terminal cell states from a single progenitor state within the developing palate contributing to either the proximal or distal palatal regions.
Results
Workflow and cluster identification of single cell RNA sequencing of the E13.5 anterior palate
We performed scRNA seq on E13.5 WT anterior palates (Fig. 1A) to investigate cell populations comprising the anterior palate at this key timepoint. Anterior palates were collected while leaving posterior palate tissue intact (Fig. 1A, S1A–B). After filtering and quality assessment of data, we kept 22,238 cells from three embryos for downstream analysis. Sample preparations were labeled as wildtype (WT)_1, WT_2, and WT_3 and yielded 7959, 6994, and 7268 cells respectively. Single cell population data is visualized in a Uniform Manifold Approximation and Projection (UMAP) wherein each individual dot represents a cell and shorter distances between dots constitute more similar transcriptomes between cells. Our sequencing produced six distinct clusters of cells (Fig. 1B). Based on gene expression (Fig. S2A–F), cell clusters were broadly defined as mesenchyme (Col1a2, Col3a1, Col1a1 expressing)34–38, proliferating (Top2a, Hist1h3c, Hist1h1b expressing)39,40, epithelial (Epcam, Igfbp5, Pdgfa expressing)21,41–43, endothelial (Pecam1, Plvap, Cdh5 expressing)44–46, immune (Fcer1g, Tyrobp, Cd52 expressing)47–49, and Schwann cells (Fabp7, Mpz, Sox10 expressing)50–53 (Fig. 1C). The most prominent cell types were mesenchyme and proliferating mesenchyme cells followed by epithelial cells (Fig. 1B, S2A–C). The endothelial, immune, and Schwann cell populations represent only a small fraction of the total cells (Fig. 1B, S2A–C). While expected cell types were easily identified by gene expression, we hypothesized that higher resolution thresholding may reveal differences within cell populations that have not been previously identified.
Figure 1:
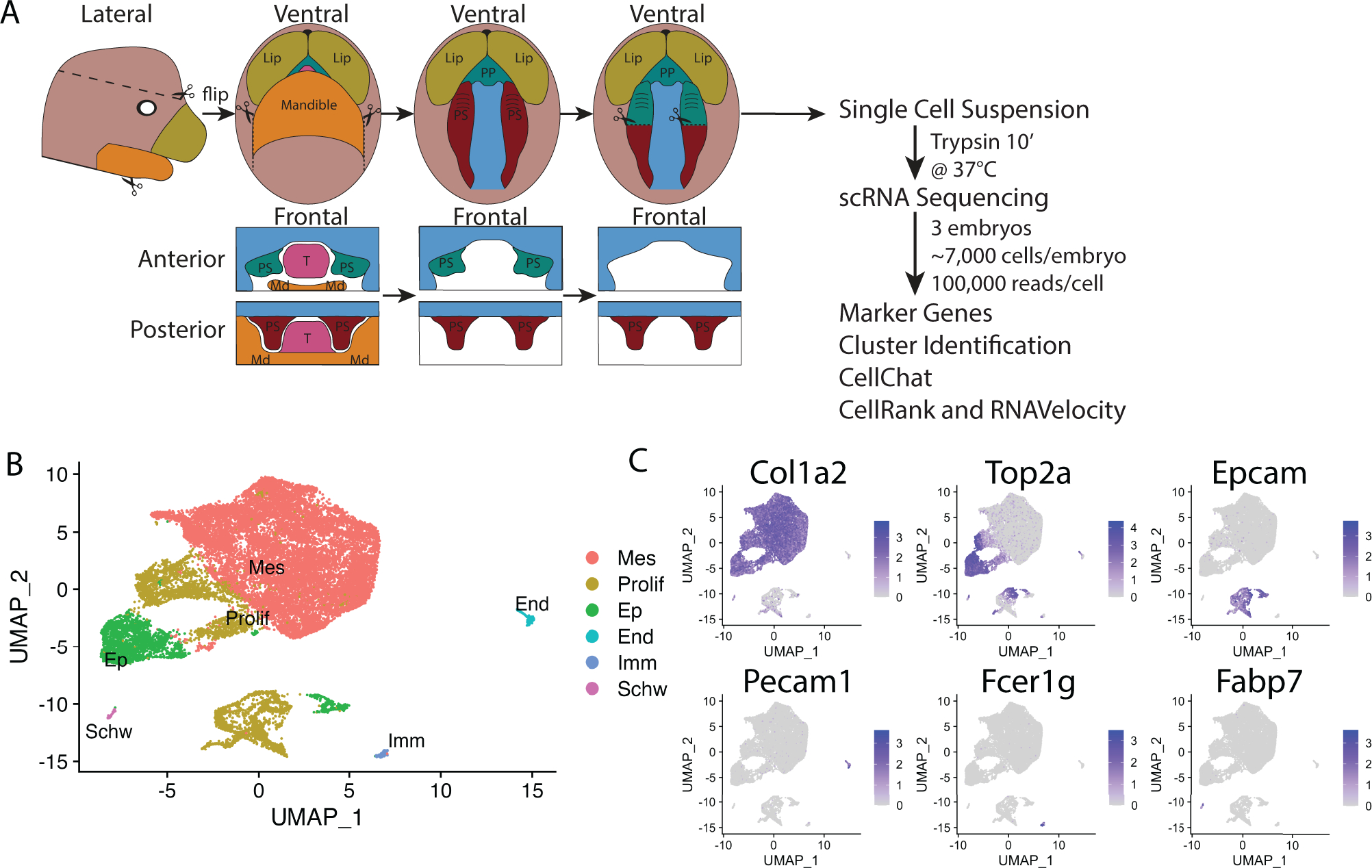
A) A schematic illustrates palate dissection and experimental workflow. Teal marks anterior palate and red marks middle and posterior palate. Schematized anterior and posterior frontal sections show boundaries of removed palatal tissue. B) A UMAP shows six cell populations recovered from scRNA sequencing. C) FeaturePlots show gene expression of prominent markers of each of the six cell populations. PS=Palatal Shelf, PP=Primary Palate, Mx=Maxilla, Md=Mandible, T=Tongue.
Single cell RNA sequencing reveals heterogeneity within the E13.5 anterior palate
Cell heterogeneity among the cells within the embryonic palate remains poorly understood. To determine if cells within the E13.5 anterior palate are a diverse population, we subclustured three main populations: mesenchyme, proliferating mesenchyme, and epithelial cells at a resolution of 0.2 shared nearest neighbor (snn) within the Seurat pipeline. This produced three proliferating clusters, two epithelial clusters, and four mesenchymal clusters (Fig. 2A–B). Cell cluster proportionality was consistent between sample preparations with all expected cell types appearing within each sample in expected ratios (Fig. S3A–B). Top 10 markers of each of these subclusters are distinct showing clear segregation of cluster identities (Fig. 2C, S4A–I). Top 15 markers of each cluster are listed in Table 1. We chose one-two distinct markers per cluster to determine spatial localization of cell populations within anterior palate tissue (Fig. 2B). Subclustering of proliferating cells demonstrated various stages of the cell cycle. Prolif_1 is marked by conserved histone H3 variant Cenpa, which determines kinetochore position (Fig. 2B–C, S4A, 3A). Prolif_2 is characterized by high levels of histones such as Hist1h3c and Hist1h1b indicating condensed chromatin (Fig. 2B–C, S4B, 3B). Prolif_3 is marked by Ccne2 which regulates G1-S progression (Fig. 2B–C, S4C, 3C). Epithelial cells cluster into two similar, but distinct groups. The top genes of Ep_1 are also highly expressed within Ep_2 with the exception of a few genes such as follistatin (Fst), insulin like growth factor binding protein 2 (Igfbp2), and transformation related protein 63 (Trp63) (Fig. 2B–C, S4D, 3D–E). Ep_2 remains more transcriptionally distinct from Ep_1 through strong expression of several keratin (Krt) genes (Krt8, Krt15, Krt17, Krt18, Krt19) (Fig. 2B–C, S4E, 3F–G).
Figure 2:
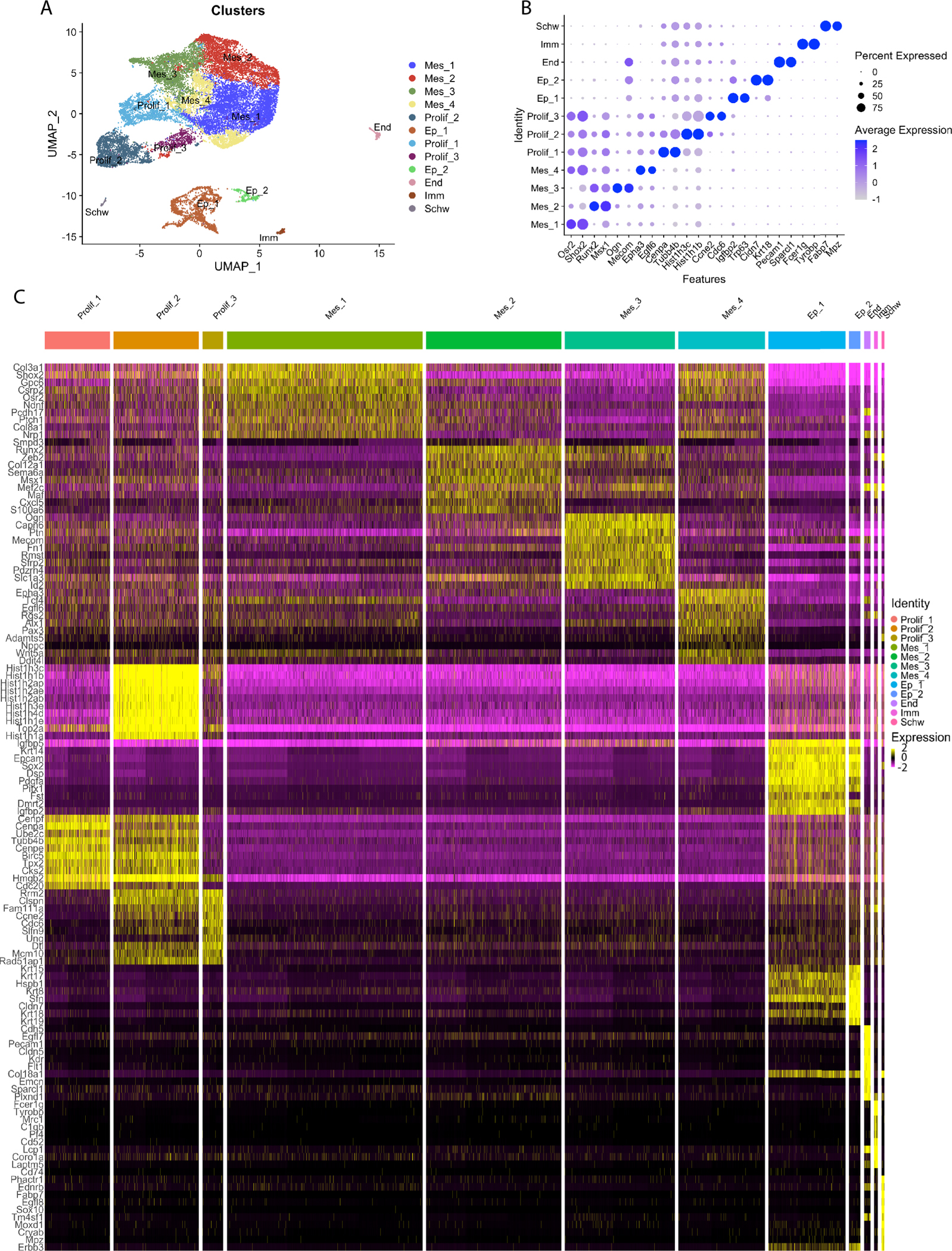
A) A UMAP of six cell populations shows subclusters of Mesenchyme, Epithelial, and Proliferating populations. B) A DotPlot shows two highly expressing genes for each subcluster. C) A heatmap of top 10 marker genes sorted by p-value per cluster shows that each cluster cell population has a distinct set of highly expressed genes. Each row represents a single gene, and each column is a single cell. Cells are grouped based on clusters, shown at the top. Prolif_1 is pink. Prolif_2 is orange. Prolif_3 is mustard yellow, Mes_1–3 are different shades of green. Mes_4 is turquoise. Ep1–2 are different shades of blue. Endothelial cells are purple. Immune and Schwann cells are bright pink. Purple delineates low expression and yellow high expression.
Table 1:
Top 15 markers genes of each cluster
| Mes_1 | Mes_2 | Mes_3 | Mes_4 | ProlifJ | Prolif_2 |
| Col3a1, Shox2, Gpc6, Csrp2, Osr2, Ndnf, Pcdh17, Ptch1, Col8a1, Nrp1, Cped1, Peg10, Meis2, Twist1, Zfhx4 | Smpd3, Runx2, Zeb2, Col12a1, Sema6a, Msx1, Mef2c, Mat, Cxcl5, S100a6, Rorb, Npnt, Emp3, Cd200, Pou3f3 | Ogn, Capn6, Ptn, Mecom, Fn1, Rmst, Sfrp2, Pdzm4, Slc1a3, Id2, Tpm1, Cdknlc, Gm26771, Rspo3, Tsc22d1 | Epha3, Tcf4, Egft6, Rgs2, Alx1, Pax3, Adamts5, Nppc, Wnt5a, Ddit4l, Prg4, Mfap4, Meisl, Crabp2, Limch1 | Cenpf, Cenpa, Ube2c, Tubb4b, Cenpe, Birc5, Tpx2, Cks2, Hmgb2, Cdc20, Ccnb2, Mki67, Nusap1, Ccnb1, Cdca8 | Hist1h3c, Hist1h1b, Hist1h2ap, Hist1h2ae, Hist1h2ab, Hist1h3e, Hist1h4d, Hist1h1e, Top2a, Hist1h1a, Hist2h2ac, Hist1h1d, Hist1h4h, Hist1h1c, 2810417H113Rik |
| Prolif_3 | Ep_1 | Ep_2 | End | Imm | Schw |
| Rrm2, Clspn, Fam111a, Ccne2, Cdc6, Slfn9, Ung, Dtl, Mem 10, Rad51ap1, Dhfr, Uhrf1, Lig1, Gmnn, Hells | Igfbp5, Krt14, Epcam, Sox2, Dsp, Pdgfa, Pitx1, Fst, Dmrt2, Igfbp2, Sival, Dapl1, Sema3d, Trp63, Fgf9 | Krt15, Krt17, Hspb1, Krt8, Dsp, Sfn, Epcam, Cldn7, Krt18, Krt19, Lgi2, Ezr, Krt14, Krt5, Ehf | Cdh5, Egfl7, Pecam1, Cldn5, Kdr, Flt1, Col18a1, Emcn, SparcH, Plxnd1, Cd34, Ecscr, S100a16, Plvap, Crip2 | Fcer1g, Tyrobp, Mrc1, C1qb, Pf4, Cd52, Lcp1, Corola, Laptm7, Cd74, C1qa, Lyz2, Ctss, Cx3cr1, C1qc | Phactr1, Ednrb, Fabp7, Egfl8, Sox10, Tm4sfl1, Moxd1, Cryab, Mpz, Erbb3, Cdh6, Foxd3, Gfra3, Npy, Slitrk2 |
Figure 3:
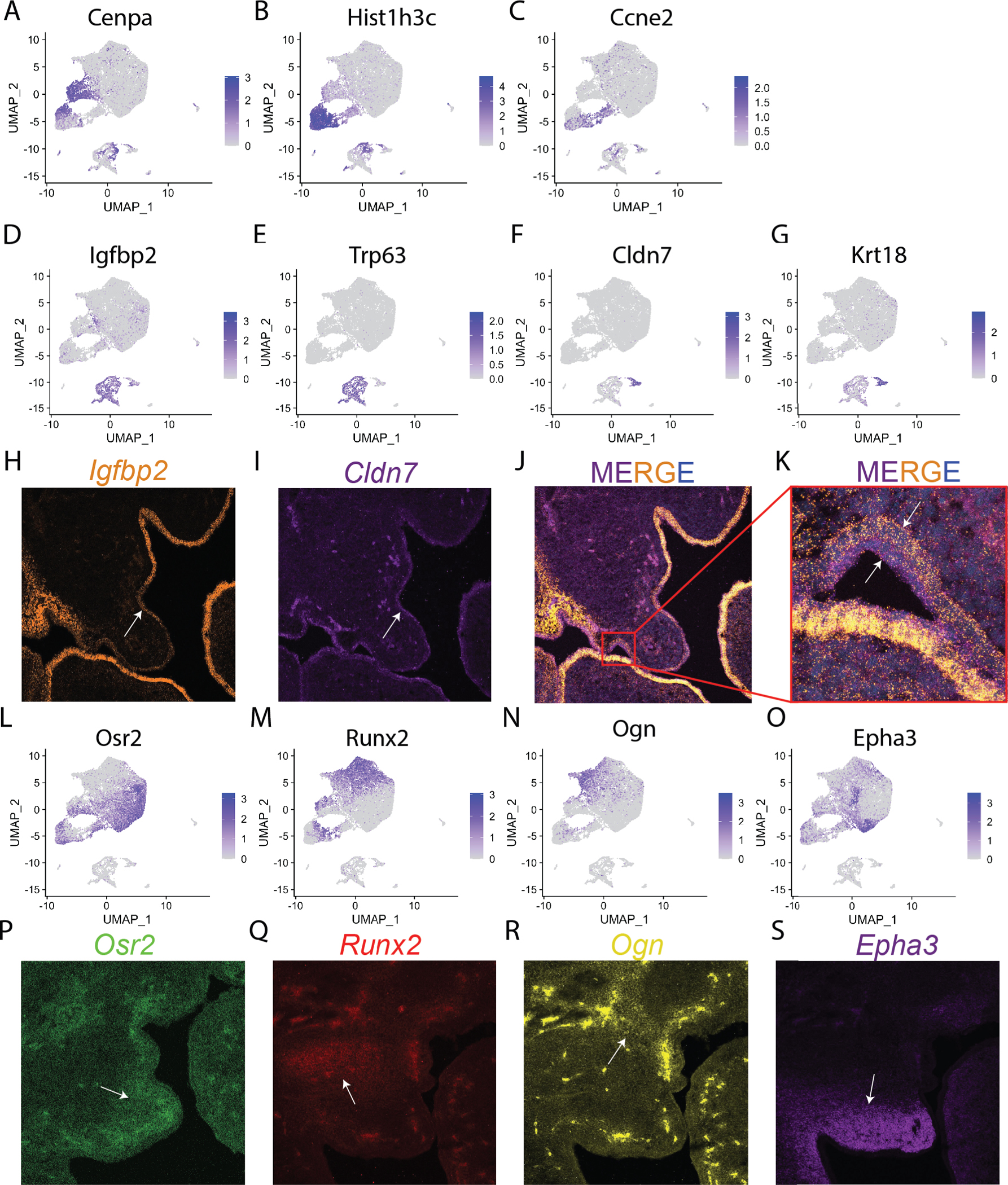
A FeaturePlot shows cells that express Cenpa (A) Hist1h3c (B) Ccne2 (C) for the three proliferating cells clusters. A FeaturePlot shows expression of epithelial cell cluster markers Igfbp2 (D), Trp63 (E), Cldn7 (F), and Krt18 (G). HCR RNA FISH shows expression localization of epithelial cell cluster markers Igfbp2 (H), Cldn7 (I), DAPI (J), and Merged (K). A FeaturePlot shows expression mesenchymal cluster marker genes Osr2 for Mes_1 (L), Runx2 for Mes_2 (M), Ogn for Mes_3 (N), and Epha3 for Mes_4 (O). HCR RNA FISH shows expression localization of mesenchymal cell cluster markers Osr2 for Mes_1 (P), Runx2 for Mes_2 (Q), Ogn for Mes_3 (R), and Epha3 for Mes_4 (S).
To determine whether epithelial cell populations are restricted to different domains or if they are interspersed, we performed hybridization chain reaction RNA fluorescence in situ hybridization (HCR RNA FISH) with probes for Igfbp2 and Cldn7, top markers for Ep_1 and Ep_2 respectively. Cells expressing Igfbp2 and Cldn7 are stacked upon on another (Fig. 3H–K). Igfbp2 cells line the inside of the epithelial layer contacting the mesenchyme while Cldn7 cells line the oral cavity laying on top of the Igfbp2 positive cells (Fig. 3K).
Mesenchymal subclustering produced four new clusters: Mes_1-Mes_4. Mes_1 is characterized by high expression of Osr and Shox2 (Fig. 2B–C, S4F, 3L). Mes_2 has high expression of Runx2 and Msx1 (Fig. 2B–C, S4G, 3M). Mes_3 has high expression of Ogn and Mecom (Pdrm3) (Fig. 2B–C, S4H, 3N). Mes_4 is characterized by high expression of Epha3 and Egfl6 (Fig. 2B–C, S4I, 3O). To determine spatial domains of each mesenchymal cell population, we performed HCR with probes for Ogn, Runx2, Osr2, and Epha3. Osr2 marks the distal tip and nasal compartment of the palate (Fig. 3P). Runx2 marks the population of mediolateral cells without significant overlap with neighboring compartments (Fig. 3Q). The Ogn population is dorsal and proximal to the Runx2 population marking the most proximal palatal mesenchyme (Fig. 3R). Epha3 overlaps in domain with Osr2 from the distal tip of the palate to the oral palatal mesenchyme (Fig. 3S). Importantly, expression of both epithelial and mesenchyme marker genes remains consistent between cells from all three embryos (Fig. S5A–B).
Proliferating subcluster markers were not independently labeled by HCR. However, it can be noted from observing expression of mesenchyme markers Osr2, Runx2, Ogn, and Epha3 on the FeaturePlots that the proliferating subclusters are comprised of all four mesenchymal subclusters (Fig 3L–O). This suggests that proliferation occurs within all mesenchymal domains at this stage.
Shox2 and Msx1 are independent populations
To determine how known markers of the anterior palate resolve at a single cell resolution, we examined expression of Msx1 and Shox2. Although both are considered markers of the anterior mesenchyme32,33,54–56, our data shows Msx1 and Shox2 expression are restricted to opposite populations within the palatal mesenchyme (Fig. 4A). Most mesenchymal clusters express both Shox2 and Msx1 to an extent; however, there is a clear delineation in their relative expression within those populations (Fig. 4A–B). Mes_1 and Mes_4 express Shox2 more strongly while Mes_2 expresses Msx1 most strongly (Fig. 4A–B). We performed HCR RNA FISH for cluster marker genes to determine if cell populations were spatially distinct from one another or if these different cell types were interspersed together. HCR revealed that Shox2 occupies the most distal nasal compartment of the palate while Msx1 is diffuse throughout the tissue with highest expression in the center of the palate shelf (Fig. 4C).
Figure 4:
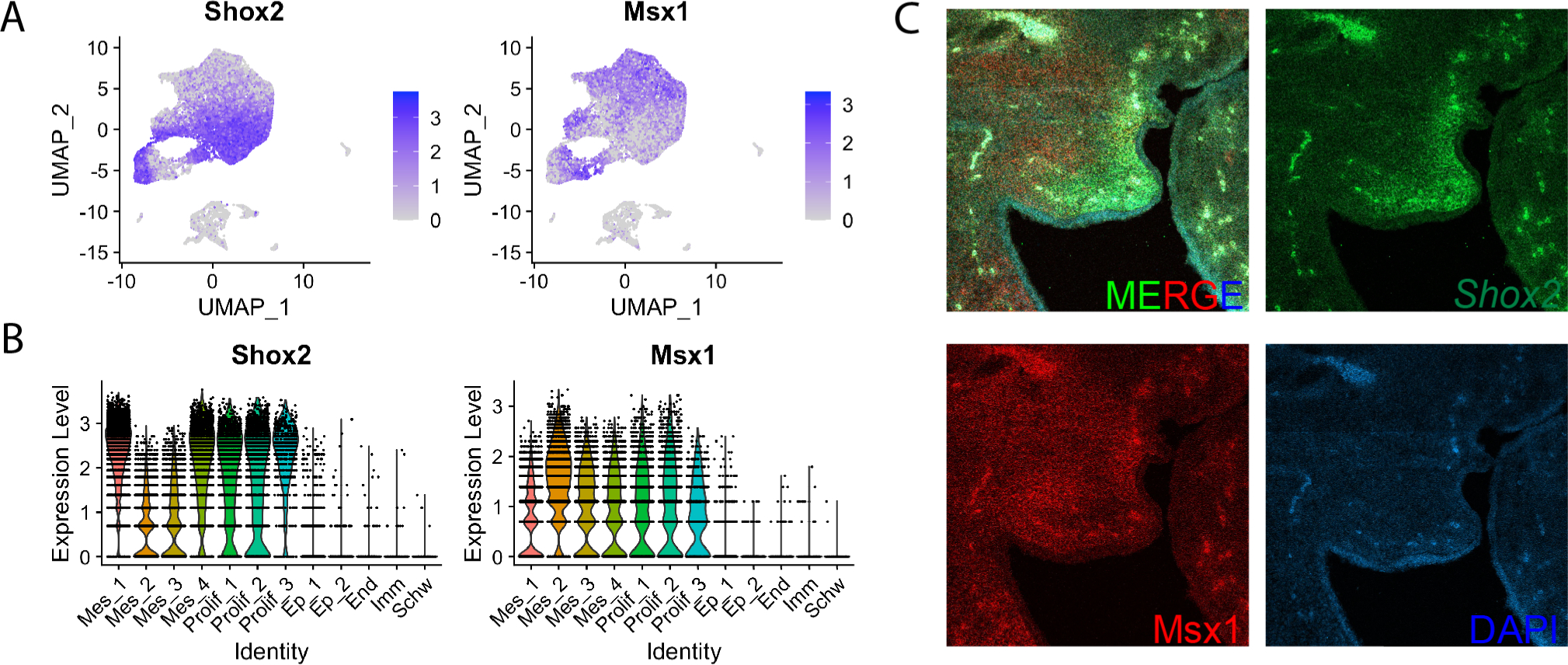
A) FeaturePlots of Shox2 and Msx1 show that they are expressed in opposite cell populations within the anterior palate. B) ViolinPlots of Shox2 and Msx1 show that cells that have high expression of Shox2 have low expression of Msx1 and cells that have high expression of Msx1 have low expression of Shox2. C) HCR RNA FISH of Shox2 and Msx1 show that Shox2 and Msx1 are expressed highly in different domains.
Combining expression data with HCR RNA FISH, it is apparent that there are distinct domains within the palate mesenchyme. Ogn (Mes_3), Runx2 (Mes_2), and Epha3 (Mes_4) can be seen marking the oral, central, and proximal domains of the palate without significant overlap (Fig. 5A). Shox2 and Osr2 span Mes_1 and Mes_4 marking the nasal mesenchyme, distal tip of the palate, and parts of the oral mesenchyme (Fig. 3P–S, 5B). Mes_4 can be distinguished from Mes_1 through expression of Epha3 which is expressed exclusively along the oral side of the palatal mesenchyme (Fig. 3S, 5A–B). Mes_1 does not express Epha3 and marks the nasal mesenchyme but not the distal tip of the palate (Fig. 2B–C, 3L, 3P). Mes_4 marks the remaining oral and distal mesenchyme (Fig. 2B, 3O, 3S, 5A, 5E). Shox2 is not highly expressed in Mes_2 or Mes_3 (Fig. 2B). Therefore, Shox2-low populations likely mark more central and proximal areas of the palatal mesenchyme. Indeed, HCR RNA FISH for Runx2 or Msx1 shows high expression in a population of cells in the center of the palate, Mes_2 (Fig. 5A, 5C). Msx1, like Shox2, marks multiple clusters (Fig. 2B, 4A–C). Ogn delineates Mes_3 from Mes_2 marking the most dorsal and proximal population of cells within the palate (Fig. 3R, 5A–B). Based on our data, we propose boundaries for these domains in a schematized palate (Fig. 5D–E).
Figure 5:
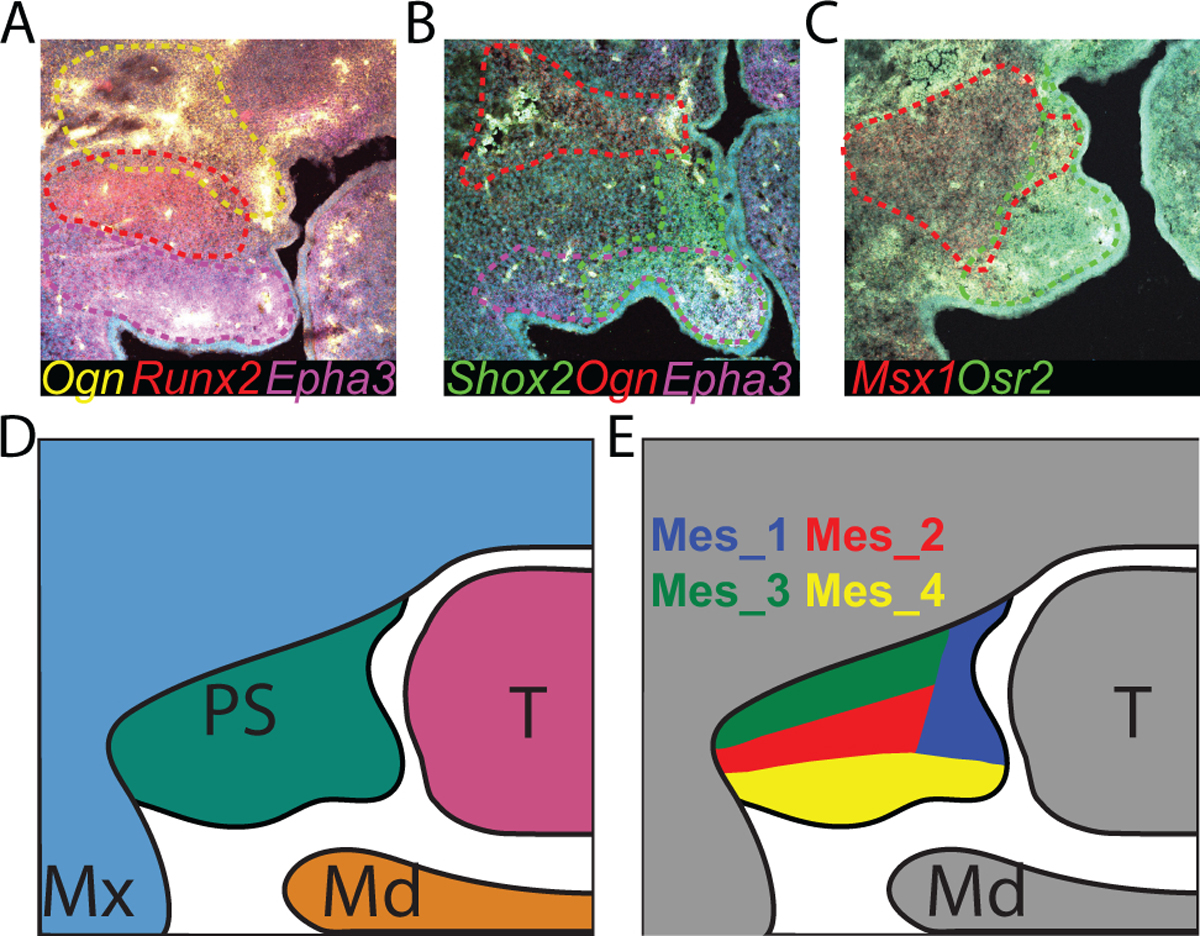
HCR RNA FISH of Ogn, Runx2, and Epha3 (A), Shox2, Ogn, and Epha3 (B), and Msx1 and Osr2 (C) shows reproducible boundaries of cluster-specific expression domains. D) A schematic shows proposed subdomains of each palate mesenchyme cell population. Mes subdomains are color coded to match clusters in UMAP in Figure 2A. PS=Palatal Shelf, Mx=Maxilla, Md=Mandible, T=Tongue.
Mes_3 has unique signaling patterns distinct from other mesenchyme
We hypothesized that there may be functional differences between mesenchymal cell clusters in the anterior palate because cell populations are spatially distinct. To determine differences in signaling capacities between clusters, we utilized CellChat cellular communication analysis to discover dominant cellular communication patterns. The CellChat database uses literature curated resources to interpret scRNA seq data and infer statistically and biologically significant cellular communications57. Visualizing the river plots of outgoing communication patterns of secreting cells, interpreted as ligand profiles of each cluster, Mes_1, Mes_2, and Mes_4 group together into Pattern 1, composed of Midkine (MK), Collagen, non-canonical Wnt (ncWnt), Ephrin-B (EPHB), Insulin-like growth factor (IGF), Periostin, Tenascin, Angiopoietin-like (ANGPTL), Activin, Semaphorin 5 (SEMA5), and Neurexin (NRXN). Mes_3 uniquely groups into Pattern 3, composed of pleiotrophin (PTN), Fibronectin 1 (FN1), Neural Cell adhesion molecule (NCAM), Myelin protein zeta (MPZ), Chemokine (C-X-C motif) ligand (CXCL), Thrombospondin (THBS), Vascular Cell adhesion protein (VCAM), Growth differentiation factor (GDF), and Neuronal growth regulator (NEGR). Both Ep clusters group together into Pattern 2, composed of Laminin, Platelet-derived growth factor (PDGF), Wnt, Cadherin (CDH), Cell adhesion molecule (CADM), Fibroblast growth factors (FGF), Agrin (AGRN), Desmosome, Notch, Junctional adhesion molecule (JAM), Semaphorin 3 (SEMA3), Nephronectin (NPNT), Hedgehog (HH), Heparin sulphate proteoglycan (HSPG), Leukemia inhibitory factor receptor (LIFR), Granulin (GRN), and Occludin (OCLN) (Fig. 6A). Exploring incoming communication patterns of target cells, interpreted as receptors present within each cluster, we once again see that Mes_1, Mes_2, and Mes_4 group together (Pattern 1) while Mes_3 demonstrates a unique receptor profile by grouping alone into Pattern 4 (Fig. 6B). Interestingly, we also see a split in receptors present between Ep_1 and Ep_2 (Fig. 6B).
Figure 6:
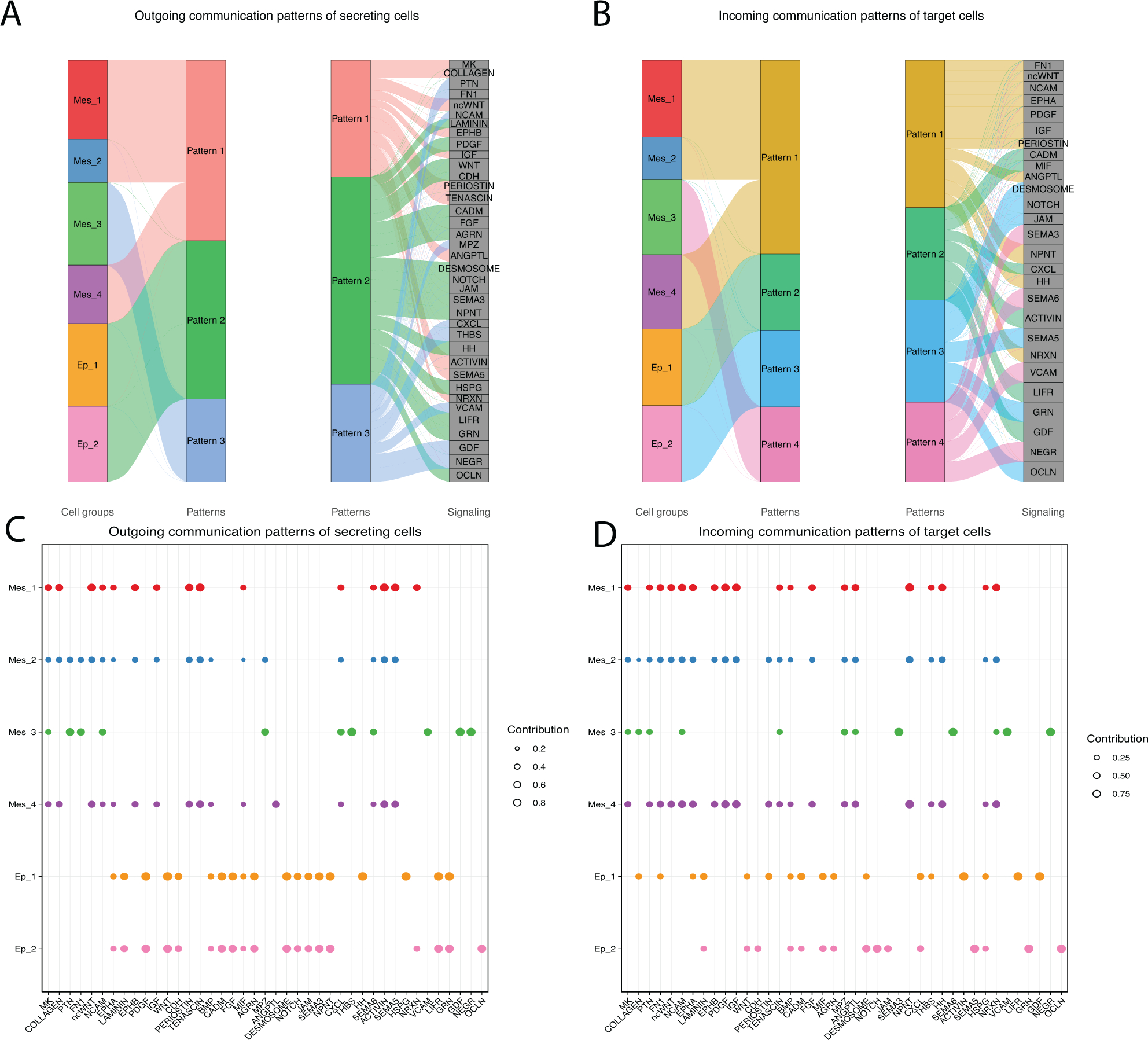
A) River plots show outgoing communication patterns of ligand expressing cells from each cell population within the anterior palate. The river plot to the left shows which clusters fall into which patterns. The river plot to the right shows the signaling compositions of each pattern. B) River plots show incoming communication patterns of target cells based on expression of receptors within a cluster. C) A DotPlot shows outgoing communication patterns of secreting cells. Clusters are shown on the y-axis and pathways are shown on the x-axis. The size of the dot represents contribution of that pathway to the communication pattern. D) A DotPlot shows incoming communication patterns of target cells.
We further delineate the compositions of these patterns through dot plots for outgoing or incoming communication patterns (Fig. 6C–D). Cells in Mes_3 specifically lack ligands associated with Collagen, ncWNT, EphA, EphB, IGF, Periostin, Tenascin, Macrophage migration inhibitory factor (MIF), SEMA6, and Activin that all other mesenchyme clusters express (Fig. 6C). Conversely, Mes_3 cells express Thrombospondin (THBS), NRXN, GDF, and NEGR ligands that the rest of the mesenchyme does not (Fig. 6C). All mesenchyme clusters except for Mes_3 express receptors associated with the FN1, ncWNT, EPHA, Laminin, PDGF, IGF, Tenascin, FGF, NPNT, THBS, HH, and HSPG pathways (Fig. 6D). Mes_3, but not the rest of the mesenchyme, expresses receptors of SEMA3, Sema6, NRXN, and NEGR (Fig. 6D). Together, these data show that one population of anterior mesenchymal palate cells sends and receives a different set of signals than the other mesenchymal cell populations.
CellRank trajectory analysis reveals two lineages within the E13.5 palate
The striking segregation of Shox2+ and Msx1+ cells within the palate led us to question whether distinct lineages can be traced within the E13.5 transcriptome. We utilized a pseudotime analysis using CellRank and RNA velocity to determine progenitor and terminal populations within the E13.5 anterior palate. Because some clusters spanned large distances over the UMAP, we first subclustered data at a higher resolution to minimize pseudotime analysis warping (Fig. 7A). RNA Velocity shows mesenchyme populations originate from the proliferating cells (Prolif_2a) and move towards two distinct terminal states, Mes_1a and Mes_2a (Fig. 7B). The initial and terminal states are highlighted (Fig. 7C). A directed Partition-based graph abstraction (PAGA) denotes CellRank probabilities at each node with arrows indicating pseudotemporal ordering (Fig. 7C). We visualize gene expression changes from initial to terminal states (i.e. from Prolif_2a to either Mes_1a or Mes_2b) using heatmaps (Fig. 7D–E). These data show that different mesenchymal clusters are distinct populations of cells rather than differentiation stages of the same population.
Figure 7:
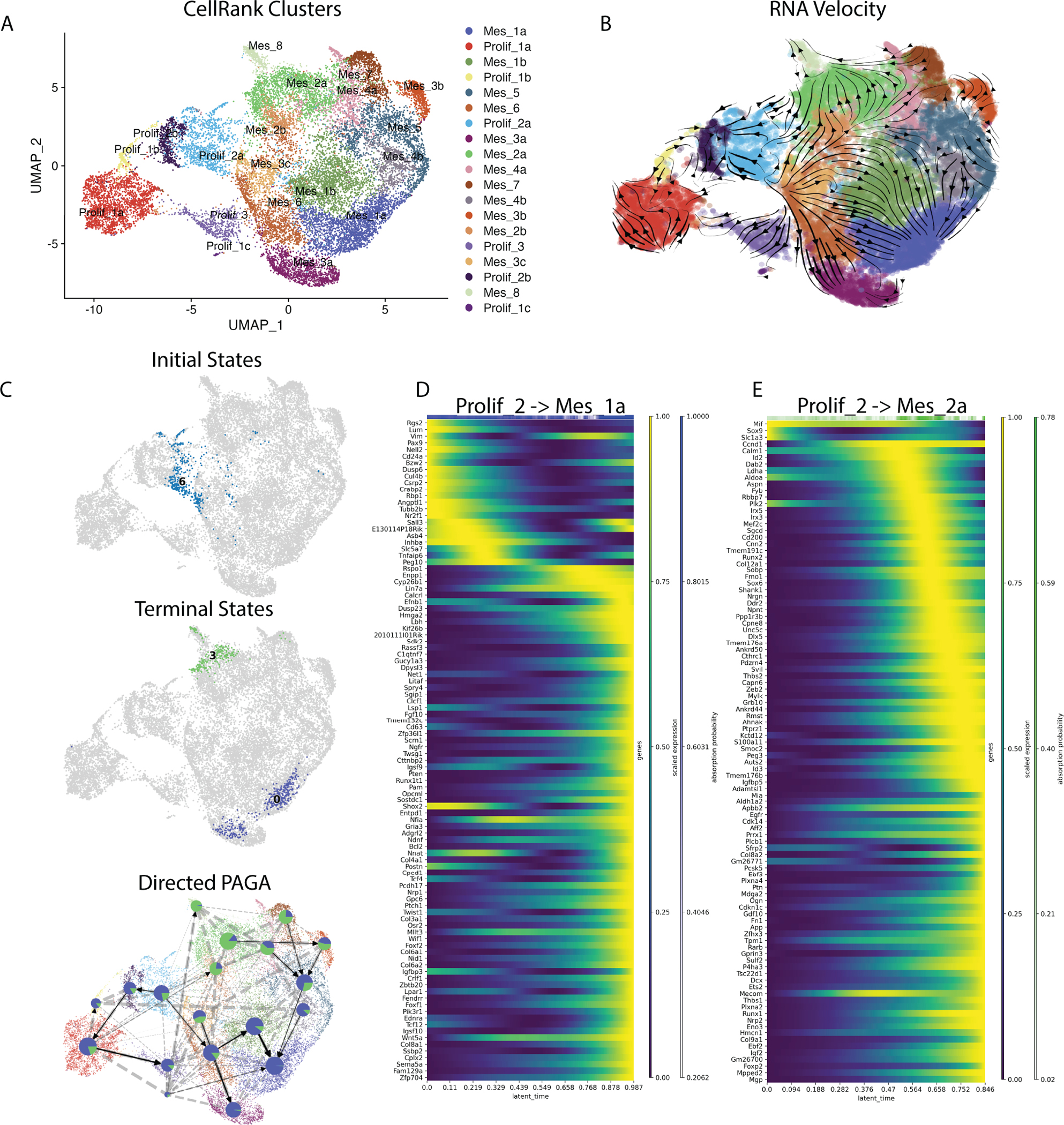
A) A UMAP shows scVelo subclustering used for CellRank trajectory analysis. B) A UMAP is overlayed with RNA velocity vectors. Vectors show the direction of RNA velocity, the time derivative of the gene expression state. C) UMAPs show initial states (Prolif_2a, blue) and terminal states (Mes_1a, purple) and (Mes_2a, green) and a directed PAGA plot showing proportion of cells within a cluster expected to contribute to either of the two terminal states. D) A temporal heatmap shows changes in gene expression over latent time from initial (Prolif_2) to terminal state one, Mes_1a. E) A temporal heatmap shows changes in gene expression over laten time from from initial (Prolif_2) to terminal state two, Mes_2a.
Discussion
Here we utilize advances in single cell sequencing and bioinformatic techniques to explore the intricacies of the E13.5 anterior palate transcriptome. Previously, excellent analysis of scRNA-seq has characterized the soft palate at E13.5, which makes up the posterior 1/3 of the structure23. We chose to focus on the anterior palate to conserve resources for increasing sequencing depth and to gain meaningful information about potentially rare transcripts within these populations. We acknowledge that the middle palate is not captured within our dataset or other published datasets and remains an area of investigation for future studies. E13.5 is a crucial point in palatal development representing a period of rapid vertical growth of the palatal shelves just prior to palatal elevation and fusion and remains understudied. Using scRNA-seq to develop an atlas of the E13.5 anterior palate, we identify expected cell types in our dataset: mesenchymal, proliferating, epithelial, endothelial, immune, and Schwann cells. These populations express genes associated with the respective cell types. This confirms accuracy of our dissections and our sequencing at capturing our desired RNAs of interest.
We subcluster mesenchymal, proliferating, and epithelial populations to reveal unexpected diversity within these populations. Even at low resolution threshold clustering, we find that these populations subcluster into distinct identities with unique marker gene profiles. These marker genes map back to the respective clusters on the UMAPs. The proliferating clusters subclustered into three populations. Prolif_3, marked by Ccne2 are cells entering the cell cycle. Prolif_2, marked by transcription of histone components, are cells with condensed chromatin. Prolif_1, marked by CenpA, represents cells preparing to divide. These proliferating populations are not specific to any one cell type, but rather represent a mixture of cells from multiple clusters that are grouped together based cell cycle stage. We did not define domains for proliferating populations because their transcriptional profiles suggest that they contribute to multiple populations and therefore could not be distinguished by HCR RNA FISH. This aligns with previous studies that show relatively consistent proliferation across the palatal shelves at E13.558,59. We chose to not regress out cell cycle in our analyses because mesenchyme is highly proliferative at this stage in development and regressing out cell cycle genes would remove valuable information from our dataset. Furthermore, retaining cell cycle information provides a resource to determine RNAVelocity and CellRank in our analysis to determine progenitor pools.
HCR RNA FISH confirms scRNA-seq results and defines the spatial location of these clusters in relationship to one another. Marker genes from our scRNA-seq show the correct cell types within the palate confirming our clustering and assignments. Cells from the two epithelial subclusters captured appear to segregate into rows. Ep_1 cells marked by Igfbp2, comprise the bulk of the tissue and line the inside of the epithelia in contact with the mesenchyme. Ep_2 cells, marked by Cldn7, lie superficial to Ep_1 in contact with the oral cavity. Although CellChat analysis demonstrates epithelial cells express similar ligands and cluster into shared signaling pathways, we observe a separation into two patterns when looking at incoming (receptor) communication signals. This suggests that these cell populations respond to different signals.
Interestingly, cells from the mesenchymal subclusters spatially segregate from one another. We show distinct expression patterns of two genes that have historically served as equivalent markers of the anterior palate, Shox2 and Msx1, demonstrating one application of how our dataset can be used as a community resource31–33. We find that these genes fall within separate clusters in our dataset and spatially segregate within the palate when viewed using HCR RNA FISH. Msx1 is expressed throughout the palatal mesenchyme, but HCR RNA FISH data clearly shows an Msx1-high expressing population, Mes_2, which lies in the middle of the palatal shelf. Conversely, Shox2 expression is restricted to the medial edge and distal tip of the palate and is not broadly expressed in other areas of the palate. Utilizing only one of these two markers may yield incomplete results when performing experiments in the anterior palate as neither completely marks the full extent of the tissue. We use HCR RNA FISH and scRNA-seq data to develop a spatial cluster map of the palate for this dataset so others can quickly identify several genes within a particular spatial domain within the anterior palate to bypass this problem. Using single cell resolution techniques, we can find improved markers of these populations to avoid confusion among researchers in the field.
Previous work has identified differences in signaling patterns between oral and lingual or medial and lateral compartments using bulk RNA-seq at E14.520. Our mesenchyme clusters segregate into oral, lingual, proximal, and central domains, our dataset can be used to identify good candidates for exploring these spatial differences at higher resolution at this earlier timepoint. Using the CellChat ligand receptor interaction explorer, we map signaling patterns of ligands and receptors expressed within each cluster. We identify that Mes_3, the proximally located cluster within the palate, has a unique ligand and receptor profile when compared to the remaining mesenchymal clusters. We used an CellRank and RNAVelocity analysis to determine the origins and terminations of cell populations. We identified that cells originated from the proliferating clusters within our dataset and differentiated into either of two fates. Our results show mesenchymal clusters are not merely different developmental stages of the same cell population and are in fact distinct cell populations. The first being the cells that occupy the distal tip of the palate shelf and the other being the unique signaling population we identified using CellChat, Mes_3. This data shows two paths for proliferating cells as they differentiate: either contribute to the rapidly proliferating tip of the palatal shelf to aid in palatal elevation or supply cells to the base of the palate. Is there a functional importance of Mes_3 cells, which express a unique signaling profile? Because these cells line the proximal border of the palatal shelf, they are not likely to be contributing to palatal outgrowth and certainly will not play a direct role during palatal fusion. CellChat data shows Mes_3 cells lack expression of ligands and receptors from key developmental signaling pathways including Bone morphogenetic protein (BMP), Platelet-derived growth factor (PDGF), Fibroblast growth factor (FGF), Insulin-like growth factor (IGF), Ephrin-A (EphA), Ephrin-B (EphB), noncanonical Wnt (ncWnt), and Hedgehog (Hh) signaling which are present in at least one of the other mesenchyme clusters. Clearly Mes_3 is not a primary signaling center like the other mesenchymal clusters have been documented to be. One possibility is that this population of cells provides guidance cues for projecting neurons. Indeed, CellChat data shows that Mes_3 cellular communication patterns are partially defined by the presence of Neuronal growth regulator (NEGR) as well as Midkine (MK) and Pleiotrophin (PTN) which are members of the Neurite growth-promoting factor (NEGF) family. Interestingly, Mes_3 signaling pathways are dominated by several glycoprotein pathways such as Thrombospondin (THBS), Fibronectin (FN1), Neural cell adhesion molecule (NCAM), and Vascular cell adhesion molecule (VCAM). Although the exact mechanisms by which the secondary palate elevates have been contentious, it is generally agreed upon that there is horizontal outgrowth and remodeling from the medial wall of palatal shelves to give rise to the elevated palatal shelves by E14.5. These rapid structural changes can apply large biophysical forces on the tissue. Perhaps the glycoprotein rich Mes_3 serves as an “anchor” domain for the palate as it undergoes this sheer stress from remodeling. Future studies will illuminate why Mes_3 functionally segregates from the rest of the palatal mesenchyme and how this population contributes to palate development.
Curation of this dataset and others allows for advancement in the understanding of the etiology of cleft palate on both clinical and research fronts. De novo mutations found in patients with cleft palate can be queried within our dataset to determine embryonic expression in the mouse. Correlation of a cleft palate-associated genetic variant and expression at the time of plate closure would provide rationale for experimentation in the mouse to determine causation. Genes of interest can then be referenced to the provided spatial map to determine where to expect expression of the gene of interest. Similarly, researchers can use this dataset for identification of genes expressed in the palate and to determine if their gene of interest is expressed in the same cells as other genes of interest. Furthermore, researchers can determine whether genes of interest are co-expressed with other molecular pathway genes and marker genes to form new hypotheses. Arguably one of the most promising uses of this dataset will come from its integration with existing and future datasets. Temporal integration and lineage tracing with earlier and later timepoints will give key insights into the origins and fates of the cells that we have defined. Additionally, comparing the formation of the anterior palate with that of the posterior palate would similarly be quite valuable. This dataset adds to a growing body of work with the goal of unveiling the molecular underpinnings of palatal development.
Experimental Procedures
Mice
C57BL/6J Mice originally obtained from The Jackson Laboratory and maintained as a colony in accordance with IACUC protocol #0139 at the University of Colorado Anschutz Medical Campus. Observation of the vaginal plug was considered embryonic day 0.5. On embryonic day 13.5, pregnant dams were euthanized by isofluorane followed by cervical dislocation and embryos were harvested. Heads were kept in ice-cold PBS for dissection. All three sequenced embryos were female.
Single cell RNA sequencing
In ice-cold PBS, heads were removed from embryos. The top of the head was cut flat, and the head was laid ventral. The secondary palate was exposed by removing the mandible and tongue. The anterior 1/3 of the palate was dissected away from the remainder of the head using forceps. Paired anterior palatal shelves were washed twice in PBS and transferred to 750 uL 0.25% Trypsin-EDTA for 10 minutes at 37C with frequent trituration. Trypsinization was stopped with addition of 750uL DMEM + 10% FBS (Invitrogen/Life Technologies). Cells were pipetted up and down 20 times to assist in cell dispersion, pelleted at 300 g for 3 min, and resuspended in PBS with 0.1% BSA before being filtered through a Flowmi 40uM cell strainer. Single cell suspensions were confirmed by microscopy and cells were counted on a hemocytometer with trypan blue staining. Single cell suspensions were submitted to the University of Colorado Anschutz Medical Campus Genomics Shared Resource for droplet derivation and library preparation with the Chromium single cell 3’ NextGen V3.1 chemistry (10X Genomics). Sequencing was performed on an Illumina NovaSeq6000 with V1.5 chemistry. 7976, 6994, and 7268 cells were collected from 3 embryos (22,238 cells in total across all embryos) and sequenced at a depth of 100,000 reads per cell. Cells from different embryos were run separately and not pooled together during sequencing.
Data analysis
RNA-sequencing analysis was performed using the Seurat pipeline60. Data was normalized using SingleCellTransform (SCT) and clustered at a resolution of 0.2 snn and integrated using the Seurat v4 integration method. Populations were identified based on marker gene expression. Heatmaps were generated based on top ten marker genes and plotted across every cell within the dataset. Global intercellular communication patterns between the cells in a subset of the integrated SeuratObject containing mesenchymal and epithelial clusters was inferred using CellChat57,61 following the tutorial. Number of outgoing and incoming communication patterns was selected based on an agreement in the drop of Cophenetic and Silhouette values. The integrated SeuratObject was subset to select WT mesenchymal and proliferating cells and converted to an h5ad object. This subset was analyzed with scVelo62 to determine RNA Velocity63 and CellRank64 to calculate the initial and terminal states based on the RNA Velocity, a directed partition-based graph abstraction (PAGA) based on the latent time inferred pseudotime, and the lineage driver genes.
In situ hybridization
E13.5 embryos were collected, and heads were fixed in 4% paraformaldehyde (PFA). Heads were dehydrated in 30% sucrose and embedded in OCT (Sakura, Tissue-Tek, Cat. 4583). Samples were sectioned at 12 μM thickness on a cryostat. Hybridization chain reaction (Molecular Instruments) was used for multiplexed RNA in situ hybridization according to manufacturer’s protocol. Briefly, sectioned tissues were allowed to thaw at room temperature, fixed in 4% PFA, dehydrated, treated with 2 μg/ml Proteinase K for 2 min, re-fixed, and hybridized with 1.6 pmol of probe at 37°C overnight. The following day slides were washed in wash buffer and SSCT per manufacturers protocol and amplified with 6 picomole snap-cooled hairpins complementary to the probes overnight at room temperature. On day three, slides were washed in SSCT and mounted for imaging. Slides were imaged on a Zeiss LSM 980 inverted confocal laser scanning microscope.
Supplementary Material
Supplemental Figure 1: A coronal section of the E13.5 anterior mouse head shows where the palate shelves were removed. B. A coronal section of the E13.5 posterior mouse head shows where the palate was left intact.
Supplemental Figure 2: Dot plots show percentage of cells and average expression level of the top ten genes expressed in each cell population (cluster) within the anterior palate.
Supplemental Figure 3: A) A segmented bar chart shows the proportion of each cell population (cluster) that comprises each sample (embryo). B) Three UMAPs of twelve cell populations shows subclusters of Mesenchyme, Epithelial, and Proliferating populations separated based on embryo of origin.
Supplemental Figure 4: Dot plots show percentage of cells and average expression level of the top ten genes expressed in each cell population (cluster), including subclustering of proliferating, mesenchyme, and epithelial populations within the anterior palate.
Supplemental Figure 5: A) FeaturePlots of ectoderm genes marking either Ep_1 (Igfbp2 and Trp63) or Ep_2 (Cldn7 and Krt18) split by embryo of origin. B) FeaturePlots of mesenchyme genes marking Mes_1 (Osr2), Mes_2 (Runx2), Mes_3 (Ogn), or Mes_4 (Epha3) split by embryo of origin.
Acknowledgements:
We would like to thank the University of Colorado Genomics Core for performing sequencing of samples and being a valuable resource.
Funding:
This work was supported by the National Institute of Dental and Craniofacial Research NIH-NIDCR-R01DE025311 to E.A.B. and NIH-T32GM141742–02S1 to Y.H.O.
Footnotes
Declarations of interest: None
Data Availability:
Raw and processed data has been made available via a NCBI GEO Submission (accession code GSE222205). Code is accessible via GitHub at https://github.com/yunusozekin/WT_E13.5_AntPalate_scRNAseq_Ozekin.git.
References
- 1.Research NIoDaC. Cleft Lip & Palate.
- 2.Deshpande AS, Goudy SL. Cellular and molecular mechanisms of cleft palate development. Laryngoscope Investig Otolaryngol. Feb 2019;4(1):160–164. 10.1002/lio2.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinelli M, Palmieri A, Carinci F, Scapoli L. Non-syndromic Cleft Palate: An Overview on Human Genetic and Environmental Risk Factors [Review]. Frontiers in Cell and Developmental Biology. 2020-October-20 2020;8. 10.3389/fcell.2020.592271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gritli-Linde A Molecular control of secondary palate development. Developmental Biology. 2007/January/15/ 2007;301(2):309–326. 10.1016/j.ydbio.2006.07.042. [DOI] [PubMed] [Google Scholar]
- 5.Gunnerbeck A, Edstedt Bonamy AK, Wikstrom AK, Granath F, Wickstrom R, Cnattingius S. Maternal snuff use and smoking and the risk of oral cleft malformations--a population-based cohort study. PLoS One. 2014;9(1):e84715. 10.1371/journal.pone.0084715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeRoo LA, Wilcox AJ, Drevon CA, Lie RT. First-trimester maternal alcohol consumption and the risk of infant oral clefts in Norway: a population-based case-control study. Am J Epidemiol. Sep 15 2008;168(6):638–46. 10.1093/aje/kwn186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ozekin YH, Isner T, Bates EA. Ion Channel Contributions to Morphological Development: Insights From the Role of Kir2.1 in Bone Development [Mini Review]. Frontiers in Molecular Neuroscience. 2020-June-09 2020;13. 10.3389/fnmol.2020.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dixon MJ, Marazita ML, Beaty TH, Murray JC. Cleft lip and palate: understanding genetic and environmental influences. Nature Reviews Genetics. 2011/March/01 2011;12(3):167–178. 10.1038/nrg2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Werler MM, Ahrens KA, Bosco JL, et al. Use of antiepileptic medications in pregnancy in relation to risks of birth defects. Ann Epidemiol Nov 2011;21(11):842–50. 10.1016/j.annepidem.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Margulis AV, Mitchell AA, Gilboa SM, et al. Use of topiramate in pregnancy and risk of oral clefts. Am J Obstet Gynecol. Nov 2012;207(5):405.e1–7. 10.1016/j.ajog.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puhó EH, Szunyogh M, Métneki J, Czeizel AE. Drug treatment during pregnancy and isolated orofacial clefts in hungary. Cleft Palate Craniofac J. Mar 2007;44(2):194–202. 10.1597/05-208.1. [DOI] [PubMed] [Google Scholar]
- 12.Vyas T, Gupta P, Kumar S, Gupta R, Gupta T, Singh HP. Cleft of lip and palate: A review. J Family Med Prim Care. Jun 2020;9(6):2621–2625. 10.4103/jfmpc.jfmpc_472_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen C, Hernandez-Boussard T, Davies SM, Bhattacharya J, Khosla RK, Curtin CM. Cleft Palate Surgery: An Evaluation of Length of Stay, Complications, and Costs by Hospital Type. The Cleft Palate-Craniofacial Journal. 2014;51(4):412–419. 10.1597/12-150. [DOI] [PubMed] [Google Scholar]
- 14.Yu K, Deng M, Naluai-Cecchini T, Glass IA, Cox TC. Differences in Oral Structure and Tissue Interactions during Mouse vs. Human Palatogenesis: Implications for the Translation of Findings from Mice. Front Physiol. 2017;8:154. 10.3389/fphys.2017.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walker BE, Fraser FC. Closure of the Secondary Palate in Three Strains of Mice. Development. 1956;4(2):176–189. 10.1242/dev.4.2.176. [DOI] [Google Scholar]
- 16.Bush JO, Jiang R. Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development. 2012;139(2):231–243. 10.1242/dev.067082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Depew MJ, Tucker AS, Sharpe PT. 19 - Craniofacial Development. In: Rossant J, Tam PPL, eds. Mouse Development. San Diego: Academic Press; 2002:421–498. [Google Scholar]
- 18.Luke DA. Cell proliferation in palatal processes and Meckel’s cartilage during development of the secondary palate in the mouse. J Anat. Aug 1989;165:151–8. [PMC free article] [PubMed] [Google Scholar]
- 19.Brown NL, Knott L, Halligan E, Yarram SJ, Mansell JP, Sandy JR. Microarray analysis of murine palatogenesis: Temporal expression of genes during normal palate development. Development, Growth & Differentiation. 2003;45(2):153–165. 10.1034/j.1600-0854.2004.00686.x. [DOI] [PubMed] [Google Scholar]
- 20.Potter AS, Potter SS. Molecular Anatomy of Palate Development. PLoS One. 2015;10(7):e0132662. 10.1371/journal.pone.0132662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, Jones KL, Hooper JE, Williams T. The molecular anatomy of mammalian upper lip and primary palate fusion at single cell resolution. Development. 2019;146(12). 10.1242/dev.174888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gu R, Zhang S, Saha SK, et al. Single-cell transcriptomic signatures and gene regulatory networks modulated by Wls in mammalian midline facial formation and clefts. Development. 2022;149(14). 10.1242/dev.200533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han X, Feng J, Guo T, et al. Runx2-Twist1 interaction coordinates cranial neural crest guidance of soft palate myogenesis. eLife. 2021/January/22 2021;10:e62387. 10.7554/eLife.62387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jing J, Feng J, Yuan Y, et al. Spatiotemporal single-cell regulatory atlas reveals neural crest lineage diversification and cellular function during tooth morphogenesis. Nat Commun. Aug 16 2022;13(1):4803. 10.1038/s41467-022-32490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan Y, Loh YE, Han X, et al. Spatiotemporal cellular movement and fate decisions during first pharyngeal arch morphogenesis. Sci Adv. Dec 2020;6(51). 10.1126/sciadv.abb0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu J, Liu H, Lan Y, et al. Hedgehog signaling patterns the oral-aboral axis of the mandibular arch. eLife. 2019/January/14 2019;8:e40315. 10.7554/eLife.40315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farmer DT, Mlcochova H, Zhou Y, et al. The developing mouse coronal suture at single-cell resolution. Nat Commun. Aug 10 2021;12(1):4797. 10.1038/s41467-021-24917-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holmes G, Gonzalez-Reiche AS, Lu N, et al. Integrated Transcriptome and Network Analysis Reveals Spatiotemporal Dynamics of Calvarial Suturogenesis. Cell Reports. 2020/07/July/ 2020;32(1):107871. 10.1016/j.celrep.2020.107871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holmes G, Gonzalez-Reiche AS, Saturne M, et al. Single-cell analysis identifies a key role for Hhip in murine coronal suture development. Nat Commun. Dec 8 2021;12(1):7132. 10.1038/s41467-021-27402-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L, Tang Q, Xu J, et al. The transcriptional regulator MEIS2 sets up the ground state for palatal osteogenesis in mice. J Biol Chem. Apr 17 2020;295(16):5449–5460. 10.1074/jbc.RA120.012684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lan Y, Xu J, Jiang R. Chapter Three - Cellular and Molecular Mechanisms of Palatogenesis. In: Chai Y, ed. Current Topics in Developmental Biology. Vol 115. Academic Press; 2015:59–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.FaceBase. Gene summary for Shox2.
- 33.FaceBase. Gene summary for MSX1.
- 34.Vanyai HK, Garnham A, May RE, et al. MOZ directs the distal-less homeobox gene expression program during craniofacial development. Development. 2019;146(14). 10.1242/dev.175042. [DOI] [PubMed] [Google Scholar]
- 35.FaceBase. Gene Summary for Col3a1.
- 36.Grimaldi A, Parada C, Chai Y. A Comprehensive Study of Soft Palate Development in Mice. PLoS One. 2015;10(12):e0145018. 10.1371/journal.pone.0145018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jia S, Zhou J, Fanelli C, et al. Small-molecule Wnt agonists correct cleft palates in Pax9 mutant mice in utero. Development. 2017;144(20):3819–3828. 10.1242/dev.157750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng W, Leach SM, Tipney H, et al. Spatial and temporal analysis of gene expression during growth and fusion of the mouse facial prominences. PLoS One. Dec 16 2009;4(12):e8066. 10.1371/journal.pone.0008066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gustafson KS, Clark DP. Chapter 17 - Molecular Cytopathology. In: Tubbs RR, Stoler MH, eds. Cell and Tissue Based Molecular Pathology. Philadelphia: Churchill Livingstone; 2009:167–180. [Google Scholar]
- 40.Marzluff WF, Gongidi P, Woods KR, Jin J, Maltais LJ. The Human and Mouse Replication-Dependent Histone Genes. Genomics. 2002/November/01/ 2002;80(5):487–498. 10.1006/geno.2002.6850. [DOI] [PubMed] [Google Scholar]
- 41.He F, Soriano P. A Critical Role for PDGFRα Signaling in Medial Nasal Process Development. PLOS Genetics. 2013;9(9):e1003851. 10.1371/journal.pgen.1003851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kutejova E, Engist B, Self M, Oliver G, Kirilenko P, Bobola N. Six2 functions redundantly immediately downstream of Hoxa2. Development. 2008;135(8):1463–1470. 10.1242/dev.017624. [DOI] [PubMed] [Google Scholar]
- 43.Lee J, Rabbani CC, Gao H, et al. Hair-bearing human skin generated entirely from pluripotent stem cells. Nature. 2020/June/01 2020;582(7812):399–404. 10.1038/s41586-020-2352-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Woodfin A, Voisin M-B, Nourshargh S. PECAM-1: A Multi-Functional Molecule in Inflammation and Vascular Biology. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(12):2514–2523. 10.1161/ATVBAHA.107.151456. [DOI] [PubMed] [Google Scholar]
- 45.Guo L, Zhang H, Hou Y, Wei T, Liu J. Plasmalemma vesicle-associated protein: A crucial component of vascular homeostasis. Exp Ther Med. Sep 2016;12(3):1639–1644. 10.3892/etm.2016.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sauteur L, Krudewig A, Herwig L, et al. Cdh5/VE-cadherin Promotes Endothelial Cell Interface Elongation via Cortical Actin Polymerization during Angiogenic Sprouting. Cell Reports. 2014/October/23/ 2014;9(2):504–513. 10.1016/j.celrep.2014.09.024. [DOI] [PubMed] [Google Scholar]
- 47.Dong K, Chen W, Pan X, et al. FCER1G positively relates to macrophage infiltration in clear cell renal cell carcinoma and contributes to unfavorable prognosis by regulating tumor immunity. BMC Cancer. 2022/February/04 2022;22(1):140. 10.1186/s12885-022-09251-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haure-Mirande J-V, Audrain M, Ehrlich ME, Gandy S. Microglial TYROBP/DAP12 in Alzheimer’s disease: Transduction of physiological and pathological signals across TREM2. Molecular Neurodegeneration. 2022/August/24 2022;17(1):55. 10.1186/s13024-022-00552-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J, Zhang G, Sui Y, et al. CD52 Is a Prognostic Biomarker and Associated With Tumor Microenvironment in Breast Cancer [Original Research]. Frontiers in Genetics. 2020-November-02 2020;11. 10.3389/fgene.2020.578002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.D’antonio M, Michalovich D, Paterson M, et al. Gene profiling and bioinformatic analysis of Schwann cell embryonic development and myelination. Glia. 2006;53(5):501–515. 10.1002/glia.20309. [DOI] [PubMed] [Google Scholar]
- 51.Finzsch M, Schreiner S, Kichko T, et al. Sox10 is required for Schwann cell identity and progression beyond the immature Schwann cell stage. Journal of Cell Biology. 2010;189(4):701–712. 10.1083/jcb.200912142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liang Y, Bollen AW, Aldape KD, Gupta N. Nuclear FABP7 immunoreactivity is preferentially expressed in infiltrative glioma and is associated with poor prognosis in EGFR-overexpressing glioblastoma. BMC Cancer. Apr 19 2006;6:97. 10.1186/1471-2407-6-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jacob C, Lötscher P, Engler S, et al. HDAC1 and HDAC2 control the specification of neural crest cells into peripheral glia. J Neurosci. Apr 23 2014;34(17):6112–22. 10.1523/jneurosci.5212-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Z, Song Y, Zhao X, Zhang X, Fermin C, Chen Y. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development. 2002;129(17):4135–4146. 10.1242/dev.129.17.4135. [DOI] [PubMed] [Google Scholar]
- 55.Yu L, Gu S, Alappat S, et al. Shox2-deficient mice exhibit a rare type of incomplete clefting of the secondary palate. Development. 2005;132(19):4397–4406. 10.1242/dev.02013. [DOI] [PubMed] [Google Scholar]
- 56.Zhou J, Gao Y, Lan Y, Jia S, Jiang R. Pax9 regulates a molecular network involving Bmp4, Fgf10, Shh signaling and the Osr2 transcription factor to control palate morphogenesis. Development. 2013;140(23):4709–4718. 10.1242/dev.099028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jin S, Guerrero-Juarez CF, Zhang L, et al. Inference and analysis of cell-cell communication using CellChat. Nature Communications. 2021/February/17 2021;12(1):1088. 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu X, Gao J, Liao Y, Tang S, Lu F. Retinoic acid alters the proliferation and survival of the epithelium and mesenchyme and suppresses Wnt/β-catenin signaling in developing cleft palate. Cell Death & Disease. 2013/October/01 2013;4(10):e898–e898. 10.1038/cddis.2013.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li N, Liu J, Liu H, et al. Altered BMP-Smad4 signaling causes complete cleft palate by disturbing osteogenesis in palatal mesenchyme. Journal of Molecular Histology. February/01 2021;52:1–17. 10.1007/s10735-020-09922-4. [DOI] [PubMed] [Google Scholar]
- 60.Hao Y, Hao S, Andersen-Nissen E, et al. Integrated analysis of multimodal single-cell data. Cell. 2021/June/24/ 2021;184(13):3573–3587.e29. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vu R, Jin S, Sun P, et al. Wound healing in aged skin exhibits systems-level alterations in cellular composition and cell-cell communication. Cell Rep. Aug 2 2022;40(5):111155. 10.1016/j.celrep.2022.111155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bergen V, Lange M, Peidli S, Wolf FA, Theis FJ. Generalizing RNA velocity to transient cell states through dynamical modeling. Nature Biotechnology. 2020/December/01 2020;38(12):1408–1414. 10.1038/s41587-020-0591-3. [DOI] [PubMed] [Google Scholar]
- 63.La Manno G, Soldatov R, Zeisel A, et al. RNA velocity of single cells. Nature. 2018/August/01 2018;560(7719):494–498. 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lange M, Bergen V, Klein M, et al. CellRank for directed single-cell fate mapping. Nature Methods. 2022/February/01 2022;19(2):159–170. 10.1038/s41592-021-01346-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: A coronal section of the E13.5 anterior mouse head shows where the palate shelves were removed. B. A coronal section of the E13.5 posterior mouse head shows where the palate was left intact.
Supplemental Figure 2: Dot plots show percentage of cells and average expression level of the top ten genes expressed in each cell population (cluster) within the anterior palate.
Supplemental Figure 3: A) A segmented bar chart shows the proportion of each cell population (cluster) that comprises each sample (embryo). B) Three UMAPs of twelve cell populations shows subclusters of Mesenchyme, Epithelial, and Proliferating populations separated based on embryo of origin.
Supplemental Figure 4: Dot plots show percentage of cells and average expression level of the top ten genes expressed in each cell population (cluster), including subclustering of proliferating, mesenchyme, and epithelial populations within the anterior palate.
Supplemental Figure 5: A) FeaturePlots of ectoderm genes marking either Ep_1 (Igfbp2 and Trp63) or Ep_2 (Cldn7 and Krt18) split by embryo of origin. B) FeaturePlots of mesenchyme genes marking Mes_1 (Osr2), Mes_2 (Runx2), Mes_3 (Ogn), or Mes_4 (Epha3) split by embryo of origin.
Data Availability Statement
Raw and processed data has been made available via a NCBI GEO Submission (accession code GSE222205). Code is accessible via GitHub at https://github.com/yunusozekin/WT_E13.5_AntPalate_scRNAseq_Ozekin.git.