

Highlights
-
•
This is the first report of a germline DICER1-associated Sertoli-Leydig cell tumor (SLCT) diagnosed in pregnancy.
-
•
SLCT is linked to DICER1 pathogenic variants, but little is known about management of DICER1-associated SLCT.
-
•
There is an extended risk for metachronous SLCT in patients with germline DICER1 pathogenic variants who retain an ovary.
-
•
Prophylactic contralateral salpingo-oophorectomy may be offered with shared decision making to patients with inherited SLCT.
-
•
Genetic testing for DICER1 should be offered to all patients with moderately or poorly differentiated SLCT.
Keywords: DICER1, Genetic testing, Metachronous tumor, Post-treatment surveillance, Pregnancy, Sertoli-Leydig cell tumor
1. Introduction
Sertoli-Leydig cell tumors (SLCT) are rare sex cord-stromal tumors of the ovary that can be benign or malignant. Sex cord-stromal tumors comprise 7 % of malignant ovarian neoplasms, with SLCT accounting for less than 0.5 % (Colombo et al., 2007). SLCT exhibit testicular structures with androgen-producing capability. Given its rarity, much of the existing evidence on SLCT is based on case series. Ovarian SLCT generally develop before the age of 40 years. Survival is excellent for Stage I disease (>90 % at five years) but declines with advancing stage, tumor grade, and presence of heterologous elements (Colombo et al., 2007, Sigismondi et al., 2012). Unlike the more common granulosa cell tumor, SLCT tends to recur early. Indeed, two-thirds recur within one year and nearly 75 % manifest within two years; only 7 % of recurrences appear after five years. Notably, recurrence is associated with high mortality (Sigismondi et al., 2012).
SLCT are linked to germline and somatic DICER1 pathogenic variants (PVs) in the majority of cases. DICER1, on chromosome 14q32, encodes an RNaseIII endonuclease, which is essential in microRNA production. Germline DICER1 PVs follow an autosomal dominant inheritance pattern (Schultz et al., 2017, Frio et al., 2011) and are additionally associated with pleuropulmonary blastoma, thyroid disease, and various rare cancers (Schultz et al., 2018, Han et al., 2022). The population prevalence of germline DICER1 PVs ranges from 1/3700 to 1/1100 (Mirshahi et al., 2021), and one registry reported that 60 % of its patients with ovarian SLCT were found to have a germline DICER1 PV (Schultz et al., 2017).
The literature on SLCT associated with a germline DICER1 PV is sparse. Existing evidence is drawn from case reports and a single ovarian sex cord-stromal tumor registry (the International Ovarian and Testicular Stromal Tumor Registry) (Schultz et al., 2017). While there is reasonable data to guide management of SLCT broadly, what is unknown is how the addition of a genetic predisposition to SLCT affects residual risk following primary treatment. Therefore, in this report, we present a case of germline DICER1-associated SLCT, with consideration of surveillance and management strategies for this unique subset of patients.
2. Case report
A 36-year-old gravida 3, para 1 was noted to have an elevated alpha-fetoprotein (AFP) of 591 (19.21 MoM) at 17 5/7 weeks of gestation on routine prenatal screening. Her pregnancy had been uncomplicated up to this point, and she did not have any signs of androgen excess. Subsequent imaging showed normal fetal anatomy but demonstrated a complex solid and cystic mass of the left ovary initially measuring 10-cm by 12-cm. Given high suspicion for malignancy, she was referred to gynecologic oncology.
At 19 2/7 weeks of gestation, she underwent exploratory laparotomy, left salpingo-oophorectomy, left pelvic and para-aortic lymph node dissection, omentectomy, peritoneal biopsies, and pelvic washings. Final pathology revealed a 9-cm malignant Sertoli-Leydig cell neoplasm of the left ovary, moderately differentiated with heterologous elements, including intestinal type mucinous epithelium and carcinoid tumor (Fig. 1). Staging biopsies and pelvic washings were negative, placing her at FIGO Stage IA. After extensive counseling, she underwent four cycles of adjuvant chemotherapy with carboplatin and paclitaxel every three weeks, from 23 weeks to 32 weeks of gestation. She ultimately delivered a healthy baby boy weighing 2920 g at 39 weeks via repeat Cesarean section. Serial ultrasounds in pregnancy were used to monitor fetal growth and for the risk of fetal pleuropulmonary blastoma; growth was appropriate and there was no evidence of pleuropulmonary blastoma. The newborn had a normal chest radiograph after delivery. At six months of age, he had a surveillance computed tomography (CT) of the chest that showed two pulmonary cysts in the left lung and two pulmonary cysts in the right lung. Repeat imaging at eight months of age showed persistence of three lung lesions with some interval growth, felt to be consistent with type 1 pleuropulmonary blastoma. The infant subsequently underwent thoracotomy and removal of a 7-mm cyst from his left lung. Pathology was consistent with pleuropulmonary blastoma type 1 cyst (Coleman et al., 2016). He continues to undergo close follow up with oncology and serial imaging.
Fig. 1.
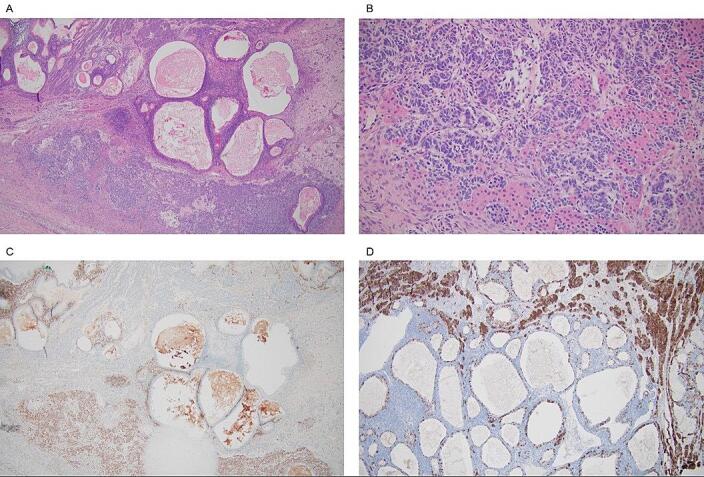
Histopathological images of the patient’s tumor. A, H&E-stained section of moderately-differentiated SLCT with adjacent mucinous and carcinoid heterologous elements. B, Higher powered view showing mixture of Sertoli and Leydig cells. C, Steroidogenic factor 1 (SF-1) immunohistochemistry highlights Sertoli-Leydig cell component. Carcinoid and mucinous components are negative for SF-1. D, Synaptophysin immunohistochemistry highlights carcinoid heterologous elements and is negative in Sertoli-Leydig cell component.
The patient’s family history was notable for a sister with moderately-differentiated SLCT at age 15 years, followed by uterine adenosarcoma and uterine polyps a decade later. Genetic evaluation of our patient detected a monoallelic germline DICER1 splice site variant c.1377-2A > T, classified as likely pathogenic. Genetic testing confirmed the same germline DICER1 PV in her two children and affected sister (Fig. 2).
Fig. 2.

Family pedigree for this patient with DICER1-associated SLCT. Our patient is indicated by the arrow. Age of cancer diagnosis is provided in parentheses. Our patient has a paternal cousin with epithelial ovarian cancer and a PV in BRCA2. Our patient does not carry this BRCA2 PV.
Postpartum, she was followed with pelvic ultrasound every four months as well as CT chest-abdomen-pelvis every four to six months following completion of adjuvant chemotherapy. All scans to date have shown no evidence of disease. Her AFP measurements have also been within normal range. At time of manuscript preparation, she was approaching two years of post-treatment surveillance.
3. Discussion
Reports of germline DICER1 PV-associated SLCT are limited in the literature. Here, we present one such case for discussion of optimal management. In primary treatment of SLCT, there are shared principles irrespective of DICER1 status. As most SLCT are Stage I at diagnosis and the overwhelming majority of SLCT are unilateral (98.5 %) (Colombo et al., 2007, Durmuş et al., 2019), surgery is the standard of care. The specific surgical procedures vary based on age, fertility desires, and extent of disease, from total hysterectomy with bilateral salpingo-oophorectomy to fertility-sparing unilateral salpingo-oophorectomy (Colombo et al., 2007, Sigismondi et al., 2012, Durmuş et al., 2019, Gui et al., 2012). Current consensus recommends adjuvant chemotherapy for patients with worse prognosis factors: greater than FIGO Stage I, tumors with moderate or poor differentiation, and tumors with heterologous elements on histology (Sigismondi et al., 2012, Durmuş et al., 2019, Gui et al., 2012). This patient was in the second trimester at diagnosis with a moderately-differentiated SLCT with heterologous elements. Given the rapid timeframe for recurrence of these cancers, waiting to deliver adjuvant therapy until after delivery was suboptimal. The patient also had a strong desire to continue her pregnancy. Paclitaxel/carboplatin is the current preferred adjuvant regimen and less teratogenic than the alternate regimens of etoposide/cisplatin +/- bleomycin (Sigismondi et al., 2012, Gui et al., 2012). Therefore, four cycles were safely administered prior to fetal maturity with appropriate maternal and fetal monitoring.
The next question for our patient with Stage IA SLCT involves managing the risk of recurrent or metachronous (second primary) tumor—specifically the risk in the retained contralateral ovary. Among SLCT broadly, recurrence after treatment is found in a minority of patients, though the mortality rate increases notably among recurrent cases. According to an ovarian sex cord-stromal tumor registry, overall recurrence for Stage IA SLCT after primary surgical treatment (for well-differentiated tumors) or surgery and adjuvant chemotherapy (for moderately- or poorly-differentiated tumors) was 16.3 %, with a mortality of 50 % among the recurrences (Schultz et al., 2017). However, the germline DICER1 PV complicates the picture. The sole detailed case report of germline DICER1-associated SLCT follows two sisters who underwent fertility-sparing surgery and adjuvant chemotherapy. Both patients experienced tumor recurrence and ultimately died within 1.5 years of primary treatment completion (Zhang et al., 2020). In contrast, the registry recurrence rate among germline DICER1-associated SLCT was only 9.1 % (2/22), with one death by five years (Schultz et al., 2017).
Interestingly, the sex cord-stromal tumor registry also demonstrates the possibility of metachronous tumors. Three metachronous SLCT developed among the 22 patients (13.6 %) with germline DICER1-associated SLCT, at 5–14 years after the initial diagnosis. The primary SLCT were all Stage IA with either intermediate or poor differentiation and had been treated with at least surgery (the addition of adjuvant chemotherapy could not be clearly deduced from the presented data). All three metachronous tumors were Stage IA and underwent successful surgical-only treatment (Schultz et al., 2017). A separate study recorded two metachronous SLCT among nine patients (22.2 %) with germline DICER1 PVs, occurring 6 years and 46 years after the primary neoplasm was treated with fertility-sparing surgery (Merideth et al., 2020). Comparison data from a non-germline DICER1-associated SLCT cohort demonstrated a relatively similar rate of metachronous SLCT (4/38, 10.5 %), though these metachronous neoplasms developed 4–48 months after the original diagnosis. The metachronous neoplasms were treated with either surgery or surgery and adjuvant chemotherapy and the individuals were alive at 51 months of follow up (Schneider et al., 2015). The apparent extended risk period for metachronous DICER1-associated SLCT makes sense intuitively in the context of a germline PV.
Existing expert consensus recommends screening pelvic and abdominal ultrasounds every 6–12 months until at least age 40 years for patients with a known germline DICER1 PV. This age cutoff was set because 95 % of SLCT are diagnosed by 40 years, though the oldest patient with germline DICER1-associated SLCT was 61 years at diagnosis (Schultz et al., 2018). Accordingly, both of the patient’s children commenced recommended screening, including for pleuropulmonary blastoma, leading to her son’s early diagnosis at age eight months. However, there is no specific guidance for surveillance after primary fertility-sparing treatment of germline DICER1-associated SLCT.
The optimal post-treatment surveillance strategy for germline DICER1-associated SLCT may be based on time from treatment rather than a specific age threshold. The greatest risk in the first two years after therapy is for recurrent disease. Thus, we elected to follow our patient during this period with close surveillance for recurrence in the contralateral ovary via pelvic ultrasounds and for systemic failure (CT scans and AFP monitoring). At 2.5 years post diagnosis, her risk of recurrent cancer is diminishing, and we are discussing the role of risk-reducing contralateral oophorectomy to address risk of a second SLCT. In the literature, the first documented metachronous SLCT occurred at post-treatment year five, while all documented metachronous tumors in non-germline DICER1 mutants had been diagnosed by two years (Schultz et al., 2017, Schneider et al., 2015). Presumably, ultrasound surveillance would be more effective for early detection of SLCT than for epithelial ovarian cancers, but data is lacking. Similarly, there is no data to inform ideal timing for preventive oophorectomy in this patient. At age 39, surgical menopause has serious consequences, but our risk-adverse patient wishes to minimize the chance of metachronous SLCT. Risk-reducing salpingo-oophorectomy is a reasonable option after individualized shared decision making, considering age, fertility desires, likelihood of compliance with hormone replacement therapy, and willingness to tolerate risk from possible metachronous and/or recurrent disease.
There are no clear guidelines for genetic testing of individuals with ovarian SLCT who lack family history of other DICER1 cancers. Germline or somatic PVs in DICER1 have been reported in up to 97 % of SLCT (Schultz et al., 2017). The International Ovarian and Testicular Stromal Tumor Registry reported on germline testing for 41 individuals with ovarian SLCT, 25 (61 %) of whom had DICER1 PVs, including three who were mosaic. An additional 11 cases had somatic DICER1 PVs (Schultz et al., 2017). The presence of DICER1 PVs is nearly universal in moderately- and poorly-differentiated SLCT while absent in well-differentiated neoplasms (Schultz et al., 2017, de Kock et al., 2017, Karnezis et al., 2019). We conclude that germline testing for DICER1 should be offered to all individuals with moderately- or poorly-differentiated ovarian SLCT, regardless of family history. The importance of genetic testing is highlighted by this case, in which the genetic results had substantial implications for management of cancer risk, both for our patient and for her two young children.
4. Consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
CRediT authorship contribution statement
Joyce Y. Wang: Investigation, Data curation, Visualization. Kimberly K. Ma: Resources, Validation, Data curation. Daniel J. Reiter: Resources, Visualization. Ana Torvie: Resources, Validation. Elizabeth M. Swisher: Conceptualization, Resources, Validation, Data curation, Visualization, Supervision.
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: EMS has received institutional support through grants funded by Clovis, Plexxicon, and GSK. EMS has also received consulting fees from Ideaya Biosciences for serving on their Scientific Advisory Board and participates in the Data Safety Monitoring Board/Advisory Board for Novartis. The remaining authors declare no conflicts of interest.
Contributor Information
Joyce Y. Wang, Email: wyjoyce@uw.edu.
Kimberly K. Ma, Email: kkma@uw.edu.
Daniel J. Reiter, Email: djreiter@uw.edu.
Ana Torvie, Email: atorvie@gmail.com.
Elizabeth M. Swisher, Email: swishere@uw.edu.
References
- Coleman A., Kline-Fath B., Stanek J., Lim F.Y. Pleuropulmonary blastoma in a neonate diagnosed prenatally as congenital pulmonary airway malformation. Fetal Diagn. Ther. 2016;39(3):234–238. doi: 10.1159/000365352. [DOI] [PubMed] [Google Scholar]
- Colombo N., Parma G., Zanagnolo V., Insinga A. Management of ovarian stromal cell tumors. J. Clin. Oncol. 2007;25(20):2944–2951. doi: 10.1200/JCO.2007.11.1005. [DOI] [PubMed] [Google Scholar]
- de Kock L., Terzic T., McCluggage W.G., Stewart C.J.R., Shaw P., Foulkes W.D., et al. DICER1 mutations are consistently present in moderately and poorly differentiated Sertoli-Leydig cell tumors. Am. J. Surg. Pathol. 2017;41(9):1178–1187. doi: 10.1097/PAS.0000000000000895. [DOI] [PubMed] [Google Scholar]
- Durmuş Y., Kılıç Ç., Çakır C., Yüksel D., Boran N., Karalök A., et al. Sertoli-Leydig cell tumor of the ovary: analysis of a single institution database and review of the literature. J. Obstet. Gynaecol. Res. 2019;45(7):1311–1318. doi: 10.1111/jog.13977. [DOI] [PubMed] [Google Scholar]
- Frio T.R., Bahubeshi A., Kanellopoulou C., Hamel N., Niedziela M., Sabbaghian N., et al. DICER1 mutations in familial multinodular goiter with and without ovarian Sertoli-Leydig cell tumors. J. Am. Med. Assoc. 2011;305(1):68–77. doi: 10.1001/jama.2010.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui T., Cao D., Shen K., Yang J., Zhang Y., Yu Q., et al. A clinicopathological analysis of 40 cases of ovarian Sertoli-Leydig cell tumors. Gynecol. Oncol. 2012;127(2):384–389. doi: 10.1016/j.ygyno.2012.07.114. [DOI] [PubMed] [Google Scholar]
- Han L.M., Weiel J.J., Longacre T.A., Folkins A.K. DICER1-associated tumors in the female genital tract: molecular basis, clinicopathologic features, and differential diagnosis. Adv Anat Pathol. 2022;29(5):297–308. doi: 10.1097/PAP.0000000000000351. [DOI] [PubMed] [Google Scholar]
- Karnezis A.N., Wang Y., Keul J., Tessier-Cloutier B., Magrill J., Kommoss S., et al. DICER1 and FOXL2 mutation status correlates with clinicopathologic features in ovarian Sertoli-Leydig cell tumors. Am. J. Surg. Pathol. 2019;43(5):628–638. doi: 10.1097/PAS.0000000000001232. [DOI] [PubMed] [Google Scholar]
- Merideth M.A., Harney L.A., Vyas N., Bachi A., Carr A.G., Hill D.A., et al. Gynecologic and reproductive health in patients with pathogenic germline variants in DICER1. Gynecol. Oncol. 2020;156(3):647–653. doi: 10.1016/j.ygyno.2019.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirshahi U.L., Kim J., Best A.F., Chen Z.E., Hu Y., Haley J.S., et al. A genome-first approach to characterize DICER1 pathogenic variant prevalence, penetrance, and phenotype. JAMA Netw. Open. 2021;4(2):e210112. doi: 10.1001/jamanetworkopen.2021.0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider D.T., Orbach D., Cecchetto G., Stachowicz-Stencel T., Brummel B., Brecht I.B., et al. Ovarian Sertoli Leydig cell tumours in children and adolescents: an analysis of the European Cooperative Study Group on Pediatric Rare Tumors (EXPeRT) Eur. J. Cancer. 2015;51(4):543–550. doi: 10.1016/j.ejca.2014.11.013. [DOI] [PubMed] [Google Scholar]
- Schultz K.A.P., Harris A.K., Finch M., Dehner L.P., Brown J.B., Gershenson D.M., et al. DICER1-related Sertoli-Leydig cell tumor and gynandroblastoma: clinical and genetic findings from the international ovarian and testicular stromal tumor registry. Gynecol. Oncol. 2017;147(3):521–527. doi: 10.1016/j.ygyno.2017.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz K.A.P., Williams G.M., Kamihara J., Stewart D.R., Harris A.K., Bauer A.J., et al. DICER1 and associated conditions: Identification of at-risk individuals and recommended surveillance strategies. Clin Cancer Res Off J Am Assoc Cancer Res. 2018;24(10):2251–2261. doi: 10.1158/1078-0432.CCR-17-3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigismondi C., Gadducci A., Lorusso D., Candiani M., Breda E., Raspagliesi F., et al. Ovarian Sertoli-Leydig cell tumors. A retrospective MITO study. Gynecol Oncol. 2012;125(3):673–676. doi: 10.1016/j.ygyno.2012.03.024. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Ren M., Hong Y., Zhong Y., Cong X., Chen C., et al. Sertoli-Leydig cell tumor in two siblings with DICER1 syndrome. Medicine (Baltimore) 2020;99(27):e20806. doi: 10.1097/MD.0000000000020806. [DOI] [PMC free article] [PubMed] [Google Scholar]