

Abstract
Comprehensive phenotypic characterization of the many mutations found in cancer tissues is one of the biggest challenges in cancer genomics. In this study, we evaluated the functional effects of 29,060 cancer-related transition mutations that result in protein variants on the survival and proliferation of non-tumorigenic lung cells using cytosine and adenine base editors and single guide RNA (sgRNA) libraries. By monitoring base editing efficiencies and outcomes using surrogate target sequences paired with sgRNA-encoding sequences on the lentiviral delivery construct, we identified sgRNAs that induced a single primary protein variant per sgRNA, enabling linking those mutations to the cellular phenotypes caused by base editing. The functions of the vast majority of the protein variants (28,458 variants, 98%) were classified as neutral or likely neutral; only 18 (0.06%) and 157 (0.5%) variants caused outgrowing and likely outgrowing phenotypes, respectively. We expect that our approach can be extended to more variants of unknown significance and other tumor types.
Analyses of tumor samples have revealed a landscape of cancer mutations1–3 along with lists of putative cancer-related genes4–7. Although these efforts have greatly enhanced understanding of cancer genomics, the phenotypic effects of most cancer-associated mutations are unknown. A method for the high-throughput functional evaluation of variants, including variants of unknown significance (VUSs) in mammalian cells, exogenous expression of 8,258 and 5,708 p53 protein variants8,9, 9,595 PPARG protein variants10 and 6,810 ERK2 protein variants11, has been performed in cells in which the corresponding endogenous target genes had previously been knocked out. In addition, yeast cells12 and fluorescent reporters13–15 have been used to determine the function or stability of thousands of exogenously expressed protein variants. However, the function of such exogenously expressed variants can differ from that of the corresponding endogenous mutant proteins encoded by genes expressed using their native promoters and enhancers. Thus, homology-directed repair (HDR) has been used as a method for introducing sequence changes into an endogenous gene16 and enabled the determination of the functional effects of 3,893 point mutations (resulting in 2,251 protein variants) in a single endogenous gene, BRCA1 (ref. 17). However, the efficiency of HDR is usually limited in mammalian cells, and the induction of a large number of variations in multiple genes using HDR is currently very challenging.
Approximately 50–70% of mutations found in cancer tissue are transition point mutations4,18. Transition mutations can be induced using base editors, which are composed of a Cas9 nickase–deaminase fusion protein and an sgRNA19–21. In this study, we used a high-throughput method to evaluate the functional effects of cancer-associated non-synonymous transition mutations on the proliferation and survival of pre-cancerous non-tumorigenic bronchial epithelial cells using cytosine base editor (CBE) and adenine base editor (ABE) and associated sgRNA libraries.
Results
Generating cancer-associated mutations using base editors.
To introduce cancer-associated transition mutations into endogenous target sequences using CBE and ABE, we first generated cell lines that express CBE or ABE in a doxycycline-responsive manner. HBEC30KT cells are immortalized non-tumorigenic bronchial epithelial cells derived from normal lung cells22. As pre-cancerous cells, we used HBEC30KT cells that lentivirally express a short hairpin RNA (shRNA) targeting TP53 (HBEC30KT-shTP53; hereafter, for brevity, P cells). Although P cells express only low levels of TP53 mRNA, gene set enrichment analysis showed that the p53 pathway was upregulated (Extended Data Fig. 1). Similarly to HBEC30KT cells, P cells require epidermal growth factor (EGF) for cell expansion and are non-tumorigenic22. We sequentially transduced lentiviral vectors expressing reverse tetracycline-controlled transactivator (rtTA) and a base editor (CBE or ABE; Fig. 1a) into P cells (Methods). The resulting cell lines, which express CBE or ABE in a doxycycline-inducible manner, were named P-C cells or P-A cells, respectively.
Fig. 1 |. Base editor-directed generation of cancer-associated transition mutations.
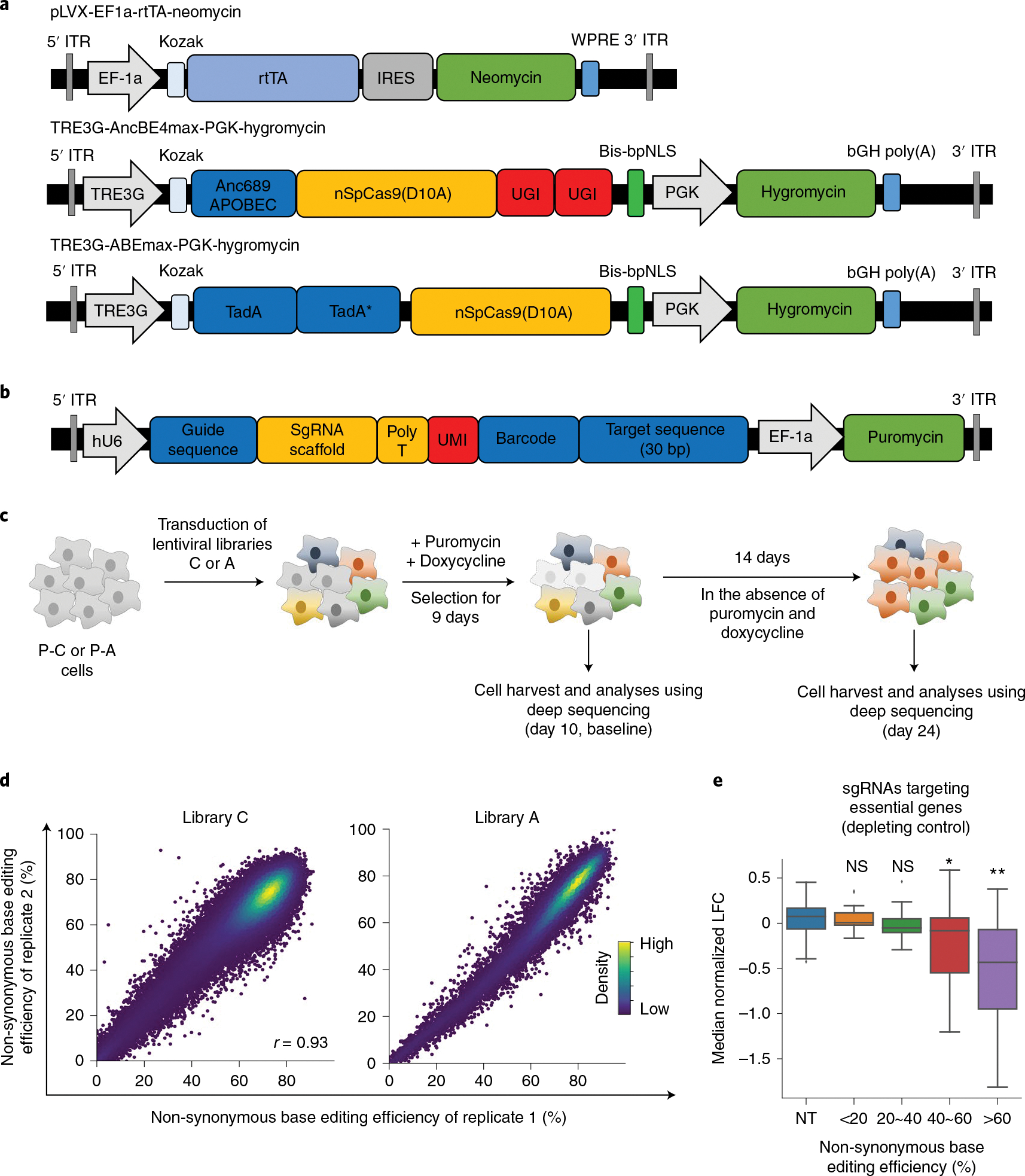
a, Maps of lentiviral vectors used for the expression of rtTA (pLVX-EF1a-rtTA-neomycin), CBE (TRE3G-AncBE4max-PGK-Hygromycin) and ABE (TRE3G-ABEmax-PGK-Hygromycin). These vectors were used to generate P-C cells and P-A cells. Anc689APOBEC, codon-optimized ancestral APOBEC1 (AncBE4max);26 TadA, tRNA adenosine deaminase;80 bis-bpNLS, biparticle nucleus localization signal at both the N- and C-termini;26 TRE3G, tetracycline response element 3G promoter. b, A map of the lentiviral vector containing the library of sgRNA-encoding sequence and surrogate target sequence pairs. c, Schematic of CBE-mediated and ABE-mediated high-throughput evaluations of variants. P-C cells and P-A cells are non-tumorigenic human bronchial epithelial cells that express CBE and ABE, respectively, in a doxycycline-dependent manner. P-C cells and P-A cells were transduced with lentiviral sgRNA libraries C and A, respectively. In different replicates, cells were transduced with different batches of the lentiviral library on different days. Untransduced cells were removed by puromycin selection, and the expression of CBE or ABE was induced for 9 days by doxycycline. Ten days after the transduction, half of the cells were harvested for analyses, and the remaining cells were cultured in the absence of puromycin and doxycycline for another 14 days, after which these cells were also analyzed. d, Correlations between non-synonymous base editing efficiencies at the integrated target sequences of biological replicates. The color of each dot was determined by the number of neighboring dots (that is, dots within a distance that is three times the radius of the dot). Pearson correlation coefficients (r) are shown. e, Distribution of median normalized LFCs of 190 sgRNAs targeting essential genes depending on the non-synonymous base editing efficiencies determined at the integrated target sequences in library C2. NT, non-targeting sgRNAs. The number of sgRNAs n = 99 (NT), 13 (<20%), 17 (20%~40%), 31 (40%~60%) and 129 (>60%). Box plots are represented as follows: center line of box indicates the median; box limits indicate the upper and lower quartiles; and whiskers show the 1.5 times interquartile range (in comparison with NT; two-sided Student’s t-test; NS, not significant, *P = 6.1 × 10−7, **P = 2.3 × 10−21).
To identify target sequences that can be modified by CBE or ABE to contain transition mutations observed in human cancer tissues, we used the Catalogue of Somatic Mutations in Cancer (COSMIC)23 and identified 84,806 C > T and G > A single-nucleotide variants (SNVs) and 23,176 A > G and T > C SNVs that can be respectively generated by CBE and ABE at high predicted efficiencies using 80,203 and 23,008 sgRNAs (Extended Data Fig. 2a and Methods). We also added two negative control groups of sgRNAs: the first group contained sgRNAs that do not target any sequences in the human genome (hereafter, non-targeting sgRNAs or NT); the second group consisted of sgRNAs that, with CBE or ABE, would induce synonymous mutations24,25. As a result of this process, we prepared 83,731 and 23,613 sgRNAs for CBE and ABE, respectively (Extended Data Fig. 2b and Supplementary Table 1). The efficiency of base editing is often limited, and base editing can result in multiple editing outcomes in addition to the intended editing19,21,26,27. To monitor base editing efficiencies and outcomes, we added corresponding surrogate target sequences to the sgRNA-encoding lentiviral vector (Fig. 1b) as we previously did to evaluate the efficiencies of CRISPR nucleases28–32, base editors27 and prime editor 2 (ref. 33).
We generated lentiviral libraries, respectively named libraries C and A, of the 83,731 (for CBE) and 23,613 (for ABE) pairs of sgRNA-encoding and target sequences described above (Fig. 1b). The frequency of shuffling of barcodes and sgRNA-encoding sequences34 would be about 4.3%32,33, which would not substantially affect the functional evaluations (Supplementary Fig. 1 and Supplementary Note 1). In addition, we added an 8-nucleotide-(nt)-long unique molecular identifier (UMI) between the sgRNA-encoding and target sequences in both libraries for tracking of transduced cells and subsequent analyses35,36. We respectively transduced libraries C and A into P-C cells and P-A cells in duplicate and supplemented the culture medium with doxycycline to induce CBE or ABE expression (Methods and Fig. 1c). When the base editing efficiencies at the integrated target sequences were measured at day 10 after the initial transduction, the efficiencies were found to be high (Extended Data Fig. 3a,b); the median efficiencies at positions 4, 5, 6 and 7 were 37%, 59%, 61% and 53% for CBE and 16%, 68%, 68% and 59% for ABE. The measured base editing efficiencies were predicted better by base editor efficiency-predicting computational models such as DeepCBE27, DeepABE27 and BE-Hive37 than by Cas9 nuclease activity-predicting models such as DeepSpCas9 (ref. 30) and Rule Set 2 (ref. 38) (Supplementary Fig. 2a). The effects of the sequence context surrounding the target nucleotide on base editing efficiencies, which are compatible with previously reported results27,37, are shown in Supplementary Fig. 2b,c. When we compared amino acid-changing or non-synonymous editing efficiencies in independent biological replicates, we observed high correlations, with Pearson correlation coefficients of 0.93 and 0.97 (Fig. 1d). Very low levels of indels were observed at the integrated target sequences (Extended Data Fig. 3c; median 0.63% and 0.45% for library C, 0.2% and 0.31% for library A), which is in line with previous results19,21. In addition, a fraction, albeit minor, of synonymous control sgRNAs also showed non-synonymous editing (median 24–29% in libraries A and C; Extended Data Fig. 3d), emphasizing the importance of monitoring base editing efficiencies and outcomes. Thus, we did not use these sgRNAs as negative controls in subsequent analysis.
Next, we investigated the relationship between the base editing efficiency at integrated target sequences and phenotypic changes. Using 190 unique sgRNAs targeting 65 curated essential genes in the C2 library, which will be described below (Methods), we found robust depletion of sgRNA-transduced cells when the non-synonymous base editing efficiency in the surrogate sequences was over 60% (Fig. 1e and Extended Data Fig. 4a). In line with this finding, receiver operating characteristic analyses revealed that essential gene-targeting sgRNAs with efficiencies higher than 60% at the integrated target sequences had better performance, with an area under curve (AUC) of 0.77 in library C2 and 0.72 in library C, than did those with efficiencies less than or equal to 60% (sgRNA efficiencies <40%, AUCs of 0.4 and 0.58 in libraries C2 and C, respectively; 40% ≤ sgRNA efficiencies ≤ 60%, AUCs of 0.64 and 0.59 in libraries C2 and C, respectively) (Extended Data Fig. 4b,c). Therefore, we assumed that sgRNAs associated with a less than 60% base editing efficiency in surrogate sequences could result in insufficient base editing at the endogenous target sites, which could mask a possible outgrowing or depleting phenotype associated with such sgRNAs. In addition, filtering out sgRNAs with a low number of UMIs improved the accuracy of functional classifications (Extended Data Fig. 4d). When we mathematically calculated the relationship between the base editing efficiencies at endogenous sites and the log-fold changes (LFCs) of the corresponding sgRNAs as a parameter for the growth phenotype, such as an increase or decrease in proliferation and survival, the LFC and base editing efficiency correlated almost, albeit not exactly, in a linearly proportional manner when the growth phenotype was relatively modest as observed in our experimental settings (Supplementary Note 2 and Supplementary Fig. 3a–c). In addition, when base editing efficiencies were lower than 60%, a larger percentage of sgRNAs inducing stop codons in essential genes were classified as neutral than when the efficiencies were higher than 60% (Supplementary Fig. 3d). Thus, we filtered out those inefficient sgRNAs and sgRNAs with an insufficient number of UMIs (<50) at day 10 from functional classifications.
Functional classification of cancer-associated mutations.
To evaluate the functional effects of the variants generated by CBE and ABE on cell proliferation and survival, we cultured these mutation-containing cell populations in the absence of doxycycline for 14 days (Fig. 1c). Genomic DNA was isolated from the cell populations at day 10 (baseline) and day 24 after the initial transduction of libraries C and A and subjected to deep sequencing to evaluate the relative frequencies of sgRNA and target sequence pairs and UMIs. We calculated median LFCs and P values for each sgRNA (Methods and Supplementary Table 2). Based on the −log10(P value)39 and the median LFC of each sgRNA, we functionally classified the sgRNAs into depleting, likely depleting, likely neutral (possibly depleting), neutral, likely neutral (possibly outgrowing), likely outgrowing and outgrowing using the distribution of control non-targeting sgRNAs (Fig. 2a,b and Supplementary Table 3). When we classified nonsense mutation-inducing sgRNAs that target common essential genes defined in DepMap40, only 47% of sgRNAs with non-synonymous base editing efficiencies lower than 60% were classified as depleting, likely depleting or possibly depleting, whereas 69% of sgRNAs with editing efficiencies higher than 60% were classified as depleting, likely depleting or possibly depleting (Supplementary Fig. 3d), underscoring the importance of base editing efficiency monitoring.
Fig. 2 |. Functional classification of cancer-associated transition mutations.
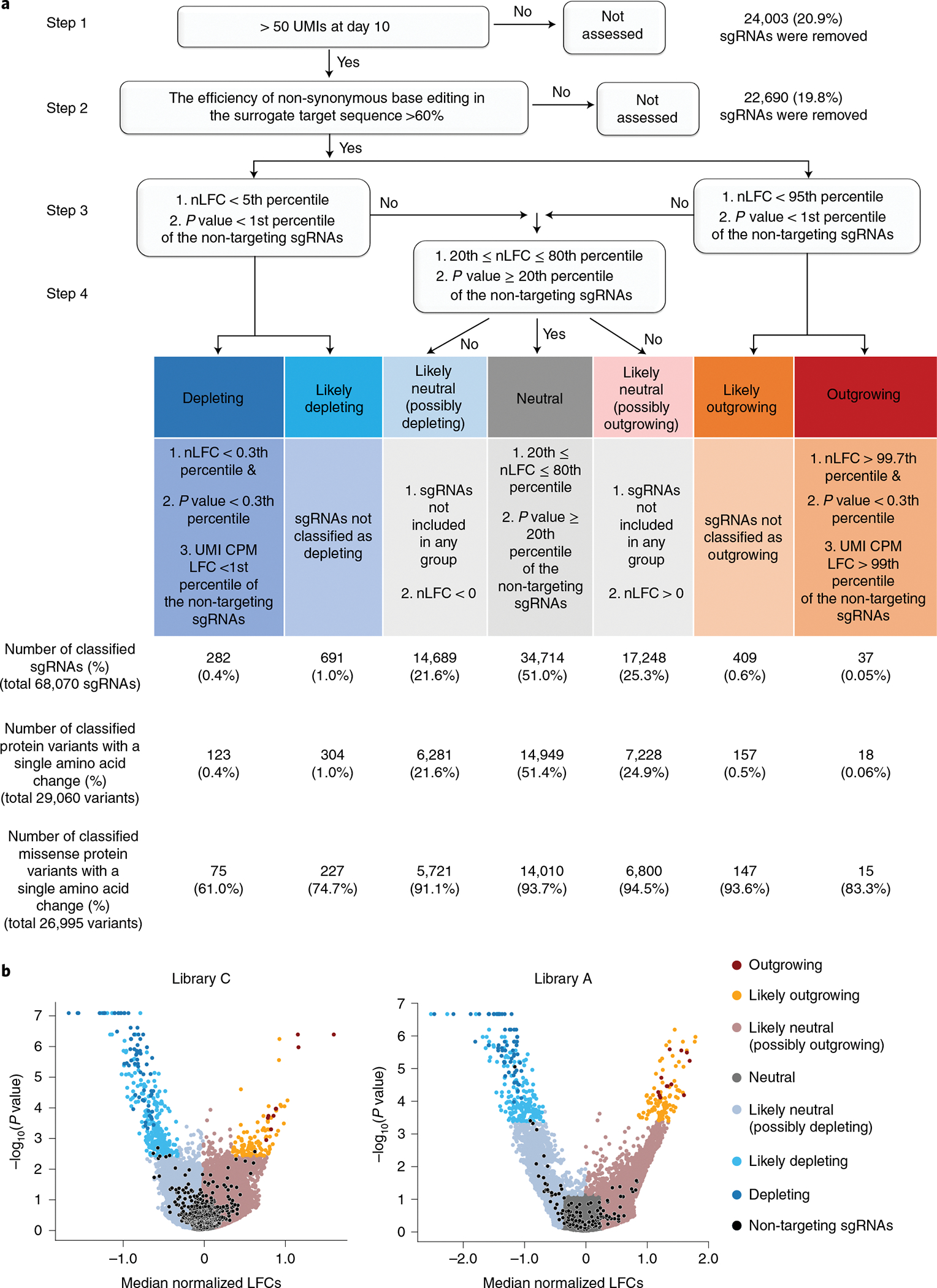
a, Functional classification process. (Step 1) sgRNAs harboring more than 50 UMIs were used as inputs for MAGeCK analysis. (Step 2) sgRNAs associated with a non-synonymous editing efficiency of less than 60% in the integrated target sequence were eliminated. (Step 3) sgRNAs were grouped depending on their nLFCs and P values obtained from MAGeCK-UMI analyses. The cutoff value was determined by the distribution of the non-targeting controls in each library. (Step 4) For the outgrowing and depleting groups, UMI CPM LFCs were further considered to prevent false classification into outgrowing and depleting groups. The number of sgRNAs and mutant proteins classified in each group are shown in the chart (integrated results based on libraries C, C1, C2, C3, A, A1, A2 and dA are shown). b, Volcano plots of nLFCs and negative logarithm of RRA P values of sgRNAs. The colors of the dots (sgRNAs) represent their functional classifications. Non-targeting sgRNAs are shown in black.
In addition to changes in the abundance of each UMI (that is, the LFC in the reads per million (RPM) of a UMI), we also used the number of UMIs for each sgRNA (that is, the LFC in the UMI counts per million (CPM)) for functional classification (Methods). The number of UMIs for each sgRNA decreases if an sgRNA-induced mutation is depleting36. Thus, the number of UMIs for each sgRNA was previously used as an additional parameter to increase the accuracy of hit calling in Cas9-based screening36. Indeed, we found that the LFC in the UMI CPM for the CBE sgRNAs targeting essential genes (depleting sgRNAs) in library C2, which is described below, as well as the median of LFCs in the RPMs of UMIs for the depleting sgRNAs, decreased over time from day 10 to day 24 (Extended Data Fig. 4e). To reduce the number of false depleting or outgrowing sgRNAs in the classifications, we classified sgRNAs that meet the criteria for depleting or outgrowing with respect to the LFC in the RPM and P value as likely depleting or likely outgrowing if the LFC in the UMI CPM did not suggest depletion or outgrowth, respectively (Fig. 2a).
Functional evaluations are reproducible at different scales.
To test whether the scale of the high-throughput evaluation can be modified and whether the classification results are reproducible using independent libraries, we prepared three smaller libraries (containing 3,261 and 3,170 unique sgRNAs for CBE and 1,595 unique sgRNAs for ABE), named C1, C2 and A1, respectively (Extended Data Fig. 5a,b and Methods). We observed high correlations between non-synonymous base editing efficiencies at the same integrated target sequences in libraries C, C1 and C2 and libraries A and A1 (Fig. 3a and Extended Data Fig. 5c). We performed functional classification of sgRNAs using the methods described in Fig. 2a (Fig. 3b) and found that the classifications of variants using the large libraries (C and A) are compatible with those using smaller libraries C1, C2 and A1 (Fig. 3c), suggesting that our high-throughput classifications are reproducible even when the scale of the experiments is reduced.
Fig. 3 |. High-throughput classifications are reproducible at different scales.
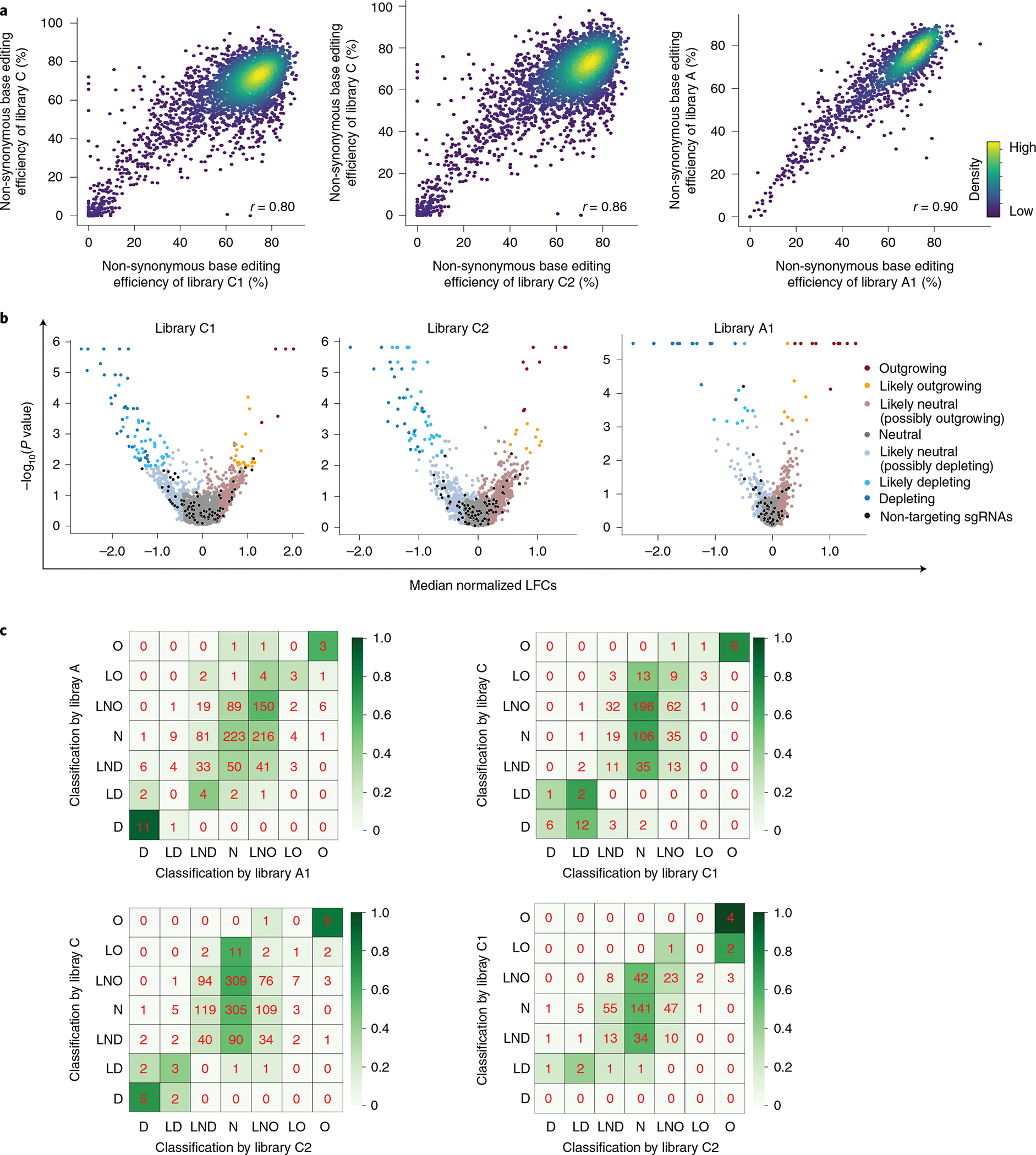
a, Correlations between non-synonymous base editing efficiencies at the integrated target sequences of libraries C and A and small libraries C1, C2 and A1. Only sgRNAs with more than 100 raw read counts for each replicate or library were included. The base editing efficiencies were determined 10 days after the initial transduction of the library. Pearson correlation coefficients (r) are shown. b, Volcano plots of nLFCs and negative logarithm of RRA P values of sgRNAs. The colors of the dots (sgRNAs) represent their functional classifications (using the same colors shown in Fig. 2b). Non-targeting sgRNAs are shown in black. c, Heat maps showing the correlations between functional classifications made using libraries C and A and small libraries C1, C2 and A1. The color intensity was determined by the relative number of variants within each cell in each row. D, depleting, LD, likely depleting, LND, likely neutral (possibly depleting), N, neutral, LNO, likely neutral (possibly outgrowing), LO (likely outgrowing), O (outgrowing).
Functional classification using three more small libraries.
Whereas most nonsense mutations lead to loss of function, the functional effects of missense mutations are more difficult to predict. Thus, we generated two more ABE libraries: one named dA (drivers for ABE) that can induce 2,797 missense transition mutations observed in 262 driver genes and another named A2 that can induce 1,468 missense transition mutations observed in 627 tumor suppressor genes (Methods). In addition, we generated another library named C3 to induce 1,080 missense transition mutations and 83 nonsense mutations observed in 116 tumor suppressor genes (Methods). These three libraries were used to determine the functional effects of variants as similarly conducted for libraries C, C1, C2, A and A1 (Supplementary Fig. 4).
Functional classification based on integrated results.
We integrated the results from libraries C, C1, C2, C3, A, A1, A2 and dA (Methods) and provide the classifications of the sgRNAs and associated protein variants (Supplementary Table 3). A total of 68,070 sgRNAs were classified as follows: 282 depleting, 691 likely depleting, 14,689 likely neutral (possibly depleting), 34,714 neutral, 17,248 likely neutral (possibly outgrowing), 409 likely outgrowing and 37 outgrowing. Analysis of the surrogate target sequences allowed us to identify which sgRNAs mainly induced a single protein variant in a highly efficient manner. In these cases, the phenotype induced by the sgRNA can be attributed to that major protein variant, which we call a ‘primary’ protein variant for the sgRNA in this study. We classified a protein variant as a primary protein variant if its relative frequency among all the base editor-generated protein variants was higher than 75% (the frequency of the primary variant was at least three times higher than the combined frequencies of the remaining base-edited variants) (Methods). Among 68,070 sgRNAs functionally classified in this study, 29,060 (43%) induced one primary mutation, enabling the pairing of that primary mutation with the cellular phenotype caused by sgRNA-induced base editing (Extended Data Fig. 6a). Thus, we provide functional classifications for 29,060 protein variants generated by transition mutations: 123 depleting, 304 likely depleting, 6,281 likely neutral (possibly depleting), 14,949 neutral, 7,228 likely neutral (possibly outgrowing), 157 likely outgrowing and 18 outgrowing (Fig. 2a and Supplementary Table 3).
Each of the remaining 39,012 functionally classified sgRNAs induced either a single primary variant with two amino acid changes (12,820 sgRNAs) or a group of variants without a primary variant (26,192 sgRNAs). Although we cannot determine with certainty the functional effects of single amino acid changes in the variants induced by the 39,012 sgRNAs, the information we have provided about the functional effects of the variant groups, together with the frequency of each variant in the variant group determined by analysis of the surrogate target sequences, could be, albeit at low accuracy, informative and should assist in predicting the functional effects of single amino acid changes, especially when the phenotypes induced by the sgRNAs are classified as neutral. Most sgRNAs (77%, 20,138) out of the 26,192 sgRNAs that induced multiple variants without a primary variant resulted in two protein variants if we count variants whose frequencies are higher than 10% (Extended Data Fig. 6b). Thus, phenotypes of these 20,138 sgRNAs could be attributable to either or both of the two corresponding protein variants. We also performed potential functional classifications of 22,690 sgRNAs that showed editing efficiencies lower than 60%. (Supplementary Table 3). However, it should be noted that the accuracy of this classification is expected to be low.
Several computational models are available to predict the functional effects of variants. We evaluated the correlations between our functional classifications and the predicted scores of 4,143 variants using CTAT-cancer4 and CHASM41 and those of 3,899 variants using SIFT42 and PolyPhen-2 (ref. 43). We found modest correlations; models tended to predict that functional effects would be caused by variants placed in non-neutral categories (such as outgrowing or depleting) by our system, in comparison with their predictions for neutral variants (Extended Data Fig. 7a,b). Previously, the accuracies of these computational models have been shown to be limited, and the necessity of functional evaluations for more accurate determination of variant functions has been emphasized44–46. In line with these previous findings, the modest correlations observed in the current study corroborate the importance of functional assays17. In addition, we also assessed correlations between depleting effects of variants in common essential genes and those predicted by computational models. We found modest correlations between LFCs and SIFT and PolyPhen-2 scores (Extended Data Fig. 7c,d), underscoring the necessity of functional evaluations.
We provide the results of this classification in Supplementary Table 3. We also provide a webtool at http://deepcrispr.info/BEvariants that researchers can use to find functional classifications of variants in a gene of interest.
Individual validations of high-throughput classifications.
To validate our classifications based on the results of the high-throughput experiments, we individually tested the effects of variants generated by base editing. We selected 28 sgRNAs used for the high-throughput evaluations (Methods and Supplementary Fig. 5) and individually delivered these sgRNAs into P-C cells or P-A cells via lentiviral transduction. The transduced cells were cultured with doxycycline for 7 days to induce base editing and incubated for an additional 14 days in the absence of doxycycline. The cells were harvested and analyzed at 6, 10, 17 and 24 days after infection to track individual allele frequencies after delivery of an sgRNA (Fig. 4a, upper panel). As expected, we observed high correlations between the frequencies of 61 base-edited alleles induced by the 20 selected sgRNAs at integrated target sequences of the high-throughput experiments and those at the endogenous target sites in the independent individual experiments (Fig. 4b; Pearson r = 0.72 and Spearman R = 0.70).
Fig. 4 |. Individual validation of sgRNas and their associated base-edited variants supports the high accuracy of high-throughput functional classifications.
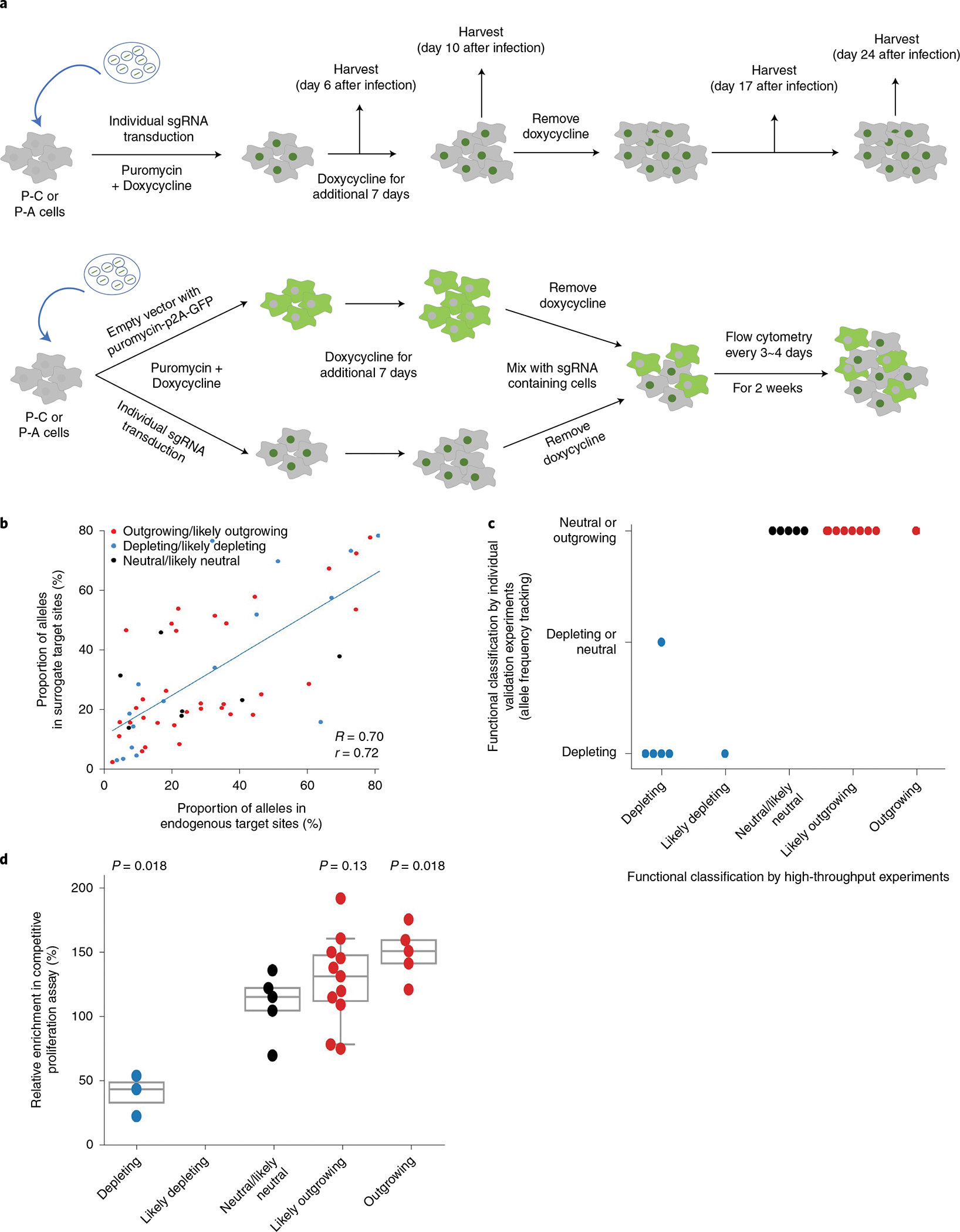
a, Schematic of experiments to validate the function of each sgRNA and variant. Competitive proliferation (top) and allele frequency tracking (bottom) assays are shown. b, Correlation between frequencies of base-edited outcome sequences induced by base editing at endogenous target sites in individual validation experiments and corresponding integrated target sequences in the high-throughput experiments. Spearman correlation (R) and Pearson correlation (r) coefficients are shown. Base editing outcomes with frequencies higher than 1% are included. The number of base editing outcome sequences n = 57. c, Correlation of phenotypes caused by sgRNA-induced base editing determined by individual allele frequency tracking experiments and high-throughput experiments. The number of sgRNAs n = 20. d, Correlation of phenotypes caused by sgRNA-induced base editing determined by individual competitive proliferation assays and high-throughput experiments. The number of sgRNAs n = 24. Statistical significances determined by comparison with the neutral/likely neutral group are shown (two-sided Mann–Whitney U-test). Box plots are represented as follows: center line of box indicates the median; box limits indicate the upper and lower quartiles; and whiskers show the 10th and 90th percentiles.
Deep sequencing showed that the frequencies of variants generated by the base editing increased, remained unchanged or decreased (Extended Data Fig. 8 and Supplementary Table 4). We classified an sgRNA as depleting if the base-edited variant frequency decreased and the wild-type sequence frequency increased over time after day 10. When the base-edited variant frequency increased and the wild-type sequence frequency decreased, the relevant sgRNA was classified as outgrowing or neutral, considering that the leaky expression of base editors even in the absence of doxycycline47,48 can, albeit slightly, increase the frequency of base-edited variants. When the frequencies of base-edited variants and wild-type sequences were unchanged over time after day 10, we classified the sgRNAs as neutral or depleting, considering the leaky expression of base editors. We found that the results of this functional classification of individual sgRNAs based on the frequencies of variant and wild-type sequences were in line with those from the high-throughput evaluations (Fig. 4c and Extended Data Fig. 8).
Because it is difficult to distinguish outgrowing and neutral phenotypes using such variant frequency tracking, we next performed competitive proliferation assays49 to compare the proliferation of sgRNA-transduced and non-transduced cells (Fig. 4a, lower panel, and Methods). We classified sgRNAs based on the enrichment or depletion of the sgRNA-transduced cells over time as compared to those transduced with non-targeting sgRNAs. Flow cytometry showed that the classifications based on this assay were compatible with those of the high-throughput evaluations (Fig. 4d and Extended Data Fig. 9), supporting the high accuracy of the high-throughput evaluations.
Evaluation of dependency on EGF signaling.
The above analyses are based on evaluations of cell proliferation and viability. Given that one of the most important hallmarks of cancer is self-sufficiency in growth signals5,50,51, we evaluated the cells’ dependency on a growth signal, EGF, which is required for the proliferation of P cells. We generated a library named eC (epidermal growth factor-CBE) to induce 3,967 transition mutations observed in 162 genes relevant to the EGF/EGF receptor (EGFR) signaling pathway (Methods). The eC library was transduced into P-C cells, and base editing was induced by the addition of doxycycline. The cell population was split into an EGF depletion arm, in which EGF was removed and 10 nM of the EGFR inhibitor afatinib was added, and an untreated control arm, after which both arms were cultured for an additional 10 days (Fig. 5a).
Fig. 5 |. Base editor-directed investigation of mutations related to resistance to an egFR tyrosine kinase inhibitor.
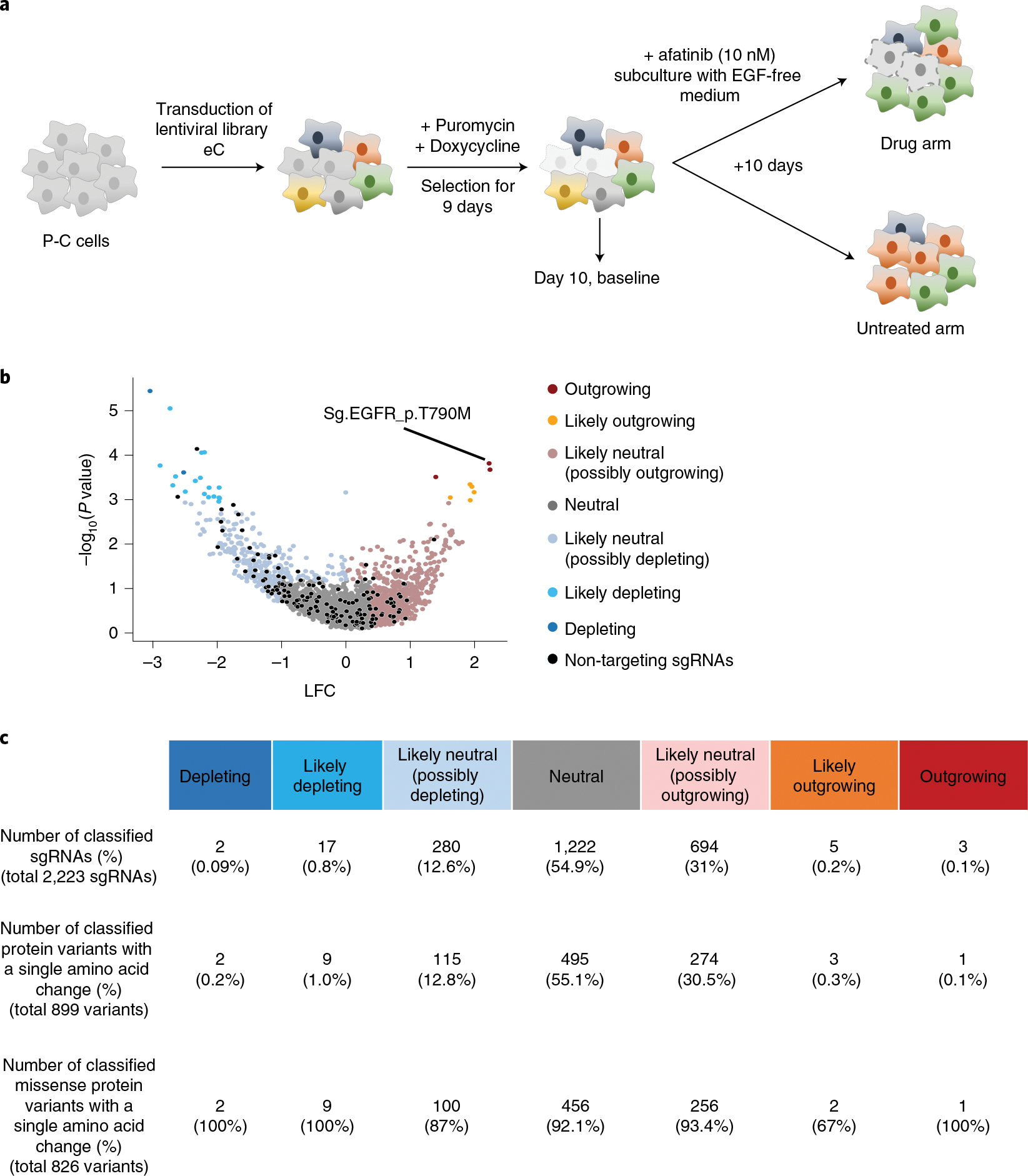
a, Schematic of CBE-mediated high-throughput evaluations of variants conferring resistance to the EGFR tyrosine kinase inhibitor afatinib. P-C cells were transduced with lentiviral sgRNA library eC in duplicate. In different replicates, cells were transduced with different batches of the lentiviral library on different days. Untransduced cells were removed by puromycin selection, and the expression of CBE or ABE was induced for 9 days by doxycycline. Ten days after the transduction, half of the cells were harvested for analyses, and the remaining cells were refreshed with EGF-free medium containing 10 nM afatinib. b, Volcano plots of nLFCs and RRA P values of sgRNAs. The colors of the dots (sgRNAs) represent their functional classifications (using the same colors shown in Fig. 2b). Non-targeting sgRNAs are shown in black. The sgRNA generating EGFR_p.T790M is indicated. c, The numbers of sgRNAs and mutant proteins classified in each group are shown.
Similarly to experiments conducted above, we functionally classified sgRNAs by comparing the number of cells in the EGF depletion group with that in the control group (Fig. 5b). After evaluation of editing outcomes at the integrated target sequences, we functionally classified a total of 899 protein variants with a single amino acid change, identifying only one outgrowing variant conferring resistance to afatinib, EGFR_p.T790M, which is a well-known gain-of-function mutation52; two depleting variants, SH3GL3_p. D169N and PIK3C2B_p.E650K; and 495 neutral variants (Fig. 5c). Our high-throughput evaluation revealed that EGFR_p.P753S, which has been a VUS, is associated with a likely depleting phenotype. Our finding is compatible with a case report that a patient with this variant showed a markedly positive response to therapy with cetuximab, an EGFR inhibitor, for treating cutaneous squamous cell carcinoma53.
Notable findings from the functional classifications.
Among the classified protein variants, most of them (28,458/29,060 = 98%) were classified as neutral (14,949, 51%) or likely neutral (13,509, 46%). This wet-experimental evidence is line with the previous expectation that most mutations found in cancer would be passengers rather than drivers7,18.
Notable gene groups related to the outgrowing phenotype included those in the Cancer Gene Census (CGC)5. Among the 68,070 functionally classified sgRNAs, 9.0% (6,119) targeted CGC genes (Extended Data Fig. 10a, top). However, the fraction of CGC genes was significantly enriched in the outgrowing and likely outgrowing sgRNA groups, to 15% (63/409) and 38% (14/37), respectively (P values = 2.8 × 10−5 and 1.9 × 10−6, respectively; Fisher’s exact test). Among the sgRNAs targeting CGC genes in these two groups, the largest fraction, 35% (27/77), targeted TP53. When we performed the same analysis using 29,060 functionally classified protein variants, we observed similar findings (Extended Data Fig. 10a, bottom). The gene most frequently containing outgrowing and likely outgrowing variants among CGC genes was TP53 (36% (10/28)). Conversely, of the 40 classified variants in TP53, one and nine were linked to outgrowing and likely outgrowing phenotypes, respectively. Although p53 protein expression is knocked down in P-C cells and P-A cells by a constitutively expressed shRNA that targets TP53 RNA, the p53 protein would still be expressed, albeit at a low level. Loss-of-function mutations in TP53 could further reduce p53 activity, enhancing cell survival and proliferation.
Notable gene groups related to the depleting phenotype included common essential genes. Among the depleting and likely depleting sgRNAs, 52% (147/282) and 27% (190/691) were associated with common essential genes40, respectively (P values = 6.9 × 10−69 and 1.5 × 10−98, respectively), whereas only 6.1% (4,153/68,070) of all functionally classified sgRNAs targeted common essential genes (Extended Data Fig. 10b, left graph). Similar enrichment of protein variants encoded by common essential genes was observed when we performed the analysis using 29,060 functionally classified protein variants instead of sgRNAs (Extended Data Fig. 10b, right graph).
Other notable findings from our study related to individual variants include the following. First, the p.Y727C mutation affecting EGFR caused an outgrowing phenotype observed in both the high-throughput analyses and individual validation experiments. Given that EGFR activation induces cell proliferation and survival54, the p.Y727C mutation, which was annotated as a VUS by ClinVar55, should be classified as a gain-of-function mutation based on our results. Another interesting finding is related to PHLDA1, a protein that inhibits Akt56. Although decreased PHLDA1 expression has been reported in several tumors57, the role of PHLDA1 mutations in cancer has not been well-evaluated. Both our high-throughput analyses and individual experiments suggest that the p.Q201* and p.Y249C variants cause an outgrowing and likely outgrowing phenotype, respectively, indicating that these mutations can increase cell survival and proliferation. IRF6 has tumor suppressor activity in squamous cell carcinomas;58 furthermore, the p.Y97C mutation in IRF6 has been proposed as the cause of Van der Woude syndrome59, characterized by a cleft lip and palate, raising the possibility that p.Y97C might not be neutral. Both our high-throughput analyses and individual experiments show that p.Y97C causes an outgrowing phenotype. CASP8 is relevant to apoptosis and has been proposed to be a tumor suppressor60. Our high-throughput analysis showed that two variants, whose functions are unknown, p.S158F and p.Y507C, cause outgrowing and likely outgrowing phenotypes, respectively. An individual validation experiment confirmed the outgrowing phenotype of p.S158F. CREBBP has been proposed to be a tumor suppressor against small cell lung carcinoma61, leukemia and lymphoma62, and the p.Y1482H variant has been reported to be a loss-of-function mutation in lymphoma62. In line with these findings, our high-throughput evaluation revealed that p.Y1482H causes an outgrowing phenotype.
Discussion
In this study, we classified 29,060 protein variants, including 26,995 missense variants (93%), encoded by a total of 11,637 genes based on their effects on non-tumorigenic bronchial epithelial cell proliferation and survival, and 899 variants, including 826 missense variants (92%), based on their effects on independency from EGF signaling. The vast majority of the variants (98%) were classified as neutral or likely neutral, which could be compatible with the expectation that most mutations found in cancer would be passengers rather than drivers7,18. Because not every driver mutation necessarily leads to a proliferation advantage on its own, we classified the variants with respect to their effects on cell proliferation and survival, using labels such as outgrowing, neutral and depleting rather than in terms of tumorigenicity, which would have involved classifying variants as drivers, passengers and tumor suppressors. Subsequent functional evaluations of the variants in vivo could provide further information about tumorigenic functions of these variants.
In addition, because the pre-cancerous cells (P cells) used in this study are derived from primary bronchial epithelial cells, the functions shown in our study could be different in other cell types. Although the distribution of lung cancer-associated mutations in each functional category was similar to that of mutations associated with other cancers (Supplementary Fig. 6a), conducting high-throughput evaluations in other cell types could provide more solid generalized conclusions. In addition, the distribution of mutations that are also listed in The Cancer Genome Atlas (TCGA) is similar to that of mutations not listed in TCGA (Supplementary Fig. 6b).
Although CBE, but not ABE, was recently used to find transition mutations with functional effects24,63, these screenings required individual validations of the potential hits because base editing efficiencies and outcomes were not monitored during the screening. We enabled high-throughput functional evaluations of transition mutations by monitoring base editing efficiencies and outcomes with surrogate target sequences and using UMIs. Very recently, a high-throughput evaluation of CBE-induced, but not ABE-induced, cancer-related variants using a similar base editing efficiency and outcome monitoring system was also reported64. However, the base editing outcomes and efficiencies at the integrated target sequences can sometimes differ from those at the corresponding endogenous sites (Fig. 4b); in these cases, the determination of a primary protein variant for each sgRNA could be less accurate. Although the direct monitoring of base editing outcomes at the endogenous sites could prevent this potential discrepancy, it is not practically possible to monitor all the endogenous sites in these large-scale, high-throughput experiments. Individual or small-scale experiments involving the direct monitoring of the endogenous sites could improve the accuracy of determining the primary protein variant.
Among 1,488,704 C•G to T•A mutations and 381,635 A•T to G•C mutations reported in COSMIC, we identified only 84,806 C•G to T•A mutations (5.7%) and 23,176 A•T to G•C mutations (6.1%) that can be efficiently generated by CBE and ABE, respectively. Most of the mutations were filtered out due to a lack of an NGG PAM or expected low base editing activities. NGG PAM-based restrictions in which sites were potentially editable were similar between CGC genes and other genes (Supplementary Fig. 7). Using base editors based on a CRISPR nuclease with different PAM compatibilities, such as SaCas9 (refs. 65,66) or Cas12a67,68, or engineered SpCas9 variants with broadened or different PAM compatibilities32,69–73, could potentially extend the list of transition mutations that can be functionally evaluated using base editors.
In addition, the relatively wide editing windows of the CBE and ABE versions used in this study often resulted in multiple protein variants as the editing outcomes, which prevented linkage between the functional phenotype associated with a given sgRNA and that of a single variant protein. Using base editors with narrower editing windows65,74 would reduce the number of editing outcomes, facilitating functional classifications of protein variants generated by base editing. In addition, the use of recently developed high-activity base editors75–77 would reduce the number of sgRNAs classified as ‘not assessed’ due to low base editing efficiencies. Furthermore, the function of transversion mutations could be similarly analyzed using transversion base editors78,79.
In summary, our high-throughput evaluations, which used UMIs and surrogate target sequences, determined the functional effects of at least 29,060 protein variants on proliferation and survival of non-tumorigenic cells. These results will help improve understanding of the functional role of mutations in the development of cancer, especially lung cancer. We envision that our results will contribute to enhancing the clinical utility of genetic testing of cancer samples, and that this approach can be extended to determine the function of more VUSs.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41587-022-01276-4.
Methods
Design of libraries C and A.
We extracted SNVs found in human cancer tissues from a recent version of the COSMIC database23 (release version 84). Mutations listed in COSMIC were accessed from the website in March 2018. From the database, we acquired 458,189 C > T SNVs and 255,580 A > G SNVs found in human cancers. To achieve a high frequency of base editing, we considered the highly active 4-base pair (bp) activity window spanning protospacer positions 4–7, numbered such that the end distal to the NGG PAM is designated as position 1 for both CBE19,27 and ABE21,27. We identified 153,425 C > T and 35,163 A > G SNVs that can be generated using CBE and ABE, respectively. Next, we filtered out all mutations with BsmBI cut sites in the sgRNA sequences and corresponding genomic target sequences. After filtering out synonymous SNVs, we attempted to remove SNVs that cannot be generated at high efficiency. Given that the base editing efficiency is usually low when the Cas9 nuclease activity is low27, we removed the 10% of the target sequences with the lowest DeepSpCas9 scores, which represent computationally predicted SpCas9 activities30. After these steps, 80,203 and 23,008 sgRNAs remained, which can induce 84,806 C > T SNVs and 23,176 A > G SNVs using CBE and ABE, respectively.
As negative controls, we added 500 sgRNAs into library C and 139 sgRNAs into library A; these sgRNAs do not target any sequence in the human genome (non-targeting control sgRNAs) and have been used as negative controls in genome-wide Cas9-induced knockout screening in human cells81,82. We also included synonymous mutation-inducing sgRNAs as another type of negative control;24,25 we included 3,028 such sgRNAs in library C and 466 sgRNAs in library A. This group of sgRNAs can induce synonymous SNVs found in the CGC of 719 genes5, which represents an expertly curated catalog of genes that have been implicated in cancer evolution.
Cell lines and culture.
HBEC30KT (RRID: CVCL-AS83) cells are normal human bronchial epithelial cells that were immortalized by the stable expression of CDK4 and hTERT. These cells exhibit intact contact inhibition of proliferation and lack tumorigenic potential83. HBEC30KT-shTP53 cells (P cells) were generated by lentiviral delivery of TP53-targeting shRNA into HBEC30KT cells, and the molecular features and properties of this cell line were previously described22. In brief, immunoblot analysis of the products of oncogenic genes, such as TP53, KRAS and LKB1 (STK11), showed that P cells resemble their normal matched control HBEC30KT cells except for reduced expression of the p53 protein.
P cells were cultured in ACL-4 medium (RPMI 1640 (Gibco, 2.05 mM L-glutamine) supplemented with 0.02 mg ml−1 of insulin, 0.01 mg ml−1 of transferrin, 25 nM sodium selenite, 50 nM hydrocortisone, 10 mM HEPES, 1 ng ml−1 of EGF, 0.01 mM ethanolamine, 0.01 mM O-phosphorylethanolamine, 0.1 nM triiodothyronine, 2 mg ml−1 of BSA and 0.5 mM sodium pyruvate) with 2% Tet System-Approved FBS (Clontech) and 1% penicillin–streptomycin (Gibco) at 37 °C with 5% CO2. HEK293T cells (American Type Culture Collection) were cultured in DMEM (Gibco) with 10% FBS (Gibco) at 37 °C with 5% CO2.
Cloning.
All primers used for cloning are listed in Supplementary Table 5. All nucleotides were purchased from Macrogen. The schematics of each viral vector are shown in Fig. 1a.
To generate pLenti-TRE3G-AncCBE4max-PGK-hygro, we combined the following six DNA fragments using Gibson assembly: (1) BamHI-NcoI digested pLVX-TRE3G (Clontech, 631187)-based lentiviral backbone; (2) sequences encoding AncAPOBEC1 and the N-terminal region of nCas9 (D10A) amplified via PCR from pCMV-AncBE4max26 (Addgene, 112094) using primer pair 1/2; (3) sequences encoding the C-terminal region of nCas9 (D10A) and 2× uracil glycosylase inhibitor amplified from pCMV-AncBE4max using primer pair 3/4; (4) the PGK promoter amplified from pLVX-TRE3G using primer pair 5/6; (5) the hygromycin resistance gene amplified from pLenti HRE Luc pGK Hygro (Addgene, 118706) using primer pair 7/8; and (6) the WPRE element amplified from pLVX-TRE3G using primer pair 9/10.
To generate pLenti-TRE3G-ABEmax-PGK-hygro, we combined the following six DNA fragments using Gibson assembly: (1) BamHI-NcoI digested pLVX-TRE3G (Clontech, 631187)-based lentiviral backbone; (2) sequences encoding ecTadA and the N-terminal region of nCas9 (D10A) amplified from pCMV-ABE4max26 (Addgen, 112098) using primer pair 1/2; (3) sequences encoding the C-terminal part of nCas9 (D10A) amplified from pCMV-ABE4max using primer pair 3/4; (4) the PGK promoter amplified from pLVX-TRE3G using primer pair 5/6; (5) the hygromycin resistance gene amplified from pLenti HRE Luc pGK Hygro (Addgene, 118706) using primer pair 7/8; and (6) the WPRE element amplified from pLVX-TRE3G using primer pair 9/10.
To generate pLenti-Guide-Puro-p2A-EGFP, we combined the following four DNA fragments using Gibson assembly: (1) FseI-NcoI digested lentiguide-puro (Addgene, 52963)-based lentiviral backbone; (2) the sequence encoding EF-1a and the puromycin resistance gene amplified from lentiguide-puro using primer pair 11/12; (3) sequences encoding p2A and enhanced green fluorescent protein (EGFP) amplified from pCMV-AncBE4max-p2A-GFP (Addgene, 112100) using primer pair 13/14; and (4) WPRE and LTR segments amplified from lentiguide-puro using primer pair 15/16.
All PCR-amplified DNA fragments were generated using Phusion High-Fidelity DNA Polymerase (NEB) with 25 cycles of amplification and an annealing temperature of 60 °C, after which they were size-selected by electrophoresis on a 1% agarose gel. The Gibson assembly reaction was performed using NEBuilder HiFi DNA Assembly Master Mix (NEB).
Lentivirus production.
HEK293T cells were seeded in 100-mm culture dishes at a density of 5 × 106 cells per dish 24 hours before transfection. On the day of transfection, the growth medium was exchanged for 10 ml of DMEM containing 25 μM chloroquine diphosphate, after which cells were cultured for 5 hours. Transfer plasmids containing the gene of interest, psPAX2, and pMD2.G were mixed at a molar ratio of 1.64:1.3:0.72 pmol and diluted into 500 μl of Opti-MEM (Life Technology). Polyethylenimine (PEI) was diluted into Opti-MEM in a total volume of 500 μl and added to the DNA mixture such that the ratio of μg DNA:μg PEI was 1:3, resulting in a total volume of 1,000 μl. The mixture was incubated for 20 minutes and added to cells. To achieve a high viral titer, caffeine (Sigma-Aldrich, C0750) was added to the culture medium, at a final concentration of 4 mM, after treatment with the PEI:DNA mixture as previously described84. At 12 hours after transfection, 10 ml of growth medium supplemented with 4 mM of caffeine was added to refresh the cells.
After 24 hours, the growth medium was harvested and centrifuged at 2,000g for 10 minutes to pellet cell debris. The supernatant was filtered through a Millex-HV 0.45-μm low protein-binding membrane (Millipore), divided into aliquots, and kept frozen at −80 °C until use.
Generation of base editor knock-in cell lines.
In total, 1.8 million P cells were plated in six-well culture plates, with 1.5 × 105 cells per well. The cells were infected with virus carrying sequences encoding the EF1a-rtTA with the neomycin resistant gene (pLVX-EF1a-Tet3G (Clontech, 631359)) supplemented with 10 μg ml−1 of polybrene (Sigma-Aldrich) at a multiplicity of infection (MOI) of 0.4. The six-well plates were centrifuged at 1,000g for 2 hours at 37 °C. After centrifugation, the cells were incubated overnight and then refreshed with growth medium containing 1.0 mg ml−1 of G418 disulfate salt (Sigma-Aldrich). After 10 days of selection, P cells containing rtTA (P-rtTA) were maintained and used for base editor knock-in.
To generate base editor knock-in cell lines, 1.8 million P-rtTA cells were plated in six-well culture plates, with 1.5 × 105 cells per well. The cells were infected with virus carrying sequences encoding a doxycycline-dependent base editor (Lenti-TRE3G-AncCBE4max-PGK-hygro or Lenti-TRE3G-ABEmax-PGK-hygro) as described above. The day after transduction, cells were refreshed with growth medium containing 80 μg ml−1 of Hygromycin B Gold (InvivoGen). After 10 days of selection, the obtained base editor-expressing cell lines (P-C cells or P-A cells) were aliquoted and used for screening.
Plasmid library construction.
Pooled 150-nt oligonucleotides for plasmid construction were array-synthesized by Twist Bioscience. Each plasmid in our library was designed to include the following elements (Extended Data Fig. 2b): (1) a 19-nt homology arm with a U6 promoter at the 3′ terminus; (2) a 20-nt sequence with a G at the 5′ terminus, followed by a 19-nt sgRNA guide sequence; (3) a random 20-nt sequence flanked by a BsmBI cut site on either side (11 nt each); (4) a 20-nt (library C, A and A1) or 19-nt (library C1 and C2) unique barcode sequence corresponding to each sgRNA; (5) a 30-nt surrogate target sequence containing a PAM (a 4 + 23-nt target sequence plus a 3-nt PAM + 3-nt), which is identical to an endogenous genomic target locus; and (6) a 20-nt homology arm.
The methods used to generate the plasmid library were previously described in detail32. In brief, the pooled oligonucleotides were amplified using primer pair 17/18 and Phusion High-Fidelity DNA Polymerase (NEB), after which they were size-selected by electrophoresis on a 2% agarose gel. The amplicons were assembled into linearized Lenti-gRNA-Puro (Addgene, 84752) after digestion with BsmBI using NEBuilder HiFi DNA Assembly Master Mix (NEB). Next, 200 ng of linearized vector and 120 ng of purified oligonucleotides were used in one Gibson assembly reaction (with a total volume of 20 μl). A total of 16 and eight reactions were performed for the CBE and ABE libraries (designated the C and A libraries), respectively. After the assembly reactions, the mixtures were pooled and concentrated using a MEGAquick-spin Total Fragment DNA Purification Kit (iNtRON Biotechnology) and used in up to 12 and eight electroporation reactions to maximize the library complexity.
An improved form of sgRNA scaffold85 and a UMI were synthesized (IDT, Primer 19) and amplified using primer pair 20/21. The resulting amplicon was digested with BsmBI and purified. A ligation reaction was then performed using 60 ng of the sticky-ended sgRNA scaffold–UMI fragments and 250 ng of the scaffoldless plasmid library generated above, also digested with BsmBI. A total of 16 and eight reactions were performed for the CBE and ABE libraries, respectively. The reaction mixtures were pooled, concentrated and used in up to 12 and eight electroporation reactions.
High-throughput evaluation of SNVs using CBE and ABE.
Twenty-four hours before transduction of lentiviral libraries C and A, 168 million P-C cells and 48 million P-A cells were seeded in duplicate, resulting in 2,000-fold coverage of the sgRNA libraries (that is, on average, 2,000 cells per sgRNA) in each replicate. In different replicates, cells were transduced with different batches of the lentiviral library on different days. The cells in each replicate were infected with lentiviral library C or A with 10 μg ml−1 of polybrene at an MOI of 0.3, such that every sgRNA was represented in approximately 600 cells. After 24 hours of infection, the medium was replaced with fresh medium containing 20 μg ml−1 of puromycin (Invitrogen) and 2 μg ml−1 of doxycycline hyclate (Sigma-Aldrich) to induce expression of CBE or ABE; cells were cultured under these conditions for an additional 9 days and were harvested at day 10 after infection with 1,000~1,500-fold coverage of the sgRNA libraries. The concentration of puromycin in the medium was unusually high because untransduced P cells express a low level of a puromycin resistance gene, which was used for the immortalization of bronchial epithelial cells22,83. The remaining cells were maintained at numbers sufficient for 2,000-fold coverage of the sgRNA libraries (that is, P-C cells, 83,731 sgRNAs × 2,000 cells per sgRNA = ~168 million cells; P-A cells, 23,613 sgRNAs × 2,000 cells per sgRNA = ~48 million cells) for an additional 14 days. At day 24 after infection, the cells were collected for genomic DNA extraction.
In experiments involving small libraries C1, C2 and A1, the same methods were used, except that the cells were maintained at numbers sufficient for 10,000-fold coverage of the sgRNA libraries throughout the experiments. In experiments involving libraries C3, A2 and dA, the same methods were used except that the cells were maintained at numbers sufficient for 3,000-fold coverage of the sgRNA libraries.
In experiments involving library eC, 24 million P-C cells were seeded in duplicate, resulting in 6,000-fold coverage of the sgRNA library in each replicate. In different replicates, cells were transduced with different batches of the lentiviral library on different days. The cells were infected with the lentiviral library as described above, after which the medium was replaced with medium containing 20 μg ml−1 of puromycin (Invitrogen) and 2 μg ml−1 of doxycycline hyclate (Sigma-Aldrich). The cells were incubated for an additional 9 days and were harvested at day 10 after infection with approximately 2,000-fold coverage of the sgRNA library. Upon removal of puromycin and doxycycline, the cell population was split into drug and untreated arms at a representation of 2,000 cells per sgRNA and maintained at numbers sufficient for at least 3,000-fold coverage of the sgRNA library. Cells in the drug arm were cultured and passaged every 3–4 days with EGF-free ACL-4 medium containing 10 nM afatinib (Santa Cruz Biotechnology) for an additional 10 days. Cells in the untreated arm were cultured with complete ACL-4 medium for 10 days.
Genomic DNA preparation and deep sequencing.
Genomic DNA was extracted using a Wizard Genomic DNA Purification Kit (Promega) according to the manufacturer’s protocol.
Using the isolated genomic DNA as template, the integrated barcode and target sequences were amplified and prepared for deep sequencing through two PCR steps using 2× Pfu PCR Smart Mix (SolGent). In the first step, genomic DNA was divided into multiple 50-μl reactions containing 2.5 μg of genomic DNA, 20 pmol of forward primer (22/23/24 primer mixture; Supplementary Table 5), 20 pmol of reverse primer (25/26/27 primer mixture) and 25 μl of PCR pre-mix. The PCR cycling parameters were as follows: an initial 2 minutes at 95 °C, followed by 30 seconds at 95 °C, 30 seconds at 60 °C and 40 seconds at 72 °C, for 24 cycles, and a final 5-minute extension at 72 °C. The total amount of genomic DNA for each experiment represented more than 1,000× coverage of the library, assuming 6.6 μg of genomic DNA per 106 cells86.
Library C: 360 separate 50-μl reactions per replicate experiment (900 μg of DNA, ~1,600× coverage)
Library A: 96 separate 50-μl reactions per replicate experiment (240 μg of DNA, ~1,500× coverage)
Library C1: 40 separate 50-μl reactions per replicate experiment (100 μg of DNA, ~3,800× coverage)
Library A1: 20 separate 50-μl reactions per replicate experiment (50 μg of DNA, ~3,800× coverage)
Library C2: 80 separate 50-μl reactions per replicate experiment (200 μg of DNA, ~7,600× coverage)
Library C3: 80 separate 50-μl reactions per replicate experiment (200 μg of DNA, ~15,000× coverage)
Library A2: 80 separate 50-μl reactions per replicate experiment (200 μg of DNA, ~15,000× coverage)
Library eC: 80 separate 50-μl reactions per replicate experiment (200 μg of DNA, ~7,500× coverage)
Library dA: 80 separate 50-μl reactions per replicate experiment (200 μg of DNA, ~7,600× coverage)
Amplicons for each experiment were pooled and concentrated with a MEGAquick-spin Total Fragment DNA Purification Kit and size-selected with agarose gel electrophoresis.
In the second PCR step, which was performed to attach sequencing adaptors and barcodes, a total of 250 ng of purified PCR product from the first step was used in eight separate 50-μl reactions for the screening libraries, and a total of 40 ng of purified PCR product from the first step was used in two separate 50-μl reactions for the focused libraries, with 20 pmol of Illumina indexing primers in each reaction (primer pair 28/29). The PCR cycling parameters were as follows: an initial 2 minutes at 95 °C, followed by 30 seconds at 95 °C, 30 seconds at 60 °C and 40 seconds at 72 °C, for eight cycles, and a final 5-minute extension at 72 °C. Amplicons for each experiment were size-selected with agarose gel electrophoresis and sequenced using a HiSeq 2500 system (Illumina) and a NextSeq 550 system (Illumina).
Design of small libraries.
We designed seven independent small libraries named C1 (containing 3,261 sgRNAs), C2 (3,170 sgRNAs), C3 (1,941 sgRNAs), A1 (1,595 sgRNAs), A2 (2,082 sgRNAs), dA (3,136 sgRNAs) and eC (4,157 sgRNAs) (Extended Data Fig. 5a,b).
Libraries C1 and A1. To generate libraries C1 and A1, 857 and 1,538 sgRNAs were randomly chosen from libraries C and A, respectively. Additionally, 2,404 and 47 sgRNAs that were not analyzed due to a low number of UMIs (<50) in libraries C and A were included (see ‘Functional classification of cancer-associated transition mutations’ for detailed filtering conditions for sgRNA analysis). Finally, 100 and 50 non-targeting sgRNAs81,82 were included in each library.
Library C2. To generate library C2, 1,710 sgRNAs were randomly chosen from library C. Additionally, 1,240 sgRNAs that were not analyzed due to a low number of UMIs in library C were included. Third, we included sgRNAs that mediate the disruption of essential genes by base editing-induced stop codon generation as previously described25,87. As the essential gene candidates, we first selected 123 genes included in both a curated set of pan-cancer core fitness genes88 and the BAGEL essential gene set89. From the 123 curated genes, we selected 65 related to essential cellular structures and processes: ribosomal proteins (39 genes), DNA replication (two genes), RNA polymerases (four genes), proteasomes (eight genes) and spliceosomes (12 genes)88. We used the CRISPR-iSTOP tool87 to design sgRNAs that induce stop codons in these genes and selected 220 sgRNAs that were predicted by DeepCBE tools27 to induce stop codons with high efficiency. Finally, 100 non-targeting sgRNAs were included in each library.
Libraries C3 and A2. To generate libraries C3 and A2, 100 and 48 sgRNAs that were highly depleted in previous screenings of libraries C/C1/C2 and A/A1, respectively, were initially chosen. Next, 290 and 163 sgRNAs were randomly chosen from previous screenings of libraries C/C1/C2 and A/A1, respectively. Third, sgRNAs designed to induce 1,151 and 1,468 SNVs in known tumor suppressor genes recorded in COSMIC (data release version 84) and the TCGA database1 (data release version 29.0) were included in libraries C3 and A2, respectively; these SNVs can be generated by CBE or ABE in the canonical activity window (positions 4–8). Finally, 400 non-targeting sgRNAs were included in each library.
Library dA. To generate library dA, we first chose 369 high-confidence driver genes with high ratios of non-synonymous to synonymous mutations (dN/dS);90 from these, we selected 2,797 SNVs that can be generated by ABE within the canonical activity window (positions 4–8). Next, 53 and 23 sgRNAs that had respectively been classified as depleting/likely depleting and outgrowing/likely outgrowing in previous library screenings were included. Finally, 263 non-targeting sgRNAs were included, resulting in 3,136 sgRNAs.
Library eC. We first selected 162 genes related to EGF/EGFR signaling pathways;91 from these, we chose 3,967 SNVs recorded in COSMIC and TCGA that can be generated by CBE within the canonical activity window (positions 4–8); sgRNAs designed to induce these SNVs were included. Next, 24 sgRNAs with various classifications (six outgrowing, four likely outgrowing, eight possibly outgrowing, one neutral and five not assessed) from previous screenings were included. Finally, 166 non-targeting sgRNAs were included, resulting in 4,157 sgRNAs.
Selection of sgRNAs for individual functional evaluations.
First, we selected six and seven of the most significantly depleting and outgrowing sgRNAs from the depleting and outgrowing groups, respectively (that is, the lowest P values), in the small libraries. All seven of the representative outgrowing sgRNAs were predicted to induce TP53-related mutations (Cg.TP53_p.Q192*, Cg.TP53_p.T155I, Cg.TP53_p.Q100*, Ag.TP53_p.R280G, Ag.TP53_p.N239D, Ag.TP53_p.K120E and Ag.TP53_p.K351E). Five out of the six most significantly depleting sgRNAs were predicted to induce mutations in common essential genes (Cg.POLR1C_p.A6V, Cg.MMS22L_p.R661*, Cg.POLR2B_p.P714L, Ag.CTCF_p.H312R and Ag.SRSF1_p.D139G). Next, we arbitrarily selected four sgRNAs that were annotated as likely outgrowing (Cg.PTPN14_p.Q110* and Cg.CDC23_p.T381M) and likely neutral (Cg.ACOX3_p.Q145* and Cg.KMT2C_p.R1906*). Additionally, we picked one TP53-related sgRNA (Ag.TP53_p.T125A) that was predicted to introduce a mutation known to destabilize the p53 protein;92 however, only library C contained the sgRNA, and it was classified as likely outgrowing in library C. Also, we picked two sgRNAs classified as likely neutral (possibly outgrowing), but predicted to introduce missense mutations in common essential genes (Ag.POLE_p.Y1889C and Ag.ACTL6A_p.T405A), for controls. Finally, we picked an additional eight sgRNAs classified as likely outgrowing or outgrowing in this study. In summary, we picked 28 sgRNAs for validation (Supplementary Fig. 5).
We individually cloned sgRNA-encoding sequences into the Lenti-Guide-Puro vector (Addgene, 52963). In total, 1.2 million base editor knock-in cells per sgRNA were seeded in 100-mm culture dishes 24 hours before transduction. The cells were infected in duplicate with lentivirus harboring sequences encoding individual sgRNAs at a low MOI (~0.4). In addition, base editor knock-in cells were seeded as above for a GFP+ control. In this case, lentivirus harboring an empty sgRNA cassette and the puromycin resistance gene-p2A-GFP fusion gene was used to infect cells at a low MOI (~0.4). The day after transduction, the medium was replaced with fresh medium containing 20 μg ml−1 of puromycin (Invitrogen) and 2 μg ml−1 of doxycycline hyclate (Sigma-Aldrich) to induce expression of the base editor; these conditions were maintained for 48 hours. After removal of puromycin, the cells were maintained for an additional 7 days with doxycycline treatment.
Competitive proliferation assay.
Ten days after infection, cells transduced with lentivirus encoding candidate hit sgRNAs (GFP−) and cells transduced with the positive control lentivirus (GFP+) were mixed and grown together in triplicate. The cells were sampled every 3 or 4 days, and the ratio of GFP+to GFP−cells in the mixture was quantified via longitudinal flow cytometry. By assuming that the cells exhibit an exponential growth rate, the number of cells (N) at times t1 and t2 can be described by the following equation, where is the absolute fitness of the reference cells and is the fitness change caused by the transduced sgRNA93.
The between a certain timepoint and the reference timepoint was obtained according to the equation:
The ratio between the number of GFP− cells and the number of GFP+ cells was obtained from the competitive growth assay, and we assumed that the relative fitness of the GFP+ cells was equal to the fitness of the reference cells . The relative enrichment between a certain timepoint and the reference timepoint (Fig. 4d and Extended Data Fig. 9b) was determined as follows:
Allele frequency tracking after transduction of an individual sgRNA.
The cells harboring an individual sgRNA and a base editor were seeded in duplicate after removal of doxycycline at 10 days after infection. These cells were cultured for an additional 2 weeks and harvested at 10, 17 and 24 days after infection. Each sgRNA-targeted genomic site was amplified using site-specific primers (Supplementary Table 5) and analyzed by deep sequencing.
The amplicons for deep sequencing were prepared through three successive rounds of PCR. The first PCR step was performed using 1 μg of genomic DNA, Q5 DNA polymerase (NEB) and 20 pmol of ‘amplifying’ primer (Supplementary Table 5) in a single 20-μl reaction. The second step was performed using 3 μl of the first-step PCR product and 20 pmol of ‘adaptor’ primer (Supplementary Table 5). The third step was performed to attach sequencing adaptors and a barcode sequence, using 2 μl of the first-step PCR product and 20 pmol of Illumina indexing primers (primer pair 28/29). In all cases, the PCR cycling parameters were as follows: an initial 2 minutes at 98 °C, followed by 30 s at 98 °C, 30 seconds at 58 °C and 1 minute and 30 seconds at 72 °C, for 20 cycles, and a final 5-minute extension at 72 °C.
Analysis of base editing outcomes in surrogate target sequences.
Deep sequencing data generated from the library were analyzed using custom Python scripts, which were used previously27. In brief, guide RNAs and corresponding surrogate sequences were extracted using the ‘sorting barcode’, including the TTTG sequence (a common 4-nt sequence for the BsmBI restriction site), the unique barcode sequences located upstream of the target sequences (20 nt in length for libraries C, A and A1; 19 nt in length for libraries C1 and C2) and the 4-nt sequence downstream of the surrogate target sequence (only for libraries C and A) (Supplementary Table 1). We considered insertions or deletions located near the 8-nt region surrounding the expected cleavage site to be indels.
For analysis of base editing efficiencies and allele frequencies, the reads were sorted by their unique barcode sequences, and reads containing indels were excluded from further analysis. For ABE and CBE, we only considered base editing of any A converted to G or any C converted to T, respectively. We filtered out any pairs with fewer than 100 reads in both replicates, and the A > G or C > T conversion efficiency at each position of each sgRNA target site (Extended Data Fig. 3a,b) was calculated as follows:27
For analysis of the proportion of bases that underwent editing, each barcode-sorted read was analyzed according to the sequence outcomes in the base editing window. We analyzed the full length of the sgRNA target site, from position 1 to 20, to exclude any possibility of unintended amino acid changes outside of the canonical base editing window (spanning positions 4–8). The proportion of base editing outcomes was calculated as follows:
Next, we transformed the outcome proportion derived from nucleotide editing to a codon based outcome proportion using an in-house Python script (see ‘Code availability’). Non-synonymous base editing efficiencies were calculated as the sum of any base editing outcomes that changed amino acid codons in the target gene (Supplementary Table 3).
Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout.
For UMI analysis, 8-nt UMI sequences were counted and analyzed according to the sorting barcode with in house Python scripts (see ‘Code availability’)94. To minimize incorrect identification of UMLs as a result of sequencing errors, we used a directional network to integrate UMk: we combined different UMIs when they varied by only one nucleotide and when their read count fold difference was three or more95. When the read count fold difference of UML that differed by one nucleotide was less than three, UML were not combined and were regarded as unique, For UMI-based MAGeCK analysis, we generated a UMI read count table that contained the read count of every UMI of each sgRNA. The UMI read count was normalized as RPM. To calculate the fold change and the statistical significance of read count changes between dyy 24 and day 10 samples, we performed Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout (MAGeCK) (MAGeCK 0.5.9.3)39 analysis. For such analysis, we excluded spRNAs containing fewer than 50 UMls at day 10 to increase the accuracy of functional chassification (Extended Data Fig-4d).
We used four internal groups of UM1-derived clones (replicateUMI) for each sgRRA to calculate the fold changes and significance of the spRNA on the basis of four internal replicates, as described previously96,97. UMIs were randomly annotated to each replicateUMI so that four replicatesUMI had the same or a similar2 number of UMIs. Then, the median RPM of the day 24 and day 10 samples in each replicate was calculated and used as input for MAGeCK analysis to derive a positive/neghative value and LFC of an sgRNA. The LFC values calculated from the MAGieCK algorithm were median subtracted to obtain a normalized LFC (nLFC). Platting the nLFC (x axis) versus the negative logarithm of the robust rank aggregation (RR.A) P value ( axis) produced a volcano plot (Figs. 2b and 3b). The P value used in the volcano plat was the selected lower value between the negative and positive value. When we combined results from replicates, the percentile ranks of the nL.FCs and values were averaged across replicates38.
UMI count analysis.
UMI count analysis was performed using the fold change of CPM, which was scaled by the total UMI counts between day 10 and day 24. To calculate the L.PC, we added a pseudo-count of 1 to all counts to handle UMI counts of 0 in the day 10 or day 24 sample.
Functional classification of sgRN/s.
For UMI-based analysis, all UMIs with fewer than five raw read counts at day 10 were excluded from further analysis. The functional classification system that we used is summarized in a flowchart in Fig. 2a. First, sgRNAs with 50 or fewer UMIs at day 10 were excluded from further analysis (Step 1). Second, we also excluded sgRNAs from further analysis if the non-synonymous base editing efficiency induced by the gRNA was lower than 60% in the surrogate target sequence (Step 2). The remaining sgRNis were elassified depending on their nLFCs and values using cutotf values determined by the distribution of the non-targeting control sgRNis in each library.
Finally, we classified sgRNAs into seven groups as follows:
Depleting: sgRNAs whose nLFC and P value were less than the corresponding values in the 0.3th percentile of the non-targeting sgRNAs and whose UMI CPM fold change was less than the corresponding values in the 1st percentile of the non-targeting sgRNAs
Likely depleting: sgRNAs whose nLFC and P value were less than the corresponding values in the 5th and 1st percentiles, respectively, of the non-targeting sgRNAs but that were not classified as depleting
Likely neutral (possibly depleting): sgRNAs whose nLFC was less than 0 and that were not classified as depleting, likely depleting or neutral
Neutral: sgRNAs whose nLFC was between the corresponding values in the 20th and 80th percentiles and whose P value was greater than the corresponding values in the 20th percentile of the non-targeting sgRNAs
Likely neutral (possibly outgrowing): sgRNAs whose nLFC was greater than 0 and that were not classified as outgrowing, likely outgrowing or neutral
Likely outgrowing: sgRNAs whose nLFC was greater than the corresponding values in the 95th percentile and whose P value was less than the corresponding values in the 1st percentile of the non-targeting sgRNAs but that were not classified as outgrowing
Outgrowing: sgRNAs whose nLFC was greater than the corresponding values in the 99.7th percentile and whose P value was less than the corresponding values in the 0.3th percentile of the non-targeting sgRNAs and whose UMI CPM fold change was greater than the corresponding values in the 99th percentile of the non-targeting sgRNAs
For sgRNAs with two barcodes, UMIs from the two barcodes were combined for subsequent analyses. When the functional classification of an sgRNA differed depending on which sgRNA library was used, we chose the classification from the library that had a higher number of UMIs (UMI CPM) for the sgRNA. When the relative frequency of the variant allele among the base-edited non-synonymous sequences in surrogate sequences was higher than 75%, we considered that the functional classification of the allele was the same as that of the corresponding sgRNA.
Statistical significance.
To compare the LFCs of the sgRNAs according to the base editing efficiency (Fig. 1e and Extended Data Fig. 4a) and the enrichment values of the target sgRNA and non-targeting sgRNA control (Fig. 4d and Extended Data Fig. 9b), we used the two-tailed Student’s t-test. Statistical significance was calculated using PASW Statistics (version 18.0, IBM). We used one-way analysis of variance followed by Dunn’s post hoc test to compare base conversion efficiency among score bins (Supplementary Fig. 2a). To determine the fraction of sgRNAs predicted to introduce mutations in common essential genes (CEGs) or CGC genes (Extended Data Fig. 10) among all classified sgRNAs, Fisher’s exact test was performed using the scipy.stats.fisher_exact function of the Python library.
Extended Data
Extended Data Fig. 1 |. Exon transcript profiles of P cells.
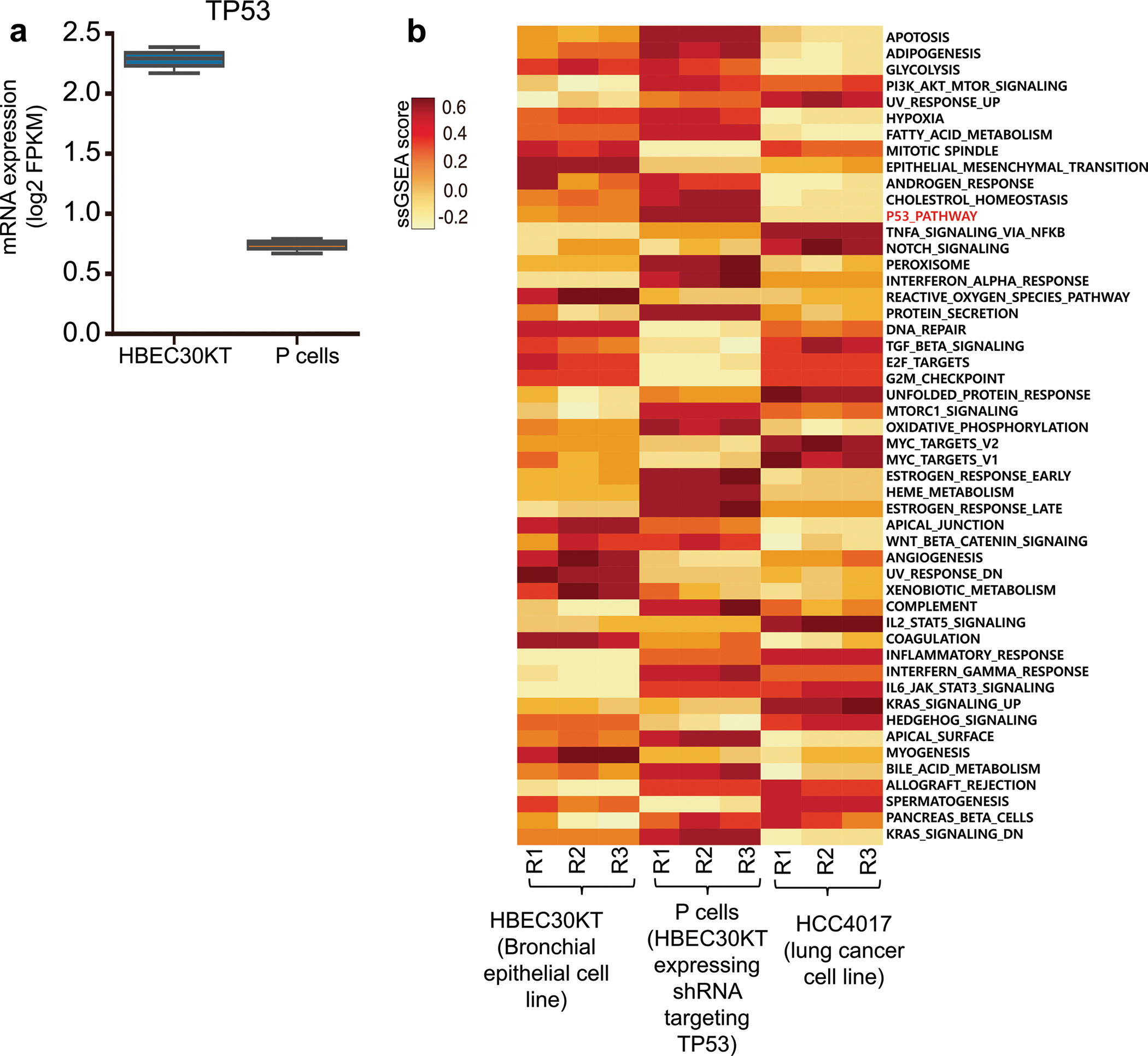
a, Expression of TP53 mRNA in P cells and HBEC30KT cells. FPKM, fragments per kilobase of transcript per million. Boxplots are represented for n = 3 biologically independent samples as follows: center line of box indicating the median, box limits indicating the upper and lower quartile; whiskers show the 1.5 times interquartile range. b, Gene set enrichment analysis (GSEA) of exon transcript profiles of HBEC30KT, P cells, and HCC4017, a lung cancer cell line. The single sample GSEA score (ssGSEA score) represents the degree to which the genes in a particular gene set are up- or down-regulated within the sample. RNA expression data were retrieved from Kim et al34.
Extended Data Fig. 2 |. Generation of libraries C and A.
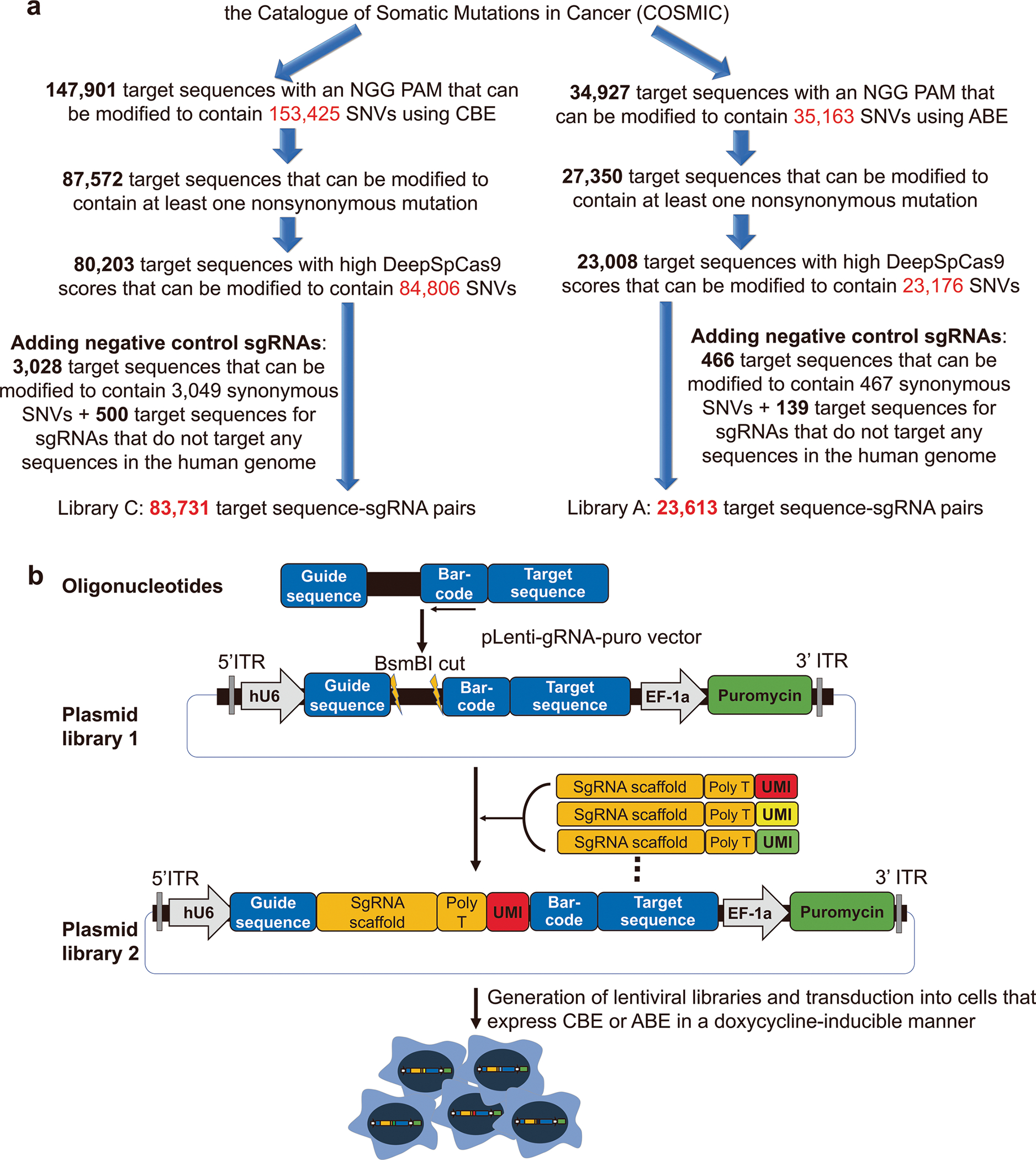
a, The process of selecting sgRNA-target pairs for the generation of libraries C and A. SNVs, single nucleotide variants; sgRNA, single guide RNA. b, Generation of lentiviral libraries of sgRNA-encoding and target sequence pairs with unique molecular identifiers (UMIs). Oligonucleotides containing a 20-nt guide sequence, and the corresponding target sequence were synthesized and cloned into the pLenti-gRNA-puro vector to create plasmid library 1. The plasmids were then digested with BsmBI restriction enzyme and ligated with fragments containing the sgRNA scaffold sequences and UMIs to create plasmid library 2. Lentiviral libraries generated from plasmid library 2 were then transduced into cells expressing cytosine base editor (CBE) or adenine base editor (ABE) in a doxycycline-inducible manner.
Extended Data Fig. 3 |. Base editing efficiencies and indel frequencies at integrated target sequences.
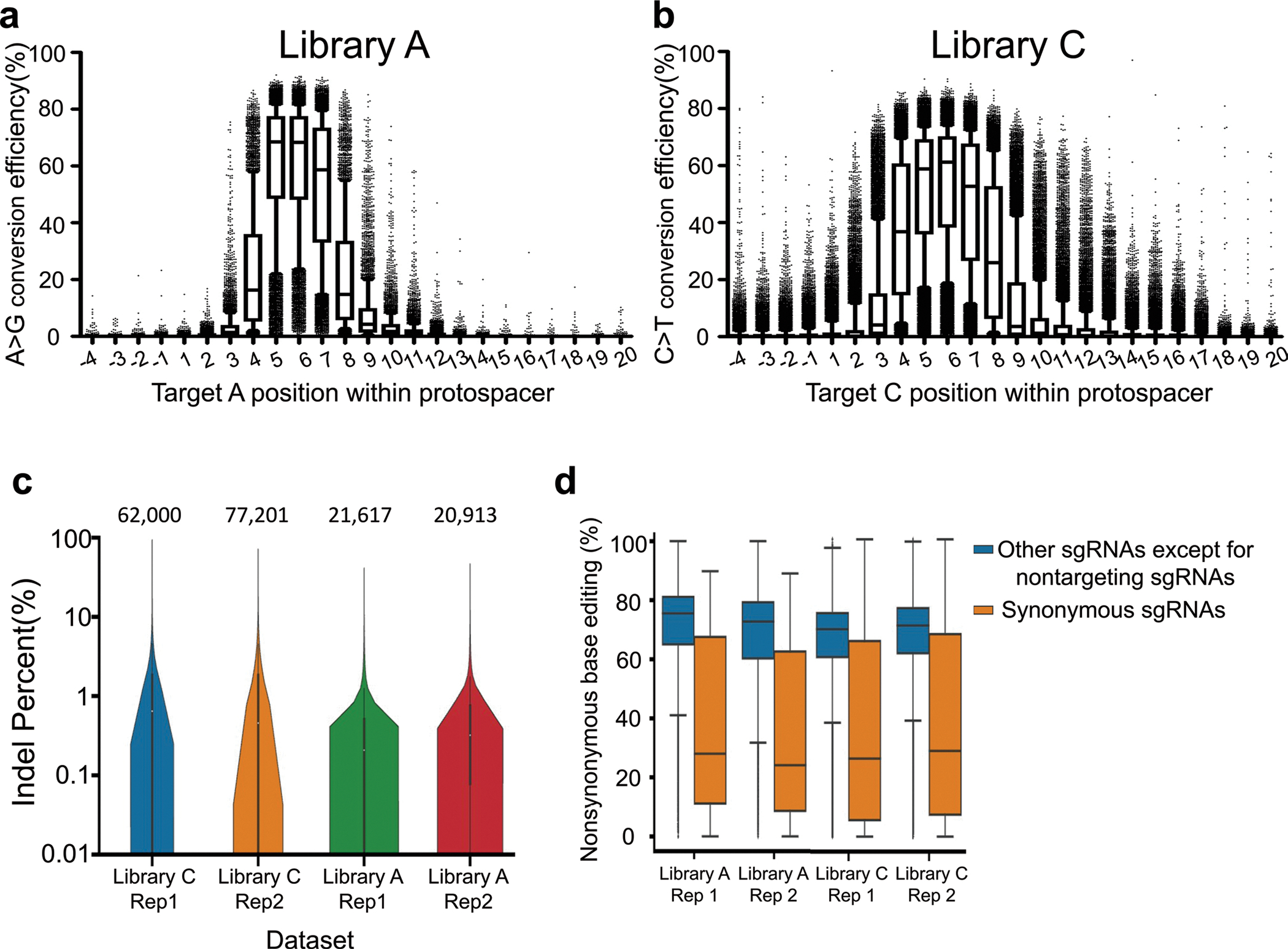
Base editing efficiencies measured at each position in the indicated region for target nucleotide Cs (a) or As (b) in integrated surrogate target sequences. Position 1 is the 5’ end of the target sequence and position 20 is immediately upstream of the NGG PAM. The numbers of analyzed target sequences (n) are as follows: n = 5,865 (position −4), 5,393 (position −3), 5,782 (position −2), 5,815 (position −1), 5,292 (position 1), 5,614 (position 2), 5,697, 6,394, 10,586, 9,382, 8,837, 5,421, 6,130, 5,339, 5,541, 5,796, 5,058, 5,723, 5,955, 5,348, 5,779, 5,437, 4,884, 5,502 (position 20) for ABE (a); n = 19,475 (position −4), 20,753 (position −3), 20,110 (position −2), 19,425 (position −1), 19,984 (position 1), 20,004 (position 2), 17,873, 24,870, 35,421, 33,186, 32,807, 19,895, 19,195, 20,227, 19,549, 18,986, 20,367, 18,793, 18,361, 20,478, 19,605, 20,975, 21,542, 22,952 (position 20) for CBE (b). Boxplots are represented as follows: center line of box indicating the median, box limits indicating the upper and lower quartile; whiskers show the 10th and 90th percentiles. Outliers are shown using dots. c, Indel frequencies measured 10 days after the transduction of sgRNA target pairs. The number of analyzed target sequence is indicated at the top of each dataset. (n = 62,000 (Library C, Replicate 1), 77,201 (Library C, Replicate 2), 21,617 (Library A, Replicate 1) and 20,913 (Library A, Replicate 2). Boxplots are represented as follows: center white dot of box indicating the median, box limits indicating the upper and lower quartile; the distributions of indel frequencies are represented with kernel densities. d, Nonsynonymous base editing efficiencies at the integrated target sequences of synonymous control sgRNAs and other sgRNAs in the given datasets. The number of synonymous and other sgRNAs are as follows; 431 and 21,055 (Library A, Replicate 1), 413 and 20,372 (Library A, Replicate 2), 2,272 and 59,390 (Library C, Replicate 1), and 2,795 and 73,691 (Library C, replicate 2), respectively. Boxplots are represented as follows: center line of box indicating the median, box limits indicating the upper and lower quartile; whiskers show the 1.5 times interquartile range.
Extended Data Fig. 4 |. Performance of high-throughput evaluations.
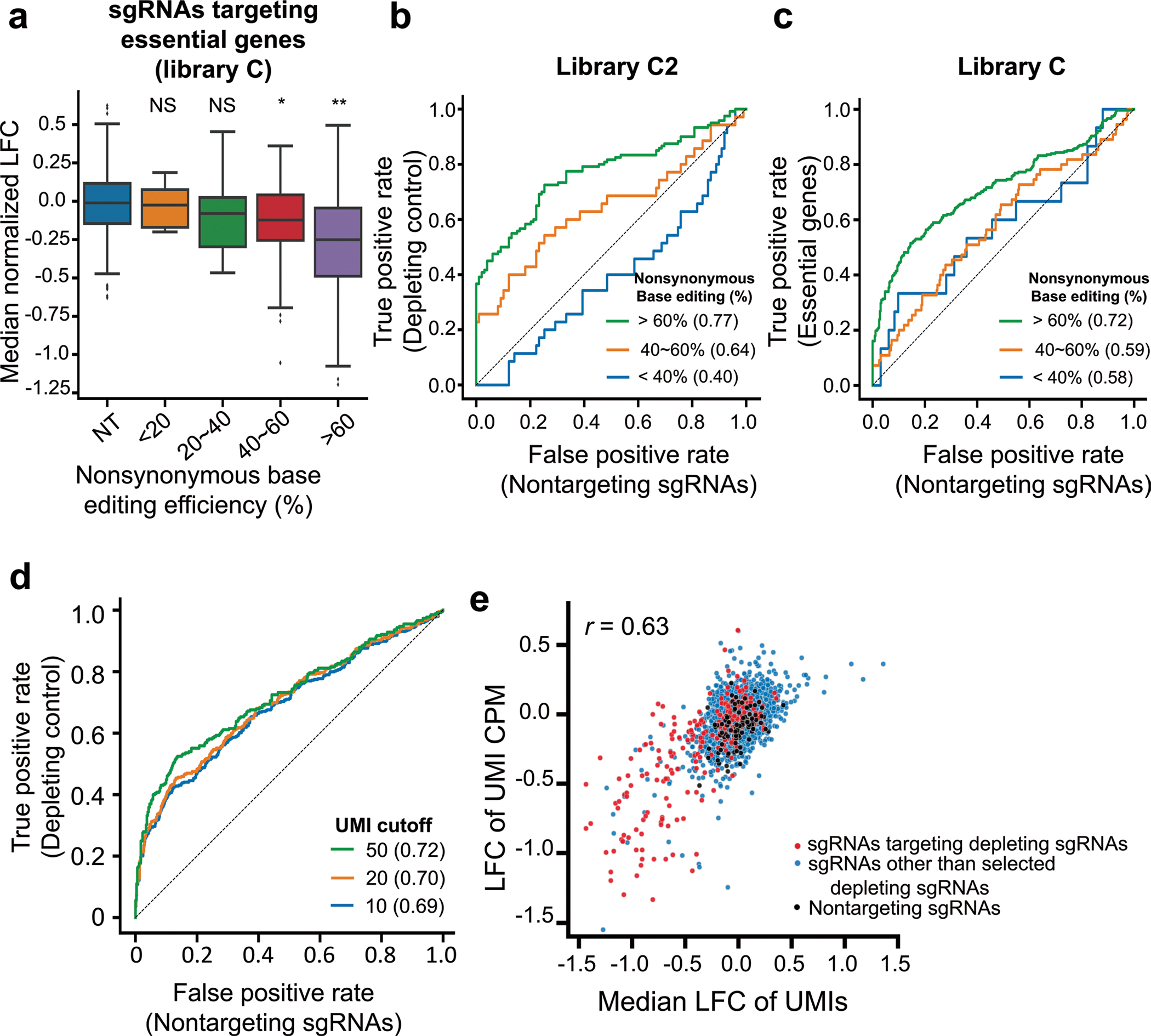
a, Distribution of median normalized log fold changes (LFCs) of 338 sgRNAs targeting essential genes depending on the nonsynonymous base editing efficiencies determined at the integrated target sequences in library C2. NT, nontargeting sgRNAs. The number of sgRNAs n = 359 (NT), 5 (<20%), 10 (20%~40%), 55 (40%~60%), 268 (>60%). Boxplots are represented as follows: center line of box indicating the median, box limits indicating the upper and lower quartile; whiskers show the 1.5 times interquartile range. (in comparison with NT, student’s t-test; NS, not significant, *P = 1.5 × 10−4, **P = 2.2 × 10−32). b,c, Receiver operating characteristic-area under the curve (ROC-AUC) analysis of LFCs for sgRNAs predicted to induce stop codons in common essential genes versus nontargeting sgRNAs in library C2 (b) and library C (c) at increasing thresholds of nonsynonymous base editing efficiencies. AUC values are indicated in parentheses. d, ROC-AUC analysis of LFCs for sgRNAs predicted to induce stop codons in common essential genes versus nontargeting controls at increasing thresholds of the number of UMIs in each sgRNA in library C. An area under curve for each UMI cutoff is shown in the parenthesis. e, Correlations between median LFCs of UMIs for sgRNAs and LFCs of UMI CPM (counts per million) for the same sgRNAs in library C2. Red dots indicate sgRNAs predicted to induce nonsense mutations in selected common essential genes. The number of sgRNAs n = 3,229 (merged), 2,913 (other sgRNAs, blue dots), 217 (sgRNAs targeting essential genes, red dots), 99 (nontargeting sgRNAs, black dots). Pearson correlation coefficients (r) are shown.
Extended Data Fig. 5 |. Design of small libraries and reproducibility of base editing efficiencies using these libraries.
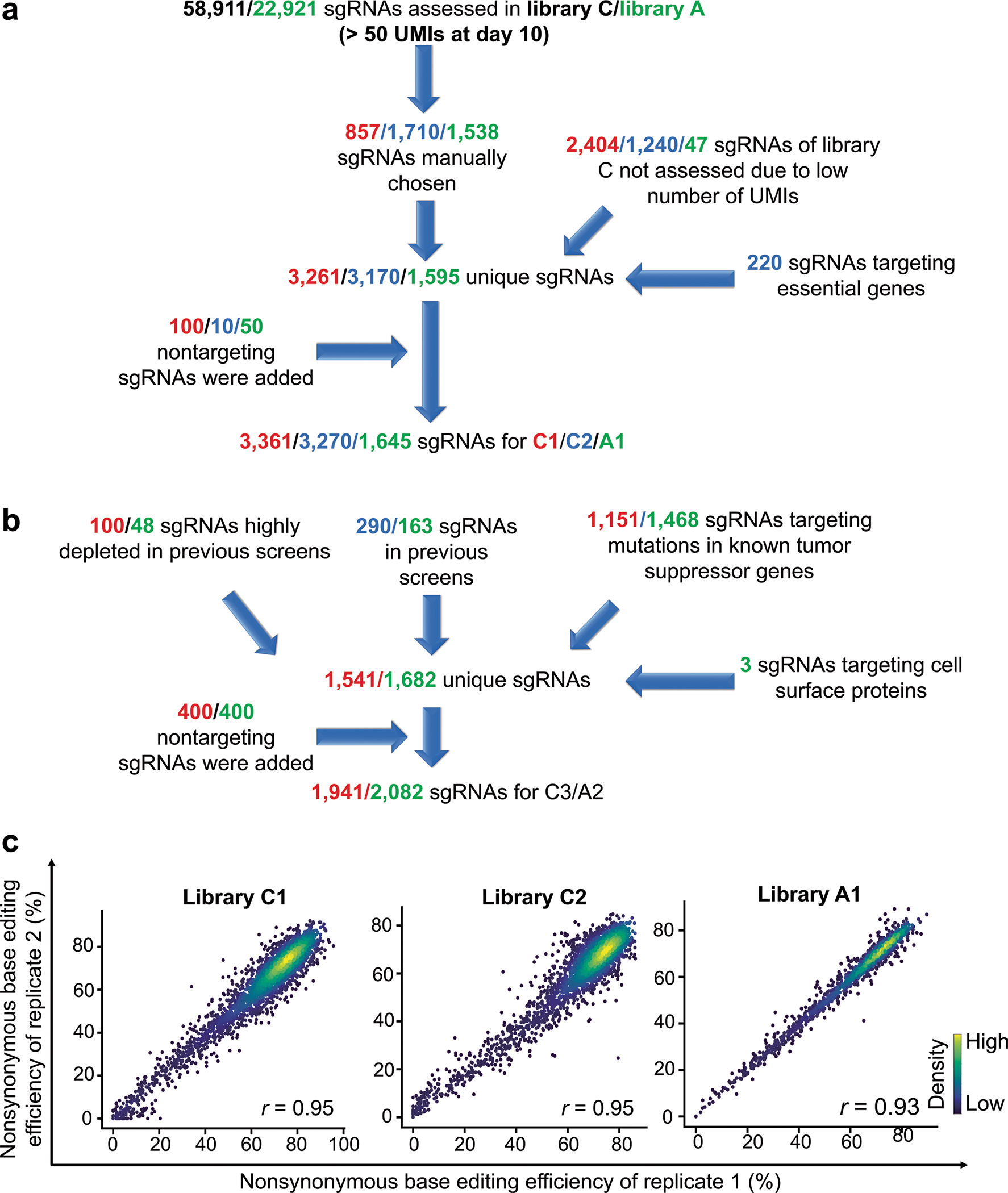
a-b, Design of small libraries C1, C2, and A1 (a) and C3 and A2 (b). UMIs, unique molecular identifiers. c, Correlations between nonsynonymous base editing efficiencies at the integrated target sequences of biological replicates. The color of each dot was determined by the number of neighboring dots (that is, dots within a distance that is three times the radius of the dot). The base editing efficiencies were determined ten days after the initial transduction of each library into P-C or P-A cells. Only sgRNAs with more than 100 raw read counts in each replicate were included. Pearson correlation coefficients (r) are shown. The number of sgRNAs n = 3,181 (library C1), 3,063 (library C2), and 1,520 (library A1).
Extended Data Fig. 6 |. The number of protein variants generated by an sgRNa.
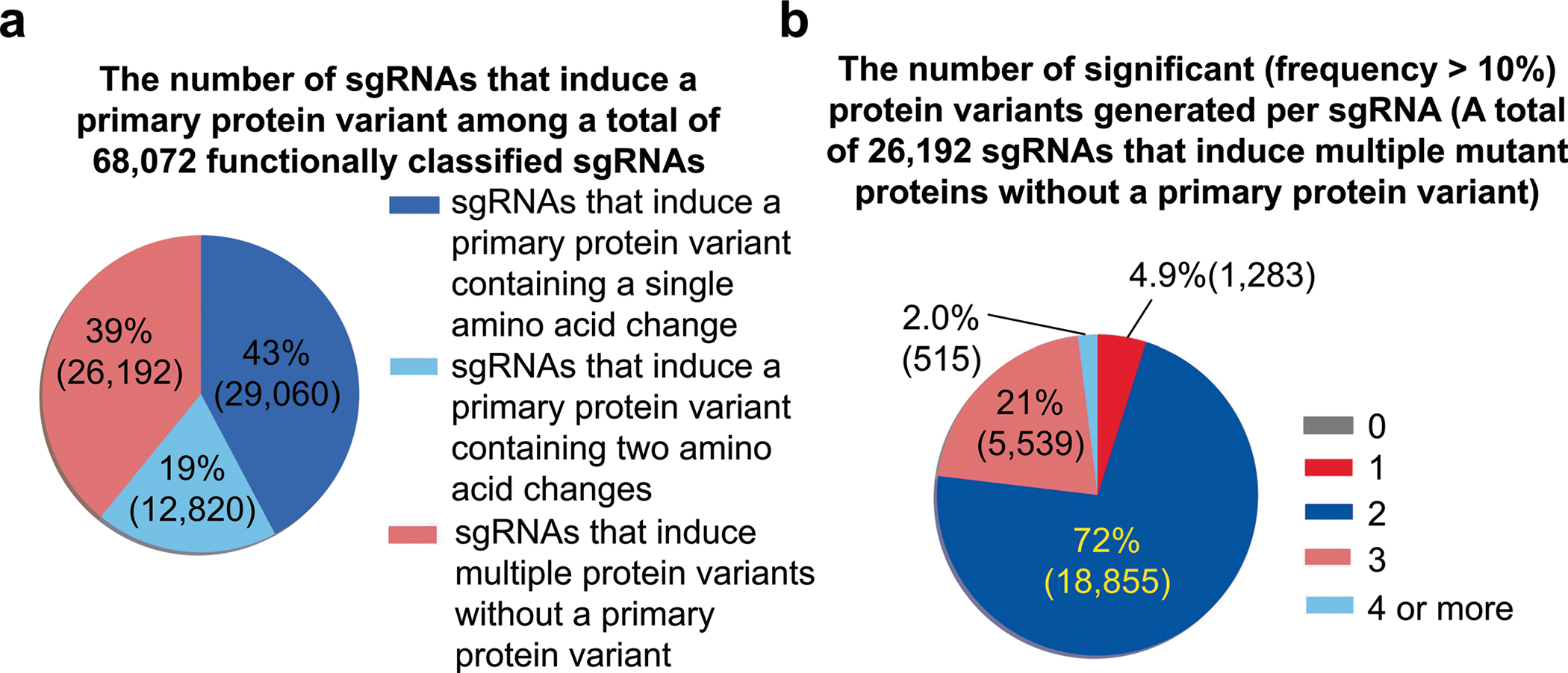
a, The proportion of sgRNAs that induce a primary protein variant. The numbers of sgRNAs are indicated in parentheses. b, The number of significant (frequency > 10%) protein variants generated by sgRNAs that induce multiple protein variants without a primary protein variant. The numbers of protein variants are indicated in parentheses.
Extended Data Fig. 7 |. Association between computationally predicted functions of variants and measured functions of variants.
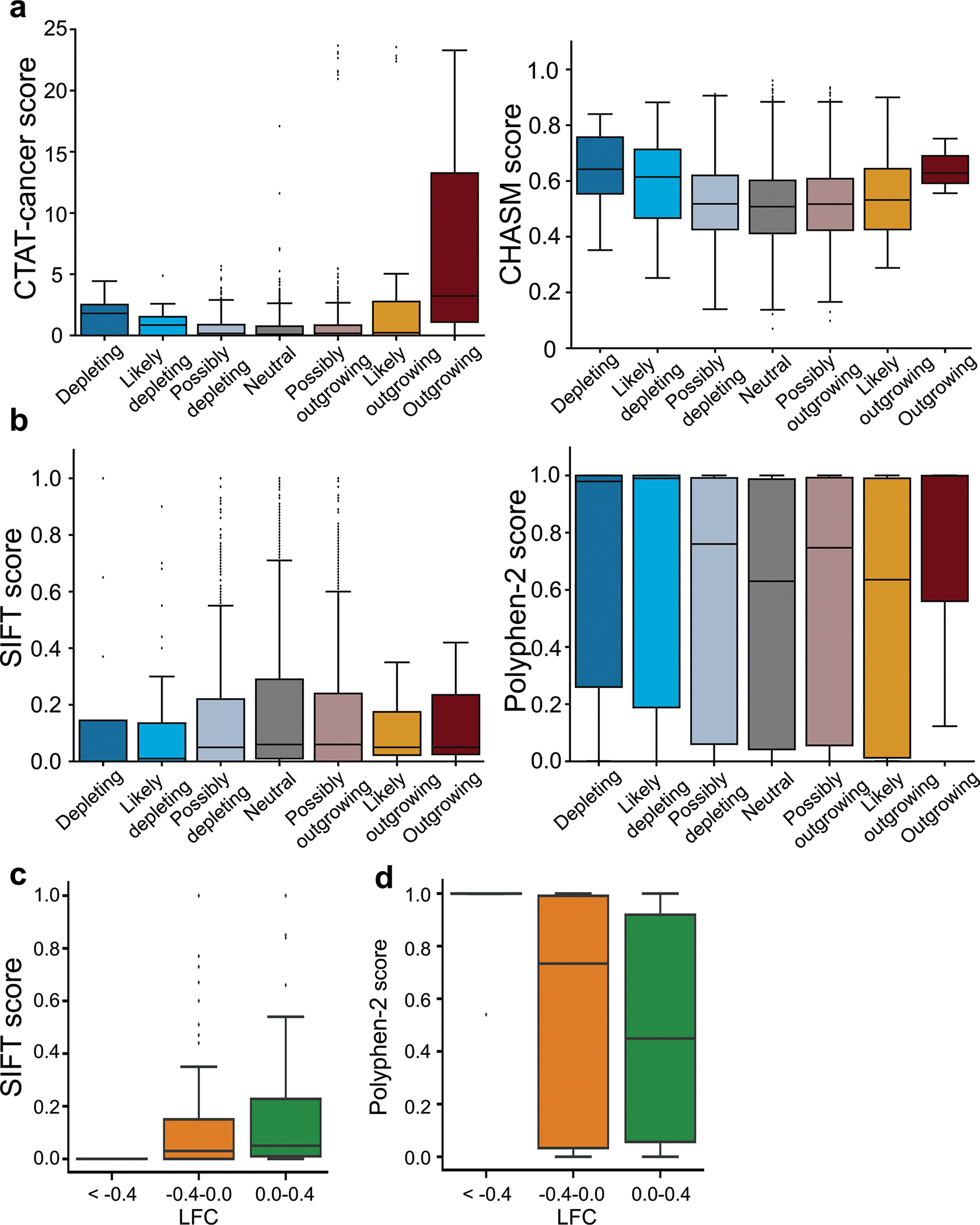
a, The scores from driver detection algorithms (CTAT-cancer and CHASM) for 4,143 protein variants. The number of variants n = 15 (depleting), 39 (likely depleting), 864 (possibly depleting), 2,141 (neutral), 1,056 (possibly outgrowing), 25 (likely outgrowing), and 3 (outgrowing). b, The scores from algorithms that predict the functional effects of variants (SIFT and PolyPhen-2) for 3,899 protein variants. The number of variants n = 12 (depleting), 38 (likely depleting), 807 (possibly depleting), 2,009 (neutral), 1,008 (possibly outgrowing), 22 (likely outgrowing), and 3 (outgrowing). c,d, Distribution of SIFT scores (c) and PolyPhen-2 scores (d) for missense variants in common essential genes according to the LFC in library C. The number of variants n = 10 (<−0.4), 65 (−0.4~0), 82 (0~0.4). Boxplots are represented as follows: center line of box indicating the median, box limits indicating the upper and lower quartile; whiskers show the 1.5 times interquartile range.
Extended Data Fig. 8 |. Allele frequency tracking after transduction of sgRNa-encoding sequences into P-C or P-a cells.
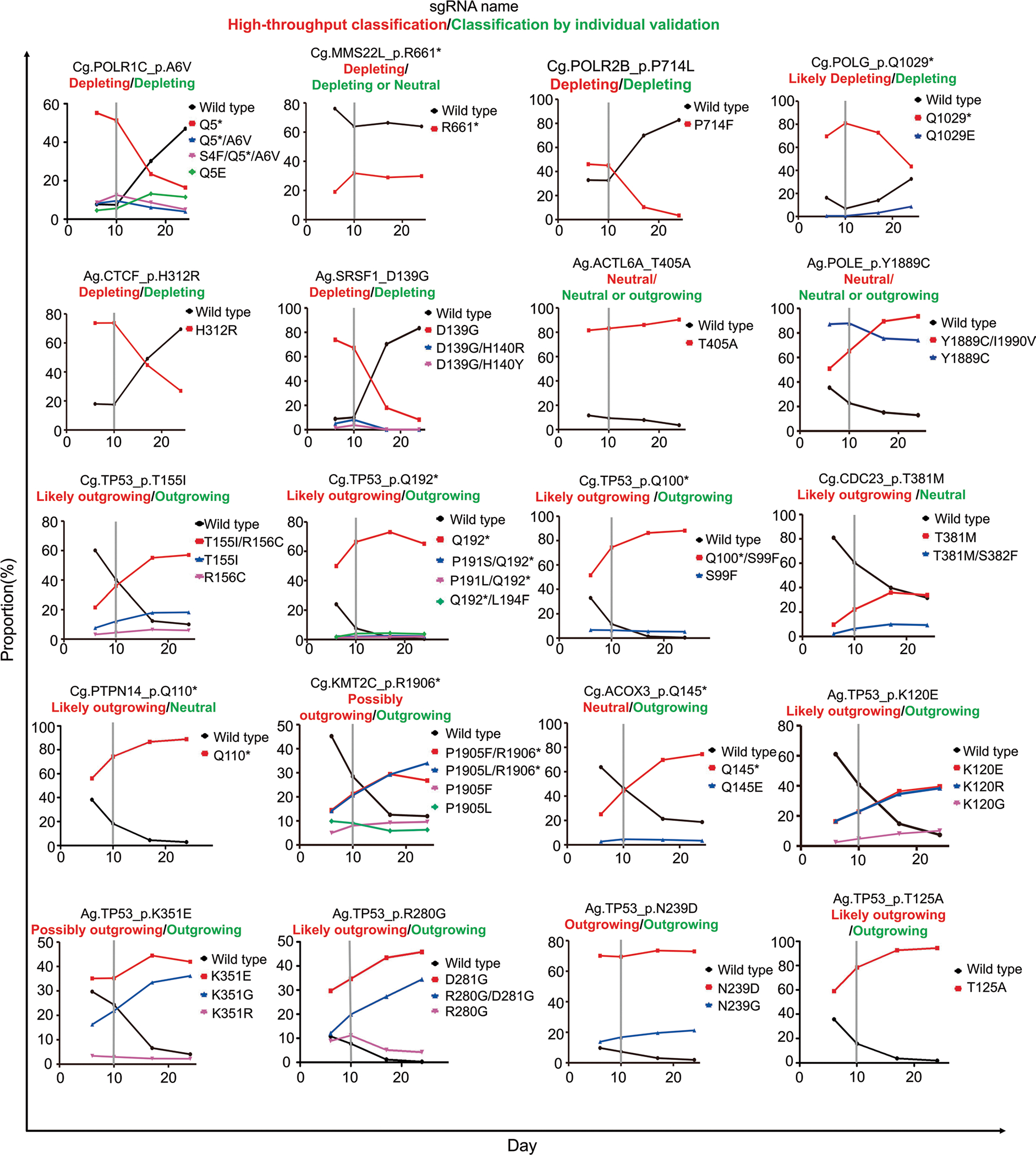
sgRNA-encoding lentivirus was transduced into P-C and P-A cells at day 0 and doxycycline was added to induce expression of CBE and ABE, respectively, and maintained until day 10, after which doxycycline was removed. The functional classification results obtained from the high-throughput experiments and those from these individual experiments are shown in red and green, respectively, on the top of each graph. The mean values of two independent samples are indicated.
Extended Data Fig. 9 |. The results of competitive proliferation assays.
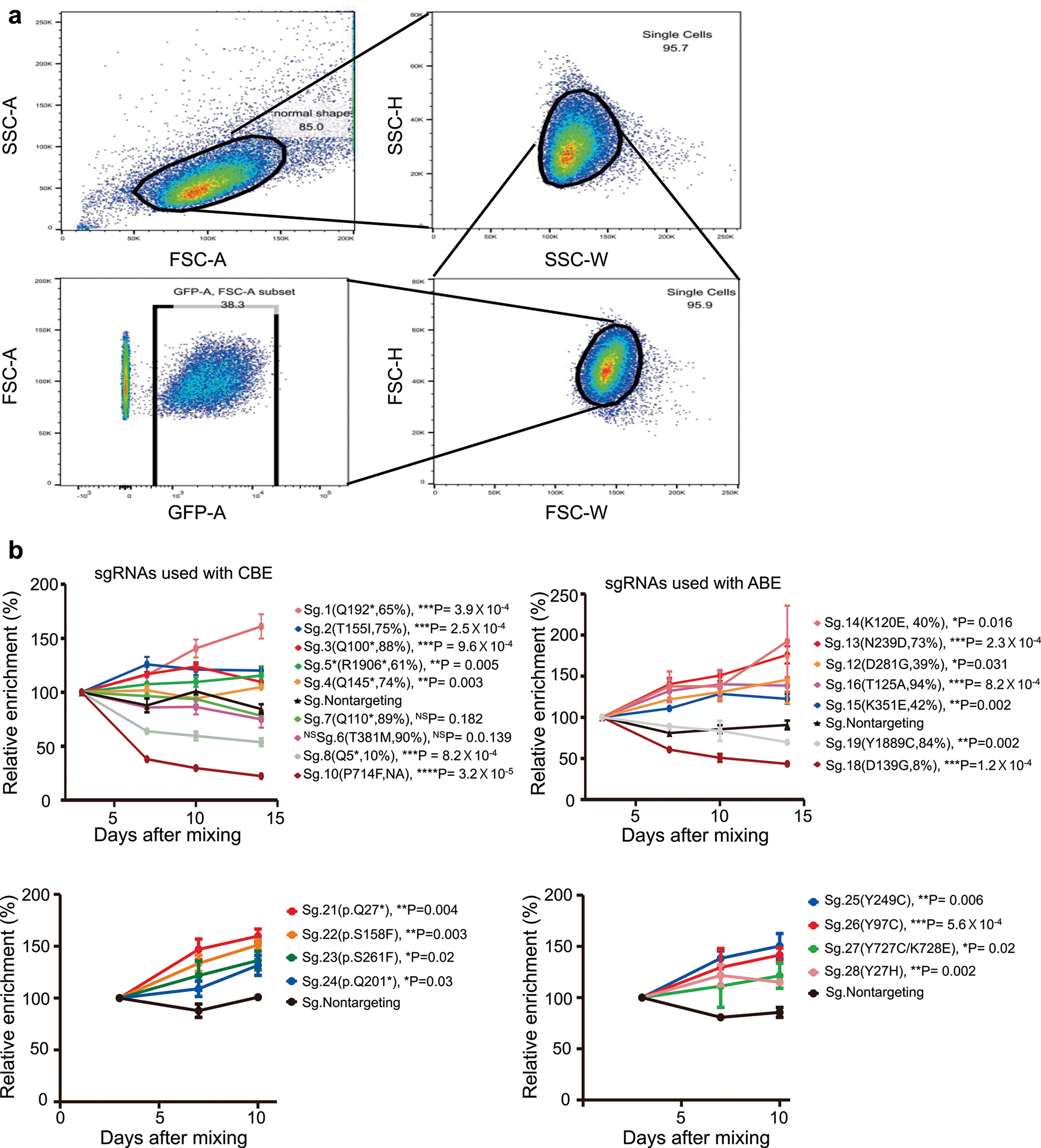
a, An example for flow cytometry gating strategy used in the competitive proliferation assays. b, Mean relative enrichment values ± standard deviation of three replicates. Student’s t test was performed under the null hypothesis that the proportions of sgRNA-transduced and nontargeting sgRNA-transduced cells would be the same. Two nontargeting sgRNAs were used as the control and the mean values of relative enrichment were used as the control.
Extended Data Fig. 10 |. Notable gene groups associated with outgrowing/likely outgrowing and depleting/likely depleting sgRNas and variants.
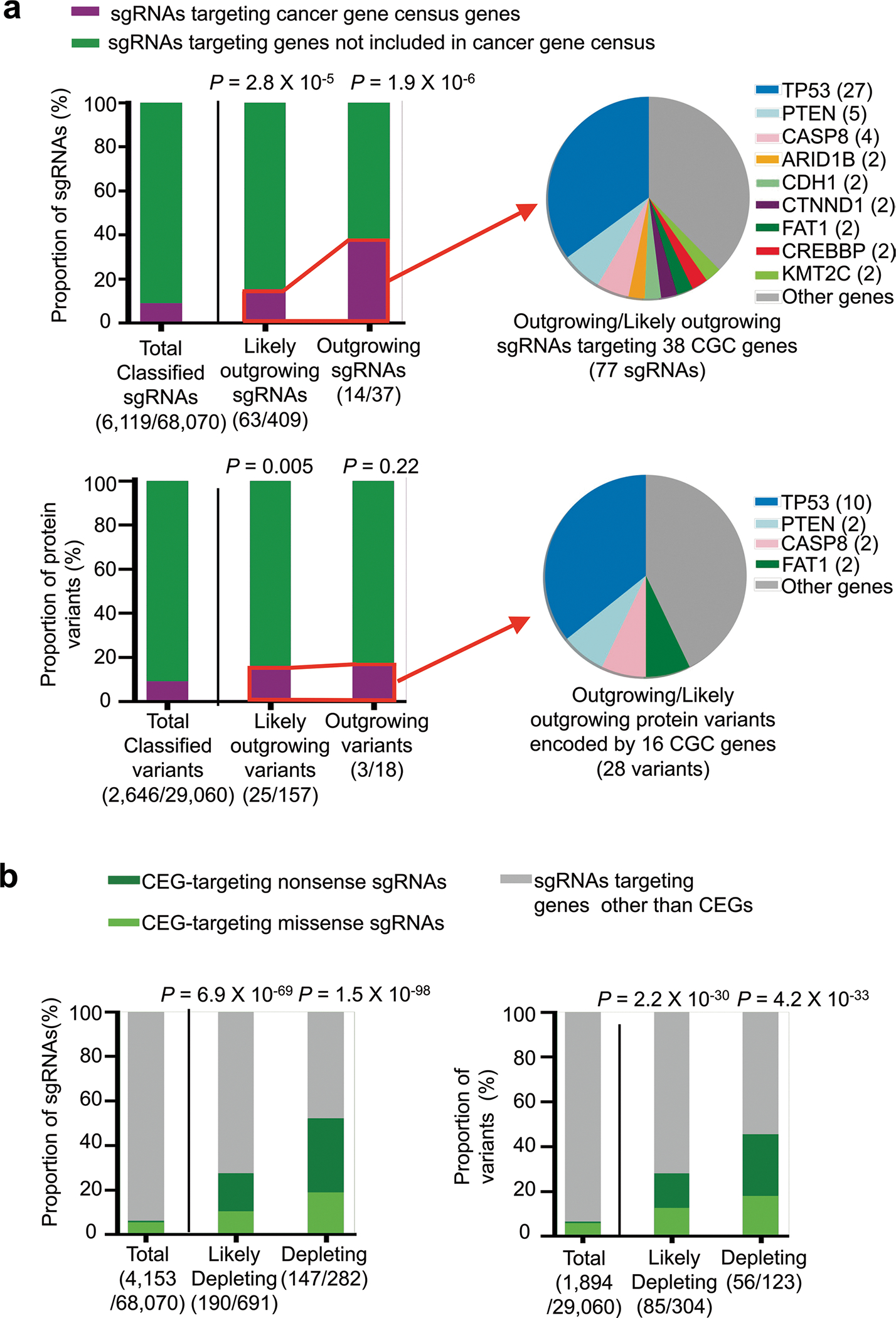
a, (Left panel) The fraction of functionally classified sgRNAs (top) targeting cancer gene census (CGC)5 genes and primary protein variants (bottom) encoded by CGC genes in the outgrowing and likely outgrowing groups. Results from all libraries except library eC were combined. P-values from two-sided Fisher’s exact test are shown. The number of sgRNAs or variants either targeting or encoded by CGC genes among all sgRNAs or variants in each group are shown on the x-axes. (Right panel) Detailed distribution of sgRNAs predicted to introduce mutations in CGC genes (top) and variants generated in CGC genes (bottom). The number of sgRNAs or variants corresponding to each gene is specified in parentheses. b, The fraction of functionally classified sgRNAs (left) targeting Depmap common essential genes (CEGs) and protein variants (right) encoded by CEGs in the depleting and likely depleting groups. Results from all libraries except library eC were combined. P-values from two-sided Fisher’s exact test are shown. The numbers of sgRNAs or variants either targeting or encoded by CEG genes among all sgRNAs or variants in each group are shown on the x-axes.
Supplementary Material
Acknowledgements
We thank J. W. Choi for assisting with computational analysis. This work was supported, in part, by the National Research Foundation of Korea (grants 2017R1A2B3004198 (H.H.K.), 2017M3A9B4062403 (H.H.K.) and 2018R1A5A2025079 (H.H.K)); the Brain Korea 21 Plus Project (Yonsei University College of Medicine); the Yonsei Signature Research Cluster Program of 2021–22-0014 (H.H.K.); a grant of the MD-PhD/Medical Scientist Training Program (S.L.) through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea; Lung Cancer SPORE P50 (CA070907; J.D.M.); and the Korean Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (grant HI21C1314 (H.H.K.)).
Footnotes
Competing interests
Yonsei University has filed a patent application based on this work, in which Y.K., S.L. and H.H.K. are listed as inventors. J.D.M. receives licensing fees from the National Institutes of Health and the University of Texas Southwestern Medical Center for distributing human cell lines. All the other authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41587-022-01276-4.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41587-022-01276-4.
Peer review information Nature Biotechnology thanks the anonymous reviewers for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
Code availability
The custom Python scripts used for the generation of the MAGeCK input file using UMIs are available on GitHub (https://github.com/oreolic/CancerLibrary).
Reporting Summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
We have submitted the deep sequencing data from this study to the National Center of Biotechnology Information’s Sequence Read Archive under accession number PRJNA667758. We have provided the datasets used in this study as Supplementary Tables 2–4 and deepcrispr.info/BEvariants.
References
- 1.McLendon R et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hudson TJ et al. International network of cancer genome projects. Nature 464, 993–998 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campbell PJ et al. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailey MH et al. Comprehensive characterization of cancer driver genes and mutations. Cell 173, 371–385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sondka Z et al. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 18, 696–705 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rheinbay E et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 578, 102–111 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stratton MR, Campbell PJ & Futreal PA The cancer genome. Nature 458, 719 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giacomelli AO et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet. 50, 1381–1387 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kotler E et al. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol. Cell 71, 178–190 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Majithia AR et al. Prospective functional classification of all possible missense variants in PPARG. Nat. Genet. 48, 1570–1575 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brenan L et al. Phenotypic characterization of a comprehensive set of MAPK1/ERK2 missense mutants. Cell Rep. 17, 1171–1183 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahler E et al. A combined approach reveals a regulatory mechanism coupling Src’s kinase activity, localization, and phosphotransferase-independent functions. Mol. Cell 74, 393–408 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matreyek KA et al. Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat. Genet. 50, 874–882 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Starita LM et al. A multiplex homology-directed DNA repair assay reveals the impact of more than 1,000 BRCA1 missense substitution variants on protein function. Am. J. Hum. Genet. 103, 498–508 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiasson MA et al. Multiplexed measurement of variant abundance and activity reveals VKOR topology, active site and human variant impact. eLife 9, e58026 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim H & Kim JS A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 15, 321–334 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Findlay GM et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature 562, 217–222 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kandoth C et al. Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishida K et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353, aaf8729 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Gaudelli NM et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim HS et al. Systematic identification of molecular subtype-selective vulnerabilities in non-small-cell lung cancer. Cell 155, 552–566 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bamford S et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 91, 355–358 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanna RE et al. Massively parallel assessment of human variants with base editor screens. Cell 184, 1064–1080 (2021). [DOI] [PubMed] [Google Scholar]
- 25.Kuscu C et al. CRISPR-STOP: gene silencing through base-editing-induced nonsense mutations. Nat. Methods 14, 710–712 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Koblan LW et al. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 36, 843–846 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song M et al. Sequence-specific prediction of the efficiencies of adenine and cytosine base editors. Nat. Biotechnol. 38, 1037–1043 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Kim HK et al. In vivo high-throughput profiling of CRISPR–Cpf1 activity. Nat. Methods 14, 153–159 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Kim HK et al. Deep learning improves prediction of CRISPR–Cpf1 guide RNA activity. Nat. Biotechnol. 36, 239–241 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Kim HK et al. SpCas9 activity prediction by DeepSpCas9, a deep learning-based model with high generalization performance. Sci. Adv. 5, eaax9249 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim HK et al. High-throughput analysis of the activities of xCas9, SpCas9-NG and SpCas9 at matched and mismatched target sequences in human cells. Nat. Biomed. Eng. 4, 111–124 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Kim N et al. Prediction of the sequence-specific cleavage activity of Cas9 variants. Nat. Biotechnol. 38, 1328–1336 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Kim HK et al. Predicting the efficiency of prime editing guide RNAs in human cells. Nat. Biotechnol. 39, 198–206 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Hill AJ et al. On the design of CRISPR-based single-cell molecular screens. Nat. Methods 15, 271–274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michlits G et al. CRISPR-UMI: single-cell lineage tracing of pooled CRISPR–Cas9 screens. Nat. Methods 14, 1191–1197 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Schmierer B et al. CRISPR/Cas9 screening using unique molecular identifiers. Mol. Syst. Biol. 13, 945 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arbab M et al. Determinants of base editing outcomes from target library analysis and machine learning. Cell 182, 463–480 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR–Cas9. Nat. Biotechnol. 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li W et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghandi M et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carter H et al. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res. 69, 6660–6667 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ng PC & Henikoff S Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adzhubei IA et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miosge LA et al. Comparison of predicted and actual consequences of missense mutations. Proc. Natl Acad. Sci. USA 112, E5189–E5198 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun S et al. An extended set of yeast-based functional assays accurately identifies human disease mutations. Genome Res. 26, 670–680 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen H et al. Comprehensive assessment of computational algorithms in predicting cancer driver mutations. Genome Biol. 21, 43 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Markusic D, Oude-Elferink R, Das AT, Berkhout B & Seppen J Comparison of single regulated lentiviral vectors with rtTA expression driven by an autoregulatory loop or a constitutive promoter. Nucleic Acids Res. 33, e63 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yi SA et al. HPV-mediated nuclear export of HP1γ drives cervical tumorigenesis by downregulation of p53. Cell Death Differ. 27, 2537–2551 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eekels JJM et al. A competitive cell growth assay for the detection of subtle effects of gene transduction on cell proliferation. Gene Ther. 19, 1058–1064 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Hanahan D & Weinberg RA The hallmarks of cancer. Cell 100, 57–70 (2000). [DOI] [PubMed] [Google Scholar]
- 52.Sequist LV et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 3, 75ra26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ganesan P et al. Epidermal growth factor receptor P753S mutation in cutaneous squamous cell carcinoma responsive to cetuximab-based therapy. J. Clin. Oncol. 34, e34–e37 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Stabile LP et al. Combined targeting of the estrogen receptor and the epidermal growth factor receptor in non-small cell lung cancer shows enhanced antiproliferative effects. Cancer Res. 65, 1459–1470 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Landrum MJ et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 48, D835–D844 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen Y et al. PHLDA1, another PHLDA family protein that inhibits Akt. Cancer Sci. 109, 3532–3542 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nagai MA Pleckstrin homology-like domain, family A, member 1 (PHLDA1) and cancer. Biomed. Rep. 4, 275–281 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Botti E et al. Developmental factor IRF6 exhibits tumor suppressor activity in squamous cell carcinomas. Proc. Natl Acad. Sci. USA 108, 13710–13715 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jobling R et al. Monozygotic twins with variable expression of Van der Woude syndrome. Am. J. Med. Genet. A 155A, 2008–2010 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stupack DG Caspase-8 as a therapeutic target in cancer. Cancer Lett. 332, 133–140 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jia D et al. Crebbp loss drives small cell lung cancer and increases sensitivity to HDAC inhibition. Cancer Discov. 8, 1422–1437 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pasqualucci L et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 471, 189–195 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cuella-Martin R et al. Functional interrogation of DNA damage response variants with base editing screens. Cell 184, 1081–1097 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sánchez-Rivera FJ et al. Base editing sensor libraries for high-throughput engineering and functional analysis of cancer-associated single nucleotide variants. Nat. Biotechnol. 10.1038/s41587-021-01172-3 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim YB et al. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 35, 371–376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ran FA et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li X et al. Base editing with a Cpf1–cytidine deaminase fusion. Nat. Biotechnol. 36, 324–327 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Zetsche B et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR–Cas system. Cell 163, 759–771 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nishimasu H et al. Engineered CRISPR–Cas9 nuclease with expanded targeting space. Science 361, 1259–1262 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hu JH et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Anders C, Bargsten K & Jinek M Structural plasticity of PAM recognition by engineered variants of the RNA-guided endonuclease Cas9. Mol. Cell 61, 895–902 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kleinstiver BP et al. Engineered CRISPR–Cas9 nucleases with altered PAM specificities. Nature 523, 481–485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Walton RT, Christie KA, Whittaker MN & Kleinstiver BP Unconstrained genome targeting with near-PAMless engineered CRISPR–Cas9 variants. Science 368, 290–296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou C et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature 571, 275–278 (2019). [DOI] [PubMed] [Google Scholar]
- 75.Thuronyi BW et al. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat. Biotechnol. 37, 1070–1079 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Richter MF et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 38, 883–891 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gaudelli NM et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 38, 892–900 (2020). [DOI] [PubMed] [Google Scholar]
- 78.Kurt IC et al. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol. 39, 41–46 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhao D et al. Glycosylase base editors enable C-to-A and C-to-G base changes. Nat. Biotechnol. 39, 35–40 (2021). [DOI] [PubMed] [Google Scholar]
- 80.Hanson G & Coller J Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 19, 20–30 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sanjana NE, Shalem O & Zhang F Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Meier JA, Zhang F & Sanjana NE GUIDES: sgRNA design for loss-of-function screens. Nat. Methods 14, 831–832 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ramirez RD et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 64, 9027–9034 (2004). [DOI] [PubMed] [Google Scholar]
- 84.Ellis BL, Potts PR & Porteus MH Creating higher titer lentivirus with caffeine. Hum. Gene Ther. 22, 93–100 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dang Y et al. Optimizing sgRNA structure to improve CRISPR–Cas9 knockout efficiency. Genome Biol. 16, 280 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shalem O et al. Genome-scale CRISPR–Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Billon P et al. CRISPR-mediated base editing enables efficient disruption of eukaryotic genes through induction of STOP codons. Mol. Cell 67, 1068–1079 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Behan FM et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 568, 511–516 (2019). [DOI] [PubMed] [Google Scholar]
- 89.Hart T, Brown KR, Sircoulomb F, Rottapel R & Moffat J Measuring error rates in genomic perturbation screens: gold standards for human functional genomics. Mol. Syst. Biol. 10, 733 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Martincorena I et al. Universal patterns of selection in cancer and somatic tissues. Cell 171, 1029–1041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kandasamy K et al. NetPath: a public resource of curated signal transduction pathways. Genome Biol. 11, R3 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang G & Fersht AR Mechanism of initiation of aggregation of p53 revealed by Φ-value analysis. Proc. Natl Acad. Sci. USA 112, 2437–2442 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhao D et al. Combinatorial CRISPR–Cas9 metabolic screens reveal critical redox control points dependent on the KEAP1–NRF2 regulatory axis. Mol. Cell 69, 699–708 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Clement K et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Smith T, Heger A & Sudbery I UMI-tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res. 27, 491–499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhu S et al. Guide RNAs with embedded barcodes boost CRISPR-pooled screens. Genome Biol. 20, 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xu P et al. Genome-wide interrogation of gene functions through base editor screens empowered by barcoded sgRNAs. Nat. Biotechnol. 39, 1403–1413 (2021). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We have submitted the deep sequencing data from this study to the National Center of Biotechnology Information’s Sequence Read Archive under accession number PRJNA667758. We have provided the datasets used in this study as Supplementary Tables 2–4 and deepcrispr.info/BEvariants.