


Abstract
Transdominant genetics using expression libraries can identify proteins and peptides that affect cell division. In conjunction with these libraries, oligonucleotide-conjugated beads and flow cytometry were used to test a strategy that potentially expands the range of such genetic studies. The experimental approach involved creation of tagged expression libraries, introduction of these libraries into cells, growth of the cultured cells for several generations and recovery on oligonucleotide-conjugated beads of sequences that encode growth-modulatory proteins or peptides. Experiments in Saccharomyces cerevisiae demonstrating the feasibility of the strategy are presented.
INTRODUCTION
Genetic mutants provide a strong link to physiology, because defined phenotypic characters are selected in cells or organisms. In certain tractable organisms such as Escherichia coli, Saccharomyces cerevisiae, Caenorhabditis elegans and Drosophila melanogaster, genetic analysis has yielded huge returns. In many other organisms, however, genetic studies have been hampered by a variety of impediments, including the inability to recover recessive mutations, the difficulty of mapping mutant loci and genome instability.
We and others have developed transdominant genetic technologies that address several of these problematic issues (1–6). Our formulation of the technology involves the use of expressed peptides or protein fragments (termed perturbagens) to disrupt specific biochemical functions within cells (1). In combination with appropriate assays or selections, perturbagen-based genetics is a powerful method for identification of components of physiological pathways in cells, and potentially for the discovery of novel drug targets. The perturbagen-based strategy has been successfully applied in model systems. For example, selections in yeast have identified a variety of peptides and protein fragments that interfere with the pheromone response. These proteinaceous agents in turn were used to define components of the pheromone response pathway.
Despite the unqualified success of these efforts, it is important to note that the experimental circumstances were extremely favorable. The selection was ‘tight’, with a background survivor rate of only 1/50 000; this permitted single colonies to be picked and retested. Perturbagen-induced colonies grew independently, in the absence of competition from other colonies; thus a large number of different perturbagen sequences were recovered with penetrances ranging from a few to 100%. This type of parallel isolation would not be possible in a mixture of competing cells, because the very strong, highly penetrant perturbagens would dominate; the weaker ones would be lost.
Here we describe a method, fluorescence-activated bead sorting (FABS), that may expand the scope of transdominant genetic selections. FABS combines oligonucleotide-conjugated particles with specially prepared, tagged expression libraries, whose clones can be tracked and isolated with the help of a flow sorter. We show that the method identifies perturbagens that are similar to those obtained in experiments where single colonies are analyzed. The technique may prove useful in experiments with other organisms, including cultured mammalian cells, where issues such as high backgrounds, colony growth and negative selections (i.e. selections for perturbagens that cause cell death or growth arrest) pose problems.
MATERIALS AND METHODS
ID tag design
The structure of the ID tags was based on a compromise between ease of synthesis, selectivity and diversity. In-house written software (Monomer), which creates all possible sequences of length N, identifies those that have either a specific base composition or melting temperature (based on mean stacking energies of dinucleotides; 7), and selects a subset which differs by at least m mismatches, was used to check Nmer sets for compatibility with ID tag constraints. We chose 8mers as the ID tag unit for several reasons. For example, if one begins with a single sequence and fixes the composition at each position, there are 219 ways to distribute 3–8 mismatches over an 8mer to satisfy the constraints (W = N!/n!m!, where N is the size of the segment, n is the number of matches and m is the number of mismatches). In contrast, there are only 42 ways to distribute at least three mismatches in a 6mer. Thus, 8mer space is less constrained than 6mer space. To obtain sufficient diversity, a combination of 8mers is preferable. Three 8mer units permit diversities over 1 000 000.
Specifically, the ID tags were composed of three 8mer units drawn from a set of 256 8mers (Fig. 2). The 8mer set was further refined by demanding that the 8mers selected for the 5′-end of the ID tag would not have mismatches exclusively located in the three 5′-most positions. This additional criterion yielded 111 possible 8mers for the 5′-end. Similarly, the 3′ 8mers were required not to have the three mismatches located exclusively at the 3′-end of the 8mer. This constraint left 107 8mers. After applying these criteria, 105 8mers remained as choices for units on either the 3′- or 5′-end of the ID tag, leaving 151 8mers for the middle unit of the ID tag. These additional sequence requirements were instituted to maximize destabilization of ID tags by demanding that the mismatches of inappropriately hybridized ID tags be located internally, instead of at the ends of the ID tag.
Figure 2.
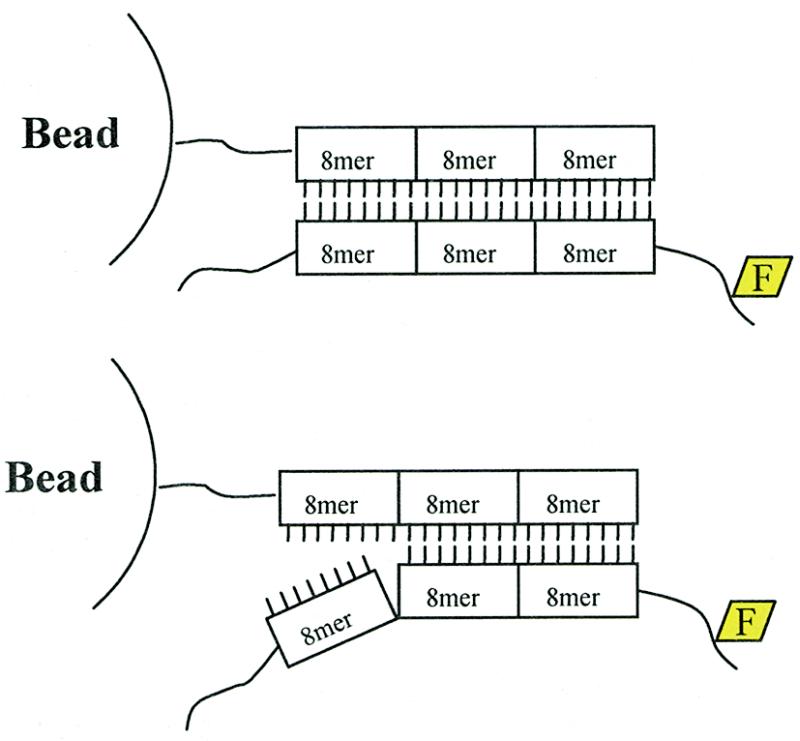
ID tag structure and hybridization specificity. ID tags were composed of three 8mer units. A minimum mismatch of three internal bases was a requirement for each 8mer. Examples of a perfectly matched and mismatched (with one non-cognate 8mer unit) ID tag after hybridization are shown.
The bead used for synthesis and flow cytometry was an amino-derivatized polystyrene particle 30 µm in diameter. Standard pool-and-split combinatorial synthesis created sets of 24mer ID tags, each built from three 8mer units. Two independent methods were used to quantify the number of functional oligonucleotides per bead. Both methods yielded a value of roughly 108–109/bead (see Estimation of oligonucleotides/bead).
Combinatorial chemistry and oligonucleotide synthesis
The Perceptive Biosytems Expedite oligonucleotide synthesizer with a Multiple Oligo Synthesis System (MOSS unit) was used to make all oligonucleotides and synthesize ID-tag libraries.
Beads. The particles (beads) were amino-derivatized Primer Support 30HL (Pharmacia). Before ID tags were synthesized on this support, the resin was conjugated to an 18 atom spacer phosphoramidite (Glen Research, Sterling, VA) and free amino groups were capped en masse using the following protocol. An aliquot of 900 mg of resin was added to 240 ml of Deblock solution 8 (Perceptive Biosystems), stirred for 1 min and removed by filtration through a 300 ml Kontes filter funnel with a hydrophilic nylon membrane (Millipore). The resin was washed thoroughly with acetonitrile. With the vacuum off, the resin was mixed with 15 ml activator solution for 1 min and removed by filtration. Six milliliters of 1:60 dilution of the 18 atom spacer phosphoramidite was added, mixed for 1 min, removed by filtration and washed with 9 ml activator and 500 ml acetonitrile. With the vacuum off, 144 ml of premixed CapA and CapB (1:1 v/v) solution was added, reacted for 15 min and removed by vacuum filtration. The particles were washed with 50 ml acetonitrile. The capping and wash steps were repeated once and 60 ml oxidizer solution was added, followed by a wash with 24 ml premixed CapA and CapB. Oxidation of the bead linkage produced an acid/base-stable phosphoramidate bond to the bead. A final wash with 100 ml acetonitrile prepared the beads for ID tag synthesis.
Combinatorial chemistry on beads. Twist columns (Glen Research) were loaded with 30 mg of resin derivatized with the 18 atom spacer. Standard phosphoramidite chemistry was employed to synthesize the ID tag on the resin using the 200 nmol synthesis scale of the Expedite oligonucleotide synthesizer. The first round of synthesis added an additional 18 atom spacer followed by a predetermined 8 nt sequence on each of 24 columns. The trityl group was left on. The columns were removed and the resin from the columns was pooled, mixed and reapportioned into 22 columns in each of which a second 8mer (a predetermined set of 8 nt sequence) was synthesized. The trityl group was again left on. The procedure was repeated, but this time 27 columns were utilized to attach the final set of 8mers. The trityl group was removed and the resin was pooled from all columns and resuspended in 20% ammonium hydroxide at room temperature (16 h) to remove the protecting groups. The resin was washed three times in PBS and 0.1% Triton. This created a bead population consisting of 14 256 different ID-tagged beads.
Estimation of oligonucleotides/bead. A homogeneous population of 100 000 oligonucleotide-conjugated beads, containing on their surfaces a single sequence type, was hybridized to saturation to a complementary oligonucleotide, labeled during synthesis with fluorescein. The beads were washed as above, and analyzed on a Coulter XL flow cytometer. The beads were compared to Molecule Equivalents of Soluble Fluorochrome (MESF) particles (Flow Cytometry Standards Corp., San Juan, PR) analyzed with identical flow cytometer settings to determine the number of fluorochromes present on the ID-tagged bead. The QuickCal program (part of the Quantum Kit) was used to determine the MESFs on the ID-tagged beads (7 ± 2 × 108), assuming one fluorochrome per hybridized oligonucleotide.
A second method, elution of bound oligonucleotides and quantitation by fluorimetry, was also used to estimate oligonucleotide density on the bead surface. Hybridized particles similar to those described above were resuspended in 1 ml of ddH2O and incubated at 100°C for 10 min. The supernatant was removed and the fluorescently labeled oligonucleotides were precipitated overnight at –20°C with ethanol and sodium acetate. The pellet was resuspended in PBS and the amount of fluorochrome present in the sample was determined using a Turner Model 450 Fluorimeter with excitation filter SC450 and emission filter NB540. A standard curve was generated using the appropriate fluorochrome to determine the concentration and moles of fluorochrome present in the sample eluted from the hybridized particles. Note that after the fluorescently labeled oligonucleotides were eluted from the ID-tagged particles, flow cytometry was used to verify that >99% of the hybridized material was eluted from the surface by analyzing the ID-tagged beads. This method of quantification determined the number of oligonucleotides able to hybridize to the ID-tagged particles as 1.1 ± 1.0 × 109/bead.
ID tags for library construction. Free ID tags were synthesized on a standard synthesis resin using the same pool-and-split synthesis technique as described above. The following minor changes were incorporated: (i) no spacer was used, instead, a leader sequence (CTCGAGG) was added to allow for cloning; (ii) ID tags complementary to those synthesized on the particles were created with a sequence (GTCAGGAACT) on the end of all ID tags to facilitate cloning. The following two oligonucleotides were synthesized for cloning the ID tag library by the splint method: 5′-GACGTCAGTCCTTGAC and 5′-GAGCT-CCTTAAG (8).
Yeast strains and media
The S.cerevisiae strain used in the selection for α-factor-resistant clones was yVT12 [MATa leu2-3,112 his3 lys2 sst2d ade2-1 HMLa HMRa mfa1::hisG mfa2::hisG ste3::Gal1 (uas)–STE3]. Yeast strains were transformed by the method of Gietz and Scheiestl (9), and plasmids were isolated as described (10).
Plasmids and libraries
In brief, pVT252 (a vector containing the perturbagen expression library superimposed on the ID-tagged library) was constructed from the parent vector pVT21 (1). pVT21 was modified in the following ways: (i) specific restriction sites were inserted at the 3′-end of GFP for cloning of DNA fragment libraries; (ii) the two oligonucleotides ovt545 (5′-GGGGCATGCTAATACGACT-CACTATAGGGCTGCAGGGGAATTCTGCATGCAAGC-TAGCTCGTACGTAGTCGACGGG) and ovt546 (5′-CCCC-GTACGATTATGCTGAGTGATATCCCGACGTCCCCTT-AAGACGTACGTTCGATCGAGCATGCATCAGCAGCCC) were hybridized together and cloned into the vector to generate a T7 polymerase promoter and restriction sites for PstI and EcoRI, located upstream of the ID tag; (iii) the ID tags were cloned into the EcoRI and PstI sites to generate 10× coverage of the 14 256 ID-tagged library; (iv) the transformants were pooled and the library plasmid isolated and prepared by Ecl136 digestion to accept library inserts; (v) sheared yeast genomic fragments were introduced into an Ecl136 site at the C-terminus of GFP using blunt end ligation. The genomic fragment library generated 2.5 × 106 clones. 500 000 clones (one ID tag/35 perturbagens) were pooled and used as described below.
Growth selection assay
Strain yVT12 was transformed with the pVT252 ID-tagged library. Yeast harboring the library were cultured briefly in selective medium supplemented with galactose and raffinose and transferred to ten 15 cm yeast extract/peptone/galactose/raffinose plates containing 10 nM α-factor (Sigma) at an initial density of 50 000 cells/plate.
Five plates were used for the following assay. Colonies forming 2–4 days after plating were patched to plates lacking uracil, then replica-plated to selective plates containing either dextrose or galactose and raffinose. After 24 h, they were replica-plated onto yeast extract/peptone/dextrose or extract/peptone/galactose/raffinose plates containing 1 µM α-factor. Individual plasmids were isolated from cells that displayed galactose-specific growth in the presence of α-factor. These plasmids were amplified in E.coli to provide sufficient plasmid to transform yVT12 to test for linkage between the plasmid and escape from α-factor arrest.
The transformants on the other set of five plates were pooled on day 3 and the plasmid sub-library isolated from them. The library was introduced into E.coli to generate enough plasmid to retransform yVT12 and repeat the assay with the yeast transformants generated with the passaged library. The clones that grew in the presence of α-factor were pooled and the plasmids isolated and expanded in E.coli. The plasmids were expanded in E.coli to generate sufficient template DNA for the generation of RNA by the following methodology. The twice-passaged library was linearized downstream of the ID tag cassette at the SalI site. The RiboMax kit (Promega) was used to generate milligram quantities of a 100 base RNA that contained the complementary ID tag sequence found on the beads. Label IT kits (Panvera) were used to label 100 µg quantities of the RNA with fluorescein.
Perturbagen penetrance assays
‘Penetrance’ of individual perturbagens was determined by plating ~500 cells on selective medium containing galactose (which induced perturbagen expression) in the presence or absence of 0.5 nM α-factor. The cells were allowed to grow on the plates for 4 days at 30°C. After 4 days, the number of colonies on α-factor-containing plates were compared to cells grown on control plates. The number of colonies present on the α-factor-containing plate divided by the total number of colonies on the control plate × 100 = percent penetrance.
Optimized conditions for RNA hybridization to ID-tagged beads
All solutions were made with DEPC-treated ddH2O just before use. 200 000 ID-tagged beads were resuspended in the hybridization solution (2× SSPE, 10% formamide, 0.5% SDS. 0.1% proteinase K) that contained 10 µg of labeled RNA generated from the twice-passaged pVT252 ID-tagged perturbagen library. The sample was incubated at 55°C for 1000 min with mixing. The samples were then washed three times with wash buffer (1× SSPE, 0.5% SDS, 0.1% proteinase K) at 55°C and resuspended in PBS, 0.5% SDS, 0.1% proteinase K.
Flow cytometry and particle sorting
A Coulter Elite ESP cell sorter equipped with a 15 mW 488 nm air-cooled argon laser was used to analyze and sort ID-tagged beads based on the fluorescence emission at 525 ± 15 nm. Single beads were sorted into PCR microtiter wells.
RT–PCR of ID tags and perturbagen recovery
Reverse transcription was used to generate single-stranded cDNA from single, sorted beads which was subjected to PCR. In brief, 1 µl of oVT600 (50 µm) was added to 10 µl of ddH2O and mixed with individual beads. The sample was incubated at 90°C for 10 min and chilled on ice before addition of 7 µl of a cocktail consisting of 4 µl of 5× buffer, 2 µl of 0.1 M DTT and 1 µl of 10 mM dNTPs. The sample was brought to 42°C and 1 µl of MMLV RT (Life Sciences) was added and incubated for 1 h. RNases A and H were added and the sample was incubated for 1 h at 37°C. One microliter of this RT reaction was added to the following PCR reaction on ice: 2.5 µl of 10× PCR buffer, 2.5 µl of 25 mM MgCl2, 1 µl of ovt586 (50 µM), 1 µl of ovt600 (50 µM), 0.5 µl of 10 mM dNTPs, 15.5 µl of dH2O. After amplification the reaction was treated with an exonuclease I/shrimp alkaline phosphatase mix (exo/sap) for 30 min at 37°C. The exo/sap mix was inactivated by heating (80°C for 15 min). The product was sequenced using oVT597 and Big Dye terminator sequencing on an automated DNA sequencer (Perkin Elmer).
Primer sequences: RT primer oVT600, 5′-TACGTACGA-GCTAGCTTGCATG; PCR primer oVT586, 5′-GAACTGC-AGGGAGGAACTG; sequencing primer oVT597, 5′-GTACGAGCTAGCTTGCATGCAG.
To identify the perturbagen associated with each ID tag from the beads, a PCR primer representing the ID tag was synthesized and used in conjunction with a common vector primer 3′ of the perturbagen insert position. The two primers were mixed with the twice-passaged library as template, and the resultant PCR product was sequenced to determine the perturbagen identity.
RESULTS
Overall scheme
We sought to combine perturbagen-based genetic methods with FABS, yielding a modified technology for growth assays (Fig. 1). The basic scheme for these experiments is as follows. A perturbagen library is constructed in an appropriate expression vector such that each clone has a unique identifier sequence, or ID tag, joined to it. The ID tags endow each library clone with a label that serves to bind it to a specific bead that contains the complementary sequence covalently linked to its surface (Fig. 2). The perturbagen–ID tag library is introduced into cells and, following a period of cell growth and selection, the library is re-isolated. The library ID tag sequences, after passage through the cells, are labeled with, for example, the fluorescein fluorochrome.
Figure 1.

Process flow for post-passage library analysis using the FABS technology. F, fluorochrome; GFP, green fluorescent protein; PT, perturbagen.
To recover growth-stimulatory sequences, the labeled library sequences are mixed and hybridized to beads that contain the entire set of complementary ID tags. The set of beads is then examined on a flow sorter and bright beads that have an excess of fluorescein are collected. These beads should correspond to perturbagen sequences that are amplified selectively in one cell type. The identity of the perturbagen-encoding nucleic acid can be determined from the sequences bound to the bead. This process of post-passage library analysis permits sequences that have positive effects on growth to be identified among the vast background of sequences that do not affect growth.
ID tag design and synthesis
ID tags are a critical component of the process because they label individual library sequences with markers that permit unambiguous identification after genetic selection. To design ID tags, we adopted a strategy similar to one suggested originally by Brenner et al. to create by combinatorial synthesis a set of minimally cross-hybridizing nucleic acid sequences (11). A set of ID tags that have similar melting temperatures was developed using a program based on the mean stacking temperature of the different base pairs (7; see Materials and Methods). The ID tags consisted of 24mers (three minimally cross-hybridizing 8mers in tandem). Combinatorial chemistry allowed us to synthesize 14 256 different ID tags from a pool of 256 different 8mers.
Library construction and synthesis of probe
A perturbagen library consisting of 500 000 clones [500 000 independent, randomly sheared, (0.4–2.5 kb, median 0.8 kb) yeast genomic DNA fragments fused downstream of the GFP coding sequence, superimposed on an ID tag library of 14 256 ID tags (1 ID tag/35 perturbagen sequences)] was constructed in a vector containing an ID tag library cassette (Fig. 3). The cassette included a T7 RNA polymerase binding site, an ID tag, RT–PCR primer sites and a restriction endonuclease site permitting creation of run-off transcripts 100 bases in length. The primer sites were used in conjunction with RT–PCR procedures to amplify material from the sorted particles that had RNA hybridized specifically to their surfaces.
Figure 3.

Expression vector for the ID-tagged perturbagen library. The GFP coding sequence was flanked by the upstream activation sequence of the GAL1 gene and a genomic insert fused to its 3′-end to form a galactose-inducible GFP–perturbagen fusion protein. The plasmid was a shuttle vector for E.coli and S.cerevisiae with the β-lactamase gene and the URA3 gene for selectable markers.
The T7 strategy was chosen after a series of optimization experiments revealed that a single-stranded 100 base RNA molecule containing the ID tag provided the highest quality, quantity and sensitivity for hybridization to and detection of oligonucleotide-conjugated particles. Probe lengths significantly greater than 100 bases did not hybridize to the particles efficiently, and probe lengths significantly shorter were inadequately labeled (data not shown).
Hybridization specificity and sensitivity
We tested sensitivity, kinetics and discrimination using a 14 256 tag complex ID tag library under a variety of conditions, arriving at an optimal protocol. For example, we found that we could separate 20% of the ID tag set, labeled with fluorochrome, from the remaining 80% using optimized conditions (Fig. 4). Quantitative analysis of the hybridization revealed that roughly 20% of the total bead population, as expected, was present in a diffuse bright peak. In the negative control in which a single ID tag sequence (not present in the 14 256 tag collection) was labeled and mixed with the bead population, the percentage of beads in the bright peak remained at the background level.
Figure 4.

Hybridization of 2851 fluorescently labeled ID tags to a 14 256 complex ID-tagged bead population. A sub-population of fluorescently labeled ID tags, whose sequences were complementary to a subset present on the beads, was generated by oligonucleotide synthesis (see Materials and Methods). The sub-population represented 20% of the 14 256 ID tags. The fluorescently labeled ID tags were hybridized to the ID-tagged beads and then analyzed. The sort gate was located one log above the mean fluorescence of the background population of unlabeled beads.
We also determined the sensitivity of the FABS system by limiting the concentration of labeled oligonucleotides in the hybridization mix and holding the hybridization time constant. Femtomolar amounts of individual ID tags within a 14,256 tag complex mixture could be detected (Fig. 5). By using different particles with lower autofluorescence on which to synthesize the complementary ID tags, we believe detection of attomolar concentrations of labeled ID tags is possible (data not shown).
Figure 5.

Hybridization specificity and sensitivity of ID tags on beads. Histograms showing populations of fluorescent beads after hybridization with various masses of labeled cRNA representing a single ID tag whose cognate beads were present at 1% of the total bead population. (A) 1.0 µg; (B) 0.1 µg; (C) 0.01 µg.
Isolation of perturbagen sequences
Post-passage library analysis using FABS was tested in the same model system as in our previous experiments with perturbagens, the yeast pheromone response pathway (1). The positive selection for pheromone resistance in yeast required that cells grow in the presence of pheromone (α-factor), which normally causes G1 arrest. We chose this system as a testing ground for FABS for three reasons: (i) the yeast selection has been applied successfully in numerous laboratories, including our own (1,5,6); (ii) the vectors to build the libraries are readily available; (iii) perturbagens previously isolated from a similar screen could function as positive controls.
Perturbagen libraries consisting of genomic DNA–GFP fusions were introduced into yeast a cells and exposed to α-factor on culture plates. Two perturbagen sequences isolated previously, pep3(c) and orf8, were doped in as positive controls at ratios of 1:10 000 (1).
Approximately 500 000 yeast transformants, representing less than single coverage of the 500 000 clone perturbagen library, were plated onto 10 agar plates (50 000 cells/15 cm plate). On half the plates, yeast survivors were allowed to form colonies. Of 193 colonies picked and retested, 82 contained clones for bona fide galactose-dependent perturbagens (Table 2A). DNA sequence analysis grouped these into 14 individual sequences, including the two positive controls. One of the 12 new clones encoded a peptide fused to GFP, the product of the α1 gene joined to the GFP coding sequence in the antisense orientation, producing a C-terminal fusion peptide 30 amino acids long. The remaining 11 perturbagens were fused in-frame to GFP. Two of these sequences, orf14 and orf18, were known previously to participate in yeast mating. One of these, orf18, produced a fragment of STE12p, a transcription factor involved in expression of pheromone-induced genes (12). orf18 presumably encoded a molecule with dominant negative activity that interfered with wild-type STE12p function. The other, orf14, was derived from the α2 locus, a gene that regulates expression of mating type-specific genes. Presumably orf14 promoted escape from pheromone by altering the phenotype of a cells such that they were unresponsive to pheromone (13). Five of the 12 novel perturbagens lacked ID tags. This result was expected from the characteristics of the expression library which contained ~40% sequences with absent or corrupted ID tags (data not shown). Penetrance of the individual clones, defined as the ability to induce escape from pheromone on a per cell basis, ranged from 11 to 100%.
Table 2. Perturbagens isolated from experiments.
| Perturbagen | Locus | Length (amino acids) | Chromosome and position | No. of times isolated | ID tag | Penetrance (%) |
|---|---|---|---|---|---|---|
| A. Using the colony formation assay on plates | ||||||
| pep3 | NA | 59 | XII: 345,284 | 57 | 14-7-4 | 90 ± 5 |
| orf8 | YBR059C | 174 | II: 357,343 | 4 | 17-11-1 | 65 ± 9 |
| orf12 | MGE | 100 | XV: 774,844 | 2 | 6-21-23 | 48 ± 3 |
| orf13 | LTV1 | 110 | XI: 177,275 | 1 | 1-22-6 | 70 ± 13 |
| orf14 | α2a | Complete gene | III: 12,685 | 1 | 10-7-24 | 69 ± 21 |
| orf15 | Ynl144c | 118 | XIV: 355045 | 1 | 6-2-6 | 19 ± 3 |
| orf16 | ZDS2 | 150 | XIII: 52,968 | 1 | 4-8-48 | 12 ± 9 |
| orf17 | YNRO42W | 37 | XIV: 701,475 | 2 | 15-4-23 | 11 ± 7 |
| orf18 | STE12 | 247 | VIII: 275,465 | 5 | 4-8-? | 69 ± 6 |
| orf19 | IQG-1 | 105 | XVI: 91,053 | 2 | None | 47 ± 4 |
| orf20 | Nam2 | 100 | XII: 884,306 | 3 | None | 11 ± 2 |
| orf21 | MDJ1 | 180 | VI: 105,122 | 1 | None | 27 ± 3 |
| orf22 | SKI2 | 104 | XII: 918,471 | 1 | None | 36 ± 4 |
| pep7 | α1 | 30 | III: 13,224 | 1 | None | 62 ± 5 |
| B. Using FABS [penetrance was measured as in Caponigro et al. (4)] | ||||||
| pep3 | NA | 59 | XII, 345,284 | 7 | 14-7-4 | 90 ± 5 |
| orf8 | YBR059C | 174 | II, 357,343 | 3 | 17-11-1 | 65 ± 9 |
| orf12 | MGE | 100 | XV, 774,844 | 7 | 6-21-23 | 48 ± 3 |
| orf23 | AGPI | 104 | III, 76,700 | 2 | 12-20-21 | 88 ± 9 |
| orf24 | α2a | Complete gene | III, 11,618 | 5 | 18-19-24 | 94 ± 3 |
| orf25 | α2a | Complete gene | III, 11,565 | 5 | 13-14-18 | 105 ± 5 |
NA, not applicable.
aClones derived from the same gene (α2), but different break points and, therefore, independent clones. The α2 clone identified in the plate assay is a fusion with GFP and is galactose-dependent. The α2 clones identified with FABS are not galactose-dependent. Clones in bold are positive controls for assay.
Cells from the other halves of the Petri dishes on which yeast were plated were pooled. Plasmids containing the ID-tagged perturbagen sequences were isolated from yeast and expanded in E.coli to provide sufficient plasmid to retransform yeast. The yeast were transformed with the sub-library and the selection was repeated. Plasmids were isolated from the yeast survivors en masse and reintroduced into E.coli to generate sufficient quantities of material for in vitro transcription. The cycling step reduced the number of clones isolated that were present in the library due to genomic mutations that rendered the clone insensitive to pheromone (see Discussion and Materials and Methods).
RNA representing the ID tags was post-transcriptionally labeled with fluorescein and hybridized to 200 000 beads encompassing the set of 14 256 ID tags. The 200 000 beads were screened using a FACS machine and single fluorescently labeled beads were sorted onto a microtiter plate. A total of 79 beads were recovered for analysis (Fig. 6). The expected background of beads (i.e. the number present in the sorting gate using non-hybridized control beads) was 40. Thus, roughly 39 (79 – 40) true positives were expected.
Figure 6.

Recovery of labeled beads by FACS. Histogram showing actual sort histograms of (A) control library beads that were never exposed to complementary RNA and (B) the beads after hybridization to fluorescently labeled cRNA with the sort gate criteria used to identify positive beads.
Nested RT–PCR was performed on the sorted beads, resulting in amplified DNA fragments from 29 beads, reasonably close to the expected number. These products were cloned to determine the ID tag sequences. The remaining 50 beads did not generate product from PCR either because of anomalous fluorescence (i.e. they fell in the sort gate even though they had no hybridized nucleic acid), missorted particles (i.e. no particle in the well), failed reverse transcription or failed PCR. The sequence analysis of independent cloned products confirmed that hybridization to individual beads was indeed specific (Table 1). PCR amplification overwhelmingly produced a single type of ID tag from each bead. Based on the sequence of the ID tags, primers specific to individual ID tags were used in PCR reactions along with a common vector primer, and the passaged perturbagen/ID tag library served as template for amplification of linked perturbagen sequences. The resulting perturbagen-encoding fragments were cloned and subjected to DNA sequence analysis for identification.
Table 1. RT–PCR amplification, cloning and sequencing of ID tags.
| Bead no. | No. of transformants sequenced | ID tag sequence type | No. of times identified | Other ID tag sequences | Non-ID tag sequencea |
|---|---|---|---|---|---|
| 1 | 10 | 18-19-24 | 8 | 0 | 2 |
| 2 | 10 | 14-7-4 | 7 | 0 | 3 |
| 3 | 10 | 12-20-21 | 9 | 0 | 1 |
| 4 | 10 | 13-14-18 | 7 | 0 | 3 |
| 5 | 10 | 17-11-1 | 5 | 0 | 5 |
| 6 | 10 | 6-21-23 | 7 | 0 | 3 |
Six individual beads that generated amplicons from the RT–PCR were cloned and 10 transformants of each RT–PCR reaction were sequenced.
aSome clones gave rise to one of two DNA sequences that were not related to ID tags; one was from the vector (i.e. no insert), while the other was unknown (except for the PCR primers), suggesting that it was an artifact of the original PCR reaction.
A total of six perturbagens, including the two positive controls, were represented among the 29 sequences amplified from the sorted beads (Table 2B). All were retested individually, and conferred resistance to pheromone with penetrances ranging from 48 to 100%. No false positives were recovered. Two of the perturbagen sequences were derived from the α2 locus and a third, a fragment of MGE1, was identical to one recovered from the colony experiment. There were no peptides among the non-control perturbagens isolated using FABS.
DISCUSSION
Several groups have reported isolation of perturbagens (protein fragments and peptides) that interfere with the pheromone response in yeast (1,5,6). In our laboratory, a total of 16 different perturbagens were recovered in a previous experiment: 11 orfs and five peptides (including two synthetic peptides). These perturbagens were characterized for strength (genetic penetrance), and the stronger ones were mapped by epistasis tests in the well-studied pheromone pathway. Based mainly on two-hybrid analysis, binding partners were assigned to roughly half of the perturbagens. Two other groups have reported isolation of peptides that promote escape from pheromone, and the basis for the activities of several peptides has been defined (5,6).
With the exception of the positive controls that were doped in, none of the perturbagens recovered in the present study was identical to any seen before. There are at least two explanations for the lack of overlap. First, the library employed here was different from those used before. For example, the STE12 fragment was not present in the library utilized in our previous experiments (sheared genomic versus Sau3A partially digested DNA) (1). Second, limited sampling of genomic sequences in these experiments and the earlier ones assured minimum overlap of isolated perturbagens. The breakpoint within a genomic fragment may be as important as the ORF from which a perturbagen is derived with regard to the phenotype associated with the fragment, i.e. the interaction with a specific cellular protein could depend on the precise breakpoint between GFP and the genomic fragment. In the case of peptide perturbagens, this is obviously true. But even with protein fragments, precise breakpoints may be of considerable importance. The library we used represents an approximately 30× coverage of the yeast genome [500 000 clones × 800 bp/12 × 106 (yeast genome size in bp)]. However, the number of possible breakpoints at the GFP–perturbagen junction is 2.4 × 107, vastly greater than the library size. Thus, the lack of overlap may reflect the large number of perturbagens potentially encoded by the yeast genome, coupled with the sparse sampling of these breakpoints in each experiment.
However, the approximate frequency with which perturbagens were recovered from the libraries in the two colony experiments was similar (~1/50 000 clones). The frequency from the FABS experiment was lower, but was reasonably close if corrected for the percentage of library clones with non-functional ID tags. It is also likely that more perturbagens would have been identified had the sort gate been set more liberally, i.e. had dimmer beads been collected. These dimmer beads might correspond to perturbagens of weaker penetrance. With the stringent setting of the sort gate, the false positive rate for the FABS experiment was 0. In contrast, of 193 colonies picked and retested, 82 contained clones for bona fide galactose-dependent perturbagens, yielding a total of 14 perturbagens, eight with intact ID tags. Thus, 111 colonies were most likely derived either from chromosomal mutations that conferred pheromone insensitivity, allowing the colony to grow independent of the plasmid-based library clone it carried, or from non-galactose-dependent perturbagens (such as α2).
FABS technology could be used for genetic experiments in microbes other than S.cerevisiae for which good genetic tools do not exist. In addition, the technology could be applied to selection in mammalian cells. In the case of mammalian cells, however, the scale of the experiments would necessarily be much greater. It is likely that libraries of at least 1 000 000 clones would be needed. This in turn requires ID tags and oligonucleotide-conjugated bead populations of higher diversity. Alternatively, a pooling strategy could be employed.
In the current set of experiments, FABS was used to screen a single-passaged library, but the real power of the post-passage library approach will be realized in applications that involve comparisons between independently passaged libraries. Libraries passaged independently through different cell types, or through cells exposed to different conditions, could identify those perturbagens over- or under-represented in one sub-library compared to the other. This in turn would permit perturbagens with selective effects (i.e. cell type- or cell state-specific effects) to be discerned. For example, a single tagged library might be introduced separately into two different cell populations, such as yeast cells grown on minimal medium and yeast grown on rich medium. After several generations, the libraries could be re-isolated and compared using FABS. One passaged library would be labeled with one fluorochrome and the second passaged library with another. After mixing the samples and hybridization to beads, particles displaying skewed ratios of the two dyes could be recovered for further analysis. These experiments would identify perturbagens that have differential effects in yeast grown on rich or minimal medium; for example, those inhibiting components of biosynthetic pathways. Such two-dye versions of FABS have worked well in reconstitution experiments (M.J.Feldhaus, unpublished results).
The present experiments involved positive selection (for sequences that promote cell division); a more challenging application of FABS involves negative selection (for sequences that promote cell death or arrest). Traditionally, mutations that are lethal have been isolated by replica-plating under non-permissive conditions (e.g. high temperature), sib selection or similar strategies. However, as FABS can reveal over-represented library sequences, it could also in principle find under-represented ones, i.e. perturbagen-encoding sequences that are eliminated due to cell type- or cell state-specific growth suppressive activity. A requirement of negative selection with FABS is that the tagged library has on average 1 ID tag per ≤1 perturbagen. For example, the library used here in the positive selection (1 ID tag/35 perturbagens) could not be used because loss of one of the 35 perturbagens (on average) that have the same ID tag could not be detected (i.e. the fluorescence signal would drop by only 1/35). We can distinguish, however, ID-tagged beads that have lost half or two-thirds of the control fluorescence, corresponding to loss of an active perturbagen sequence that shares a specific ID tag with one or two other neutral perturbagen sequences, respectively (M.J.Feldhaus, unpublished results). Thus, the sensitivity of our system places limits on the perturbagen sequence/ID tag ratio.
The population of oligonucleotide-conjugated beads resembles a dispersed microarray, as if a microarray were carved up into individual pieces, each containing unique sequence features (14). The advantage of the beads compared to microarrays is that bound sequences can be readily recovered by flow sorting. Oligonucleotide-conjugated beads have been synthesized previously, either for exploratory purposes, for labeling beads on which other chemicals are attached or for diagnostic applications (15–17). However, such beads have not been used in the context of genetic experiments as described here. For these passaged library experiments, it is not feasible to use native sequences (e.g. the sequence of the clone itself), because most sequences may be represented multiple times in different contexts, nearly all of which may be genetically inert. For instance, only one clone with specific breakpoints may act as a perturbagen, while other closely related clones may not. In addition, it may be desirable to use random oligonucleotides that encode peptides for perturbagen libraries; these must also be labeled for recovery. Here, conventional microarrays are not an option. However, microarrays designed to contain ID tags could be used, and we are testing this possibility. Such ID tag arrays would have the advantage of eliminating the RT–PCR step in the process.
Other methods besides FABS could potentially be used for post-passage library comparisons. Various forms of PCR-based subtraction, for instance, have been implemented to identify sequences that are over- or under-represented in nucleic acid samples (18,19). These methods have been successful in identification of a variety of tissue-specific mRNAs. However, the techniques are not quantitative and suffer from the vagaries of PCR (e.g. differences in amplification efficiencies between longer and shorter fragments).
In summary, transdominant genetics has a variety of advantages, especially in organisms ill-suited to traditional genetic manipulation. We have explored the use of FABS with the hope that the range of experimental possibilities for transdominant approaches may be broadened. A test case involving positive selection in S.cerevisiae was successful, yielding multiple perturbagens. The most useful applications of FABS probably lie in the areas of: (i) two-dye experiments to compare libraries passaged through two cell populations; (ii) negative selection; and (iii) mammalian cells.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to A. Gaglio for assistance with flow cytometry, G. Caponigro and M. Abedi for comments on the manuscript, I. Peterson for DNA sequencing, and C. Wang for programming.
REFERENCES
- 1.Caponigro G., Abedi,M.R., Hurlburt,A.P., Maxfield,A., Judd,W. and Kamb,A. (1998) Proc. Natl Acad. Sci. USA, 95, 7508–7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holzmayer T.A., Pestov,D.G. and Roninson,I.B. (1992) Nucleic Acids Res., 20, 711–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gudkov A.V., Kazarov,A.R., Thimmapaya,R., Axenovich,S.A., Mazo,I.A. and Roninison,I.B. (1994) Proc. Natl Acad. Sci. USA, 91, 3744–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whiteway M., Dignard,D. and Thomas,D.Y. (1992) Proc. Natl Acad. Sci. USA, 89, 9410–9414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geyer C.R., Colman-Lerner,A. and Brent,R. (1999) Proc. Natl Acad. Sci. USA, 96, 8567–8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Norman T.C., Smith,D.L., Sorger,P.K., Drees,B.L., O’Rourke,S.M., Hughes,T.M., Roberts,C.J., Friend,S.H., Fields,S. and Murray,A.W. (1999) Science, 285, 591–595. [DOI] [PubMed] [Google Scholar]
- 7.Ornstein R.L., Rein,R., Breen,D.L. and MacElroy,R.D. (1978) Biopolymers, 17, 2341–2360. [DOI] [PubMed] [Google Scholar]
- 8.Cwirla S.E., Peters,E.A., Barrett,R.W. and Dower,W.J. (1990) Proc. Natl Acad. Sci. USA, 87, 6378–6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geitz R.D. and Schiestl,R.H. (1995) Methods Mol. Cell. Biol., 5, 255–269. [Google Scholar]
- 10.Hoffman C.S. and Winston,F. (1987) Gene, 57, 267–272. [DOI] [PubMed] [Google Scholar]
- 11.Brenner S. and Lerner,R.A. (1992) Proc. Natl Acad. Sci. USA, 89, 5381–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pi H., Chien,C.T. and Fields,S. (1997) Mol. Cell. Biol., 17, 6410–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bardwell L., Cook,J.G., Inouye,C.J. and Thorner,J. (1994) Dev. Biol., 166, 363–379. [DOI] [PubMed] [Google Scholar]
- 14.Marshall A. and Hodgson,J. (1998) Nature Biotechnol., 16, 27–31. [DOI] [PubMed] [Google Scholar]
- 15.Needels M.C., Jones,D.G., Tate,E.H., Heinkel,G.L., Kochersperger,L.M., Dower,W.J., Barrett,R.W. and Gallop,M.A. (1993) Proc. Natl Acad. Sci. USA, 90, 10700–10704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maskos U. and Southern,E.M. (1992) Nucleic Acids Res., 20, 1679–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fulton R.J., McDade,R.L., Smith,P.L., Kienker,L.J. and Kettmen,J.R.,Jr (1997) Clin. Chem., 43, 1749–1756. [PubMed] [Google Scholar]
- 18.Liang P. and Pardee,A.B. (1992) Science, 257, 967–971. [DOI] [PubMed] [Google Scholar]
- 19.Lisitsyn N., Lisitsyn,N. and Wigler,M. (1993) Science, 259, 946–951. [DOI] [PubMed] [Google Scholar]