

Significance
SARS-CoV-2 infections can result in diverse clinical outcomes, including severe disease. Monoclonal antibodies (mAbs) have been used therapeutically to treat infection, but the emergence of variants has compromised their efficacy. Thus, identifying mAbs that are more durable in the face of SARS-CoV-2 evolution is a pressing need. Here, we describe four mAbs isolated from a Delta-breakthrough infection that can potently neutralize diverse variants, including multiple Omicron variants. In addition, one mAb shows broader activity against coronaviruses. The breadth of these mAbs is due to their focus on highly conserved regions of the viral protein antigen, including regions that are required for the virus to enter the cell. These properties make them promising candidates for therapeutic use.
Keywords: SARS-CoV-2, monoclonal antibodies, spike glycoprotein, variants of concern
Abstract
The antiviral benefit of antibodies can be compromised by viral escape especially for rapidly evolving viruses. Therefore, durable, effective antibodies must be both broad and potent to counter newly emerging, diverse strains. Discovery of such antibodies is critically important for SARS-CoV-2 as the global emergence of new variants of concern (VOC) has compromised the efficacy of therapeutic antibodies and vaccines. We describe a collection of broad and potent neutralizing monoclonal antibodies (mAbs) isolated from an individual who experienced a breakthrough infection with the Delta VOC. Four mAbs potently neutralize the Wuhan-Hu-1 vaccine strain, the Delta VOC, and also retain potency against the Omicron VOCs through BA.4/BA.5 in both pseudovirus-based and authentic virus assays. Three mAbs also retain potency to recently circulating VOCs XBB.1.5 and BQ.1.1 and one also potently neutralizes SARS-CoV-1. The potency of these mAbs was greater against Omicron VOCs than all but one of the mAbs that had been approved for therapeutic applications. The mAbs target distinct epitopes on the spike glycoprotein, three in the receptor-binding domain (RBD) and one in an invariant region downstream of the RBD in subdomain 1 (SD1). The escape pathways we defined at single amino acid resolution with deep mutational scanning show they target conserved, functionally constrained regions of the glycoprotein, suggesting escape could incur a fitness cost. Overall, these mAbs are unique in their breadth across VOCs, their epitope specificity, and include a highly potent mAb targeting a rare epitope outside of the RBD in SD1.
While truly remarkable progress has been made in preventing and treating SARS-CoV-2 infections, success has been eroded by viral variation and immune escape. This is true for vaccines as well as for antibody-based therapeutic approaches to treat SARS-CoV-2 infections. Novel SARS-CoV-2 variants of concern (VOCs) harbor mutations in most of the epitopes of neutralizing monoclonal antibodies (mAbs) identified to date. Consequently, the first-generation mAbs authorized for the treatment of COVID-19 are now ineffective against circulating Omicron VOCs (1), resulting in limited choices for treating and preventing Omicron infections with antibody-based therapies. Many of these same epitopes are likely to be targets of the responses elicited by immunization because vaccine efficacy is similarly compromised against VOCs with these mutations (2–5). These issues highlight the need to identify antibodies that target conserved epitopes and show greater breadth across VOCs than those isolated to date to define the best approaches for long-term protection in the face of viral immune escape.
The main viral target of interest for vaccines and antibodies against SARS-CoV-2 is the entry glycoprotein spike, which is a trimer of heterodimers comprised of two subunits, S1 and S2, that are proteolytically cleaved at the S1/S2 boundary (6). The S1 subunit contains an N-terminal domain (NTD), a receptor-binding domain (RBD), and the C-terminal subdomain (variably called CTD and SD). The receptor-binding motif (RBM) within the RBD binds the angiotensin-converting enzyme 2 (ACE2) receptor on host cells, leading to a series of changes in the S2 subunit that drives viral–host membrane fusion (7–11). Neutralizing mAbs targeting the SARS-CoV-2 RBD have been the main focus of vaccine strategies and antibody therapies, as they collectively contribute the majority of the neutralization activity in serum from vaccinated or convalescent individuals (12–19), and many have been shown to potently block virus entry in cell culture (20–23) and prevent infection or disease in animal models (24–26). RBD-targeting mAbs have been grouped into several classes (Class 1 to 4) based on the contact residues and accessibility of the epitope described through structural studies (27) and deep mutational scanning (DMS) methods that define escape pathways (28). Although RBD epitopes are a continuum (29–31), these defined classes have been useful in predicting potential escape mutations in future variants and determining how different mAbs could act in combination to limit escape compared to single antibodies (32).
The RBD is one of the most variable domains of the spike glycoprotein, and antibody escape within RBD is a particular issue with Omicron VOCs that currently drive the pandemic. The first Omicron VOC (B.1.1.529 or BA.1) had 15 mutations in the RBD that collectively altered binding across all of the RBD mAb classes (33), resulting in reduced activity of the majority of RBD-targeting mAbs (29, 32, 34–36). Similarly, serum from individuals immunized with the first generation of the SARS-CoV-2 vaccines have reduced neutralization potency against Omicron VOCs (2–5, 37–40) as predicted given the high proportion of neutralizing activity focused on RBD epitopes. mAbs that received emergency authorization to treat infections with the ancestral Wuhan-Hu-1 (WH-1) strain and early VOCs all target variable RBD epitopes, and they are no longer effective against current VOCs (41–44). The last remaining mAb for the treatment of SARS-CoV-2 infections, LY-CoV1404 (i.e., bebtelovimab) (45), which binds proximal to the ACE2-binding site on the RBD surface (28), has broad and highly potent neutralization activity against dominant Omicron VOCs up through BA.4/BA.5 (41, 43). However, as predicted by DMS-based escape profiles (46), mutations to the K444 and G446 sites present in several recently emerged VOCs (BQ.1.1 and XBB, respectively) largely abolish its activity in pseudotyped virus assays (44, 47), and it is unlikely to be effective in individuals infected with those variants. As a result, LY-CoV1404 is also no longer authorized for treating SARS-CoV-2 infections in the United States, leaving no option for antibody-based treatments at present. Together, these findings suggest that for vaccines and antibody-based therapies to be most effective long term, they will need to target more conserved epitopes and have greater breadth than that of currently available mAbs while retaining potency. To date, mAbs that target more conserved epitopes outside of RBD with similar potency as the best RBD mAbs have not yet been identified.
In general, a single mAb is unlikely to be the optimal option for long-term treatment of SARS-CoV-2 given the demonstrated potential for immune escape. To date, combination approaches have focused on the more immunodominant types of epitope classes of RBD because these tend to be the most potent against WH-1 (32, 48). However, it is clear that a more creative strategy for combining mAbs with different pathways of escape will be critical for retaining efficacy against emerging VOCs (1–5, 37). Combining mAbs that target more diverse epitopes, specifically in regions that are conserved and functionally constrained both within and outside of the RBD, may create a more complex path to viral escape. This is true for therapeutic applications of mAbs and is also very relevant to vaccine responses, where a polyclonal response is important for avoiding continued pressure on a few epitopes that tolerate mutation–a situation that could lead to new VOCs. Thus, antibody-focused approaches to preventing SARS-CoV-2 infection and disease require a more comprehensive understanding of the spectrum of functionally constrained and conserved epitopes to spike.
In this study, we describe four mAbs with broad and potent SARS-CoV-2 neutralization activity across VOCs including recent Omicron VOCs. In addition, one of these mAbs neutralizes SARS-CoV-1. These mAbs target diverse, highly conserved epitopes: three within RBD and one in a rare immunogenic epitope in the SD1 domain of spike. Thus, these diverse spike-targeting mAbs are attractive candidates for therapeutics for the current pandemic and potentially future coronavirus outbreaks as well.
Results
Identification of Monoclonal Antibodies from an Individual (C68) with SARS-CoV-2 Delta VOC Breakthrough.
To identify antibodies against SARS-CoV-2 with breadth and potency against VOCs, we isolated spike-specific mAbs from a participant enrolled in the Hospitalized or Ambulatory Adults with Respiratory Viral Infections (HAARVI) cohort who had a breakthrough infection with the Delta VOC in July 2021, two months after completion of a two-dose regimen of the Pfizer-BioNTech mRNA vaccine. This individual was one of the first confirmed Delta-breakthrough cases enrolled into the cohort and was of interest because they experienced a heterologous antigen exposure of WH-1 spike through vaccination and Delta VOC spike through the infection. Memory B cells (CD3−, CD14−, CD16−, CD19+, IgM−, IgD−) that bound to labeled recombinant Delta spike glycoprotein or spike S2 baits (PE+ and APC+ double positive) were isolated from peripheral blood mononuclear cells (PBMCs) collected 30 days post symptom onset (dpso) and single-cell sorted into 96-well plates. An optimized pipeline for recovery of variable regions of the VH and VL chain immunoglobin genes in individual wells was used as described previously (49–53). For wells containing a productive, in-frame pair of VH and VL variable regions, we cloned gene fragments into the appropriate IgG gamma, kappa, or lambda constructs and produced full-length mAbs for functional characterization.
C68 mAbs Have Broad Recognition to SARS-CoV-2 VOC Spike Glycoproteins.
We screened the mAbs for binding to a panel of prefusion-stabilized SARS-CoV-2 spike glycoproteins using enzyme-linked immunosorbent assays (ELISA) to identify mAbs that exhibited binding breadth across SARS-CoV-2 VOCs. We identified four mAbs (C68.3, C68.13, C68.59, and C68.61) with significant breadth across the recombinant spike glycoproteins (Fig. 1A and SI Appendix, Fig. S1). The EC50s for these mAbs for the WH-1 spike were 8 to 10 ng/mL and for Delta VOC spike 10 to 17 ng/mL. The EC50s were comparable to the EC50s of commercial mAbs, previously used therapeutically, against the WH-1 spike glycoprotein (EC50s = 12 to 58 ng/mL). These four mAbs had strong binding to Omicron VOC spikes, including BA.1, BA.2, and BA.4/BA.5, with similar EC50s to WH-1 (EC50 = 7 to 37 ng/mL). Three of the four mAbs (C68.3, C68.59, and C68.61) also bound spike glycoproteins from the more recently circulating Omicron VOCs XBB and BQ.1.1 (EC50 = 11 to 30 ng/mL) (Fig. 1A and SI Appendix, Fig. S1). C68.13 did not bind the spike glycoprotein from XBB and very weakly bound BQ.1.1 spike (EC50 = 899 ng/mL). Overall, C68.59 exhibited the highest binding with EC50s ≤16 ng/mL for all of the SARS-CoV-2 spike glycoproteins tested, which was slightly better than the binding of several previously authorized therapeutic mAbs that were tested in parallel (Fig. 1A). Binding of C68 mAbs (IgGs) to WH-1 spike was also analyzed by biolayer interferometry (BLI). The IgGs exhibited extremely tight binding (SI Appendix, Fig. S2), with notably slow off rates; this made quantification of the binding kinetics unreliable but demonstrated that the antibodies formed stable complexes with the WH-1 spike.
Fig. 1.
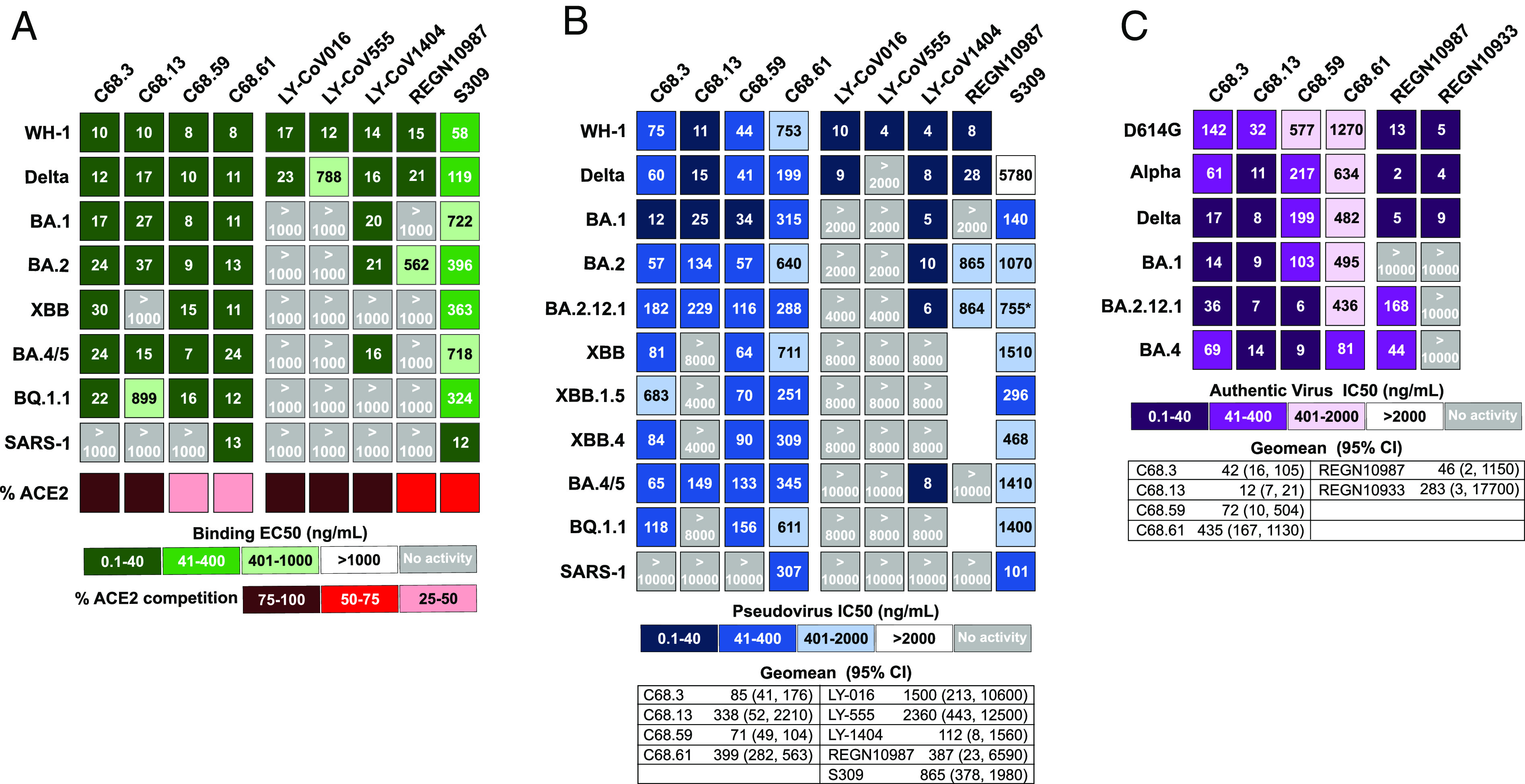
Binding and neutralization measures for four C68 SARS-CoV-2 mAbs and previously authorized therapeutic mAbs. Each column in the heatmaps represents a different antibody with the C68 mAbs on the left and the therapeutic mAbs on the right. (A) Antibody binding to recombinant spike trimers from SARS-CoV-2 VOCs and SARS-CoV-1 labeled in each row. EC50 values (ng/mL), the mAb concentration that binds the spike glycoproteins at half the maximal binding signal, were calculated by nonlinear regression analysis and are the average of two independent experiments each with technical replicates. The values for the therapeutic mAbs are the average of technical replicates in one experiment. Percent inhibition of each mAb on ACE2 binding (red) assessed by competition ELISAs averaged over two independent experiments with technical replicates. (B) Neutralization of spike-pseudotyped lentiviruses by mAbs is shown. The SARS-CoV-2 VOC or SARS-CoV-1-pseudotyped viruses are labeled in each row. IC50 values (ng/mL) were calculated by nonlinear regression analysis from the average of at least three independent experiments with technical replicates tested over two different pseudotyped virus batches. Therapeutic mAbs were tested in one to two independent experiments each with two technical replicates. Geomean (95% CI) = geometric mean of the IC50s and 95% CI across SARS-CoV-2 VOCs tested (SARS-CoV-1 not included). IC50 values greater than the highest tested concentration were set to the highest concentration for this calculation. (C) Neutralization of authentic SARS-CoV-2 viruses by mAbs is shown. IC50s (ng/mL) were calculated by nonlinear regression averaged across three to four replicates in one to two independent experiments with Geomean (95% CI) = geometric mean indicated. For the EC50s and IC50s values, the smaller the value, the darker the shade, the more activity measured. Light gray boxes denote mAbs with no measured activity in the concentration range of the assay.
In addition, we measured binding of the mAbs to the SARS-CoV-1 spike glycoprotein, which shares ~75% identity to the WH-1 spike of SARS-CoV-2, and one of the mAbs, C68.61, had similarly high binding to the SARS-CoV-1 spike (EC50 = 13 ng/mL) as to WH-1 spike (EC50 = 8 ng/mL) (Fig. 1A and SI Appendix, Fig. S1), suggesting a highly conserved epitope across the SARS coronaviruses. In this assay, binding of C68.61 was similar to that of mAb S309 (EC50 = 12 ng/mL), which was isolated from a convalescent individual post-SARS-CoV-1 infection in 2003 (21).
C68 mAbs Exhibit Broad and Potent Neutralization to SARS-CoV-2 VOCs.
The C68 mAbs were tested for neutralization potency using two assays: a spike-pseudotyped lentivirus neutralization assay (54, 55) and an authentic SARS-CoV-2 virus assay (56–58). All the four mAbs neutralized WH-1 and Delta viruses in the pseudotyped virus assay (IC50 = 11 to 753 ng/mL) (Fig. 1B and SI Appendix, Fig. S3). These mAbs showed breadth and potency to the earlier SARS-CoV-2 Omicron VOCs tested (BA.1 IC50 = 12 to 315 ng/mL; BA.2 IC50 = 57 to 640 ng/mL; BA.2.12.1 IC50 = 116 to 288 ng/mL) and retained potency to Omicron VOC BA.4/BA.5 (IC50 = 65 to 345 ng/mL) (Fig. 1B and SI Appendix, Fig. S3). When we assessed neutralization of pseudotyped viruses with spikes from more recently circulating Omicron VOCs, XBB and subvariants XBB.1.5 and XBB.4, and BA.5 lineage subvariant BQ.1.1, we observed continued neutralization of all variants with three of the C68 mAbs (C68.3, C68.59, and C68.61). C68.13 was no longer able to neutralize XBB subvariants and BQ.1.1, as expected based on reduced binding to XBB and BQ.1.1 spike glycoproteins as measured by ELISA (Fig.1A and SI Appendix, Fig. S1). C68.59 and C68.61 maintained consistent potency across all VOCs tested (Fig. 1B and SI Appendix, Fig. S3). There was reduced potency for C68.3 against XBB.1.5 (~5 to 10-fold reduction), but the mAb continued to neutralize this VOC (IC50 = 683 ng/mL) and potently neutralized all other VOCs (IC50 = 12 to 182 ng/mL). To quantify the overall potency of the mAbs against SARS-CoV-2, we calculated the geometric mean IC50 (GM) across all SARS-CoV-2 pseudotyped viruses tested. C68.59 had the highest overall potency [GM (95% CI) = 71 (49, 104) ng/mL], followed by C68.3 [GM (95% CI) = 85 (41, 176) ng/mL], and C68.61 [GM (95% CI) = 399 (282, 563) ng/mL]. For C68.13, we set the IC50 for those viruses with no neutralization activity to the highest mAb concentration tested. Due to its high potency against the other VOCs, the GM was 338 ng/mL; however, its lack of breadth against the dominant Omicron VOCs makes it a poor candidate for use against current variants.
We also directly compared neutralization potency of the C68 mAbs against several mAbs that had been previously authorized for the treatment of SARS-CoV-2 infections. Against WH-1 and Delta, the most potent C68 mAbs were ~5 to 20-fold less potent than the therapeutic SARS-CoV-2 mAbs (Fig. 1B). However, while most of the therapeutic mAbs lost activity against Omicron VOCs in our assay (IC50s > 2,000 to 10,000 ng/mL), mAbs C68.3, C68.59, and C68.61 did not, and they performed much better than the SARS-CoV-1 mAb S309 (21), which had neutralization activity against all VOCs tested and was previously authorized for therapeutic use against WH-1 and early Omicron VOCs (Fig. 1B). One of the C68 mAbs, C68.61, also potently neutralized SARS-CoV-1 with an IC50 = 307 ng/mL (Fig. 1B and SI Appendix, Fig. S3). It was more potent than CR3022 (IC50 = 958 ng/mL) but less potent than S309 (IC50 = 101 ng/mL) in the same assay (SI Appendix, Fig. S3), both mAbs were isolated from individuals with SARS-CoV-1 infections (21, 59).
Using the same spike-pseudotyped lentivirus assay, we evaluated the neutralization activity of the matched 30-dpso plasma to see whether the same breadth and potency would be observed. The plasma showed considerable neutralization potency against the original WH-1 vaccine and Delta strains (NT50, reciprocal plasma dilution resulting in 50% neutralization titer, = 2,318 and 2,613, respectively) (SI Appendix, Fig. S4), but there was reduced potency against the first Omicron VOC BA.1 (NT50 = 875), with continued erosion of neutralization potency against the later Omicron VOCs. Neutralization was weak but detectable against Omicron BA.2 (NT50 = 146) and Omicron BA.4/BA.5 (NT50 = 40) and undetectable against the XBB and BQ.1.1 VOCs tested (NT50 < 10). This individual also showed low but detectable neutralization of SARS-CoV-1 (NT50 = 152) (SI Appendix, Fig. S4). Overall, while there was some breadth and potency in the plasma from the same collection timepoint as the PBMCs used to isolate the mAbs described above, the plasma activity was not at a level where we could have predicted the magnitude of the breadth and activity we observed for this small subset of mAbs isolated from memory B cells.
We next examined the activity of these broad and potent C68 mAbs in an authentic virus neutralization assay (56–58). C68.3, C68.13, C68.59, and C68.61 all showed broad activity against the ancestral (WH-1+D614G) virus, Delta, and Omicron BA.1, BA.4, and BA.2.12.1 VOCs, as well as the Alpha VOC (Fig. 1C and SI Appendix, Fig. S5). These mAbs showed greater breadth than the two previously approved therapeutic mAbs (REGN10933 and REGN10987) tested in parallel for comparison. The most broadly potent mAb against this set of viruses, C68.13, had IC50s ranging from 7 to 14 ng/mL against the VOCs tested, which is comparable to the IC50 values of therapeutic mAbs against the WH-1+D614G virus in this same assay (IC50 = 5 to 13 ng/mL). C68.59 performed better against the more evolved Omicron VOCs, BA.4 and BA.2.12.1, compared to Omicron BA.1 and other earlier variants, with IC50s <10 ng/mL. We again calculated the geometric mean of the IC50s (GM) across the authentic SARS-CoV-2 viruses to evaluate the overall potency of these broad mAbs. C68.13 was the most potent across this panel of viruses [GM (95%CI) = 12 (7 to 21) ng/mL], followed by C68.3 [GM (95% CI) = 42 (16 to 105) ng/mL], C68.59 [GM (95% CI) = 72 (10 to 504) ng/mL], and C68.61 [GM (95% CI) = 435 (167 to 1130) ng/mL]. Notably, these mAbs were not tested against authentic viruses of XBB or BQ.1.1 variants, and based on our other data, C68.13 would likely no longer neutralize those viruses. The results of the authentic virus assay and the pseudotyped virus assay were correlated, but there were some cases where virus–mAb pairs showed several fold differences and this was true in both directions. Of note, C68.59 showed ~15 to 20-fold better potency in the authentic virus assay against Omicron BA.4 and BA.2.12.1, but it was less potent against the other VOCs compared to the pseudotyped virus assay. Overall, in both assays, neutralization activity was retained by three of the C68 mAbs against all VOCs tested.
Broadly Neutralizing C68 mAbs Target Diverse Epitopes in Spike Glycoprotein.
To identify the regions of the spike glycoprotein where the C68 mAbs bound, we tested for binding to various recombinant spike subdomain proteins. Three of the mAbs bound to WH-1 RBD (SI Appendix, Fig. S6A), whereas C68.59 did not bind to any of the tested subdomains (RBD, NTD, and S2) even at mAb concentrations of 50 μg/mL (SI Appendix, Fig. S6B). To further refine the epitopes of the three mAbs that bound to RBD, we performed competition ELISAs using several commercial mAbs representing the RBD classes that have been defined for RBD antibodies on the basis of structural analyses (27) and DMS (28). These mAbs included the following: LY-CoV016 (Class 1), LY-CoV555 (Class 2), S309 (Class 3), REGN10987 (Class 3), LY-CoV1404 (Class 2/3), CR3022 (Class 4). C68.3 competed most strongly with LY-CoV555, suggesting that it is a Class 2 RBD mAb, and C68.13 competed with LY-CoV1404 and S309 straddling the Class 2/3 classes, similar to LY-CoV1404 (SI Appendix, Fig. S7A). In the case of C68.61, we observed competition with the Class 3 mAb S309 but only in one direction when S309 was the blocking mAb but not when C68.61 was blocking (SI Appendix, Fig. S7B). These results suggest that C68.61 has a partially but not completely overlapping epitope with S309 within the RBD core region. Overall, these results suggest that this individual produced broad and potent antibodies to various RBD epitopes.
We also performed competition ELISAs to determine whether the C68 mAbs interfere with ACE2 binding (Fig. 1A and SI Appendix, Fig. S7C). C68.3 and C68.13 blocked ACE2 binding over 95% at concentrations less than 1 μg/mL on par with what was seen for LY-CoV-1404. C68.61 weakly blocked ACE2 binding, comparable to CR3022 and S309, which are known to bind RBD distant from the ACE2-binding site (21, 60). Finally, C68.59 interfered with ACE2 binding moderately despite not directly binding to RBD. From these data, we can surmise that C68.3 and C68.13 have epitopes that overlap with the ACE2-binding region, whereas C68.61 likely binds in the RBD core. The mechanism by which the non-RBD mAb C68.59 interferes with ACE2 is likely due to allosteric changes or spike destabilization.
C68.3, C68.13, and C68.61 Target Constrained RBD Epitopes with Distinct Escape Pathways.
To further identify the epitopes for the RBD-binding mAbs (C68.3, C68.13, C68.61) and simultaneously map how single amino acid mutations would affect the binding of the mAbs to RBD, we performed DMS using a yeast display library of mutant RBDs as previously described (61). With this FACS-based assay, we quantified the fraction of yeast cells harboring RBD mutations that drive mAb binding escape (gating scheme shown in SI Appendix, Fig. S8A). This system has been used to map the epitope and escape pathways of numerous RBD-specific mAbs (28, 31, 35, 62, 63), including those used previously as therapeutics (31, 32, 46, 48, 64). Using a WH-1 RBD background, we observed that C68.3 had a very focused epitope centered around residues A475 and G476 (Fig. 2 A, Top), which are in the RBM or the ACE2-contact region of RBD as shown by the red shaded areas on the space-filled RBD structure (Fig. 2 B, Top). These sites have been conserved across the major circulating VOCs over time including recent Omicron VOCs BQ.1.1 and XBB.1.5. (Fig. 2C). There was some binding escape observed in the G485/F486 patch as well, although selection for escape was modest, particularly compared to position A475. We also mapped the escape profile of C68.3 in two Omicron libraries, BA.1 and BA.2, to compare how the mutations effect C68.3 binding in the context of different VOC backgrounds. The conserved residues of A475 and G476 were again key binding sites in the epitope, as evidenced by the strong selection for escape particularly in A475 (Fig. 2 A, Middle and Bottom). However, we observed additional regions with escape in the Omicron backgrounds including F456, Y473, and increased escape mutations in the 485/486/487 region (Fig. 2 A and B, Middle and Bottom). The broadening of the escape pathways in pre-Omicron-elicited antibody responses with Omicron VOCs compared to WH-1 has been observed in other studies (46, 65) and could provide additional flexibility for escape.
Fig. 2.

Profiles of mutations that escape C68.3 binding in three SARS-CoV-2 RBD libraries. (A) Line plots (Left) identify sites of binding escape (quantified as the sum of the escape fractions) at each site in the Wuhan-Hu-1 (Top), Omicron BA.1 (Middle), Omicron BA.2 (Bottom) RBDs. Escape fractions were averaged across two replicate experiments performed in independently generated libraries. Sites that have strong escape mutations (high escape fractions) in at least one of the three backgrounds are marked with pink boxes under the plots and are represented in the logo plots (Right). The logo plots show the mutations that confer escape at each of these sites where the size of the amino acid letters is scaled according to the contribution to the overall escape fraction, and the color of the mutations indicates the effect of that mutation on ACE2 binding in the specified background determined from previously published data (46, 61). A yellow mutation signifies a deleterious effect that decreases ACE2-binding affinity and dark red signifies no effect of that mutation on ACE2 binding compared to the wild-type amino acid. (B) Sites of escape for C68.3 mapped on the RBD structure (space-filled) bound to ACE2 (ribbons). The intensity of the red coloring is scaled according to the magnitude of the mutational escape fraction at each residue, with white representing no change in binding between the wild-type and mutant amino acids at that site. Sites with the highest mutation escape fractions (darkest red) indicate key binding residues in the mAb epitope. (C) Sequence alignment of a region within RBD (sites 450 to 490) across SARS-CoV-2 VOCs and SARS-CoV-1. Residues are colored based on percentage similarity at each site across the listed sequences, with darker color indicating residues invariant across the sequences. Sites with the highest escape fractions for C68.3, indicative of the binding residues across the three backgrounds, are marked with yellow arrows and boxes.
Next, we investigated the specific amino acid changes at each of these key sites to predict the likelihood of escape from C68.3 binding. Also, to evaluate whether the escape mutations we observed in the C68.3 epitope would be predicted to impact ACE2 binding and incur a fitness cost, we coupled our data with previously published DMS profiles of the effect of mutations on ACE2 binding to RBD using the same yeast display assay (46, 61). In the case of C68.3, most of the escape mutations identified also suffer severe deficits in ACE2 binding of an order of magnitude or more (shown by the scaled color in the logo plots in Fig. 2A), particularly at the key binding residues A475/G476 and F456, Y473, G485, and N487. These sites have remained conserved across the major VOCs (Fig. 2C). Mutations have become more prevalent at the F486 residue in recently circulating VOCs including Omicron BA.2.75.2 and XBB, which have a F486S mutation, and Omicron BA.5 and BQ.1.1, which have a F486V mutation (Fig. 2C). These specific amino acid changes were not observed to drive binding escape for C68.3 in the DMS profiles (Fig. 2A), which is consistent with the continued, potent neutralization of XBB and BQ.1.1 pseudotyped viruses. One of the newest, dominant circulating VOCs XBB.1.5 has selected for an additional mutation at 486 going from a serine in the XBB parental sequence to a proline. The proline at site 486 was enriched in the DMS profiling of C68.3, suggesting that mutation led to reduced binding (Fig. 2A). S486P resulted in a ~four-fold affinity loss for ACE2, and it was the most permissive mutation selected for escape in terms of altering ACE2 binding. Accordingly, when we evaluated the neutralization activity of C68.3 against XBB.1.5 spike–pseudotyped virus, activity was retained but reduced ~5 to 10-fold compared to the other pseudotyped viruses. Overall, these data indicate that C68.3 has largely focused its epitope onto highly constrained residues within the ACE2-binding surface that would be predicted to reduce viral entry if mutated (62).
We mapped C68.61-binding residues in the Omicron BA.2 yeast display library background and observed localized escape at sites K462, E465, R466, and I468 (Fig. 3A). This epitope is distant from the ACE2-binding region in the RBD core (Fig. 3B) and is highly conserved across major SARS-CoV-2 VOCs and SARS-CoV-1 (Fig. 3C). The amino acid at site K462 is not conserved between SARS-CoV-2 and SARS-CoV-1 (K to R, respectively). However, an R was not expected to drive escape based on the mapping profile (Fig. 3A; logo plot), which is consistent with the potent neutralization of SARS-CoV-1 pseudotyped virus by C68.61 (Fig. 1B and SI Appendix, Fig. S3). Although the escape mutations we identified are not constrained with respect to mutational effects on RBD affinity for ACE2, as shown by the dark red shading of the mutations in the logo plot (Fig. 3A), they are packed at a quaternary interface in closed spike trimers that likely constrain mutations over viral evolution. Therefore, mAb C68.61 could represent a broad and potent mAb for SARS-CoV-2 VOCs and other sarbecoviruses.
Fig. 3.

Profiles of RBD mutations that escape C68.61 in Omicron BA.2 RBD. (A) Line plot (Left) of the summed escape fraction for mutations across the Omicron BA.2 RBD sites. Escape fractions were averaged across two replicate experiments performed in independently generated libraries. The individual mutants that confer binding escape from C68.61 at those key sites are shown in the logo plots (Right) where the color of the mutations indicates the effect of that mutation on ACE2 binding (46, 61). (B) Sites of escape for C68.61 are mapped onto the RBD structure (space-filled) bound to ACE2 (ribbons). The intensity of the red coloring is scaled according to the mutational escape fraction at each residue. (C) Sequence alignment of region of RBD (WH-1 sites 450-490) across SARS-CoV-2 VOCs and SARS-CoV-1. Dark blue shows sites invariant across the sequences. Sites with the highest escape fractions for C68.61 are marked with yellow arrows and boxes. The details for this figure are as described in Fig. 2.
Finally, for C68.13, the mAb only bound to the yeast display library in the WH-1 background despite strong binding of this mAb to various recombinant spike VOC glycoproteins in an ELISA (Fig.1A and SI Appendix, Fig. S1). With DMS in the WH-1 background, we were able to broadly localize the epitope to a region within a glycan ridge in RBD throughout the residues located in WH-1 (339 to 346), which have an N-glycosylation site at N343, and residues 437 to 444 (SI Appendix, Fig. S9). However, because of the wide escape profile, we were not able to precisely identify escape mutations. These wide escape profiles are indicative of weak binding of the mAb to the yeast-displayed RBDs, which could be due to differences in yeast and mammalian glycans, as noted previously (31, 62). The C68.13 epitope defined by DMS was nonetheless consistent with the results of competitive binding to other RBD mAbs described above (SI Appendix, Fig. S7A), which suggested overlap between C68.13 and class 2/3 mAbs.
C68.59 Targets a Rare, Conserved Epitope Downstream of RBD in SD1.
To elucidate the epitope of C68.59, which does not bind RBD and thus is not a candidate for the yeast RBD display system, we used a pseudotyped lentivirus DMS system with a library covering functionally tolerated mutations across the entire spike glycoprotein (66). In this assay, escape is measured by comparing the mutant pseudotyped viruses that infect in the presence versus absence of the antibody. Using the SARS-CoV-2 Omicron BA.1 spike–pseudotyped lentivirus system, we mapped escape mutants for C68.59 to five key residues (in WH-1 numbering): E554, K558, R577, E583, and L585 (Fig. 4A and interactive plot at https://dms-vep.github.io/SARS-CoV-2_Omicron_BA.1_spike_DMS_C68.59/C68.59_escape_plot.html). These sites cluster in the region just downstream of RBD within the SD1 domain, also referred to as the CTD region (Fig. 4B), and they are invariant across all VOCs in our comparison and the SD1 domain overall is highly conserved (Fig. 4C). The observation that C68.59 targets an invariant epitope across VOCs is consistent with the broad SARS-CoV-2 neutralization we observed in both the pseudotyped and authentic virus neutralization assays (Fig. 1 B and C). Notably, we did not observe neutralization of SARS-CoV-1 by C68.59 (Fig. 1B and SI Appendix, Fig. S3), which differs at two of the predicted binding residues; E554 and K558 in WH-1 sequence are P and R, respectively, in SARS-CoV-1.
Fig. 4.
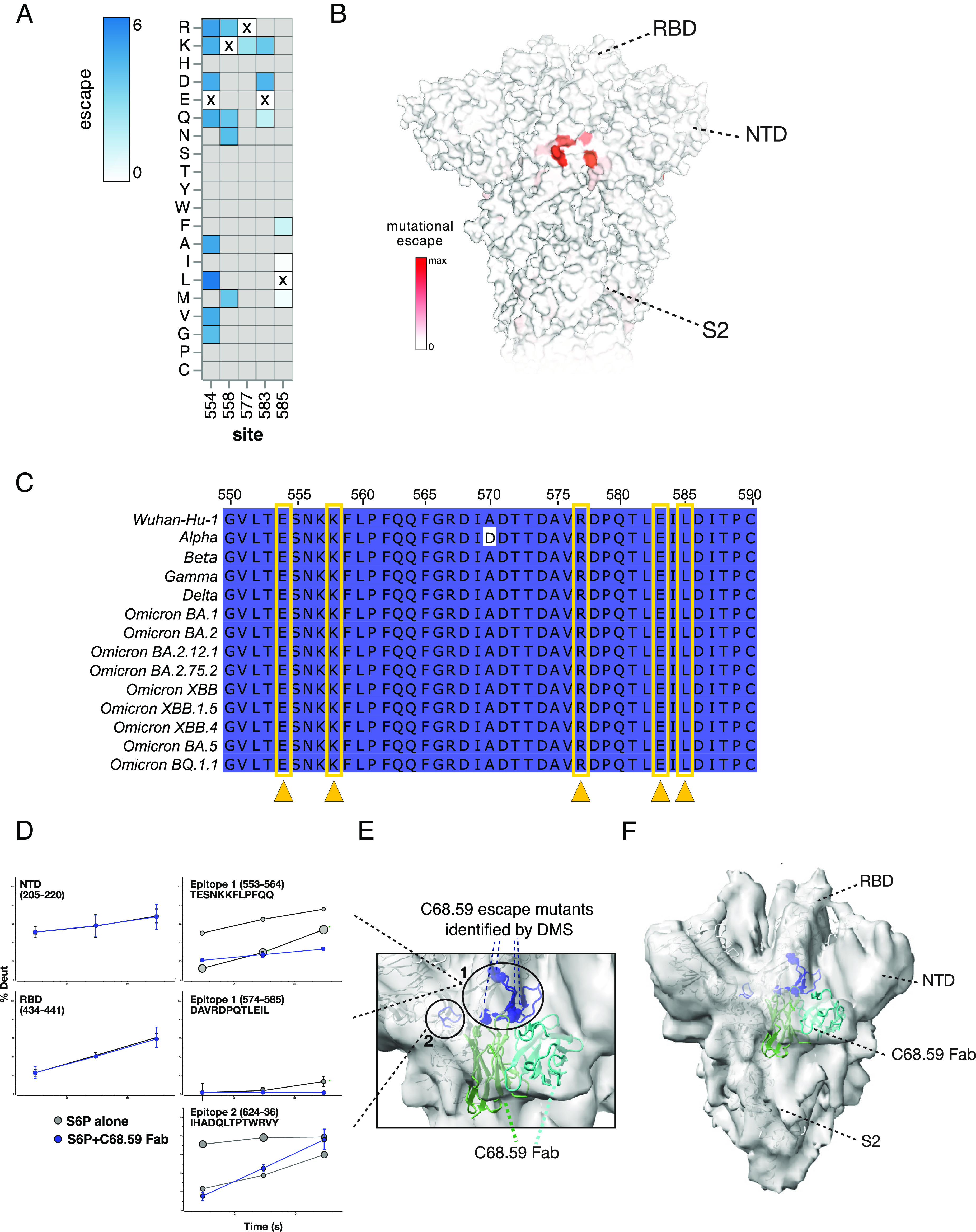
Mapping C68.59 epitope and neutralization escape mutations in SD1 by DMS in an Omicron BA.2 background and structural methods: HDX-MS and cryo-EM. (A) Heatmap showing the magnitude of spike-pseudotyped lentivirus neutralization escape at key sites (measured by mutation escape scores) for the mutations tested. Darker blue = more escape. Residues marked with an X are the wild-type amino acid at that site in Omicron BA.2. Gray mutations were not tested in the library. Escape scores are averaged across two replicate experiments in independently generated libraries. (B) Surface representation of spike colored by the mean escape score per site, with darker red indicating greater escape. PDB ID: 68R8 (C) Sequence alignment of region of SD1 (WH-1 sites 550 to 590) between SARS-CoV-2 VOCs, with darker color indicating residues invariant across the sequences. The key sites of escape for C68.59 are marked (yellow arrows/boxes). (D–F) Structural mapping of the C68.59-binding epitope by HDX-MS and cryo-EM. (D) In response to C68.59 Fab binding, regions including the NTD and RBD do not change in their local structural ordering as reported by HDX-MS, demonstrating that those sites are not targeted by the antibody. By contrast, two adjacent sites consisting of peptide segments 553 to 564, 574 to 585, and 624 to 636 show dramatic increase in local ordering and quenching of conformational sampling when C68.59 Fab is bound to the S6P trimeric spike. Bubble plots show the uptake rates of the two distinct populations at the unbound epitope sites (gray), with the size of the bubble indicating the population fraction. (E and F) Cryo-EM reconstruction of C68.59 Fab bound to S6P structure (PDB ID 7SBP) where C68.59 Fab structure is calculated by AlphaFold fitted into map. The zoomed view of the epitope (E) and the overall architecture of the C68.59 Fab bound to S6P spike (F) are shown. Experimental electron density shown in gray, S6P structural model shown in white, C59.68 Fab shown in two shades of green to distinguish the heavy and light chains. Binding-site peptides showing protection by HDX-MS (residues 553 to 568, 575 to 585, 624 to 636) are colored blue in the cartoon structure. Residues exhibiting escape mutations by DMS are circled.
To further examine the epitope targeted by C68.59, we carried out two complementary structural methods: hydrogen deuterium exchange-mass spectrometry (HDX-MS) and cryogenic electron microscopy (cryo-EM) structure determination. In both cases, the C68.59 Fab was bound to stabilized WH-1 spike containing the “HexaPro” mutations (S6P). HDX-MS identifies the binding interface for protein–ligand interactions as well as any resulting allosteric conformational changes induced by antibody Fab binding by mapping changes in backbone amide solvent exchange under native solution conditions. HDX-MS analysis of C69.59 identified two sites that had increases in structural ordering with Fab bound, consistent with changes one would expect for antibody binding to an epitope footprint (Fig. 4D). The first site spanned residues 553 to 564 and 574 to 585, including residues associated with escape from neutralization identified by DMS (Fig. 4E). Additionally, a second site exhibited significant increases in protection consistent with involvement in a binding interface. This site was adjacent in the three-dimensional structure to the first site and spanned residues 624 to 636 (Fig. 4 D and E). Intriguingly, in the unliganded trimer spike, both sites exhibit bimodal exchange kinetics (shown as “bubble plots” in Fig. 4D, with the size of the bubble scaled for the amount of that state), indicating that the region samples multiple structural states. Upon Fab binding, these two states collapsed into a single heavily protected state, suggesting that the antibody quenched local conformational sampling at those sites. In addition to these binding footprints, HDX-MS also revealed a dramatic destabilization of significant portions of S2 (SI Appendix, Fig. S10).
Single-particle cryo-EM analysis of C68.59 Fab–bound spike trimers allowed determination of a moderate-resolution reconstruction (6.7 Å) revealing Fab docking to both sites identified by HDX-MS and consistent with the DMS data (Fig. 4 E and F). The local resolution of the reconstructed map varied across subdomains of spike (6.7 to 10 Å), with α-helices clearly resolved in the central S6P core, and lower resolution distally in RBD, NTD, and S2 domains as well as the Fab density (Fig. 4F). The Fab density clearly bridges both sites identified by HDX-MS and DMS, being centered over the site identified by DMS and HDX-MS (residues 553 to 568 and 575 to 585) and contacting the secondary site only identified by HDX-MS (residues 624 to 636) (Fig. 4E).
To sort out heterogeneity in conformation and occupancy, 3D-classification was performed revealing two additional major classes of particles (SI Appendix, Fig. S11 and Table S1). A subset of the particles did not contain Fab density, allowing reconstruction of unbound spike (6.7 Å). In comparison to the Fab-bound class, the unbound spike exhibited higher local resolution throughout the reconstructed map, including in the S2 subunit and RBD. These data support the HDX-MS results that indicate C68.59 increases dynamics and conformational heterogeneity in these regions. Finally, the third class exhibited extensive disruption of the native spike structure, particularly around the S2 subunit and RBDs. This class likely represents a range of conformations and was consequently reconstructed to a lower resolution than that of the other classes (9.2 to 12 Å). Together, these data localize the C68.59 epitope to the SD1 region of the spike glycoprotein, consistent with DMS profiling, and suggest dramatic allosteric changes to spike after binding of the mAb.
Discussion
In this study, we identified four mAbs with broad and potent neutralization activity to SARS-CoV-2 VOCs from a breakthrough infection case. Three of these mAbs target different regions of RBD and one mAb targets an epitope outside of the RBD in SD1. Their epitopes are in highly conserved and/or functionally constrained regions of the spike glycoprotein, suggesting that escape from these antibodies could incur a fitness cost to the virus. Not only do these mAbs show breadth across SARS-CoV-2 VOCs, including potency to recent widely circulating VOCs BQ.1.1 and XBB.1.5 for three of the mAbs, one also potently neutralizes SARS-CoV-1 at levels comparable to antibodies isolated from cases of SARS-CoV-1 infection. Collectively, this study gives further evidence that some individuals can elicit broad and potent antibodies to SARS-CoV-2, even against VOCs not yet circulating.
These mAbs were elicited after primary vaccination with the two-shot WH-1 mRNA vaccine followed by a breakthrough infection with a Delta VOC. Even though this individual had not yet been exposed to Omicron VOCs, the post-Delta infection mAbs were able to potently neutralize Omicron VOCs, including recently circulating BA.4/BA.5, BQ.1.1, and XBB.1.5. The elicitation of broadly neutralizing antibodies to Omicron variants prior to Omicron emergence has also been described in both plasma responses (67–69) and for mAbs, most notably in the case of LY-CoV1404 (45). Similarly, despite no known previous exposure to SARS-CoV-1, C68 developed an mAb that could neutralize SARS-CoV-1 as potently as it neutralized the SARS-CoV-2 vaccine and breakthrough strains, as previously observed (69). Multiple antigen exposures, whether by repeated vaccination doses or vaccination plus infection, have been shown to boost serum neutralization potency across SARS-CoV-2 VOCs (16, 69–72). Heterologous antigen exposures with different spikes, as was the case for C68, seem to contribute to broader, more potent antibody responses as compared to the responses following homologous exposures (68, 72–74). What remains to be understood is the ideal combination of spikes and the sequencing of those exposures to elicit optimal responses especially in the face of continued viral evolution. Understanding the factors that can drive this kind of broad and potent response in individuals could be important for developing superior vaccine strategies.
Despite the high mutational frequency that has been observed in the RBD region of spike across VOCs, especially in the RBM, we have identified RBD-targeting mAbs that maintain high neutralization potency in both pseudotyped virus and authentic virus assays with multiple VOCs, including recent Omicron VOCs. The previously authorized therapeutic mAbs, all of which target RBD, have reduced or completely abolished neutralization activity against novel variants (29, 32, 34–36), a finding further verified in this study when these mAbs were tested in parallel to the C68 mAbs. LY-CoV1404 (bebtelovimab) (45), which had retained potent neutralization activity through Omicron BA.4/BA.5, BA.2.12.1, and BA.2.75.2, is now no longer authorized for the treatment of SARS-CoV-2 infections in the United States because of loss of activity against BQ.1.1 and XBB sublineages (44, 47), which are as of this writing the dominant strains. These two VOCs encode critical mutations within the LY-CoV1404 epitope, including a K444T mutation in BQ.1 and BQ.1.1, and a G446S in XBB, both of which were identified as escape mutations by DMS (46). Overall, escape profiles defined by DMS have predicted instances where the mutations that emerged in VOCs, especially mutations in the RBM, resulted in loss of activity for all authorized therapeutic mAbs (28, 31, 32, 46, 48, 64).
DMS-based mapping and escape profiles of the C68 mAbs suggest that their epitopes are in functionally constrained regions of the spike glycoprotein. C68.3, one of the potent mAbs described here, has a focused epitope in the RBM, but most of the mutations that result in binding escape also exact a functional cost on ACE2 binding. Recent Omicron VOCs have focused selection of mutations to the F486 residue, which is a potential site of escape for C68.3. Site 486 is an ACE2-binding residue as well and many of the possible mutations at this site would reduce ACE2 affinity, likely resulting in a fitness cost (61). The most recent dominating Omicron VOC in the United States at the time of this writing is XBB.1.5, which harbors a F486P mutation, and a proline was identified as a potential escape mutation for C68.3 by DMS. As predicted, XBB.1.5 was able to partially escape neutralization by C68.3 in a pseudotyped virus assay (~5 to 10-fold reduced mAb potency). However, activity was not completely lost, indicating that total escape might require mutations at multiple sites or mutations that would result in a significant fitness cost to the virus.
The cross-sarbecovirus mAb C68.61 binds to the RBD core, which is highly conserved across SARS-CoV-2 VOCs and SARS-CoV-1. The key binding sites we identified by DMS are closely located at a quaternary interface in closed spike trimers, which could constrain mutations in this region over viral evolution. Studies have shown that mAbs that bind the RBD core tend to have more sarbecovirus breadth but at the expense of neutralization potency compared to RBM mAbs (21, 31, 75), with S309 being a notable exception (21). Here, we show that C68.61 demonstrates breadth across SARS-CoV-2 VOCs and SARS-CoV-1, while maintaining a relatively high potency compared to other RBD mAbs in the same class including S309 and CR3022 when compared head to head in the same assay. Another recently described mAb, S2H97, which binds to the same surface of RBD as C68.61, demonstrated broad binding across the entire breadth of bat SARS–related coronaviruses and protects hamsters from SARS-CoV-2 challenge (31). C68.61 had comparable breadth and neutralizing potency as S2H97 against WH-1 pseudovirus and had no apparent drop in potency over the VOCs or SARS-CoV-1, making it an attractive mAb for development as a pan-coronavirus therapy to prepare for future coronavirus pandemics.
The third RBD mAb we describe C68.13 also appears to target an epitope that could be critical for viral entry. DMS profiling, while low resolution for this mAb, localizes the epitope of C68.13 to the “glycan ridge” region of RBD that contains the N343 N-glycosylation site. This N-glycan has been proposed to be involved in RBD opening to the up position (76) and is essential for SARS-CoV-2 entry into target cells (77), thereby driving some functional constraint at this epitope. However, we observed complete escape from C68.13 with BQ.1.1 and the XBB subvariants. Similar to LY-CoV1404, K444T appears to be a part in the C68.13 epitope, which is a key mutation in BQ.1.1. In the XBB lineage, some potential mutations driving C68.13 escape include N440K and R346T, the latter of which is also seen in BQ.1.1. Despite the high potency and breadth of C68.13 against early VOCs, including Omicron VOCs BA.4/BA.5, the loss of activity against XBB and BQ.1.1 highlights the continued ability of SARS-CoV-2 to evolve and escape antibody responses and the need for mAbs targeting functionally constrained regions.
In this study, we also describe a unique mAb, C68.59, that binds to a rare epitope in the SD1 region downstream of RBD that has high neutralization potency and breadth across SARS-CoV-2 VOCs. RBD-targeting mAbs comprise the majority of the neutralization activity in vaccinated and convalescent serum (12–19), and while some other neutralizing epitopes of spike have been described (13, 15, 17, 19, 58, 78–84), usually the potency of mAbs targeting these other epitopes pales in comparison to RBD mAbs. Compared to neutralizing mAbs that target the S2 region outside of RBD, C68.59 is anywhere from ~10 to 1000-fold more potent (82–85). In authentic virus neutralization assays, C68.59 was ~10-fold less potent than the previously authorized therapeutic RBD mAbs against WH-1 or Delta when run head to head, but it was more potent against Omicron VOCs BA.4 and BA.2.12.1, with IC50s comparable to the first-generation therapeutic mAbs against the original WH-1 strain. Notably, C68.59 retained potency against recent, dominant VOCs BQ.1.1 and XBB.1.5 in spike-pseudotyped virus assays, which evade neutralization by the most potent and broad previous therapeutic mAb LY-CoV1404. Two studies have described mAbs that bind to the SD1 domain within spike: one study of mAbs isolated from an engineered mRNA scFv library (86) and a second study of naturally elicited mAbs from SARS-CoV-2 infection or vaccination (87). These SD1 mAbs and C68.59 have some overlap in their epitopes as they are focused on the loop 3 region of SD1 (residues 553 to 564), but they have very different neutralization potencies. The engineered mAbs neutralized weakly or did not reach 100% neutralization in an authentic virus assay (86). The most potent naturally elicited mAb, P008_60, had good breadth across VOCs including Omicron BA.1, but it neutralized ~100-fold less potently than C68.59 in a similar pseudotyped virus assay (87). These previously described SD1 mAbs were not able to bind the prefusion-stabilized versions of the spike trimer, whereas C68.59 was able to bind stabilized spike, which might account for the improved neutralization potency.
The SD1 region is in the hinge of the spike glycoprotein, located in between RBD and the S2 subunit, and has been proposed to facilitate the transition of RBD between up and down orientations (88). Structural studies of the other SD1 mAbs suggest allosteric and destabilizing effects throughout the spike trimer following mAb binding especially in the S2 subunit. Consistent with this, the HDX-MS analysis of C68.59 showed significant destabilization of S2 after binding. We speculate that in native spike that lacks the stabilizing 2P or 6P mutations and S1/S2 cleavage site knockout changes, C68.59 binding could induce S1 shedding due to spike destabilization. The strong neutralization potency in combination with the high degree of sequence conservation in the C68.59 epitope across SARS-CoV-2 VOCs in this region supports the further exploration of C68.59.
We acknowledge that there are several potential limitations to our study. First, we have used results from previous DMS experiments to predict fitness costs of possible escape mutations acquired in the context of antibody selection but have not reconstructed authentic viruses with individual mutations to validate the costs. Second, the current cryo-EM analysis was only moderate resolution; however, coupled with the DMS and HDX-MS data, we were able to verify the main contacts of the C68.59 epitope. Third, this study does not include in vivo studies to describe the protective efficacy of these mAbs in humans. There is growing evidence for SARS-CoV-2 that pseudotyped virus and authentic virus neutralization assays can be used as correlates of protection for mAbs in a therapeutic setting as authorization/deauthorization decisions have been largely driven by these types of data for the previously authorized mAbs. However, we acknowledge that the highest bar for evaluating protective efficacy would be in clinical trials. Finally, the mAbs we describe were isolated only 30 d after Delta infection and 90 d after the second vaccine dose, which is still relatively soon after antigen exposures. Therefore, the mAbs we describe have limited affinity maturation and more mature mAbs may exist at later timepoints, which we can explore in future studies.
Finding durable therapeutics against SARS-CoV-2 and improving vaccine efficacy are both pressing public health priorities. Not only is there an urgent and continuing need to improve and understand responses to SARS-CoV-2, but there is also a need to prepare for future coronavirus pandemics, given the emergence of SARS-CoV-1 and SARS-CoV-2 in the recent past and evidence that other SARS variants may have entered the human population as well (89). Collectively, this study gives further evidence that some individuals are able to elicit a broad, polyclonal antibody response to SARS-CoV-2, even against VOCs not yet circulating. However, our results suggest that identifying these mAbs may not always coincide with detection of remarkable plasma activity as a notably broad response was not observed in contemporaneous plasma from the individual examined here. One feature that drives the breadth of the mAbs isolated from this individual is their focus on diverse, functionally constrained regions in the Spike protein, which makes them candidates for development as combination therapeutics and suggests improved durability against future VOCs than most mAbs isolated to date.
Materials and Methods
Study Participant and Specimens.
Paired plasma and PBMCs were collected 30-days post symptom onset (pso) from SARS-CoV-2 infection from a 27-y-old individual enrolled in the Hospitalized or Ambulatory Adults with Respiratory Viral Infections (HAARVI) research study (90). Written, informed consent was obtained when the individual was enrolled in July 2021. The study was approved by Institutional Review Boards at University of Washington and Fred Hutchinson Cancer Center.
Single-Cell Sorting for Spike-Specific Memory B Cells.
Memory B cells expressing receptors encoding spike-specific antibodies were sorted from 30-day pso PBMCs using standard methods (51, 91) and see SI Appendix, Supplemental Methods. Spike-specific B cells were selected for using a “bait” approach by staining with a pool of APC/PE-labeled Delta HexaPro spike protein and the spike S2 subunit (Acro Biosystems, cat. S2N-C52E8). The gating strategy used for this B cell sort is shown in SI Appendix, Fig. S12.
Reconstruction of Antibodies.
Antibody gene sequences were recovered from the sorted B cells using established methods (51, 91) and described in SI Appendix, starting with one 96-well plate. From the first set of 96 sorted B cell wells, we identified 43 with paired, productive heavy and light chains that were able to produce antibodies in vitro. These 43 antibodies were screened for binding to SARS-CoV-2 spike glycoproteins and 37 were found to bind to WH-1 spike, and six antibodies did not bind SARS-CoV-2 spikes. Eight of the 37 spike-binding mAbs were able to neutralize WH-1 pseudotyped virus. The four mAbs described here were selected from these eight for further study based on better neutralization potency and binding breadth. The sequences for the variable regions of the heavy and light chain genes for C68.3, C68.13, C68.59, and C68.61 are shown in SI Appendix, Supplementary Methods.
Binding and Neutralization Assays.
Binding of the mAbs to recombinant, stabilized spike trimers was assessed by direct ELISA as previously described (54, 78). Competition ELISAs to map epitopes of RBD-specific mAbs or determine ACE2-binding interference were performed as previously described, with some modifications (52). Data were analyzed and plotted using GraphPad Prism (v9).
Spike-pseudotyped lentiviruses were produced and their infectious titers determined as previously described (55, 78). Codon-optimized plasmids with spike genes specific for WH-1, Delta, Omicron VOCs BA.1, BA.2, BA.4/BA.5, BA.2.12.1, XBB, XBB.1.5, XBB.4, BQ.1.1, and SARS-CoV-1 were individually transfected with lentiviral helper plasmids into HEK293T cells for virus production. Neutralization of the pseudoviruses by the plasma and mAbs was performed as previously described (54, 55). HEK293T-ACE2 cells were used as the target cells to measure infection and neutralization. Data were analyzed and plotted using GraphPad Prism (v9).
The authentic SARS-CoV-2 microneutralization assay was performed as previously described in a BSL3+ lab (56) with Vero E6-TMPRSS2 cells. Authentic SARS-CoV-2 viruses of WH-1+D614G, Alpha, Delta, BA.1, BA.4, or BA.2.12.1 virus were obtained from Laboratoire de santé publique du Québec. Data were analyzed and plotted using GraphPad Prism (v9).
Detailed experimental and analysis methods and reagent sources for all assays are shown in SI Appendix, Supplementary Methods.
DMS.
Yeast-display libraries containing virtually all single amino acid mutations in the Wuhan-Hu-1, Omicron BA.1, and Omicron BA.2 RBDs were used to identify escape mutations exactly as previously described (61). Two independent mutant libraries were generated, and each was sorted against the antibody binding and sequenced in parallel. The final escape fractions were averaged across the two replicates.
Full BA.1 spike–pseudotyped lentivirus-based DSM libraries were made as described previously (66). The libraries were designed to contain all functionally tolerated mutations at every position in spike. To identify mutations in BA.1 spike that would escape C68.59 antibody neutralization, we used the approach described in ref. 66. This experiment was run in replicate in two independently generated libraries.
The computational analysis and GitHub links are shown in SI Appendix, Supplemental Methods.
BLI.
BLI was performed using Wuhan-1 6P (S6P) (produced as described in SI Appendix, Supplemental Methods) on a ForteBio OctetRed 96 instrument in Tris-buffered saline containing 1 mg/mL BSA and 0.02% Tween 20. Anti-human Fc tips were hydrated and loaded with IgG at 5 µg/mL, then dipped into serial dilutions of spike. Data were analyzed using ForteBio Data Analysis software.
Hydrogen Deuterium Exchange-Mass Spectrometry (HDX-MS).
Equal volumes of S6P spike glycoprotein (~9 μM) and C68.59 Fab (~18 μM) or HBS were mixed and incubated at room temperature for 1 h prior to exchanges. Fourteen microliters of protein was diluted with 86 µL deuterated HBS and incubated at room temperature (~22 °C) for the indicated time. The reaction was quenched by mixing with 100 µL cold 0.1M KH2PO4, 0.2M tris(2-carboxyethyl)phosphine (TCEP), 4M guanidine-HCl, and 0.2% formic acid to a final pH of 2.5, vortexed, and flash-frozen in liquid nitrogen.
Frozen samples were thawed on ice and injected on a custom-built refrigerated LC system (92). The protein was passed through an immobilized Nepenthesin II column (Affipro) kept at 15 °C before trapping and separation at 1 °C on a C18 column (Waters). Mass spectrometry was performed on a Waters SYNAPT G2 with ion-mobility separation. Data were analyzed using HDExaminer (v2, Sierra Analytics).
Cryogenic-Electron Microscopy (Cryo-EM).
Cryo-EM sample preparation and data collection.
Purified S6P Spike (4.5 μM) and C59.68 Fab (17 μM) in HBS buffer were mixed and incubated for 30 min on ice prior to application of 3 uL to Quantifoil R 2/2 plunge freezing grids (EMS) that were glow discharged (negative charge) under a current of 25 mA for 30 s under vacuum. Sample was incubated on grids for 1 min prior to plunge freezing on a Vitrobot Mark IV (ThermoFisher). Plunge freezing was performed at 100% humidity, blot force 0, and 4 s blot time. Grids were imaged on a 300 kV Titan Krios (ThermoFisher) equipped with a K3 direct electron detector (Gatan) and a postspecimen energy filter. Data collection was performed using SerialEM (93). Dose-fractionated movies were collected with a total exposure of 2.397 s and 0.03 s frames, with a total electron dose of 50 electrons/Å2 at a magnification of 105 kx in superresolution mode (pixel size 0.417 Å/pixel). Movies were collected between −0.7 and −1.2 μM defocus. The Cryo-EM data processing is described in SI Appendix, Supplemental Methods. The cryo-EM structures have been deposited in the EM Data Resource (https://www.emdataresource.org) under the following accession codes: Class 1: EMDB-29053 (94), Class 2: EMDB-29054 (95), Class 3: EMDB-29052 (96).
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Marceline Côté (University of Ottawa) and Amit Sharma (Ohio State University) for generously providing spike variants of concern plasmids and the Laboratoire de santé publique du Québec for the stocks of authentic SARS-CoV-2 viruses. We would also like to thank the participants and the study staff of the Hospitalized or Ambulatory Adults with Respiratory Viral Infections study. We also thank David Veesler (University of Washington) for providing Delta spike trimer to use as bait to capture spike-specific B cells. The SARS-CoV-2 S 6P expression vector was a gift from Jason McLellan (Addgene plasmid # 154754). This work has been supported by grant AI 38709 to J.O. T.N.S. was supported by NIH/NIAID K99AI166250. The work of J.D.B. was supported by NIH/NIAID grant R01AI141707 and the Howard Hughes Medical Institute. The Finzi lab was supported by the Canadian Institutes of Health Research grant nos. 352417 and 177958 and by an Exceptional Fund COVID-19 from the Canada Foundation for Innovation #41027 to A.F. A.F. is the recipient of a Canada Research Chair on Retroviral Entry # RCHS0235 950-232424. K.K.L. was supported by grant R01AI165808. J.G., M.L., C.I.D., V.C., F.R., and M.S.K. were all researchers at Fred Hutch Cancer Center in the Overbaugh group. K.N.L and J.T.C. are researchers at the University Washington with the Lee group. S.D. is a researcher in the Finzi group at the Centre de Recherche du CHUM.
Author contributions
J.G. and J.O. designed research; J.G., M.L., T.N.S., B.D., K.N.L., J.T.C., C.I.S., V.C., S.D., F.R., and M.S.K. performed research; A.F., J.D.B., and H.Y.C. contributed new reagents/analytic tools; J.G., M.L., T.N.S., B.D., K.N.L., J.T.C., C.I.S., S.D., F.R., K.K.L., and J.O. analyzed data; H.Y.C. led the HAARVI study Provided scientific guidance; K.N.L. provided scientific guidance; and J.G. and J.O. wrote the paper.
Competing interests
J.O. consults for Aerium Therapeutics. T.N.S. consults for Apriori Bio on deep mutational scanning. J.D.B. serves as a scientific advisor to Apriori Bio and Oncorus. Subsequent to the completion of the research described in this manuscript, he also began to serve as a scientific advisor to Aerium Therapeutics and the Vaccine Company. H.Y.C. consults with Ellume, Merck, Abbvie, Pfizer, Medscape, Vindico, and the Bill and Melinda Gates Foundation. J.O. and J.G. are on a patent (22-173-US-PSP2) for the C68 antibodies.
Footnotes
Preprint server: Manuscript was submitted as a preprint to bioRxiv.org.
Reviewers: M.S.D., Washington University in St. Louis School of Medicine; and P.L.M., University of the Witwatersrand Johannesburg.
Data, Materials, and Software Availability
Computational data, code, intermediate data analysis files, computational pipeline from yeast-display DMS have been posted on GitHub: https://github.com/jbloomlab/SARS-CoV-2-RBD_Omicron_MAP_Overbaugh_v1/blob/main/results/summary/summary.md (97); https://github.com/jbloomlab/SARS-CoV-2-RBD_Omicron_MAP_Overbaugh_v1/blob/main/results/supp_data/Wuhan_Hu_1/all_mAbs_WH1_raw_data.csv (98); https://github.com/jbloomlab/SARS-CoV-2-RBD_Omicron_MAP_Overbaugh_v1/blob/main/results/supp_data/Omicron_BA1/all_mAbs_BA1_raw_data.csv (99); and https://github.com/jbloomlab/SARS-CoV-2-RBD_Omicron_MAP_Overbaugh_v1/blob/main/results/supp_data/Omicron_BA2/all_mAbs_BA2_raw_data.csv. Raw sequencing data for yeast-display DMS experiments are on the NCBI SRA under BioProject PRJNA770094 and BioSample SAMN34381850 (100–102). Data and analysis files from the spike-pseudotyped virus DMS assay have been posted on GitHub: https://github.com/dms-vep/SARS-CoV-2_Omicron_BA.1_spike_DMS_C68.59 (103); https://dms-vep.github.io/SARS-CoV-2_Omicron_BA.1_spike_DMS_C68.59/ (104). cryo-EM structures have been deposited in the EM Data Resource: https://www.emdataresource.org [For cryo-EM structures (Class 1: EMDB-29053 (94), Class 2: EMDB-29054 (95), Class 3: EMDB-29052 (96)].
Supporting Information
References
- 1.National Institutes of Health, SARS-CoV-2 variants and susceptibility to Anti-SARS-CoV-2 monoclonal antibodies (2022). https://www.covid19treatmentguidelines.nih.gov/tables/variants-and-susceptibility-to-mabs/. Accessed 6 March 2023.
- 2.Collier D. A., et al. , Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature 593, 136–141 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garcia-Beltran W. F., et al. , Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell 184, 2372–2383.e9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang P., et al. , Increased resistance of SARS-CoV-2 variant P.1 to antibody neutralization. Cell Host. Microbe. 29, 747–751.e4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatterjee D., et al. , SARS-CoV-2 Omicron Spike recognition by plasma from individuals receiving BNT162b2 mRNA vaccination with a 16-week interval between doses. Cell Rep. 38, 110429 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walls A. C., et al. , Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281–292.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia S., et al. , Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 30, 343–355 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walls A. C., et al. , Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 114, 11157–11162 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belouzard S., Chu V. C., Whittaker G. R., Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl. Acad. Sci. U.S.A. 106, 5871–5876 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shang J., et al. , Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. U.S.A. 117, 11727–11734 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan X., Cao D., Kong L., Zhang X., Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nat. Commun. 11, 3618 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao Y., et al. , Potent neutralizing antibodies against SARS-CoV-2 Identified by high-throughput single-cell sequencing of convalescent patients’ B cells. Cell 182, 73–84.e16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chi X., et al. , A neutralizing human antibody binds to the n-terminal domain of the spike protein of sars-cov-2. Science 369, 650–655 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ju B., et al. , Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 584, 115–119 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Jennewein M. F., et al. , Isolation and characterization of cross-neutralizing coronavirus antibodies from COVID-19+ subjects. Cell Rep. 36, 109353 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stamatatos L., et al. , mRNA vaccination boosts cross-variant neutralizing antibodies elicited by SARS-CoV-2 infection. Science 372, 1413–1418 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu L., et al. , Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 584, 450–456 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Piccoli L., et al. , Mapping neutralizing and immunodominant sites on the SARS-CoV-2 spike receptor-binding domain by structure-guided high-resolution serology. Cell 183, 1024–1042.e1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suryadevara N., et al. , Neutralizing and protective human monoclonal antibodies recognizing the N-terminal domain of the SARS-CoV-2 spike protein. Cell 184, 2316–2331.e5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang C., et al. , A human monoclonal antibody blocking SARS-CoV-2 infection. Nat. Commun. 11, 2251 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pinto D., et al. , Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 583, 290–295 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Wec A. Z., et al. , Broad neutralization of SARS-related viruses by human monoclonal antibodies. Science 369, 731–736 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wan J., et al. , Human-IgG-neutralizing monoclonal antibodies block the SARS-CoV-2 infection. Cell Rep. 32, 107918 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rogers T. F., et al. , Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 369, 956–963 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hassan A. O., et al. , A SARS-CoV-2 infection model in mice demonstrates protection by neutralizing antibodies. Cell 182, 744–753.e4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zost S. J., et al. , Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature 584, 443–449 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barnes C. O., et al. , SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 588, 682–687 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greaney A. J., et al. , Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat. Commun. 12, 4196 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao Y., et al. , Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 602, 657–663 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dejnirattisai W., et al. , The antigenic anatomy of SARS-CoV-2 receptor binding domain. Cell 184, 2183–2200.e22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Starr T. N., et al. , SARS-CoV-2 RBD antibodies that maximize breadth and resistance to escape. Nature 597, 97–102 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Starr T. N., Greaney A. J., Dingens A. S., Bloom J. D., Complete map of SARS-CoV-2 RBD mutations that escape the monoclonal antibody LY-CoV555 and its cocktail with LY-CoV016. Cell Rep. Med. 2, 100255 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu H., Wei P., Kappler J. W., Marrack P., Zhang G., SARS-CoV-2 variants of concern and variants of interest receptor binding domain mutations and virus infectivity. Front. Immunol. 13, 825256 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greaney A. J., et al. , Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host. Microbe. 29, 463–476.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greaney A. J., et al. , Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host. Microbe. 29, 44–57.e9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wibmer C. K., et al. , SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat. Med. 27, 622–625 (2021). [DOI] [PubMed] [Google Scholar]
- 37.VanBlargan L. A., et al. , An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat. Med. 28, 490–495 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carreno J. M., et al. , Activity of convalescent and vaccine serum against SARS-CoV-2 Omicron. Nature 602, 682–688 (2022). [DOI] [PubMed] [Google Scholar]
- 39.Andrews N., et al. , Covid-19 vaccine effectiveness against the Omicron (B.1.1.529) variant. N. Engl. J. Med. 386, 1532–1546 (2022), 10.1056/NEJMoa2119451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidt F., et al. , Plasma neutralization of the SARS-CoV-2 Omicron variant. N. Engl. J. Med. 386, 599–601 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cao Y., et al. , BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 608, 593–602 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takashita E., et al. , Efficacy of antibodies and antiviral drugs against Covid-19 Omicron variant. N. Engl. J. Med. 386, 995–998 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takashita E., et al. , Efficacy of antibodies and antiviral drugs against Omicron BA.2.12.1, BA.4, and BA.5 subvariants. N. Engl. J. Med. 387, 468–470 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cao Y., et al. , Imprinted SARS-CoV-2 humoral immunity induces convergent Omicron RBD evolution. Nature 614, 521–529 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Westendorf K., et al. , LY-CoV1404 (bebtelovimab) potently neutralizes SARS-CoV-2 variants. Cell Rep. 39, 110812 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Starr T. N., et al. , Deep mutational scans for ACE2 binding, RBD expression, and antibody escape in the SARS-CoV-2 Omicron BA.1 and BA.2 receptor-binding domains. PLoS Pathog. 18, e1010951 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anonymous, Fact sheet for healthcare providers: Emergency use authorization for bebtelovimab, U.S. Food and Drug Administration, Ed. (2022).
- 48.Starr T. N., et al. , Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 371, 850–854 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doepker L. E., et al. , Diversity and function of maternal HIV-1-specific antibodies at the time of vertical transmission. J. Virol. 94, e01594-19 (2020), 10.1128/jvi.01594-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Williams K. L., et al. , Superinfection drives HIV neutralizing antibody responses from several B cell lineages that contribute to a polyclonal repertoire. Cell Rep. 23, 682–691 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simonich C. A., et al. , HIV-1 neutralizing antibodies with limited hypermutation from an infant. Cell 166, 77–87 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Williams K. L., et al. , Identification of HIV gp41-specific antibodies that mediate killing of infected cells. PLoS Pathog. 15, e1007572 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Williams K. L., et al. , HIV-specific CD4-induced antibodies mediate broad and potent antibody-dependent cellular cytotoxicity activity and are commonly detected in plasma from HIV-infected humans. EBioMedicine 2, 1464–1477 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Itell H. L., et al. , SARS-CoV-2 antibody binding and neutralization in dried blood spot eluates and paired plasma. Microbiol. Spectr. 9, e0129821 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crawford K. H. D., et al. , Protocol and reagents for pseudotyping lentiviral particles with SARS-CoV-2 spike protein for neutralization assays. Viruses 12, 513 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ding S., VE607 stabilizes SARS-CoV-2 spike in the “RBD-up” conformation and inhibits viral entry. iScience 25, 104528 (2022). 10.1016/j.isci.2022.104528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gasser R., et al. , Major role of IgM in the neutralizing activity of convalescent plasma against SARS-CoV-2. Cell Rep. 34, 108790 (2021), 10.1016/j.celrep.2021.108790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beaudoin-Bussieres G., et al. , A Fc-enhanced NTD-binding non-neutralizing antibody delays virus spread and synergizes with a nAb to protect mice from lethal SARS-CoV-2 infection. Cell Rep. 38, 110368 (2022), 10.1016/j.celrep.2022.110368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.ter Meulen J., et al. , Human monoclonal antibody combination against SARS coronavirus: Synergy and coverage of escape mutants. PLoS Med. 3, e237 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yuan M., et al. , A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 368, 630–633 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Starr T. N., et al. , Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 182, 1295–1310.e20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park Y. J., et al. , Antibody-mediated broad sarbecovirus neutralization through ACE2 molecular mimicry. Science 375, 449–454 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tortorici M. A., et al. , Broad sarbecovirus neutralization by a human monoclonal antibody. Nature 597, 103–108 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dong J., et al. , Genetic and structural basis for SARS-CoV-2 variant neutralization by a two-antibody cocktail. Nat. Microbiol. 6, 1233–1244 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Witte L., et al. , Epistasis lowers the genetic barrier to SARS-CoV-2 neutralizing antibody escape. Nat. Commun. 14, 302 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dadonaite B., et al. , A pseudovirus system enables deep mutational scanning of the full SARS-CoV-2 spike. Cell 186, 1263–1278.e20 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kitchin D., et al. , Ad26.COV2.S breakthrough infections induce high titers of neutralizing antibodies against Omicron and other SARS-CoV-2 variants of concern. Cell Rep. Med. 3, 100535 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Walls A. C., et al. , SARS-CoV-2 breakthrough infections elicit potent, broad, and durable neutralizing antibody responses. Cell 185, 872–880.e3 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.He W. T., et al. , Targeted isolation of diverse human protective broadly neutralizing antibodies against SARS-like viruses. Nat. Immunol. 23, 960–970 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wratil P. R., et al. , Three exposures to the spike protein of SARS-CoV-2 by either infection or vaccination elicit superior neutralizing immunity to all variants of concern. Nat. Med. 28, 496–503 (2022). [DOI] [PubMed] [Google Scholar]
- 71.Cameroni E., et al. , Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature 602, 664–670 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Park Y. J., et al. , Imprinted antibody responses against SARS-CoV-2 Omicron sublineages. Science 378, 619–627 (2022). [DOI] [PubMed] [Google Scholar]
- 73.Jia J. Z., et al. , Priming conditions shape breadth of neutralizing antibody responses to sarbecoviruses. Nat. Commun. 13, 6285 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tauzin A., et al. , Strong humoral immune responses against SARS-CoV-2 Spike after BNT162b2 mRNA vaccination with a 16-week interval between doses. Cell Host. Microbe. 30, 97–109.e105 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rappazzo C. G., et al. , Broad and potent activity against SARS-like viruses by an engineered human monoclonal antibody. Science 371, 823–829 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sztain T., et al. , A glycan gate controls opening of the SARS-CoV-2 spike protein. Nat. Chem. 13, 963–968 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zheng L., et al. , SARS-CoV-2 spike protein receptor-binding domain N-glycans facilitate viral internalization in respiratory epithelial cells. Biochem. Biophys. Res. Commun. 579, 69–75 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Garrett M. E., et al. , High-resolution profiling of pathways of escape for SARS-CoV-2 spike-binding antibodies. Cell 184, 2927–2938.e11 (2021), 10.1016/j.cell.2021.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen Y., et al. , Broadly neutralizing antibodies to SARS-CoV-2 and other human coronaviruses. Nat. Rev. Immunol. 23, 189–199 (2022), 10.1038/s41577-022-00784-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Voss W. N., et al. , Prevalent, protective, and convergent IgG recognition of SARS-CoV-2 non-RBD spike epitopes. Science 372, 1108–1112 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brouwer P. J. M., et al. , Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 369, 643–650 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Low J. S., et al. , ACE2-binding exposes the SARS-CoV-2 fusion peptide to broadly neutralizing coronavirus antibodies. Science 377, 735–742 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pinto D., et al. , Broad betacoronavirus neutralization by a stem helix-specific human antibody. Science 373, 1109–1116 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sun X., et al. , Neutralization mechanism of a human antibody with pan-coronavirus reactivity including SARS-CoV-2. Nat. Microbiol. 7, 1063–1074 (2022). [DOI] [PubMed] [Google Scholar]
- 85.Dacon C., et al. , Broadly neutralizing antibodies target the coronavirus fusion peptide. Science 377, 728–735 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tanaka S., et al. , Rapid identification of neutralizing antibodies against SARS-CoV-2 variants by mRNA display. Cell Rep. 38, 110348 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Seow J., et al. , A neutralizing epitope on the SD1 domain of SARS-CoV-2 spike targeted following infection and vaccination. Cell Rep. 40, 111276 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Henderson R., et al. , Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 27, 925–933 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Oude Munnink B. B., et al. , Transmission of SARS-CoV-2 on mink farms between humans and mink and back to humans. Science 371, 172–177 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Crawford K. H. D., et al. , Dynamics of neutralizing antibody titers in the months after SARS-CoV-2 infection. J. Infect. Dis. 223, 197–205 (2020), 10.1093/infdis/jiaa618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Simonich C. A., et al. , A diverse collection of B cells responded to HIV infection in infant BG505. Cell Rep Med. 2, 100314 (2021), 10.1016/j.xcrm.2021.100314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Watson M. J., et al. , Simple platform for automating decoupled LC-MS analysis of hydrogen/deuterium exchange samples. J. Am. Soc. Mass. Spectrom. 32, 597–600 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mastronarde D. N., Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 94.Croft J. T., Lee K. K., EMDB-29053 SARS-CoV-2 Spike Hexapro - C59.68 Fab (Class 1 - No Fab bound). EMDataResource. https://www.emdataresource.org/EMD-29053. Deposited 9 December 2022.
- 95.Croft J. T., Lee K. K., EMDB-29054 SARS-CoV-2 Spike Hexapro - C59.68 Fab (Class 2 - Fab bound). EMDataResource. https://www.emdataresource.org/EMD-29054. Deposited 9 December 2022.
- 96.Croft J. T., Lee K. K., EMDB-29052 SARS-CoV-2 Spike Hexapro - C59.68 Fab (Class 3 - Disordered). EMDataResource. https://www.emdataresource.org/EMD-29052. Deposited 9 December 2022.
- 97.Starr T. N., SARS-CoV-2-RBD_Omicron_MAP_Overbaugh_v1. Summary. Github. https://github.com/jbloomlab/SARS-CoV-2-RBD_Omicron_MAP_Overbaugh_v1/blob/main/results/summary/summary.md. Deposited 26 November 2022.
- 98.Starr T. N., SARS-CoV-2-RBD_Omicron_MAP_Overbaugh_v1. WH1 raw data. Github. https://github.com/jbloomlab/SARS-CoV-2-RBD_Omicron_MAP_Overbaugh_v1/blob/main/results/supp_data/Wuhan_Hu_1/all_mAbs_WH1_raw_data.csv. Deposited 26 November 2022.
- 99.Starr T. N., SARS-CoV-2-RBD_Omicron_MAP_Overbaugh_v1. Github. https://github.com/jbloomlab/SARS-CoV-2-RBD_Omicron_MAP_Overbaugh_v1/blob/main/results/supp_data/Omicron_BA1/all_mAbs_BA1_raw_data.csv. Deposited 26 November 2022.
- 100.Starr T. N., SARS-CoV-2-RBD_Omicron_MAP_Overbaugh_v1. Github. https://github.com/jbloomlab/SARS-CoV-2-RBD_Omicron_MAP_Overbaugh_v1/blob/main/results/supp_data/Omicron_BA2/all_mAbs_BA2_raw_data.csv. Deposited 26 November 2022.
- 101.Starr T. N., SAMN34381850 Illumina barcode sequencing from mutational antigenic profiling of C68 donor mAbs, v1. NCBI SRA. https://www.ncbi.nlm.nih.gov/biosample/?term=SAMN34381850. Deposited 26 April 2023. [Google Scholar]
- 102.Starr T. N., PRJNA770094 Deep mutational scanning of the RBD from SARS-CoV-2 variant lineages using yeast display of barcoded libraries. NCBI SRA. https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA770094. Deposited 10 October 2021. [Google Scholar]
- 103.Dadonaite B., SARS-CoV-2_Omicron_BA.1_spike_DMS_C68.59 raw data & code. GitHub. https://github.com/dms-vep/SARS-CoV-2_Omicron_BA.1_spike_DMS_C68.59. Deposited 29 November 2022.
- 104.Dadonaite B., SARS-CoV-2_Omicron_BA.1_spike_DMS_C68.59 Summary & Data Analysis. GitHub. https://dms-vep.github.io/SARS-CoV-2_Omicron_BA.1_spike_DMS_C68.59/. Deposited 29 November 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
Computational data, code, intermediate data analysis files, computational pipeline from yeast-display DMS have been posted on GitHub: https://github.com/jbloomlab/SARS-CoV-2-RBD_Omicron_MAP_Overbaugh_v1/blob/main/results/summary/summary.md (97); https://github.com/jbloomlab/SARS-CoV-2-RBD_Omicron_MAP_Overbaugh_v1/blob/main/results/supp_data/Wuhan_Hu_1/all_mAbs_WH1_raw_data.csv (98); https://github.com/jbloomlab/SARS-CoV-2-RBD_Omicron_MAP_Overbaugh_v1/blob/main/results/supp_data/Omicron_BA1/all_mAbs_BA1_raw_data.csv (99); and https://github.com/jbloomlab/SARS-CoV-2-RBD_Omicron_MAP_Overbaugh_v1/blob/main/results/supp_data/Omicron_BA2/all_mAbs_BA2_raw_data.csv. Raw sequencing data for yeast-display DMS experiments are on the NCBI SRA under BioProject PRJNA770094 and BioSample SAMN34381850 (100–102). Data and analysis files from the spike-pseudotyped virus DMS assay have been posted on GitHub: https://github.com/dms-vep/SARS-CoV-2_Omicron_BA.1_spike_DMS_C68.59 (103); https://dms-vep.github.io/SARS-CoV-2_Omicron_BA.1_spike_DMS_C68.59/ (104). cryo-EM structures have been deposited in the EM Data Resource: https://www.emdataresource.org [For cryo-EM structures (Class 1: EMDB-29053 (94), Class 2: EMDB-29054 (95), Class 3: EMDB-29052 (96)].