
Figure 3: Adenosine Metabolism and Epileptogenesis.
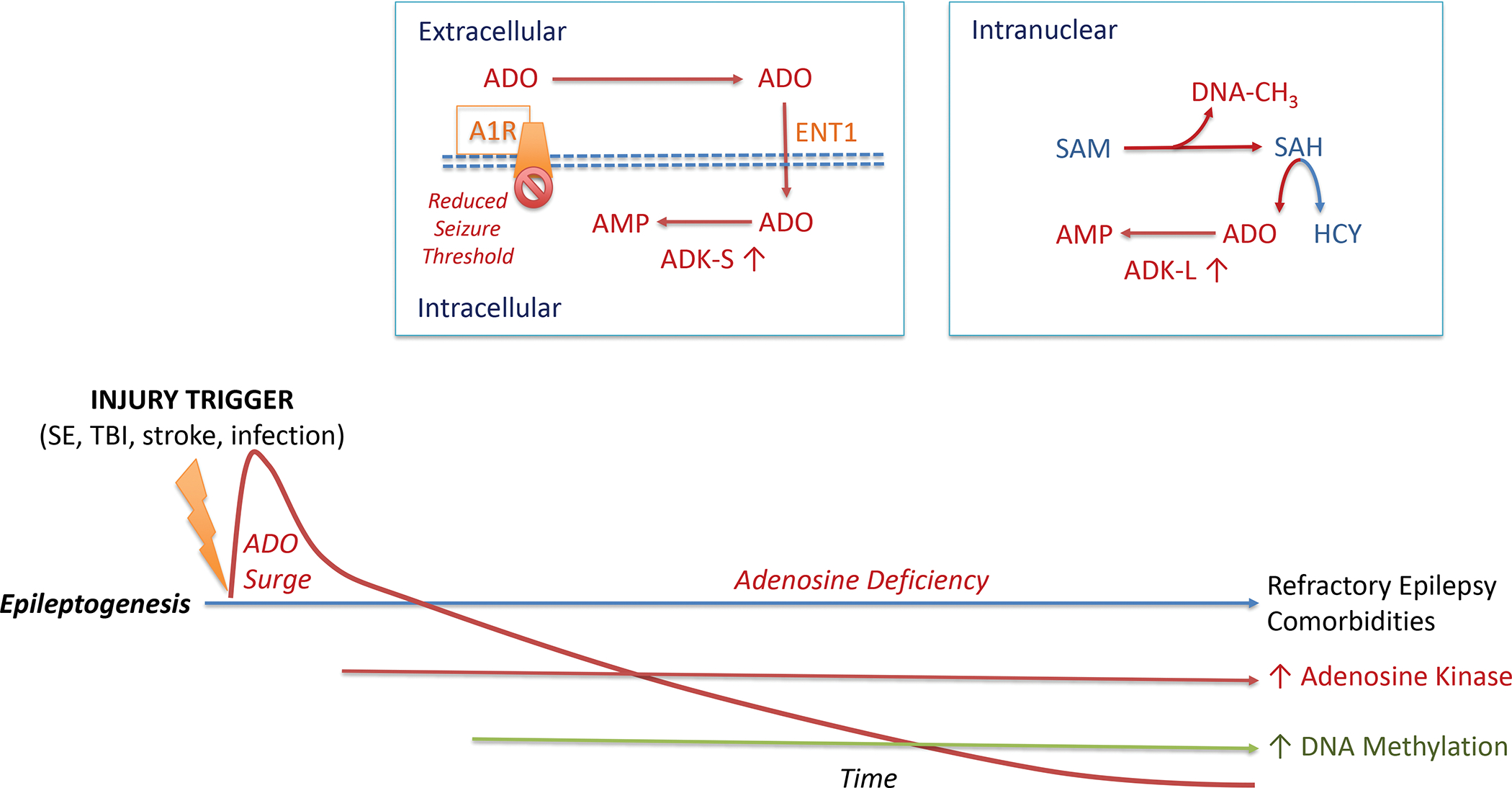
The epileptogenic process leading to acquired epilepsy can be initiated through a variety of injurious triggers including status epilepticus (SE), traumatic brain injury (TBI), stroke, or an infection of the brain. Such precipitating events lead to a biphasic response of the adenosine system: an acute neuroprotective adenosine (ADO) surge followed by progressive adenosine deficiency during epileptogenesis. Chronic adenosine deficiency is driven by maladaptive overexpression of the key adenosine metabolizing enzyme (ADK), which also drives an increase in DNA methylation, an epigenetic hallmark of acquired epilepsies. Extracellular adenosine is regulated by intracellular metabolism of the cytoplasmic isoform of adenosine kinase (ADK-S), which drives the uptake of adenosine through the equilibrative nucleoside transporter 1 (ENT1)). Metabolism-driven cellular uptake of adenosine leads to reduced adenosine A1 receptor (A1R) binding, which is a mechanistic explanation why a pathological increase in ADK-S reduces seizure thresholds. In the cell nucleus, adenosine is coupled to the S-adenosylmethionine (SAM)-dependent transmethylation pathway, which determines DNA methylation (DNA-CH3). The reaction product S-adenosylhomocysteine (SAH) is hydrolized into homocysteine (HCY) and adenosine. Metabolism of adenosine to AMP via the nuclear isoform of adenosine kinase (ADK-L) drives the flux of methylation reactions through this pathway. Thereby, increased ADK-L drives increased DNA methylation. It is important to note that this figure is not drawn to scale. The acute adenosine surge takes place during the first 24 hours after an injurious event, whereas all subsequent steps of the epileptogenic cascade occur over a time-span of days to weeks to months (or longer) after a precipitating event.