


Abstract
The Intronerator (http://www.cse.ucsc.edu/~kent/intronerator/ ) is a set of web-based tools for exploring RNA splicing and gene structure in Caenorhabditis elegans. It includes a display of cDNA alignments with the genomic sequence, a catalog of alternatively spliced genes and a database of introns. The cDNA alignments include >100 000 ESTs and almost 1000 full-length cDNAs. ESTs from embryos and mixed stage animals as well as full-length cDNAs can be compared in the alignment display with each other and with predicted genes. The alt-splicing catalog includes 844 open reading frames for which there is evidence of alternative splicing of pre-mRNA. The intron database includes 28 478 introns, and can be searched for patterns near the splice junctions.
INTRODUCTION
In recent years a tremendous amount of sequence data has become available for Caenorhabditis elegans, including the full genomic sequence (1), >100 000 expressed sequence tags (ESTs) (2) and almost 1000 full-length complementary DNAs (cDNAs) (3). Our laboratory was interested in using this data to look for instances of alternative splicing of pre-mRNA, and, in particular, regulated alternative splicing. AceDB (4), a tool for browsing the C.elegans genome, provided a very useful starting point, but we desired a more detailed, more up-to-date display of cDNA alignments. Therefore, to increase the ability of scientists to access information about splicing and potential alternative splicing in C.elegans, we designed the Intronerator. Intronerator is a database in which every C.elegans EST and full-length cDNA sequence in GenBank (3) is aligned to the genomic sequence and displayed in such a way that splicing patterns are easily seen. Based on these alignments we have generated a database of 844 alternatively spliced genes as well as a listing of >28 000 cDNA-confirmed introns. The Intronerator is a general purpose tool of potential use to anyone studying gene structure in C.elegans. It is based largely on existing alignment algorithms and observations about splice sites (4–8), but it contains a few new approaches as well. The interface was designed to work over the WWW and is accessible at http://www.cse.ucsc.edu/~kent/intronerator/
DESCRIPTION
The Tracks Display (Fig. 1) is the heart of the Intronerator and can be entered directly by following the ‘Tracks Display’ link in the home page. You can type in the name of a gene, open reading frame (ORF) or cosmid, and the Intronerator will display a graphic alignment of cDNAs and predicted genes within the specified region of the genome. Clicking on a particular gene or intron of interest from other pages in the Intronerator will also take you to the Tracks Display. The Tracks Display contains three main parts: a row of buttons for scrolling and zooming, a graphic containing the cDNA alignments and C.elegans Sequencing Consortium gene predictions (1), and a control panel which lets you view and modify which part of a chromosome is displayed or jump to another gene or cosmid directly. The graphical display is also interactive. Clicking on a splicing diagram retrieves the DNA sequence for that section of the chromosome with the predicted coding exons in upper case. Figure 1 shows the Tracks Display for the predicted C.elegans ORF ZK1127.9, which is one of two C.elegans homologs of the yeast splicing factor prp40 (9). By comparing the cDNA alignments with the predicted gene in the Tracks Display, two main differences are seen. First, an exon between predicted exons 1 and 2 is used in six of the cDNAs covering this region but skipped in the rest. This provides strong evidence for the presence of an alternative exon. In addition, the only EST to span predicted exon 3, yk327h11.5, does not include this exon. This limited amount of data indicates that predicted exon 3 may not be real.
Figure 1.

Tracks Display of the region around the predicted gene ZK1127.9. Alignments of ESTs deriving from mixed stage animals (black) and embryos (green) are not in complete agreement with the Genefinder prediction (blue) and provide evidence for an alternative exon between predicted exons 1 and 2. The parts of the alignments drawn in lighter colors are regions of the ESTs with sequencing errors that do not correspond 100% with the genomic sequence.
Clicking on a cDNA alignment brings up a detailed view of the alignment of the cDNA with the genomic sequence. Figure 2 demonstrates what happens when the alignment diagram for the EST yk507g11.5 in Figure 1 is clicked. The detailed alignment view contains three main parts. On top is the cDNA sequence displayed with bases that match the genomic DNA colored blue. The ends of aligned blocks are colored a lighter blue. In the middle is the genomic sequence with bases that match the cDNA-colored blue. Parts of genomic DNA that are predicted to be coding regions are in upper case. One can quickly compare the agreement of the gene predictions with the cDNA alignments by seeing if the areas in upper case (predicted exons) are the same as the areas in blue (regions aligning to the selected cDNA). On the bottom of the page is also a detailed alignment view in a more traditional display, similar to the output of BLAST (not shown in Fig. 2) (5).
Figure 2.
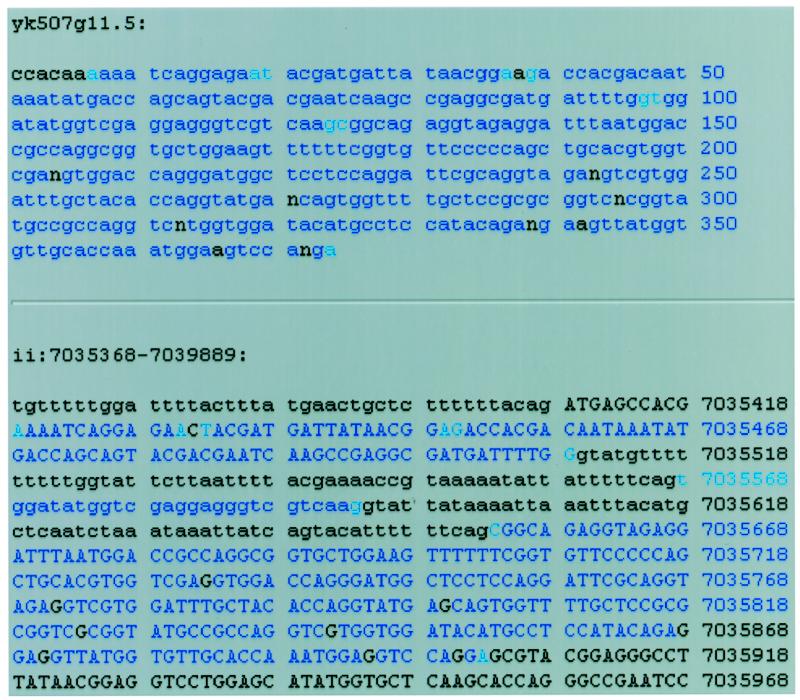
Detailed display of alignment between the EST yk507g11.5 and the area of the genome around ZK1127.9 obtained by clicking this EST’s graphic display in Figure 1. In the upper part of the display, the blue areas represent nucleotides that match perfectly between cDNA and genomic DNA. The lighter blue areas represent the ends of matching areas. In the second area of the display, regions that are predicted to be coding exons as annotated in AceDB are in upper case. Though the EST sequence contains some sequencing errors, the alternatively spliced exon is apparent.
While the Tracks Display is useful for browsing around a chromosome, the Alt-Splicing Catalog can take you directly to potential alternatively spliced genes. The Alt-Splicing Catalog is a table currently containing 844 genes for which there is cDNA evidence for alternative splicing of pre-mRNA. In some cases the evidence is quite compelling—supported by a half dozen cDNAs for each isoform. Each entry in the catalog contains information which loosely categorizes the type of alternative splicing. Information which allows the user to roughly assess the degree of evidence for alternative splicing of each gene is also provided. In most cases it is worthwhile to follow the hyperlink to the Tracks Display, where the quality of evidence for alternative splicing is more readily apparent to the eye (as in Fig. 1).
Our laboratory has been examining the hypothesis that regulated splicing events may be associated with departures from the consensus sequence at intron end points (10,11). To help test this notion, a database of 28 478 introns confirmed by cDNA alignments was assembled. This intron database can be searched for introns with particular sequences in exons or introns near the splice donor or acceptor sites. It returns a list of genes containing introns matching the query, which are in turn hyperlinked to the Tracks Display, where the intron will be highlighted. Underneath the list of ORFs, the database displays the frequency of occurrence of each base at each position near the splice junction for this family of introns. The logic of the search query can be controlled so that searches for introns that do not contain a certain nucleotide at a key position can also be done. This makes it an easy matter to view the 768 introns which depart from the gt..ag intron start and stop consensus sequence. As with the Alt-Splicing Catalog, the amount of evidence for any particular item in the database varies. The database sorts the introns that match a particular query so that those with the most supporting cDNAs appear at the top. An interesting example of the type of gene that can be discovered with this approach is shown in Figure 3. This gene was identified by searching for genes containing an intron that begins with GC instead of the canonical GU. By clicking on the hyperlink for C36F7.3 in that list we come to the Tracks Display for this gene in which the intron beginning with the non-canonical GC is highlighted in blue and in which evidence is shown that it defines an alternative exon.
Figure 3.

Display of an ORF in which an unusual intron—one which starts with GC instead of the canonical GU—is highlighted in light blue. EST evidence for alternative splicing of this gene is also shown.
A more detailed description of the methods used to generate the various parts of the Intronerator can be found by following the Algorithms hyperlink on the home page. The home page also includes a number of small but useful tools for molecular biology. These include: WormAlign, which aligns a single piece of DNA against the worm genome; FuzzyFinder, for small scale alignments between two DNA sequences; DNADuster, which is useful for adding or stripping numbers and spaces from DNA sequences and for translation; and PrimeMate, a convenient tool for generating PCR primers.
FUTURE DIRECTIONS
We are planning a number of enhancements to the Intronerator in the near future, many of which will be straightforward because of the direct comparison of genomic and mRNA sequence that Intronerator allows. These include a catalog of mRNAs for which evidence of possible RNA editing is present, a catalog of gene operons in C.elegans and alignments of homologous Caenorhabditis briggsae genomic regions. In the future we plan to extend Intronerator to cover the Drosophila, human and mouse genomes as those sequences become available.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank David Haussler, Mark Diekhans, David Kulp, Ewan Birney and Lincoln Stein for their advice, encouragement and help with the computational biology and web programming. We would also like to thank members of the Zahler lab and Manny Ares for helpful discussions on the biology. This work was supported by grant #1R01GM52848 from the National Institutes of Health and a grant from the University of California Cancer Research Coordinating Committee, both to A.M.Z. In addition, a training grant from the University of California Biotechnology Program provided support.
REFERENCES
- 1.The C.elegans Sequencing Consortium (1998) Science, 282, 2012–2018.9851916 [Google Scholar]
- 2.Fields S., Kohara,Y. and Lockhart,D.J. (1999) Proc. Natl Acad. Sci. USA, 96, 8825–8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benson D.A., Boguski,M.S., Lipman,D.J., Ostell,J., Ouellette,B.F., Rapp,B.A. and Wheeler,D.L. (1999) Nucleic Acids Res., 27, 12–17. Updated article in this issue: Nucleic Acids Res. (2000), 28, 15–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eeckman F.H. and Durbin R. (1995) Methods Cell Biol., 48, 583–605. [PubMed] [Google Scholar]
- 5.Altschul S.F., Madden,T.L., Schäffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolfsberg T.G. and Landsman,D. (1997) Nucleic Acids Res., 25, 1626–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gelfand M.S., Dubchak,I., Dralyuk,I. and Zorn,M. (1999) Nucleic Acids Res., 27, 301–302. Updated article in this issue: Nucleic Acids Res. (2000), 28, 296–297.9847209 [Google Scholar]
- 8.Black D.L. (1995) RNA, 1, 763–771. [PMC free article] [PubMed] [Google Scholar]
- 9.Kao H.Y. and Siliciano,P.G. (1996) Mol. Cell Biol., 16, 960–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopez A.J. (1998) Annu. Rev. Genet., 32, 279–305. [DOI] [PubMed] [Google Scholar]
- 11.Chabot B. (1996) Trends Genet., 12, 472–478. [DOI] [PubMed] [Google Scholar]