
Fig. 2 |. KRAS* synergizes with co-occurring mutations to direct metabolism.
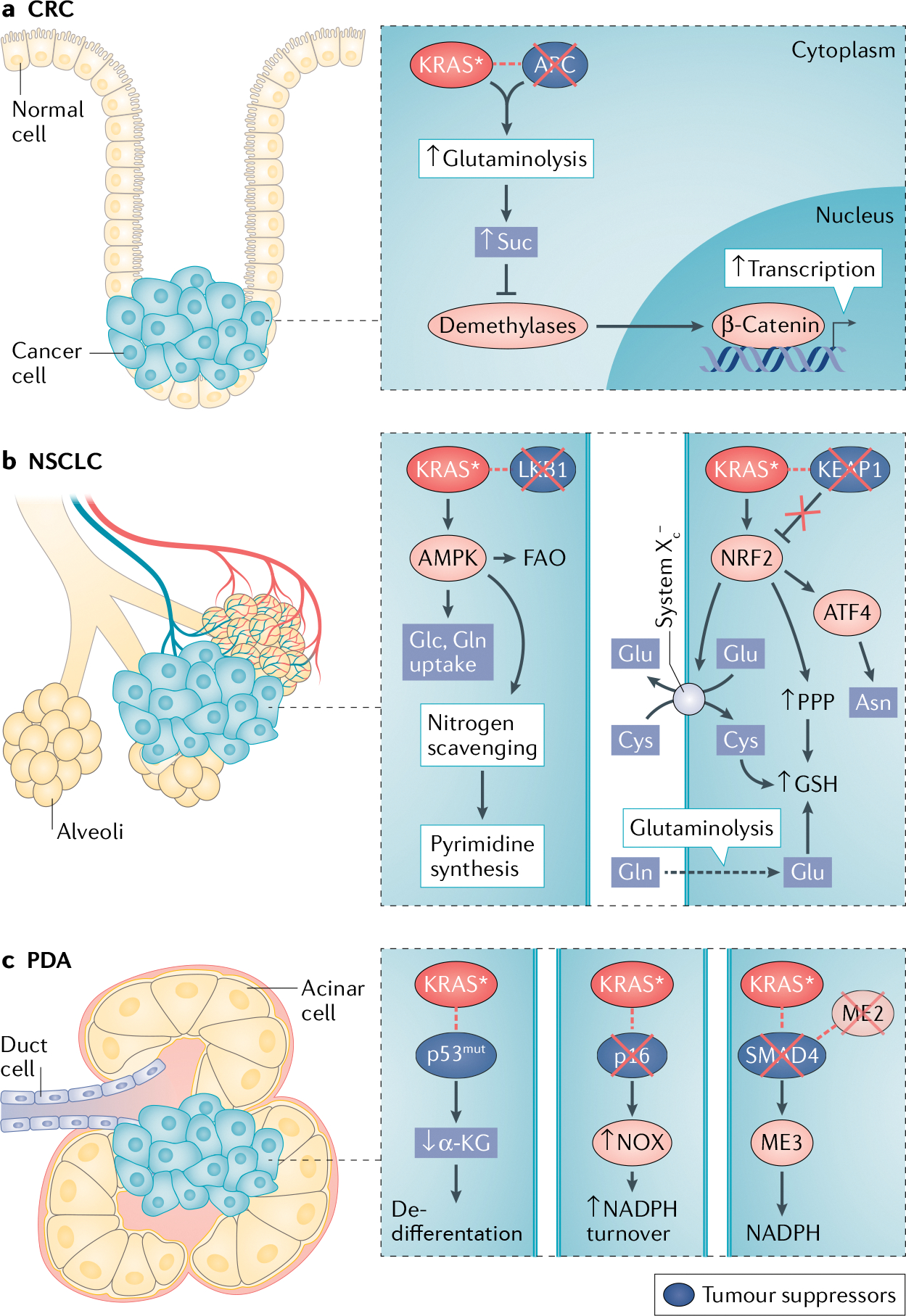
a | In colorectal carcinoma (CRC), mutant KRAS (KRAS)*-mediated glutamine (Gln) catabolism increases levels of the tricarboxylic acid cycle intermediate succinate (Suc), which inhibits the activity of α-ketoglutarate (αKG)-dependent demethylases. Hypermethylation of Wnt pathway target genes promotes their expression and cooperates with β-catenin transcriptional activity for maximal activation downstream of loss of adenomatous polyposis coli (APC). b | In non-small-cell lung cancer (NSCLC), aberrant activity of adenosine 5′-monophosphate-acivated kinase (AMPK), due to loss of liver kinase B1 (LKB1), increases fatty acid oxidation (FAO) and uptake of glucose (Glc) and glutamine (left). Scavenging of ammonia from the urea cycle supplies nitrogen needed for pyrimidine synthesis. Loss of Kelch-like ECH-associated protein 1 (KEAP1) activates the nuclear factor erythroid 2-related factor 2 (NRF2) antioxidant transcriptional programme. Increased glutamate (Glu) produced from glutamine through glutaminolysis is exchanged for extracellular cystine (cysteine dimer; Cys–Cys) through system XC−, which is also upregulated by NRF2. Cysteine is incorporated into glutathione (GSH) for protection from reactive oxygen species. NRF2 also stimulates production of GSH through the pentose phosphate pathway (PPP) and regulates asparagine (Asn) production through activating transcription 4 (ATF) activity (right). c | In pancreatic ductal adenocarcinoma (PDA), mutations of p53 (p53mut) lead to reduced αKG levels and impaired demethylase activity, affecting epigenetic programmes and cell state (left). Loss of the tumour suppressor p16 leads to increased NADH oxidase (NOX) expression and activity, which is important for nicotinamide adenine dinucleotide phosphate (NADPH) turnover and redox balance (middle). Loss of the SMAD4 genetic locus coincides with loss of malic enzyme 2 (ME2), rendering cells dependent on compensatory ME3 activity for NADPH production (right).