

Abstract
Recessive variants in the oxidoreductase PYROXD1 are reported to cause a myopathy in 22 affected individuals from 15 families. Here, we describe two female probands from unrelated families presenting with features of a congenital connective tissue disorder including osteopenia, blue sclera, soft skin, joint hypermobility and neuromuscular junction dysfunction in addition to known features of PYROXD1 myopathy including respiratory difficulties, weakness, hypotonia and oromotor dysfunction. Proband AII:1 is compound heterozygous for the recurrent PYROXD1 variant Chr12(GRCh38):g.21452130A>G;NM_024854.5:c.464A>G;p.(N155S) and Chr12(GRCh38):g.21462019_21462022del;NM_024854.5:c.892_895del;p.(V298Mfs*4) and proband BII:1 is compound heterozygous for Chr12(GRCh38):g.21468739-21468741del;NM_024854.5:c.1488_1490del;p.(E496del) and Chr12(GRCh38):g.21467619del;NM_024854.5:c.1254+1del. RNA studies demonstrate c.892_895del;p.(V298Mfs*4) is targeted by nonsense mediated decay and c.1254+1delG elicits in-frame skipping of exon-11. Western blot from cultured fibroblasts shows reduced PYROXD1 protein levels in both probands. Testing urine from BII:1 and six individuals with PYROXD1 myopathy showed elevated levels of deoxypyridinoline, a mature collagen crosslink, correlating with PYROXD1-disorder severity. Urine and serum amino acid testing of the same individuals revealed no reportable changes. In contrast to PYROXD1 knock-out, we find no evidence for disrupted tRNA ligase activity, as measured via XBP1 splicing, in fibroblasts expressing PYROXD1 variants. In summary, we expand the clinical spectrum of PYROXD1-related disorders to include an overlapping connective tissue and myopathy presentation, identify three novel, pathogenic PYROXD1 variants, and provide preliminary evidence that elevated urine DPD crosslinks may provide a clinical biomarker for PYROXD1 disorders. Our results advocate consideration of PYROXD1 variants in the differential diagnosis for undiagnosed individuals presenting with a connective tissue disorder and myopathy.
Introduction
Myopathies are a heterogenous group of muscle disorders with >300 known genetic causes (1), variable clinical presentations and severity, and overlapping histological features. Recessive variants in PYROXD1 are associated with a congenital myopathy (2–4) and an adult-onset limb-girdle muscular dystrophy (5–7). PYROXD1 is a ubiquitously expressed oxidoreductase enzyme with cell essential roles (8–10). Although the precise function of PYROXD1 remains unknown, previous studies support its enzymatic function contributing to the maintenance of cellular redox balance (2,3,6), mitochondrial function (5) and regulation of the tRNA ligase complex (11).
Reported clinical features of PYROXD1 myopathy include generalized weakness and poor muscle bulk, feeding and respiratory difficulties, and in a subset of patients, hypernasal speech, long fingers, joint hypermobility and decreased bone mineral density (2–7). With only 22 individuals affected with PYROXD1 myopathy thus far reported worldwide, the phenotypic spectrum associated with PYROXD1 variants is yet to be fully elucidated. A recurrent c.464A>G;(p.N155S) variant has been identified on at least one allele in 19 of 22 known affected individuals. Individuals homozygous for c.464A>G;(p.N155S) appear to have the mildest presentation (2–7), while moderate to severe clinical presentations manifest in individuals with N155S in trans with a loss-of-function variant or with compound heterozygosity of other missense, deletion and splicing variants (2–4,6). Currently, there are five pathogenic coding (2–7) and three non-coding PYROXD1 variants disrupting mRNA splicing (2,3) described, including a deep intronic variant activating inclusion of a 110 nucleotide pseudoexon between exons 4 and 5 encoding a premature termination codon inherited in trans with c.116G>C;(pQ372H) (3).
Here, we describe two female probands from unrelated families who received differential diagnoses of connective tissue disorders with associated muscle weakness and were not initially screened for PYROXD1 variants, which at the time had only recently been identified (2) and was not yet included on neuromuscular gene panels. Both individuals carry biallelic variants in PYROXD1 and provide important evidence that the phenotypic spectrum of PYROXD1 disorders extends to classical connective tissue features of osteopenia, blue sclera, soft skin and marked joint hypermobility in addition to muscle weakness.
We sought to identify a biomarker of PYROXD1 function. Given the recently published role of PYROXD1 in regulation of the tRNA ligase complex (11), we explored whether individuals with PYROXD1 variants manifest an inborn error of metabolism of any amino acid, which relies on the tRNA ligase complex for intron removal, or if cytoplasmic function of the tRNA ligase complex was altered as measured by XBP1 splicing. Since these yielded no observable differences, we further investigated excreted collagen crosslinks due to the emerging connective tissue presentation associated with PYROXD1 variants.
We, therefore, advocate for PYROXD1 screening in individuals with features overlapping a connective tissue disorder and myopathy for which a genetic cause has not been identified and raise the potential of urine deoxypyridinoline (DPD) levels to be used as a biomarker of PYROXD1 function.
Results
Clinical history
AII:1 is the first child of unrelated parents of European Greek descent born at term following an uncomplicated pregnancy (Fig. 1A). Her birth weight was 3.64 kg (68th centile), length 51 cm (52nd centile) and head circumference 33 cm (15th centile). She required nasogastric feeds from day 3 of life because of a poor suck and swallow; a modified barium swallow showed aspiration of fluids and reduced pharyngeal motility. She sat independently and rolled from tummy to back at age 7 months and was alert and interactive. Review at age 10 months showed generalized hypotonia and proximal muscle weakness, oromotor dysfunction, incoordinate swallow with aspiration, mild gross motor delay and subtle facial features. Craniofacial features include dolichocephaly, blue sclerae, long eyelashes, dystopia canthorum, a broad nasal bridge, a slightly bulbous nasal tip, a short columella, an open-mouthed posture, full lips, a well-defined philtrum, protuberant ears, a high narrow palate, prominent cheeks, micrognathia and mild translucency of the facial skin, with no rash. At the time, a differential diagnosis of the connective tissue disorder Loeys–Dietz syndrome was considered, although there was no evidence of bifid uvula or aortic dissection on echocardiogram.
Figure 1.
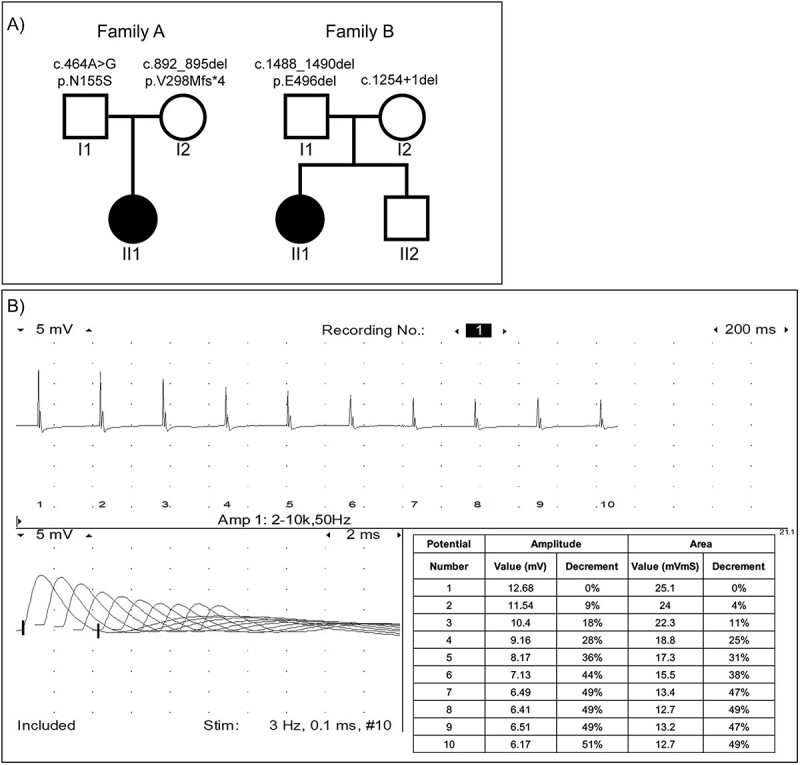
(A) Family pedigrees for AII:1 and BII:1 showing PYROXD1 variants. (B) Repetitive stimulation at 3 Hz in the right abductor digiti minimi muscle showing decrement in AII:1. Single fiber EMG was not performed.
AII:1 crawled at 2 and 3 years of age was able to stand and walk with moderate support. She had continuing significant dysarthria and dysphagia. Her cognitive function was normal and fine motor control was reasonable, with no tremor. During her third year of life, she had three episodes of severe pneumonia requiring hospitalization, each associated with significant weight loss and regression of gross motor skills. She was unable to weight-bear independently thereafter, despite extensive rehabilitation. By age 4 years, she had diffusely osteopenic bones and had sustained a fracture of the right femur. Bone densitometry confirmed low bone mineral density (z scores < −2.5) and X-ray showed mild bilateral hip dysplasia. Episodes of pneumonia requiring hospitalization with associated weight and strength loss have continued.
When examined at age 4 years her skin was soft and velvety but not hyperelastic. She had a Beighton score (12) of 8 for joint hypermobility, a positive thumb sign and prominent heels. Upper limb strength was 3/5 in all muscle groups, hip flexors were 2+/5 bilaterally, knee extension was 3/5 and foot dorsiflexion was 1/5. Reflexes were present in upper limbs and depressed in lower limbs and marked dysarthria continued. She had 5-degree knee contractures which progressed to 25–30% by age 8.
Following an episode of pneumonia at age 4, polysomnography showed mild hypoventilation with a baseline CO2 of 50–51 mmHg (normal < 45 mmHg) which was treated with nocturnal non-invasive ventilation (BiPAP) until age 7 but then restricted to periods of illness since the use of BiPAP worsened dysarthia and open bite.
At age 4 months, nerve conduction studies and electromyography (EMG) of deltoid and vastus lateralis were normal. Nerve conduction studies showed normal sensory studies with low-amplitude compound muscular action potentials in the upper and lower extremities. Slow (3 Hz) repetitive stimulation of the median nerve, recorded over abductor digiti minimi, showed a 36% decrement (normal range < 10%) (Fig. 1B), consistent with neuromuscular junction dysfunction (NMJ) and myopathic changes. Pyridostigmine and salbutamol were subsequently sequentially trialled without clinical efficacy.
At 8 years of age, AII:1 was dependent upon use of a wheelchair, required maximum assistance for daily living, was unable to smile with progressive dysarthria, had dysphagia with risk of aspiration and was in the 4th percentile for weight (19 kg) requiring a gastrostomy tube to maintain weight.
BII:1 is the first child of unrelated parents of European descent (Fig. 1A) born at full term with a birth weight of 2.92 kg and a length of 52 cm. She was first reported to be floppy with hypotonia at age 1–2 months but sat at 6 months of age before becoming progressively weaker.
BII:1 had a myopathic face, with protruding ears, micrognathia and blue sclera. She had joint laxity, and soft skin but with no rash and no abnormal scarring. Nerve conduction studies at 2 years of age showed small CMAP amplitudes but were otherwise normal. Repetitive stimulation (2 Hz) of the right ulnar nerve showed no clear CMAP amplitude decrement although testing was technically complicated by patient movement. At this time, a differential diagnosis of Ehlers–Danlos syndrome musculocontractural type 2 was considered.
Due to prominent bulbar symptoms, pyridostigmine (1 mg/kg three times daily) was empirically prescribed with some reported improvements in facial muscle tone, pencil grip, sleeping, ability to support self and use of arms.
At the age of 3 years and 4 months, BII:1 was 14.4 kg (45th centile) and 104 cm (96th centile). At age 5 years, she was non-ambulatory and relied on the use of a wheelchair, with continued generalized severe hypotonia and normal cognition. She had respiratory insufficiency with a history of hospital admissions for pneumonia and was treated with nocturnal non-invasive ventilation (BiPAP).
Diagnostic investigations and findings
AII:1 had extensive clinical testing including gene panels (neuromuscular and brittle bone), myotonic dystrophy DNA testing, uniparental disomy studies, eye examination, echocardiogram, TORCH serology, creatine kinase (CK) and extensive biochemical and hormonal screens; all yielded normal results. Initial trio whole exome sequencing (WES) in 2017 was uninformative since variants in PYROXD1 had only recently been identified as a novel congenital myopathy-associated disease gene and at the time was not considered a phenotypic match. Repeat trio whole genome sequencing performed in 2019 identified compound heterozygous variants in PYROXD1 in AII:1 (Fig. 1A); the previously reported missense Chr12(GRCh38):g.21452130A>G;NM_024854.5(PYROXD1):c.464A> G;p.(N155S) variant (2–7) in exon 5 (paternal allele) and a 4 bp deletion Chr12(GRCh38):g.21462019_ 21462022del;NM_024854.5 (PYROXD1):c.892_895del;p.(V298Mfs*4) in exon 9 (maternal allele). Both variants were confirmed by Sanger sequencing of gDNA from cultured fibroblasts from AII:1 (Fig. 2Ai) and segregation studies confirmed each parent carries one heterozygous variant (see Fig. 1A). The maternal c.892_895del;(p.V298Mfs*4) was found in 14/281622 alleles (allele frequency 0.00004971) in the Genome Aggregation Database (gnomAD v2.1.1) (13) with no reported homozygotes, and to our knowledge, has not been reported previously as a disease-causing variant.
Figure 2.

(A) Sanger sequencing chromatograms from (i) AII:1 and (ii) BII:1 confirming PYROXD1 variants on gDNA. (B) RNA studies. (i) Schematic of exons 3–12 of PYROXD1 mRNA with location of variants denoted by magenta (AII:1) or blue (BII:1) asterisks, and primers by arrows. (ii) and (iv) Agarose gel electrophoresis for all primer pairs described, including no template control (NTC) for each. Sanger sequencing chromatograms for (iii) AII:1 showing the detection of two alleles following CHX inhibition of NMD and for (v) BII:1 showing normal splicing of the p.E496del bearing allele (upper band) and in-frame exon 11 skipping from the c.1254+1delG allele (lower band) that is not subject to NMD.
BII:1 had multiple genetic investigations including chromosomal microarray, a spinal muscular atrophy panel, neuromuscular disease panel screen, and heritable disorders of connective tissue panel which identified several variants of uncertain significance, including a maternal chromosomal deletion in 16p13.3, and heterozygous changes in BIN1, FKRP, TTN, COL3A1 and TNXB with no second variants found, and insufficient evidence to attribute pathogenicity. Exome sequencing reported in 2020 identified likely pathogenic compound heterozygous variants in PYROXD1, Chr12(GRCh38):g.21468739-21468741del; NM_024854.5:c.1488_1490del;p.(E496del) (paternal allele, not reported in gnomAD v2.1.1) and Chr12(GRCh38):g21467619del; NM_024854.5:c.1254+1del (maternal allele, found in 1/247 898 alleles in gnomAD v2.1.1 with no homozygotes). Both variants were confirmed by Sanger sequencing of gDNA from cultured fibroblasts from BII:1 (Fig. 2Aii) and segregation studies confirmed each parent carries one heterozygous variant (Fig. 1A).
RT-PCR and western blot studies
RT-PCR was used to interrogate the effect of the variants at the RNA level. RNA was extracted from cultured fibroblasts from AII:1, BII:1 and age-matched healthy controls with and without treatment with cycloheximide (CHX) to inhibit nonsense mediated decay (NMD). Figure 2Bi shows the position of primers used for PCR of synthesized cDNA.
For AII:1 primers produced amplicons spanning exons 3–6 (to detect NM_024854.5:c.464A>G) and exons 7–10 (to detect NM_024854.5:c.892_895del). An additional primer pair was designed to specifically amplify cDNA derived from the maternal c.892_895del allele in AII:1 by positioning the forward primer at the site of the deletion. The deletion-specific primer pair only amplified a product in AII:1 fibroblasts treated with CHX and did not amplify a product in control or DMSO control-treated AII:1 cells (Fig. 2Bii), suggesting that mRNA derived from the maternal c.892_895del;(p.V298Mfs*4) allele is effectively degraded by NMD. Active NMD is also supported by Sanger sequencing of the exon 7–10 amplicon, which showed CHX-mediated rescue of transcripts from the c.892_895del allele (Fig. 2Biii, see overlapping trace files +CHX and only a single trace file −CHX). Rescue of NMD-targeted isoforms of SRSF1 (14) was used as a positive control confirming the successful inhibition of NMD with CHX treatment (Fig. 2Bii, bottom row).
For BII:1, primers produced amplicons spanning exons 7–12 (to detect the effect of NM_024854.5 c.1254+1delG) and exons 11–12 (to detect NM_024854.5:c.1488_1490del) (Fig. 2Biv). Using primers amplifying exons 7–12 revealed two PCR products, one corresponding to a normally spliced PYROXD1 transcript (bearing the c.1488_1490del) and the other a transcript with in-frame exon 11 skipping (c.1254+1del, Fig. 2Bv), which is detected both with and without CHX treatment, indicating that this allele is not subject to NMD. Using a forward primer within exon 11 and reverse in the 3′UTR detected only the c.1488_1490del allele, consistent with c.1254+1del resulting in loss of PYROXD1 transcripts with canonical splicing of exons 10–12.
Figure 3A shows the position of all currently reported PYROXD1 variants within a schematic of the gene-protein structure, with the majority of variants affecting the predicted oxidoreductase domain (gray bar). The variants identified herein are coloured pink (Family A) and blue (Family B). Western blot from cultured fibroblasts (muscle biopsy was not available for either proband) showed PYROXD1 protein levels in AII:1 were ~60%, and in BII:1 were ~35% compared with two healthy controls (C1 and C2), with three other individuals with PYROXD1 biallelic variants (P3–P5) also demonstrating reduced protein expression (Fig. 3B). Variation in levels of PYROXD1 protein between the five individuals tested likely reflects the collective consequence of different variants on mRNA levels and stability of the encoded PYROXD1 protein. In contrast to a previous study (3), we did not observe differences in glutathione reductase (GR) levels in fibroblasts from five PYROXD1 patients relative to two controls (Fig. 3C), suggesting that GR is not up-regulated in response to PYROXD1 dysfunction.
Figure 3.
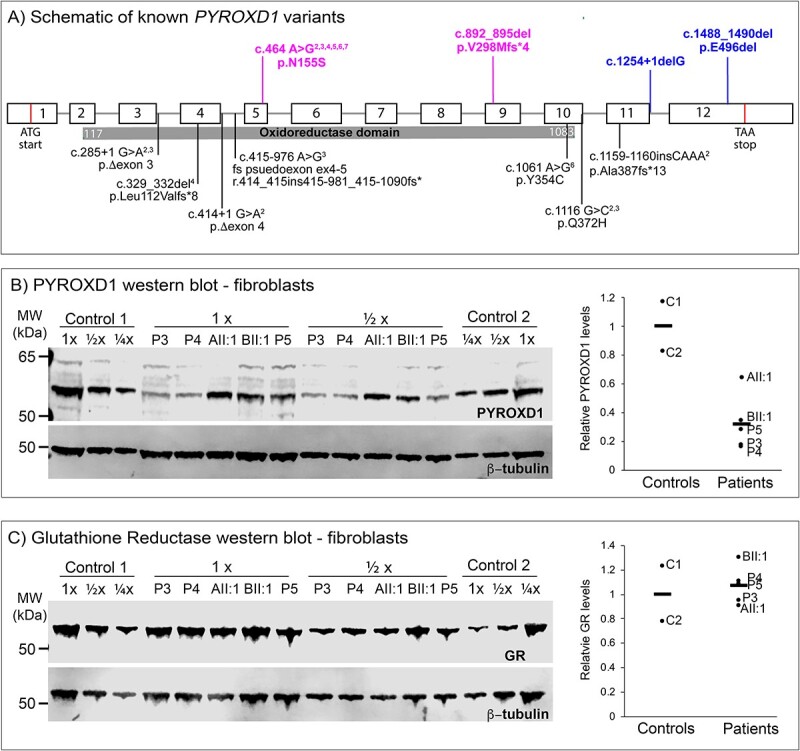
(A) Schematic of genomic location of known PYROXD1 variants (black text below schematic) with the region encoding the functional oxidoreductase domain shown by the grey bar. Exons represented by boxes, introns by lines. AII:1 variants in magenta, BII:1 variants in blue above the line. (B) Western blot of PYROXD1 levels in cultured fibroblasts and corresponding quantification. (C) Western blot of glutathione reductase (GR) levels in cultured fibroblasts and corresponding quantification. (B and C) P3 and P4 are individuals with PYROXD1 myopathy and P5 is an individual with biallelic variants in PYROXD1 including one VUS currently under investigation by our group.
Clinical testing for biomarker of PYROXD1 function
We explored avenues to identify a reliable, non-invasive biomarker of human PYROXD1 function that could be useful for two purposes: (1) to enable monitoring of disease progression in a similar way to the use of creatine kinase in muscular dystrophies and (2) to provide additional diagnostic evidence supporting likely pathogenicity of PYROXD1 genetic variants that would otherwise be classified as variants of uncertain significance, precluding a molecular diagnosis. As a previous study has demonstrated defects in the tRNA ligase complex associated with acute knock-down of PYROXD1 (11), we tested for an inborn error of metabolism by measuring urine and serum amino acid levels and measured the cytoplasmic function of the tRNA ligase complex via XBP1 splicing (15) in cultured fibroblasts (Fig. 4A). Amino-acid testing of urine and serum from seven PYROXD1 patients (including BII:1) revealed no reportable abnormalities. We found no evidence of abnormal tRNA ligase activity in fibroblasts from eight PYROXD1 patients, including AII:1 and BII:1 (shown for five patients in Fig. 4A) which showed normal induction of the unfolded protein response measured via XBP-1 splicing.
Figure 4.
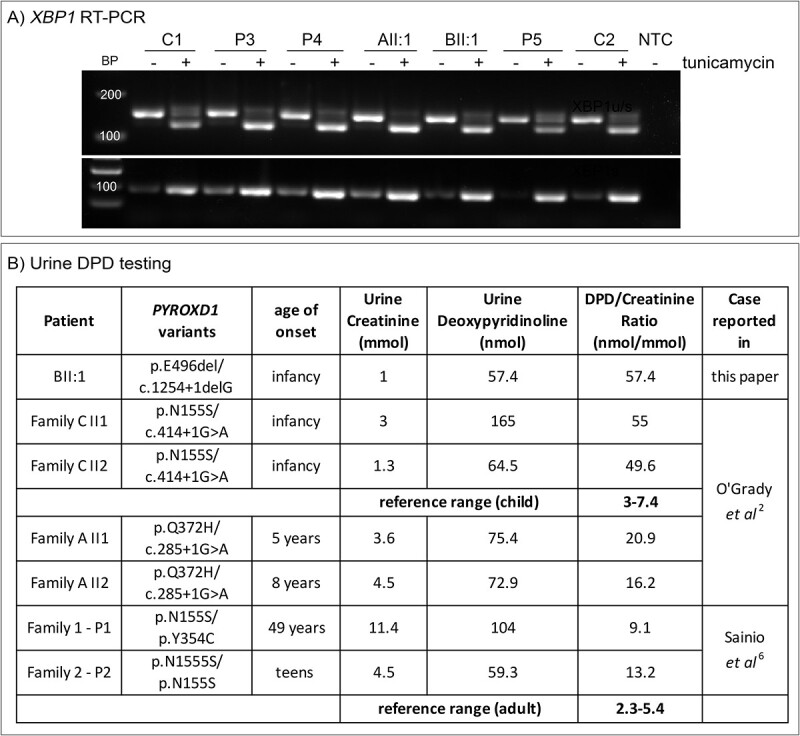
(A) XBP1 RT-PCR from cultured fibroblasts with or without tunicamycin treatment with primers common to the spliced and unspliced transcript (upper) or specific for the spliced transcript only (lower). P3 and P4 are individuals with PYROXD1 myopathy and P5 is an individual with biallelic variants in PYROXD1 including one VUS currently under investigation by our group. (B) Urine DPD and reference creatinine testing for BII:1 and six individuals with PYROXD1 myopathy. Normal relevant reference range for children or adults in bold.
Primary connective tissue disorders can be diagnosed clinically by testing levels of urinary collagen cross-links (16,17) that are excreted during bone and collagen turnover. Given the connective tissue phenotype of AII:1 and BII:1, we tested urine DPD levels (compared to creatinine to normalize for urine concentration) in BII:1 and six other individuals affected with PYROXD1-disorders described previously (2,6) for whom we had frozen urine archived (Fig. 4B). All urine specimens from individuals with PYROXD1-disorders had significantly elevated DPD/Creatinine ratios compared with the age-relevant reference range, with the degree of elevation correlating with disease severity, such that patients with an earlier onset of disease had more significantly elevated DPD/Creatinine ratios.
Discussion
We describe two female probands with the congenital presentation of recessive PYROXD1 disease associated with hypotonia, weakness and notable connective tissue features. Subtle connective tissue features have been described in other individuals with PYROXD1 myopathy (2,3); however, early and pronounced connective tissues features resulted in a differential diagnosis of Loeys–Dietz syndrome for AII:1 and Ehlers–Danlos syndrome musculocontractural type 2 for BII:1, deflecting scrutiny away from genes associated with a primary myopathy. There is substantial phenotypic overlap between some myopathies and inherited connective tissue disorders; for example, collagen-6 related disorders, with features common to both including muscle weakness, reduced muscle bulk, hypotonia, exercise intolerance, easy fatiguability, joint hypermobility and/or contractures, scoliosis and obstructive sleep apnoea.
In AII:1, the decremental response on low-frequency repetitive nerve stimulation was indicative of a myopathy and NMJ dysfunction as is also seen in other congenital onset myasthenic and limb-girdle myasthenic syndromes (e.g. GMPPB, GFPT1). Normal sensory responses and the absence of neuropathic changes on EMG were not in favour of a neuropathic process. Reduced CMAP amplitudes on nerve conduction studies are described in two other individuals with congenital-onset PYROXD1-disorders (2), though were not identified in an adult with a later-onset PYROXD1-disorder who showed normal nerve conduction studies and an abnormal EMG, consistent with a myopathic process (6). It remains to be established how common abnormalities on nerve conduction or repetitive nerve stimulation studies are in individuals with PYROXD1-disorders, and whether this is influenced by age-of-onset and/or genotype. While not sufficient for diagnosis, we propose repetitive nerve stimulation studies could be a useful adjunct investigation for individuals with suspected PYROXD1-related connective tissue and/or myopathy presentations.
With so few PYROXD1 myopathy cases described, it is difficult to determine the prevalence or breadth of phenotypic features, which currently include muscle, connective tissue, nerve and cardiac involvement. Consistent with multi-system involvement, transcriptomic analyses show PYROXD1 is expressed ubiquitously, with PYROXD1 knock-out incompatible with cellular or animal life (8–10). Accordingly, there are no individuals identified with two loss-of-function PYROXD1 variants, or who show deficiency of PYROXD1 protein, which is presumed lethal.
Currently, two cell essential pathways are linked to PYROXD1 activity: the mitochondrial respiratory chain (2,5) and regulation of the tRNA ligase complex (11). Abnormal mitochondrial distribution is a prominent feature of muscle biopsies from individuals with early-onset PYROXD1-disorders (2,3) and mitochondrial function is perturbed in cells following PYROXD1 knockdown (5). Muscle biopsies are not available for AII:1 or BII:1, through detailed analyses of their fibroblast lines forms part of a larger proteomic and metabolomic study in progress by our group. Asanović et al. recently identify PYROXD1 as a key regulator of tRNA ligase complex activity (11), but we did not observe a defect in the cytoplasmic function of this complex as measured by UPR-activated XBP-1 splicing in fibroblasts from individuals with PYROXD1 disorders, including AII:1 and BII:1. Importantly, knock-down or knock-out of PYROXD1 expression is a different biological context to expression of variant forms of PYROXD1 enzyme with partial function in patient fibroblasts. Nevertheless, at this point, we are unable to link a defect in tRNA ligase complex function to PYROXD1 dysfunction in fibroblasts from individuals with PYROXD1 disorders. Further, in contrast to Lornage et al. (3), we do not find evidence for a measurable alteration in glutathione reductase levels associated with reduced PYROXD1 protein levels in human fibroblasts expressing PYROXD1 variants.
Age of onset, disease progression and ambulation status vary greatly among individuals affected with PYROXD1 myopathy. While there is insufficient published data for accurate prognostications based on genotype, individuals with one loss-of-function variant (frameshift, nonsense, splicing) appear to be more severely affected than those homozygous for the recurrent p.(N155S) variant with milder symptomology. Common features are weakness of the upper and lower limbs and respiratory and feeding difficulties; while many patients have facial weakness, including ptosis, this has not been a consistent finding. Herein, we show that blue sclera, facial features, soft skin and osteopenia, symptoms often associated with connective tissue disorders linked to collagen defects, can also be associated with biallelic PYROXD1 variants. Though preliminary, we provide data indicating that urine DPD levels are elevated in individuals with PYROXD1 disorders though to lower levels than seen in primary connective tissue disorders such as Ehlers–Danlos syndrome (16). We acknowledge that additional testing of prospective patient samples is required to validate DPD testing for PYROXD1 disease, though propose this could represent an adjunct clinical test to assess PYROXD1 function or an orthogonal test to assist in interpretation of likely pathogenicity of novel PYROXD1 variants. Importantly, our findings suggest a potential role for PYROXD1 in the regulation of a collagen biosynthetic enzyme and we are exploring this hypothesis in our cell and animal models of PYROXD1 disease.
To the best of our knowledge, this is the first report of PYROXD1 myopathy associated with early and significant connective tissue involvement, in addition to myasthenic features and myopathy. While we specifically describe two individuals here, connective tissue features of joint laxity and blue sclera have been anecdotally reported to us by other individuals with PYROXD1 myopathy. Considering only 22 patients are currently known to be diagnosed with PYROXD1 variants, the additional two individuals described here significantly expand the recognized phenotypic spectrum associated with this rare autosomal recessive condition. Increased awareness of PYROXD1 as a disease-causing gene associated with myopathic, neurological and/or connective tissue features is important to identify additional PYROXD1 cases within genetically undiagnosed cohorts presenting with complex phenotypes. A more comprehensive picture of the full clinical spectrum of PYROXD1 myopathy will enable timely and targeted genetic diagnoses in the future. Significant increases in patient diagnoses from wider targeted genetic screening may warrant the future inclusion of PYROXD1 on connective tissue disorder gene panels.
Materials and Methods
This study was approved by the Children’s Hospital at Westmead Human Research Ethics Committee (Biospecimen Bank_10/CHW/ 45 and 2019/ETH11736) with informed, written consent. For AII:1 WES and PCR-free, WGS was performed and analyzed at the Broad Institute of Harvard and MIT as previously described (18). For BII:1, clinical XomeDxPlus WES was performed.
RNA was isolated from cultured fibroblasts and RT-PCR performed as previously described (19).
PYROXD1 RT-PCR primers used; PYROXD1-Ex7F 5′-AGTGCATTG GGACCAGATTG-3′ with PYROXD1-Ex10R 5′-TTTCAGGCCACCATCT TCTC-3′, PYROXD1-WTSpecificF 5′-TGATACAGAGATGTGGCCTGT CT −3′ with Ex10R, PYROXD1-DeletionSpecificF 5′-TGATACAGAGA TGTGGCCTATGT-3′ with Ex10R, PYROXD1-Ex3F 5′-TGGCGTAAAGC AACTGAAGA-3′ with PYROXD1-Ex6R 5′-GCTCCTGCATCGAAGAAA GT-3′ with cycling conditions of 94°C for 180 s, then 30 cycles of 94°C 20 s, 58°C 20s, 72°C 30 s, then 72°C 7 min. Sf2-Ex3F 5′- CACTGGTGTCGTGGAGTTTGTACGG-3′ with Sf2-Ex4R 5′- GGGCAGGAATCCACTCCTATG-3′ with cycling conditions of 95°C 5 min, then 35 cycles of 95°C 30 s, 60°C 30 s, 72°C 2 min 30 s, then 72°C 5 min.
Human fibroblasts from healthy controls and PYROXD1 patients were maintained in DMEM (Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum (GE HyClone), 5% Amniomax II complete medium (Life Technologies) and 50 μg/ml gentamicin (Life Technologies). Cell pellets were harvested, protein concentration determined, and western blotting performed as previously described (19). Antibodies used were against PYROXD1 (kind gift of Professor Javier Martinez, MFPL and MedUni Wien), GAPDH (MAB374, Merck Millipore) and Glutathione Reductase (ab16801, Abcam). For inhibition of NMD, cells were treated with 100 μg/ml CHX for 5 h before harvesting for RNA extraction and RT-PCR.
XBP1 RT-PCR was performed as in (20, 21) using XBP1u/sF 5′- CCTGGTTGCTGAAGAGGAGG-3′ with XBP1u/sR 5′- ATCCATGGGGA GATGTTCTGG-3′, XBP1sF 5′- TGCTGAGTCCGCAGCAG GTG-3′ with XBP1sR 5′- GCTGGCAGGCTCTGGGGAAG-3′ with cycling 94°C 3 min, 30 cycles of 94°C 20 s, 58°C 20 s, 72°C 30 s then 72°C 7 min.
Urine organic acid profiling was performed by liquid phase extraction and analyzed by gas chromatography/mass spectrometry on a Shimadzu QP2010 (Shimadzu, Kyoto, Japan). Briefly, organic acids were extracted from acidified, salt-saturated urine into ethylacetate. The organic extract was evaporated to dryness under air and the dried extract was derivatized with BSTFA (N,O-Bis (trimethylsilyl)trifluoroacetamide) to form trimethylsylyl (TMS) esters of carboxyl groups and ethers of hydroxyl groups. The derivatized metabolites were separated on a HP5MS capillary column using temperature programming. The column eluent was fed directly into a mass spectrometer operating in full-scan mode. The resulting spectra was analyzed by GCMS PostRun software (Shimadzu).
Urine amino acids were analyzed by liquid chromatography-mass spectrometry on a XEVO TQS analyzer (Waters, UK). Urine was diluted to a creatinine concentration of 1 mmol/l, dried and butylated. Amino acids were separated with an Acquity UPLC BEH C18 column using a water/acetonitrile solvent gradient and were detected in a positive ion mode by looking at specific mass to charge transitions. Quantitation was performed using TargetLynx software (Waters).
Serum amino acids were analyzed on a Waters Acquity Amino Acid Analyzer (Waters). Samples were deproteinized using s-sulphosalicylic acid precipitation and the amino acids in the resulting supernatant were derivatised with 6-aminoaquinolyl-N-hydroxysuccinimidyl carbamate. The derivatized amino acids were separated with a reverse phase BEH C18 column and water/acetonitrile solvent gradient. The amino acids were detected by UV absorption at 260 nm and the resulting chromatography was analyzed with Empower 3 chromatography data (Waters).
Acknowledgements
We thank the families involved in this study for their invaluable contributions to this research, and the clinicians and healthcare workers involved in their assessment and management. We thank Professor Javier Martinez and team for the kind gift of the PYROXD1 antibody, Professor R. Bryan Sutton and Isaac Scott for the kind gift of recombinant PYROXD1 protein, and Professor Carsten Bönnemann and Professor Henna Tyynismaa for sharing patient fibroblast cell lines and coordinating urine sample collection. DPD clinical testing was performed as a paid service through NSW Health Pathology, Sydney Australia. Sequencing and analysis were provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG)
Conflict of Interest statement. S.T.C. is director of Frontier Genomics Pty Ltd (Australia). S.T.C. receives no remuneration (salary or consultancy fees) for this role. Frontier Genomics Pty Ltd has no current financial interests that will benefit from publication of this data. S.T.C. is a named inventor on intellectual property owned jointly by the University of Sydney and Sydney Children’s Hospitals Network. This IP relates to splicing variant detection and interpretation and is licensed by Frontier Genomics Pty Ltd. The remaining co-authors declare no competing interests.
Contributor Information
Frances J Evesson, Kids Neuroscience Centre, Kids Research Institute, The Children’s Hospital at Westmead, Westmead, NSW, Australia; Functional Neuromics, Children's Medical Research Institute, The University of Sydney, Westmead, NSW, Australia.
Gregory Dziaduch, Kids Neuroscience Centre, Kids Research Institute, The Children’s Hospital at Westmead, Westmead, NSW, Australia; Functional Neuromics, Children's Medical Research Institute, The University of Sydney, Westmead, NSW, Australia.
Samantha J Bryen, Kids Neuroscience Centre, Kids Research Institute, The Children’s Hospital at Westmead, Westmead, NSW, Australia; Discipline of Child and Adolescent Health, Faculty of Medicine and Health, The University of Sydney, Westmead, NSW, Australia.
Francesca Moore, NSW Biochemical Genetics Service, The Children’s Hospital at Westmead, Westmead, NSW, Australia.
Sara Pittman, Department of Neurology, Washington University School of Medicine, Saint Louis, MO, USA.
Beena Devanapalli, NSW Biochemical Genetics Service, The Children’s Hospital at Westmead, Westmead, NSW, Australia.
Leigh B Waddell, Kids Neuroscience Centre, Kids Research Institute, The Children’s Hospital at Westmead, Westmead, NSW, Australia; Discipline of Child and Adolescent Health, Faculty of Medicine and Health, The University of Sydney, Westmead, NSW, Australia.
Monique M Ryan, Royal Children’s Hospital, Murdoch Children’s Research Institute and University of Melbourne, Melbourne, Australia.
Manoj P Menezes, Discipline of Child and Adolescent Health, Faculty of Medicine and Health, The University of Sydney, Westmead, NSW, Australia; Department of Neurology, Children's Hospital at Westmead, Westmead, NSW, Australia.
Conrad C Weihl, Department of Neurology, Washington University School of Medicine, Saint Louis, MO, USA.
Adviye Ayper Tolun, Discipline of Child and Adolescent Health, Faculty of Medicine and Health, The University of Sydney, Westmead, NSW, Australia; NSW Biochemical Genetics Service, The Children’s Hospital at Westmead, Westmead, NSW, Australia.
Craig Zaidman, Department of Neurology, Divisions of Child Neurology and Neuromuscle, Washington University in St. Louis School of Medicine, St. Louis, MO, USA.
Helen Young, Department of Clinical Genetics, The Children’s Hospital at Westmead, Sydney, Australia.
Lesley C Adès, Discipline of Child and Adolescent Health, Faculty of Medicine and Health, The University of Sydney, Westmead, NSW, Australia; Department of Clinical Genetics, The Children’s Hospital at Westmead, Sydney, Australia.
Sandra T Cooper, Kids Neuroscience Centre, Kids Research Institute, The Children’s Hospital at Westmead, Westmead, NSW, Australia; Functional Neuromics, Children's Medical Research Institute, The University of Sydney, Westmead, NSW, Australia; Discipline of Child and Adolescent Health, Faculty of Medicine and Health, The University of Sydney, Westmead, NSW, Australia.
Funding
National Health and Medical Research Council of Australia (APP1048816, APP1136197 and APP1080587 to S.T.C.); Muscular Dystrophy Association of the USA (Development Grant to F.J.E.); Take Part Foundation; National Human Genome Research Institute; National Eye Institute; National Heart, Lung and Blood Institute grant (UM1 HG008900); National Human Genome Research Institute grant (R01 HG009141).
Ethical Approval
This study was approved by the Children’s Hospital at Westmead Human Research Ethics Committee (Biospecimen Bank_10/CHW/ 45 and 2019/ETH11736) with informed, written consent.
References
- 1. Benarroch, L., Bonne, G., Rivier, F. and Hamroun, D. (2023) The 2023 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul. Disord., 33, 76–117. [DOI] [PubMed] [Google Scholar]
- 2. O’Grady, G.L., Best, H.A., Sztal, T.E., Schartner, V., Sanjuan-Vazquez, M., Donkervoort, S., Abath Neto, O., Sutton, R.B., Ilkovski, B., Romero, N.B. et al. (2016) Variants in the oxidoreductase PYROXD1 cause early-onset myopathy with internalized nuclei and Myofibrillar disorganization. Am. J. Hum. Genet., 99, 1086–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lornage, X., Schartner, V., Balbueno, I., Biancalana, V., Willis, T., Echaniz-Laguna, A., Scheidecker, S., Quinlivan, R., Fardeau, M., Malfatti, E. et al. (2019) Clinical, histological, and genetic characterization of PYROXD1-related myopathy. Acta Neuropathol. Commun., 7, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Daimagüler, H.-S., Akpulat, U., Özdemir, Ö., Yis, U., Güngör, S., Talim, B., Diniz, G., Baydan, F., Thiele, H., Altmüller, J. et al. (2021) Clinical and genetic characterization of PYROXD1-related myopathy patients from Turkey. Am. J. Med. Genet. Part A, 185, 1678–1690. [DOI] [PubMed] [Google Scholar]
- 5. Saha, M., Reddy, H.M., Salih, M.A., Estrella, E., Jones, M.D., Mitsuhashi, S., Cho, K.A., Suzuki-Hatano, S., Rizzo, S.A., Hamad, M.H. et al. (2018) Impact of PYROXD1 deficiency on cellular respiration and correlations with genetic analyses of limb-girdle muscular dystrophy in Saudi Arabia and Sudan. Physiol. Genomics, 50, 929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sainio, M.T., Välipakka, S., Rinaldi, B., Lapatto, H., Paetau, A., Ojanen, S., Brilhante, V., Jokela, M., Huovinen, S., Auranen, M. et al. (2019) Recessive PYROXD1 mutations cause adult-onset limb-girdle-type muscular dystrophy. J. Neurol., 266, 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Woods, J.D., Khanlou, N., Lee, H., Signer, R., Shieh, P., Chen, J., Herzog, M., Palmer, C., Martinez-Agosto, J. and Nelson, S.F. (2020) Myopathy associated with homozygous PYROXD1 pathogenic variants detected by genome sequencing. Neuropathology, 40, 302–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blomen, V.A., Majek, P., Jae, L.T., Bigenzahn, J.W., Nieuwenhuis, J., Staring, J., Sacco, R., van Diemen, F.R., Olk, N., Stukalov, A. et al. (2015) Gene essentiality and synthetic lethality in haploid human cells. Science, 350(80), 1092–1096. [DOI] [PubMed] [Google Scholar]
- 9. Hart, T., Chandrashekhar, M., Aregger, M., Durocher, D., Steinhart, Z., Brown, K.R., Macleod, G., Mis, M., Zimmermann, M., Fradet-Turcotte, A. et al. (2015) High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell, 163, 1515–1526. [DOI] [PubMed] [Google Scholar]
- 10. Wang, T., Birsoy, K., Hughes, N.W., Krupczak, K.M., Post, Y., Wei, J.J., Lander, E.S. and Sabatini, D.M. (2015) Identification and characterization of essential genes in the human genome. Science, 350, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Asanović, I., Strandback, E., Kroupova, A., Pasajlic, D., Meinhart, A., Tsung-Pin, P., Djokovic, N., Anrather, D., Schuetz, T., Suskiewicz, M.J. et al. (2021) The oxidoreductase PYROXD1 uses NAD(P)+ as an antioxidant to sustain tRNA ligase activity in pre-tRNA splicing and unfolded protein response. Mol. Cell, 81, 2520–2532.e16. [DOI] [PubMed] [Google Scholar]
- 12. Beighton, P., Solomon, L. and Soskolnet, C.L. (1973) Articular mobility in an African population. Ann. Rheum. Dis., 32, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alföldi, J., Wang, Q., Collins, R.L., Laricchia, K.M., Ganna, A., Birnbaum, D.P. et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun, S., Zhang, Z., Sinha, R., Karni, R. and Krainer, A.R. (2010) SF2/ASF autoregulation involves multiple layers of post-transcriptional and translational control. Nat. Struct. Mol. Biol., 17, 306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jurkin, J., Henkel, T., Nielsen, A.F., Minnich, M., Popow, J., Kaufmann, T., Heindl, K., Hoffmann, T., Busslinger, M. and Martinez, J. (2014) The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J., 33, 2922–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pasquali, M., Dembure, P.P., Still, M.J. and Elsas, L.J. (1994) Urinary pyridinium cross-links: a noninvasive diagnostic test for Ehlers-Danlos syndrome type VI. N. Engl. J. Med., 331, 132–133. [DOI] [PubMed] [Google Scholar]
- 17. Borisova, N.V., Pokrovskay, A.Y., Zakharova, E.Y. and Krasnopolskaya, K.D. (1994) Analysis of collagen hydroxypyridinium crosslinks in samples of tissues and urine of patients with inherited connective tissue disorders. Connect. Tissue Res., 30, 177–190. [DOI] [PubMed] [Google Scholar]
- 18. Cummings, B.B., Marshall, J.L., Tukiainen, T., Lek, M., Donkervoort, S., Foley, A.R., Bolduc, V., Waddell, L.B., Sandaradura, S.A., O’Grady, G.L. et al. (2017) Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci. Transl. Med., 9(386), eaal5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bryen, S.J., Ewans, L.J., Pinner, J., MacLennan, S.C., Donkervoort, S., Castro, D., Töpf, A., O’Grady, G., Cummings, B., Chao, K.R. et al. (2020) Recurrent TTN metatranscript-only c.39974–11T>G splice variant associated with autosomal recessive arthrogryposis multiplex congenita and myopathy. Hum. Mutat., 41, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oslowski, C.M. and Urano, F. (2011) Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol., 490, 71–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van Schadewijk, A., Van’T Wout, E.F.A., Stolk, J. and Hiemstra, P.S. (2012) A quantitative method for detection of spliced X-box binding protein-1 (XBP1) mRNA as a measure of endoplasmic reticulum (ER) stress. Cell Stress Chaperones, 17, 275–279. [DOI] [PMC free article] [PubMed] [Google Scholar]