

Abstract
In recent years, immune checkpoint blockades (ICBs) have made great progress in the treatment of cancer. However, most ICBs have not yet been observed to be satisfactory in the treatment of osteosarcoma. Herein, we designed composite nanoparticles (NP–Pt-IDOi) from a reactive oxygen species (ROS) sensitive amphiphilic polymer (PHPM) with thiol-ketal bonds in the main chain to encapsulate a Pt(IV) prodrug (Pt(IV)–C12) and an indoleamine-(2/3)-dioxygenase (IDO) inhibitor (IDOi, NLG919). Once NP-Pt-IDOi enter the cancer cells, the polymeric nanoparticles could dissociate due to the intracellular ROS, and release Pt(IV)–C12 and NLG919. Pt(IV)–C12 induces DNA damage and activates the cGAS-STING pathway, increasing infiltration of CD8+ T cells in the tumor microenvironment. In addition, NLG919 inhibits tryptophan metabolism and enhances CD8+ T cell activity, ultimately activating anti-tumor immunity and enhancing the anti-tumor effects of platinum-based drugs. NP-Pt-IDOi were shown to have superior anti-cancer activity in vitro and in vivo in mouse models of osteosarcoma, providing a new clinical paradigm for combining chemotherapy with immunotherapy for osteosarcoma.
Keywords: Indoleamine-(2/3)-dioxygenase inhibitor, Platinum, STING, Osteosarcoma, Immunotherapy
Graphical abstract
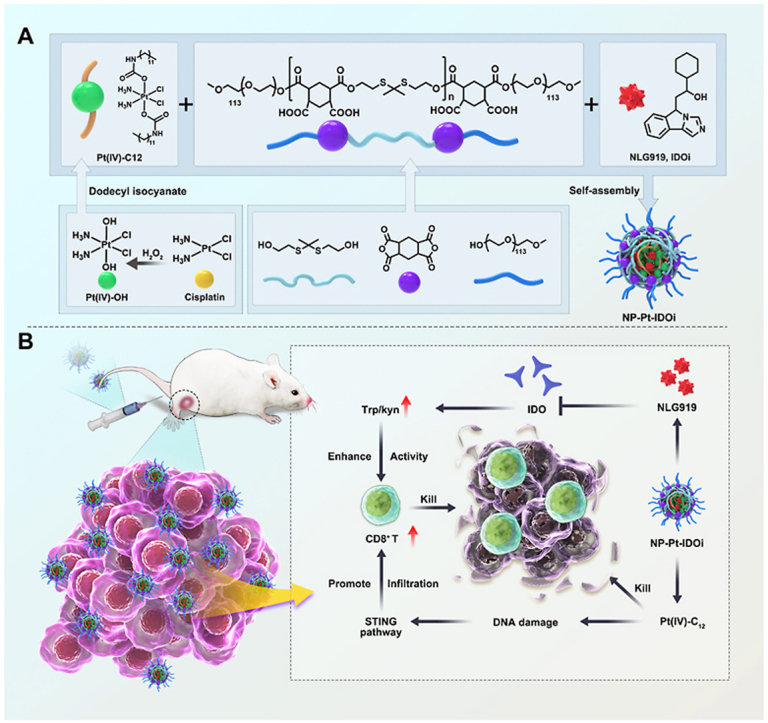
Osteosarcoma is a rare and aggressive bone cancer that occurs in the long tubular bones of adolescents [1,2]. It has a high propensity to metastasize and requires intensive chemotherapy before and after surgery [3]. The prolonged and excessive use of anti-cancer drugs increases the risk of harmful side effects and cancer cell resistance, creating a hostile and immune-suppressive environment around the tumor [[4], [5], [6], [7]]. Reprogramming the immunosuppressive microenvironment shows promise in improving the efficacy of combined chemotherapeutic agents and immunotherapy [[8], [9], [10]].
Stimulator of interferon genes (STING) is a protein embedded in the membrane of the endoplasmic reticulum [11,12]. When the STING protein is activated, it triggers a powerful immune response against tumors, turning "cold" tumors with low immunity into "hot" tumors with high immunity [13,14]. Therefore, activating STING pathway could enhance the anti-tumor effect with powerful immune response [15,16], and STING agonists have attracted much attention for cancer immunotherapy [17,18]. Platinum-based anti-cancer drugs, such as cisplatin (Cis), oxaliplatin and carboplatin, are the first-line anti-tumor agents in osteosarcoma chemotherapy regimens [2,19]. Cis exerts its anti-cancer effect by cross-linking with the DNA in cancer cells and causing DNA damage [20,21]. Research has shown that the damaged DNA fragments in cancer cells can escape into the cytoplasm and be detected by cGAS, which produces cGAMP from ATP and GTP [22,23]. Subsequently, cGAMP could bind to STING in the cancer cells and activate the STING pathway [[24], [25], [26]]. Hence, novel drug delivery systems for platinum drugs can reduce Cis's systemic toxicity and enhance its tumor-targeted accumulation [[27], [28], [29]], as well as improve the immunosuppressive microenvironment and prevent cancer cells from evading the immune system [30,31].
Indoleamine 2,3-dioxygenase (IDO) is an enzyme that can oxidize tryptophan into kynurenine in immune cells [32]. However, tumor cells can overexpress IDO and deplete tryptophan in the tumor microenvironment, which suppresses the function and activity of tryptophan-sensitive T cells [33,34]. Therefore, inhibiting IDO activity and expression can effectively enhance the activity of tumor-infiltrating T cells induced by STING activation, and thus enhance the activation of the immune system to kill tumor cells [35,36]. However, STING agonists and IDO inhibitor (IDOi) need to reach the tumor cells effectively to exert their full potential [37,38]. Previous studies have shown that drug delivery systems can enhance drug accumulation in tumors [39,40]. Therefore, developing drug delivery systems can efficiently transport STING agonists to the tumor site and cancer cells is crucial for activating the STING pathway and improving the immunotherapy outcomes [41,42].
To this end, a platinum (IV) prodrug (Pt(IV)–C12) with two hydrophobic aliphatic chains in the axial direction was developed from Cis [43]. Moreover, a new polymer with thiol-ketal bonds in the main chain (P1) that can react with ROS was also designed [44]. Subsequently, P1, Pt(IV)–C12 and IDOi (NLG919) were self-assembled by nanoprecipitation into composite nanoparticles (NP–Pt-IDOi, Scheme 1) that not only inhibited tryptophan metabolism in cancer cells, but also induced DNA damage and activated the STING pathway. The inhibition of cancer cells by NP-Pt-IDOi was evaluated on several osteosarcoma cell lines in vitro. Furthermore, a mouse tibia in situ tumor model based on K7M2-LUC cells was constructed, and the inhibition of osteosarcoma growth by NP-Pt-IDOi in vivo was investigated. We demonstrated that NP-Pt-IDOi exhibited the synergistic effect of chemotherapy and immunotherapy and could facilitate the translational application to treat osteosarcoma patients with poor sensitivity to platinum drugs (Scheme 1).
Scheme 1.

NP-Pt-IDOi activated the STING pathway in vivo and enhanced immune responses for efficient cancer therapy.
1. Results and discussion
1.1. Preparation, characterization, and intracellular uptake of NP-Pt-IDOi
NLG919 is a highly effective IDO inhibitor with poor water solubility, which limits its use in the clinic [45,46]. In order to deliver NLG919 for rapid drug release in cancer cells, an oxidation-sensitive polymer with thiol-ketal bonds in the main chain (PHPM) was synthesized [47,48]. A Cis(IV) prodrug, namely, Pt (IV)–C12 with two hydrophobic aliphatic chains in the axial positions was synthesized [49,50]. Pt(IV)–C12 loaded nanoparticles (NP–Pt) were prepared by a nano-precipitation method. Using the same method, PHPM was co-assembled with Pt(IV)–C12 and NLG919 in a certain ratio (2.5:1) to form nanoparticles named NP-Pt-IDOi. NP-Pt and NP-Pt-IDOi were homogeneous with complete spherical structures observed by the transmission electron microscope (TEM) (Fig. S1, Fig. 1A), with an average particle size of 71.5 ± 7.6 nm and 92.3 ± 9.5 nm. The average hydrodynamic particle sizes of NP-Pt and NP-Pt-IDOi were around 95.3 ± 10.2 nm (Fig. S2) and 113.2 ± 3.6 nm (Fig. 1B) by dynamic light scattering (DLS), respectively. In addition, the zeta potentials of NP-Pt and NP-Pt-IDOi were −18.9 and −10.1 mV (Fig. 1C), respectively, due to the carboxy groups in the polymer. The polydispersity index (PDI) values of NP-Pt and NP-Pt-IDOi were 0.194 and 0.157, and their size and PDI remained stable when stored at 4°C for 7 days, indicating good dispersivity and stability in aqueous solution (Fig. 1D, Fig. S3). Subsequently, in order to demonstrate the successful preparation of NP-Pt-IDOi, the elemental composition of NP-Pt-IDOi and the atomic valence states of Pt, Cl, S were further analyzed by X-ray photoelectron spectroscopy (XPS) (Fig. S4). Moreover, the encapsulation efficiency of Pt(IV)–C12 and IDOi were determined by inductively coupled plasma-mass spectrometry (ICP-MS) and high performance liquid chromatography (HPLC), the results were 8.5% and 6.3%, respectively (Fig. S5).
Fig. 1.

Preparation, characterization, and intracellular uptake of NP-Pt-IDOi. A) Representative transmission electron microscopy (TEM) image of NP-Pt-IDOi. Scale bar = 100 nm. B) Particle size distribution of NP-Pt-IDOi measured by DLS. C) Zeta potential of NP-Pt and NP-Pt-IDOi measured by DLS. D) PDI of NP-Pt and NP-Pt-IDOi measured by DLS. E) The particle size changes of NP-Pt-IDOi after 24 h of exposure to 1 mM H2O2. F) The release of NP-Pt-IDOi measured by high-performance liquid chromatography (HPLC) in the presence of 1 mM H2O2 at different times. G) The uptake of NP-Pt-IDOi@Cy5.5 (5 μM Pt) by HOS cells observed by CLSM at 1 h, 4 h and 7 h. Blue: DAPI of the stained nucleus; Red: Cy5.5 in NP-Pt-IDOi@ Cy5.5; Green: Alexa-488 stained cytoskeleton. Scale bar = 30 μm. H) Intracellular uptake and fluorescence intensity in HOS cells treated with NP-Pt-IDOi@ Cy5.5 (5 μM Pt) at 1 h, 4 h and 7 h measured by FCM. I) Semi-quantitative analysis of the fluorescence intensity in (H). The statistical difference between the two groups was calculated by one-way ANOVA. ∗∗∗p < 0.001, and ns indicates no statistical significance. Data were shown as mean ± standard deviation (SD) from n independent experiments (n = 3).
The dissociation process of nanoparticles (NP–Pt-IDOi) was explored under different conditions (1 mM H2O2, pH = 5.0, DMEM, 37 °C) for 24 h, and the particle size changes were measured by DLS. Two bimodal curves with two peaks (920.2 nm and 18.6 nm, 396.5 nm and 18.2 nm) were observed (Fig. S6, Fig. 1E), suggesting the breakdown of thiol ketal bonds and dissociation of NP-Pt-IDOi triggered by H2O2 and acid environment. The peak of large particle size may be attributed to the aggregates of hydrophobic polymer fragments, while the peak of small size may be due to the aggregates of hydrophilic polymer fragments. The release of NLG919 from NP-Pt-IDOi was further monitored by HPLC. The results showed that the release of NLG919 reached as high as 78% after treatment (Fig. 1F), demonstrating the oxidation-sensitive drug release behavior of NP-Pt-IDOi. It is reported that there is abnormal redox homeostasis in tumor microenvironment of osteosarcoma with high levels of ROS and GSH [51]. We demonstrated that NP-Pt-IDOi could release the encapsulated NLG919 through the degradation of polymeric carriers and the dissociation of nanoparticles in the presence of H2O2 (1 mM), indicating that the NP-PT-IDOi could degrade in the osteosarcoma cancer cell.
To quantitatively evaluate the uptake of NP-Pt-IDOi by cancer cells, a Cy5.5 dye (red) was encapsulated to label NP-Pt-IDOi (NP-Pt-IDOi@Cy5.5). Later, NP-Pt-IDOi@Cy5.5 was used to treat the human osteosarcoma (HOS) cells for 1 h, 4 h, and 7 h, respectively. The red fluorescence of cells was observed at different time by confocal laser scanning microscopy (CLSM). The results showed that the intracellular red fluorescence intensity of HOS cells treated with NP-Pt-IDOi@Cy5.5 increased with time, indicating the increase of NP-Pt-IDOi@Cy5.5 uptake with longer incubation time (Fig. 1G). Subsequently, flow cytometry (FCM) was used to quantitatively monitor the uptake of NP-Pt-IDOi@Cy5.5 by HOS cells. The results showed that the intracellular red fluorescence intensity at 7 h was nearly 2.3-fold higher than that at 1 h (Fig. 1H and I). The above results indicated that NP-Pt-IDOi could be efficiently taken up by cancer cells.
To dive deeper into the underlying cellular uptake mechanism that NP-Pt-IDOi could actively enter into cancer cells, we further investigated this process by an endocytosis inhibition assay. The cellular uptake mechanism of the nanoparticles was investigated by pretreating the cells with various endocytosis inhibitors, 5% dextrose and at energy blockade 4 °C individually. Endocytosis inhibitors used in this assay include chlorpromazine (clathrin-mediated inhibitor), Sa-Sodium azide (energy-dependent inhibitor), genistein (caveolae-mediated inhibitor), and wortmannin (macropinocytosis inhibitor). Flow cytometry analysis revealed that the cellular uptake of NP-Pt-IDOi@Cy5.5 was almost completely inhibited (6.1%) at 4 °C, which was 14.1-fold and 12.4-fold lower than that of PBS and 5% dextrose (37 °C), and Sa-Sodium azide pretreatment also resulted in significant uptake suppression (46.7%). Moreover, the results showed that cellular uptake of NP-Pt-IDOi@Cy5.5 was almost not affected by the caveolae-mediated endocytotic inhibitors (genistein 68.4%) and was only partially (60.8%) impeded by the macropinocytosis inhibitors (wortmannin). Nonetheless, clathrin-mediated endocytosis inhibitors (chlorpromazine) substantially reduced cellular uptake level by 32% (Fig. S7). The above results suggested that NP-Pt-IDOi@Cy5.5 were ingested by HOS cells primarily through clathrin-mediated endocytosis.
1.2. Anti-cancer activity of NP-Pt-IDOi in vitro
To investigate the anti-cancer activity of NP-Pt-IDOi in vitro, four osteosarcoma cell lines (HOS, 143B, K7M2, MG63) were selected for MTT toxicity test. The results showed that NP-Pt-IDOi had the lowest IC50 among all the four osteosarcoma cell lines compared with Cis, IDOi, Cis + IDOi, and NP-Pt groups (Fig. 2A and B), indicating the highest cytotoxicity of NP-Pt-IDOi. Specifically, NP-Pt-IDOi had a 3-fold lower IC50 against the MG63 cell line compared with Cis, suggesting that the NP-Pt-IDOi improved the anti-cancer activity of platinum drug against Cis-insensitive cell lines. Interestingly, the IC50 of Cis + IDOi was higher than Cis group for the four cell lines due to NLG919, while the NP-Pt-IDOi with Cis and IDOi in a nanoparticle formulation showed higher cytotoxicity than the Cis group. The reason for the highest cytotoxicity of NP-Pt-IDOi could be that the polymeric nanoparticles increased endocytosis of drugs by the four cell lines (Fig. 1G–I).
Fig. 2.

In vitro anti-cancer effect of NP-Pt-IDOi. A) The cell viability of HOS, 143B, K7M2 and MG63 after 48 h treatment with Cis, IDOi, Cis + IDOi, NP-Pt and NP-Pt-IDOi by MTT assay. B) Statistical table of IC50 values against four osteosarcoma cell lines after 48 h treatment with drugs. C) The apoptosis ratio of HOS cells treated with different drugs (5 μM Pt, 10 μM NLG919) measured by FCM. D) Semi-quantitative analysis of apoptosis ratio. E) The colony formation of HOS cells treated with different drugs (0.5 μM Pt, 1 μM NLG919) for 10 days via crystal violet staining. F) Live and dead assay of 3D tumor spheroid after 48 h treatment of PBS, Cis, IDOi, Cis + IDOi, NP-Pt, NP-Pt-IDOi (5 μM Pt, 10 μM NLG919) by CLSM. The 3D tumor spheroids were stained with Calcein-AM (green, living cells) and PI (red, dead cells). Scale bar = 50 μm. The data were shown as mean ± SD from n independent experiments (n = 3). The statistical difference between the two groups was calculated by one-way ANOVA. ∗∗p < 0.01.
Next, the apoptosis of HOS cells treated with different drugs for 48 h was studied by an Annexin V-FITC and propidium iodide (PI) double staining assay. The results showed that the apoptotic ratio of cells treated with NP-Pt-IDOi was 23.62 ± 2.91% (Fig. 2C and D), which was 8.6 times that of cells treated with PBS (2.75 ± 1.51%). This apoptosis ratio was also higher than that of cells treated with Cis at 13.41 ± 5.72% and NP-Pt at 18.46 ± 6.63%. To observe the anti-cancer effect more intuitively, the photo of the cell colonies after the treatment of NP-Pt-IDOi was taken (Fig. 2E). The results showed that the number of cell colonies treated with NP-Pt-IDOi was significantly less than those treated with other drugs, indicating that NP-Pt-IDOi could effectively inhibit the growth of HOS cells in vitro. Finally, in order to mimic the physiological conditions in vivo, 3D HOS spheroids were established to evaluate the anti-cancer ability of NP-Pt-IDOi by a live/dead assay (living cells are green, and the apoptotic cells are red) (Fig. 2F). The results showed that the number of red cells in 3D cell spheroids treated with NP-Pt-IDOi was higher than that treated with other drugs. Taken together, NP-Pt-IDOi could inhibit the cancer cell growth in vitro, and promote cancer cell apoptosis with higher anti-cancer activity than Cis + IDOi, IDOi and NP-Pt groups.
1.3. NP-Pt-IDOi increased DNA damage to activate the STING pathway in vitro
To explore the ability of NP-Pt-IDOi to enhance DNA damage in vitro, HOS cells were treated with Cis, IDOi, Cis + IDOi, NP-Pt and NP-Pt-IDOi (5 μM Pt, 10 μM NLG919) for 48 h. First, DNA damage marker, γ-H2AX, was immunostained (green) to study the DNA damage of tumor cells. The green fluorescence intensity in the nuclei of cells treated with NP-Pt-IDOi was found to be significantly higher than that of cells treated with other drugs by CLSM (Fig. 3A), indicating the higher expression of γ-H2AX in cells treated with NP-Pt-IDOi. Similarly, the expression of γ-H2AX protein in cancer cells treated with various drugs was studied by western blot (WB) and quantified by Image J software. The results showed that the expression of γ-H2AX was significantly increased in the cells treated with NP-Pt and NP-Pt-IDOi (Fig. 3B and D). Second, the results showed that the expression of IDO protein decreased in cells treated with NP-PT-IDOi, indicating that NP-PT-IDOi had the strongest inhibitory effect on IDO (Fig. 3B and D). Next, in order to explore whether NP-Pt-IDOi could activate the STING pathway, the expression of several related proteins (STING, TBK-1, IRF-3, p-STING, p-TBK1, p-IRF3) were detected in HOS cells treated with different drugs by WB. The gray value of protein expression was quantitatively analyzed through Image J software. The results showed that the expression of STING, TBK-1 and IRF-3 protein in cells treated with IDOi, NP-Pt, and NP-Pt-IDOi were similar (Fig. 3B, 3 E-3J). Surprisingly, the expression of p-STING, p-TBK1 and p-IRF3 in cells treated with NP-Pt-IDOi were 1.6, 2.1, and 1.3 times higher than those in cells treated with NP-Pt, respectively. Among these, p-STING protein was the most significantly up-regulated, suggesting that NP-Pt-IDOi were more prominent in activating the proteins related to the STING pathway. These results indicated that NP-Pt-IDOi could efficiently activate the STING pathway in osteosarcoma cells.
Fig. 3.

NP-Pt-IDOi could increase DNA damage to activate STING pathway in vitro. A) CLSM images of the DNA damage-related protein (γ-H2AX) in HOS cells treated with different drugs (5 μM Pt, 10 μM NLG919; blue, DAPI; green, γ-H2AX). Scale bar = 30 μm. B) DNA damage-related proteins and STING pathway related proteins were measured by WB. The cells were treated with PBS, Cis, IDOi, Cis + IDOi, NP-Pt and NP-Pt-IDOi (5 μM Pt, 10 μM NLG919) after 48 h. Tubulin was the internal reference protein. C-J) The semi-quantitative analyses of gray values of IDO, γ- H2AX, STING, TBK-1, IRF-3, p-STING, p-TBK-1, p-IRF-3 proteins detected by WB. The data were shown as mean ± SD from n independent experiments (n = 3). The statistical difference between the two groups was calculated by one-way ANOVA. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ns indicates no statistical significance.
1.4. Bio-distribution and anti-tumor activity of NP-Pt-IDOi in vivo
First of all, the biosafety of nanoparticles is the premise of their application in vivo. In order to explore the biosafety of NP-Pt-IDOi in vivo, the toxicity of NP-Pt-IDOi was systematically studied in 6-week-old healthy KM mice. The mice were divided into six groups and injected intravenously for three consecutive days with PBS, Cis, IDOi, Cis + IDOi, NP-Pt and NP-Pt-IDOi. The body weight of mice in each group was recorded every two days after the first injection. After 14 days of monitoring, the blood samples were taken, and six biochemical indicators of serum were tested. The body weight of mice treated with Cis, Cis + IDOi decreased significantly (Fig. S8), while there was no statistically significant difference between the weight of mice treated with PBS, NP-Pt and NP-Pt-IDOi, indicating that NP-Pt-IDOi has low systemic toxicity. Moreover, the biochemical indexes also were relatively normal in all groups (Fig. S9). In summary, the aforementioned results indicated the excellent biocompatibility of the NP-Pt-IDOi.
Next, to explore the accumulation of NP-Pt-IDOi in the tumor site, K7M2-LUC cells (1 × 106 cells/0.1 mL) were implanted into the tibia of 4-week-old female mice to establish an in-situ osteosarcoma model (Fig. 4A). Later, NP-Pt-IDOi labeled with a fluorescent dye Cy7.5 (NP-Pt-IDOi@Cy7.5) were administered to mice by tail vein injection. The distribution of NP-Pt-IDOi@Cy7.5 was tracked through in vivo imaging system (IVIS) after 3 h, 6 h, 12 h, 24 h, 36 h and 48 h of administration in K7M2-LUC tumor-bearing mice (Fig. 4B). The quantitative analysis showed that the fluorescence intensity in the tumors of mice gradually increased with time, and reached the peak at 36 h, indicating the accumulation of NP-Pt-IDOi@Cy7.5 in the tumor (Fig. 4C). The mice were then sacrificed at 48 h, and their major organs (heart, liver, spleen, lung, and kidney) and tumors were taken out. Imaging of various organs of mice showed that there was strong fluorescence in the tumor and liver (Fig. 4D). Further quantitative analysis demonstrated that the fluorescence intensity in the tumor was the highest among all organs (Fig. 4E), which was consistent with the living animal imaging. The high fluorescence intensity in the liver may be due to drug metabolism in the liver. These results indicated that NP-Pt-IDOi could accumulate and retain in the tumor.
Fig. 4.

Biodistribution and anti-tumor activity of NP-Pt-IDOi in vivo. A) The timeline of establishment and treatment of the orthotopic model of osteosarcoma based on K7M2-LUC cells in the tibia of mice. B) Biodistribution of NP-Pt-IDOi@Cy7.5 on K7M2-LUC tumor bearing mice at different time imaged by IVIS. C) The fluorescence intensity at different time was quantitatively analyzed. D) The mice were sacrificed 48 h later, and the major organs (heart, liver, spleen, lung, and kidney) and tumors were taken out for fluorescence imaging. E) The fluorescence intensity of major organs and tumors was analyzed semi-quantitatively. F) Bioluminescence imaging of the mice by IVIS on day 0, 7, 14 and 20 to track the anti-tumor effect of each drug. G) The anti-tumor effect of the drugs shown by quantitative analysis of bioluminescence. H) Body weight changes of mice after 20 days of drug treatment. I) The tumor morphological changes by H&E staining after 20 days of drug treatment. Scale bar = 200 μm.
Finally, the anti-tumor activity in vivo was investigated. An orthotopic osteosarcoma model based on K7M2-LUC cells was established. Then mice were treated with saline (i.v. injection), IDOi (oral administration), Cis (i.v. injection), Cis + IDOi (i.v. injection + oral administration), NP-Pt (i.v. injection) or NP-Pt-IDOi (i.v. injection) at an identical Pt dose of 3.5 mg kg−1 and NLG919 dose of 25 mg kg−1 for three times after tumor formation (Fig. 4A). To monitor the tumor growth, bioluminescence imaging of the mice was conducted by IVIS at 0, 7, 14 and 20 days. Fig. 4F showed that the bioluminescence in mice treated with NP-Pt-IDOi was the weakest at day 20 compared with the other groups. The quantitative analysis of the fluorescence intensity demonstrated the slowest increase of bioluminescence in mice treated with NP-Pt-IDOi (Fig. 4G). The average bioluminescence after 20 days of NP-Pt-IDOi treatment was 3.50 × 109 p/s/cm2/sr, which was about one fifth of the bioluminescence of mice without drug treatment (1.72 × 1010 p/s/cm2/sr), and about half of the mice treated with NP-Pt (6.12 × 109 p/s/cm2/sr). The results demonstrated that NP-Pt-IDOi had the best anti-tumor effect. In addition, within 20 days of treatment, there was no significant difference in weight change between mice treated with various drugs, indicating that these anti-tumor drugs had good performance in biosafety (Fig. 4H). Next, the mice were sacrificed, and the tumors were collected for hematoxylin and eosin (H&E) staining. The results showed extensive nuclear shrinkage and disappearance in the tumor tissues of mice treated with NP-Pt-IDOi (Fig. 4I). Taken together, the above results demonstrated that NP-Pt-IDOi could effectively inhibit tumor growth in vivo.
1.5. NP-Pt-IDOi activated STING pathway to reprogram tumor microenvironment (TME)
In order to further study the anti-tumor mechanism of NP-Pt-IDOi in vivo, we explored the activation of STING pathway by NP-Pt-IDOi and the changes in the immune microenvironment in tumor tissues, spleen and lymph nodes of mice. FCM was used to determine the proportion of changes in several immune cells in mice treated with each drug, including matured DCs (CD11c+CD80+CD86+) in lymph nodes (Fig. S10), matured DCs in tumor tissue (Fig. S11), central memory T cells (CD3+CD8+CD44+CD62L+, TCM) in spleen (Fig. 5A), CD3+CD4+/CD3+CD8+ T cells in tumor tissue (Fig. 5B), Tregs cells (CD3+CD4+Foxp3+) in tumor tissue (Fig. 5C), TCM in lymph nodes (Fig. S12), and M2-like macrophages (F4/80+CD80−CD206+) to M1-like macrophages (F4/80+CD80+CD206-) transformation in tumor tissue (Figs. S13 and S14). Quantitative analysis was then carried out, including matured DCs in lymphoid tissue (Fig. 5D), matured DCs in tumor tissue (Fig. 5E), TCM cells in the spleen (Fig. 5F), TCM cells in lymphoid tissue (Fig. 5G), and CD4+ cells in tumor tissue (Fig. 5H), CD8+ cells in tumor tissue (Fig. 5I), M2-like to M1-like transformation in tumor tissue (Fig. 5J), and Tregs cells in tumor tissue (Fig. 5K). Compared with the other four drugs, the DCs stained with CD11c+/CD80+/CD86+ in lymph nodes and tumor tissues of mice treated with NP-Pt-IDOi were 40.6% and 18.4%, respectively, which were 1.76 times and 2.25 times that of mice treated with PBS (Fig. 5D and E). The results also showed that NP-Pt-IDOi could significantly promote the maturation of DCs in tumor-bearing mice compared with IDOi and Cis + IDOi.
Fig. 5.

NP-Pt-IDOi activated STING pathway to improve tumor immune microenvironment. A) The proportion of memory T cells (TCM) in the spleen of mice treated with various drugs including Cis, IDOi, Cis + IDOi, NP-Pt and NP-Pt-IDOi by FCM. B) The proportion of T cells stained with CD4+/CD8+in tumors of mice treated with various drugs by FCM. C) The proportion of Tregs cells in tumors of mice treated with various drugs by FCM. D) Quantitative analysis of mature DCs in lymph nodes of mice. E) Quantitative analysis of mature DCs in mice tumor tissues. F) Quantitative analysis of TCM cells in mouse spleen. G) Quantitative analysis of TCM cells in lymph nodes of mice. H) Quantitative analysis of CD4+ cells in mice tumor tissues. I) Quantitative analysis of CD8+ cells in mice tumor tissues. J) Quantitative analysis on the transformation of M2-like to M1-like in mice tumor tissues. K) Quantitative analysis of Tregs cells in mouse tumor tissues. The data were shown as mean ± SD from n independent experiments (n = 5). The statistical difference between the two groups was calculated by one-way ANOVA. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
In addition, the TCM in the spleen and lymph node of mice treated with NP-Pt-IDOi were significantly increased. Specifically, the ratio of TCM in the spleen and lymph node of the mice treated with NP-Pt-IDOi was 21.7% and 24.1%, respectively, which was 2.35 times and 3.08 times that of the mice treated with PBS (Fig. 5A, F, 5G). These results demonstrated that NP-Pt-IDOi could significantly promote the transformation of CD8+ T cells in the spleen and lymph node to TEM cells. Next, the proportion of CD4+ and CD8+ cells in tumor tissues of mice treated with NP-Pt-IDOi were 30.7% and 35.6%, respectively, which were 2.32 and 2.96 times that of mice treated with PBS (Fig. 5B, H, 5I). Furthermore, the infiltration of CD4+/CD8+ effector T cells in tumors of mice treated with NP-Pt-IDOi was significantly increased compared with the other four drugs. Therefore, the results demonstrate that NP-Pt-IDOi could transform the "cold tumors" with low immune responses into the "hot tumors" with high immune responses in vivo by increasing the number of TCM cells in spleen and lymph node tissues, mature DCs in lymph nodes and tumor tissues, and CD4+/CD8+ effector T cells in tumor tissues.
Tumor immune escape plays an important role in treatment failure and drug resistance of chemotherapeutic drugs. To study whether NP-Pt-IDOi can improve the low tumor responses to chemotherapy drugs by regulating immune cells with tumor immune escape ability, the polarization ratio of M2-like to M1-like in tumor associated macrophages and the infiltration ratio of CD4+/Foxp3+ immunosuppressive regulatory T cells (also known as Tregs cells) were quantitatively analyzed by FCM. Generally speaking, most tumor-associated macrophages (TAMs) in osteosarcoma are the M2-like phenotype, which plays an important role in inhibiting the activation of cytotoxic T cells [52,53]. The results showed that the proportion of M1/M2 in tumors of mice treated with NP-Pt-IDOi was higher than that of mice treated with Cis, IDOi and Cis + IDOi (Fig. 5J). Specifically, the number of M1 phenotype cells in tumors of mice treated with NP-Pt-IDOi was 17.8%, which was 2.66 times that of mice treated with PBS. However, the M2-like phenotype cells of mice treated with PBS were 24.8%, which was 1.48 times that of mice treated with NP-Pt-IDOi. In addition, recent studies showed that Tregs cells could promote tumor immune escape by inhibiting the activation of CD4+ and CD8+ T cells. To further investigate the regulatory ability of NP-Pt-IDOi on Tregs cells in vivo, the Tregs cells in tumors of mice treated with different drugs were analyzed by FCM. The results showed that the proportion of Tregs in tumor tissues of mice treated with NP-Pt-IDOi was the lowest among mice treated with all drugs (Fig. 5C and K). Specifically, the average proportion of Tregs in tumors of mice treated with NP-Pt-IDOi was 6.48%, while the average proportion of Tregs in tumors of mice treated with PBS was 21.7%. The results suggested that NP-Pt-IDOi down-regulated the proportion of Tregs in tumor tissues by 3.35 times. In conclusion, NP-Pt-IDOi could activate the STING pathway in the tumor cells in vivo, which could effectively reshape the TME [54], transform the "cold tumor" with low immune responses into the "hot tumors" with high immune responses, thus enhancing the anti-tumor effect of platinum drugs [55,56]. NP-Pt-IDOi are a promising strategy for tumor chemo-immunotherapy.
2. Conclusion
In this work, we developed polymeric nanoparticles to deliver the platinum drug and STING agonist for the synergistic chemotherapy and immunotherapy for osteosarcoma. Oxidation-sensitive amphiphilic polymers with thiol-ketal bonds in the main chain were synthesized and self-assembled into polymeric nanoparticles (NP–Pt-IDOi) to encapsulate Pt(IV) prodrug (Pt(IV)–C12) and IDO inhibitor (NLG919). NP-Pt-IDOi could degrade and disassociate to release NLG919 in 1 mM H2O2 environment. The uptake of NP-Pt-IDOi by HOS cells was observed, and NP-Pt-IDOi exhibited the highest cytotoxicity against four osteosarcoma cell lines compared with Cis, IDOi, Cis + IDOi, and NP-Pt. NP-Pt-IDOi accumulated in the tumor and effectively inhibited the tumor progression in an orthotopic osteosarcoma mouse model. The activation of STING pathway in the cancer cells was demonstrated in vitro and in vivo. We further demonstrated that NP-Pt-IDOi could reverse the low immune response of osteosarcoma and reprogram the TME by promoting the maturation of DCs, increasing infiltration of CD8+ T cells, and decreasing the M2-like TAMs and Tregs in the TME. NP-Pt-IDOi provide a new strategy to improve chemotherapy by the activation of STING pathway and are promising for chemo-immunotherapy for osteosarcoma in the future.
Credit author statement
Wenzhi Bi and Meng Xu designed and directed the study. Dongquan Xiang, Xinli Han, and Jianxiong Li performed experiments, wrote, and prepared the manuscript. Dongquan Xiang, Jiabin Zhang, Haihua Xiao, Ting Li, Xuelin Zhao and Hejian Xiong provided materials and technical supports and offered the constructive proposals. All authors read and approved the final manuscript for publication. Dongquan Xiang, Xinli Han, and Jianxiong Li: Methodology, Investigation, Writing - Original Draft, Writing - Review & Editing. Jiabin Zhang: Methodology. Haihua Xiao: Methodology. Ting Li: Visualization. Xuelin Zhao: Formal analysis. Hejian Xiong: Visualization.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81972901) and Foundation Strengthening Program Technical Field Fund Project (2019-JCJQ-JJ-147). All animal experiments reported herein were performed under the guidelines evaluated and approved by the ethics committee of Animal Center of Chinese PLA General Hospital.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.mtbio.2023.100675.
Contributor Information
Hejian Xiong, Email: Hejian.Xiong@utdallas.edu.
Meng Xu, Email: profxum301@163.com.
Wenzhi Bi, Email: biwenzhi@sina.com.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- 1.Anwar M.A., El-Baba C., Elnaggar M.H., Elkholy Y.O., Mottawea M., Johar D., Al Shehabi T.S., Kobeissy F., Moussalem C., Massaad E., Omeis I., Darwiche N., Eid A.H. Novel therapeutic strategies for spinal osteosarcomas. Semin. Cancer Biol. 2020;64:83–92. doi: 10.1016/j.semcancer.2019.05.018. [DOI] [PubMed] [Google Scholar]
- 2.Gill J., Gorlick R. Advancing therapy for osteosarcoma. Nat. Rev. Clin. Oncol. 2021;18:609–624. doi: 10.1038/s41571-021-00519-8. [DOI] [PubMed] [Google Scholar]
- 3.Kansara M., Teng M.W., Smyth M.J., Thomas D.M. Translational biology of osteosarcoma. Nat. Rev. Cancer. 2014;14:722–735. doi: 10.1038/nrc3838. [DOI] [PubMed] [Google Scholar]
- 4.Viswanathan V.S., Ryan M.J., Dhruv H.D., Gill S., Eichhoff O.M., Seashore-Ludlow B., Kaffenberger S.D., Eaton J.K., Shimada K., Aguirre A.J., Viswanathan S.R., Chattopadhyay S., Tamayo P., Yang W.S., Rees M.G., Chen S., Boskovic Z.V., Javaid S., Huang C., Wu X., Tseng Y.Y., Roider E.M., Gao D., Cleary J.M., Wolpin B.M., Mesirov J.P., Haber D.A., Engelman J.A., Boehm J.S., Kotz J.D., Hon C.S., Chen Y., Hahn W.C., Levesque M.P., Doench J.G., Berens M.E., Shamji A.F., Clemons P.A., Stockwell B.R., Schreiber S.L. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–457. doi: 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang C., Pu K. Recent progress on activatable nanomedicines for immunometabolic combinational cancer therapy. Small Structures. 2020;1:1039–1059. [Google Scholar]
- 6.Corre I., Verrecchia F., Crenn V., Redini F., Trichet V. The osteosarcoma microenvironment: a complex but targetable ecosystem. Cells. 2020;9:976–1001. doi: 10.3390/cells9040976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsvetkova D., Ivanova S. Application of approved cisplatin derivatives in combination therapy against different cancer diseases. Molecules. 2022;27:2466–2517. doi: 10.3390/molecules27082466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J., Huang D., Saw P.E., Song E. Turning cold tumors hot: from molecular mechanisms to clinical applications. Trends Immunol. 2022;43:523–545. doi: 10.1016/j.it.2022.04.010. [DOI] [PubMed] [Google Scholar]
- 9.Shi C., Liu T., Guo Z., Zhuang R., Zhang X., Chen X. Reprogramming tumor-associated macrophages by nanoparticle-based reactive oxygen species photogeneration. Nano Lett. 2018;18:7330–7342. doi: 10.1021/acs.nanolett.8b03568. [DOI] [PubMed] [Google Scholar]
- 10.Tang L., Mei Y., Shen Y., He S., Xiao Q., Yin Y., Xu Y., Shao J., Wang W., Cai Z. Nanoparticle-mediated targeted drug delivery to remodel tumor microenvironment for cancer therapy. Int. J. Nanomed. 2021;16:5811–5829. doi: 10.2147/IJN.S321416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang R., Ning Q., Zhao J., Zhao X., Zeng L., Yi Y., Tang S. Targeting STING for cancer immunotherapy: from mechanisms to translation. Int. Immunopharm. 2022;113 doi: 10.1016/j.intimp.2022.109304. [DOI] [PubMed] [Google Scholar]
- 12.Gao X., Lei G., Wang B., Deng Z., Karges J., Xiao H., Tan D. Encapsulation of platinum prodrugs into PC7A polymeric nanoparticles combined with immune checkpoint inhibitors for therapeutically enhanced multimodal chemotherapy and immunotherapy by activation of the STING pathway. Adv. Sci. 2022 doi: 10.1002/advs.202205241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du J.M., Qian M.J., Yuan T., Chen R.H., He Q.J., Yang B., Ling Q., Zhu H. cGAS and cancer therapy: a double-edged sword. Acta Pharmacol. Sin. 2022;43:2202–2211. doi: 10.1038/s41401-021-00839-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galon J., Bruni D. Approaches to treat immune hot, altered and cold tumors with combination immunotherapies. Nat. Rev. Drug Discov. 2019;18:197–218. doi: 10.1038/s41573-018-0007-y. [DOI] [PubMed] [Google Scholar]
- 15.Wang F., Su H., Xu D., Dai W., Zhang W., Wang Z., Anderson C.F., Zheng M., Oh R., Wan F., Cui H. Tumor sensitization via the extended intratumorally release of a STING agonist and camptothecin from a self-assembled hydrogel. Nat. Biomed. Eng. 2020;4:1090–1101. doi: 10.1038/s41551-020-0597-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hines J.B., Kacew A.J., Sweis R.F. The development of STING agonists and emerging results as a cancer immunotherapy. Curr. Oncol. Rep. 2023;25:189–199. doi: 10.1007/s11912-023-01361-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Samson N., Ablasser A. The cGAS-STING pathway and cancer. Nat. Can. (Ott.) 2022;3:1452–1463. doi: 10.1038/s43018-022-00468-w. [DOI] [PubMed] [Google Scholar]
- 18.Tian Z., Zeng Y., Peng Y., Liu J., Wu F. Cancer immunotherapy strategies that target the cGAS-STING pathway. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.996663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaikh A.B., Li F., Li M., He B., He X., Chen G., Guo B., Li D., Jiang F., Dang L., Zheng S., Liang C., Liu J., Lu C., Liu B., Lu J., Wang L., Lu A., Zhang G. Present advances and future perspectives of molecular targeted therapy for osteosarcoma. Int. J. Mol. Sci. 2016;17:506–527. doi: 10.3390/ijms17040506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang X., Yu Y., Huang X., Chen Q., Wu H., Wang R., Qi R., Miao Y., Qiu Y. Delivery of platinum (II) drugs with bulky ligands in trans-geometry for overcoming cisplatin drug resistance. Mat. Sci. Eng. C-mater. 2019;96:96–104. doi: 10.1016/j.msec.2018.10.092. [DOI] [PubMed] [Google Scholar]
- 21.Chen H., Wang Y., Liu Y., Tang L., Mu Q., Yin X., Zheng L., Chen Y., Liu C. Delivery of cationic platinum prodrugs via reduction sensitive polymer for improved chemotherapy. Small. 2021;17(45) doi: 10.1002/smll.202101804. [DOI] [PubMed] [Google Scholar]
- 22.Zheng J., Mo J., Zhu T., Zhuo W., Yi Y., Hu S., Yin J., Zhang W., Zhou H., Liu Z. Comprehensive elaboration of the cGAS-STING signaling axis in cancer development and immunotherapy. Mol. Cancer. 2020;19:133–152. doi: 10.1186/s12943-020-01250-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang M., Chen P., Wang L., Li W., Chen B., Liu Y., Wang H., Zhao S., Ye L., He Y., Zhou C. cGAS-STING, an important pathway in cancer immunotherapy. J. Hematol. Oncol. 2020;13:81–92. doi: 10.1186/s13045-020-00916-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du H., Xu T., Cui M. cGAS-STING signaling in cancer immunity and immunotherapy, Biomed. Pharma. 2021;133 doi: 10.1016/j.biopha.2020.110972. [DOI] [PubMed] [Google Scholar]
- 25.Vashi N., Bakhoum S.F. The evolution of STING signaling and its involvement in cancer. Trends Biochem. Sci. 2021;46:446–460. doi: 10.1016/j.tibs.2020.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L., Shang K., Li X., Shen M., Lu S., Tang D., Han H., Yu Y. Reduction sensitive polymers delivering cationic platinum drugs as STING agonists for enhanced chemo-immunotherapy. Adv. Funct. Mater. 2022;32 [Google Scholar]
- 27.Yu Y., Zhang L., Qin Z., Karges J., Xiao H., Su X. Unraveling and overcoming platinum drug-resistant cancer tumors with DNA nanostructures. Adv. Funct. Mater. 2022;33 [Google Scholar]
- 28.Chen S., Peng A., Chen M., Zhan M. Nanomedicines targeting activation of STING to reshape tumor immune microenvironment and enhance immunotherapeutic efficacy. Front. Oncol. 2022;12 doi: 10.3389/fonc.2022.1093240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiong G., Huang D., Lu L., Luo X., Wang Y., Liu S., Chen M., Yu S., Kappen M., You C., Lu S., Yu Y., Lu J., Lin F. Near-infrared-II light induced mild hyperthermia activate cisplatin-artemisinin nanoparticle for enhanced chemo/chemodynamic therapy and immunotherapy. Small Methods. 2022;6(9) doi: 10.1002/smtd.202200379. [DOI] [PubMed] [Google Scholar]
- 30.Luo K., Li N., Ye W., Gao H., Luo X., Cheng B. Activation of stimulation of interferon genes (STING) signal and cancer immunotherapy. Molecules. 2022;27:4638–4655. doi: 10.3390/molecules27144638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li R., Chen Z., Li J., Dai Z., Yu Y. Nano-drug delivery systems for T cell-based immunotherapy. Nano Today. 2022;46 [Google Scholar]
- 32.Abd El-Fattah E.E. Ido/kynurenine pathway in cancer: possible therapeutic approaches. J. Transl. Med. 2022;20:347–360. doi: 10.1186/s12967-022-03554-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujiwara Y., Kato S., Nesline M.K., Conroy J.M., DePietro P., Pabla S., Kurzrock R. Indoleamine 2,3-dioxygenase (Ido) inhibitors and cancer immunotherapy. Cancer Treat Rev. 2022;110 doi: 10.1016/j.ctrv.2022.102461. [DOI] [PubMed] [Google Scholar]
- 34.Dolsak A., Gobec S., Sova M. Indoleamine and tryptophan 2,3-dioxygenases as important future therapeutic targets. Pharmacol. Ther. 2021;221 doi: 10.1016/j.pharmthera.2020.107746. [DOI] [PubMed] [Google Scholar]
- 35.Gargaro M., Manni G., Scalisi G., Puccetti P., Fallarino F. Tryptophan metabolites at the crossroad of immune-cell interaction via the aryl hydrocarbon receptor: implications for tumor immunotherapy. Int. J. Mol. Sci. 2021;22:4644–4658. doi: 10.3390/ijms22094644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heidari F., Ramezani A., Erfani N., Razmkhah M. Indoleamine 2, 3-dioxygenase: a professional immunomodulator and its potential functions in immune related diseases. Int. Rev. Immunol. 2020;41:346–363. doi: 10.1080/08830185.2020.1836176. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y., Luo J., Alu A., Han X., Wei Y., Wei X. cGAS-STING pathway in cancer biotherapy. Mol. Cancer. 2020;19:136–152. doi: 10.1186/s12943-020-01247-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gan Y., Li X., Han S., Liang Q., Ma X., Rong P., Wang W., Li W. The cGAS/STING pathway: a novel target for cancer therapy. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.795401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kwon J., Bakhoum S.F. The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov. 2020;10:26–39. doi: 10.1158/2159-8290.CD-19-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou J., Ventura C.J., Fang R.H., Zhang L. Nanodelivery of STING agonists against cancer and infectious diseases. Mol. Aspect. Med. 2022;83 doi: 10.1016/j.mam.2021.101007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pu F., Chen F., Liu J., Zhang Z., Shao Z. Immune regulation of the cGAS-STING signaling pathway in the tumor microenvironment and its clinical application. OncoTargets Ther. 2021;14:1501–1516. doi: 10.2147/OTT.S298958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang M., Zou Y., Zhou X., Zhou J. Inhibitory targeting cGAS-STING-TBK1 axis: emerging strategies for autoimmune diseases therapy. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.954129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Q., Wang X., Kuang G., Yu Y., Zhao Y. Photopolymerized 3D printing scaffolds with Pt(IV) prodrug initiator for postsurgical tumor treatment. Research. 2022;2022 doi: 10.34133/2022/9784510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mirzaei S., Hushmandi K., Zabolian A., Saleki H., Torabi S.M.R., Ranjbar A., SeyedSaleh S., Sharifzadeh S.O., Khan H., Ashrafizadeh M., Zarrabi A., Ahn K.S. Elucidating role of reactive oxygen species (ROS) in cisplatin chemotherapy: a focus on molecular pathways and possible therapeutic strategies. Molecules. 2021;26:2382–2419. doi: 10.3390/molecules26082382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abd El-Fattah E.E. Ido/kynurenine pathway in cancer: possible therapeutic approaches. J. Transl. Med. 2022;20:347–360. doi: 10.1186/s12967-022-03554-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fujiwara Y., Kato S., Nesline M.K., Conroy J.M., DePietro P., Pabla S., Kurzrock R. Indoleamine 2,3-dioxygenase (Ido) inhibitors and cancer immunotherapy. Cancer Treat Rev. 2022;110 doi: 10.1016/j.ctrv.2022.102461. [DOI] [PubMed] [Google Scholar]
- 47.Peng X., Zhao Z., Liu L., Bai L., Tong R., Yang H., Zhong L. Targeting indoleamine dioxygenase and tryptophan dioxygenase in cancer immunotherapy: clinical progress and challenges. Drug Des. Dev. Ther. 2022;16:2639–2657. doi: 10.2147/DDDT.S373780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Q., Kuang G., Yu Y., Ding X., Ren H., Sun W., Zhao Y. Hierarchical microparticles delivering oxaliplatin and NLG919 nanoprodrugs for local chemo-immunotherapy. ACS Appl. Mater. Interfaces. 2022;14:48527–48539. doi: 10.1021/acsami.2c16564. [DOI] [PubMed] [Google Scholar]
- 49.Zhong Y., Jia C., Zhang X., Liao X., Yang B., Cong Y., Pu S., Gao C. Targeting drug delivery system for platinum(Ⅳ)-Based anti-tumor complexes. Eur. J. Med. Chem. 2020;194 doi: 10.1016/j.ejmech.2020.112229. [DOI] [PubMed] [Google Scholar]
- 50.Wei D., Huang Y., Wang B., Ma L., Karges J., Xiao H. Photo-reduction with NIR light of nucleus-targeting Pt(IV) nanoparticles for combined tumor-targeted chemotherapy and photodynamic immunotherapy. Angew. Chem., Int. Ed. Engl. 2022;61 doi: 10.1002/anie.202201486. [DOI] [PubMed] [Google Scholar]
- 51.Wang L., Yu Y., Wei D., Zhang L., Zhang X., Zhang G., Ding D., Xiao H., Zhang D. A systematic strategy of combinational blow for overcoming cascade drug resistance via NIR-light-triggered hyperthermia. Adv. Mater. 2021;33 doi: 10.1002/adma.202100599. [DOI] [PubMed] [Google Scholar]
- 52.Zhou W., Ma X., Wang J., Xu X., Koivisto O., Feng J., Viitala T., Zhang H. Co-delivery CPT and PTX prodrug with a photo/thermo-responsive nanoplatform for triple-negative breast cancer therapy. Smart Medicine. 2022;1 doi: 10.1002/SMMD.20220036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arora L., Pal D. Remodeling of stromal cells and immune landscape in microenvironment during tumor progression. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.596798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuang G., Zhang Q., Yu Y., Ding X., Sun W., Shen X., Zhao Y. Lyophilization-inactivated cancer cells composited Janus scaffold for tumor postoperative immuno-chemotherapy. Chem. Eng. J. 2023;455 [Google Scholar]
- 55.Chen G., Zhang H., Wang H., Wang F. Immune tolerance induced by immune-homeostatic particles. Engineered Regeneration. 2021;2:133–136. [Google Scholar]
- 56.Wang Y., Ma X., Zhou W., Liu C., Zhang H. Reregulated mitochondrial dysfunction reverses cisplatin resistance microenvironment in colorectal cancer. Smart Medicine. 2022;1 doi: 10.1002/SMMD.20220013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.