

Abstract
Tandem duplications (TDs) of the UBTF gene have been recently described as a recurrent alteration in pediatric acute myeloid leukemia (AML). Here, by screening 1946 newly diagnosed adult AML, we found that UBTF-TDs occur in about 3% of patients aged 18–60 years, in a mutually exclusive pattern with other known AML subtype-defining alterations. The characteristics of 59 adults with UBTF-TD AML included young age (median 37 years), low bone marrow (BM) blast infiltration (median 25%), and high rates of WT1 mutations (61%), FLT3-ITDs (51%) and trisomy 8 (29%). BM morphology frequently demonstrates dysmyelopoiesis albeit modulated by the co-occurrence of FLT3-ITD. UBTF-TD patients have lower complete remission (CR) rates (57% after 1 course and 76% after 2 courses of intensive chemotherapy [ICT]) than UBTF-wild-type patients. In patients enrolled in the ALFA-0702 study (n = 614 patients including 21 with UBTF-TD AML), the 3-year disease-free survival (DFS) and overall survival of UBTF-TD patients were 42.9% (95%CI: 23.4–78.5%) and 57.1% (95%CI: 39.5–82.8%) and did not significantly differ from those of ELN 2022 intermediate/adverse risk patients. Finally, the study of paired diagnosis and relapsed/refractory AML samples suggests that WT1-mutated clones are frequently selected under ICT. This study supports the recognition of UBTF-TD AML as a new AML entity in adults.
Subject terms: Acute myeloid leukaemia, Cancer genomics
Introduction
The upstream binding transcription factor (UBTF) gene, located at 17q21, encodes a ubiquitously expressed nucleolar protein. UBTF is a key component of the pre-initiation complex mediating the recruitment of RNA polymerase I to ribosomal DNA promoter regions but is also enriched at polymerase II-transcribed genes throughout human genomes [1–3]. UBTF is a member of the high mobility group (HMG)-box protein family, which contains six conserved HMG box DNA binding domains [4, 5]. UBTF dysregulation have been linked to different diseases including childhood neurodegeneration (due to germline gain-of-function missense mutations) [6] and cancers through various mechanisms. These include UBTF upregulation in solid tumors such as melanoma [7], lung [8] and colon cancers [9], or the oncogenic gene fusions UBTF::ETV4 in prostate cancer [10] and UBTF::ATXN7L3 in B-cell precursor acute lymphoblastic leukemia [11].
Recently, tandem duplications of UBTF (UBTF-TDs) have been described as a recurrent alteration in pediatric acute myeloid leukemia (AML), accounting for 4% of newly-diagnosed cases and 9% of relapse cases [12–15]. In pediatric AML, UBTF-TDs are associated with distinct genetic features including the frequent co-occurrence of FLT3-internal tandem duplications (FLT3-ITDs) and WT1 mutations, normal karyotype or isolated trisomy 8, mutual exclusivity with known AML subtype-defining lesions (i.e. NPM1 mutations and recurrent fusions) and activation of the HOXA/HOXB cluster genes [13]. UBTF-TDs were associated with poor outcome and measurable residual disease (MRD) positivity after induction chemotherapy. Together, these data suggest that UBTF-TD defines a new entity of high-risk pediatric AML [12–14, 16].
Here, we studied the prevalence of UBTF-TD in adult AML by screening 1946 newly diagnosed AML from 3 prospective trials. We describe the clinical, biological, genetic, and prognostic features associated with this alteration in a cohort of 59 AML patients with UBTF-TD (including 21 with prospectively collected survival data in the ALFA-0702 study) receiving intensive chemotherapy (ICT) usually followed by hematopoietic stem cell transplantation (allo-HCT).
Methods
Patients and samples
A total of 1946 available AML diagnostic samples from patients registered in 3 French ICT prospective trials were retrospectively screened for UBTF-TD. DNA samples were stored in the Tumor bank of the Acute Leukemia French Association (ALFA) in Lille Hospital (certification NF 96900-2014/65453-1). Patients were aged 18 to 60 years old in the ALFA-0702 (n = 614; NCT00932412) [17] and BIG1 (n = 895; NCT02416388) trials, and 60 years or older in the ALFA-1200 trial (n = 437; NCT01966497) [18].
Thirteen additional patients with UBTF-TDs identified in 2 centers from routine sequencing of AML samples (Saint-Louis Hospital AP-HP and CHU Lille) were also included to provide further insights into the disease description (Supplementary Fig. 1). All of them were registered in the Hauts-de-France (HDF)-AML observatory (Commission Nationale de l’Informatique et des Libertés identifier 2214454v0) and/or ALFA-PPP registry (NCT04777916). The study was conducted in accordance with the Declaration of Helsinki and French ethics regulations.
UBTF-TD screening
Screening for UBTF-TD was performed by polymerase chain reaction (PCR) on genomic DNA extracted from bone marrow (BM) or peripheral blood (PB) using 6-FAM-labeled primers designed on exons 12 and 14 (Supplementary Table 1) of the UBTF gene. The Expand™ Long Template PCR System (Roche) with a touchdown PCR program (start from 65 °C to 62 °C with a decrease of 0.5 °C per cycle, followed by 28 cycles at 62 °C) was used because of off-target priming within the UBTFL8 (UBTF-like 8) pseudogene in standard conditions. Then, PCR was followed by high resolution fragment analysis (4 h migration) on an Applied Biosystems™ 3730 DNA Analyzer 48-capillary array (ThermoFisher) using the GeneScan™ 1200 LIZ™ dye Size Standard (ThermoFisher).
Targeted DNA sequencing
All samples with positive screening were further studied by captured-based next generation sequencing (NGS) with a custom panel of 154 genes including UBTF (Supplementary Table 2). Details about library preparation, sequencing and annotations are provided in the Supplemental Appendix.
WGS, WES and RNA-sequencing
Whole genome sequencing (WGS), whole exome sequencing (WES) and whole transcriptome RNA sequencing (WTS) were performed in one patient (#L220L8879S) with refractory AML. Details are provided in the Supplemental Appendix.
Statistical analysis
Variables are reported as numbers and percentages or median and interquartile range (IQR). Comparisons of categorical and continuous variables were made with Fisher exact and Mann-Whitney U tests, respectively. P-values were corrected for multiple testing by using the Benjamini-Hochberg procedure (q-values) [19]. As baseline features and outcome for UBTF-wt patients enrolled in the BIG1 trial were not available at time of this work, UBTF-TD patients were compared to the 593 UBTF-wt patients from the ALFA-0702 study only. Considering the rarity of UBTF-TDs in AML patients over 60 years, comparisons were done between patients aged 18 to 60 years only. Complete remission (CR)/CR with incomplete platelet recovery (CRp) rates were compared using the Fisher exact test. Multivariate analysis of CR/CRp was done with logistic regressions accounting for age, white blood cell (WBC) count and European LeukemiaNet (ELN) 2022 risk stratification [20]. Overall survival (OS) and OS from CR/CRp were defined from inclusion in the ALFA-0702 trial or time of CR/CRp achievement, respectively, until death or last follow-up. Disease-free survival (DFS) was defined from the date of CR/CRp to the date of relapse or death (whichever came first) or until last follow-up. Follow-up was estimated by the reverse Kaplan–Meier method. Survival was analyzed with the Kaplan–Meier method. Impact of UBTF-TD was estimated by the log-rank test for OS and DFS. The impact of allo-HCT on DFS and OS from CR/CRp was studied considering allo-HCT as a time-dependent variable in a univariable Cox model. All tests were two-sided, statistical significance was defined as a p-value or q-value <0.05, and statistical analyses were performed with R software 4.1.2 (cran.r-project.org).
Results
Prevalence of UBTF-TD and molecular characteristics
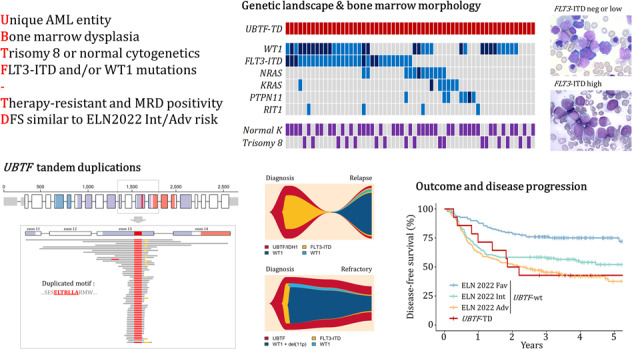
Presence of UBTF-TD was screened by fragment analysis and confirmed by targeted NGS (Fig. 1A–B) in 1946 patients with AML enrolled in 3 ICT trials. Somatic UBTF-TDs were identified in 3% of AML patients aged 18–60 years (21/614 [3.4%] and 23/895 [2.6%] patients from the ALFA-0702 and BIG1 cohorts respectively) but only 0.5% of AML patients 60 years or older (2/437 patients in the ALFA-1200 cohort). Together with patients prospectively identified by NGS, we report here a total of 59 adult AML patients with somatic UBTF-TDs.
Fig. 1. UBTF-TDs in adult AML.

A Fragment analysis of mutant (665 bp) and wild-type (617 bp) alleles of UBTF in a patient with a 48-bp tandem duplication. B Integrative Genomics Viewer (IGV) visualization showing the soft-clipped reads and increased coverage in UBTF exon 13. C UBTF gene structure and location of UBTF-TDs. The common minimal duplicated motif in exon 13 is highlighted in red.
UBTF-TDs were highly variable in size, ranging from 39 to more than 900 nucleotides. Most frequent TDs were 48 (n = 18), 51 (n = 10) and 54 (n = 6) base pairs (bp) in size, accounting together for 58% of all UBTF-TDs. All TDs led to in-frame insertions within exon 13 of UBTF with a common minimal duplicated region of 27 nucleotides (corresponding to nucleotides 1306 to 1326, using the NM_014233.3 transcript version as reference) shared by all but one patient. At the amino-acid level, this region encodes the leucine-rich ELLTRLA motif (Glu436-Leu437-Leu438-Thr439-Arg440-Leu441-Ala442) of the HMG4 domain (Fig. 1C, Supplementary Table 3). In the last patient not sharing this motif, the mutation was a 42-bp insertion encoding another leucine rich motif (GLCLRFNQLDLDQA). It should be noted that the size of very large duplications was not necessarily a multiple of 3 but was assumed to lead to in-frame insertions after RNA splicing. This hypothesis was verified by WTS in one patient (#L220L8879S) who harbored a large duplication (598 bp) spanning exons 12 to 14 finally leading to an exon13-exon13 fused RNA transcript (Supplementary Fig. 2).
As previously noticed in pediatric cases [13], we observed that most cases did not harbor perfect duplications. Insertions of non-templated nucleotides and deletions were frequent alongside the duplication. Fifteen patients (25%) had small deletions (mainly involving Trp445, Asn446 and Asp447) within the duplication. This feature was more frequent in large TDs >100 bp (64% vs. 13% in TDs <100 bp). In addition, while the variant callers used in our study (Vardict, Mutect2) generally failed to correctly annotate UBTF-TDs larger than 80 bp, this mismatch-creating feature enhanced our ability to suspect UBTF alterations with standards NGS algorithms. In all samples screened as positive, the variant callers identified at least one mismatch in UBTF (in contrast to UBTF-wt samples; data not shown). Because of difficulties to assess properly VAF of large insertions, VAFs for UBTF-TDs were measured using mismatches and data coverage. Across the 59 diagnostic samples, the median VAF of UBTF-TD was 45% (IQR 37–48) suggesting it was an early/founding event in leukemogenesis.
Baseline features of adult AML patients with UBTF-TDs
Among 59 adult AML patients with somatic UBTF-TD, 39 were males (sex ratio 2/1). Median age at AML diagnosis was 37 years (IQR 23–47) and only 2 were over 60 years old. Median WBC count was 3.6 × 109/L (IQR 2–26), and median BM blast infiltration was 25% (IQR 20–60) (Supplementary Table 4). Karyotype was normal in 39 patients (66%). The most frequent cytogenetic abnormality was trisomy 8 in 17 patients (29%, possibly found as a subclone). At least one additional mutation was found in 56 patients (95%) (median number of additional mutations: 3, range 0–9) (Fig. 2A). The most frequent co-occurring alterations were WT1 mutations in 36 patients (61%) and FLT3-ITDs in 30 (51%). Twenty (33%) had both WT1 and FLT3-ITD. Other mutations frequently involved signaling genes (NRAS, KRAS, PTPN11, RIT1), especially in cases lacking FLT3-ITD (55% of FLT3-ITD-negative patients presented at least one mutation in this set of genes). By contrast, mutations in epigenetic-related or RNA-splicing genes were rare. Finally, no other additional candidate gene was found by WGS/WES in patient #L220L8879S (Supplementary Table 5, Supplementary Table 6).
Fig. 2. Baseline biological features associated with UBTF-TDs.

A Mutational landscape of AML with UBTF-TD. Only genes with at least one mutation in one patient are shown. The full list of screened genes is given in the Supplementary Table 2. B BM blast infiltration and (C) WBC count according to UBTF and NPM1 status in AML patients aged 18–60 years. The black bars indicate the median values. (D) Associations among gene and cytogenetic abnormalities in AML patients aged 18–60 years. (E) BM blast infiltration, (F) WBC count and (G) BM morphology classification according to FLT3-ITD status in UBTF-TD AML. The FiLT3r algorithm was used for detection and precise quantification of FTL3-ITDs [23]. H BM morphology in patient #L171D0708G (FLT3-ITD with VAF < 5%) and (I) patient #L151D4757X (FLT3-ITD negative) showing characteristic dyserythropoiesis and blasts with rare Auer rods (red arrow). J BM morphology in patient #L221T6545N (FLT3-ITD with VAF > 70%) showing massive blast infiltration.
Among patients younger than 60 years, AML patients with UBTF-TD (n = 57) were significantly younger (median 36 years vs. 47 years; q-value <0.001), had lower BM blast infiltration (median 25% vs. 60%, q-value <0.001)and higher rates of WT1 mutations (63% vs. 8%, q-value <0.001), FLT3-ITDs (53% vs. 22%, q-value <0.001) and trisomy 8 (28% vs. 8%, q-value <0.001) compared to UBTF-wt AML patients in the ALFA-0702 study (Fig. 2B–D, Table 1, Supplementary Table 7). Conversely, UBTF-TD was mutually exclusive with NPM1 mutations, CEBPA in-frame bZIP mutations, TP53 mutations, adverse cytogenetics and recurrent fusion transcripts and less frequently associated with myelodysplasia-related mutations [20] than UBTF-wt (9% vs. 29%, q-value = 0.003). The vast majority UBTF-TD AML were assigned to the ELN 2022 intermediate risk group. Only 5 were assigned to the adverse group because of myelodysplasia-related mutations (RUNX1, n = 2; BCOR, n = 1; SF3B1, n = 1; U2AF1, n = 1).
Table 1.
Characteristics of AML patients (18–60 y) according to UBTF status.
| Parameters | UBTF-TD AML 18–60 y | UBTF wild-type AML 18–60 y (ALFA-0702) |
|---|---|---|
| No. of patients | 57 | 593 |
| Age (y), median (IQR) | 36 (24–45) | 47 (37–54) |
| WBC (×109/L), median (IQR) | 3.55 (2–27.4) | 8.3 (2.6–32.9) |
| BM blasts (%), median (IQR) | 25 (20–63) | 60 (39–82) |
| BM morphology, n (%) | ||
| M0 | 0 | 28/467 (6%) |
| M1/M2 | 15/33 (45%) | 254/467 (54%) |
| M4/M5 | 0 | 169/467 (36%) |
| M6 | 14/33 (42%) | 14/467 (3%) |
| M7 | 0 | 2/467 (0%) |
| MRC | 4/33 (12%) | |
| Cytogenetics | ||
| Normal, n (%) | 38/57 (67%) | 329/563 (58%) |
| Trisomy 8, n (%) | 16/57 (28%) | 46/545 (8%) |
| Monosomy 5/del(5q), n (%) | 0 | 33/545 (6%) |
| Monosomy 7/del(7q), n (%) | 0 | 47/545 (9%) |
| Monosomy 17/del(17p), n (%) | 0 | 22/545 (4%) |
| Del(20q), n (%) | 0 | 14/545 (3%) |
| Del(12p), n (%) | 0 | 14/545 (3%) |
| Complex, n (%) | 0 | 67/545 (12%) |
| WT1 mutations, n (%) | 36/57 (63%) | 48/572 (8%) |
| Signaling mutations | ||
| FLT3-ITD, n (%) | 30/57 (53%) | 127/572 (22%) |
| FLT3-TKD, n (%) | 4/57 (7%) | 72/572 (13%) |
| NRAS, n (%) | 13/57 (23%) | 126/572 (22%) |
| KRAS, n (%) | 7/57 (12%) | 43/572 (8%) |
| PTPN11, n (%) | 6/57 (11%) | 32/572 (6%) |
| RIT1, n (%) | 6/57 (11%) | 19/572 (3%) |
| DNA-methylation gene mutations | ||
| DNMT3A, n (%) | 3/57 (5%) | 162/572 (28%) |
| TET2, n (%) | 5/57 (9%) | 74/572 (13%) |
| IDH1, n (%) | 3/57 (5%) | 53/572 (9%) |
| IDH2, n (%) | 3/57 (5%) | 74/572 (13%) |
| NPM1 mutations, n (%) | 0 | 208/572 (36%) |
| CEBPA double mutations, n (%) | 0 | 30/572 (5%) |
| TP53 mutations, n (%) | 0 | 39/572 (7%) |
| MDS-related gene mutations*, n (%) | 5/57 (9%) | 168/572 (29%) |
| ELN 2022 risk | ||
| Favorable | 0 | 143/548 (26%) |
| Intermediate | 52/57 (91%) | 157/548 (29%) |
| Adverse | 5/57 (9%) | 248/548 (45%) |
*MDS-related genes: ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1, and/or ZRSR2.
These data refer to only 57 of the 59 patients with UBTF-TDs (the 2 patients over 60 years of age were excluded for comparisons).
BM bone marrow, IQR interquartile range, MRC myelodysplastic-related changes, WBC white blood cell count.
BM morphology of UBTF-TD AML usually showed myelodysplastic features with trilineage dysmyelopoiesis including specific dyserythropoiesis (megaloblastosis, giant erythroblasts multinuclearity or nuclear lobulation) and megakaryocytic features (micromegakaryocytes). Blasts displayed myeloid features with myeloperoxidase positivity and rare Auer rods. Overall, 60% of cases showed a BM morphology compatible with the diagnosis of AML-M6 according to the FAB classification [21] or AML with myelodysplastic-related changes [22]. Interestingly, the co-occurrence of FLT3-ITD, especially at high VAF (≥20%), was associated with a specific AML presentation with higher WBC counts, massive BM blast infiltrations with disappearance of more mature cells and a dominance of the AML-M1/M2 morphological subtypes (Fig. 2E–J, Supplementary Fig. 3, Supplementary Table 8) [23].
Disease history and evolution in UBTF-TD AML
Fifty-eight of the 59 patients with UBTF-TD AML received cytarabine-anthracycline based intensive induction chemotherapy (without FLT3 inhibitors). Twenty-five (43%) failed to achieve CR/CRp after the first induction course among whom 10 finally achieved CR/CRp after a second salvage induction. This led to an overall CR rate of 57% after 1 course and 76% after 2 courses of ICT. The quantification of WT1 transcript was performed at diagnosis and at early evaluation for 31 patients [24, 25]. As expected, WT1 remained overexpressed after induction in all tested patients with refractory AML (n = 12). Among 19 patients in CR after the first induction course, 9 (47%) still had persistent WT1 overexpression (Supplementary Fig. 4).
Figure 3 shows the disease history in 35 patients with available follow-up. Among those 35 patients, with a median follow-up of 2.7 years, 21 relapsed including 9 after allo-HCT and 12 before any allo-HCT. Median time from CR achievement to relapse in patients without allo-HCT was 3 months (range 1.2–21.6). The median time from allo-HCT to post-allo-HCT relapse was 22.8 months (range 3.6–79.2).
Fig. 3. Swimmer plot graph of adult AML patients with UBTF-TDs.

Each bar represents one patient in the study. Only patients with available follow-up (n = 35) are shown.
Impact of UBTF-TD on CR achievement in AML patients 18–60 y treated with ICT
To explore the clinical impact of UBTF-TD on outcome, we compared the 21 UBTF-TD patients (all assigned to the ELN 2022 intermediate risk group) to the 593 UBTF-wt patients from the ALFA-0702 study. Among these 614 patients of the ALFA-0702 trial, 522 (85.0%) achieved CR/CRp after 1 or 2 courses of ICT. The proportion of UBTF-TD patients obtaining CR/CRp after 2 courses was 66.7% (n = 14) compared to 85.7% (n = 508) in UBTF-wt patients (p-value = 0.026) and 87.9% (n = 138) in ELN 2022 intermediate UBTF-wt patients (p-value = 0.018).
In multivariable logistic regression, UBTF-TD predicted lower rates of CR/CRp (OR = 0.23, 95%CI: 0.08–0.70, p-value = 0.007) independently of ELN 2022 risk stratification, WBC count and age (Table 2). UBTF-TD still predicted lower rates of CR/CRp (OR = 0.27, 95%CI: 0.08–0.88, p-value = 0.027) when restricted the analysis to patients in the ELN 2022 intermediate risk group, independently of WBC count and age. While it represents a small number of patients, among UBTF-TD patients enrolled in the ALFA-0702 study, WT1 mutations were associated with lower CR/CRp rates (n = 4/10 [40%] vs. n = 10/11 [91%], p-value = 0.024). Conversely, among WT1-mutated patients in the ALFA-0702 study, UBTF-TD was associated with lower CR/CRp rates (n = 4/10 [40%] vs. n = 38/48 [79%], p-value = 0.020).
Table 2.
Multivariate logistic regression for CR/CRp achievement in ALFA-0702 cohort.
| CR/CRp OR (95%CI) | p-value | |
|---|---|---|
| UBTF wild-type | 1 | |
| UBTF-TD | 0.23 (0.08–0.70) | 0.007 |
| Age (per 10-years of age) | 0.96 (0.77–1.18) | 0.69 |
| Log10(WBC) | 0.69 (0.47–1.01) | 0.06 |
| ELN 2022 Intermediate risk | 1 | |
| ELN 2022 Favorable risk | 3.64 (1.41–11.26) | 0.01 |
| ELN 2022 Adverse risk | 0.37 (0.20–0.67) | <0.0001 |
Prognostic significance of UBTF-TD in AML patients 18–60 y treated with ICT
We next studied the prognostic value of UBTF-TD on DFS and OS in the ALFA-0702 cohort. Among 548 UBTF-wt patients for whom the ELN 2022 risk stratification was available, 26% were favorable (n = 143), 29% were intermediate (n = 157) and 45% were adverse (n = 248). The 3-year DFS and OS were 42.9% (95%CI: 23.4–78.5%) and 57.1% (95%CI: 39.5–82.8%) in UBTF-TD patients (n = 21) compared to 57.6% (95%CI: 53.4–62.1%; p-value = 0.5) and 60.9% (95%CI: 57.0–64.9%; p-value = 1.0) in UBTF-wt patients. When considering ELN 2022 risk stratification, DFS of UBTF-TD patients was significantly lower than DFS in UBTF-wt favorable risk patients (3y-DFS: 76.0%, 95%CI: 69.2–83.5%, p-value = 0.008) but not significantly different from DFS in UBTF-wt intermediate (3y-DFS: 58.4%, 95%CI: 50.7–67.3%, p-value = 0.6) and adverse risk patients (3y-DFS: 45.4%, 95%CI: 38.8–53.1%, p-value = 0.8). Similarly, OS of UBTF-TD patients was lower than OS in UBTF-wt favorable risk patients (3y-OS: 82.3%, 95%CI: 76.3–88.9%, p-value = 0.01) but not significantly different from OS in UBTF-wt intermediate (3y-OS: 59.0%, 95%CI: 51.7–67.2%, p-value = 0.9) and adverse risk patients (3y-OS: 48.7%, 95%CI: 42.8–55.3%, p-value = 0.3)(Fig. 4A–B). Furthermore, although this only refers to a limited number of patients, the OS of UBTF-TD AML tended to worsen with the cooccurrence of WT1 mutations (3y-OS: 20% [95%CI: 6–69%] vs. 91% [95%CI: 75–100%], p-value = 0.002) or FLT3-ITD (3y-OS: 25% [95%CI: 8–83%] vs. 77% [95%CI: 57–100%], p-value = 0.008)(Supplementary Fig. 5).
Fig. 4. Clinical outcome of UBTF-TD AML 18–60 y.

A Disease-free survival in patients achieving CR or CRp after induction and (B) overall survival according to UBTF status and ELN 2022 risk stratification. Study restricted to patients enrolled in the ALFA-0702 trial with available ELN 2022 risk stratification.
According to the design of the ALFA-0702 study, all UBTF-TD patients (n = 21) were assigned to the intermediate risk group, and were thus eligible to allo-HCT. Among 14 UBTF-TD patients who achieved CR/CRp, 9 underwent allo-HCT in first CR/CRp (64.3%) and 3 after relapse (21.4%). Among this limited number of patients, allo-HCT in first CR/CRp seemed to be associated with prolonged DFS (2y-DFS: 77.8% vs. 0%, p = 0.00976) but not with prolonged OS from CR/CRp (2y-OS 100% vs. 65.6%; p = 0.521) (Supplementary Fig. 6).
Clonal architecture and disease evolution of AML with UBTF-TD
To better understand the clonal architecture of UBTF-TD AML and investigate the co-occurrence and order of acquisition of somatic mutations, 12 cases were studied both at diagnosis and during evolution (relapsed AML, n = 10 or refractory AML, n = 2)(Fig. 5) [26]. The clonal evolution was inferred from bulk NGS data considering VAF as a surrogate measure of clonal abundance. VAFs were corrected with copy number analysis to account for biallelic alterations. Patient #L220L8879S was shown to carry both WT1 mutation and deletion. Patient #L171D0708G was shown to carry a homozygous WT1 mutation probably due to an acquired uniparental disomy [27]. In most cases, UBTF-TD was present with the highest VAF at both timepoints (except for patients #L171D0708G and #L200D5231D for whom UBTF-TD probably occurred after or concomitantly with DNMT3A and IDH1 mutations). WT1 and FLT3-ITD mutations appeared to involve only a subset of leukemic cells and were frequently subject to clonal evolution during progression, suggesting they are late cooperative events (Supplementary Fig. 7).
Fig. 5. Patterns of clonal evolution in UBTF-TD AML.

Fishplots are imputed from VAFs obtained by bulk NGS at AML diagnosis and during disease progression (relapsed or refractory AML). Twelve patients with available matched samples are presented: (A–J) Relapsed AML; (K–L) Refractory AML. Disappearance of all mutations (including UBTF-TD) in CR was verified by NGS (1% threshold) in patients A, B, C, and G and was assumed in the others. Details about patient history are provided in the Fig. 3. Figure made with the Fishplot package for R (version 4.2.0) [26].
Analysis of paired diagnostic-relapse cases revealed a preferential pattern of disease progression likely driven by WT1 mutations. All relapse AML cases (n = 10) harbored at least one WT1 mutation with high VAF compatible with clonal dominance (Supplementary Fig. 8). Moreover, 6 of the 10 relapse AML cases carried multiple WT1 mutants, with VAFs suggesting that 2 mutants coexisted in the same clone in 5 cases (patients referred as F to J in the Fig. 5 and Supplementary Fig. 8). Another one (patient E) was shown to carry a homozygous WT1 mutation at relapse. Overall, biallelic WT1 alterations (WT1bi_alt) were found in 6/10 (60%) relapse samples. The WT1bi_alt clone driving relapse was already present at diagnosis at low levels in 2 cases (E and J), derived from a WT1 monoallelic clone (WT1mono_alt) in 3 cases (F, H and I), or fully emerged at relapse (or was present below the sensitivity threshold [VAF 1%] at diagnosis) in 1 case (patient G). Other mutations could coexist within the WT1bi_alt clone at relapse, frequently involving the RAS pathway (NF1, PTPN11, KRAS, and NRAS in patients F, H, I and J). In addition, patient K who was the only studied patient with a dominant WT1bi_alt clone (mutation + deletion) at AML diagnosis was refractory to induction, suggesting the resistance of this clone to intensive chemotherapy. Consistent with these observations, 21/35 (60%) WT1-mutated patients (regardless of the number or VAF of the mutation) receiving ICT were refractory to the first course of ICT, compared with 4/23 (17%) WT1 wild-type patients.
Another pattern of disease relapse was observed in patients A and B for whom the relapse appeared to be driven by a WT1mono_alt arising in a preexisting FLT3-ITD clone. While FLT3-ITD is generally associated with resistance and disease progression in AML [28], FLT3-ITDs did not seem to confer clonal advantage in UBTF-TD AML when not associated with WT1 mutation. Indeed, some FLT3-ITDs detected at diagnosis were frequently lost during progression (patients A, D and E). Similarly, patient L who was refractory to induction chemotherapy (without FLT3 inhibitor) demonstrated the rapid disappearance of FLT3-ITD and the selection of a preexisting WT1mono_alt/NF1-comutated clone.
Finally, 2 patients in the cohort received a FLT3 inhibitor (FLT3i; quizartinib) at relapse, one of whom (patient A) experienced a second relapse that was documented by sequencing. The post-quizartinib relapse was still positive for FLT3-ITD and characterized by the emergence of 2 distinct FLT3-ITD/FLT3-TKD (D835H/Y) mutants, which have been described as a recurrent mechanism of resistance to FLT3 inhibitors (Supplementary Fig. 9).
Discussion
Molecular and cytogenetic analyses are routinely used to identify AML subtype-defining structural variants and mutations and have become critical for risk stratification and treatment guidance. Despite 15 years of genomics research since the first AML genome publication and large studies like The Cancer Genome Atlas (TCGA), some AML cases remain genetically unclassifiable with current knowledge [29–31]. Notably, a subset of AMLs with FLT3-ITD and/or WT1 mutation were known to lack a known initiating event, while being associated with a poor outcome in both adults and children [27, 32]. The aggregation of some of these AMLs under a single entity, namely UBTF-TD AML, paves the way for their detection in routine practice and future development of new therapies. Overall, it is likely that UBTF-TDs have been underestimated in previous studies due to limitations in detecting this type of aberration with many of the current bioinformatic approaches used in genetics laboratories [13]. This may be due to the difficulty to align tandem duplications-containing reads to the reference genome but also to the complexity of the UBTF sequence which includes repetitive motifs and frequent homopolymers complicating variant calling.
All TDs identified in the present study led to in-frame insertions within exon 13 of UBTF with a common minimal duplicated region encoding the ELLTRLA motif of the HMG4 domain of the UBTF protein, as previously described in pediatric cases [13]. TDs ranged from 39 to more than 900 nucleotides with frequent insertions of non-templated nucleotides and deletions alongside the duplication. This feature has been already observed in other in frame duplications such as FLT3-ITD [33]. The question of a common pathophysiologic background involved in the genesis of these 2 anomalies deserves to be explored. Also, the consequences of the exon 13 duplication on UBTF function and leukemogenesis are still unknown. Umeda et al have shown that UBTF-TD expression in CD34 + cells was sufficient to induce a proliferative advantage, increases clonogenic activity and activates the HOXB gene cluster, recapitulating the transcriptional signature observed in UBTF-TD AML patients [13].
Here we found that UBTF-TDs occur in about 3% of AML in patients aged 18–60 years. However, given the variability of TDs, we cannot exclude that some perfect and large duplications were missed by the screening method. Adult AML patients with UBTF-TDs were significantly younger and had lower BM blast infiltration with a BM morphology frequently demonstrating myelodysplastic features including severe dyserythropoiesis. It should be noted that UBTF-TD AML were associated with the M6 morphology subtype [34] according to the FAB classification (especially in cases without FLT3-ITD or low ratio) and the diagnosis of AML could thus be made by counting the proportion of BM blasts among the non-erythroid cells according to the FAB criteria [21]. It is likely that some patients may have been diagnosed with myelodysplastic syndrome with increased/excess blasts using current classifications [35, 36].
UBTF-TDs were mutually exclusive with other known AML subtype-defining alterations. Sequencing data revealed UBTF-TDs were always clonal and stable during disease progression, in agreement with an early/initiating event. The most frequent co-occurring somatic aberrations were by far WT1 mutations, FLT3-ITD (high ratios being associated with increased WBC count and BM blast infiltration) and trisomy 8 (85% of cases carried at least one of these aberrations and 47% had at least two). Patients with UBTF-TDs displayed high rates of induction failure.
Prognostic analyses in patients enrolled in the ALFA-0702 study (including 21 UBTF-TD AML) showed that DFS and OS of UBTF-TD patients were not significantly different from those of UBTF-wt ELN 2022 intermediate or adverse risk patients. Although long-term remissions were observed after allo-HCT, post-allo-HCT relapse remained common and could occur after a significant delay. This shows that surveillance as well as MRD and chimerism monitoring should be repeated and would potentially allow for preemptive measures (e.g. donor lymphocyte infusion, immunomodulation). However, considering the limited number of patients studied, additional data are required to confirm the prognosis of UBTF-TD AMLs and assess the benefits of allo-HCT in this context.
Interestingly, the study of paired diagnosis and relapsed/refractory AML samples showed that WT1 mutations are likely to play a major role in disease progression. Especially, WT1bi_alt or WT1mono_alt/FLT3-ITD co-mutations were observed in most relapses, whereas patients with these features at AML diagnosis were more likely to be refractory to induction chemotherapy. Together, these results suggest strong cooperation within the UBTF-TD/WT1/FLT3-ITD trio in establishing the leukemic phenotype and determining the clinical outcome. Also, these results emphasize the need to evaluate new therapies in combination to first-line treatment or in maintenance. FLT3 inhibitors have been shown to improve outcome in FLT3-ITD-positive AML but it should be noted that WT1 mutations have been identified as contributing to drug resistance [37, 38]. The mechanism of WT1 mutations in leukemogenesis remains elusive. WT1 has a functional duality since it may act, depending on the cellular context, as a transcriptional activator or repressor through its DNA-binding zinc-finger domain and interactions with Tet enzymatic proteins that regulate DNA hydroxymethylation [39, 40]. Given this epigenetic role, the use of hypomethylating agents such as azacytidine should also be considered as a potential strategy in these cases [41]. Finally, given the high expression of HOX cluster genes in UBTF-TD AML, the use of menin inhibitors is also a promising therapeutic approach that deserves to be studied in the future [42].
Overall, our study revealed that UBTF-TDs can be found in about 3% of adult AML aged 18–60 years and define a distinct subtype of AML with specific biological and clinical features and lower CR rates. Together with pediatric data, this study supports the recognition of UBTF-TD AML as a new AML entity to be included in disease classifications, a prerequisite for the development of future guidelines and therapies, especially in young patients.
Supplementary information
Acknowledgements
The authors thank all Acute Leukemia French Association (ALFA) investigators and French Innovative Leukemia Organization (FILO) investigators. The work of all clinical research assistants is also acknowledged.
Author contributions
ND and CP conceived and planned the experiments. ND, RK, MP, LL, LF, AM-R, MC, ED, JS, EC and CP interpreted the molecular data. ALH, SG, NH, performed experimentations and developed tools for analysis. ND and PL performed morphological analysis. LA, DL, CB, JL, EM, JB-M, CG, HD, CR, AP, MH and RI managed patients and provided clinical data. KC-L supervised data management. ND and LV performed statistical analysis. ND wrote the manuscript. All authors reviewed and approved the manuscript.
Funding
This study was supported by the French Cancer National Institute (InCa – PHRC 2007/1911 and PRTK TRANSLA10-060). This research was made possible through access to the data generated by the France Genomic Medicine Plan 2025. ND is supported by grants from the Ligue Contre le Cancer (AAP 2021 du Septentrion, Comité du Pas-de-Calais) and the CHU of Lille (AAP 2021 du Fonds Hospitalier d’aide à l'émergence).
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41375-023-01906-z.
References
- 1.Kwon H, Green MR. The RNA polymerase I transcription factor, upstream binding factor, interacts directly with the TATA box-binding protein. J Biol Chem. 1994;269:30140–6. doi: 10.1016/S0021-9258(18)43788-X. [DOI] [PubMed] [Google Scholar]
- 2.Sanij E, Diesch J, Lesmana A, Poortinga G, Hein N, Lidgerwood G, et al. A novel role for the Pol I transcription factor UBTF in maintaining genome stability through the regulation of highly transcribed Pol II genes. Genome Res. 2015;25:201–12. doi: 10.1101/gr.176115.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stefanovsky VY, Moss T. The splice variants of UBF differentially regulate RNA polymerase I transcription elongation in response to ERK phosphorylation. Nucleic Acids Res. 2008;36:5093–101. doi: 10.1093/nar/gkn484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stros M, Launholt D, Grasser KD. The HMG-box: a versatile protein domain occurring in a wide variety of DNA-binding proteins. Cell Mol Life Sci CMLS. 2007;64:2590–606. doi: 10.1007/s00018-007-7162-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ueshima S, Nagata K, Okuwaki M. Internal associations of the acidic region of upstream binding factor control its nucleolar localization. Mol Cell Biol. 2017;37:e00218–17. doi: 10.1128/MCB.00218-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Edvardson S, Nicolae CM, Agrawal PB, Mignot C, Payne K, Prasad AN, et al. Heterozygous De Novo UBTF gain-of-function variant is associated with neurodegeneration in childhood. Am J Hum Genet. 2017;101:267–73. doi: 10.1016/j.ajhg.2017.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J, Zhang J, Liu W, Ge R, Gao T, Tian Q, et al. UBTF facilitates melanoma progression via modulating MEK1/2-ERK1/2 signalling pathways by promoting GIT1 transcription. Cancer Cell Int. 2021;21:543. doi: 10.1186/s12935-021-02237-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie ZC, Tang RX, Gao X, Xie QN, Lin JY, Chen G, et al. A meta-analysis and bioinformatics exploration of the diagnostic value and molecular mechanism of miR-193a-5p in lung cancer. Oncol Lett. 2018;16:4114–28. doi: 10.3892/ol.2018.9174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsoi H, Lam KC, Dong Y, Zhang X, Lee CK, Zhang J, et al. Pre-45s rRNA promotes colon cancer and is associated with poor survival of CRC patients. Oncogene. 2017;36:6109–18. doi: 10.1038/onc.2017.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barros-Silva JD, Paulo P, Bakken AC, Cerveira N, Løvf M, Henrique R, et al. Novel 5’ fusion partners of ETV1 and ETV4 in prostate cancer. Neoplasia N. Y N. 2013;15:720–6. doi: 10.1593/neo.13232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Passet M, Kim R, Gachet S, Sigaux F, Chaumeil J, Galland A, et al. Concurrent CDX2 cis-deregulation and UBTF::ATXN7L3 fusion define a novel high-risk subtype of B-cell ALL. Blood. 2022;139:3505–18. doi: 10.1182/blood.2021014723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stratmann S, Yones SA, Mayrhofer M, Norgren N, Skaftason A, Sun J, et al. Genomic characterization of relapsed acute myeloid leukemia reveals novel putative therapeutic targets. Blood Adv. 2021;5:900–12. doi: 10.1182/bloodadvances.2020003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Umeda M, Ma J, Huang BJ, Hagiwara K, Westover T, Abdelhamed S, et al. Integrated genomic analysis identifies UBTF tandem duplications as a recurrent lesion in pediatric acute myeloid leukemia. Blood Cancer Disco. 2022;3:194–207. doi: 10.1158/2643-3230.BCD-21-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noort S, Oosterwijk J van, Ma J, Garfinkle EAR, Nance S, Walsh M, et al. Analysis of rare driving events in pediatric acute myeloid leukemia. Haematologica. 2023;108:48–60. [DOI] [PMC free article] [PubMed]
- 15.Kaburagi T, Shiba N, Yamato G, Yoshida K, Tabuchi K, Ohki K, et al. UBTF-internal tandem duplication as a novel poor prognostic factor in pediatric acute myeloid leukemia. Genes Chromosomes Cancer. 2022 10.1002/gcc.23110. Online ahead of print. [DOI] [PubMed]
- 16.Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371–6. doi: 10.1038/nature25795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomas X, de Botton S, Chevret S, Caillot D, Raffoux E, Lemasle E, et al. Randomized phase II study of clofarabine-based consolidation for younger adults with acute myeloid leukemia in first remission. J Clin Oncol. 2017;35:1223–30. doi: 10.1200/JCO.2016.70.4551. [DOI] [PubMed] [Google Scholar]
- 18.Gardin C, Pautas C, Fournier E, Itzykson R, Lemasle E, Bourhis JH, et al. Added prognostic value of secondary AML-like gene mutations in ELN intermediate-risk older AML: ALFA-1200 study results. Blood Adv. 2020;4:1942–9. doi: 10.1182/bloodadvances.2019001349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57:289–300. [Google Scholar]
- 20.Döhner H, Wei AH, Appelbaum FR, Craddock C, DiNardo CD, Dombret H, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140:1345–77. [DOI] [PubMed]
- 21.Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposed revised criteria for the classification of acute myeloid leukemia. a report of the French-American-British cooperative group. Ann Intern Med. 1985;103:620–5. doi: 10.7326/0003-4819-103-4-620. [DOI] [PubMed] [Google Scholar]
- 22.Vardiman J, Reichard K. Acute myeloid leukemia with myelodysplasia-related changes. Am J Clin Pathol. 2015;144:29–43. doi: 10.1309/AJCP58RSMFRHLHHH. [DOI] [PubMed] [Google Scholar]
- 23.Boudry A, Darmon S, Duployez N, Figeac M, Geffroy S, Bucci M, et al. Frugal alignment-free identification of FLT3-internal tandem duplications with FiLT3r. BMC Bioinforma. 2022;23:448. doi: 10.1186/s12859-022-04983-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lambert J, Lambert J, Thomas X, Marceau-Renaut A, Micol JB, Renneville A, et al. Early detection of WT1 measurable residual disease identifies high-risk patients, independent of transplantation in AML. Blood Adv. 2021;5:5258–68. doi: 10.1182/bloodadvances.2021004322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cilloni D, Renneville A, Hermitte F, Hills RK, Daly S, Jovanovic JV, et al. Real-time quantitative polymerase chain reaction detection of minimal residual disease by standardized wt1 assay to enhance risk stratification in acute myeloid leukemia: a European leukemianet study. J Clin Oncol. 2009;27:5195–201. doi: 10.1200/JCO.2009.22.4865. [DOI] [PubMed] [Google Scholar]
- 26.Miller CA, McMichael J, Dang HX, Maher CA, Ding L, Ley TJ, et al. Visualizing tumor evolution with the fishplot package for R. BMC Genomics. 2016;17:880. doi: 10.1186/s12864-016-3195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaidzik VI, Schlenk RF, Moschny S, Becker A, Bullinger L, Corbacioglu A, et al. Prognostic impact of WT1 mutations in cytogenetically normal acute myeloid leukemia: a study of the German-Austrian AML study group. Blood. 2009;113:4505–11. doi: 10.1182/blood-2008-10-183392. [DOI] [PubMed] [Google Scholar]
- 28.Thiede C, Steudel C, Mohr B, Schaich M, Schäkel U, Platzbecker U, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99:4326–35. doi: 10.1182/blood.V99.12.4326. [DOI] [PubMed] [Google Scholar]
- 29.Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl J Med. 2013;368:2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duncavage EJ, Schroeder MC, O’Laughlin M, Wilson R, MacMillan S, Bohannon A, et al. Genome sequencing as an alternative to cytogenetic analysis in myeloid cancers. N. Engl J Med. 2021;384:924–35. doi: 10.1056/NEJMoa2024534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bolouri H, Farrar JE, Triche T, Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. 2017;24:103–12. doi: 10.1038/nm.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borrow J, Dyer SA, Akiki S, Griffiths MJ. Terminal deoxynucleotidyl transferase promotes acute myeloid leukemia by priming FLT3-ITD replication slippage. Blood. 2019;134:2281–90. doi: 10.1182/blood.2019001238. [DOI] [PubMed] [Google Scholar]
- 34.Iacobucci I, Wen J, Meggendorfer M, Choi JK, Shi L, Pounds SB, et al. Genomic subtyping and therapeutic targeting of acute erythroleukemia. Nat Genet. 2019;51:694–704. doi: 10.1038/s41588-019-0375-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140:1200–28. [DOI] [PMC free article] [PubMed]
- 36.Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36:1703–19. doi: 10.1038/s41375-022-01613-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alotaibi AS, Yilmaz M, Kanagal-Shamanna R, Loghavi S, Kadia TM, DiNardo CD, et al. Patterns of resistance differ in patients with acute myeloid leukemia treated with Type I versus Type II FLT3 inhibitors. Blood Cancer Disco. 2021;2:125–34. doi: 10.1158/2643-3230.BCD-20-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33:299–312. doi: 10.1038/s41375-018-0357-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pronier E, Bowman RL, Ahn J, Glass J, Kandoth C, Merlinsky TR, et al. Genetic and epigenetic evolution as a contributor to WT1-mutant leukemogenesis. Blood. 2018;132:1265–78. doi: 10.1182/blood-2018-03-837468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rampal R, Alkalin A, Madzo J, Vasanthakumar A, Pronier E, Patel J, et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014;9:1841–55. doi: 10.1016/j.celrep.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rampal R, Figueroa ME. Wilms tumor 1 mutations in the pathogenesis of acute myeloid leukemia. Haematologica. 2016;101:672–9. doi: 10.3324/haematol.2015.141796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gundry MC, Goodell MA, Brunetti L. It’s all about MEis: Menin-MLL inhibition eradicates NPM1-mutated and MLL-rearranged acute leukemias in mice. Cancer Cell. 2020;37:267–9. doi: 10.1016/j.ccell.2020.02.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.