

Abstract
Fruiting bodies (sporocarps, sporophores or basidiomata) of mushroom-forming fungi (Agaricomycetes) are among the most complex structures produced by fungi. Unlike vegetative hyphae, fruiting bodies grow determinately and follow a genetically encoded developmental program that orchestrates their growth, tissue differentiation and sexual sporulation. In spite of more than a century of research, our understanding of the molecular details of fruiting body morphogenesis is still limited and a general synthesis on the genetics of this complex process is lacking. In this paper, we aim at a comprehensive identification of conserved genes related to fruiting body morphogenesis and distil novel functional hypotheses for functionally poorly characterised ones. As a result of this analysis, we report 921 conserved developmentally expressed gene families, only a few dozens of which have previously been reported to be involved in fruiting body development. Based on literature data, conserved expression patterns and functional annotations, we provide hypotheses on the potential role of these gene families in fruiting body development, yielding the most complete description of molecular processes in fruiting body morphogenesis to date. We discuss genes related to the initiation of fruiting, differentiation, growth, cell surface and cell wall, defence, transcriptional regulation as well as signal transduction. Based on these data we derive a general model of fruiting body development, which includes an early, proliferative phase that is mostly concerned with laying out the mushroom body plan (via cell division and differentiation), and a second phase of growth via cell expansion as well as meiotic events and sporulation. Altogether, our discussions cover 1 480 genes of Coprinopsis cinerea, and their orthologs in Agaricus bisporus, Cyclocybe aegerita, Armillaria ostoyae, Auriculariopsis ampla, Laccaria bicolor, Lentinula edodes, Lentinus tigrinus, Mycena kentingensis, Phanerochaete chrysosporium, Pleurotus ostreatus, and Schizophyllum commune, providing functional hypotheses for ~10 % of genes in the genomes of these species. Although experimental evidence for the role of these genes will need to be established in the future, our data provide a roadmap for guiding functional analyses of fruiting related genes in the Agaricomycetes. We anticipate that the gene compendium presented here, combined with developments in functional genomics approaches will contribute to uncovering the genetic bases of one of the most spectacular multicellular developmental processes in fungi.
Citation: Nagy LG, Vonk PJ, Künzler M, Földi C, Virágh M, Ohm RA, Hennicke F, Bálint B, Csernetics Á, Hegedüs B, Hou Z, Liu XB, Nan S, M. Pareek M, Sahu N, Szathmári B, Varga T, Wu W, Yang X, Merényi Z (2023). Lessons on fruiting body morphogenesis from genomes and transcriptomes of Agaricomycetes. Studies in Mycology 104: 1–85. doi: 10.3114/sim.2022.104.01
Keywords: cell wall remodelling, comparative genomics, development, fruiting body morphogenesis, functional annotation, mushroom, transcriptome
TABLE OF CONTENTS
- INTRODUCTION ............................................................................................................................................................................................. 3
- What are fruiting bodies and how do they develop ............................................................................................................................................................................................. 3
- The evolution of Agaricomycete fruiting bodies: from simple to complex morphogenesis ............................................................................................................................................................................................. 4
- Cellular and regulatory processes involved in fruiting body development ............................................................................................................................................................................................. 5
- Genomic and transcriptomic resources ............................................................................................................................................................................................. 6
DESCRIPTION OF THE APPROACHES USED IN THIS PAPER ............................................................................................................................................................................................. 6
- METHODS ............................................................................................................................................................................................. 7
- Bioinformatic analyses of RNA-Seq data ............................................................................................................................................................................................. 7
- Orthology based on reciprocal best hits ............................................................................................................................................................................................. 8
- Identification of CDE orthogroups ............................................................................................................................................................................................. 8
- Phylogenetic analyses ............................................................................................................................................................................................. 8
- RESULTS: DEVELOPMENTALLY EXPRESSED GENE CLASSES IN THE AGARICOMYCETES ............................................................................................................................................................................................. 8
- Cell division, proliferation and growth ............................................................................................................................................................................................. 9
- Meiotic and mitotic gene expression show two distinct expression peaks ............................................................................................................................................................................................. 9
- Ribosomal genes ............................................................................................................................................................................................. 11
- Growth by cell expansion and turgor manipulation - a biphasic model of fruiting body development ............................................................................................................................................................................................. 12
- Acetyl-CoA production by central carbon metabolism ............................................................................................................................................................................................. 13
- Lipid metabolism ............................................................................................................................................................................................. 16
- Beta oxidation ............................................................................................................................................................................................. 16
- Ergosterol biosynthesis ............................................................................................................................................................................................. 16
- Sterol transport ............................................................................................................................................................................................. 17
- Phospholipid and fatty acid biosynthesis ............................................................................................................................................................................................. 17
- Linoleic acid producing fatty acid desaturases ............................................................................................................................................................................................. 18
- Other potentially membrane lipid related genes ............................................................................................................................................................................................. 20
- Storage carbohydrate metabolism ............................................................................................................................................................................................. 20
- Transcriptional regulators ............................................................................................................................................................................................. 23
- Transcription factors ............................................................................................................................................................................................. 23
- RNA binding proteins ............................................................................................................................................................................................. 24
- RNA interference, Small RNA ............................................................................................................................................................................................. 25
- Chromatin regulators ............................................................................................................................................................................................. 25
- Histone gene expression correlates with fruiting body initiation and sporulation ............................................................................................................................................................................................. 26
- Other chromatin related genes ............................................................................................................................................................................................. 27
- Alternative splicing and spliceosome components ............................................................................................................................................................................................. 28
- Signal transduction systems and communication ............................................................................................................................................................................................. 28
- Oxylipins ............................................................................................................................................................................................. 29
- Pheromone and pheromone receptor pathways ............................................................................................................................................................................................. 30
- MAPK, TOR and cAMP pathways ............................................................................................................................................................................................. 31
- G-protein coupled receptors ............................................................................................................................................................................................. 32
- Other signal transduction related families ............................................................................................................................................................................................. 32
- Light sensing systems and light receptors ............................................................................................................................................................................................. 33
- Cell wall biosynthesis and modification ............................................................................................................................................................................................. 34
- Cell wall biosynthesis ............................................................................................................................................................................................. 35
- Chitin biosynthesis (GT2) ............................................................................................................................................................................................. 35
- β-glucan biosynthesis ............................................................................................................................................................................................. 35
- β-1,6-glucan (Kre9/Knh1, GH16, GH63) ............................................................................................................................................................................................. 35
- β-1,3-glucan (GT48) ............................................................................................................................................................................................. 36
- Cell wall remodelling ............................................................................................................................................................................................. 36
- Chitin-active enzyme families (GH18, GH20, X325, AA9, AA14, CBM5/12, CBM50, CE4) ............................................................................................................................................................................................. 36
- Glucan-linked families (GH3, GH5, GH16, GH17, GH30, GH55, GH71, GH128, GH152, Cdc43, WSC) ............................................................................................................................................................................................. 39
- Miscellaneous cell wall remodelling (GH27, GH72, GH76, GH79, GH88, GT18, pectinesterases) ............................................................................................................................................................................................. 41
- Multicopper oxidases and cupredoxins ............................................................................................................................................................................................. 42
- Expansins and cerato-platanins ............................................................................................................................................................................................. 42
- Miscellaneous cell wall related families (HTP, DyP, AA3, AA5, GH76, GT8, Slt2, Cfs1, Eln3, peptidases) ............................................................................................................................................................................................. 44
- Cell surface and cell wall proteins ............................................................................................................................................................................................. 45
- Hydrophobins ............................................................................................................................................................................................. 46
- Lectins ............................................................................................................................................................................................. 46
- Other cell surface proteins (HsbA, Con6, PriA, AA7, CFEM, wax synthases, fasciclins, peptidases, intradiol ring cleavage dioxygenase) ............................................................................................................................................................................................. 47
- Defence ............................................................................................................................................................................................. 49
- Secondary metabolism and natural product biosynthesis ............................................................................................................................................................................................. 52
- Cytoskeleton ............................................................................................................................................................................................. 53
- Ubiquitin-proteasome system ............................................................................................................................................................................................. 54
- Cop9 signalosome ............................................................................................................................................................................................. 54
- Protein ubiquitination ............................................................................................................................................................................................. 55
- Other gene groups ............................................................................................................................................................................................. 56
- Transporters ............................................................................................................................................................................................. 56
- Stress response genes ............................................................................................................................................................................................. 57
- Unannotated genes ............................................................................................................................................................................................. 58
- Functionally poorly known genes ............................................................................................................................................................................................. 59
- Ferric reductase superfamily proteins and iron metabolism ............................................................................................................................................................................................. 60
- A SYNOPTIC MODEL OF FRUITING BODY DEVELOPMENT ............................................................................................................................................................................................. 60
- Early events: primordium formation and cell proliferation ............................................................................................................................................................................................. 60
- Transition to growth by expansion ............................................................................................................................................................................................. 62
- Late events: meiosis and spore production ............................................................................................................................................................................................. 63
OPEN QUESTIONS RELATED TO FRUITING BODY MORPHOGENESIS ............................................................................................................................................................................................. 63
CONCLUSIONS ............................................................................................................................................................................................. 64
ACKNOWLEDGEMENTS ............................................................................................................................................................................................. 64
REFERENCES ............................................................................................................................................................................................. 65
INTRODUCTION
Fungi are one of the five main extant lineages in which complex macroscopic structures emerged have evolved over the course of evolution. The best-known macroscopic structures of fungi are sexual fruiting bodies, which enclose spore-producing cells in a three-dimensional protective environment and facilitate spore dispersal (Nagy et al. 2018). Sexual fruiting bodies are complex multicellular structures based as defined by Knoll (2011) and show evidence of repeated evolution across the fungal tree of life (Stajich et al. 2009, Merényi et al. 2020). They occur in at least eight lineages, with the best-known representatives belonging to the Pezizomycotina (e.g., morels, truffles), Agaricomycotina (e.g., agarics, boletes), but spectacular examples have also evolved in several other lineages, such as Endogone spp. in the Mucoromycota (Chang et al. 2019b) or the enigmatic genus Neolecta in the Taphrinomycotina (Nagy 2017, Nguyen et al. 2017). Fruiting bodies reached highest complexity in the Agaricomycetes, which include developmentally integrated morphologies that follow a spatially and temporally tightly regulated developmental program (Hibbett 2004, Virágh et al. 2022). In terms of complexity level, certain agaricomycete fruiting bodies approach the complexity of simple animals and plants (Taylor & Ellison 2010, Nagy et al. 2018).
Fruiting body morphogenesis has been a subject of intense research, however, despite tremendous efforts in this area, morphogenesis of Basidiomycota fruiting bodies is quite poorly understood. In comparison, fruiting body development and its underlying genetics are much better studied in the Ascomycota (see e.g., Pöggeler et al. 2006). This may be due to a mixture of factors, including the complexity of developmental programs in the Basidiomycota, the lack of easy-to-use model systems or the relative difficulty of testing hypothesis through genetic manipulation (e.g., due to the low frequency of homologous recombination (De Jong et al. 2010). Recent research on agaricomycete fruiting body morphogenesis has largely relied on comparative -omics techniques, which provided insights into the gene repertoire and the regulation of gene expression during fruiting body development. This adds to the rich body of literature on functionally characterised genes in Coprinopsis cinerea (summarised in Kües 2000), Schizophyllum commune and other species. There are several recent milestones in fruiting body research such as the publication of key genome sequences (Ohm et al. 2010, Stajich et al. 2010, Morin et al. 2012) or comparative transcriptomic datasets (e.g., Krizsán et al. 2019, Merényi et al. 2020, 2022). An emerging driving force on the study of fruiting body morphogenesis is the mushroom industry, which uses sexual fruiting bodies as food or as a source of bioactive compounds (Royse et al. 2017, Ma et al. 2018). Fruiting body development has been a subject of several excellent reviews, surprisingly, however, the last gene- centred review on fruiting body development in mushroom-forming fungi was published by (Whiteford & Thurston 2000).
The aim of this paper is to systematically catalogue genes and genetic processes related to fruiting body morphogenesis in the Agaricomycetes, based on both literature reviews and a meta-analysis of published developmental transcriptomes. We provide a handbook-like inventory of gene families and their putative functions based on expression data and functional predictions of genes borrowed from well-researched model systems. Because a negligible proportion of morphogenesis genes has so far been functionally characterised in the Agaricomycetes, it is currently impossible to build a comprehensive picture based on mechanistic studies alone, as has recently been done for hypha morphogenesis (Riquelme et al. 2018). Instead, we derive functional hypotheses for genes based on comparative expression data across multiple species. Our starting point is that genes which are developmentally regulated in the majority of species for which transcriptome data are available, should belong to the core pathways of fruiting body morphogenesis, whereas genes that show species-specific expression patterns should be responsible for sculpting species-specific morphologies or represent transcriptional or technical noise. Finally, we incorporate novel and previous observations into a new synthesis on processes active during fruiting body morphogenesis.
What are fruiting bodies and how do they develop
Fruiting bodies may be some of the best-known structures of fungi. They are sexual reproductive organs that facilitate spore dispersal. While in simple Basidiomycota spores are born on naked basidia, mushroom-forming fungi evolved complex three-dimensional structures that offer tremendous advantages in spore dispersal efficiency. Here, we refer to such structures as fruiting bodies, for consistency with recent literature, but note that other terms, such as “basidiome”, “basidiocarp” or “sporophore” could also be used.
Developmental events that take place in fruiting bodies have been investigated previously in several species, of which Coprinopsis/Coprinellus spp. and S. commune are the two most widely used groups of models. The development of fruiting bodies in these species has been described in previous reviews (Kües 2000, Kües & Liu 2000, Palmer & Horton 2006, Kües & Navarro-González 2015), therefore, we here only provide a short introduction, and describe the process in more detail (combining new results) in Chapter “A synoptic model of fruiting body development”. Briefly, fruiting body development is initiated on sexually competent mycelia as the reprogramming of hyphal branching patterns, which results in the interlacing of hyphae to form a 3-dimensional fruiting body initial (Kües & Liu 2000, Kües & Navarro-González 2015). Rather than relying on zygotic (e.g., maternal) mRNA cargo as animals and plants do, fungal hyphae undergo transcriptional reprogramming during fruiting body initiation. This reprogramming is influenced and triggered by a mixture of - often species-specific – environmental factors, such as starvation, light or temperature changes (reviewed by Sakamoto 2018). Once appropriate internal and external signals converge on the initiation of fruiting, morphogenesis starts with an undifferentiated initial (hyphal knot, aggregate), which undergoes tissue differentiation to form primordia. In subsequent late primordium and young fruiting body stages rapid growth (stipe elongation and cap expansion) and meiosis happen in a coordinated manner, which ultimately results in species-specific mature morphologies and culminates in spore formation and release.
Development of the fruiting bodies is a tightly regulated process that has a high degree of autonomy. That is, once started, the developmental program is not interrupted by even large perturbations, such as injuries, or removal of the fruiting body from the supporting colony at or after certain stages (Moore 2013). In fruiting bodies not only morphogenetic events, but also several internal processes (e.g., nutritional and sexual) are developmentally regulated. For example, in C. cinerea, stipe elongation starts immediately after meiosis takes place, after which basidiospore production, maturation and cap expansion follow in a tightly choreographed chronology (Kües 2000, Moore 2013). For detailed description of the process and molecular summaries of current knowledge on fruiting body morphogenesis the reader is referred to previous reviews (Kües 2000, Kües & Navarro-González 2015, Virágh et al. 2021) and the following detailed chapters of this paper.
The evolution of Agaricomycete fruiting bodies: from simple to complex morphogenesis
How fruiting body complexity evolved has interested mycologists for decades and has received considerable attention recently. Therefore, we provide only a brief introduction here and refer the reader to recent reviews on the topic (e.g., Kües & Navarro-González 2015, Nagy et al. 2018, Virágh et al. 2021). Phylogenetic studies have made tremendous progress in uncovering broad patterns of morphological transformation and trends in fruiting body evolution (e.g., Hibbett 2004, Sánchez-García et al. 2020, Varga et al. 2019). Fruiting body morphologies show a great diversity in the Basidiomycota, from naked lawns of basidia, to resupinate, coralloid or highly complex, or the so called pileate-stipitate fruiting bodies of well-known agarics (e.g., C. cinerea) (Fig. 1).
Fig. 1.

Species used in this study. Top row left to right: Agaricus bisporus, Cyclocybe aegerita, Armillaria ostoyae, Auriculariopsis ampla; Middle row: Coprinopsis cinerea, Laccaria bicolor, Lentinula edodes, Lentinus tigrinus; Bottom row: Mycena kentingensis, Pleurotus ostreatus, Phanerochaete chrysosporium, Schizophyllum commune. Scale bars = 1 cm.
Agaricomycete fruiting bodies might have emerged in or around the last common ancestor of Dacrymycetes and Agaricomycetes over 400 million years ago (Varga et al. 2019, Prasanna et al. 2020), although there is some uncertainty around ancestral states. This is partly due to the presence of jelly-like telia reminiscent of fruiting bodies in the Pucciniomycotina, uncertainties in the branching order of Basidiomycota subphyla and reductive evolution in early-diverging clades (Bartheletiomycetes, Wallemiomycetes) of the Agaricomycotina subphylum (reviewed in Prasanna et al. 2020). Nevertheless, all Agaricomycetes share a single, fruiting body-forming ancestor, which suggests there should be a core, deeply conserved genetic toolkit of fruiting body development. Phylogenetic comparative studies have provided sound evidence that this ancestor had corticioid/resupinate/jelly like fruiting bodies (e.g., Phanerochaete chrysosporium) and that more complex forms derived from such morphologies repeatedly (Hibbett 2004, Varga et al. 2019). Within the Agaricomycetes, tissues that protect and enclose basidia became thicker and more complex, eventually leading to the mushroom fruiting body structures we know today. From the perspective of this paper, it is important to mention that phylogenetic studies have also established that fruiting body evolution is highly convergent, that is, many of the characteristic traits (e.g., cap, gills) evolved independently several times. Therefore, an evolutionary framework is important for understanding how fruiting bodies evolved and for establishing homologies among tissues. Gills, caps and stipes, for example, although superficially similar, are not homologous across Polyporales and Agaricales and that these structures evolved independently in the two orders and thus may reflect different gene expression profiles. This implies that such, convergent forms (e.g., gills of Lentinus tigrinus in the Polyporales and that of C. cinerea in the Agaricales) are not homologous across lineages and their development should not be expected to follow the same genetic principles. On the other hand, basidia, the sexual processes that happen therein and the development of 3-dimensional fruiting body tissue, are homologous across all mushroom-forming fungi (i.e., they have a single origin in the Basidiomycota), providing a reliable framework for analysis.
These examples illustrate how the diversity of fruiting bodies can complicate the identification of core components of their morphogenesis. Ideally, we are interested in developmental processes and underlying genetic components that uniformly characterise fruiting bodies of all mushroom-forming fungi, i.e., those that can be traced back to the first fruiting body-forming Agaricomycete ancestor.
Therefore, building ‘minimal models’, which encompass the minimum set of genes necessary to build a fruiting body are highly relevant. Such minimal models could be, among others, species that produce flat, crust-like ‘resupinate’ fruiting bodies (e.g., Ph. chrysosporium) (Krizsán et al. 2019). Recently, Cryptococcus neoformans (Tremellomycetes, Basidiomycota) proved to be a useful model organism, because it produces simple lawns of basidia, sometimes referred to as fruiting bodies, in which basidium differentiation, meiosis and spore formation take place (Liu et al. 2018). Therefore, C. neoformans ‘fruiting bodies’ encompass all processes needed for Basidiomycota sexual reproduction but lack features essential to the definition of fruiting bodies such as three-dimensional organization, tissue differentiation and others. Merényi et al. recently sorted developmental genes of Pleurotus ostreatus and seven other complex species into groups that were developmentally regulated both in C. neoformans and mushroom-forming fungi and those in which developmental expression was observed only in mushroom-forming fungi (Merényi et al. 2022). ‘Shared’ orthogroups comprised most mitosis/meiosis related genes, but also some previously thought to be specific for complex morphogenesis (e.g., fasciclins). This indicates that sexual morphogenesis, even at the very simple forms that exist in C. neoformans, involves a morphogenetic program. Comparing genes that are developmentally regulated in fruiting bodies to those regulated in C. neoformans, thus, can help constructing more precise genetic models of complex fruiting body morphogenesis.
Cellular and regulatory processes involved in fruiting body development
Fruiting body development is an incredibly complex process and this complexity is reflected in the underlying cellular and genetic processes. Several cellular processes are emerging as important regulatory mechanisms underlying fruiting body morphogenesis, whereas some others that proved developmentally relevant in animals and plants seem to be of limited importance in fungi (e.g., gene body methylation, Borgognone et al. 2018, Wen et al. 2019). This chapter summarises the most important - mostly transcription related - processes described in fruiting bodies recently. Splicing and RNA interference are discussed below under chapters “RNA interference, small RNA” and “Alternative splicing and spliceosome components”, respectively.
Probably the most influential and most commonly assayed mechanisms of fruiting body development are transcriptional reprogramming and gene expression changes. Gene expression changes naturally associate with developmental processes and are informative regarding the cellular processes that take place during fruiting. In silico functional analyses of differentially or developmentally expressed genes have been a primary tool for mycologists to understand fruiting body functions. Developmentally dynamic gene expression patterns form the primary data source for this paper and the main part of the paper is devoted to discussing genes with conserved expression patterns. A significant unresolved question is how gene expression changes are regulated in various stages of fruiting body development. It is not known currently if transcriptional reprogramming events (e.g., during fruiting body initiation) are mediated by broad regulation of chromatin accessibility, by regulating the expression and activity of regulatory genes (e.g., transcription factors), or a mixture of these. A potentially significant regulatory mechanism is post-translational modification of transcription factors or molecular switches in intracellular signalling pathways, as demonstrated for the Hom2 or the Ras1 gene of S. commune (Knabe et al. 2013, Pelkmans et al. 2017). Another exciting avenue of future research is the regulatory potential of selective post-translational protein modification by the ubiquitin/proteasome system. Although direct evidence for its role in fruiting body development has not yet been published, indirect evidence was provided by the remarkable expansion of genes encoding F-box, BTB/POZ and RING-type zinc finger proteins in the genomes of mushroom-forming fungi (Krizsán et al. 2019). These gene families provide the substrate-specificity of ubiquitin ligases and have been shown, in other lineages, to be involved in transcriptional regulation (Kipreos & Pagano 2000).
Natural antisense transcripts (NATs) are RNA transcripts that show complementarity to transcripts of protein coding genes. NATs occur ubiquitously across eukaryotes and have been detected in mushroom-forming fungi too (Ohm et al. 2010, Muraguchi et al. 2015, Merényi et al. 2021). Because NATs do not encode proteins, they often evolve very fast and are hard to link to specific functions. Nevertheless, their expression is developmentally dynamic in all species examined to date, suggesting that they may be involved in regulating development. Muraguchi et al. reported that an antisense transcript of the stipe elongation related AspE gene is transcribed just before stipe elongation (Muraguchi et al. 2015). However, it should also be kept in mind that many NATs may result from random transcription initiation and may therefore be transcriptional noise in fruiting body transcriptomes (Merényi et al. 2022).
Allele-specific expression (ASE) refers to the imbalance of gene expression from two divergent alleles in diploid or dikaryotic organisms. Fungal dikaryons almost always harbour two allelically different monokaryons (heterokaryons), which can lead to intermediate, A- or B-dominated expression. ASE has so far been detected in fruiting body transcriptomes of A. bisporus (Gehrmann et al. 2018) and P. ostreatus (Merényi et al. 2021), but may be widespread in the Agaricomycetes. In A. bisporus, Gehrmann et al. suggested that ASE may facilitate the division of labour between nuclei in a dikaryon (Gehrmann et al. 2018). Another study, based on P. ostreatus, suggested that, at macroevolutionary timescales, ASE may be a neutrally arising phenomenon that can nevertheless generate adaptive gene expression variation (Merényi et al. 2022). It was found to be mostly characteristic of evolutionarily young genes, consistent with neutral evolution. Allele-specific expression has agriculturally broad consequences in hybrid genetics of plants (Bartoš et al. 2019) and could become a similarly central question in the mushroom industry.
RNA editing has recently been detected in fruiting body transcriptomes of several Ascomycota species (Liu et al. 2016, Liu et al. 2017, Teichert et al. 2017) and subsequently reported in Basidiomycota also (Zhu et al. 2014). Like alternative splicing, RNA editing provides a (post-)transcriptional mechanism for diversifying transcripts encoded by a single gene, by enzymatically modifying certain bases of the primary RNA transcript. A critical analysis recently found no evidence for RNA editing in fruiting body transcriptomes of P. ostreatus (Merényi et al. 2022). This study raised the possibility that, because allele-specific expression and RNA editing can generate similar signatures in RNA-Seq data, ASE or read mapping around splice sites may have been mistaken for RNA editing in some prior Basidiomycota analyses and that more research is needed to prove a role of RNA editing in mushroom development.
Several other exciting phenomena may be involved in the morphogenesis of Basidiomycota fruiting bodies, including regulatory processes that may yet to be discovered. For example, epigenetic regulation represents a potentially widespread but largely underexplored regulatory mechanism for fruiting body morphogenesis (Vonk & Ohm 2021). Similarly, upstream open reading frames (uORFs), which are open reading frames located in the 5’ untranslated region of an mRNA, can regulate (e.g., via encoded peptides) or interfere with the transcription of the gene (Hood et al. 2009). uORFs are known to be important in the asexual development of Ascomycota (Han & Adams 2001).
Genomic and transcriptomic resources
Genomic resources are also available for most model species of fruiting body development (e.g., C. cinerea (Schaeff.) Redhead, Vilgalys & Moncalvo, S. commune Fr., C. aegerita (V. Brig.) Vizzini) (Ohm et al. 2010, Stajich et al. 2010, Muraguchi et al. 2015, Gupta et al. 2018, Almási et al. 2019) and those used by the mushroom industry, including Hericium erinaceus (Bull.) Pers. (Gong et al. 2020), Ganoderma spp. (Chen et al. 2012), Flammulina spp. (Park et al. 2014, 2019, Chen et al. 2020), L. edodes (Berk.) Pegler (Chen et al. 2016, Shim et al. 2016, Sakamoto et al. 2017), Pleurotus spp. (Borgognone et al. 2018, Wang et al. 2018, Dai et al. 2019; Lee et al. 2021), Agaricus spp. (Morin et al. 2012, O’Connor et al. 2019, Sonnenberg et al. 2020), Tricholoma matsutake (S. Ito & S. Imai) Singer (Min et al. 2020), Sparassis crispa (Wulfen) Fr. (Kiyama et al. 2018), Hypsizygus marmoreus (Peck) H.E. Bigelow (Min et al. 2018), among others. More broadly, in the Agaricomycetes, draft genome sequences have been proliferating at a steady pace, due to interest in biodiversity, lignocellulose decomposition (Martinez et al. 2004, 2009a, Floudas et al. 2012, Riley et al. 2014) and mycorrhiza-formation (Martin et al. 2008, Kohler et al. 2015, Miyauchi et al. 2020). This trend has virtually eliminated genome data being the bottleneck in biological discovery in fungi. This is further supported by the spread of third generation sequencing techniques, long-read sequencing data, which allow more and more complete (telomere-to-telomere) assemblies and a range of analyses that draft genomes did not permit.
The continuous development of technologies has also influenced the identification of genes related to fruiting body morphogenesis. While early studies isolated fruiting body-specific proteins/genes using mutant analyses, hybridization techniques (among others), these were later substituted by RT-PCR and microarray studies, then by high throughput sequencing analyses, such as 5’-serial analysis of gene expression (SAGE) (Chum et al. 2008, Cheng et al. 2013) and most recently RNA-Seq. As of today, RNA-Seq datasets are available for a wide range of species and conditions. These include C. cinerea (Plaza et al. 2014, Muraguchi et al. 2015, Krizsán et al. 2019, Xie et al. 2020), Flammulina spp (Yan et al. 2019, Liu et al. 2020), L. edodes (Park et al. 2017, Sakamoto et al. 2017, Song et al. 2018, Wang et al. 2018, Yoo et al. 2019, Kim et al. 2020), Agaricus spp (Gehrmann et al. 2018, Lu et al. 2020, O’Connor et al. 2021), Pleurotus ostreatus (Jacq.) P. Kumm. (Wen et al. 2019, Merényi et al. 2022), H. marmoreus (Zhang et al. 2015b), C. aegerita (Orban et al. 2021), Phanerochaete chrysosporium Burds. (Krizsán et al. 2019), Lentinus tigrinus (Bull.) Fr. (Krizsán et al. 2019), Mycena kentingensis Y.S. Shih, Chi Y. Chen, W.W. Lin & H.W. Kao (Ke et al. 2020), Rickenella mellea (Singer & Clémençon) Lamoure (Krizsán et al. 2019), Armillaria ostoyae (Romagn.) Herink (Sipos et al. 2017), Pisolithus microcarpus (Cooke & Massee) G. Cunn (de Freitas Pereira et al. 2017), Laccaria bicolor (Maire) P.D. Orton (Ruytinx et al. 2021), S. commune (Ohm et al. 2010) and Auriculariopsis ampla (Lév.) Maire (Almási et al. 2019). Many of these datasets build the foundation of this study, by providing windows into developmentally expressed genes in different fungal species.
A general observation we made during the analyses we present hereafter is that the resolution of the transcriptomic data is key to identifying expression dynamics and patterns. Both tissue-wise and temporal resolution of the data allowed the discovery of more developmentally relevant expression patterns. We found several cases where tissue-resolved transcriptomes provided clear signal for conserved, tissue-specific expression of a gene, whereas in non-resolved transcriptomes the same gene showed a more or less flat expression curve. This is an inherent nature of bulk RNA-Seq data: spatial expression patterns average out in samples that contain large populations of multiple cell types.
Beyond gene expression, a broad array of other types of information could be leveraged for understanding fruiting body development. Notable recent examples include assays of the overrepresentation of certain gene families in the genomes of Agaricomycetes species (Krizsán et al. 2019), a population genomics study combined with selection analyses and gene expression profiling in L. edodes (Zhang et al. 2021c) to identify genes involved in fruiting body development (Zhang et al. 2021c), or ChIP-Seq analyses for assaying promoter occupancy and thus transcriptional activity (Vonk & Ohm 2021).
DESCRIPTION OF THE APPROACHES USED IN THIS PAPER
In this study, we address the puzzle of how a fruiting body develops and what novel insight can be gleaned from systematic comparisons of developmental transcriptomes. We aim at providing a comprehensive listing of putatively development-related gene families in the Agaricomycetes. We provide, for each discussed functional group or gene family, information on their expression, orthology to well-established model organisms (Saccharomyces cerevisiae, Schizosaccharomyces pombe, Aspergillus nidulans, Neurospora crassa) and to key mushroom-forming fungi as tables. We also annotate several conserved pathways in C. cinerea, based on strict 1-to-1 orthology to known pathway members in well-researched model systems (see Methods for details).
Each chapter is structured so that it starts with a literature review of the given gene family followed by a discussion of novel findings (if any) based on our meta-analysis of transcriptomes. Our main units of investigation are conserved developmentally expressed (CDE) orthogroups, which are groups of single-copy genes in which the majority (>65 %1) of the genes were developmentally expressed (see Methods and Merényi et al. 2022). For simplicity CDE orthogroups are considered to be groups of genes sharing similar functions (see limitations below) in fruiting body development.
We performed a meta-analysis of 12 developmental transcriptomes (Agaricus bisporus (Gehrmann et al. 2018), C. aegerita (Orban et al. 2021), A. ostoyae (Sipos et al. 2017), A. ampla (Almási et al. 2019), C. cinerea (Krizsán et al. 2019), L. bicolor (Ruyntinx et al. 2021), L. edodes (Zhang et al. 2021d), L. tigrinus (Krizsán et al. 2019), M. kentingensis (Ke et al. 2020), Ph. chrysosporium (Krizsán et al. 2019), P. ostreatus (Krizsán et al. 2019), S. commune (Almási et al. 2019) (Fig. 1). These species belong to two orders (Agaricales, Polyporales), which span an estimated 200 million years of evolution (Varga et al. 2019), and represent diverse ecologies and morphological adaptations, such as simple resupinate (Ph. chrysosporium), pileate-stipitate (e.g., C. cinerea) and cyphelloid (A. ampla, S. commune) forms. The most common fruiting body form among the examined species is pileate stipitate, consisting of a cap and a stalk, like most ‘stereotypical’ mushroom-forming fungi. Simpler species include cyphelloid ones, which are simplified, pendant cup-shaped fruiting bodies, that are considered to have evolved by secondary simplification (Virágh et al. 2021). Another simpler type is represented by resupinate forms, which are flat, crust-like fruiting bodies that presumably represent the ancestral fruiting body morphology in the Agaricomycetes (Varga et al. 2019).
Each of these species and their ecological adaptations show several unique aspects (e.g., fruiting body ecotypes) which could be reflected in gene expression patterns. However, we are here interested in only the shared aspects of fruiting body development, and therefore use the diversity of the compared species in focusing our attention to widely conserved fruiting body genes which should help define the core building blocks required for fruiting body morphogenesis.
The comparative approach we follow is paramount to separate ‘wheat from chaff’, i.e., developmentally regulated genes that are relevant for the development of fruiting bodies in general from genes with taxonomically restricted or species-specific developmental roles. This builds on the assumption that a gene involved in core fruiting body functions should be conserved across Agaricomycetes and should show similar expression dynamics in all species. Therefore, in our approach we focus on conservation of both sequence and expression. A comparative approach is also desirable because it is now well-known that gene expression is associated with ‘biological noise’, which may influence the detection of differentially expressed or developmentally regulated genes (Merényi et al. 2022) in any single species, whereas across-species comparisons effectively cancel the effects of gene expression noise.
This study, like all others, has limitations. First, it should be noted that evidence for gene function remains, in an overwhelming majority of cases, circumstantial, inferred from expression patterns or extrapolated from the functions of orthologous genes in model fungi. At the moment we rely on these types of evidence due to the scarcity of developmental genetic and in-depth functional studies in most Agaricomycetes. Despite the inferential nature of the study, many orthologs display very conserved expression patterns across Agaricomycetes species (e.g., aquaporins in stipe tissues, see below), allowing us to make confident predictions on the function of such genes. Second, a perfect comparative transcriptomic study would compare corresponding developmental stages across species. In the Agaricomycetes establishing homology relationships among individual species is complicated (see above). Furthermore, the developmental transcriptomes we used differ in the sampled time points and resolution (e.g., tissue-wise or bulk) and do not follow a unified nomenclature for developmental stages (e.g., aggregate in S. commune vs. hyphal knot in C. cinerea). Therefore, we here focused broadly on comparing expression dynamics and the shape of the expression curve, rather than attempting to find 1-to-1 correspondence between stages of different species. Finally, in the present study, we focus on genes with developmentally dynamic expression. However, not all morphogenetically relevant genes show expression dynamics, constantly expressed genes can often be important for development, for example, if regulated post-transcriptionally (e.g., alternative splicing isoforms, Gehrmann et al. 2016, Krizsán et al. 2019) or post-translationally (e.g., phosphorylation, ubiquitylation etc. (Knabe et al. 2013, Pelkmans et al. 2017).
METHODS
Bioinformatic analyses of RNA-Seq data
The raw RNA-Seq data of 12 previously published Basidiomycota species were reanalysed following Merényi et al. (2022). These species were chosen because they have published, high quality (tissue and time resolved) expression data available. The following reference genomes were used: C. cinerea (AmutBmut pab1-1 v3.0, Muraguchi et al. 2015), A. ostoyae (C18/9 (Sipos et al. 2017), A. ampla (NL-1724 v1.0, Almási et al. 2019), S. commune (H4-8a and H4-8b, Ohm et al. 2010), L. bicolor (v2.0, Martin et al. 2008), L. edodes (Le (Bin) 0899 ss11 v1.0, Zhang et al. 2021c), L. tigrinus (RLP-9953-sp, Wu et al. 2018), Ph. chrysosporium (RP-78 v2.2, Ohm et al. 2014), P. ostreatus (PC15 v2.0, Riley et al. 2014), M. kentingensis (Ke et al. 2020), A. bisporus (var. bisporus H97, Morin et al. 2012), C. aegerita (AAE-3, Gupta et al. 2018). To remove adaptors, ambiguous nucleotides and any low quality read ends, reads were trimmed using bbduk.sh and overlapping read pairs were merged with bbmerge.sh (part of BBMap/BBTools; http://sourceforge.net/projects/bbmap/) with the following parameters: qtrim=rl trimq=25 minlen=40. A two-pass STAR alignment (Veeneman et al. 2016) was performed against reference genomes with the same parameters as in our previous study (Krizsán et al. 2019) except that the maximal intron length was reduced to 3000 nt. Reads were counted and summarised for each exon with FeatureCounts (Liao et al. 2014) taking into account the mode of sequencing (paired-end, single-end). Since the C. aegerita transcriptome was obtained with the Quantseq method, reads were counted only in the -100 - +400 region of the 3’ ends of the genes. Read count data were normalized using EdgeR (Robinson et al. 2010). Expression levels were calculated as fragments per kilobase of transcript per million mapped reads (FPKM).
Developmentally regulated genes were identified as before (Krizsán et al. 2019, Merényi et al. 2021), based on a stricter four-fold or a more permissive two-fold change cutoff between adjacent developmental stages or tissue types. Genes showing a 4-fold upregulation in the first primordium stage relative to vegetative mycelium were termed ‘FB-init’, and considered regulated during fruiting body initiation. It should be noted that the data we analyse is noisy, based on different chemistries, different labs and protocols, etc., which introduces a certain amount of data loss in our analyses. Nevertheless, the transition to fruiting body development is such a dramatic change that we expect similar gene expression changes to be discoverable across species, despite the heterogeneity of the data. This is supported by previous comparative transcriptomic studies (Plaza et al. 2014, Krizsán et al. 2019, Merényi et al. 2020, 2022) as well as the biologically meaningful conclusions obtained here.
Orthology based on reciprocal best hits
Proteins of each species were searched against the proteomes of other species using the reciprocal best hit (RBH) module of MMSeqs2 (Steinegger & Söding 2017). To remove spurious RBHs, a bidirectional 50 % coverage and at least 1e-6 e-value were required. Proteins were clustered into single copy clusters using a connected component clustering algorithm in the igraph package, based on reciprocal best hits. This step clustered 74.3 % of the proteins in the examined species. The other 25.7 % of proteins form clusters with duplication in any or more species. For these 1:1 orthogroups were circumscribed using the ‘ortholog coding’ algorithm of the COMPARE pipeline (Nagy et al. 2014). In the case of terminal duplications, orthogroup membership was decided based on connectivity; the protein which showed more hits to other members of the orthogroup was selected while the other(s) moved to separate orthogroup(s). Complete orthology relationships of genes across all 12 species are given in this paper are presented in Supplementary Table S1.
This approach is suitable for identifying moderately to very conserved gene groups, whereas for highly volatile genes (i.e., those that show frequent duplication/loss), a more liberal approach building on orthologous groups (like in Krizsán et al. 2019) is more appropriate. For fast-evolving genes, approaches looking at sequence conservation might not work. Therefore, in the case of certain gene families, such as defence genes or cell wall remodelling CAZymes, we relied on primary gene family classification instead of reciprocal best hit based orthogroups. In the case of CAZymes, we followed the cazy.org classification (Levasseur et al. 2013).
Identification of CDE orthogroups
The central unit of discussion in this paper is referred to as ‘conserved developmentally expressed (CDE) orthogroup’, which refers to groups of single-copy genes (one gene per species) in which the majority of genes is developmentally regulated either at fold change >2 or >4. As a principal rule, we considered an orthogroup to be a CDE orthogroup if >65 % of the genes in it were developmentally regulated, and at least 8 species were represented. Deviations from this rule were allowed in unique cases (e.g., taxonomically restricted but important gene family) and are transparently presented in tables and supplementary tables under each of the following chapter.
Phylogenetic analyses
The linoleate diol synthase family has been examined using phylogenetic analyses of amino acid sequences. For these sequences were aligned using mafft-linsi v. 170427 (Katoh & Standley 2013). A maximum likelihood phylogenetic tree was inferred using RAxML v. 7.1.2 (Kozlov et al. 2019) under the WAG model with gamma distributed rate heterogeneity and one hundred bootstrap replicates.
RESULTS: DEVELOPMENTALLY EXPRESSED GENE CLASSES IN THE AGARICOMYCETES
In this paper we synthesized literature reviews with a meta-analysis of developmental transcriptomes to identify conserved gene families involved in fruiting body development. For the latter we reanalysed data for 12 species and identified developmentally regulated genes, i.e., those that showed >2- or >4-fold changes between successive developmental stages or adjacent tissue types (Supplementary Table S2). Statistics on developmentally expressed genes are provided in Supplementary Table S2/b. Numbers of developmentally regulated genes were similar to those reported by previous studies, (Almási et al. 2019, Krizsán et al. 2019, Merényi et al. 2020, 2022) therefore, we here do not discuss these in detail, rather focus on those that are conserved across species.
We organized developmentally regulated genes into strict 1-to-1 orthogroups following the logic of Merényi et al. (2022). Orthogroups in which >65 % of the genes were developmentally regulated were designated as conserved developmentally expressed (CDE) orthogroups at fold change values of >2 or >4. We identified 921 CDE orthogroups across the twelve Agaricomycetes species; these form the basis of the discussion presented in the rest of this paper. Statistics on the number of species which had developmentally regulated genes in these orthogroups are shown on Fig. 2A. The transcriptome data of A. bisporus was an outlier in several aspects. We currently do not know if this is due to biological differences of this species or technical reasons, nevertheless this transcriptome was assigned lower weight and discussed rarely in the following chapter. By relying on conserved developmentally expressed orthogroups, we here take shared expression dynamics as a proxy for conservation of function. It should be mentioned that this approach may be, in several cases, only a crude approximation of real conservation of function, but facilitates comparative discussion of gene families and their speculated functions in fruiting body morphogenesis.
Fig. 2.

The distribution of conserved developmentally regulated orthogroups across species. A.The number of orthogroups as a function of the number of species in which the orthogroup is developmentally regulated at fold change >2 (top) and fold change >4 (bottom). Horizontal axis represent the number of species. B. Numbers of genes in each of the functional categories discussed in the paper.
We functionally characterised CDE orthogroups and arranged them into broader groups based on function inferred by orthology to genes in well-known model systems (S. cerevisiae, Schizosaccharomyces pombe, A. nidulans, N. crassa) and/or their Gene ontology and InterPro annotations. We defined 17 broader functional groups, cell division/proliferation, defence, transcriptional regulators, signal transduction, cell wall biosynthesis and remodelling, cell surface proteins, secondary metabolism, cytoskeleton, basic metabolism and acetyl-CoA production, lipid metabolism, storage carbohydrate metabolism, ubiquitin/proteasome system, transporters, functionally poorly known genes, unannotated genes, stress response related genes and ferric reductases (Fig. 2B). The main categories were further subdivided into finer groups to facilitate discussions of putative functions. We categorized developmental genes manually to the best of our knowledge based on inferred gene function, though the subdivision is probably not impeccable. For example, overlaps exist between chromatin remodelling and DNA replication related regulatory factors or between the latter and cytoskeleton related genes (e.g., kinesins involved in moving chromatids during mitosis).
The two largest categories were functionally poorly characterised and unannotated genes, these contained 182 and 176 CDE orthogroups, respectively (Fig. 2B). The first, ‘functionally poorly characterised genes’ contains orthogroups which we could not confidently link to any fruiting body functions. It is possible that this category could be subdivided and with further manual annotation more orthogroups could be explained in the context of fruiting body development. On the other hand, unannotated genes are those that contain no existing Pfam or InterPro domain signatures and lack all kinds of functional annotation. The largest functional group for which a function could be determined was related to cell proliferation (DNA replication, DNA repair, mitosis, meiosis) followed by cell wall biosynthesis and remodelling. Transcriptional regulators (including transcription factors, chromatin-related and RNA-binding regulatory proteins), transporters and Acetyl-CoA production related genes were also represented by a considerable number of genes. Certain functional categories, such as ‘defence’ and ‘secondary metabolism’ contained only a small number of CDE orthogroups. In these cases, these low numbers reflect the lack of conservation of gene sequences, rather than the lack of a role of these gene families in fruiting bodies.
Our discussion is guided here by conservation of developmentally regulated families. However, there are gene families that do not form conserved orthogroups, but are nevertheless very important for fruiting body development. These can be either multigene families that undergo frequent duplication and thus orthology relationships are intricate (e.g., hydrophobins), or fast-evolving families in which divergence quickly erases orthology relationships (e.g., F-Box proteins, defence-related proteins). Therefore, in addition to gene families/pathways identified through CDE orthogroups, we discuss broader, fast-evolving gene families based on annotations at the gene family level only (i.e., without orthogroups) and performed manual annotations to identify broader containing gene families or cellular pathways. Accordingly, in addition to the 921 CDE orthogroups, we discuss further 558 gene groups which belong to broad functionalities involved in fruiting body development. For some processes we were able to reconstruct (nearly) complete pathways, such as in the case of mitosis/meiosis related genes or fatty acid biosynthesis, whereas in other cases we only document the widespread developmental regulation and expression profile conservation of a gene family, but without information on the containing pathway of broader cellular function. For some gene groups, such as cytoskeletal genes, GPCRs or MAP kinase pathways, we provide annotations despite their limited expression dynamics during fruiting body development. Such gene families may be important in fruiting body development despite their low expression dynamics, therefore, we decided to annotate them in order to facilitate comparative discussion across mushroom-forming fungi and more thoroughly investigated Ascomycota model systems.
In the following chapter we discuss each of the more important gene groups in detail, with emphasis on expression profiles, putative function and role in fruiting body development. Besides well-known ones, we intentionally focus on lesser-known gene families with widespread developmental expression, to potentially distil new insights into fruiting body development. We provide catalogues and references to recent reviews for well-researched gene families as well (e.g., hydrophobins or laccases).
Cell division, proliferation and growth
Meiotic and mitotic gene expression show two distinct expression peaks
Mitosis and meiosis are key to the development and growth of multicellular organisms. Mushroom fruiting bodies are no exception: mitosis leads to cell proliferation early in fruiting body development, whereas meiosis happens in basidia, which are localized to gills, to produce spores.
Transcriptomic data provided clear evidence for expression peaks of genes related to mitotic or meiotic cell division, as well as associated processes in DNA replication, repair, chromosome dynamics/movement etc., during fruiting body development. The enrichment of meiotic/mitotic genes during fruiting body development and sporulation was noted in C. cinerea (Burns et al. 2010, Muraguchi et al. 2015, Cheng et al. 2015, Krizsán et al. 2019), Pisolithus microcarpus (Pereira et al. 2017), Tricholoma matsutake (Tang et al. 2020) and L. edodes (Song et al. 2018), Agaricus blazei (Lu et al. 2020) as well as in a six-species comparison published by Krizsán et al. (2019), among others. Similar signal for meiotic gene upregulation has been detected in Ascomycota fruiting bodies as well (Rodenburg et al. 2018). C. cinerea has been used as a model system of Basidiomycota meiosis, in particular in so called white cap mutants that have meiosis-associated defects (Lu et al. 2003). These works revealed many conserved and some unique aspects of the meiotic process as well as resulted in meiosis being one of the best understood processes in this species (Seitz et al. 1996, Stassen et al. 1997, Li et al. 1999, Gerecke & Zolan 2000, Nara et al. 2001, Cummings et al. 2002, Lu et al. 2003, Namekawa et al. 2004, Muraguchi et al. 2008a, Many et al. 2009).
Probably the most detailed such study was performed by Burns et al. (2010), who investigated by microarray the transcriptional events that happen in C. cinerea gills during a time course that encompasses the meiotic events. They found that 2 721 genes exhibited changing probe intensity during the six time points around meiosis. Using dikaryotic mycelium as reference, they identified 886 genes that were expressed in gill tissue only; these genes included core meiotic components. Like in other species, genes were induced in successive waves during the meiotic process; these waves were arranged into 9 clusters based on expression trajectories. The clusters separated genes into functional groups, e.g., those required for prophase I or sporulation in early and late in the time series, respectively. Early genes further included ones related to DNA replication, cytoskeleton organization and regulation. Late induced genes in their study are mostly related to spore formation and show overlaps with genes related to ascospore formation in S. cerevisiae (Chu et al. 1998, Primig et al. 2000) and Sch. pombe (Mata et al. 2002). Ribosomal protein coding gene expression was found to be high up to karyogamy, at which point it starts to decrease and does not lift up again. This might reflect the shutting down of protein synthesis as the fruiting body enters the sporulating phase.
Pereira et al. (2017) examined sporogenesis in Pisolithus, an ectomycorrhizal fungus in the Boletales, and found that meiotic genes (annotated by KEGG) showed dynamics throughout the development of basidiospore-containing peridioles. In this species spores are produced in peridioles, lentil-shaped to globular compartments within the fruiting body. Several genes that were differentially expressed in peridioles of different maturation stages were annotated as meiotic/cell cycle or cell division related, most of which showed an expression peak in unconsolidated, young, and mature peridioles. These observations are consistent with microscopic observations on the sporogenesis of P. microsporus (Campos & Costa 2010).
We annotated 180 conserved orthogroups as involved in cell proliferation, such as mitosis, meiosis, DNA replication, DNA repair or the cell cycle, based on expression conservation 141 of these qualified as CDE orthogroup (Supplementary Table S3). These included previously functionally characterised genes such as ku70, rad50 and Mre11 of C. cinerea (Many et al. 2009, Nakazawa et al. 2011), DMC1 (=Lenedo1_1211634) of L. edodes (Sakamoto et al. 2009), ku80 of S. commune (De Jong et al. 2010) and Msh4 of P. ostreatus (Lavrijssen et al. 2020). Most of these genes showed developmental regulation and similar expression profiles in most of the species. This is not surprising, given that both mitosis and meiosis have well-documented roles in sculpting fruiting bodies. Nevertheless, to our knowledge, these genes represent the most comprehensive, although surely not complete, list of cell proliferation related genes to date. We tentatively subdivided CDE orthogroups into ones related to DNA replication, DNA repair, meiosis as well as both mitosis and meiosis; these contained 87, 26, 13, 54 genes, respectively (Supplementary Table S3). These include members of several well-characterised protein complexes described from S. cerevisiae, such as Ku, SMC, MCM, Ndc80, Gins and cohesin complexes (Supplementary Table S3). Orthologs of C. cinerea arp9, which was described as a putative actin-related protein involved in chromatin remodelling (Nakazawa et al. 2016), were also found to follow similar expression profiles, although at moderate dynamics (Supplementary Table S3).
These genes showed characteristic expression profiles in most species. The expression of meiotic genes shows a distinct peak in gill tissues (Fig. 3, Supplementary Fig. S1) or in stages that contain meiotic tissue. Meiotic gene expression precedes early phases of sporulation and was clearly discernible in C. neoformans or P. ostreatus in previous transcriptomic studies (Liu et al. 2018, Merényi et al. 2022). Because the onset of meiosis is tightly regulated in most species (among mushrooms best known in C. cinerea, Kües 2000) meiotic gene expression also provides a landmark to calibrate developmental chronologies among species where this may be hard to do because of morphological divergence. It should be noted that some species develop in ways so that we do not expect a clear peak. For example, cell proliferation and spore production is not synchronized in S. commune, where it appears that sporulation progresses from the basal, older parts and progresses towards the edges of the fruiting body (Kües & Liu 2000). In S. commune, this may be explained by the highly derived morphology of the fruiting bodies, consisting of an assemblage of cyphelloid fruiting bodies. Therefore, we would not expect one clear peak of meiotic gene expression, unless we sample different zones of the fruiting body separately.
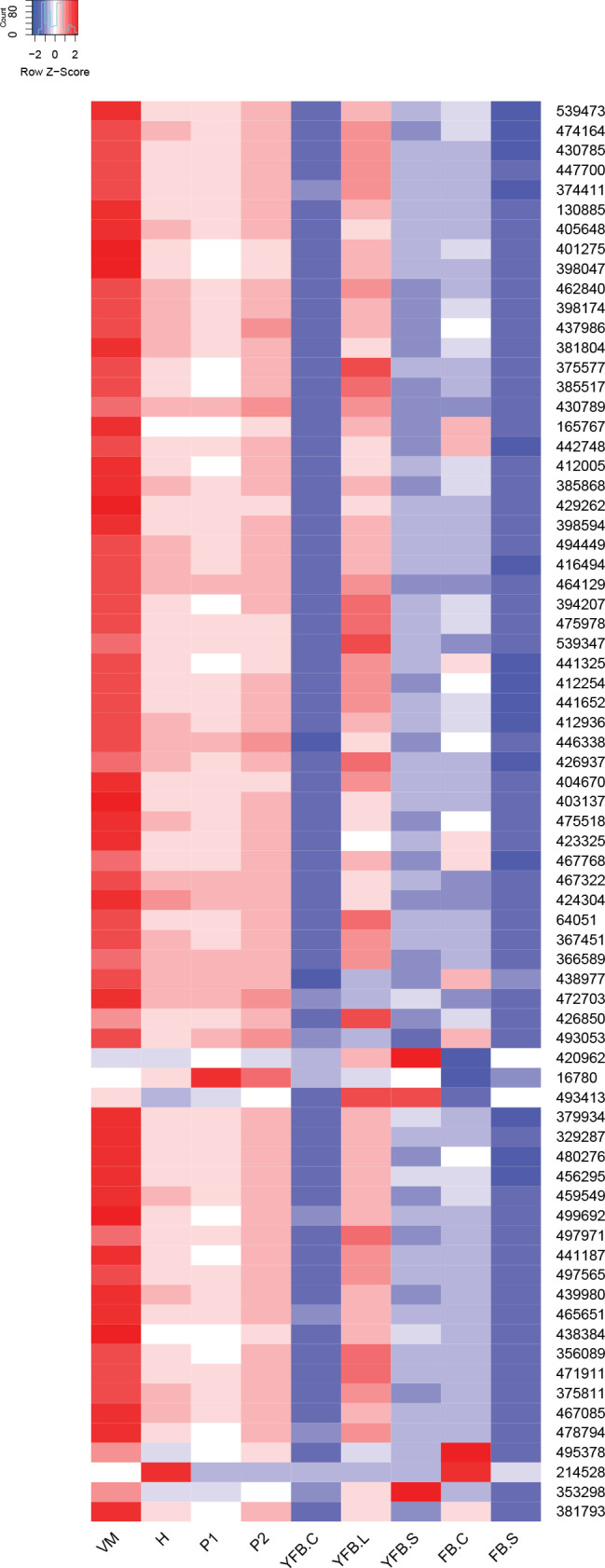
Fig. 3.

Expression heatmap of DNA replication, repair, mitosis and meiosis related genes in C. cinerea. A model expression trajectory, illustrating the two peaks characteristic of most examined species, is shown above the left panel. Well-delimited complexes mentioned in the paper are shown separately. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively. Developmental stages are abbreviated as follows: VM – vegetative mycelium, H – hyphal knot, P1 – stage 1 primordium, P2 – stage 2 primordium, YFB.C – young fruiting body cap, YFB.L – young fruiting body gills, YFB.S – young fruiting body stipe, FB.C – mature fruiting body cap (including gills), FB.S – mature fruiting body stipe.
On the other hand, mitotic, DNA replication and repair genes showed a clear peak associated with primordium stages, and their expression levelled off in mature fruiting bodies (Fig. 3, Supplementary Fig. S1). This probably reflects intense cell proliferation in primordia, which establishes the main tissues and developmental modules of the mushroom. The expression of these genes later decreases to low levels in young fruiting body stipes and caps (also high in meiotic tissues where one round of mitosis happens) and in mature fruiting bodies. This peak was also detected in previous studies on Agaricus blazei (Lu et al. 2020) and P. ostreatus (Merényi et al. 2022) and probably relates to the biphasic development of fruiting bodies, where the first phase is concerned with cell proliferation and tissue formation whereas the second with growth by cell expansion without much change in cell numbers. Our data suggest that this pattern should be conserved across agarics, but potentially missing or less clear in species that follow different developmental patterns (e.g., polypores).
Cryptococcus neoformans recently proved to be a particularly useful model for teasing apart morphogenetic processes (Merényi et al. 2021). This species forms simple fruiting bodies composed of lawns of aerial hyphae bearing basidia. Liu et al. pointed out in Cryptococcus, that genes co-induced with meiotic genes might be related to basidium morphogenesis, a process that greatly overlaps with meiosis (Liu et al. 2018). Based on transcriptome data it may be hard to separate such genes from meiotic genes, because of the temporal & spatial overlap of their expected expression. Nevertheless, one of the genes reported by Liu et al., C. neoformans Csa1 (C. cinerea protein ID: 471238, see Supplementary Table S13), which has been implicated in basidium development (and to a lesser extent meiosis (Liu et al. 2018), is developmentally expressed in 10/10 species in our data and shows expression peaks that overlap with that of meiotic genes.
An orthogroup that includes the serine-threonine kinases Sch. pombe Ran1 and S. cerevisiae Sks1 (represented by C. cinerea 456276, Supplementary Table S3) showed marked peaks in gills of mature fruiting bodies in all agaricoid species (except C. cinerea) and in mature fruiting bodies in species with no separable gills or caps (S. commune, A. ampla). Ran1 is a negative regulator of Sch. pombe meiosis (with Mei2 as target, Caligiuri et al. 1997), whereas Sks1 is involved in the adaptation to low glucose concentrations and pseudohyphal growth in yeast. Despite this apparent strong contradiction, the characteristic expression peaks in meiotic tissues and developmental stages in mushroom-forming fungi suggest roles in sporulation-related processes, possibly the repression of meiotic processes during spore morphogenesis.
In summary, mitotic and meiotic (including associated processes such as DNA replication and repair) gene expression showed conserved patterns across the examined mushroom-forming fungi. This probably reflects, on one hand, intense cell proliferation during primordium development and, on the other hand, meiosis that precedes sporulation in basidia. These gene expression patterns are easily recognizable in transcriptomic data and provide landmarks for calibrating developmental series. The downregulation of mitosis related genes in the second half of fruiting body development coincides with ceasing cell proliferation and the transition of the mushroom to growth by cell expansion.
Ribosomal genes
Ribosomes produce proteins, the necessary building blocks for living cells. They are composed of four RNA subunits and up to 80 different proteins. The number of ribosomes, and thus ribosomal protein expression, in a cell is proportional to its protein synthesis demand (Kraakman et al. 1993, Jorgensen et al. 2002). Therefore, ribosomal protein expression has been used as a proxy for the proliferative activity of a cell or a tissue and we expect it can be similarly informative in mushrooms too.
We observed a preponderance of ribosomal protein genes among CDE orthogroups, especially at FC>2 and to a lesser extent at FC>4. To look globally at structural proteins of ribosomes, we identified ribosomal protein genes based on previously reported ribosomal compositions, excluding mitochondrial ribosomal proteins and ribosome biogenesis-related genes. The 74 orthogroups identified this way (Supplementary Table S4) showed very characteristic expression dynamics, albeit at low fold change values, in most species. Ribosomal genes were highly expressed in the early developmental stages of most species followed by gradual shutting down in young and mature fruiting bodies. A second, sharp expression peak was observed in gill tissues of P. ostreatus, A. ostoyae, A. bisporus and C. cinerea (Fig. 4, Supplementary Fig. S2). Like in the case of mitotic and meiotic genes, we hypothesize that these two peaks correspond to an early, proliferative stage of development and the protein synthesis demand of spore production/meiosis. Ribosomal genes seem to be co-expressed with genes involved in cell division and proliferation (meiotic, DNA replication and repair, see above) in both primordia and in the gills. This could signify the need for increased protein production for cell proliferation in primordia and for meiosis and spore production, although other processes happening in gills requiring a large amount of protein cannot be ruled out. The characteristic expression of ribosomal genes is a conspicuous pattern and was detected in partial or complete form in several transcriptomic papers in both Basidiomycota (Cheng et al. 2013, Zhou et al. 2014, Zhang et al. 2015b, Song et al. 2018, Krizsán et al. 2019, Liu et al. 2020, Lu et al. 2020) and Ascomycota (Rodenburg et al. 2018, Tong et al. 2020).
Fig. 4.
Expression heatmap of ribosomal protein encoding genes in C. cinerea. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively. Developmental stages are abbreviated as follows: VM – vegetative mycelium, H – hyphal knot, P1 – stage 1 primordium, P2 – stage 2 primordium, YFB.C – young fruiting body cap, YFB.L – young fruiting body gills, YFB.S – young fruiting body stipe, FB.C – mature fruiting body cap (including gills), FB.S – mature fruiting body stipe.
The vegetative mycelium generally showed high ribosomal protein-coding gene expression, though not in all species, which we hypothesize depended on whether samples for RNA-Seq were harvested from older, inactive or actively growing (e.g., colony edge) parts of the mycelium.
A ribosome-bound Hsp70 chaperone protein formed a CDE orthogroup developmentally regulated in 9 species (FC>2) (C. cinerea protein ID: 438977) (Supplementary Table S4). This gene was also detected previously as upregulated in primordia of C. cinerea (Cheng et al. 2013). It is orthologous to S. cerevisiae Ssb2, a Hsp70-family chaperone that is bound by the ribosome and escorts the folding of nascent polypeptides (Peisker et al. 2010).
In summary, a characteristic expression trajectory of ribosomal genes is characteristic of most mushroom-forming fungi, which likely reflects high protein demands of intensely growing tissue in primordial stages and gills and supports a bi-phasic model of fruiting body development (see below).
Growth by cell expansion and turgor manipulation - a biphasic model of fruiting body development
A key difference in fruiting bodies and hyphae is that in the latter growth and expansion takes place at the tip, whereas in the former, cell expansion can vary between apical and isotropic. Already de Bary established, by studying Mycena vulgaris, that mushroom fruiting bodies grow mostly by cell expansion not by cell proliferation (Bary et al. 1887). This appears to be a rather widespread feature of fruiting bodies and shows broad similarity to growth by cell expansion in plant fruits (Krizsán et al. 2019). More precisely, fruiting body development seems to be divided into a first proliferative stage, during which cell division, differentiation as well as tissue formation takes place, and a growth phase, which is primarily concerned with cell expansion to achieve the mature shapes and sizes of the fruiting bodies (Moore et al. 1979, Kües 2000, Straatsma et al. 2013, Kües & Navarro-González 2015, Krizsán et al. 2019, Liu et al. 2021). Of the better investigated species, this two-phased development is probably most spectacular in C. cinerea, which is adapted to very rapidly lifting cap above ground and expanding it for spore dispersal (Kües 2000).
It should be noted that the two-phase development is not fully universal among fungi and does not always mean a complete separation of proliferative and expansive phases. For example, Craig et al. (1977) showed that the top regions of A. bisporus stipes elongated up to 9-fold, whereas cell lengths increased up to 2.2-fold, and from these data derived that cell division also occurs to achieve this elongation rate. In S. commune, it appears that outer parts of the fruiting body are still growing (by cell proliferation) while older parts are already sporulating (Kües & Liu 2000).
Whether for the entire fruiting body or only parts of it, growth by cell expansion seems to be a widespread trait during the second half of development of mushrooms. How cell expansion is achieved, in general, is poorly known, but it probably requires water supply, turgor manipulation (by osmolytes), cell wall elasticity and new cell wall material deposition. Water supply may be realized by the regulated expression of aquaporin genes (see below). On the other hand, the identity of the osmolytes has not been resolved yet. In the Ascomycota ions, ion channels, glycerol and sugars were reported as main players of turgor manipulation (Fischer et al. 2004). In Basidiomycota fruiting bodies, sugars (mannitol, trehalose) offer themselves as straightforward candidates, but Tan & Moore (1994) speculated that high molecular weight sugars are unlikely to be the osmolytes that drive stipe expansion. Trehalose and polyols have been excluded as osmolytes (Kües 2000, Trail et al. 2005), whereas potassium oxaloacetate has been hypothesized, which would be consistent with the upregulation of oxalate metabolism (see below), but not confirmed (Knoll 2011, Kües 2000). On the other hand, ions would either need to be transported to the fruiting body from the mycelium, or released locally.
Factors modulating cell wall elasticity and remodelling are better known. During fruiting body growth, cell wall deposition occurs throughout the cell surface, and not only in the tip region as in hyphae, which was termed diffuse extension (Kües 2000). In Agaricus bisporus larger proportion of β-1,6-linked glucose side branches in a β-1,3-glucan backbone was observed in the rapidly elongating top segment of the stipe (Mol & Wessels 1990). This causes hydrogen-bonds among glucan chains to be weaker in rapidly elongating stipe compared with mature stipe and vegetative hyphae. It has been hypothesized that these weak bonds are disrupted and reformed by turgor pressure rather than hydrolysing enzymes (Mol et al. 1990). The architecture of cell wall also regulates stipe elongation; in primordial stages chitin microfibrils are arranged isotropically, which is remodelled to helical arrangement during development (Kamada et al. 1991). The turgor pressure acting on the cell wall and insertion of new chitin microfibrils allow elongation and in that dynamic rearranges of microfibrils chitin synthases, chitinases and glucanases play the major role in C. cinerea (Kamada et al. 1991). Niu et al. observed differences in structure of chitin microfibrils in elongating and mature stipe regions (Niu et al. 2015). Extension of the cell wall happens by insertion of newly synthesized microfibrils. In contrast, studies in F. velutipes and C. cinerea suggested that stipe cell wall extension is mediated by endogenous expansin-like proteins, not turgor pressure or cell wall glycoside hydrolases (Fang et al. 2014, Zhang et al. 2014). Plant expansins mediate Turgor-mediated cell expansion (Baccelli 2015) and several other morphogenetic processes (e.g., organ abscission, fruit softening, Sampedro & Cosgrove 2005). The auxin-driven expansion of plant cells without changes in cell number is known as acid growth (Rayle & Cleland 1992, Arsuffi & Braybrook 2018), because low pH is required by cell wall modifying proteins (e.g., expansins) to loosen the cell wall. In fungi, it is not known if an analogous process exists, but the acidifying effect of external CO2 has been hypothesized as a factor in stipe cell elongation (Liu et al. 2021) and there is evidence for expansins participating in the process (see below). Accordingly, high external CO2 during mushroom cultivation was speculated to cause malformed, long-stipe fruiting bodies by acidifying the extracellular space and activating cell wall modifying proteins (Liu et al. 2021).
Acetyl-CoA production by central carbon metabolism
We found evidence for widespread developmental expression of multiple acetyl-coenzyme A (Ac-CoA) producing enzyme families. Ac-CoA is a key intermediate in central carbon metabolism as well as other processes (biosynthesis of lipids and certain amino acids (Chen et al. 2015). Under normal carbon metabolic conditions, the generated Ac-CoA, which comes from either glycolysis or fatty acid beta-oxidation enters the TCA cycle, where it is oxidized to CO2 for energy (ATP) production. However, Ac-CoA is required also for the production of biological membranes (Strijbis & Distel 2010, Chen et al. 2015), and our results point in the direction of multiple basic metabolic pathways converging on excess Ac-CoA production for membrane biosynthesis. Combined with gene expression profiles of these genes, we hypothesize that excess Ac-CoA is being channeled into ergosterol and fatty acid production during fruiting body development, as detailed below.
For this work, we started from Ac-CoA related CDE orthogroups detected in the transcriptomes of the examined species and developmental stages, which belonged to several pathways. We reasoned that if there is an increased demand for Ac-CoA at certain developmental stages of fruiting bodies, then this should be reflected in the regulation of Ac-CoA-producing pathways. Thus, we annotated the TCA cycle, the glyoxal cycle, the fatty acid beta-oxidation, ethanol utilization, glycolytic as well as pyruvate decarboxylation pathways of C. cinerea, based on protein orthology to corresponding pathways of S. cerevisiae (Fig. 5, Supplementary Fig. S3, Table 3). These led to a model we put forth on Fig. 5, with the additional note that it reflects one possible synthesis of the observations into a mechanistic model compatible with what we know about fruiting body development, but that other interpretation may also be viable.
Fig. 5.

Basic metabolic pathways of C. cinerea. Genes in the pathways are denoted by the S. cerevisiae gene name, followed by the protein ID of the C. cinerea ortholog and, in parentheses the number of species in which the gene was developmentally regulated at fold change >2/>4. Pathways are colored differently for clarity. The branch to oxalate accumulation during wood-decay was suggested by Munir et al. (2001).
First of all, we found that oxaloacetase (oxaloacetate lyase) genes were developmentally expressed in 11 species (FC>4, C. cinerea 499734). Oxaloacetases produce oxalate and acetate from oxaloacetate, which is an intermediate of both the TCA and the Glyoxylate cycles. In S. cerevisiae acetate is converted to Ac-CoA by acetyl-CoA synthases (Acs1), orthologs of which were developmentally regulated in 11 and 4 species at fold change of 2 and 4, respectively (represented by C. cinerea 500719). We also found a CDE orthogroup comprising putative orthologs of the S. cerevisiae Ac-CoA hydrolase Ach1 (represented by C. cinerea 442787, FC>2/4: 7/5). Ach1 is a relatively poorly known gene, which was recently shown to be responsible for the conversion of Ac-CoA to acetate, which can then be easily transferred from the mitochondrion to the cytoplasm (Chen et al. 2015). Cytoplasmic acetate is then converted to cytoplasmic Ac-CoA, a starting material for lipid synthesis, suggesting that Ach1 mediated Ac-CoA supply for lipid synthesis. Remarkably, Ach1 orthologs are present in only 8 species and completely missing in A. bisporus (missing even in telomere-to-telomere assemblies, Sonnenberg et al. 2020), M. kentingensis, Ph. chrysosporium, L. tigrinus, Pt. gracilis. This patchiness might indicate that the role of these genes is dispensable and probably others can take over their roles.
Acetate is also an intermediate of the ethanol utilization pathway, in which two enzymes form a linear pathway that terminates in the production of Acetyl-CoA (Flipphi et al. 2003). Aldehyde dehydrogenases orthologous to AldA of A. nidulans and Ald4 of S. cerevisiae formed a CDE orthogroup (C. cinerea protein ID: 500663, 9/7); the encoded enzymes also contribute to acetate production and, in C. cinerea, showed peak expression in young fruiting body caps. Ald4/AldA converts the toxic compound acetaldehyde to acetate (Felenbok et al. 2001). Although the ethanol utilization pathway is mostly discussed in the context of extracellular ethanol (a nonfermentable carbon source) uptake and degradation, as well as anaerobic fermentation, acetaldehyde can also be converted to ethanol (Fig. 5), if it is present in excess amounts in the cells. This may explain the developmental regulation of orthologs of S. cerevisiae Adh3 and A. nidulans AlcA/AlcC (represented by C. cinerea 500663), which were developmentally expressed in 11/7 species (FC>2/4, Supplementary Table S5). Enzymes of the alcohol utilization pathway were recently detected in proteomics data of fruiting bodies of Guyanagaster necrorhiza, where their presence was interpreted as evidence for anaerobic fermentation within the fruiting body, as part of an intriguing mutualism with N2 fixing bacteria (Koch et al. 2021).
The last step of glycolysis is catalysed by the pyruvate decarboxylase Pdc1 in S. cerevisiae, which converts pyruvate to acetaldehyde. Pdc1 generates cytoplasmic Ac-CoA, which is essential for the biosynthesis of lipids (Chen et al. 2015). Pdc1 orthologs (represented by C. cinerea 422402) were developmentally expressed in 11/8 species (FC>2/4), whereas genes encoding other enzymes in glycolysis showed much smaller dynamics (Supplementary Fig. S3). Together, these observations suggest a widespread upregulation of acetaldehyde and from that either acetate or ethanol producing enzyme genes in fruiting bodies.
Upstream of oxaloacetases, enzymes responsible for oxaloacetate production in the TCA (yeast Mdh1, C. cinerea 500663, 11/3) and glyoxylate cycles (yeast Mdh2, C. cinerea 449845, 9/3) were also quite widely developmentally regulated. In the glyoxylate cycle we found orthologs of S. cerevisiae Mls1 (malate synthase, C. cinerea 530292) and Icl1 (isocitrate lyase, C. cinerea 495730) to form CDE orthogroups developmentally regulated in 12/11 and 12/7 species (FC>2/4), respectively. This suggests that the glyoxylate cycle is strongly induced in fruiting bodies. On the other hand, members of the TCA cycle showed lower developmental dynamics (Fig. 5).
What happens to the oxalate that is produced by oxaloacetases? It is possible that the resulting oxalate is a waste product, but it may also be used somehow by the fungus, e.g., as a developmental signal molecule (Palmieri et al. 2019) or as an electron donor, as has been observed in wood-decay (Hastrup et al. 2012, Zhu et al. 2016, Grąz et al. 2017), where it acts as an electron donor for oxidative enzymes. One hypothesis for the role of oxalate is that oxidative enzymes, which might participate in the modification of the fungal cell wall during development (see above), utilize it as an electron donor, as do enzymes involved in wood-decay. On the other hand, if oxalate is a by-product, excess amounts may need to be broken down actively. We found evidence for the upregulation of oxalate catabolism in the compared species. The two-oxalate degradation pathway in fungi comprises oxalate decarboxylase, which converts oxalate to formate and CO2 and formate dehydrogenase, which breaks format down to CO2 (Mäkelä et al. 2014). We detected two CDE orthogroups of oxalate decarboxylases (C. cinerea protein ID: 361522, 8/6, Schco3_2751877, 7/6) and one of format dehydrogenases (C. cinerea protein ID: 452378, 7/6, orthologous to Fdh1 of S. cerevisiae) (Supplementary Table S5), which were upregulated in fruiting bodies widely, suggesting that fruiting bodies utilize enzymatic mechanisms for the neutralization of oxalate. An alternative route of oxalate degradation in S. cerevisiae is catalysed by PCS60, an oxalyl-CoA synthetase (Foster & Nakata 2014). Orthologs of this gene were found in only a subset of species (represented by C. cinerea 474928), but were developmentally regulated in most of them. In addition, a CDE orthogroup of format transporters, orthologous to the Postia placenta gene reported in Martinez et al. (2009), was developmentally expressed in 8/5 species (FC>2/4, represented by Schco3_2738153, missing in C. cinerea).
Building on the evidence for intense Ac-CoA production, we annotated and analysed the Coenzyme-A producing pathway as well as two other sources of Ac-CoA, the fatty acid beta-oxidation pathway and the cellular cycle for pyruvate decarboxylation to Ac-CoA. These pathways were reconstructed and annotated based on corresponding pathways in S. cerevisiae (Cherry et al. 2012). Members of these pathways did not show remarkable developmental dynamics (Supplementary Table S5).
Several genes connected to acetate metabolism and transport also formed CDE orthogroups. Orthologs of S. cerevisiae Ady2, an acetate transporter (Paiva et al. 2004), are widely developmentally regulated (represented by C. cinerea 441597, 7/6), and were strongly induced in young fruiting body caps of C. cinerea. A CDE orthogroup of putative monocarboxylate (lactate, acetate, succinate, etc.) transporters (C. cinerea protein ID: 375404 7/5) (Supplementary Table S5) orthologous to S. cerevisiae Jen1 (Casal et al. 1999) were also highly induced in YFB-caps in C. cinerea. We also identified putative orthologs of S. cerevisiae Yat1 (represented by C. cinerea 443703, 10/5), a carnitine acetyltransferase that shuttles Ac-CoA across the mitochondrial membrane. A CDE orthogroup of aquaglyceroporins orthologous to S. cerevisiae Fps1, which is involved in the uptake of acetate (C. cinerea protein ID: 373141 7/6), were developmentally regulated in 7/6 species. This gene encodes a protein that has been associated with either glycerol influx/efflux (Luyten et al. 1995) or acetate uptake (Mollapour & Piper 2007, Watcharawipas et al. 2018), which leaves some uncertainty on which role it fulfils in fruiting bodies. Orthologues of S. cerevisiae Sym1 also formed a CDE orthogroup, although only at a fold change cut-off of 2 (C. cinerea protein ID: 147126, 9/3) (Supplementary Table S5). This gene is said to be involved in ethanol utilization and stress response, but mechanistic details are not known (Reinhold et al. 2012, Gilberti et al. 2018). Similarly, orthologs of yeast Sfc1, a succinate/fumarate antiporter that transports C4 succinate (which is produced from acetate (Bojunga et al. 1998), to the mitochondria, are developmentally regulated in several species (represented by C. cinerea 374148, 12/7). The Sfc1 ortholog is also strongly induced late in the development of C. neoformans (Liu et al. 2018). Finally, a CDE orthogroup of putative malate symporters orthologous to Sch. pombe Mae1, which is involved in the uptake of extracellular malic acid, was detected as developmentally regulated in 9/5 species (FC>2/4, represented by C. cinerea 534888, Supplementary Table S5). Whether these genes are related to the above discussed observation of acetate/Ac-CoA related gene upregulation, or how cell surface transporters may be related remains to be determined. The induction of these genes does not happen in the same or similar developmental stage in all species (although there is consistency within species, see the upregulation of several genes in YFB-caps of C. cinerea), nevertheless, their induction suggests differential acetate concentrations and an important role for acetate during in fruiting body development.
CO2 produced by the TCA cycle may be fixed into oxaloacetate by the conversion of CO2 to hydrogencarbonate by carbonic anhydrases, then conversion of HCO3- and pyruvate into oxaloacetate by pyruvate carboxylases (Elleuche & Pöggeler 2010) (Pyc1 in S. cerevisiae). We found that several carbonic anhydrases (2–9 genes in Agaricomycetes genomes) are developmentally regulated in mushroom forming fungi and they form at least one CDE orthogroup (represented by C. cinerea 450047) (Supplementary Table S5). C. cinerea 450047 shows an upregulation in young fruiting bodies, coincident with the induction of Ac-CoA producing enzymes. This observation raises the possibility that CO2 produced by the TCA cycle in fruiting bodies may be fixed into oxaloacetate, which can further be converted to Ac-CoA (Fig. 5). Of note, carbonic anhydrases are involved in the regulation of sexual development in the Ascomycota (Elleuche & Pöggeler 2010).
Taken together, we observed developmental regulation of key enzymes in several pathways that seem to converge on increased acetyl-CoA production, either directly, or indirectly (through acetate or oxaloacetate). Several connected pathways, including oxalate degradation, the ethanol degradation pathway, the terminal reaction of glycolysis as well as evidence from the TCA and glyoxylate cycles were pointing in this direction. The expression of these genes tended to be upregulated late during development and showed good consistency within, but not necessarily across, species. A possible interpretation of these data is that increased Ac-CoA production is needed for growth by cell expansion late in development, as a starting material for lipid and sterol biosynthesis. This is underpinned by the fact that biosynthetic pathways of ergosterol and other membrane components are also upregulated during late developmental stages (e.g., the young fruiting body stage in C. cinerea), probably for membrane assembly needed for rapid cell growth. This interpretation would be consistent with the two-phased development of several mushroom-forming species, which consists of a first stage of cell division and differentiation and a second, during which cell numbers hardly change but their volumes increased considerably via Turgor-mediated expansion. Such a reprogramming of the fungus’s basic metabolism from using Ac-CoA for energy production (when citrate is in the mitochondrion) to using it for lipid metabolism (when citrate is in the cytoplasm) seems particularly well-suited for fruiting body formation, which requires tight regulation of lipid metabolism. It should be mentioned that Ac-CoA is also required for melanin production (Lee et al. 2019), although it is unlikely to be the driving force of our observation, because pigment production is not conserved across the examined species. A previously reported reprogramming of the glyoxylate cycle in Fomitopsis palustris for the production of oxalate (Munir et al. 2001) seems related, however, in that case, the main product in demand was hypothesized to be oxalate that facilitates wood-decay, whereas in our case we hypothesize the product in demand is acetate and finally Ac-CoA. Both variants of the metabolic pathways nevertheless pass-through oxaloacetate (Munir et al. 2001). The excess Ac-CoA production may rely on, or be connected to carbohydrate reserves that are deposited in the subhymenial part of the cap of the mushroom (Ji & Moore 1993, Muraguchi et al. 2015) during primordium development. However, there may be alternative interpretations and the topic is certainly of interest for detailed, further research that integrates more functional data beyond gene expression observations.
Lipid metabolism
Lipid metabolism is pivotal to very basic organismal processes such as growth, differentiation, cell division, energy storage and cell membrane composition, to name a few. Therefore, in depth understanding of gene expression changes associated with lipid metabolism, catabolism and transport can be expected to provide novel insights into fruiting body development. Indeed, our meta-analysis of Agaricomycetes transcriptomes revealed several CDE orthogroups related to lipid metabolism. We discuss these below, complemented with annotations of major related pathways (but no attempt is made to comprehensively annotate all pathways) and processes. We base our annotations on data from well-studied model organisms, such as S. cerevisiae, A. nidulans and Sch. pombe, with orthology relationships to Agaricomycetes genes established on the basis of 1-to-1 reciprocal best Blast hits.
Beta oxidation
We reconstructed the beta-oxidation pathway (Supplementary Table S6) in C. cinerea based on the yeast beta-oxidation pathway (van Roermund et al. 2003) (Fig. 5). The expression of genes comprising the beta-oxidation pathway in general does not appear to be widely dynamic in fruiting bodies. We found orthologs of yeast CTA1, a catalase A, to form a CDE orthogroup. However, this catalase in yeast is only a marginal component of the beta-oxidation pathway, and may be involved in a wide range of other functions (e.g., signalling). The more widespread developmental expression of ACS1 orthologs is remarkable (11/4 species at FC>2/4), for a possible connection with sterol biosynthesis (see above). ACS1 produces Acetyl-CoA, which is the starting point for ergosterol and membrane lipid biosynthesis.
Ergosterol biosynthesis
We reconstructed the ergosterol biosynthesis pathway based on orthology to known pathway member genes in S. cerevisiae (Cherry et al. 2012) (Supplementary Table S7). ERG pathway genes of Agaricomycetes are generally higher expressed in fruiting bodies than in vegetative mycelia (VM/P1>1 in 20 out of 21 genes in C. cinerea), with expression peaking in young fruiting bodies of C. cinerea (Fig. 6, Supplementary Table S7). Expression peaks at similar stages in other species too, in primordium 3/young fruiting body in P. ostreatus and young fruiting bodies/mature fruiting bodies stages in A. ostoyae (Supplementary Fig. S4). Similar pictures were obtained for other species. It should be noted that although the ergosterol biosynthesis pathway shows clear expression trends, the dynamics of expression is relatively low, mostly between 2<FC<4. Therefore, ergosterol biosynthesis genes mostly do not form CDE orthogroups, nevertheless, the expression patterns are distinctive so they warrant discussion here.
Fig. 6.
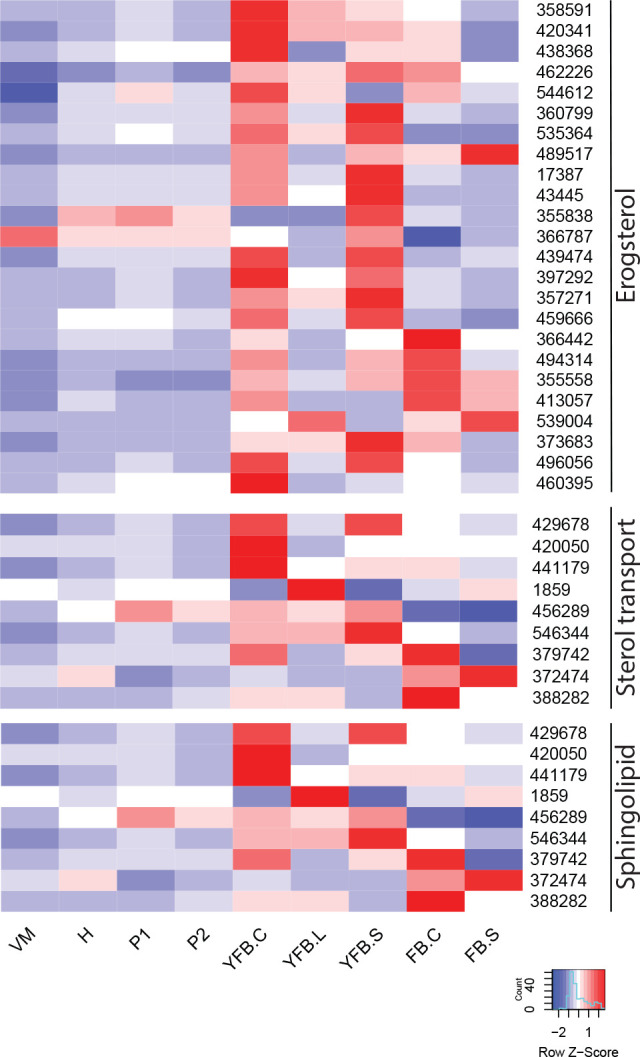
Expression heatmap of ergosterol and sphingolipid biosynthesis related genes in C. cinerea. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively. Developmental stages are abbreviated as follows: VM – vegetative mycelium, H – hyphal knot, P1 – stage 1 primordium, P2 – stage 2 primordium, YFB.C – young fruiting body cap, YFB.L – young fruiting body gills, YFB.S – young fruiting body stipe, FB.C – mature fruiting body cap (including gills), FB.S – mature fruiting body stipe.
In these species, expression peaks seem to coincide with intense growth by cell expansion in fruiting bodies. Above we showed that mitotic, ribosomal and histone gene expression level off in late primordium stages (P2 in C. cinerea, see Fig. 3), after which growth by cell expansion takes place. This might create an increased demand for membrane constituents during rapid cell expansion/growth by turgor, which is what we might see in the expression patterns of genes in the ergosterol biosynthesis pathway (Fig. 6, Supplementary Fig. S4). This hypothesis would also explain why there is no clear peak in C. neoformans and Pt. gracilis, where sudden growth by cell expansion is lacking.
Similarly, a homolog of the Sterol-Regulatory Element Binding Proteins (SREBP, Bien & Espenshade 2010), similar to the S. cerevisiae transcription factor HMS1 shows a very similar expression pattern: in C. cinerea (C. cinerea protein ID: 488426) expression peak in YFB-S and YFB-C (Fig. 6), whereas in P. ostreatus it peaks in mature fruiting body tissues (PleosPC15_2_174910). This suggests, based on expression data, that these putative SREBPs might be regulating ergosterol biosynthesis genes also in the Agaricomycetes.
A connection with the above-mentioned Ac-CoA producing basic metabolic pathways, where certain, but not all, components were found widely upregulated, is possible here. These pathways produce Acetyl-CoA, which is the starting point of ergosterol biosynthesis. This raises the possibility that the upregulation of the ethanol utilization pathway components (see above, Fig. 5) and certain components of the glyoxylate cycle reflect increased Ac-CoA production which is required as input to ergosterol biosynthesis and eventually membrane assembly. A competing hypothesis was put forth in P. microcarpus, where upregulation of glyoxylate shunt genes was observed and suggested that it underlies the production of storage materials for spores (Pereira et al. 2017). Using our more resolved transcriptome data and based on the observation of ergosterol pathway expression dynamics, we propose that the connection to the glyoxylate shunt might represent a link to membrane constituent synthesis, although more data would be needed to verify this hypothesis.
The upregulation of the ergosterol synthesis pathway was observed in F. velutipes, where most ergosterol biosynthesis genes were upregulated in the young fruiting body stage (Wang et al. 2019) (approximately in agreement with our definition of young fruiting bodies) and showing intense growth. Sterol metabolism genes were also found to be more abundant in fruiting bodies of H. erinaceus compared to vegetative mycelia (Zeng et al. 2018). It was reported that orthologs of the lanosterol-alpha-demethylase Erg11 of S. cerevisiae, a component of the ergosterol biosynthesis pathway (corresponding C. cinerea ortholog: 397292), was upregulated in fruiting bodies of Antrodia cinnamomea (Lu et al. 2014) and P. ostreatus (Sunagawa & Magae 2005). This is consistent with the observations made in this study.
We also checked the sphingolipid biosynthesis pathway based on orthology to pathway members in S. cerevisiae. Sphingolipids, along with sterols are important in membrane rafts and microdomains and form key components of fungal cell membranes. Although some yeast genes did not have clear orthologs in Agaricomycetes, genes involved in sphingolipid biosynthesis showed a similar expression profile in C. cinerea (Fig. 6, Supplementary Table S7). This may underscore our hypothesis on the link between growth by cell expansion and membrane assembly.
A characteristic CDE orthogroup of putative C14-sterol reductase genes was developmentally expressed in 12/7 species (represented by C. cinerea 539004) (Supplementary Table S7, Fig. 6). These proteins are not part of the ergosterol biosynthesis pathway, but show a high similarity to A. nidulans and C. albicans membrane phospholipid metabolism proteins AN2040 and Erg24, respectively, which are involved in ergosterol metabolism. Genes in this orthogroup show peak expression in late development, coincident with rapid expansion of fruiting bodies. In several species, they were also upregulated in primordia relative to vegetative mycelium samples. These expression patterns resemble that of ergosterol biosynthesis genes, and suggest that this orthogroup may be linked to ergosterol metabolism.
Sterol transport
We detected several CDE orthogroups that were annotated as related to ergosterol transport and showed similar upregulation in late development as ergosterol biosynthesis genes did. We therefore inventoried sterol transport related genes of the families Niemann-Pick type C protein family (S. cerevisiae Npc2), the yeast Ncr1 family, the nonspecific sterol transport protein (Scp2) family, StAR-related lipid-transfer (START) proteins as well as members of the Oxysterol Binding Protein Homologue (OSH) protein family (Schulz & Prinz 2007).
Of these, the Ncr1, Npc2, Scp2 and START gene families are single-copy families in mushroom-forming fungi, whereas the OSH family is represented by 5 members in all mushroom-forming fungi (except S. commune which has 6, Supplementary Table S8). Npc2 (ortholog C. cinerea 475807), Scp2 (ortholog C. cinerea 478741) and START proteins as well as one member of the OSH family were (C. cinerea protein ID: 427232) (Supplementary Table S8) upregulated during the second half of development, at or after the young fruiting body stage. This is similar to the expression patterns seen in ergosterol biosynthesis genes. One member of the OSH family orthogroup (C. cinerea protein ID: 378276) showed a clearly stipe-specific expression, with peaks in stipes of young fruiting bodies or fruiting bodies of C. cinerea, A. ostoyae, M. kentingensis, A. bisporus and P. ostreatus, albeit mostly with low fold change values (2<FC<4). Interestingly, the corresponding gene was cap-upregulated in L. bicolor (Lacbi2_456536), whereas in simpler species (S. commune, A. ampla), it showed diverse expression peaks. Ncr1 orthologs (represented by C. cinerea 494709) showed consistently higher expression in fruiting bodies than in vegetative mycelia. Fold changes were generally low in these gene families, but nevertheless the patterns are phylogenetically very consistent.
These expression patterns resemble those of ergosterol biosynthesis genes, which also showed upregulation in mid- to late development, coincident with cell expansion and growth. This suggests that ergosterol transport, in addition to biosynthesis, is an important process in the second half of the development of mushroom fruiting bodies. Like synthesis, transport of sterols might be required to fulfil the needs of rapid cell expansion, which obviously coincides with cell- and membrane surface expansion too.
Although marginally related, we mention here the CAP domain/Pry1 protein family, which includes known sterol-binding proteins. The family has been associated with diverse functionalities, including cell adhesion, pathogenicity (pathogenicity-related PR proteins belong here), morphogenesis, endocrine and paracrine functions, among others (Gibbs et al. 2008, Schneiter & Pietro 2013). This protein family includes S. cerevisiae Pry1, a lipid and sterol binding protein involved in sterol export (Darwiche et al. 2017). To our best knowledge, there are no previous reports of this family in fruiting body development, whereas, their role in morphogenesis in mammals is well-established (Gibbs et al. 2008). We detected, on average 2–5 copies of CAP proteins in the Agaricomycetes, with expansions up to 9 in Pt. gracilis, L. bicolor and A. ostoyae as well as a CDE orthogroup of Pry1-related CAP-domain proteins (represented by C. cinerea 470387, FC>2/4: 7/6, Supplementary Table S8, Fig. 6). Members of this orthogroup showed peak expression late in development, at or after the young fruiting body stage in most species. The sterol transporting ability and expression patterns suggest that this family may have a role in sterol transport during the second half of development in Agaricomycetes fruiting bodies. It should be noted that neither Polyporales species (Ph. chrysosporium and L. tigrinus) had developmentally regulated genes in this family, indicating that their role, if any, may not be universal.
Phospholipid and fatty acid biosynthesis
The phospholipid and fatty acid biosynthesis pathways are conserved in Agaricomycetes, with most genes showing clear 1-to-1 orthologs to described pathway members of S. cerevisiae (Supplementary Table S9). Because active cell growth and expansion requires an excess amount of membrane components (ergosterol, phospholipids), we examined pathways involved in membrane phospholipid and triacylglycerol synthesis. We reconstructed these pathways based on well-known reaction steps in S. cerevisiae and Yarrowia lipolytica (Carman & Han 2011, Fakas 2017). The first half of the pathway, which starts from glycerol-3-phosphate and leads to the production of phosphatidic acid, shows upregulation in young and mature fruiting bodies of C. cinerea and A. ostoyae (Fig. 7). Phosphatidic acid sits in a branching point of the pathway, with one branch leading to membrane phospholipids (CDP-DAG pathway), whereas the other leads to triacylglycerol (TAG), a major storage material. The CDP-DAG pathway shows a general upregulation in the second half of development (young and mature fruiting bodies) in C. cinerea, A. ostoyae, M. kentingensis, A. bisporus (Supplementary Fig. S5). These patterns are consistent with increased membrane lipid biosynthesis in these stages, possibly serving the needs of increasing cell volumes during rapid growth. On the other hand, TAG pathway members appear upregulated somewhat later, mostly in mature fruiting bodies in most species (Fig. 7, Supplementary Fig. S5), which coincides with spore formation and could be related to storage material synthesis in spores. In P. ostreatus, both the CDP-DAG and TAG pathways show remarkably uniform upregulation in primordium stages (Fig. 7), which is quite different from the pattern seen in other species. This pattern in P. ostreatus is also observable in fatty acid biosynthesis pathways (see below).
Fig. 7.
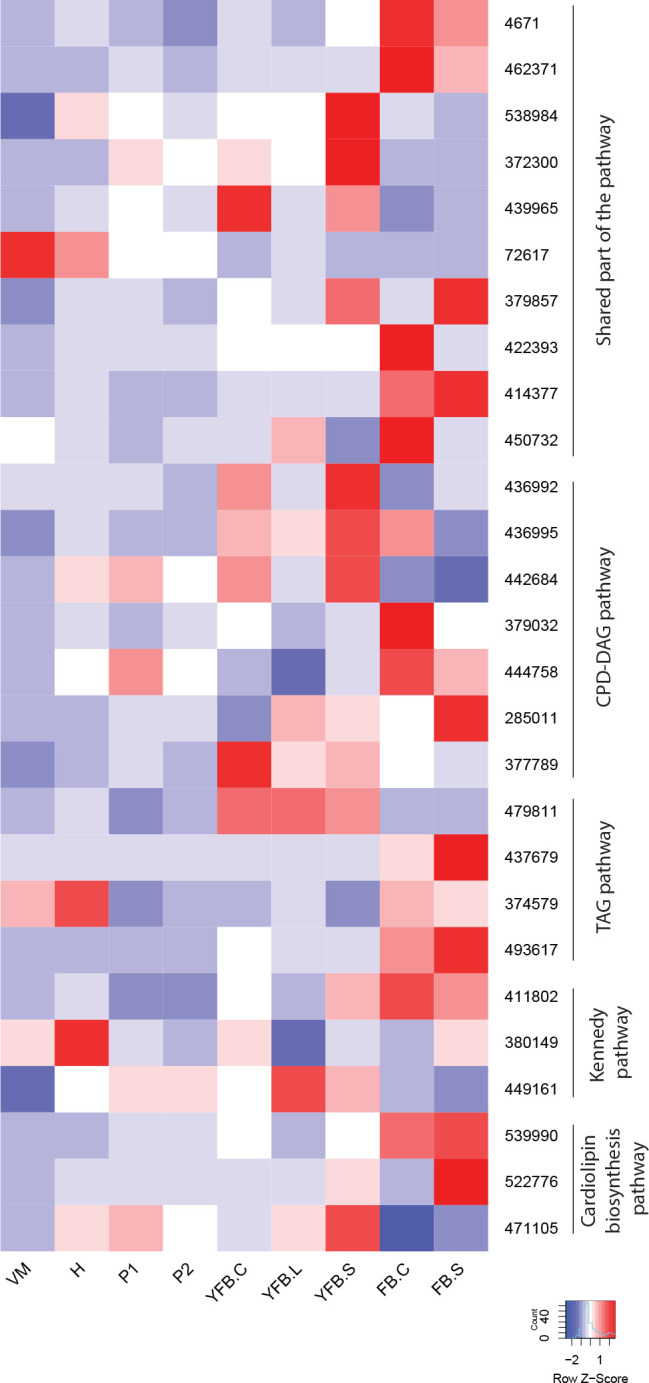
Expression heatmap of membrane phospholipid and fatty acid biosynthetic genes in C. cinerea. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively. Developmental stages are abbreviated as follows: VM – vegetative mycelium, H – hyphal knot, P1 – stage 1 primordium, P2 – stage 2 primordium, YFB.C – young fruiting body cap, YFB.L – young fruiting body gills, YFB.S – young fruiting body stipe, FB.C – mature fruiting body cap (including gills), FB.S – mature fruiting body stipe.
Similar to the CDP-DAG and TAG pathways, fatty acid biosynthesis (Supplementary Table S9) shows a signal for upregulation in young fruiting body tissues, with C. cinerea orthologs of S. cerevisiae Acc1, Fas2, Elo2 and Faa1 showing peak expression in the YFB stage. Similar patterns exist in A. ostoyae, M. kentingensis and to some extent in A. bisporus (Fig. 7). In P. ostreatus, fatty acid biosynthesis genes are co-expressed with the other phospholipid biosynthesis genes, showing an upregulation in primordia.
Both phospholipid synthesis and fatty acid biosynthesis require Acetyl-CoA as a starting point. For the former, it is needed for acylation reactions in the conversion of lysophosphatidic acid to phosphatidic acid (last element of the shared part of the pathway) whereas in the latter, Ac-CoA is the starting point of fatty acid synthesis (Fakas 2017). This provides another link with the dynamics of Ac-CoA biosynthesis genes noted above. As in the case of ergosterol biosynthesis pathway members, phospholipid and fatty acid metabolism genes showed low expression dynamics (and hence did not form CDE orthogroups), but their expression patterns were consistent across stages and species.
The expression of mitochondrial membrane components (cardiolipin and phosphatidylglycerol) is also peaking in young fruiting bodies, mostly in stipe, but not very characteristic (Fig. 7).
Linoleic acid producing fatty acid desaturases
Several CDE orthogroups of fatty acid desaturases (FAD) were detected among conserved developmentally expressed genes. Members of these orthogroups show high similarity to C. albicans Fad2 and Fad3 and A. nidulans OdeA, which are involved in the synthesis of alpha-linoleic acid. Linoleic acid is a major membrane component in fungi, which, as an unsaturated fatty acid, regulates membrane fluidity. It has also been speculated to be the starting point for the biosynthesis of volatiles, such as 1-octen-3-ol (Orban et al. 2020). Linoleic acid showed increased proportions in fruiting bodies and reproductive structures in general in both the Asco- and Basidiomycota (Shaw 1967, Goodrich-Tanrikulu et al. 1998, Sakai & Kajiwara 2003, Gessler et al. 2017, Song et al. 2018). Linoleic acid is also a precursor of oxylipins, key developmental signalling molecules that are actively produced by reproductive cells (Combet et al. 2006, Gessler et al. 2017, Orban et al. 2021). Accordingly, in the Ascomycota, several fatty acid desaturase gene mutations were reported to asexual/sexual defects (Pöggeler et al. 2006). The higher proportion of linoleic acid in fruiting bodies of Agaricomycetes was already noted in 1967 (Shaw 1967) with several similar reports (Sakai & Kajiwara 2003, Song et al. 2018) later. The proportion of unsaturated fatty acid (which include linoleic acid) increases during fruiting body formation (Sakai & Kajiwara 2003) in L. edodes. Three fatty acid desaturases, two in L. edodes (Le.Fad1 and Le.Fad2) (Sakai & Kajiwara 2005) and one in C. cinerea (Cc.odeA) (Sakai & Kajiwara 2003, 2005, Zhang et al. 2007) have been shown to be capable of producing linoleic acid. Fad genes were found to be differentially expressed in fruiting bodies of A. bisporus previously (Morin et al. 2012).
We found 7–16 fatty acid desaturase genes in the examined Agaricomycetes, many of which were developmentally regulated, although clear orthology of these genes to experimentally characterised Ascomycota genes is missing. Each species had at least 2–3 FAD encoding genes upregulated in primordia relative to vegetative mycelium, suggesting that their functions are key to the initiation of fruiting body development. FAD genes formed five CDE orthogroups (Supplementary Table S10). One of these (C. cinerea protein ID: 363743) was FB-init in most species (C. cinerea, A. ostoyae, A. ampla, M. kentingensis, P. ostreatus, S. commune, C. aegerita, L. bicolor), showed high expression in primordium stages, then expression levels dropped significantly in later developmental stages. The CDE orthogroup containing Le.FAD1 (Lenedo1_1050661) was upregulated in primordia of most species relative to mycelium samples (C. aegerita, A. ampla, S. commune, L. edodes. L. bicolor, L. tigrinus, M. kentingensis, but not A. ostoyae, P. ostreatus and A. bisporus, ortholog missing in C. cinerea), although only slightly (FC = 1.5–2x, except in L. edodes and Pt. gracilis which showed >4-fold upregulation). The orthogroup encompassing Cc.odeA (C. cinerea protein ID: 422393) and Le.Fad2 (orthogroup represented by S. commune 2511002) was upregulated in primordia relative to vegetative mycelia of several species (A. ampla, C. aegerita, S. commune, C. cinerea, L. edodes, L. tigrinus and P. ostreatus, but not in A. ostoyae, M. kentingensis, Pt. gracilis and A. bisporus). Our expression data are consistent with previous reports of transcripts/protein abundance in case of both Le.FAD1 and Le.FAD2 in L. edodes (Sakai & Kajiwara 2003, 2005). The other two FAD orthogroups showed less conservation in expression (C. cinerea protein ID: 471478, 422976, Fig. 8), including peaks in later developmental stages, such as young fruiting body caps.
Fig. 8.

Expression heatmap of putatively linoleic acid biosynthesis related fatty acid desaturase genes in C. cinerea. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively. Developmental stages are abbreviated as follows: VM – vegetative mycelium, H – hyphal knot, P1 – stage 1 primordium, P2 – stage 2 primordium, YFB.C – young fruiting body cap, YFB.L – young fruiting body gills, YFB.S – young fruiting body stipe, FB.C – mature fruiting body cap (including gills), FB.S – mature fruiting body stipe.
The linoleic acid-producing ability of the proteins encoded by these genes suggests that the detected CDE orthogroups and expression dynamics relate to linoleic acid production. It follows from our observations of expression dynamics that linoleic acid production is upregulated in the primordia of several species. This is consistent with previous reports of higher linoleic and unsaturated fatty acid proportions in fruiting bodies of Agaricomycetes (see above). There may be at least three explanations for this observation, including (i) regulation of membrane fluidity in response to temperature changes, (ii) the same being genetically encoded in fruiting body development, or (iii) fulfilling the requirements of oxylipin production. We argue that hypothesis (i) is unlikely, however, we can’t speculate on hypotheses (ii) and (iii).
Regulation of membrane fluidity by increasing the proportion of unsaturated fatty acids is a common mechanism across organisms (Sakai & Kajiwara 2005). Therefore, a question arises whether the higher inferred proportion of linoleic acid and corresponding changes in gene expression are genetically coded traits of fruiting bodies or simply a consequence of lower temperature required by several species for fruiting. At least for the transcriptomes produced by Sipos et al. (2017), Krizsán et al. (2019), Almási et al. (2019), Merényi et al. (2022), we know that vegetative mycelium samples were harvested at the same temperature as fruiting body samples for all species, which rules out the possibility that higher linoleic acid proportion is simply a response to lower fruiting temperatures. Rather, this suggests that higher linoleic acid concentration is a genetically encoded trait in fruiting bodies. Similar conclusions were reached in the context of unsaturated fatty acids in L. edodes (Sakai & Kajiwara 2003). From an evolutionary point of view, such a genetic adaptation could go back to the exposed nature of fruiting bodies; in the natural environments (soil, wood) the vegetative mycelium is in more stable conditions than fruiting bodies are, which might therefore need to be adapted to colder temperatures by increasing the proportion of unsaturated fatty acids in fruiting body tissues. At the same time, due to their large surfaces and continuous evaporation, fruiting bodies cool themselves (on average ~2.5 °C colder than their environments, Dressaire et al. 2016), which may explain why genetic encoding of higher linoleic acid proportions is beneficial for their development. It should be noted that linoleic acid production is also required for the synthesis of several oxylipin species (Combet et al. 2006), which might be involved in cell-to-cell communication.
Other potentially membrane lipid related genes
We detected several CDE orthogroups, which are not parts of the above-mentioned pathways, yet might be related to lipid metabolism, membrane organization or assembly. In many cases the functions of these orthogroups is not clear, but their widespread developmental expression suggests they might be interesting players in fruiting body development. Therefore, we discuss them below, with details on potential functions where available.
Glycerophosphodiester phosphodiesterases - A CDE orthogroup of glycerophosphodiester phosphodiesterases (represented by C. cinerea 20997) (Supplementary Table S11) was developmentally expressed in 11 species (FC>4), which makes it one of the most widely developmentally expressed families. However, it is hard to make functional inferences as these proteins show only limited similarity to experimentally characterised fungal proteins (highest to Pgc1 of S. cerevisiae). S. cerevisiae Pgc1 (Phosphatidylglycerol phospholipase C) negatively regulates phosphatidylglycerol degradation, leading to increased concentrations in deletion mutants (Šimočková et al. 2008). The glycerophosphodiester phosphodiesterase family (IPR030395), is represented by three copies in most examined Agaricomycetes (except A. ampla, L. tigrinus and S. commune, which have 4), of which only one is developmentally expressed in all species. These form a CDE orthogroup in which most genes are FB-init (C. aegerita, A. ostoyae, A. ampla L. bicolor, L. tigrinus, P. ostreatus, S. commune), whereas expression had different dynamics in C. cinerea, L. edodes, M. kentingensis, Ph. chrysosporium). These expression patterns suggest that the role of this orthogroup, although unknown at the moment, may be key to the initiation of fruiting body formation.
We noticed that the detected glycerophosphodiester phosphodiesterases showed strongly correlated expression with members of another CDE orthogroup (represented by C. cinerea 380149, Fig. 7) in six species (data not shown). This contains putative orthologs of S. cerevisiae Cpt1, a choline/ethanolamine phosphotransferase that catalyses the last step (formation of phosphatidylcholine) of the CPD-choline (Kennedy) pathway in S. cerevisiae. The strongly correlated expression in the case of six species (Pearson r >0.8) suggests that the two genes are linked and possibly form a linear pathway. However, their role in the context of fruiting body formation requires further research.
Marvel protein family - We also detected two CDE orthogroups (represented by C. cinerea 444998, 462849) (Supplementary Table S11) of Marvel proteins showing low similarity to A. nidulans mrvA, C. albicans Mrv2 and S. cerevisiae Fhn1. The gene family seems to be generally poorly characterised. In S. cerevisiae, it may be involved in nonclassical protein secretion, might act as a sensor of sphingolipids or be related to membrane organization (Loibl et al. 2010). In metazoans, marvel proteins are related to sterol-rich membrane region positioning (Sánchez-Pulido et al. 2002). Members of the first orthogroup (C. cinerea protein ID: 444998) were FB-init in several species (A. ostoyae, M. kentingensis, P. ostreatus, S. commune, L. edodes), suggesting that they fulfil key functions in fruiting body initiation. The second orthogroup (C. cinerea protein ID: 462849) showed less characteristic expression patterns, with upregulation mostly late in development in most species.
Sphingolipid C9-methyltransferases - We found two CDE orthogroups of sphingolipid C9-methyltransferases (C. cinerea protein ID: 395241, 365330) (Supplementary Table S11) that were FB-init in nearly all species. One of the orthogroups (C. cinerea protein ID: 395241) contains orthologs of Mts1 of C. albicans, which is involved in glucosylceramide biosynthesis. Glucosylceramides are glycosphingolipids that form structural components of the fungal cell membrane, with diverse roles (cell division, spore germination, pathogenicity, lipid rafts (Poeta et al. 2014), although little mechanistic detail to date.
PAP2 enzyme family - A CDE orthogroup of diacylglycerol pyrophosphate phosphatases (C. cinerea protein ID: 380219) (Supplementary Table S11) was FB-init in most species in which the gene was present. The encoded proteins show high similarity (but not 1-to-1 orthology) to A. nidulans and C. albicans membrane phospholipid metabolism proteins AN1671 and Dpp3, respectively. The latter is required also for farnesol biosynthesis (Nickerson et al. 2006). These genes belong to the broader PAP2 family, which produce diacylglycerol and are key in the synthesis of phospholipids, triacylglycerol and the generation of signalling molecules with a lipid nature. The upregulation of these genes in fruiting bodies might be related to increased membrane material production associated with cell proliferation during early development.
Caleosins - Caleosins are well-characterised in plants and indirect data indicate they may be involved in oxylipin signalling, lipid metabolism, reproduction (Rahman et al. 2018), though exact functional knowledge seems to be missing. In fungi, caleosins are involved in sporulation, pathogenicity and lipid storage (Fan et al. 2015). A previous study found that the caleosin family is present in ~30 % of fungal genomes (Rahman et al. 2018), whereas we found that it is present in most of the examined Agaricomycetes, mostly as a single-copy gene. Members of a detected caleosin CDE orthogroup (C. cinerea protein ID: 376602) (Supplementary Table S11) were FB-init in most species and consistently had peak expression late in development, which is compatible with presumed roles in fruiting body initiation and sporulation, respectively.
Storage carbohydrate metabolism
Fungal colonies accumulate and mobilize various carbohydrates as reserve material and for energy storage. Previous research has revealed that there are very specific and tightly regulated accumulation and mobilization patterns of glycogen, trehalose and mannitol within fungal fruiting bodies (Kües 2000). Storage carbohydrate metabolism and distributions in the mycelium and fruiting bodies have been studied in a number of economically important mushroom-forming species, including F. velutipes (Kitamoto et al. 2001), Pleurotus spp. (Han et al. 2003, Zhou et al. 2016a, Zhu et al. 2019), Pisolithus microcarpus (Campos & Costa 2010), A. bisporus (Wells et al. 1987, Hammond et al. 2008, Patyshakuliyeva et al. 2013), L. bicolor (Deveau et al. 2008) and described to a high precision/resolution in C. cinerea in a series of papers by Moore and colleagues (Kuhad et al. 1987, Ji & Moore 1993, Moore 2013), including links with cAMP levels. Briefly, these studies provided evidence on tightly regulated glycogen, trehalose and mannitol levels in mycelia and various tissues and developmental stages of fruiting bodies. They also revealed that the identity and quantities of major sugars in fruiting bodies differ from species to species; trehalose dominates in L. bicolor (Deveau et al. 2008) and C. cinerea (Kües 2000), mannitol in A. bisporus (Morton et al. 1985) and L. edodes (Tan & Moore 1994), and both present in similar quantities in P. ostreatus (Jai-Sik & Tae-Young 1988, Zhou et al. 2016).
More recently, genome sequences allowed the reconstruction of carbohydrate metabolism pathways based on gene orthology information to better studied model systems such as S. cerevisiae. Deveau et al. (2008) found that most of the genes in known fungal pathways related to glycogen, trehalose and mannitol metabolism were found in the genome of the ectomycorrhizal L. bicolor and similar patterns can be expected in these very conserved pathways upon examination of other Agaricomycetes genomes. Transcriptomic studies further allowed previous biochemical observations to be interpreted in the light of gene expression patterns, although this has not yet been systematically performed in any species. In the following sections, we review information on the metabolism of glycogen, trehalose and mannitol gleaned from transcriptomic studies of mushroom development. Many of these studies highlighted that shared pattern of gene expression related to storage carbohydrate metabolism exist among mushroom-forming species. For example, Merényi et al. (2022) showed that dynamic glycogen accumulation/breakdown is shared between C. neoformans basidium development and complex fruiting body morphogenesis, two extremes of Basidiomycota development along the complexity gradient. On the other hand, trehalose concentrations were found to react to heat shock in fruiting bodies of Flammulina filiformis (Liu et al. 2016). Our discussion hereafter is based on well-described pathways of storage carbohydrate metabolism in yeast (François & Parrou 2001, Cherry et al. 2012).
Glycogen metabolism - Glycogen production and mobilization take place in various tissues and developmental stages and follows tight regulation that may differ from species to species (reviewed in Ji & Moore 1993, Kües 2000, Xie et al. 2020, see also Cecarolli et al. 2011 for Ascomycota). In the Agaricomycetes, this is best known in C. cinerea and A. bisporus, whereas mechanistically glycogen metabolism is best known in S. cerevisiae (François & Parrou 2001), which can be used to reconstruct corresponding pathways in mushroom-forming fungi. Glycogen synthesis encompasses nucleation by glycogenins and elongation and branching by glycogen synthases. Glycogenins (orthologs of S. cerevisiae Glg1) are upregulated in young fruiting body gills and/or mature fruiting body gills of most compared species (Supplementary Table S12), respectively. This likely corresponds to intense glycogen synthesis during basidiospore maturation (Kües 2000). Consistent with this, orthologs of yeast Gac1, a phosphatase that positively regulates the activity of glycogen synthases, is upregulated in gills and mature fruiting bodies of multiple species. Gac1 in S. cerevisiae regulates glycogen synthases together with Glc7. The latter has orthologs in mushroom-forming fungi, but these are not developmentally regulated. In S. cerevisiae, a number of other CBM21 domain proteins (Pig1, Pig2) also participate in the regulation of glycogen synthase activity, however, these have no clear orthologs in mushroom-forming fungi. On the other hand, the glycogen branching enzyme Glc3 orthologs (represented by C. cinerea 365699) show more diverse expression peaks, and orthologs are upregulated mostly in stipe tissues (in C. cinerea, L. bicolor, P. ostreatus and A. ostoyae) (Fig. 9, Supplementary Fig. S6), indicating that linear glycogen chain production is transcriptionally decoupled from the addition of branched side chains to the glycogen backbone.
Fig. 9.

Expression heatmap of storage carbohydrate metabolism genes in C. cinerea. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively. Developmental stages are abbreviated as follows: VM – vegetative mycelium, H – hyphal knot, P1 – stage 1 primordium, P2 – stage 2 primordium, YFB.C – young fruiting body cap, YFB.L – young fruiting body gills, YFB.S – young fruiting body stipe, FB.C – mature fruiting body cap (including gills), FB.S – mature fruiting body stipe.
Genes that encode proteins upstream of glycogen synthesis, for example orthologs of the uridinephosphoglucose pyrophosphorylase Ugp1 (C. cinerea protein ID: 359312) and the phosphoglucomutase PGM1 (C. cinerea protein ID: 497782) of S. cerevisiae, are developmentally regulated in multiple species (Supplementary Table S12). They show some overlap in expression patterns, in particular, a tendency for upregulation late in development, but their pattern of expression is quite unique and show lower expression fold changes.
It should be noted that the glycogen synthase kinase 3 (GSK-3) (Chang et al. 2019a) gene in C. cinerea (C. cinerea protein ID: 362899), despite its name, it functionally not relevant to glycogen metabolism, it is an orthologue of S. cerevisiae Rim11, a pleiotropic kinase involved in cell cycle regulation.
Glycogen mobilization happens through three primary genes in S. cerevisiae (François & Parrou 2001) - all these have orthologs in mushroom-forming fungi (Supplementary Table S12). Orthologs of the yeast glycogen phosphorylase Gph1 (represented by C. cinerea 495987) mostly show high expression in vegetative mycelia or early development, whereas those of the glycogen debranching enzyme Gdb1 are upregulated also in mature fruiting bodies in all species (Fig. 9). This probably indicates differential usage of phosphorolysis and debranching activities in different stages of development. Sga1 is a sporulation specific glucoamylase in S. cerevisiae. In our species orthologs (represented by C. cinerea 473268) of this gene do not show expression peaks in gills, the tissues in which spore development takes place. Rather, in C. cinerea, this gene is upregulated in young fruiting body cap tissues. How this upregulation corresponds to patterns of glycogen metabolism remains to be understood. Large-scale postmeiotic utilization of glycogen accumulated in subhymenial tissues was reported in C. cinerea (Kües 2000), however, subhymenial tissues were part of gill samples in previous RNA-Seq experiments, which excludes the possibility that Sga1 orthologs were involved in that process. In other species, expression patterns were different, ranging from upregulation in mature fruiting bodies in A. ampla, to peak expression in vegetative mycelium (P. ostreatus and M. kentingensis). We also detected a CDE orthogroup of alpha-amylases (represented by C. cinerea 495249), which find A. nidulans AmyA as best hit and are probably involved in glycogen catabolism. In C. cinerea, this gene showed upregulation in primordia and peak expression in young fruiting body caps, similar to other genes related to glycogen mobilization.
In addition to known members of the glycogen catabolism pathways, we identified two CBM21 domain containing CDE orthogroups that were developmentally regulated in 11/7 and 11/6 species (C. cinerea protein ID: 544130 and 455149) (Supplementary Table S12). In most species, this gene had the highest expression in vegetative mycelia, but showed significant expression dynamics also during fruiting body morphogenesis. This seems to be a Basidiomycota-specific orthogroup, with no significantly similar proteins in model Ascomycota. The CBM21 domain mostly occurs in starch-binding proteins (glucoamylase and α-amylase, Ashkari et al. 1986) whereas in this orthogroup, the CBM21 domain occurs alone, providing no specific clues to function. Therefore, our placement of this CDE orthogroup under this chapter should be viewed tentatively until functional studies prove or disprove the role of the family in glycogen metabolism.
Beyond these gene families, glycogen utilization patterns in fruiting bodies raise a number of exciting research avenues, such as the mode of transport within the fruiting body and the identity of transporters involved in that process.
Trehalose metabolism - Out of the at least five pathways known to be involved in trehalose synthesis, two have been found in Agaricomycetes (Deveau et al. 2008). The first, canonical pathway comprises the action of a protein complex formed by four proteins in S. cerevisiae (Tps1 - C. cinerea 478544, Tps2 - 473004, Tsl1 and Tps3 - no ortholog, François & Parrou 2001). Of these, Tsl1 (C. cinerea protein ID: 503099) and Tps3 are paralogs that arose from a recent whole genome duplication and thus there is only one corresponding gene in filamentous fungi. The three genes showed widespread developmentally regulated expression in examined Agaricomycetes (Supplementary Table S12), with upregulation mostly late in development with notable co-regulation between the three genes, as expected for members of a protein complex (Fig. 9).
A second, noncanonical pathway involving trehalose phosphorylase (TP) exists in fungi and was detected in multiple mushroom-forming species (Saito et al. 1998, Eis & Nidetzky 1999, Wannet et al. 1999, Deveau et al. 2008, Thammahong et al. 2017). Trehalose phosphorylase gene formed a CDE orthogroup (C. cinerea protein ID: 540817) in the Agaricomycetes we examined (Merényi et al. 2021) and show upregulation in stipes and in late stages of development in most species (Supplementary Table S12), although deviations from this pattern exist e.g., in C. cinerea). For example, the gene showed peak expression in primordium stages of A. ampla, S. commune and M. kentingensis (Almási et al. 2019, Krizsán et al. 2019, Ke et al. 2020).
For catabolizing trehalose, fungi exhibit two types of trehalose hydrolysing activities, one termed acid trehalase and the other called neutral trehalase (François & Parrou 2001). Agaricomycetes species possess clear orthologs of neutral trehalases orthologous to Nth1 of S. cerevisiae. On the other hand, and in contrast to Deveau et al. (2008), we did not find proteins in the genomes of Agaricomycetes showing significant sequence similarity (based on BLAST) to acid trehalase. A single neutral trehalase gene was found in Agaricomycetes, this formed a CDE orthogroup at FC>2 (C. cinerea protein ID: 437212, Supplementary Table S12) and showed varied expression patterns in the species examined.
Orthologs of previously described fungal trehalose transporters were not detectable in Agaricomycetes genomes, based on sequence similarity, in agreement with a previous study on L. bicolor (Deveau et al. 2008). However, it should be noted that fungal trehalose transporters are poorly known and current knowledge likely misses a large proportion of genes with this activity (Thammahong et al. 2017).
Mannitol metabolism - Mannitol accumulates in high concentrations in fruiting bodies of certain species, possibly as an osmolyte assisting fruiting body growth (Chakraborty et al. 2004, Zhou et al. 2016b) (see also opposing evidence above). A NADP-dependent mannitol 2-dehydrogenase was reported from A. bisporus (Stoop & Mooibroek 1998, Hörer et al. 2001). In agreement, in L. bicolor Deveau et al. (2008) reported a single mannitol 2-dehydrogenase-dependent pathway of mannitol biosynthesis, which works via the direct reduction of fructose. Using the A. bisporus NADP-dependent mannitol 2-dehydrogenase gene (Stoop & Mooibroek 1998) as a query, we identified an orthogroup (represented by C. cinerea 429272) of short-chain dehydrogenase/reductase genes that contained only A. bisporus, L. bicolor, C. cinerea, and P. ostreatus, but was missing from other species (formed multiple orthogroups probably due to duplications in A. bisporus). However, members of this orthogroup were developmentally regulated in all species (Supplementary Table S12); they were upregulated at the initiation of fruiting and showed expression peaks in gills of C. cinerea and P. ostreatus, young and mature caps of L. bicolor, as well as a marked upregulation in the stipe tissues of A. bisporus. These observations agree with mannitol metabolism being enhanced in fruiting bodies of L. bicolor (Deveau et al. 2008).
In summary, the genomes of Agaricomycetes contain clear orthologs of experimentally characterised members of glycogen, trehalose and mannitol metabolism pathways. Many of the genes in these pathways are developmentally regulated in fruiting bodies, often showing conserved expression peaks in multiple species, reflecting a conservation of storage carbohydrate mobilization strategies within fruiting bodies. Interestingly, in L. bicolor trehalose and glycogen metabolism was upregulated in both fruiting bodies and mycorrhizae, providing links between fruiting bodies and ECM (Deveau et al. 2008, Martin et al. 2008).
Transcriptional regulators
Transcription factors
Transcription factors are proteins that modulate gene expression by binding to specific DNA sequences and subsequently activating or inhibiting transcription. Transcription factors play a major role in the regulation of development across the tree of life, and mushroom-forming fungi are no exception. They are involved in many stages of development, ranging from the establishment of a dikaryon to various aspects of mushroom development.
Mating transcription factors - The best studied are the homeodomain transcription factors of the MatA mating-type locus that are essential for the establishment of a fertile dikaryon, which is required for fruiting body formation in heterothallic basidiomycetes (Raudaskoski & Kothe 2010, Vonk & Ohm 2018). During mating the homeodomain transcription factors expressed from compatible MatA loci dimerize and activate dikaryon-specific transcriptional pathways, including the formation of clamp connections. The mechanism of mate recognition is very similar to that found in the yeast S. cerevisiae (Haber 2012) and is present across the fungal kingdom, although many ascomycetes do not use homeodomain transcription factors for mating (Houbraken & Dyer 2015). In some Agaricales, like C. cinerea and S. commune, the MatA locus has undergone expansion, improving the chances of finding a compatible mate (Freihorst et al. 2016).
TFs acting downstream of mating - Several transcription factors have been identified that act downstream of the MatA homeodomain transcription factors. Deletion of the transcription factor genes hom2, tea1 and wc2 in S. commune resulted in strains that did not show dikaryotic development beyond clamp formation (Ohm et al. 2011, 2013, Pelkmans et al. 2017). Generally, the resulting colonies remained symmetrical and showed no sign of mushroom development. Hom2 is highly conserved in mushroom-forming fungi and features four putative phosphorylation sites. Disruption of these phosphorylation sites leads to a constitutively active Hom2 and the inhibition of vegetative growth and monokaryotic fruiting, indicating that Hom2 is post-translationally regulated (Pelkmans et al. 2017).
WC2 is part of the blue light sensing white collar complex first identified in N. crassa. In N. crassa, WC1 and WC2 interact through PAS domains and WC1 contains a LOV domain that binds a FAD chromophore when exposed to blue light. This results in conformation changes in the white-collar complex and activates transcription factor domains on both WC1 and WC2 (Corrochano 2019). Orthologs of WC1 (dst1 in C. cinerea) and WC2 (Cc.wc2 in C. cinerea) in S. commune and C. cinerea fulfil a similar role and deletion (S. commune wc1 and wc2, C. cinerea wc2) or mutation (C. cinerea wc1) of either wc1 or wc2 prevents the formation of mature fruiting bodies and is called a blind phenotype (Terashima et al. 2005, Kuratani et al. 2010, Nakazawa et al. 2011, Ohm et al. 2013). In S. commune this results in a phenotype where no dikaryotic development is observed and in increased sensitivity to intense light. However, in C. cinerea a blind phenotype leads to the so called ‘dark stipe’ phenotype (Kamada et al. 2010). In this phenotype, the primordial shaft is elongated, but pileus and stipe development cannot be completed. In S. commune, WC1 does not contain a DNA-binding domain, unlike WC1 in N. crassa, making transcriptional activity dependent on WC2 (Ohm et al. 2010). Orthologs of white-collar complex members with the same function have been reported in L. edodes (Sano et al. 2009) and P. ostreatus (Qi et al. 2020). Furthermore, developmental expression of wc members was reported for F. filiformis (Liu et al. 2020).
Mutants of the gene tea1, which is downregulated in Δwc1, Δwc2, Δhom2, Δbri1 and Δfst4 dikaryons, also did not form mushrooms (Pelkmans et al. 2017). It should be noted that in tea1 deletion mutants, dikaryotic colonies sporadically produced clusters of mushrooms, indicating that Tea1 is not essential for fruiting body formation and may act as a “switch” that can be bypassed. The ARID/BRIGHT DNA binding domain transcription factor Bri1 was initially also thought to play a major role in fruiting development (Ohm et al. 2011). However, later characterization of bri1 deletion strains revealed that inactivation leads to significant growth defects that result in a delay in fruiting (Pelkmans et al. 2017). The fungal specific transcription factor fst4 has a very similar phenotype where no mushrooms are formed, but deletion strains of this transcription factor do form asymmetrical colonies after mating, which is the first step in mushroom development (Kües & Liu 2000, Ohm et al. 2010). In P. ostreatus, mutations in gat1 resulted in a strain that did not fruit (Nakazawa et al. 2019). The missense mutation inside the zinc-finger binding domain resulted in a dominant gain of function mutation, which suggests that the mutated transcription factor has an altered DNA-binding specificity. This is in line with disruption of the ortholog gat1 in S. commune, where the phenotype was not dominant and resulted in the formation of more, but smaller mushrooms (Ohm et al. 2011). A general overview of Zn (II)2Cys6 zinc cluster transcription factors, including expression patterns in P. ostreatus fruiting bodies, was published by Hou et al. (2020). In Ganoderma lucidum, silencing of the C2H2-type transcription factor pacC also resulted in the absence of fruiting bodies, as well as altered mycelial growth (Wu et al. 2016). Interestingly, PacC is conserved across the fungal kingdom and activates genes under alkaline conditions, and inhibits genes under acidic conditions (Peñalva et al. 2008). Therefore, it is unclear if the absence of fruiting can be attributed to fruiting-specific transcription or general homeostasis. In L. edodes, PriB was described as a primordium-upregulated transcription factor (Endo et al. 1994). Finally, silencing of hada-1 resulted in delayed fruiting, as well as multiple growth defects, in Hypsizygus marmoreus (Zhang et al. 2021).
Downstream in development, S. commune c2h2, fst1 and zfc7 gene disruption resulted in arrested fruiting body formation, where primordia were formed that did not develop further into mature fruiting bodies (Ohm et al. 2011, Vonk & Ohm 2021). Interestingly, overexpression of c2h2 could increase the rate of fruiting in A. bisporus, indicating that c2h2 expression is a limiting step in fruiting body development (Pelkmans et al. 2016b). The transcription factors Fst1 and Zfc7 were identified due to enrichment of histone 3 lysine 4 (H3K4) dimethylation in their genes during fruiting body development after 4 days, which indicates increased genetic accessibility and transcription during this developmental stage (Vonk & Ohm 2021). Further downstream, the C. cinerea high-mobility group transcription factor Exp1 regulates pileus expansion and autolysis to complete fruiting (Muraguchi et al. 2008b). Notably, this transcription factor is conserved and developmentally regulated in all studied mushroom-forming fungi, except for A. ampla, Pt. gracilis and S. commune, whose fruiting bodies do not feature a pileus.
Putative repressors of fruiting - Instead of promoting fruiting body development, several transcription factors are involved in promoting vegetative growth or repressing fruiting. These include F. velutipes lfc1 and hmg1, where silencing increased the rate of fruiting (Wu et al. 2020a, Meng et al. 2021). Additional transcription factors are P. ostreatus Pofst3 and S. commune fst3, gat1 and hom1 (Ohm et al. 2011, Qi et al. 2019). These genes are differentially regulated in the majority of mushroom forming species, and conserved in all of them. When the genes are deleted (S. commune) or silenced (P. ostreatus) it results in the formation of more, smaller mushrooms. Finally, premature stop codons in pcc1 in C. cinerea (natural variation, truncated from 561 to 210 amino acids, HMG-box not disrupted) and P. ostreatus (CRISPR/Cas9-assisted gene mutagenesis, truncated from 471 to 58 amino acids, HMG-box disrupted) resulted in monokaryotic fruiting and the formation of pseudoclamps (Murata et al. 1998, Boontawon et al. 2021). In C. cinerea these pseudoclamps were formed during monokaryotic growth, indicating a relation between the MatA homeodomain transcription factors and Pcc1. In P. ostreatus however, dikaryotic strains formed pseudoclamps, suggesting a role in clamp formation.
Many transcription factors are conserved across fungi or specifically in mushroom-forming fungi. Both wc2 and pacC are conserved in both ascomycetes and basidiomycetes and except for hmg1 (F. velutipes), lfc1 (F. velutipes), pdd1 (F. velutipes), pcc1 (C. cinerea and P. ostreatus) and zfc7 (S. commune) all characterised transcription factors are at least conserved in mushroom-forming fungi. Cag1 (C. cinerea protein ID: 466216) and Cc.Tup1 (C. cinerea protein ID: 369655) of C. cinerea are further examples of highly conserved transcription factors (Masuda et al. 2016). They are homologs of the highly conserved general transcriptional co-repressor Tup1 of S. cerevisiae, which can form repressive complexes with a wide range of sequence-specific transcription factors. Deletion of Cag1 resulted in a cap-growthless phenotype, which, upon closer inspection turned out to be caused by developmental defects of gills (Masuda et al. 2016). However, the expression patterns are generally not conserved during fruiting body development (Almási et al. 2019, Krizsán et al. 2019). For example, the developmental stage with the highest expression level of the ortholog can vary greatly between species. This can indicate either that transcriptional regulation of fruiting body development is poorly conserved between species, or that transcription factors are regulated post-translationally. The MatA mating type genes are an example of post-translational regulation, where the transcription factors are constitutively expressed, but only become activated when heterodimers are formed between compatible homeodomain transcription factors. Similarly, in N. crassa WC2 is activated by structural changes in its interaction partner WC1 when blue light is detected. Furthermore, Hom2 in S. commune is constitutively expressed, but post-translationally phosphorylated to become inactive (Pelkmans et al. 2017).
CDE orthogroups of transcription factors - While many expression patterns of transcription factors are not conserved in mushroom-forming fungi, there are several interesting and conserved transcription factors. We identified 51 orthogroups of TFs that were developmentally regulated in a significant number of species (Supplementary Table S13). Among the functionally characterised transcription factors, orthologs of Dst1 (=WC1, C. cinerea protein ID: 369621) and Cc.wc2 (C. cinerea protein ID: 8610) of C. cinerea formed CDE orthogroups developmentally regulated in 10/4 and 10/6 (FC>2/4) species, respectively. Orthologs of dst1 and Cc.wc2 showed varied expression profiles in the examined species. An orthogroup of homeodomain transcription factors (C. cinerea protein ID: 497620) is developmentally regulated during fruiting body formation in all mushroom forming fungi, except for Pt. gracilis, which is known for its simplified fruiting body structure. The detected CDE orthogroups also included exp1 of C. cinerea (represented by C. cinerea 421729), which were developmentally regulated in 9/5 species (FC>2/4). In accordance with its reported function in C. cinerea (Muraguchi et al. 2008b), it showed peak expression in young fruiting body caps. However, the A. ostoyae, L. bicolor and M. kentingensis orthologs were upregulated in stipe tissues. In P. ostreatus we found the highest expression in cap cuticle. Furthermore, a C2H2-type transcription factor (C. cinerea protein ID: 494418) is conserved and expressed in all mushrooms except for A. ampla, Pt. gracilis and S. commune. It may therefore play a similar role to Exp1 in C. cinerea (Muraguchi et al. 2008b), which is also conserved and developmentally regulated in most fungi except for A. ampla, Pt. gracilis and S. commune. While many other transcription factor orthogroups are conserved in all studied mushroom-forming fungi, their expression patterns are not strongly conserved. Therefore, proteomics or functional analyses are required to further elucidate the role of transcription factors in fruiting body formation.
RNA binding proteins
Pumilio family – This family comprises evolutionarily conserved sequence-specific RNA-binding proteins that bind 3’ UTR regions of specific mRNAs. This family has diverse roles in fungi, including the regulation of meiotic and sexual processes (e.g., Mcp2 of S. pombe) (Wilinski et al. 2017, Liu et al. 2018). Pumilio proteins were found to be active during fruiting body formation in both the Asco- and Basidiomycota (Krizsán et al. 2019, Merényi et al. 2020). With a few exceptions, all Agaricomycetes contain six Pumilio protein encoding genes. These formed two CDE orthogroups (Supplementary Table S14), one of which (C. cinerea protein ID: 378537) contains orthologs of the Sch. pombe meiotic coiled-coil protein Mcp2 and shows expression dynamics reminiscent of mitotic and meiotic genes (see chapter “Meiotic and mitotic gene expression”), and another, comprising orthologs of Sch. pombe Puf3 and C. neoformans Pum1 (C. cinerea protein ID: 354109). Both orthogroups are developmentally regulated in a large number of species (8/7 and 7/6 at FC>2/4, respectively). In C. neoformans, pum1 has been associated with basidium development and morphogenesis, suggesting that its orthologs may have similar roles in Agaricomycetes too.
RRM family - We identified five very conserved CDE orthogroups of RNA recognition motif (RRM) family proteins. One of these (C. cinerea protein ID: 471238) were annotated as related to mitosis and meiosis and is discussed in chapter “Meiotic and mitotic gene expression”. The RRM family comprises proteins that bind diverse RNA types and shows considerable diversity in fungi, with 68–88 genes in the Agaricomycetes. Overall, relatively few members of the family were developmentally expressed, however, the orthogroups we detected showed a conserved expression pattern. One of them (C. cinerea protein ID: 357717) was developmentally regulated in all 12 species (Supplementary Table S14) and was upregulated in the second half of fruiting body development (young fruiting body and mature fruiting body in C. cinerea), in particular in gills (A. ostoyae, P. ostreatus, M. kentingensis, C. cinerea) where gill tissue was sampled separately, or caps (L. bicolor, L. edodes). It was also upregulated considerably in primordia relative to vegetative mycelia. A less characteristic, but similar and very conserved late upregulation was observed also in the second CDE orthogroup (C. cinerea protein ID: 356106), which contains reciprocal best hits of Sch. pombe Vip1.
The third CDE orthogroup (C. cinerea protein ID: 193545, 11/11 species at FC>2/4) contains C. neoformans Csa2. Members of this orthogroup showed strong induction in the first primordium stage in C. cinerea, A. ostoyae, C. aegerita, L. bicolor, L. edodes, P. ostreatus, S. commune, A. ampla, M. kentingensis. In C. neoformans it shows a strong peak 24h after the induction of sexual development, which, combined with evidence for regulation by pum1, led Liu et al. to conclude it is involved in basidium development and sporulation (Liu et al. 2018). It’s an orthologue of Sch. pombe Mei2, which is related to meiosis. The other three basidium-related genes reported by Liu et al. (2018) (Fad1, Fas1, CNA00260) do not have clear orthologs in Coprinopsis.
The CDE orthogroup represented by C. cinerea 424800 (Supplementary Table S14) was consistently upregulated in gills and sporulating stages (in A. ampla, S. commune) in all species, suggesting roles in hymenium and/or spore formation. In model Ascomycota, proteins in this orthogroup find Pab1 of both Sch. pombe and S. cerevisiae as their best Blast hits, which are related to mRNA export and the control of poly (A) tail length.
Finally, the CDE orthogroup comprising C. cinerea 368527 includes best reciprocal hits of S. cerevisiae Nam8, a meiosis-specific splicing factor. Our expression data are consistent with a meiosis-specific role: the gene is developmentally regulated in 6/4 species (FC>2/4, Supplementary Table S3) and shows a peak expression in meiotic tissues in all species (gills, cap+gills or mature fruiting bodies). Based on these data, we hypothesize that this gene also participates in meiosis-specific splicing events in the basidia during fruiting body formation.
RNA interference, Small RNA
RNA interference is a conserved eukaryotic gene-silencing mechanism that acts via base pairing of small noncoding RNAs of ~20-30 nucleotides to DNA/RNA targets. RNAi is key to developmental and differentiation processes, genome defence against viruses or transposons. There is very limited knowledge on the role of RNAi in mushroom-forming fungi (but see Lau et al. 2018). In contrast, these processes are extensively studied in the Ascomycota (in particular N. crassa and Sch. pombe, Dang et al. 2011), where RNAi has well-documented roles in defence against viruses, transposons, meiotic silencing of unpaired DNA, translational repression and even inter-organismal pathogenic interactions (Weiberg et al. 2013).
A fully functional RNAi machinery exists in the Basidiomycota. Based on orthology to N. crassa proteins, Lau et al. annotated dicer-like proteins as Dcl-1 (C. cinerea protein ID: 444647), Dcl-2 (C. cinerea protein ID: 366047) and Dcl-3 (C. cinerea protein ID: 438955) (Lau et al. 2018), whereas Stajich et al. annotated the argonaute family proteins AGO (C. cinerea protein ID: 355234), an AGO-like protein (C. cinerea protein ID: 473372) and Qde-2 (C. cinerea protein ID: 363313) (Stajich et al. 2010) (Supplementary Table S15). We identified 3–16 genes encoding argonaute and argonaute-like proteins in the examined Agaricomycetes. Furthermore, fungal RNA interference involves RNA-dependent RNA polymerases, which are required for generating double-stranded RNA or for amplifying sRNA signals (Dang et al. 2011). The genomes of the examined Agaricomycetes contain 5–13 RNA-dependent RNA polymerases; of these, the orthogroup represented by C. cinerea 498671 is most similar to the RDP1 protein of S. pombe, which was shown to be integral to RNA interference (Carmichael et al. 2004). Other RNA-dependent RNA polymerases (Supplementary Table S13) might participate in alternative RNAi pathways or signal amplification (Calo et al. 2012). Interestingly, we found no clear orthologs of SAD-1, a meiotic silencing protein of N. crassa (Borkovich et al. 2004), in the examined genomes.
RNAi components show some expression dynamics during fruiting body development. Genes encoding Dcl-2, Dcl-3 and AGO-like proteins were upregulated in C. cinerea primordia relative to vegetative mycelium (Lau et al. 2018). Similarly, AGO-like and Qde-2 were found developmentally regulated in C. cinerea by Krizsán et al. (2019), although Dcl-1–3 genes were not. All of the seven RNA-dependent RNA polymerases were developmentally regulated in the data of Krizsán et al. (2019). In our meta-analysis, the above-listed components did not show significant developmental expression dynamics (Supplementary Table S15). The only noteworthy orthogroup (represented by C. cinerea 502116), was developmentally regulated in 9/6 species (FC>2/4) and contains Argonaute-like proteins that share highest similarity with A nidulans RsdA and Sch. pombe Ago1, the first of which is involved in RNA-silencing (Hammond et al. 2008) whereas the second has a large suite of described functions, from RNA silencing to chromosome segregation and cell cycle arrest (Carmichael et al. 2004). We found no conserved expression pattern in this orthogroup across the examined species.
The most direct evidence for the role of microRNA-like RNAs (milRNA) in mushroom development was reported by (Lau et al. 2018), who compared small RNA transcriptome profiles of vegetative mycelium and primordium stages and detected 22 milRNA in C. cinerea, several of which were differentially expressed between vegetative mycelia and primordia. These results point to the potential roles of milRNA in mushroom development, although more studies are needed to understand its roles.
RNA interference has been exploited as a tool of genetic manipulation in mushroom-forming fungi, often to compensate for the lack of reliable reverse genetics tools in these organisms. Gene knockdown has been demonstrated in S. commune (de Jong et al. 2006), C. cinerea (Namekawa et al. 2005, Wälti et al. 2006, Heneghan et al. 2007), L. bicolor (Kemppainen et al. 2009), Ph. chrysosporium (Matityahu et al. 2008), P. ostreatus, A. bisporus (Costa et al. 2009), Grifola frondosa (Sato et al. 2015) and L. edodes (Nakade et al. 2011), among others, demonstrating the utility of knockdown techniques in cases/species when gene knockout was not feasible (particularly in the pre-CRISPR times).
Chromatin regulators
Chromatin remodelling is a fundamental and general mode of epigenetic gene expression regulation. Dynamic chromatin states are generated primarily via chromatin remodelling complexes and histone modifications in fungi, in response to environmental or internal signals. Importantly, chromatin remodelling often provides the basis for broad changes in gene expression, by modulating the accessibility of DNA for transcription, as opposed to sequence-specific transcription factors, which regulate only specific genes. Epigenetic changes often associate with developmental processes; these receive increasing attention in multiple lineages, including fungi. Accordingly, studies addressing the role of chromatin remodelling in mushroom development, although quite few at the moment, revealed fundamental roles of chromatin states in fruiting-related gene transcription. For example, deleting Cc.snf5 (C. cinerea protein ID: 365798, see Supplementary Table S16), a member of the SWI/SNF chromatin remodelling complex, leads to the failure to initiate fruiting body development in C. cinerea (in addition to defects in dikaryon formation) (Ando et al. 2013). In C. cinerea two further genes identified in mutant analyses, ich1 (Muraguchi & Kamada 1998) and Cc. rmt1 (Nakazawa et al. 2010), may be related to chromatin regulation. Recent ChIP-Seq analyses of epigenetic states revealed substantial changes in transcriptional activity (with H3K4me2 as a proxy) during the transition to fruiting (Vonk & Ohm 2021), whereas histone acetyltransferase complex members were found in quantitative trait loci in L. edodes (Zhang et al. 2021d). The presence of SET domain methyltransferases in the Basidiomycota is also worth mentioning (Freitag 2017). Although evidence at the moment is scanty, the few available studies and extrapolation from other organisms suggests that chromatin remodelling may be a significant factor in fruiting body formation. Therefore, in the following chapter we annotate and discuss expression patterns of genes related to chromatin maintenance and remodelling based on homology to known chromatin remodelling components in S. cerevisiae and N. crassa.
Histone gene expression correlates with fruiting body initiation and sporulation
Histones - Histones are highly conserved proteins that assemble into octameric nucleosomes and condense DNA into chromatin in a tightly regulated manner. Shuffling of histone variants can generate nucleosome variants, which, combined with diverse posttranslational modifications of histones (acetylation, methylation, phosphorylation) can generate a finely diversified histone code contributing to fine-tuned regulation of chromatin states, which, in turn, impact transcriptional patterns of genes (Borkovich et al. 2004). Histone modifications are often used to make readouts of transcriptional states associated with development (Vonk & Ohm 2021). Agaricomycetes have 14–40 histone and histone-variant encoding genes. They show a very similar overall expression pattern in all species that resembles the expression of meiotic/mitotic and ribosomal genes: upregulation in primordium stages as well as meiotic tissues (gills or caps+gills) coincident with sporulation (Fig. 10, Supplementary Fig. S7). Though overall very conserved, there are slight variations to this general pattern: A. ampla shows a peak only in primordia, whereas in C. cinerea and L. tigrinus a meiosis/sporulation associated peak is better visible, while the primordium-peak is modest. We detected four CDE orthogroups of histones and histone variants (Supplementary Table S16). These include a basidiomycetes-specific histone H2A component (C. cinerea protein ID: 363625), which is developmentally expressed in 8/6 species (FC>2/4) and show considerable upregulation in primordia relative to vegetative mycelium in most species. Other CDE orthogroups also showed primordium- and gill-upregulation. A straightforward interpretation of the similar expression patterns of histones and cell proliferation related genes is a mutual link to DNA repair. H2A histone phosphorylation is associated with DNA repair (Downs et al. 2000, Borkovich et al. 2004), providing a potential mechanistic explanation for the observed expression patterns. Somewhat contradicting this is the fact that relatives of the Mec1 kinase (C. cinerea protein ID: 19106), which phosphorylates H2A.Z in S. cerevisiae (Downs et al. 2000), are not developmentally regulated in Agaricomycetes. On the other hand, histone upregulation in primordia and meiotic tissues may also be interpreted as evidence for intense transcriptional reprogramming, which has been hypothesized for the mycelium-fruiting body transition (Krizsán et al. 2019, Vonk & Ohm 2021). Alternatively, intense DNA replication may require increased supply of histones in proliferating tissues.
Fig. 10.

Expression heatmap of chromatin remodelling related genes (histones, histone chaperones, histone deacethylases) in C. cinerea. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively. Developmental stages are abbreviated as follows: VM – vegetative mycelium, H – hyphal knot, P1 – stage 1 primordium, P2 – stage 2 primordium, YFB.C – young fruiting body cap, YFB.L – young fruiting body gills, YFB.S – young fruiting body stipe, FB.C – mature fruiting body cap (including gills), FB.S – mature fruiting body stipe.
Histone chaperone - We detected a histone chaperone CDE orthogroup containing homologs of S. cerevisiae Chz1 (represented by C. cinerea 498885) (Supplementary Table S16), which forms a single-copy gene family in the Agaricomycetes. Members of this orthogroup are involved in histone replacement during transcription and showed the same expression pattern as histones (peaking in primordia and meiotic tissues), so we speculate they are involved in the same cellular processes during fruiting body initiation and sporulation.
We next inventoried histone acetyltransferases, histone deacetylases, histone methyltransferases, histone ubiquitylases, histone kinases and histone ADP-ribosylases based on homology to corresponding N. crassa genes described by Borkovich et al. (2004) as well as diagnostic conserved domains.
Histone acetyltransferase (HAT) - We found one HAT gene per species in the examined Agaricomycetes (represented by C. cinerea 420113), in contrast to Ascomycota, that have multiple histone acetyltransferase genes (Borkovich et al. 2004). This family showed no developmental expression dynamics in our species. However, we detected several protein N-acetyltransferases, which resemble HATs, but lack orthology to known HATs and do not contain diagnostic conserved domains of HATs. They share an identical domain composition with Sch. pombe Ard1, a Gcn5-related N-acetyltransferase (GNAT), which acetylates diverse proteins, including transcriptional regulators. A CDE orthogroup of GNATs (C. cinerea protein ID: 457726) was developmentally expressed in 8/4 species (FC>2/4, Supplementary Table S16), however, given the lack of clear histone link we only tentatively discuss this family here, noting that functional analysis is needed to confirm their functional annotation.
Histone deacetylases (HDACs) - we distinguished two groups of HDACs, one related to S. cerevisiae Hda1 and the Sirtuin family, which were detected in 6–7 (11 in A. ostoyae) and 4–9 copies in the examined Agaricomycetes, respectively. Neither group showed considerable developmental expression dynamics, except for one CDE orthogroup comprising sirtuins (represented by C. cinerea 455597), which was developmentally expressed in 9/9 species (FC>2/4, Supplementary Table S16). This family is orthologous to Hst4 of Sch. pombe and Hst3 of S. cerevisiae, which regulate DNA damage response and repair by regulating histone H3K56 acetylation (Konada et al. 2018). Consistent with their presumed role, the expression of this gene family followed closely that of mitosis/meiosis and DNA repair related genes in all species: they showed a small expression peak in primordia and a larger one in meiotic tissues coinciding with sporulation (Fig. 10).
Histone ADP-ribosylases are poly (ADP-ribose) polymerases (PARPs) and are among the least understood factors in epigenetic regulation; they attach (poly-)ADP-ribose moieties to proteins, including histones and are thought to regulate histone turnover and DNA damage response. In N. crassa the family is represented by a single gene, parp (Borkovich et al. 2004). The examined Agaricomycetes genomes harbour 3–9 copies of PARPs, of which two formed CDE orthogroups (C. cinerea protein ID: 459336, 384835), which were developmentally expressed in 8/4 and 6/5 species (FC>2/4, Supplementary Table S16), respectively. Their expression patterns did not follow that of histones, suggesting that their actions are independent of those.
Histone methyltransferases and histone kinases showed no noticeable expression dynamics in fruiting bodies (Supplementary Table S16), suggesting that they are equally important in fruiting bodies and vegetative mycelia.
Other chromatin related genes
Ich1 gene family - The only methyltransferase that might be connected to chromatin regulation was the gene underlying the ‘Ichijiku’ mutant, which was first identified by (Muraguchi & Kamada 1998), as a mutant that produced malformed, barrel-shaped primordia, in which cap development fails. The corresponding ich1 gene encodes a nuclear methyltransferase that contains a winged-helix DNA binding domain and was reported to be upregulated in primordia (Muraguchi et al. 2015). Our results agree and extend these observations: Ich1 orthologs form a CDE orthogroup (represented by C. cinerea 545797) that is developmentally expressed in 11/9 species (FC>2/4) (Supplementary Table S16) and shows a marked upregulation in primordia relative to vegetative mycelium in C. cinerea, A. ostoyae (weakly), A. ampla, S. commune, A. bisporus, M. kentingensis and P. ostreatus. The exact mechanism of how Ich1 regulates tissue formation is not known, however, this gene represents a very interesting target for detailed studies, especially given its putative role in chromatin regulation and/or methylation. On a related note, Cc.rmt1 (C. cinerea 485296), an arginine methyltransferase which was suggested to participate in histone methylation and to be required for hyphal knot and fruiting body formation (Nakazawa et al. 2010), forms an orthogroup which is developmentally regulated in nine species (at FC>2) (Supplementary Table S16). The exact role of Cc.rmt1 is not known so its role in histone methylation remains preliminary.
Histone isomerases - We found a CDE orthogroup that was widely developmentally expressed (represented by C. cinerea 493226) (Supplementary Table S16). Ascomycota orthologs of these genes are histone proline isomerases in the peptidyl-prolyl cis-trans isomerase family, which contribute to nucleosome assembly and thus regulate gene expression. This orthogroup shows upregulation in gills of C. cinerea, A. ostoyae and P. ostreatus (all species in which gills were sampled separate) and in cap tissues of L. edodes, M. kentingensis, L. tigrinus. These expression patterns are consistent with a role in sporulation. Genes in this orthogroup also show upregulation in primordia relative to vegetative mycelia, although this is usually modest (2–3x, 10x in P. ostreatus).
HMGA and AT-hook protein - We detected a CDE orthogroup (C. cinerea protein ID: 473747) (Supplementary Table S16) that includes primarily stipe-upregulated genes (in C. cinerea, A. ostoyae, L. tigrinus, P. ostreatus, M. kentingensis, but not L. bicolor) that shows expression peaks also in YFB cap of C. cinerea and FB cap of P. ostreatus. We found no expression dynamics in Schizophyllaceae species, which lack stipes. The orthogroup belongs to the High mobility group A family, which comprises AT-rich DNA binding proteins with an important role in chromatin organization. This family appears to be patchily distributed in the examined species, but nevertheless seems to be important for fruiting body development.
SRA-YDG domain proteins - We detected a CDE orthogroup (C. cinerea protein ID: 511209) (Supplementary Table S16) of proteins containing an SRA-YDG (IPR003105) domain, which occurs in histone and chromatin binding proteins and may be associated with the cell cycle. Proteins with this domain show expression patterns similar to those of histones. They are upregulated in primordia and in meiotic stages (gills, caps with gills) in C. cinerea, A. ostoyae, P. ostreatus and L. edodes. In the Schizophyllaceae as well as P. ostreatus, the gene family is upregulated in primordia relative to vegetative mycelium samples. Based on their expression patterns, we hypothesize they are related to cell proliferation, the cell cycle or chromatin modification, but available data do not allow us to further narrow down their putative function among these categories.
Alternative splicing and spliceosome components
Alternative splicing is a post-transcriptional mechanism for regulating transcript identity and abundances. mRNA variants generated by splicing can increase the diversity of proteins transcribed from a single gene, but can also destine transcripts to the nonsense-mediated decay pathway and thus reduce transcript abundance posttranscriptionally (Krizsán et al. 2019). Transcriptomes allow us to describe alternative splicing complexity of organisms at an unprecedented depth. Several studies looked at alternative splicing in fungi (Zhao et al. 2013, Grützmann et al. 2014, Jin et al. 2017), including Agaricomycetes model systems (Lugones et al. 1999a, Gehrmann et al. 2016, Krizsán et al. 2019). The Basidiomycota and, in particular, the Agaricomycetes displayed a higher percentage of spliced genes than did other groups of fungi (Grützmann et al. 2014, Krizsán et al. 2019). Results of these studies highlighted that, of the several possible splicing events, intron retention (IR) is the most common in fungi while exon-skipping, which most amply contributes to generating protein isoforms, accounts for only 0.5–2.5 % of fungal splicing events. Most research on splicing focuses on the latter, however, it appears that exon skipping-rich transcriptomes represent an animal innovation (Grau-Bové et al. 2018). IR events have been considered either as transcriptional noise or as a regulatory mechanism (Middleton et al. 2017). IR can introduce premature stop codons in transcripts or new functional elements for RNA binding proteins. Premature stop codons can activate nonsense-mediated decay, a surveillance mechanism that results in the degradation of the mRNA transcript. Recent results suggest that IR may not only regulate transcript levels, but can be a key driver of posttrancriptional processes (Middleton et al. 2017). There is evidence that in the Basidiomycota IR exerts its effect on transcripts independent of the nonsense-mediated decay pathway (Gonzalez-Hilarion et al. 2016).
Functionally characterised examples of developmentally relevant splicing events are known in the Ascomycota. For example, alternative splicing of the blue light receptor wc-1 gene was observed in Phaeosphaeria spp. and it was hypothesized that alternative splicing (AS) may control various light-dependent reactions (Chiu et al. 2010). In Neurospora wc-1 transcription is controlled by multiple distinct cis-regulatory regions (termed different promoters), which responded differently (not, weakly or strongly) to light and resulted in alternative protein isoforms (Káldi et al. 2006). These observations, although probably just scratching the surface, implicate AS as an important contributor to the development of fruiting bodies.
Alternative transcript isoforms affect a considerable portion of genes during fruiting body development. IR-generated transcript isoforms with a dynamic expression are abundant in fruiting bodies (Gehrmann et al. 2016, Krizsán et al. 2019), affecting 20–46 % of expressed genes, pointing towards potential important regulatory roles. In S. commune, 69 % of the alternative spliced transcripts had altered predicted functional properties or subcellular localization (Gehrmann et al. 2016). They reported the largest number of splicing events at the fruiting body initiation stage in S. commune, whereas Krizsán et al. (2019) did not find substantial differences in the number of splicing events across developmental stages.
Splicing can influence transcript levels significantly during fruiting body development. A recent study found 425 genes in S. commune for which an alternative transcript replaced the highest expressed transcript in some stage of development. Many such events happened at fruiting body initiation (referred to as the aggregate stage), pinpointing the magnitude of transcriptome reprogramming at this developmental transition. A conceptually similar scenario, genes whose expression do not vary considerably during fruiting body development, but have splicing isoforms that vary in their expression significantly, was detected for ~160–1 300 genes in six Agaricomycetes (Krizsán et al. 2019).
Taken together, recent studies on Agaricomycetes suggest that splicing, in particular intron retention, is an important process in fruiting body development, contributing to posttranscriptional regulation of transcript levels or generates functional diversity in proteins translated from alternative transcripts. The prevalence of intron retention in fungal transcriptomes suggests that the main role of splicing in fungi might be posttranscriptional transcript level regulation rather than the expansion of the proteome space, although mechanistic or proteome-level studies are missing at the moment.
We here annotated spliceosome components and splicing-associated proteins in C. cinerea based on knowledge in S. cerevisiae and Sch. pombe (Käufer & Potashkin 2000, Cherry et al. 2012). We annotated 50 orthogroups associated with splicing, however, none of them showed remarkable expression dynamics in fruiting bodies (Supplementary Table S17). These included the gene described as Le.cdc5 and Cc.cdc5 in L. edodes and C. cinerea, respectively (Miyazaki et al. 2004), a pre-mRNA splicing factor.
Signal transduction systems and communication
Sensing external signals, either coming from neighbouring cells or the environment is key for all multicellular organisms to orchestrate their development and behaviour. Accordingly, signal transduction and cell-to-cell communication systems have expanded during the evolution of several clades of multicellular organisms (Knoll 2011, de Mendoza et al. 2014). Fungal research focused more on environment sensing and self/nonself recognition (Bloemendal et al. 2013, Fischer & Glass 2019) and molecular pathways of cell-to-cell communication are quite poorly understood. There is evidence that various volatile compounds (e.g., oxylipins), pheromones, and conserved intracellular signalling pathways of kinases (e.g., MAP kinases), and cell surface receptors may participate in developmental processes in fungi. In this chapter we therefore take a data driven approach and discuss gene families which formed CDE orthogroups and may be involved in developmental signalling. In addition, we annotated and analysed conserved MAPK, GPCR and pheromone sensing pathways to address their role in fruiting body morphogenesis.
Oxylipins
In fungi, oxylipins are responsible for a range of signalling functionalities and act as paracrine hormone-like substances or as so-called ‘infochemicals’ (Combet et al. 2006, Holighaus & Rohlfs 2019). Ascomycota species produce diverse oxylipin compounds (reviewed in Gessler et al. 2017) and changes in their levels influence development. For example, oxylipins can regulate the yeast-to-hypha transition in dimorphic fungi (Tsitsigiannis & Keller 2007, Brodhun & Feussner 2011), as well as immune evasion (Rennemeier et al. 2011) and communication with the host in pathogens, while a well-known group of oxylipins called psi (precocious sexual inducer) factors regulates sexual and asexual development in Ascomycota (Tsitsigiannis & Keller 2007). Basidiomycota oxylipins are less known. The best-known oxylipin of mushroom-forming fungi is 1-octen-3-ol, an enantiomeric compound that is mainly produced as (R)-(–)-1-octen-3-ol by widely-known species (Zawirska-Wojtasiak 2004). On the one hand, it acts as an interkingdom-signalling molecule. It may attract or repel fungivorous arthropods (Fäldt et al. 1999, Holighaus & Rohlfs 2019) or derogate insect larval development (Yin et al. 2015). In combination with the equally prominent octan-3-one that may attract specialist fungivores, it has also been shown to attract predators of such fungivores (Fäldt et al. 1999). Furthermore, 1-octen-3-ol has been shown to retard plant seed germination and vegetative growth (Hung et al. 2014). On the other hand, 1-octen-3-ol shows a quorum-sensing like action as it inhibits colony growth (Yin et al. 2015, Kües et al. 2018), conidiation (Fäldt et al. 1999, Yin et al. 2015), fruiting initiation (Noble et al. 2009, Eastwood et al. 2013) and germination where spores are crowded (Chitarra et al. 2004, Combet et al. 2006).
The composition of the oxylipin cocktail emitted by intact mushroom tissue changes in the course of mushroom development while measurement of the most abundant oxylipins during that process crucially depends on the sampling method: disruption of fungal tissue has been shown to trigger the release of oxylipins that are not emitted when fungal material is intact (Fäldt et al. 1999, Combet et al. 2006). The changing specific oxylipin bouquet emitted by a developing mushroom may not solely reflect adaptation to insect attraction for spore dispersal and repellence of fungivores. It is also possible that, e.g., octan-3-one and 1-octen-3-ol are produced by the fruiting body to inhibit on-colony spore germination (Combet et al. 2006) or colonization by mycoparasites (Berendsen et al. 2013). During fruiting body development, octan-3-one emission either positively (Combet et al. 2009) or negatively (Zhang et al. 2008, Orban et al. 2020, 2021) correlates with the amount of 1-octen-3-ol produced by (not yet) sporulating tissue samples of certain species.
The biosynthesis pathway of 1-octen-3-ol has interested mycologists for a long time (Combet et al. 2006, Kües et al. 2018, Orban et al. 2021), but remained unresolved to date. The process starts by oxygenation of polyunsaturated fatty acids (PUFAs) with linoleic acid as the predominant PUFA in Asco- and Basidiomycota (Combet et al. 2006, Brodhun & Feussner 2011). It was suggested that lipoxygenases (LOXs), linoleate diol synthases (LDSs that combine a dioxygenase – DOX – and an isomerase activity), hydroperoxide lyases (HPLs) and alcohol dehydrogenases (ADHs) participate in 1-octen-3-ol biosynthesis (Combet et al. 2006). Still, it is possible that 1-octen-3-ol can be produced by incomplete beta-oxidation or by multiple pathways (Combet et al. 2006, Brodhun & Feussner 2011). Orban et al. recently attempted to identify genes involved in oxylipin production based on correlations between 1-octen-3-ol concentrations and gene expression patterns (Orban et al. 2021) including a gene of an ene-reductase for octan-3-one formation from ADH-mediated conversion of 1-octen-3-ol.
Two main enzyme classes have been implicated in fungal oxylipin production, LOXs and LDSs (Combet et al. 2006, Orban et al. 2021). Both act on linoleic acid as the starting point, from which various compounds are produced by first an oxidation reaction, then by further modification (Combet et al. 2006). We found LOXs to be highly patchy in Agaricomycetes, whereas LDSs (basically the Ppo family of proteins) are widely conserved. Incomplete beta-oxidation has also been suggested as a possible source of oxylipins (Venter et al. 1997, Brodhun & Feussner 2011), this hypothesis is compatible with the lack of certain enzymes in many species.
Lipoxygenases (LOXs) - Out of the examined species, we only found lipoxygenases in C. aegerita, P. ostreatus, L. bicolor and L. edodes, which have 1–5 copies. These did not form conserved orthogroups and their expression patterns mostly did not vary substantially during development. Exceptions include the gene previously described as PoLOX1 (Plagemann et al. 2013, Tasaki et al. 2013) (PleosPC15_2_1068582). PoLOX1 displays a remarkable upregulation (1 880-fold, expression level 94 000 fragment per kilobase per million) in fruiting bodies relative to vegetative mycelium. The gene showed consistently higher expression in stipes compared to caps, which confirms the previous report on the gene (Tasaki et al. 2013). A lipoxygenase gene (CaeLOX4, gene ID AAE3_04864 / Genbank accession no. CAA7262624) of C. aegerita also showed a strong upregulation (~150-fold) during late development (post sporulation stage) in this species, as noted by Orban et al. (2021). Notably, two other LOXs show strong phase-specific expression increase in fruiting stage tissue: on the one hand, CaeLOX5 (AAE3_07753) shows an expression pattern quite congruent with the maturation-associated increase of the 1-octen-3-ol levels in the headspace of the C. aegerita culture. On the other hand, CaeLOX1 (AAE3_00896) shows transcriptional upregulation before fruiting body maturation sets in. Such patterns may imply stage-specific action or (partial) functional redundancy among these genes that must, prospectively, be tested by functional characterization studies.
Linoleate diol synthases (LDSs) - The other well-known gene family involved in the production of oxylipins is the one of linoleate diol synthases, which makes up to PpoA–C family in A. nidulans. The main products of the Ppo family are the so-called psi factors, which regulate diverse developmental processes from yeast-to-hypha transition and cell aggregation (reviewed in Tsitsigiannis & Keller 2007). We identified 1–4 LDS genes in Agaricomycetes with most species having two copies. Although several genes have reciprocal best hits among Ascomycota (e.g., C. cinerea 423716 to PpoC of A. nidulans), a gene tree analysis showed that these are not true orthologs but likely resulted from a duplication event that predated the split of Asco- and Basidiomycota. Therefore, it is likely that Basidiomycota linoleate diol synthases do not have the psi factor producing activity that A. nidulans PpoA–C genes and their homologs have (Tsitsigiannis & Keller 2007).
Linoleate diol synthases grouped into three conserved orthogroups (Supplementary Table S18), of which two (C. cinerea protein ID: 398037, 423716) were developmentally expressed in a considerable number of species. One of these (C. cinerea protein ID: 423716) includes Ustilago maydis Ssp1 (Ustma2_2_11710), which was found to be specifically expressed in teliospores (Huber et al. 2002). Genes within these orthogroups seem to show diverse expression patterns, without noticeable conservation of expression across species. In the absence of functional information of these genes in Basidiomycota, it is impossible to speculate on their mechanistic role in fruiting body formation. They are likely to be involved in oxylipin production (Orban et al. 2021), but further studies are necessary to elucidate their functions.
Alcohol dehydrogenases – The alcohol dehydrogenase family participates in a broad range of cellular processes, including oxylipin biosynthesis (Orban et al. 2021). We identified a CDE orthogroup containing C. aegerita Adh1-1 (represented by C. cinerea 363127) (Supplementary Table S18), which showed partial conservation of expression: in most species it was upregulated during late development (C. aegerita, A. ostoyae, L. edodes, L. tigrinus, C. cinerea). In addition, in S. commune, Ph. chrysosporium, A. ostoyae and L. edodes it was also upregulated >4x in primordia relative to vegetative mycelium. In M. kentingensis and A. ampla the gene was not developmentally regulated, whereas L. bicolor and P. ostreatus did not have orthologs. Based on a previous report of C. aegerita Adh1-1 (Orban et al. 2021), it is possible that this orthogroup of alcohol dehydrogenases participate in oxylipin production.
Pheromone and pheromone receptor pathways
The structure and evolution of mating systems, including MAT loci-homeodomain (HD) transcription factor genes (A locus), pheromone receptors and pheromone precursor genes (B locus) is among the best-known aspects of agaricomycete biology (e.g., Kües 2000, 2015, James 2015). The primary role of pheromone/receptor systems is regulating dikariotization, i.e., the compatibility of monokaryons, nuclear movement and other downstream events. Hyphal fusion is believed to occur ubiquitously in the Basidiomycota, therefore, pheromone/receptor systems may regulate post-fusion behaviour (Raudaskoski & Kothe 2010, Jones & Bennett 2011). However, pheromone/receptor systems are involved in the regulation of sexual events too. For example, mating type loci are involved in fruiting body formation in C. cinerea (Kües et al. 1998, 2002). Basidiomycete mating systems, pheromone and pheromone receptor genes have been reviewed recently in detail (e.g., Raudaskoski & Kothe 2010, Jones & Bennett 2011, James 2015, Kües 2015). We here annotate components of the agaricomycete mating systems, with special emphasis on copy number variation across the twelve species examined in this paper and expression dynamics during fruiting body development.
Pheromone precursor genes - Basidiomycetes produce mating pheromones that are structurally analogous to S. cerevisiae a-factor (Jones & Bennett 2011). Mating pheromones are short peptides that undergo a multi-step maturation process and regulate intercellular communication and sexual development in a wide range of fungi (Jones & Bennett 2011). Copy numbers of pheromone precursor genes vary across the Agaricomycetes and are often found in the vicinity of corresponding pheromone GPCRs (Raudaskoski & Kothe 2010). Four pheromone precursor genes were found in L. edodes (Wu et al. 2013), P. eryngii (Kim et al. 2014), A. bisporus (Foulongne-Oriol et al. 2021), Moniliopthora roreri (Díaz-Valderrama & Aime 2016), five in Ph. chrysosporium (James et al. 2011), seven in Pycnoporus cinnabarinus (Levasseur et al. 2014), two in P. touliensis (Gao et al. 2018), five in C. cinerea (Riquelme et al. 2005) and eight S. commune (Specht 1995). Synthetic pheromone administration affected priA (a primordium-expressed gene) expression in L. edodes, indicating the involvement of mating pheromones in fruiting body formation (Ha et al. 2018). One of the pheromone precursor genes was expressed in glucose-limited conditions following blue light induction of fruiting in C. cinerea (Sakamoto et al. 2018).
In the genomes in our dataset, we found up to nine pheromone-precursor encoding genes, however, we did not find any in A. ostoyae, A. bisporus, M. kentingensis, Ph. chrysosporium and L. tigrinus. The explanation for this may be that they are simply missing from the genome annotations. Pheromone precursors are short ~50 amino acid peptides and several genome annotation protocols omit predicted proteins that are <50 or <100 amino acids long. For this same reason, it is likely that our inventories of pheromone-precursor genes in other examined species is incomplete. Nevertheless, we found pheromone precursors in C. cinerea, A. ampla, L. bicolor, L. edodes, S. commune and P. ostreatus; these genes were often developmentally regulated, implying that they may participate in developmental processes, possibly as signal molecules. While knowledge on the role of pheromones in mating and dikaryon formation is abundant, whether and how they participate in fruiting body development requires further research.
Pheromone modification - Pheromone precursor proteins are post-translationally modified during their maturation process. This involves prenylated, carboxymethylation as well as proteolytic cleavage to remove N-terminal sequence fragments (Jones & Bennett 2011).
Post-translational prenylation (mostly farnesylation) on CAAX motifs is performed by RAM proteins in S. cerevisiae (Caldwell et al. 1995). Based on orthology with S. cerevisiae pheromone prenylating enzymes, we identified two genes as putatively involved in pheromone prenylation. The first is orthologous to S. cerevisiae RAM1 (C. cinerea protein ID: 491483) and shows very modest expression dynamics (Supplementary Table S19), whereas the second is an ortholog of RAM2 (C. cinerea protein ID: 473005), encoding a general prenylation enzyme that also modifies other peptides, including proteins related to cell wall glucan biosynthesis. Following prenylation, the S. cerevisiae a-factor is cleaved by Rce1 (ortholog: C. cinerea 377901) and Ste24 (ortholog: C. cinerea 419650). Orthologs of these genes showed constant expression during fruiting body development (Supplementary Table S19).
On the other hand, we found evidence for the developmental expression of carboxymethylating enzymes. Two CDE orthogroups of protein-S-isoprenylcysteine O-methyltransferases (homologous to S. cerevisiae Ste14) were detected among conserved developmentally expressed genes. Ste14 mediates methylation of the isoprenylated C-terminal CAAX motif in the mating pheromone a-factor (Romano & Michaelis 2001). Whether these homologs are related to mating pheromone maturation in mushroom-forming fungi remains unclear, therefore, we only tentatively classified these as signal transduction-related genes. Two Ste14 homologs of C. cinerea were previously found to be expressed after blue light exposure (Sakamoto et al. 2018). We detected two CDE orthogroups of Ste14 homologs (Supplementary Table S19). One of the orthogroups (represented by C. cinerea 152667) is present in nine species and upregulated in primordia of C. cinerea, A. ostoyae, A. bisporus and P. ostreatus but interestingly shows the reverse pattern, high in vegetative mycelium and no expression in any fruiting body stage in M. kentingensis. The other orthogroup (represented by C. cinerea 354499) was upregulated in primordia of C. cinerea, A. ostoyae, Pt. gracilis and S. commune (but not in A. ampla and P. ostreatus). More broadly, members of this gene family were developmentally regulated in several species, although they were not conserved across species. On the other hand, 1-to-1 orthologs of S. cerevisiae Ste14 (represented by C. cinerea 153902), show constant expression in all species.
As a final step of a-factor maturation, Ste24 and Axl1/Ste23 proteolytically cleave the N-terminus in two steps (Jones & Bennett 2011). The corresponding agaricomycete orthologs did not show developmentally dynamics expression (C. cinerea protein ID: 419650 and 358206, respectively).
Pheromone sensing GPCRs - Seven-transmembrane domain G-protein coupled receptors have been studied in multiple Agaricomycetes (Kües 2000, Raudaskoski & Kothe 2010, Jones & Bennett 2011, Wirth et al. 2021). Pheromone-sensing GPCRs are located in the B mating type locus in Basidiomycota, together with pheromone precursor genes. Pheromone sensing GPCRs transduce signals intracellularly to a mitogen-activated protein kinase (MAPK) cascade (see next chapter on MAPK pathways). Homology relationships and copy numbers of Basidiomycete pheromone-sensing GPCRs vary significantly across species, but all Basidiomycete pheromone GPCRs are homologous to yeast Ste3, which is involved in mating (reviewed by Raudaskoski & Kothe 2010, Fraser et al. 2007). For example, four pheromone-like GPCR genes have recently been annotated in S. commune, of which one was suggested to participate in mating, whereas the others were proposed to have a role in directing growth in vegetative mycelia (Wirth et al. 2021).
We identified 4–19 pheromone GPCR-encoding genes in the examined species. These genes in general showed low expression dynamics in fruiting bodies (Supplementary Table S19), although several genes were upregulated slightly (FC<4) in stipes in certain species.
A-factor export proteins - We identified a CDE orthogroup (represented by C. cinerea 540181) (Supplementary Table S19) of plasma membrane ATPases which are orthologous to S. cerevisiae Ste6, the protein that exports farnesylated a-factor from the cell (Michaelis 1993). It should be noted that this orthology is based on 1-to-1 reciprocal best hits and does not necessarily reflect functional conservation, as seen in other aspects of mating systems too. This orthogroup showed partial conservation of expression across the examined species. In A. ostoyae, L. bicolor, L. edodes and M. kentingensis it was upregulated in stipe tissues relative to caps, whereas in C. cinerea, S. commune, A. ampla (FC>4) and C. aegerita (though only at FC>2) the gene was upregulated in primordia relative to vegetative mycelium.
MAPK, TOR and cAMP pathways
MAP kinase pathways - The mitogen-activated protein kinase (MAPK) cascades are evolutionary conserved intracellular signal transduction pathways, which regulate important cellular processes (Román et al. 2007). The extracellular signal is sensed by membrane proteins or receptors, which is relayed by sequential phosphorylation and activation of three kinases (MAPKKK, MAPKK, MAPK) to transcription factors that induce or repress specific target genes. MAPK cascades usually consist of further components, like various adaptors, docking and scaffold proteins which are implicated in the spatial and temporal regulation of MAP kinase signalling (Frawley & Bayram 2020).
While in Ascomycota MAPK members have been extensively examined (e.g., Teichert et al. 2014), relatively little information is available on the expression and role of MAPK pathways (except the pheromone, see above) in Agaricomycetes. In Pleurotus tuoliensis pheromone-dependent and starvation-dependent MAPK signalling pathway members were reported to be up-regulated during fruiting (Fu et al. 2017). Le.MAPK, a MAP kinase of L. edodes (Lenedo1_1058304) was reported to be developmentally regulated and one of its interacting partners was characterised (Szeto et al. 2007). In this paper, we annotated Agaricomycetes MAPK signalling pathways based on the four main MAPK pathways known in S. cerevisiae: the peptide pheromone pathway (Fus3), the starvation responding pathway (Kss1), the high osmolarity glycerol stress pathway (Hog1) and the cell wall integrity (Slt2/Mpk1) pathway (Supplementary Table S20). We found relatively few clear orthologs in C. cinerea (Supplementary Table S20). We performed transitive ortholog and domain-based search to find further putative S. cerevisiae orthologs in C. cinerea. MAPK pathway components do not show dynamic expression patterns during fruiting body development.
TOR pathway - The target of rapamycin (TOR) is an evolutionary conserved serine/threonine kinase that is a key regulator in multiple cellular events (Tatebe & Shiozaki 2017). In fungi, the TOR pathway plays a role in the response to nutrients (especially sugar and nitrogen) but is also involved in stress responses, such as osmotic and oxidative stresses. TOR signalling pathways are well-studied in yeasts, but data about their functional analysis in multicellular Agaricomycetes is rare. In G. lucidum TOR signalling was reported to play a role in the regulation of chitin and β-1,3-glucan synthesis and hence of cell wall thickness in an SLT2-MAPK dependent manner (Chen et al. 2019). Orthologs of Ph. chrysosporium were previously annotated (Nguyen et al. 2020). According to a 5’-SAGE study of C. cinerea, TOR pathway members are constantly expressed during the fruiting body formation, except Rheb (C. cinerea protein ID: 493094) a Ras-like small GTPase, which is able to prevent FKBP12 inhibition of TOR (Cheng et al. 2013).
Here we annotated TOR pathway components in mushroom-forming fungi and could find orthologs for all but three yeast genes reported to participate in the pathway (Supplementary Table S20). Consistent with previous reports, TOR pathway components showed modest expression dynamics during fruiting body development. Nevertheless, based on the broad role of the pathway in fungal physiology it will be interesting to investigate its connection with fruiting body morphogenesis in the future.
cAMP pathway - Adenylyl cyclase dependent pathway (cAMP-pathway) has diverse roles in fungi, including cell growth, metabolism, stress resistance and nutrient-sensing (especially glucose) or starvation in fungi (D’Souza & Heitman 2001). Accordingly, the cAMP pathway has been intensely researched in mushroom-forming fungi (D’Souza & Heitman 2001, Tamaki 2007). Genetic manipulation of several cAMP and connected pathway components in S. commune resulted in altered fruiting characteristics, either repression or complete lack of fruiting body formation (Yamagishi et al. 2002, 2004, Knabe et al. 2013, Pelkmans 2016). The cAMP pathway has also been linked to morphogenesis in F. velutipes: the deletion of a Gβ-like protein CPC-2 completely impaired fruiting body formation, but the addition of cAMP or its analog 8-Bromo-cAMP (also a PKA-activator) into the medium restored fruiting in Fv.cpc2 knockdown mutants (Wu et al. 2020b). Ras and G-protein - subunits and their regulators (e.g., Thn1) might also be connected to the cAMP pathway (Yamagishi et al. 2002, Palmer & Horton 2006, Knabe et al. 2013) and were repeatedly reported to affect morphogenesis of fruiting bodies (e.g., Schubert et al. 2006).
In general, cAMP is a known inducer of fruiting in several basidiomycetes, such as C. macrorhizus (Uno & Ishikawa 1973), C. cinerea (Swamy et al. 1985), S. commune (Schwalb 1974) and Ph. chrysosporium (Gold & Cheng 1979). In the latter, cAMP was speculated to induce fruiting by overriding the repressive effect of glucose (i.e., mimicked starvation in the fungus, Gold & Cheng 1979). More recent studies in S. commune reported a repressing effect of cAMP on fruiting, and suggested this was because cAMP is able to mimic high CO2 levels (Pelkmans 2016). Genes of the cAMP pathway were annotated in S. commune and the Pde2 phosphodiesterase (Schco3_2636760), which can degrade cAMP was functionally analysed (Pelkmans 2016). The overexpression of pde2 results in fruiting body development even at elevated CO2 levels. These results were interpreted as evidence for CO2 being sensed via the cAMP pathway (Pelkmans 2016).
We annotated members of the cAMP pathway with C. cinerea as reference (Supplementary Table S20). Consistent with data in S. commune (Pelkmans 2016), we could identify most members of the cAMP pathway in the examined species. Our expression data revealed that cAMP pathway members show relatively little expression dynamics in mushroom-forming fungi (Supplementary Table S20). Nevertheless, this does not preclude an important role of cAMP signalling in fruiting body formation.
G-protein coupled receptors
G-protein coupled receptors (GPCRs) comprise the largest receptor family in fungi. GPCRs are seven-transmembrane cell surface receptors, which transduce signals from a wide range of external stimuli by a G-protein heterotrimer (Xue et al. 2008). Fungal GPCRs originally divided into six classes based on their structural similarity and homology (Xue et al. 2008), however, this classification was later expanded with additional classes (reviewed in Brown et al. 2018).
Pheromone sensing GPCRs are among the most widely studied in connection with fruiting body development. In Flammulina filiformis six pheromone receptor-like genes were characterised whereby four were highly expressed in the fruiting body, while two exhibited the maximal expression level in the mycelia (Meng et al. 2020). Six pheromone-like GPCRs were characterised also in S. commune (bar3, bbr2, brl1, brl2, brl3 and brl4); it was reported that the overexpression of brl2, brl3 and brl4 enhanced mating or fruiting body formation in this species (Wirth et al. 2021). We annotated seven mating-type pheromone receptors in C. cinerea (Supplementary Table S19). Most of the pheromone receptors are highly expressed during the fruiting process, but do not have orthologs in other species. In addition, five further GPCR genes were detected, which, similarly to pheromone-sensing GPCRs mostly did not have orthologs in other species (Supplementary Table S21). This may suggest that fungal species have a specialized GPCR repertoire that is hard to examine using orthology-based approaches.
While GPCRs are not conserved, we detected three CDE orthogroups that may be related to GPCR signalling and were developmentally regulated across several species. An RGS (regulator of G-protein signalling) domain containing protein family (C. cinerea protein ID: 479562) was developmentally regulated in 11/7 species (FC>2/4) (Supplementary Table S21). RGS proteins are involved in signal transduction by regulating G-protein activity and rapidly switching on or off G-protein coupled receptors pathways. We also found a CDE orthogroup containing putative Ran-binding proteins (C. cinerea protein ID: 472370), which are members of the Ras superfamily that regulates all receptor-mediated transport between the nucleus and the cytoplasm. Finally, we detected two arrestin superfamily orthogroups (C. cinerea protein ID: 473465, S. commune: Schco3_2490312) (Supplementary Table S21). Arrestins are involved in signal transduction through the inactivation of G protein-coupled receptors, but some members of the superfamily have been reported in receptor endocytosis, cell cycle progression and pH sensing as well also (Herranz et al. 2005, Telzrow et al. 2019).
Other signal transduction related families
Kinases - The majority of CDE orthogroups classified as signal transduction were kinases (Supplementary Table S18). We found 13 conserved and developmentally expressed kinase orthogroups, however, most of these proved difficult to functionally characterise, likely due to the high rate of evolution in kinase genes and the lack of orthology to well-known genes in model species. One exception is the CDE orthogroup (C. cinerea protein ID: 451611) that contains orthologs of S. cerevisiae Rck2, which is a member of the high osmolarity glycerol (HOG) MAPK pathway. The HOG pathway is involved in responding to oxidative and osmotic stress. In most species, Rck2 orthologs showed early upregulation, with highest expression values in primordia, though its functional significance is not known. Rck2 orthologs were differentially expressed between dikaryons and A and B type monokaryons of C. cinerea as well (Stajich et al. 2010), and it was concluded that RCK2 genes in C. cinerea must serve different functions from that of S. cerevisiae orthologs. The remaining kinase orthogroups are functionally uncharacterised and may be interesting targets for functional studies, their dynamic expression patterns indicate potentially conserved and important functions in fruiting body development.
Velvet complex - The velvet complex has been identified as a central regulatory hub in fungal development and secondary metabolism (Bayram et al. 2008). The complex was characterised in the Ascomycota, where it consists of VeA, LaeA, VelB as well as VelC and VosA, which assemble into condition-specific (including light-responsive) complexes. It has been shown that the velvet domain, an ~200 amino acid fold, binds DNA and resembles the structure of the metazoan transcription factors NF-κB (Ahmed et al. 2013). Proteins containing a velvet domain exist outside the Pezizomycotina as well, however, these proteins did not appear to be orthologous to members of the Velvet complex in the aspergilli (Ojeda-López et al. 2018); accordingly, how they function and whether they form complexes also in the Basidiomycota and early-diverging fungi is not known.
The first evidence on a potential role of Velvet proteins in Basidiomycota fruiting body development was provided by (Plaza et al. 2014). They detected the upregulation of a velvet domain containing protein in primordia of C. cinerea relative to vegetative mycelia. Following these trails and combining detailed mechanistic knowledge from the velvet regulon of A. nidulans (Krijgsheld et al. 2008), they could show that some, albeit a minor proportion, of orthologs of the A. nidulans velvet-regulated genes are upregulated in fruiting bodies in C. cinerea, L. bicolor and S. commune (Plaza et al. 2014) also. This suggested some extent of conservation of function in the Dikarya, although the lack of orthology between Asco- and Basidiomycota proteins casts some uncertainty on how these data should be interpreted. Velvet protein-encoding genes were reported to be developmentally regulated in multiple Agaricomycetes species in the comparative analyses of Krizsán et al. (2019).
Our current analysis indicated that the velvet domain occurs in 5–6 proteins (only 4 in P. ostreatus) encoded in the genomes of mushroom-forming fungi. Surprisingly, the yeast-like C. neoformans also possess 6 genes with a velvet domain, in contrast to Ascomycota yeasts, which have lost velvet genes (Nagy et al. 2014). Interestingly, Ascomycota on average have 4 genes that encode velvet-domain containing proteins, indicating an expansion in the Basidiomycota. Consistent with results of Ojeda-López et al. (2018), we found that Basidiomycota velvet proteins lack clear orthology to Ascomycota genes. Only VelB and VosA of A. nidulans have reciprocal best hits in C. cinerea (C. cinerea protein ID: 365674 and 374867, respectively).
In terms of expression, observed patterns vary widely among species. For example, all six genes are developmentally regulated in C. cinerea, but none in A. ostoyae (both FC>4), indicating considerable expression plasticity through evolution. We identified three orthogroups whose members were developmentally regulated in a notable number of species (Supplementary Table S22). Of these, genes in one orthogroup (represented by C. cinerea 385561) were mostly upregulated in stipe tissues in the pileate-stipitate species C. cinerea, A. ostoyae, M. kentingensis, L. edodes (but only in mature FBs of L. bicolor). The gene was not developmentally regulated in L. tigrinus and P. ostreatus, possibly indicating the distant relationship of these species from ones in which the gene is stipe-upregulated. The second most interesting CDE orthogroup (C. cinerea protein ID: 496341) was developmentally regulated in 10 species (FC>2), but expression peaks varied from primordia (A. ampla, S. commune, A. ostoyae), to young fruiting body caps (C. cinerea) and stipes (M. kentingensis).
Based on the above data, velvet protein-encoding genes represent an interesting target for phylogenetic and functional studies of sexual fruiting bodies in the Agaricomycetes; we expect their analyses will reveal interesting insights into fruiting body development.
STRIPAK complex - The striatin-interacting phosphatase and kinase, STRIPAK (known as Far complex in S. cerevisiae (Kück et al. 2019), is a widely conserved multi-protein complex that can be found from yeast to metazoans (Reschka et al. 2018). Its function in metazoans is unknown, whereas in Ascomycota model systems (e.g., Sordaria macrospora) it is involved in hyphal fusion and fruiting body development. It was first discovered in 2004 in fungi (Pöggeler & Kück 2004) and its function has been increasingly clarified in a series of papers since then (see Reschka et al. 2018, Riquelme et al. 2018). Mechanistically, the STRIPAK complex provides links between multiple signalling pathways in eukaryotic cells (reviewed in Kück et al. 2019), although many of the details of its interactions with other proteins are not known. Its role in fruiting body formation in the Basidiomycota has not yet been investigated.
We queried the C. cinerea genome with members of the STRIPAK complex described from S. macrospora and S. cerevisiae (listed in Reschka et al. 2018). C. cinerea seems to have a fully functional STRIPAK complex, with all genes having clear orthologs, except for Far3/7 which only had weak hits. Further, the C. cinerea proteins were members of conserved single-copy Agaricomycetes orthogroups, which indicated that the complex is conserved in all the examined Agaricomycetes species. Members of this complex did not show significant expression dynamics (Supplementary Table S23). The conservation of the complex in the Agaricomycetes suggests it might have important functions, however, its role in fruiting body development remains unclear at the moment and requires further research.
Light sensing systems and light receptors
Fungi adapt developmental processes to light signals in time and space to ensure that sporulating cells can maximize spore dispersal efficiency (Yu & Fischer 2018). The photobiology and its connection to developmental events are quite well-known in both the Asco- and Basidiomycota. In the last decade, the molecular architectures and regulatory networks of sensing and responding to blue, red and green light have been uncovered in multiple species (Corrochano 2011, Fuller et al. 2015). In this chapter we discuss light sensing systems in Agaricomycetes and analyse their expression patterns in fruiting bodies. We note that the best known such system, the white-collar complex has transcription factor activity and is discussed under chapter “Transcription factors”.
Cryptochromes - Fungal genomes include DASH-type cryptochromes, photolyase genes that function as blue light photoreceptors and are involved in DNA repair (Fuller et al. 2015). Cryptochromes are regulated by the white-collar complex in N. crassa (Froehlich et al. 2010) and regulate fruiting body development in certain Ascomycota (e.g., Cordyceps militaris, Wang et al. 2017), but to our best knowledge, no such information is available in the Basidiomycota. Our analysis suggests that cryptochromes are single copy genes in the Agaricomycetes, except in L. edodes, which has two genes. The CDE orthogroup containing Agaricomycetes cryptochromes (represented by C. cinerea 492962) (Supplementary Table S24) was developmentally regulated in 9/7 species (FC>2/4), and showed strong induction in primordia relative to vegetative mycelium in A. ostoyae, M. kentingensis, L. edodes, P. ostreatus, Ph. chrysosporium and at FC>2 in C. aegerita and C. cinerea. The gene was constantly expressed in A. ampla and S. commune and was missing in L. bicolor. Based on these data, cryptochromes may be relevant to fruiting body formation in the Agaricomycetes.
Phytochromes are red- and far-red receptors that influence sexual and asexual sporulation in fungi. The first circumscribed fungal phytochrome was the fphA gene in A. nidulans, which represses sexual development under red light (Blumenstein et al. 2005). Since then, the repression of sexual development has been proven for N. crassa too and several details of the process (e.g., interaction with WCC and velvet factors) have been clarified (Idnurm & Heitman 2005, Wang et al. 2016, Yu et al. 2016). In the Basidiomycota, the few reports of phytochromes include a systematic mutant library in C. neoformans, which circumscribed the gene encoding TCO3 (Lee et al. 2016), as well as observations of phytochrome upregulation in browning mycelium of L. edodes (Tang et al. 2013) and in rhizomorphs of A. ostoyae (Sipos et al. 2017). In U. maydis red and blue light receptors have been shown to be required for the development of fruiting body-like structures (Sánchez-Arreguin et al. 2020).
Phytochromes seem to be single-copy genes in the Agaricomycetes examined here. They formed a conserved orthogroup (represented by C. cinerea 470504, Supplementary Table S24), however, genes in this orthogroup did not show significant expression dynamics during fruiting body formation, except in A. ostoyae and C. cinerea. In A. ostoyae the expression of the phytochrome gene was ~10x higher in stipes than caps in stage 2 primordia and young fruiting bodies. Similarly, in C. cinerea peak expression was observed in elongating stipes in the young fruiting body stage. However, other species did not display this expression profile, indicating that phytochrome expression is not conserved across mushroom-forming fungi. Although expression patterns did not seem to be conserved in fruiting bodies, it is possible that red light sensing phytochromes are part of a photoreceptor system that signals position in open air, which is required for sporulation and fruiting body formation for the majority of species (Rodriguez-Romero et al. 2010, Fuller et al. 2015).
Fungal light sensing systems also include rhodopsins, seven-transmembrane domain proteins involved in sensing green light (Bieszke et al. 1999). We detected rhodopsins only in L. tigrinus and Ph. chrysosporium, but none of the Agaricales in our dataset.
Genes related to blue light receptors - Blue light is one of the most important signals for fungi and responses as well as molecular aspects of the fungal blue light response are among the best known aspects of fungal photobiology in both the Asco- and Basidiomycota. The most important receptors of blue light are the white-collar complex (WCC), which comprises the photoreceptors/transcription factors WC2 and WC1. These have been discussed under chapter “Transcription factors” (see above). We here only discuss putative accessory proteins similar to VIVID of N. crassa. These proteins harbour a single PAS domain and act as repressors of the WCC in N. crassa and were suggested to provide a mechanism for adapting to changing light intensities during the day (Fuller et al. 2015). Although evidence for that is missing in the Agaricomycetes, a CDE orthogroup of PAS domain proteins (represented by C. cinerea 500409) was developmentally regulated in 9/7 species (FC>2/4) and showed strong induction in primordia relative to vegetative mycelia in A. ostoyae, C. aegerita, C. cinerea, L. bicolor, L. tigrinus, M, kentingensis, Ph. chrysosporium but not in A. ampla and P. ostreatus (and missing from L. edodes and S. commune). It will be interesting to see if this or similar genes are involved in light responses in the Agaricomycetes.
Dst2 family - We found that orthologs of the dst2 gene of C. cinerea formed a CDE orthogroup (represented by C. cinerea 361584). DST 2 has been described as a FAD-binding and Berberine-like domain containing protein that, if mutated, results in a blind dark stipe phenotype similar to what is observed in mutations on the white-collar complex (Kamada et al. 2010, Kuratani et al. 2010). Based on these observations, it has been hypothesized that dst2 represents a novel photoreceptor (Kuratani et al. 2010). The typical expression profile of these genes (seen in A. ostoyae, C. aegerita, C. cinerea, L. edodes and P. ostreatus) involved upregulation in primordia relative to mycelium and, in later stages, higher expression in stipes compared to caps. Variations to this pattern exist, such as only being FB-init in L. bicolor and S. commune, or being only stipe upregulated but not FB-init in M. kentingensis and L. tigrinus. We hypothesize that dst2 genes might be related to stipe development and, potentially, photomorphogenesis.
Cell wall biosynthesis and modification
The cell wall is an organelle that functions as an extracellular matrix in fungi. It provides integrity, rigidity to cells, defines cell shape, harbours extracellular proteins, represents a channel of communication with the environment, and provides the first line of physical defence, to name just a few of its major roles in fungal physiology. Structural roles are particularly important for the Agaricomycetes that form above ground fruiting bodies that should be rigid yet plastic enough to withstand environmental forces and support spore dispersal. In fruiting bodies, the cell wall must serve structural and defence roles but must also be sufficiently plastic to allow changes in shape and size during development. Although mechanisms of cell shape determination are not known in mushroom-forming fungi, regulated cell wall deposition and remodelling by hydrolytic enzymes residing in the wall itself (Verdín et al. 2019), are undoubtedly of key importance.
There is plenty of evidence for cell wall remodelling and differential cell wall compositions in fruiting bodies of various species (e.g., Almási et al. 2019, Buser et al. 2010, Krizsán et al. 2019), although most of that is based on transcriptome rather than functional studies. For example, stipe growth may be realized by the breaking of chitin-glucan cross-links by hydrolytic enzymes (Liu et al. 2021). Similarly, there are cross-linking (transglycosidases) and lytic (e.g., glycoside hydrolases) enzymes that rigidify and make the cell wall more plastic, respectively (Verdín et al. 2019). Mannosylation might also rigidify the cell wall and mannoproteins are often N-glycosylated, which contributes to water retention and adhesion (Lesage & Bussey 2006). In the Agaricomycetes, Buser et al. reported a fruiting body-specific N-glycan from C. cinerea (Buser et al. 2010). The regulated interplay of these enzymes might result in the formation of stage-, tissue-, and species-specific cell wall architectures in fruiting bodies.
In this chapter we review current knowledge on cell wall related genes in mushroom-forming fungi and analyse CDE orthogroups that are likely linked to cell wall synthesis or modification. We will not review cell wall composition in fruiting bodies, but refer to earlier reviews (Kües 2000). Cell wall functioning is quite well-researched in the Ascomycota (e.g., in S. cerevisiae and N. crassa) (Verdín et al. 2019), and to a much smaller extent in the Basidiomycota. We therefore base much of our functional hypotheses on knowledge in the Ascomycota, noting that the process may be different and/or significantly more complex in the Basidiomycota. For example, while some chitin-glucan cross linking enzyme families appear similarly important both in Asco- and Basidiomycota (e.g., GH16/GH17), there are some (e.g., GH72 and GH76) which are involved in cell wall remodelling in the Ascomycota, but apparently not in the Basidiomycota. Hereafter we discuss cell wall related CDE orthogroups broken down into synthetic and remodelling enzymes as well as by substrate (chitin/glucan).
From these analyses a few broad observations emerge, which we discuss here. Most importantly, the diversity of enzymes that show developmental regulation in fruiting bodies suggests that the fruiting body cell wall may be significantly more complex than we think, and we still know quite little about its exact structure and composition. A general pattern that also arises is that more tissue- and stage-resolved transcriptomes reveal proportionately more developmentally expressed genes, which indicates that cell wall remodelling and, consequently, cell wall architectures, likely show high tissue-specificity in fruiting bodies. Based on our results, it appears that fungal cell wall (FCW) remodelling and crosslinking genes show significantly more expression dynamics than genes encoding chitin and glucan synthases. In line with this observation, we found that intracellular components of cell wall biosynthesis (chaperones, glucan synthase complex, pathways involved in glucan monomer synthesis) also show little expression dynamics.
Comparisons across species revealed limited conservation in FCW related gene expression patterns across species, which suggests it diverges rapidly during evolution. This lends support to the quick evolution of cell wall architectures and corresponding enzyme profiles in fruiting bodies. Another observation related to this is that FCW-related genes show complex gene duplication/loss patterns, which complicates the inference of well-conserved groups of 1-to-1 orthologous genes. Thus, these genes might be more appropriately analysed in the context of broader orthogroups (e.g., based on Orthofinder, Emms & Kelly 2015) than based on the strict 1-to-1 orthogroups we used. However, for the sake of uniformity, we stick to the latter, with notes on overall prevalence of developmental expression in a given family.
An interesting observation is that several FCW active genes upregulated in fruiting bodies are also expressed during mycorrhiza establishment in ECM fungi such as L. bicolor (Martin et al. 2008, Veneault-Fourrey et al. 2014). Such signals provide clues to shared morphogenetic mechanisms between ectomycorrhizal development and fruiting bodies.
Finally, an important consideration for cell wall biosynthesis/ remodelling genes is whether they are upregulated in fruiting bodies is the vegetative mycelium sample that was used as control in the RNA-Seq. Non-growing inactive hyphae (where cell wall synthesis rate is low) as control of actively growing fruiting bodies can yield more gene expression differences than an actively growing mycelium sample. Although we do not think this has significantly influenced our comparisons, because we do not know the sampling conditions for all species, this possibility should be kept in mind in the interpretation of results.
We attempted to as comprehensively analyse cell wall related gene expression as possible, at least with consideration of genes that show some expression conservation at the gene family level. Nevertheless, it is certain that several important, especially species-specific genes are left out from our inventory. For example, mannosylation, a potentially interesting process contributing to cell wall functioning, is not discussed here due to the lack of orthology to experimentally characterised mannosylation components in the Ascomycota (Lesage & Busey 2006). These aspects will need to be covered in future studies.
Cell wall biosynthesis
Chitin biosynthesis (GT2)
Chitin is one of the most important, although usually not the most abundant structural polymer of the fungal cell wall. This is true for fruiting bodies as well, in which, although cell wall composition is altered relative to vegetative mycelia (Novaes-Ledieu & Garcia Mendoza 1981, Mol & Wessels 1990, Kües 2000), chitin is an important component that confers rigidity. Chitin is also a scaffold for the modification of cell wall structure either by cross-linking, incorporation of glycoproteins or chemical modifications to the chitin polymers, among others (Verdín et al. 2019). Cell wall chitin assembly involves the action of the UDP-N-acetylglucosamine biosynthesis pathway (in S. cerevisiae Gfa1p, Gna1p, Pcm1p, Qri1p), chitin synthases (Chs1–3) which elongate the growing chitin polymer as well as chitin deacetylases, which modify the chitin on the cell surface (Shahinian & Bussey 2000, Klis et al. 2006, Verdín et al. 2019) (the latter is discussed in chapter “Chitin active enzyme families”). While yeasts have up to three chitin synthases, filamentous fungi have additional chitin synthase genes (on average up to seven, Verdín et al. 2019), including highly processive ones that harbour a myosin motor domain and that were lost in yeasts (Takeshita et al. 2006).
We examined chitin synthesis and modification related genes based primarily on functional annotations and recent reviews (Shahinian & Bussey 2000, Klis et al. 2006, Verdín et al. 2019). Clear orthologs of the S. cerevisiae UDP-N-acetylglucosamine biosynthesis pathway could be identified in all examined Agaricomycetes species (Supplementary Table S25). We identified 8–13 (17 in L. bicolor) chitin synthases of the GT2 family, 2–3 chitin synthase export chaperones as well as orthologs of S. cerevisiae SKT5 (activator of chitin synthase 3) in the examined Agaricomycetes. Interestingly, UDP-N-acetylglucosamine biosynthesis pathway genes showed more expression dynamics than most chitin synthase genes (Supplementary Table S25). Among chitin synthases, although many formed conserved orthogroups, only a single appeared developmentally regulated in a wide array of species. Nevertheless, each species had developmentally regulated and/or FB-init (Krizsán et al. 2019) chitin synthases, though these did not always belong in conserved orthogroups. The most interesting orthogroup (represented by C. cinerea 359180) was developmentally regulated in 12/10 (at fold change 2/4) species and showed upregulation and high expression in primordia relative to vegetative mycelium in several species (C. cinerea, A. ampla, P. ostreatus, S. commune, C. aegerita and L. edodes). Comparative genomic data suggests that this orthogroup of chitin synthases is specific to mushroom-forming fungi or the Basidiomycota, suggesting that it may be a fruiting body-specific chitin synthase. A second CDE orthogroup of GT2 chitin synthases (C. cinerea protein ID: 519767) was also detected (Supplementary Table S25).
A chitin synthase regulator described as Csr2 from C. neoformans (Banks et al. 2005) and Skt5 from S. cerevisiae (Kawamoto et al. 1993), is conserved in the Agaricomycetes (orthogroup represented by C. cinerea 538650), but did not show significant expression dynamics (Supplementary Table S25).
β-glucan biosynthesis
β-1,6-glucan (Kre9/Knh1, GH16, GH63)
In addition to β-1,3-glucan, β-1,6-glucan is also found in fungal cell walls and is hypothesized to serve gluing purposes by covalently linking β-1,3-glucan to chitin and mannoproteins (Lesage & Bussey 2006). Its synthesis builds on UDP-glucose produced by the corresponding pathway in S. cerevisiae. We find that most members of the yeast beta-1,6-glucan biosynthesis pathway have no clear 1-to-1 ortholog in the Agaricomycetes (Kre9, Knh1, Dfg5, Dcw1, Kre5, Kre1). Only Kre6 (C. cinerea protein ID: 447635), Rot1 (C. cinerea protein ID: 241938), Rot2 (C. cinerea protein ID: 359282) and CWH41 (C. cinerea protein ID: 486333) had clear orthologs. However, at the family level, all known members of the β-1,6-glucan synthesis machinery are represented in the Agaricomycetes (Supplementary Table S26), except Kre1, which seems missing.
Kre9/Knh1 - In yeasts and filamentous Ascomycota, the Kre9/Knh1 and GH16 families include the main glucan synthases (Lesage & Bussey 2006). Several additional well-characterised proteins in the endoplasmic reticulum are necessary for β-1,6-glucan synthesis (e.g., Big1, Rot1, Rot2, Cwh41). It should be noted that there is a strong overlap between genes participating in β-1,6-glucan synthesis and remodelling, therefore, separating these two processes is harder than it is in the case of chitin or β-1,3-glucan.
The Kre9/Knh1 family of cell surface proteins are involved in β-1,6-glucan assembly in the cell wall of fungi and have been characterised in a number of Ascomycota species (Brown & Bussey 1993, Dijkgraaf et al. 1996, Nagahashi et al. 1998, Costachel et al. 2005), predominantly yeasts. A member of this family has been independently reported as a developmentally regulated gene, DRMIP, of L. edodes (Szeto et al. 2007), which was reported to be primarily expressed in primordia. Reports of this family in fruiting body transcriptomes just started to emerge recently (Almási et al. 2019, Krizsán et al. 2019, Liu et al. 2020), and no mechanistic study proved yet a link to cell wall in fruiting bodies. The family is overrepresented in Agaricomycetes (Krizsán et al. 2019), suggesting it might have experienced duplications specific to mushroom-forming fungi.
We identified 5–15 Kre9/Knh1 genes in mushroom-forming fungi. Of these 3–4 are developmentally regulated in each species. Notably, each species has 2–3 genes in this family that show a considerably higher expression in all fruiting body tissues than in vegetative mycelia, suggesting that these genes are involved in generating fruiting-body specific cell walls. For example, in C. cinerea we found three FB-init Kre9/Knh1 genes (C. cinerea protein ID: 470185, 359649, 465604), whereas in L. edodes, there was one FB-init gene, in addition to DRMIP, which was highly expressed and two-fold upregulated in all fruiting body tissues compared to vegetative mycelium. The family displayed more expression dynamics in more resolved transcriptomes (e.g., A. ostoyae, C. cinerea), suggesting that it has considerable tissue-specificity. In addition, each species showed for some genes an expression peak in vegetative mycelium, which is consistent with a partitioning of fruiting body and mycelium-specific roles. Taken together, the Kre9/Knh1 family is likely involved in β-1,6-glucan assembly in the cell wall, with several genes showing fruiting body-specific expression. We detected five CDE orthogroups of Kre9/Knh1 genes (represented by C. cinerea 359649, 465604, 470186, 417746, S. commune 2557059) (Supplementary Table S26).
GH16 - The other major β-1,6-glucan synthase family is GH16, which includes both glucan synthases (e.g., Kre6 of S. cerevisiae, = C. cinerea 447635) and glucan-chitin cross-linking enzymes that participate in cell wall remodelling (Patel & Free 2019). We discuss this family in detail under chapter “Cell wall remodeling”.
Several ER-localized β-1,6-glucan synthesis related proteins have been described in yeasts and filamentous Ascomycota (Lesage & Bussey 2006, Orlean 2012, Essen et al. 2020), including Kre5, Big1, Rot1, Rot2 and Cwh41. Rot2 and Cwh41 are also involved in protein N-glycosylation (Lesage & Bussey 2006). These are conserved in the Agaricomycetes, mostly forming single-copy families with low expression dynamics during fruiting body development. The GT24 (glycoprotein α-glucosyltransferase) family includes the Kre5 gene in S. cerevisiae and C. albicans. Kre5 has an indirect role in β-1,6-glucan synthesis and may be involved in the refolding of misfolded N-glycoproteins in the ER, in particular β-1,6-glucan synthases (Lesage & Bussey 2006). GT24 is present as a single-copy family in Agaricomycetes (Supplementary Table S26); their expression is approximately constant in fruiting bodies. Big1 is an ER membrane protein of unknown function that is required for β-1,6-glucan synthesis in S. cerevisiae (Lesage & Bussey 2006). It is a single-copy gene family (represented by C. cinerea 432992) in the Agaricomycetes, without developmentally relevant expression patterns (Supplementary Table S26). Rot1 is a chaperone involved in protein folding (in general) located in the ER. It is required for cell wall synthesis and N-glycosylation and O-mannosylation in S. cerevisiae (Lesage & Bussey 2006). Rot1 orthologs are single copy genes (C. cinerea protein ID: 241938) in the Agaricomycetes that showed no developmental dynamics in the species we examined (Supplementary Table S26). Rot2 is an ER-localized alpha-glucosidase involved in protein N-glycosylation, required for normal levels of β-1,6-glucan in the cell wall of S. cerevisiae (Lesage & Bussey 2006). It belongs to the GH31 family, which is present in 3–8 copies in Agaricomycetes. They form two CDE orthogroup (FC>2/4: 8/6 species, represented by C. cinerea 499280 and FC>2/4: 6/5 species, represented by S. commune 2543909) (Supplementary Table S26), which were developmentally regulated in 6/5 and 5/4 species, however, expression patterns of these orthogroups did not show marked conservation. 1-to-1 orthologs of S. cerevisiae Rot2 (represented by C. cinerea 359282) showed very small gene expression dynamics.
The GH63 family includes alpha-glucosidases, such as Cwh41 of S. cerevisiae, involved in cell wall beta-1,6-glucan assembly in the ER. The family is represented by 2 copies in all but one of the examined species (L. edodes has 5 genes). GH63 genes form two conserved orthogroups, of which one (C. cinerea protein ID: 486333), containing orthologs of S. cerevisiae Cwh41) did not appear to be interesting, whereas members of the other (C. cinerea protein ID: 494484) showed marked expression peaks coinciding with spore formation in all species (Supplementary Table S26). In C. neoformans, the gene showed upregulation at 72h, also coinciding with sporulation. Whether this GH63 orthogroup is needed for spore wall assembly or other processes that temporally overlap with sporulation cannot be determined based on these data, nevertheless, the family seems to be interesting in the context of fruiting body development.
β-1,3-glucan (GT48)
In S. cerevisiae β-1,3-glucan is synthesized by the FKS (Fks1–3) and the GAS (Gas1–4 families as well as a few accessory proteins, such as Smi1 (C. cinerea protein ID: 181364) and Rho1 (C. cinerea protein ID: 528086) (Lesage & Bussey 2006, Verdín et al. 2019). Of the glucan synthases only FKS1 and Gas1 had clear 1-to-1 orthologs is the Agaricomycetes (C. cinerea protein ID: 446030 and 543646, respectively). The glucan synthase complex, which is formed by FKS1, Rho1, Smi1 (Verdín et al. 2019), seems to be conserved in the Agaricomycetes.
Most Agaricomycetes species have two beta-1,3-glucan synthase genes of the GT48 family (except M. kentingensis and L. tigrinus, which have 5 and 7 respectively). Somewhat surprisingly, GT48 genes showed very modest expression dynamics and were mostly not developmentally regulated. Similarly, the Gas1 family of glucanosyltransferases is present in 1–2 copies in the examined species (L. bicolor has 7 and M. kentingensis has 4) and they were not developmentally regulated. Accessory members of the glucan synthase complex (Smi1, Rho1) showed constant expression in fruiting bodies. Collectively, these data suggest that β-1,3-glucan synthesis is present, but it is not dynamically regulated in fruiting bodies.
Cell wall remodelling
Chitin-active enzyme families (GH18, GH20, X325, AA9, AA14, CBM5/12, CBM50, CE4)
GH18 - Glycoside hydrolase family 18 contains chitinases (both endo- and exochitinases). Chitinases remodel the chitin structure by cleaving β-1,4 linkage of chitin or modify oligosaccharides (Verdín et al. 2019) (e.g., cleaving chitobiose from the cell wall). Chitinases are one of the most straightforward players in fruiting body development and accordingly have been reported in several previous genomic (Chen et al. 2012, Park et al. 2014), transcriptomic (Patyshakuliyeva et al. 2013, Sakamoto et al. 2017, Xie et al. 2018, Krizsán et al. 2019) and mechanistic (Niu et al. 2016, Zhou et al. 2016c) studies. Despite the clear prediction of their role in morphogenesis, this has not been mechanistically shown in the Basidio- or Ascomycota (Alcazar-Fuoli et al. 2011), possibly because multiple paralogous genes in each species’ genome complicate functional studies.
In the Basidiomycota, Zhou et al. (2016c) reported chiB1 (C. cinerea protein ID: 368217) (Supplementary Table S27), a class V exochitinase purified from C. cinerea and suggested to be involved in cap autolysis (Zhou et al. 2016c). Expression data agree with this, the corresponding gene shows a very strong induction in mature fruiting body caps. Similarly, ChiIII (Niu et al. 2016) of C. cinerea (C. cinerea protein ID: 354859) was reported to have both endo- and exochitinase activity, and to be expressed mostly in fruiting bodies, with peak expression in mature fruiting bodies (which is confirmed by our expression data). Most recently, Zhou et al. assayed the role of chitinases in stipe cell wall extension (Zhou et al. 2019) and named six further chitinases in C. cinerea, ChiE1, (C. cinerea protein ID: 543586), ChiE2 (C. cinerea protein ID: 520359), ChiEn1 (C. cinerea protein ID: 91051), ChiEn2 (C. cinerea protein ID: 90984), ChiEn3 (C. cinerea protein ID: 470416), and ChiEn4 (C. cinerea protein ID: 358869). Chitinases have also been examined in the context of autolysis of the model mushrooms C. cinerea and Coprinellus congregatus (Lim & Choi 2009).
GH18 genes are found in 8–12 copies in Agaricomycetes (up to 20 in Polyporales, see (Chen et al. 2012)), some of which harbour carbohydrate binding modules (CBMs), but their function has not been examined. Most chitinases are developmentally expressed in fruiting bodies. Mycelium-upregulated chitinases are also found in most species; these, although display peak expression in vegetative mycelia (often ~10x that of fruiting bodies), often also show >4-fold expression dynamics within fruiting bodies. We detected seven CDE orthogroups (Supplementary Table S27), which included C. cinerea ChiB1, ChiE2, ChiEn1 and ChiEn3. In general, although, the detected chitinase orthogroups were developmentally regulated in many species, they did not show conserved expression patterns across species, which could indicate quick divergence of function or the failure of our reciprocal-best-hit based approach to detect functionally related genes (in the latter case phylogenetic analyses might help). One orthogroup (represented by C. cinerea 520359) shows gill-specific expression peaks in the species in which separate RNA-Seq data are available for gills (A. ostoyae, P. ostreatus) or late upregulation in Pt. gracilis, C. aegerita, L. edodes, L. bicolor, Ph. chrysosporium, A. ampla and S. commune, suggesting it may have something to do with processes that take place in the hymenium late in development, possibly sporulation.
GH20 and chitin degradation products - A chitin-connected GH family is GH20, which has β-1,6-N-acetylglucosaminidase activity and thus can cleave chitobiose released by chitinases further into N-acetylglucosamine (GlcNac). GlcNac may be taken up by the cell for repurposing of cleaved cell wall components as nutrient source (López-Mondéjar et al. 2009, de Oliveira et al. 2018) or, to trigger upregulation of chitinolytic genes in a transcriptional feedback-loop (Langner & Göhre 2015). The latter hypothesis seems more plausible for cases when the fungus feeds on chitin rather than modifies it for developmental purposes. In previous reports, GH20 genes have been identified in the post-harvest transcriptome of L. edodes (Sakamoto et al. 2017). We identified 2–8 GH20 genes in the examined Agaricomycetes, some of which were developmentally regulated (including the single gene in C. neoformans), but did not form conserved CDE orthogroups. The role of this gene family in fruiting body development may be worth examining further.
If GlcNac is indeed taken up and metabolized in the cell, then two of the detected CDE orthogroups may provide clues to its mechanistic bases. An orthogroup of GlcNac transporters, which contains reciprocal best hits of C. albicans NGT1, was developmentally regulated in 9/6 species (C. cinerea protein ID: 464567, Supplementary Table S27). C. albicans NGT1 encodes a specific transporter of GlcNac (Alvarez & Konopka 2007, Zhang et al. 2020). We speculate that this orthogroup may be involved in the uptake of GlcNac monomers generated by GH20 enzymes extracellularly.
In fungi, the GlcNac catabolic pathway is best described in C. albicans (Kumar et al. 2000, Yamada-Okabe et al. 2001), however, only a few of the corresponding genes are conserved in Agaricomycetes. Only the GlcN6P deaminase (Nag1, C. cinerea 356467 FC>2/4: 7/4 species) and a GlcNac permease Nag4 (C. cinerea protein ID: 544873, FC>2/4: 6/4) (Supplementary Table S27) genes have clear reciprocal best hits in Agaricomycetes, whereas we could not identify clear orthologs of N-acetylglucosamine kinase (Nag5) or N-acetylglucosamine deacetylase (Nag2). Our annotations identified a different gene as N-acetylglucosamine deacetylase, however (C. cinerea protein ID: 446427), which was developmentally regulated in 8/6 species (FC>2/4) and may be involved in intracellular GlcNac catabolism.
In summary, the evidence presented here on GlcNac uptake and catabolism in mushroom forming-fungi is circumstantial, at best, however, provides clues for further research on this topic. GlcNac (and any other breakdown intermediate) produced during cell wall modification may be precious compounds in the fruiting body, which is otherwise dependent on distal supply of nutrients from the mycelium. This might have prompted the evolution of efficient repurposing mechanisms in fruiting bodies to mitigate its dependence on mycelial nutrient supply.
CE4 chitin deacethylases - Chitin deacetylases (CDA) are involved in chitosan production, a minor but key constituent of the cell wall. Chitosan, a more soluble polymer than chitin, occurs in both mycelia and fruiting bodies of basidiomycetes, although its role in fruiting body formation is thought to be limited (Novaes-Ledieu & Garcia Mendoza 1981, Crestini et al. 1996, Baker et al. 2007). It is produced by the synergistic activities of chitin synthases and CDAs, where the former synthesizes and the latter deacetylate chitin in various spatial or temporal patterns and to varying degrees (Sebastian et al. 2020). CDAs belong to the carbohydrate esterase 4 family (CE4) in the CAZy classification and are homologous to bacterial nodB factors that produce signal molecules in the rhizobia-legume symbiosis (Buhian & Bensmihen 2018). Deacetylation of chitin to chitosan makes it resistant to chitinases, which might provide a mechanism for regulating the extent of chitinase-mediated wall plasticity and thus cell expansion (e.g., in stipe elongation (Liu et al. 2021). Fungal CDAs have a potential in biotechnology, as chitosan is an additive in food, cosmetic and pharmaceutical products (Morin-Crini et al. 2019). CDAs have poorly known roles in Ascomycota fruiting body development, with both N. crassa CDAs, nevertheless, being highly expressed during perithecium development, suggesting roles in sexual morphogenesis (Patel & Free 2019). It should be noted that CE4 also contains acetyl xylan esterases, which are able to break down the xylan backbone of hemicellulose during wood decay (Liu & Ding 2009). Therefore, some genes of the family in the Agaricomycetes may be related to wood decay too.
CDA genes have been detected in several RNA-Seq studies focusing on fruiting body development (Morin et al. 2012, Sipos et al. 2017, Xie et al. 2018, Krizsán et al. 2019). Three chitin deacetylases have been experimentally characterised in C. cinerea, CDA1 (C. cinerea protein ID: 42776), CDA2 (C. cinerea protein ID: 502602), and CDA3 (C. cinerea protein ID: 464952) (Supplementary Table S27), all of which were reported to have high expression in the elongating region of the stipe (Wang et al. 2018, Bai et al. 2020). Another chitin deacetylase, primarily expressed in the fruiting bodies, was investigated in F. velutipes (Yamada et al. 2008).
Searching the genomes of fruiting body forming fungi reveals 5–18 chitin deacetylase genes, several of which formed conserved single-copy orthogroups. The previously investigated CDA1-3 genes of C. cinerea formed species-specific orthogroups. The majority of CDA genes in the examined species were developmentally regulated in fruiting bodies, indicating key functions in fruiting body development. Six out of 17 C. cinerea CDA genes (Supplementary Table S27) formed CDE orthogroups. These showed diverse expression peaks with no apparent trend in their expression. However, several genes were upregulated in stipes of young or mature fruiting bodies. One of them (C. cinerea protein ID: 355445) comprise proteins representing reciprocal best hits of S. cerevisiae CDA1, which is involved in the biosynthesis of the ascospore wall, required for spore wall rigidity. Expression of genes in this orthogroup did not peak during spore development, suggesting that this specific gene orthogroup is not involved in basidiospore wall synthesis. On the other hand, the expression of one and three genes were highest in young fruiting body gills and mature fruiting body gills and caps, respectively, which might be involved in producing chitosan in spore walls.
On a related note, we could not detect chitosanases, which degrade chitosan, in the genomes of mushroom-forming fungi, suggesting that chitosan is a terminal cell wall carbohydrate, not modified or cleaved further.
Carbohydrate binding modules (CBMs) - Carbohydrate binding modules, as their name suggests, bind various carbohydrates, and thus can frequently be attached to cell wall modifying enzymes. Chitin binding ability has been shown for at least 9 CBM families (Sánchez-Vallet et al. 2015). Of the most widely known chitin-binding CBMs (1, 5/12, 14, 18, 19, 50), only CBM1, 5/12, 18 and 50 families are conserved in the Agaricomycetes we examined. The CBM1 family, which primarily binds cellulose and according to some reports chitin, does not appear to be involved in fruiting body development: most genes that encode CBM1 containing proteins have virtually no expression in fruiting bodies. Some such proteins show significant expression dynamics (e.g., C. cinerea 363583, 499390), but are not conserved across species. The CBM18 family occurs in GH16 proteins, which have been described above.
CBM5/12 - The CBM5/12 family comprises chitin-binding modules, which assist the action of cell wall modifying enzymes in fungal development (Hartl et al. 2012). The CBM5/12 family is sometimes considered a lectin due to its carbohydrate binding ability (Ismaya et al. 2020). Most CBM5/12 modules occur in combination with GH18 domains, where their chitin binding ability is believed to increase the efficiency of the chitinase (Hartl et al. 2012). We identified 3–6 CBM5/12 domains in the examined Agaricomycetes. All but one CBM5/12 modules are attached to GH18 chitinases and have thus been discussed above. The single CBM5/12 module that is not attached to a chitinase occurs on proteins that also harbour a domain of unknown function (DUF3421) and a DM9 repeat as well. Such proteins occur in single copy in Agaricomycetes genomes and comprise a CDE orthogroup (C. cinerea protein ID: 413423) that show high expression in vegetative mycelia and mature stipe tissues of most species (A. ostoyae, P. ostreatus, M. kentingensis, C. cinerea, L. tigrinus, L. bicolor, L. edodes) (Supplementary Table S27). The protein is functionally uncharacterised. Both the DUF3421 and the DM9 repeat occur primarily in insects and other animals (but are found also in early-diverging fungi), but their activity is not known. Their developmental expression and the presence of a CBM5/12 domain suggests they may be linked to the chitin content in the cell wall, however, how exactly they function remains unclear.
CBM50 - Also known as LysM domain, this family binds chitin and can occur in a number of chitin-active proteins or on its own (Akcapinar et al. 2015). It has been reported to show developmentally dynamic expression in previous studies of fruiting body development (Sakamoto et al. 2017, Sipos et al. 2017, Krizsán et al. 2019, Huang et al. 2020). LysM domain proteins are widely known as effectors of plant pathogenic fungi, in which solitary secreted CBM50 modules can mask chitin residues and thus can dampen the immune response of plants (Akcapinar et al. 2015). Solitary CBM50 modules expressed in fruiting bodies could be involved in defence against insects (Künzler 2015).
The examined Agaricomycetes contain 3–9 CBM50/LysM domain encoding genes (except A. ostoyae, which has 18, but its expansion may be related to the pathogenic nature of Armillaria (Sipos et al. 2017). The CBM50 module occurs almost exclusively on its own in the examined Agaricomycetes, only C. aegerita has proteins that harboured combinations of CBM50 and chitinase domain signatures. Interestingly, CBM50 encoding genes showed relatively little expression dynamics within fruiting bodies with 0–2 developmentally expressed genes per species. This, and reports on the induction of one of the CBM50 genes of C. cinerea in response to bacterial and nematode challenge (Kombrink et al. 2019, Tayyrov et al. 2018, 2019) could indirectly support a role in defence. However, some genes had high and constant expression during fruiting body development (e.g., C. cinerea 448942), suggesting chitin-linked functions.
AA9 family (Lytic polysaccharide monooxygenases) - The family has been reported only recently to have fruiting body specific expression patterns (Almási et al. 2019, Krizsán et al. 2019). AA9 proteins were initially considered to be endoglucanases and classified in glycoside hydrolase family 61, but turned out to be lytic polysaccharide monooxygenases (LPMOs) that cleave polysaccharide chains using a distinct oxidative mechanism (Vaaje-Kolstad et al. 2010).
The examined Agaricomycetes possess 7–34 AA9 encoding genes, of which 1–10 were developmentally regulated. A part of this copy number variation can be explained by lifestyle, as wood-decay species are known to have expanded, while biotrophic or mycorrhizal species have contracted AA9 repertoires (Miyauchi et al. 2020). AA9 LPMOs might be active on β-glucan or on both glucan and cellulose as well, providing them with the activity required to remodel the fungal cell wall (Krizsán et al. 2019). The identified AA9 genes hardly formed conserved orthogroups, which may be a consequence of high duplication/loss rates.
Several species showed interesting LPMO expression patterns, which were quite consistent within, but different between species. For example, in C. cinerea several genes had a late upregulation, suggesting that LPMOs might be involved in autolysis. On the other hand, in Ph. chrysosporium many genes showed high expression in vegetative mycelium and mature fruiting bodies but low in young fruiting bodies, creating U-shaped expression curves. In L. tigrinus several genes showed a marked peak in cap and gill samples of mature fruiting bodies. The single AA9 gene (Cryne_JEC21_1_562) of C. neoformans had a strong (45-fold) induction in 24h samples, coincident with basidium formation (Liu et al. 2018). In general, all species displayed several genes with high expression in vegetative mycelium, but negligible (<5 FPKM) expression in any of the fruiting body tissues, suggesting the existence of separate vegetative mycelium-specific and fruiting body-specific AA9 subgroups. Yet other AA9 genes displayed uniformly low (<5 FPKM) expression; these LPMO-s might be specific for the plant cell wall, and thus not induced during development. Nevertheless, the developmentally dynamic expression of several AA9 genes suggests that this family is involved in fungal cell wall remodelling during fruiting body development.
AA14 family - We found a conserved orthogroup (represented by C. cinerea 362412) that contained members of the recently circumscribed AA14 family of lytic xylan oxidases (Couturier et al. 2018). These proteins require copper as their native cofactor and were speculated to unlock xylan-coated cellulose fibers in the plant cell wall. We identified members of the AA14 family based on BlastP (query Pycnoporus coccineus AUM86167 and AUM86166): we find that blastp with e-value cutoff of 10-10 yielded copy numbers that match perfectly those reported by Couturier et al. (2018) (Suppl Data Set 1). Fruiting body forming fungi contained 2–5 copies of the family, whereas yeast-like fungi (U. maydis and C. neoformans) contained none. Gene belonging to this family did not display characteristic expression peaks across species. However, as with other GH and AA families, their developmental expression in fruiting bodies suggests they may be active on some component of the fungal cell wall as well or have other extracellular roles.
X325 family - We also detected the recently described X325 family of copper-containing, LPMO-like proteins (Labourel et al. 2020). As a new gene family, the only functional knowledge available currently is that it displays a fold similar to fungal LPMOs, may be involved in copper homeostasis (Garcia-Santamarina et al. 2020), is upregulated in several mycorrhizal fungi and resides on the surface (via a GPI anchor) of the fungal cell wall (Labourel et al. 2020). A morphogenetic role for the family has been speculated. We report here that members of this family form a CDE orthogroup (represented by C. cinerea 454418) and are developmentally regulated in a wide range of species (9/6 at FC>2/4), although expression patterns differ between species. Additional developmentally regulated X325 genes from other species were segregated into other orthogroups. The L. bicolor gene is slightly upregulated in primordia relative to free living mycelium (FC=1,8) and has a peak expression in young fruiting body stipes.
AA10 and AA11, two well-known chitinolytic LPMO families (Langner & Göhre 2015, Polonio et al. 2021), are missing in the examined Agaricomycetes, so they are unlikely to be contributors to fruiting body development.
Glucan-linked families (GH3, GH5, GH16, GH17, GH30, GH55, GH71, GH128, GH152, Cdc43, WSC)
Cdc43 - We detected orthologs of S. cerevisiae Cdc43 (C. cinerea protein ID: 376392) as a CDE orthogroup in this study (Supplementary Table S28). Cdc43, together with Ram2, forms the type I geranylgeranyl transferase of S. cerevisiae, which transfers a geranyl-geranyl moiety to proteins having a CaaX sequence on their C-terminal (Finegold et al. 1991). One of the targets in S. cerevisiae is Rho1p, a rho-type GTPase, which is the regulatory subunit of beta-1,3-glucan synthase (Inoue et al. 1999). Cdc43 orthologs were developmentally expressed in 11/6 (C. cinerea protein ID: 376392, FC>2/4) (Supplementary Table S28) species as well as C. neoformans. They showed a marked upregulation from vegetative mycelium to primordia in A. ampla, A. ostoyae, C. aegerita, L. bicolor, L. edodes, C. cinerea, S. commune, and more or less constant expression in M. kentingensis, Pt. gracilis and P. ostreatus. On the other hand, orthologs of Ram2 (C. cinerea protein ID: 473005) as well as those of the main substrate, Rho1 (C. cinerea protein ID: 528086) did not show strong expression dynamics. We hypothesize that the expression dynamics of Cdc43 mirrors the dynamics of cell wall glucan biosynthesis in fruiting bodies.
GH17 - Enzymes belonging to the GH17 family are known to crosslink β-1,3-glucan in the cell wall of yeast species and presumably N. crassa too (Patel & Free 2019). The family has been reported in a transcriptomic study of mushroom development (Krizsán et al. 2019) (as endo-β-1,3-glucanosyltransferase), and it was suggested to be involved in β-1,3-glucan crosslinking, although direct evidence for this is lacking at the moment. The examined Agaricomycetes possess 1–4 GH17 encoding genes. C. cinerea has four GH17 genes, of which only one appear to be clear orthologs of Ascomycota genes: C. cinerea 389921 is reciprocal best hit to SCW10 of S. cerevisiae a soluble cell wall protein with a poorly known function. GH17-s formed 2 CDE orthogroups (Supplementary Table S28). One of the CDE orthogroups (represented by C. cinerea 476698) showed clear tissue-specific expression (cap- or stipe upregulation), but interestingly, this was not fully consistent across species.
GH16 - The GH16 family shows glycosylhydrolase and glycosyltransferase activity and contain glucan synthase and crosslinking enzymes, which are active in cell wall synthesis (e.g., Kre6 and Skn1 of S. cerevisiae) and remodelling. GH16s were shown to crosslink β-1,3-glucan and β-1,6-glucan to chitin fibers in Candida spp and N. crassa (Patel & Free 2019). Chitin-glucan crosslinking is key to morphogenesis in fungi (Arroyo et al. 2016). GH16s comprise the CRH (Congo Red Hypersensitive) family, three members of which (Crh1p, Crh2p and Crr1p) were originally circumscribed in S. cerevisiae. Of these, only Crr1 has a clear 1-to-1 ortholog in Agaricomycetes (C. cinerea protein ID: 449960), the other two do not. At the transcriptome level, the upregulation of GH16 genes have been reported before in a comparative study (Krizsán et al. 2019), in A. bisporus (Morin et al. 2012, Patyshakuliyeva et al. 2013), Leucocalocybe mongolica (Duan et al. 2021), Pleurotus spp (Xie et al. 2018, Ye et al. 2021) as well as during post-harvest development on L. edodes (Sakamoto et al. 2009, 2017). Mlg1 is a functionally characterised member of the GH16 family in L. edodes assayed for its glucanase activity during postharvest senescence (Sakamoto et al. 2009).
The species in this study have 21–36 GH16 encoding genes, which is consistent with a previous report on L. edodes (Sakamoto et al. 2017). About half of the GH16 genes in each species also carry a ‘Beta-glucan synthesis-associated Skn1’ domain, which signifies relationship with the Kre6/Skn1 family, which is involved in β-1,6-glucan synthesis (Garcia-Rubio et al. 2020). We found five GH16 CDE orthogroups (C. cinerea protein ID: 502321, 381870, 411452, 169822, S. commune 2686544) (Supplementary Table S28), that are not orthologous to functionally characterised GH16 genes in the Ascomycota. Although most genes do not form conserved orthogroups, possibly because of volatility (frequent duplication/loss), it is noteworthy that each species has 1–3 genes which show a marked upregulation in the primordium stage and remain highly expressed in the fruiting body relative to vegetative mycelium. These might be involved in the formation of 3-dimensional fungal tissues. Another interesting observation is that eight out of 21 GH16 genes in C. neoformans are also developmentally regulated, with several of them showing peak expression at 24h, coincident with basidium development.
GH30 - Glycoside hydrolase family 30 (GH30) includes enzymes with diverse activities, which have been mostly discussed for their role as endo-xylanases in wood decay in the fungal literature (Vanden Wymelenberg et al. 2009, Katsimpouras et al. 2019). However, GH30 also includes β-1,6-glucanases, which were found to be upregulated in L. bicolor ectomycorrhizae (Veneault-Fourrey et al. 2014), raising the possibility of morphogenetic roles. GH30 members were also upregulated during postharvest development of L. edodes (Sakamoto et al. 2017). Of the 1–3 (6 in L. bicolor) GH30 genes in the examined Agaricomycetes, at most 1–2 are developmentally expressed in fruiting bodies and only a single orthogroup (represented by C. cinerea 381637) was so in several species (Supplementary Table S28). Genes in this orthogroup were most often upregulated in stipe tissues relative to pileus in fruiting bodies. GH30 genes of C. cinerea have been proposed to facilitate stipe elongation by in the stipe base may contribute to replacing β-1,6-glucan to β-1,3-glucan crosslinking in the stipe base, which may reduce wall extensibility, leading to the cessation of growth (Liu et al. 2021). Further, the shared upregulation of GH30 genes in multiple fruiting body transcriptomes and in ectomycorrhizae provide strong evidence for its role in FCW remodelling during morphogenesis in the Basidiomycota.
GH3 - The GH3 family comprises enzymes with diverse activities, including exo-β-1,3-glucanase activity, and has been suggested to participate in cell wall remodelling in Ascomycetes (Patel & Free 2019). The upregulation of GH3 genes in fruiting bodies has previously been noted in a number of species (Zhou et al. 2018, Krizsán et al. 2019), raising the possibility that they participate in cell wall remodelling during fruiting body development. A GH3 family member, that was upregulated in fruiting bodies of Auricularia heimuer, was recently cloned and characterised (Sun et al. 2020). GH3 encoding genes are present in 7–20 copies in examined Agaricomycetes. Four CDE orthogroups belonging to the GH3 family (C. cinerea protein ID: 369876, 405828, 373418, S. commune Schco3_2629974,) were detected (Supplementary Table S28), genes in these orthogroups showed diverse expression patterns in the fruiting bodies of different species. It should be mentioned that some GH3 enzymes, mostly in bacteria, possess β-N-acetylhexosaminidase activity, which allows the encoded enzymes to be involved in chitin utilization, by cleaving N-acetylglucosamine dimers into monomeric GlcNac (de Oliveira et al. 2018). Most such enzymes are bacterial, with sporadic reports of β-N-acetylhexosaminidase activity in fungi (Yang et al. 2014). Therefore, whether GH3 upregulation is linked to chitin utilization or glucan remodelling cannot be determined here.
GH5 - The GH5 family is a very diverse group that includes β-1,4-glucanases, as well as several other activities. It includes classic cellulases involved in wood decay (Floudas et al. 2012), whereas Ascomycota GH5 genes, such as Exg1, Exg2 and Exg3 of S. pombe, or Exg1 of S. cerevisiae, are well-known components of the cell wall glucan remodelling machinery (Dueñas-Santero et al. 2010). GH5s were shown to not only hydrolyse β-1,3-glucan, but also to act as transglycosylases, and thus contribute to cell wall crosslinking (Arroyo et al. 2016). Based on expression patterns, GH5s were speculated to be active on beta-glucan in fruiting body cell walls also (Sakamoto et al. 2005, Veneault-Fourrey et al. 2014, Almási et al. 2019, Krizsán et al. 2019). Krizsán et al. reported multiple subfamilies (GH5_7, GH5_9, GH5_15, GH5_49) with endo-β-1,4-mannanase, endo-β-1,6- and exo-β-1,3-glucanase activities to be upregulated in fruiting bodies (Krizsán et al. 2019). The family includes the exo-beta-1,3-glucanase Exg1 from L. edodes, which was reported to be fruiting body specific (Sakamoto et al. 2005). Our re-analysis confirms that Exg1 (Lenedo1_1176007) has consistently higher expression in stipe than in cap tissues.
GH5 is a large family with 16–20 (25) copies in the examined Agaricomycetes. Consistent with their diversity, we detected five CDE orthogroups in our data (C. cinerea protein ID: 546300, 505989, 496411, 414471, 373700) (Supplementary Table S28). One of these (C. cinerea protein ID: 546300), contains reciprocal best hits of Sch. pombe Exg3, a cytoplasmic β-1,6-glucanase with unknown cellular function. All species in which this orthogroup is developmentally regulated showed upregulation in mature fruiting bodies. This could indicate a link to sporulation, or other late developmental events. Another orthogroup (C. cinerea protein ID: 414471) contains 1-to-1 orthologs of Sch. pombe and S. cerevisiae Exg1, which is involved in cell expansion and sporulation, picturing potentially similar roles in Agaricomycetes too. Members of yet another orthogroup (C. cinerea protein ID: 373700), are consistently higher expressed in all fruiting stages (primordia to mature FB) than in vegetative mycelia in A. ostoyae, C. aegerita, A. ampla, C. cinerea, P. ostreatus, S. commune and to some extent in L. bicolor and L. edodes (but not in A. bisporus and Pt. gracilis).
GH55 - This family contains exo- or endo-β-1,3-glucanases that have been characterised functionally in fruiting bodies of two Agaricomycetes. Exg2 was found to be expressed higher in the stipe than in the cap of L. edodes (Sakamoto et al. 2005). The GH55 family has also been reported to be upregulated during cap expansion of Volvariella volvacea and the corresponding gene was named Exg2 (Tao et al. 2013). We detected on average two GH55 genes in the examined Agaricomycetes (5 in A. ostoyae). One of these formed a CDE orthogroup (represented by C. cinerea 492834), which included L. edodes Exg2 and showed very strong induction in young fruiting body caps and stipes of C. cinerea, caps of A. ostoyae and stipes of M. kentingensis. Overall, these data are consistent with the suggested role of GH55 in cap expansion in V. volvacea. In L. edodes, the expression of Exg2 was higher in stipes than in cap tissues. Interestingly, the second GH55 gene of C. cinerea (C. cinerea protein ID: 454337) (Supplementary Table S28) also shows this characteristic induction and high expression in young fruiting body caps and stipes. In species with simpler morphology (S. commune, A. ampla) the family did not show characteristic expression dynamics. A third GH55 gene (Armosto1_267056) in A. ostoyae showed an induction in primordia relative to vegetative mycelium.
GH128 - GH128 has been first circumscribed in the context of fruiting body development, based on a fruiting body expressed β-1,3-glucanase, Glu1, in L. edodes (Sakamato et al. 2011). Since its discovery, some reports have mentioned upregulation of GH128 genes in fruiting bodies (Krizsán et al. 2019, Duan et al. 2021, Merényi et al. 2022), with a single report also in connection with wood decay (Mäkinen et al. 2019). On average, the GH128 family has 2–8 members in Agaricomycetes. We detected two CDE orthogroups (represented by C. cinerea 463000, 413757) which were expressed in the majority of species (Supplementary Table S28) and several GH128 genes which were developmentally expressed in a species-specific way. In C. cinerea, protein 413757 represents the best hit of L. edodes GLU1. Available data here and in previous publications suggest that GH128 might be a primarily fruiting body-related gene family, worthy of further functional examination.
GH71 - Four orthogroups containing developmentally regulated GH71 alpha-1,3-glucanases and beta-glucuronidases were detected (C. cinerea protein ID: 22054, 464399, 456054, 492754) (Supplementary Table S28). GH71 genes (also called mutanases) are involved in fungal cell wall remodelling in a wide range of species, both yeasts and filamentous fungi (Dekker et al. 2004, Veneault-Fourrey et al. 2014, Mélida et al. 2015). A well-characterised member of this family is the Sch. pombe endo-1,3-alpha-glucanase Agn1, which facilitates cell fission via the degradation of the septum material (Dekker et al. 2004). In previous studies of mushroom development, this family was reported only by Krizsán et al. (2019), Park et al. (2014), and Morin et al. (2012). In Pleurotus eryngii, GH71 genes were found downregulated in primordia after blue light stimuli (Xie et al. 2018). GH71 genes are present in 3–12 copies in the examined Agaricomycetes. One of the CDE orthogroups (represented by C. cinerea 456054) is upregulated in primordia relative to vegetative mycelium in 7 of the 10 species (C. cinerea, A. ostoyae, C. aegerita, L. tigrinus, L. bicolor, Ph. chrysosporium, P. ostreatus) in which it is present.
GH152 - The GH152 family, also known as thaumatin-like proteins, which include both putatively defence-related antimicrobial peptides and β-1,3-glucanases. They show homology to plant thaumatin-like proteins. Because the literature reports both antimicrobial and glucanase activity for this family (Künzler 2015), we do not attempt to subdivide them and refer to them as GH152/thaumatin proteins. The GH152/thaumatins include an experimentally characterised gene, Tlg1 in L. edodes (Lenedo1_561169), which was detected in a post-harvest screen of cell wall and lentinan degrading genes (Sakamoto et al. 2006). Lentinan is a glucan-like cell wall component with important medicinal properties in L. edodes. The expression of the gene was detected only after harvest, raising questions as to what the physiological role of the gene might be under natural circumstances. Our analysis of the data from Zhang et al. did not reveal developmentally dynamic expression for Tlg1 using RNA-Seq data (Zhang et al. 2021c). Structure prediction for an A. ostoyae developmentally expressed protein indicated an acidic cleft in the 3D structure, characteristic of antimicrobial members of the family (Krizsán et al. 2019). A new report of antifungal activity of the thaumatin-like protein Le.TLP1 in L. edodes further supports putative defence roles for the GH152 family (Ma et al. 2021).
The family comprises 2–8 (19 in Pt. gracilis) copies in examined Agaricomycetes, many of which are poorly conserved. Therefore, while several genes are developmentally expressed in each species, only a single CDE orthogroup was detected (represented by C. cinerea 496344) (Supplementary Table S28). The role of GH152/thaumatins in fruiting body development is unclear at the moment, it could be involved in defence against microbes, cell wall glucan remodelling, or both (Künzler 2015).
Additional beta-glucan active families (GH1, GH6, GH12) were detected in the broader orthology-based approach of Krizsán et al. (2019), which were not detected in this study, probably because of the strict orthology definition we use.
WSC family - This family of lectin-like proteins comprises a widely conserved but poorly known group of fungal proteins. The family was originally described as a putative stress receptor in S. cerevisiae (Lodder et al. 1999) and as a domain in an extracellular β-1,3-exoglucanase in Trichoderma harzianum (Cohen-Kupiec et al. 1999), and later demonstrated to be a carbohydrate binding module, similar to CBMs (Oide et al. 2019). The best-characterised member in the Basidiomycota seems to be involved in beta-glucan binding/FCW remodelling during plant-fungal interaction in Serendipita indica (Wawra et al. 2019). Whether this is true for other WSC family members too, is not known, but is indicative of a potential role in adhesion/ remodelling at the cell surface.
The examined Agaricomycetes harboured 2–12 copies of WSC-domain encoding genes. Of these, several showed strong induction in primordia and fruiting bodies in A. ostoyae, C. aegerita, C. cinerea, L. bicolor, L. edodes, L. tigrinus, Ph. chrysosporium, P. ostreatus and S. commune (but not in A. ampla and M. kentingensis). Genes with a predicted WSC domain in their encoded proteins were typically primordium-upregulated in all species. In C. cinerea these genes had high expression in early (proliferative) development (hyphal knots to stage 2 primordia), followed by very low expression in most genes in late developmental stages. The WSC domain can occur on its own or in combination with other enzymes (most often GH71 and glyoxal oxidase) and uncharacterised domains, indicating that it often fulfils a binding role in larger proteins. For example, WSC domain is present on GH71 proteins that form a 10/8 orthogroup (represented by C. cinerea 464399), which we discussed above.
Miscellaneous cell wall remodelling (GH27, GH72, GH76, GH79, GH88, GT18, pectinesterases)
In addition to chitin- and glucan-active families, several further CAZyme groups showed widespread developmental regulation in fruiting bodies, for which the substrates in fruiting bodies are less clear. These make very interesting groups indicating potentially novel cell wall components; however, they are hard to place in context of fruiting body development as of now.
GH79 and 88 - An orthogroup containing GH79 β-glucuronidases (C. cinerea protein ID: 470689) (Supplementary Table S29), showed developmental expression in several species, consistent with reports of Krizsán et al. (2019). Low amounts of glucuronic acid were reported in N. crassa cell walls (Verdín et al. 2019), but the role of GH79 enzymes in cell wall biosynthesis or remodelling are unknown. On a related note, an orthogroup of GH88 glucuronyl hydrolases was detected (C. cinerea protein ID: 384105) (Supplementary Table S29). GH88 genes are generally linked to pectin degradation by wood-decay fungi (van den Brink & de Vries 2011); their potential role in fruiting bodies is not known.
GH27 - Similarly, two CDE orthogroups (represented by Armosto1_254810 and Armosto1_269656) of GH27 enzymes with alpha-galactosidase or α-N-acetylgalactosaminidase activity were detected (Supplementary Table S29). This family is discussed mostly in the context of lignocellulose degradation (Kabel et al. 2017), to our knowledge it has not been reported in fruiting body transcriptomes before. As a tendency, GH27 encoding genes were often upregulated late in the development of the containing species. Their role in fruiting body formation is not known.
GT18 - The GT18 family, which contains α-1,3 (6)-mannosylglycoprotein β-1,6-N-acetyl-glucosaminyltransferases showed a patchy distribution in the examined species, being completely missing from the genomes of C. cinerea and A. ostoyae. However, it showed a strong upregulation in primordia relative to vegetative mycelium and constant high expression in fruiting bodies of A. bisporus, A. ampla, S. commune, C. aegerita and P. ostreatus (not in M. kentingensis and L. bicolor) (Supplementary Table S29, S. commune protein ID: Schco3_2633734). This indicates fruiting body specific functions for this family, possibly in chitin synthesis, although its exact role remains unknown. To our best knowledge, the role of the GT18 family in modifying the fungal cell wall has not been described in any species.
GH72 - Contrary to our expectations, members of the GH72 family did not appear to be developmentally relevant in Agaricomycetes. This family contains cell wall transglycosylases that crosslink glycoproteins to cell wall components in the Ascomycota (Kar et al. 2019), and therefore we expected it to be developmentally expressed in Basidiomycota as well. Their expression is considerable but largely constant in Agaricomycetes fruiting bodies. This could indicate a difference between Asco- and Basidiomycota cell wall composition, or that they act during fruiting body development irrespective of their constant expression.
GH76 - In the Ascomycota, GH76 endo-1,6-alpha-mannosidases (Dfg5 and Dcw1 in S. cerevisiae) are required for normal cell wall synthesis. Their exact role may not be known with certainty, but they were hypothesized to be involved in the proper positioning of glycosylphosphatidylinositol-anchored cell wall proteins (Orlean 2012). To our best knowledge, the GH76 family has not yet been suggested to participate in fruiting body development in the Agaricomycetes. It is present 0–10 copies in the Agaricomycetes and shows predominantly mycelium-specific expression. We only found scattered expression dynamics in fruiting bodies, such as a peak in stipes of P. ostreatus (PleosPC15_2_1064904), but most genes are not developmentally regulated. Given that the family is missing in some species completely and the predominantly vegetative mycelium-specific expression, we hypothesize that the GH76 family does not participate, or only to a small extent, in cell wall assembly in Agaricomycete fruiting bodies.
Pectinesterases - We detected a CDE orthogroup (represented by Schco3_2614029) that contains genes generally linked to pectin degradation. It contains pectinesterases that are developmentally regulated in 7/6 species (FC 2/4) (Supplementary Table S29). Because pectin is not a known cell wall component in Agaricomycetes, speculating about the function of these genes is hard and may require more research.
Multicopper oxidases and cupredoxins
Laccases catalyze the oxidation of diverse phenolic and non-phenolic compounds, which makes them suited to diverse (extra)cellular roles, from lignin degradation, pigment production to dye decolorization or fruiting body related functions (Baldrian 2006). Their biochemical properties provide laccases with significant potential in industrial applications. Despite very intense research on laccases (mostly in the context of lignocellulose decomposition), we still have very limited information on the precise role of laccases in fruiting body development.
Several reports of upregulation of laccase, or multicopper oxidase (MCO, the broader family in which laccases belong) genes or laccase activity in fruiting bodies of Agaricomycetes (Zhao & Kwan 1999, Ohga et al. 2000, Chen et al. 2003, Wang & Ng 2005, Madhavan et al. 2014, Sakamoto et al. 2015, Sipos et al. 2017b, Nagy et al. 2018) are available (reviewed in detail by Kües et al. 2011). Schizophyllum commune was reported to have 6 MCO genes (two of which are laccases), of which 3 were reported to be upregulated in fruiting bodies (Madhavan et al. 2014) (Mco2, Mco3, Lcc1) by RT-PCR, which is consistent with later RNA-Seq data (Krizsán et al. 2019). Of these, Lcc1 showed the highest similarity to Fet3-type ferroxidases. Similarly, P. ostreatus laccases were reported to be expressed in a developmental stage-specific manner, with Lacc5 and Lacc12 specifically expressed in fruiting bodies and primordia, respectively, while other laccases had even or mycelium-biased expression patterns (Park et al. 2015, Jiao et al. 2018). Stage-specific MCO expression was reported in L. bicolor with specific laccase genes induced in fruiting bodies (Lcc2), ectomycorrhizae and free-living mycelium (Courty et al. 2009). These studies have linked laccase expression to two major functions, pigment production in either spores or fruiting bodies and fungal cell wall modification or cell-cell adhesion (Kües & Ruhl 2011). Laccases were speculated to polymerize polyphenols in the intercellular spaces and thus contribute to the binding of neighbouring hyphae to each other. The hypothesis of laccases being related to adhesion comes from Leatham & Stahmann (1981), but, to our knowledge, was never experimentally verified. Although not relevant to fruiting body formation, an interesting inverse relationship between mycelial laccase expression and flushes of fruiting body development was reported in several species, possibly related to stopping mycelium growth or nutrient acquisition when nutritional competence for fruiting is achieved (reviewed in Kües & Ruhl 2011).
Developmental studies indicated laccase function in fruiting body formation in Pleurotus spp. (Das et al. 1997) and L. edodes (Nakade et al. 2011). In P. florida laccase gene silencing abolished fruiting body development and caused slower vegetative growth, whereas in L. edodes aerial hyphae formation was also reduced and hyphae developed thinner, non-layered cell walls upon the silencing of the Lcc1 laccase gene. Zhang et al. reported faster vegetative growth and earlier fruiting in laccase (Lcc1) overexpressing strains of H. marmoreus compared to wild type strains (Zhang et al. 2015a), although no clear hypothesis on how overexpression of a single structural gene could result in these phenotypes was presented.
We here present annotations for multicopper oxidases, which include laccases, ascorbate oxidases and ferroxidases (Kües & Ruhal 2011). The twelve genomes of Agaricomycetes examined here had 5–26 multicopper oxidases, a considerable proportion of which were developmentally regulated, for example, 13 out of 17 in C. cinerea. We found three CDE laccase orthogroups; these comprised C. cinerea Lcc8, Lcc2 and their orthologs as well as reciprocal best hits of the S. cerevisiae ferroxidase Fet3. [Of note, we detected reciprocal best hits of the ferroxidase Fet3 in C. cinerea, whereas a previous report, based on phylogenetic analyses did not identify Fet3-type MCOs in this species (Kües & Ruhl 2011); we note that this may arise from the limitations of our reciprocal best hit based search and that accurate classification of MCOs requires the examination of specific catalytic residues. Orthologs of C. cinerea Lcc2 (C. cinerea protein ID: 368135) are also involved in morphogenesis in L. edodes (as Lcc1), and differentially expressed during Cryptococcus basidium formation (Liu et al. 2018, Merényi et al. 2021). We found that Lac4 from Volvariella volvacea, which was implicated in fruiting body development (Chen et al. 2003) showed highest similarity to the CDE orthogroup that comprised L. edodes Lcc1 (represented by C. cinerea 368135). In the examined Agaricomycetes, this group of genes is upregulated in caps (P. ostreatus, C. cinerea), stipes (M. kentingensis) or primordia (A. ampla), or shows flat expression curves (other species). The detected ferroxidase orthogroup was present in 7 species, of which it was developmentally regulated in A. bisporus, A. ostoyae, C. cinerea, L. edodes and P. ostreatus (FC>2). A Fet3-like gene was also upregulated in S. commune (Madhavan et al. 2014), but this gene did not group into the detected orthogroup in our analysis. Ferroxidases are membrane proteins that catalyse the oxidation of Fe2+ to Fe3+ for iron uptake and have only weak laccase activities (Larrondo et al. 2003). Finally, C. cinerea 440170 is the ortholog of C. neoformans Lac1, which has been shown to be involved in melanin biosynthesis (Lee et al. 2019). This gene shows a strong expression peak in young fruiting body gills, suggesting that it is involved in producing spore pigments in C. cinerea.
In summary, experimental and gene expression evidence on the role of multicopper oxidases in fruiting body development is abundant and -omics data reveal marked developmental stage-specific expression of laccases. Hypotheses on their role either in pigment production or cell wall crosslinking/adhesion have been put forth, however, the available data do not allow us at the moment to select between these hypotheses, until more developmental studies and gene knockout phenotypes become available.
In addition to bona fide MCOs, we identified four CDE orthogroup of proteins which belong to the Cupredoxin family, like laccases, but do not contain the typical multicopper oxidase domain. These might be highly diverged MCO genes or distant relatives of true MCOs with unknown functions. The five orthogroups are conserved across Agaricomycetes (C. cinerea protein ID: 356026, 451850, 441442, 353997, 497434) (Supplementary Table S30). One of these (C. cinerea protein ID: 356026) is FB-init in A. ostoyae, C. cinerea and P. ostreatus (not in A. ampla, S. commune and M. kentingensis), whereas a second (C. cinerea protein ID: 451850) was characteristically late upregulated in most species (A. ampla, M. kentingensis, P. ostreatus, Pt. gracilis). Currently, it is impossible to speculate about the role of these proteins in fruiting body development.
Expansins and cerato-platanins
Fungal expansins (or expansin-like proteins) show similarity to plant expansins, proteins that modify the plant cell wall but do not show hydrolytic activity towards its polymers. Rather, they exert their action by loosening the cell wall structure, hence the name loosenins (Quiroz-Castañeda et al. 2011) and swollenins (Saloheimo et al. 2002) for some members of the family. Fungal expansins with activities towards either the plant or fungal cell wall have been reported. Certain fungal expansins can bind chitin and facilitating the action of hydrolytic enzymes by separating cell wall chitin microfibrils. Two experimentally characterised Basidiomycota expansins are Loos1 from Bjerkandera adusta (Quiroz-Castañeda et al. 2011) and ScExlx1 from S. commune (Tovar-Herrera et al. 2015). Both were shown to bind chitin and facilitate the action of chitinases; therefore, it was concluded that they modify, but not hydrolyse the chitin polymer structure. Recent studies suggested that expansin-like proteins are involved in stipe cell elongation in F. velutipes (Huang et al. 2018) and C. cinerea (Niu et al. 2015) and were shown to have a role in stipe elongation, by loosening the chitin polymer structure (reviewed by Liu et al. 2021).
Several expansins show expression patterns compatible with a role in fruiting body development in previous transcriptome-based studies (de O Scale barsottini et al. 2013, Sipos et al. 2017, Huang et al. 2018, Almási et al. 2019, Krizsán et al. 2019). Based on analyses of 201 fungal genomes, Krizsán et al. reported that expansins are enriched in the genomes of Agaricomycetes (Krizsán et al. 2019). The examined mushroom-forming fungi possess 9–27 expansin genes, with C. cinerea having 13. Given the dual specificity of expansins, it is likely that several of these are involved in cellulose degradation. However, a significant proportion (19–85 %) of expansin genes are developmentally regulated in fruiting bodies, indicating roles in morphogenesis too. Expansins are a highly volatile group of genes that did not group into conserved orthogroups in our analysis (except two CDE orthogroups, see below), therefore, comparative analysis of their expression patterns across species is hardly possible. Within C. cinerea expansins show diverse expression peaks, in gills, cap or stipes with some genes highest expressed in vegetative mycelia too (Fig. 11). This could indicate tissue- and stage-specific roles for expansins in fruiting body formation. For example, four genes had a strong induction in stipes (C. cinerea protein ID: 451456, 464389, 359952, 461698), which is consistent with previous reports of their participation in stipe elongation (Liu et al. 2021). Three C. cinerea genes showed significant increase in expression from the vegetative mycelium to hyphal knots (C. cinerea protein ID: 461698, 286849, 496836). Despite the volatility of the family, we detected three putative expansin orthogroups (represented by C. cinerea 496836, 467907 and 461698) which were developmentally regulated in 12/10, 11/10 and 10/5 species (at FC>2/4) (Supplementary Table S31).
Fig. 11.

Expression heatmap of expansin-encoding genes in C. cinerea. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively. Developmental stages are abbreviated as follows: VM – vegetative mycelium, H – hyphal knot, P1 – stage 1 primordium, P2 – stage 2 primordium, YFB.C – young fruiting body cap, YFB.L – young fruiting body gills, YFB.S – young fruiting body stipe, FB.C – mature fruiting body cap (including gills), FB.S – mature fruiting body stipe.
Cerato-platanins - Although initially thought to be related to hydrophobins, cerato-platanins share a common origin with expansins (Scale barsottini et al. 2013, Gaderer et al. 2014, Baccelli 2015, Luti et al. 2020). First described from the ascomycete Ceratocystis platani, they were initially and generally considered to be related to pathogenesis (Gaderer et al. 2014, Baccelli 2015, Chen et al. 2017, Luti et al. 2020), however, there is piling evidence, so far only from transcriptome studies, that they might be important in fruiting body development as well. In such studies, cerato-platanin genes showed extensive expression dynamics in fruiting bodies, suggesting an activity on the fungal cell wall (Plaza et al. 2014, Sipos et al. 2017, Almási et al. 2019, Krizsán et al. 2019, Liu et al. 2020), or in defence (Künzler 2015). Structure-based biochemistry studies in Moniliophthora perniciosa showed that certain cerato-platanins are able to bind chitin and are active on the fungal cell wall (Scale barsottini et al. 2013). Like expansins, cerato-platanins are strongly enriched in the genomes of mushroom-forming fungi (Chen et al. 2017, Krizsán et al. 2019). There is, unfortunately, little information on their mechanistic role in fruiting body development, except the study by de Scale barsottini (Scale barsottini et al. 2013), which provides proof that cerato-platanins can be active on chitin and the fungal cell wall, providing a biochemical link to their potential role in fruiting body development.
Our analyses identified 1–8 cerato-platanins in the genomes of examined mushroom-forming fungi, which is in line with copy numbers reported recently (Chen et al. 2017). Like expansins, cerato-platanins are quite volatile and we identified only two conserved CDE orthogroups (C. cinerea protein ID: 439071, 544218) (Supplementary Table S31), with most genes scattered in species-specific or smaller groups. The first showed opposing expression patterns in different species: in C. cinerea (protein ID: 544218) it was upregulated in caps relative to stipes, whereas in A. ostoyae and M. kentingensis it was upregulated in stipes relative to caps. In P. ostreatus and M. kentingensis it showed the highest expression in vegetative mycelia. In A. ampla and S. commune it showed a very strong induction (>1 000-fold) in mature fruiting bodies.
Expression data across species indicates that developmentally dynamic cerato-platanin expression was observed only in certain species, while not in others. For example, no developmentally regulated cerato-platanins were found in Ph. chrysosporium, A. bisporus, only a single in M. kentingensis, but several in A. ostoyae, C. cinerea, L. tigrinus, etc. This indicates that developmental expression of cerato-platanins may be a species-specific, rather than a conserved feature in mushroom-forming fungi.
Miscellaneous cell wall related families (HTP, DyP, AA3, AA5, GH76, GT8, Slt2, Cfs1, Eln3, peptidases)
We identified several CDE gene families which are potentially linked to the fungal cell wall; however, evidence is lacking on their mechanistic roles and/or they could not be confidently ascribed to either chitin or glucan, and/or are not enzymatic proteins. These are discussed in this chapter with noting that the widely observed developmental expression makes these protein families interesting, however, their assignment to the cell wall is preliminary.
HTPs - Heme-thiolate peroxidases (HTPs) are fungal-specific proteins that were first described in C. aegerita (as haloperoxidase) and use H2O2 to modify substrates (Ullrich et al. 2004). While their enzymatic properties have been quite thoroughly studied (Hofrichter et al. 2015), their biological roles are poorly known. A remarkable expansion of the family was noted in A. bisporus which, combined with the upregulation of HTPs in Agaricus substrates led to the hypothesis that HTPs are linked to the litter decomposer lifestyle (Morin et al. 2012). In the context of fruiting bodies, HTPs were reported only in A. ostoyae (Sipos et al. 2017b) so far. We detected two CDE orthogroups of HTPs (represented by C. cinerea 479688, 442333) (Supplementary Table S32) as well as several other species-specific developmentally regulated HTP genes. The examined Agaricomycetes have 2–10 genes with an expansion to 23 genes in both M. kentingensis and A. bisporus as reported earlier (Morin et al. 2012). Most of the species had at least one HTP encoding gene that is strongly upregulated (up to 50-fold) from vegetative mycelium to the first primordium stage. These observations establish HTPs as a potentially important morphogenetic gene family in fruiting body development with currently unknown function. We tentatively discuss this gene family under the chapter on cell wall modification, however, it should be mentioned that this is based on an assumption of a potential role of this family.
DyPs - Dye decolorizing peroxidases (DyPs) are a group of fungal peroxidases that belong to the broader heme-containing peroxidase family. They are mostly discussed in the context of plant cell wall degradation by fungi (Floudas et al. 2012); to our knowledge, this is the first report of its potential role in fruiting body morphogenesis. We detected 0–5 copies (missing in A. ampla, S. commune and A. bisporus) and a patchy distribution in Agaricomycetes. However, in species that have DyP genes, these are often developmentally expressed. DyP genes show significant upregulation from vegetative mycelium to the first primordium stages in A. ostoyae, C. cinerea, C. aegerita, L. bicolor and P. ostreatus. In M. kentingensis all DyP genes show a marked stipe expression peak, whereas in Pt. gracilis, expression curves are flat. DyP genes grouped into a single CDE orthogroup (represented by C. cinerea 407604, Supplementary Table S32). While some of the copy number variation is explained by the lignin-degrading ability of fungi (Almási et al. 2019), the developmental expression of DyP genes suggests morphogenetic roles as well, possibly in remodelling the cell wall, although this is based on assumptions distilled from the general activity of the family. It would be interesting to investigate if, similar to observations during lignin degradation, H2O2-generating enzyme gene expression is co-regulated with peroxidase expression patterns.
AA5_1 (GLOX) - Glyoxal oxidases (GLOX) in the AA5_1 family of auxiliary activities are H2O2 generating enzymes that facilitate the oxidative cleavage of lignin by peroxidases (Floudas et al. 2012). Surprisingly, GLOXs have also been reported to be upregulated in fruiting bodies in 6 species of Agaricomycetes (Krizsán et al. 2019) and were detected in fruiting bodies of Auricularia polytricha by protein mass spectrometry (Jia et al. 2017). Since lignin is obviously missing from fruiting bodies, they were speculated to contribute to cell wall remodelling (Krizsán et al. 2019). We found 1–22 GLOX genes in the examined Agaricomycetes, the fewest in the Schizophyllaceae (A. ampla, S. commune) possibly because of their reduced morphology or reduced lignin-degrading ability (Almási et al. 2019). Two CDE orthogroups (C. cinerea protein ID: 263775 and 440836 Supplementary Table S32), and several species-specific developmentally regulated GLOX genes were found in our data. We found GLOX genes significantly upregulated in primordia relative to vegetative mycelium (FB-init) in A. ostoyae, A. ampla, C. cinerea, L. tigrinus, L. bicolor, S. commune, Ph. chrysosporium, P. ostreatus and C. aegerita (not in M. kentingensis, L. edodes and A. bisporus). Two of the three C. neoformans GLOX genes also showed significant upregulation during basidium development. These data indicate that GLOXs may be important morphogenetic factors in the Basidiomycota.
AA3_1 - Cellobiose dehydrogenases in the AA3_1 family (GMC oxidoreductases) are known to donate electrons to LPMOs during lignocellulolysis in wood-decay fungi (Floudas et al. 2012). There are only two reports on the potential role of CDHs in fruiting body development, one is a comparative transcriptomics study (Krizsán et al. 2019) and the other is a report of the interaction between CDH and laccases in the formation of cinnabarinic acid, the red fruiting body pigment of Pycnoporus cinnabarinus (Temp & Eggert 1999). A CDE orthogroup of cellobiose dehydrogenase (CDH) genes (C. cinerea protein ID: 469460) (Supplementary Table S32) was developmentally regulated in all species in which it was present. Additional CDH genes (2–8 genes in Agaricomycetes) were developmentally regulated in a species-specific manner in several species, although expression levels were low in many cases. The biological significance of CDH in fruiting body development is currently unknown; based on previous studies it may be related to cell wall remodelling or pigment production.
Slt2 kinase family - A serine-threonine kinase orthologous to the MAP kinase involved in the cell wall integrity pathway of S. cerevisiae (Slt2) was detected as a CDE orthogroup (C. cinerea protein ID: 413665). Slt2 is a terminal kinase in the pathway that phosphorylates downstream transcription factors (Rlm1 and Swi4/6 in yeast) (Levin 2005). Although the gene is developmentally regulated in several species (Supplementary Table S32), expression patterns are not consistent across taxa. Given the potentially pleiotropic role of this kinase, it is hard to guess its role in fruiting bodies, but extrapolations from its role in S. cerevisiae would suggest links to cell wall integrity, morphogenesis or the cell cycle.
Cfs1 family - The orthogroup containing C. cinerea Cfs1 (C. cinerea protein ID: 447925), a cyclopropane fatty acid synthase, was developmentally regulated in 10/7 (FC>2/4) species (Supplementary Table S32). Cfs1 was identified in a mutant screen of strains with developmental defects; its mutation leads to failures in the progression of hyphal knot development (Liu et al. 2006) in C. cinerea (stalled at primary hyphal knot). The corresponding gene is highly induced 1 h after blue light exposure (Sakamoto et al. 2018). We discuss this gene under the cell wall, although some evidence suggests it may be more closely related to plasma membrane assembly. Structurally, Cfs1 and related proteins are mycolic acid cyclopropane synthases, a family that occurs in 1–6 copies in the examined Agaricomycetes. Their function is not known, but presumably they are involved in the synthesis of long fatty acids (mycolic acid is not known to occur in fungi). In most examined species (A. ampla, M. kentingensis, C. cinerea, P. ostreatus, S. commune), the gene is upregulated in primordia relative to vegetative mycelium, consistent with its phenotypic role in hyphal knot development.
Eln3 family - This family is based on the Eln3 gene of C. cinerea, which was found in a REMI mutagenesis screen. The mutant strain displayed defects in stipe elongation and mutant stipe cells were disorganized and there were large gaps among stipe cells, whereas in the wild type strain stipe cells align closely with each other (Liu 2005). It has been hypothesized that Eln3 is involved in cell-cell connections, possibly adhesion.
We found that C. cinerea Eln3 (C. cinerea protein ID: 380322) formed a small CDE orthogroup present in 7 species and was developmentally regulated in 6 species (Supplementary Table S32). In these species the gene was predominantly FB-init and stipe upregulated, in agreement with a previously reported role in stipe elongation (Arima et al. 2004). It should be noted that, in many species, upregulation in primordia is easy to confuse with stipe-specific upregulation, because primordia often comprise predominantly stipe tissue. In general, we found 7–8 genes the examined Agaricomycetes (only 4 in A. bisporus) with identical domain composition to Eln3. This does not guarantee identical function, nevertheless is indicative of the diversity of this family in mushrooms. These genes were often FB-init and showed stipe or cap-specific upregulation depending on the gene and the species. For example, in C. cinerea we found that 5 out of 6 genes were stipe-upregulated. In A. ostoyae we found four cap- and two stipe-upregulated, whereas in L. bicolor two cap- and one stipe-upregulated gene. The widespread developmental regulation of eln3 and its homologs suggest morphogenetic functions, possibly in association with the cell wall. Based on its domain composition, we hypothesize it represents a cell wall active, possibly new CAZyme family not yet recognized by Cazy.org.
GT8 - The GT8 family has 1–7 (most frequently 5) copies in the Agaricomycetes. The GT8 family includes both N-acetylglucosaminyltransferases linked to the modification of N-linked glycans in the Golgi, and glycogenins involved in glycogen metabolism. Buser et al. (2010) hypothesized that fruiting body-specific N-glycans generated by GT8 enzymes may contribute to adhesion in C. cinerea.GT8 genes formed one CDE orthogroup, represented by C. cinerea 461263 FC>2/4: 7/3 species, Supplementary Table S12), which is closest to S. cerevisiae Glg1 glycogenin (discussed under carbohydrate metabolism). Of the other GT8 genes C. cinerea (C. cinerea protein ID: 453097, =CcGnt1) (Buser et al. 2010), A. ostoyae, L. tigrinus, P. ostreatus, A. ampla and S. commune had one FB-init gene each, however, these did not form CDE orthogroups. CcGnt2 (C. cinerea protein ID: 412828) was not expressed. Other species did not have developmentally regulated genes. Due to the variability of the family, it is hard to determine whether these are closer to glycogenins or N-acetylglucosaminyltransferases.
N-glycosylation - Mannosylation and N-glycosylation of proteins in the ER is a key process for cell wall synthesis. Berends et al. provided a detailed view of the N-glycosylation machinery in S. commune, C. cinerea and L. bicolor based on orthology to the S. cerevisiae N-glycosylation pathway members and expression data in S. commune fruiting bodies and monokaryotic mycelium (Berends et al. 2009). They conclude that Agaricomycetes have the gene repertoire to produce oligomannose structures only, in contrast to some Ascomycota which can produce very complex modifications to proteins. Our expression results broadly agree with theirs in that we detected very low dynamics of corresponding genes (not shown) in the fruiting body transcriptomes analysed here.
Cell surface and cell wall proteins
Secreted cell surface proteins fulfil diverse functions in the life of fungi, ranging from signalling, interactions with the environment, adhesion, cell wall modifications, but also communication with hosts in mutualistic or pathogenic interactions (Pellegrin et al. 2015, Essen et al. 2020). The fungal cell surface proteome and secretome profile of each species in a given environment is unique, making constituents of fungal communities recognizable by their extracellular protein profiles (Campbell et al. 2015). Secreted proteins have been the subject of intense research, mostly in the context of interactions between fungi and other organisms, adhesion (e.g., via GPI-anchored adhesins) of fungal cells to various surfaces (Essen et al. 2020, Samalova et al. 2020). Fruiting bodies, as environmentally exposed, 3-dimensional multicellular structures, can be expected to display unique cell-surface attributes. In fact, cell surface proteins include some of the best-known fruiting body-associated proteins (e.g., hydrophobins, lectins, see below) or proteins related to defence (see these in the next chapter). Conceivably, adhesion and/or cell-to-cell communication, two of the most important multicellular functions (Knoll 2011), could be realized via the action of secreted cell surface proteins in fruiting bodies, to just name a few of the diverse ways mushroom-forming fungi might make use of their cell surface protein repertoire. A significant open question in fruiting body biology is the nature and role of the extracellular matrix and the biochemical and communication events that take place in the interhyphal space. Cell surface proteins are undoubtedly important players in these processes, and therefore represent interesting targets of future research.
While some cell surface proteins are the best examined fruiting body proteins (e.g., hydrophobins), the following inventory of conserved developmentally expressed genes shows that there is plenty of new insight to be learned about the cell surface proteome of Agaricomycetes fruiting bodies. In this chapter, we discuss cell surface protein families in the context of CDE orthogroups detected in our meta-analysis of fruiting body transcriptomes.
Hydrophobins
Hydrophobins are some of the best-known proteins in fruiting bodies. They confer hydrophobicity to hyphae, which they achieve by self-assembling into a water-repellent rodlet layer on the cell surface. Hydrophobins were hypothesized to line air channels in fruiting bodies (Lugones et al. 1999b), but also contribute to water sealing, which is important in fruiting bodies as they rely on water transported to them from the vegetative mycelium (Nehls & Dietz 2014). The family has extensively literature, in general (Wösten 2001, Linder et al. 2005, Bayry et al. 2012), in relation to fruiting body development (Ohm et al. 2010, Kües & Navarro-González 2015, Kim et al. 2016, Sammer et al. 2016, Tao et al. 2019) or on potential uses in bioengineering (Wösten & Scholtmeijer 2015). We therefore concentrate on novel aspects detected in our data that have not or only rarely been reported before.
Basidiomycota hydrophobins mostly belong to class I hydrophobins (Sammer et al. 2016). On average 11–44 hydrophobin genes (only 6 in M. kentingensis) were detected in the examined Agaricomycetes, a significant proportion of which were developmentally regulated. They can be arranged into distinct groups based on expression patterns. Previous studies reported a near complete turnover of hydrophobin genes from vegetative mycelium to fruiting body, indicating that two distinct sets of hydrophobins exist (Ma et al. 2007, Chum et al. 2008, Cheng et al. 2013). We also observed that mycelium-expressed and fruiting body-expressed hydrophobins form distinct groups. For example, the numbers of hydrophobins showing mycelium- vs. fruiting body specific expression was 6/5, 1/9, 10/4, 23/3 and 5/20 (at fold change 4/2) in Ph. chrysosporium, S. commune, P. ostreatus, A. ostoyae and C. cinerea, respectively. A. ostoyae further has two rhizomorph-specifically expressed hydrophobins, indicating morphogenesis-related diversification of the family. This is in line with observations in F. velutipes, where most hydrophobin genes were found to be primordium-upregulated (Kim et al. 2016). We also detected hydrophobin genes expressed during early development, but not later, indicating functions linked to primordium development, which are turned off during later development. These observations further substantiate the functional diversification, probably driven by morphogenesis and complexity.
Despite the large diversity and developmental expression of hydrophobins, only 2 CDE orthogroups were detected (C. cinerea protein ID: 99661, 419467) (Supplementary Table S33), which were present in only a subset (11 and 8 species, respectively) of the examined species. This is due to the frequent duplication of hydrophobin genes (Mgbeahuruike et al. 2017), which quickly erases clear orthology relationships. CDE orthogroups did not show conserved expression patterns. Interestingly, M. kentingensis has only 6 hydrophobin genes, none of which were developmentally expressed in our reanalysis of data from Ke et al. (2020). A possible explanation for this is that this species produces hygrophanous, rather than hydrophobic fruiting bodies, which absorb rather than repel water. The relatively low number of developmentally regulated hydrophobin genes in A. ostoyae may also be explained by the hygrophanous caps of this species. Hygrophanicity is a widespread trait in the Agaricales and might explain some of the differences in hydrophobin diversity and expression.
Taken together, hydrophobins, as already established by previous research, are one of the best-known fruiting body-related structural cell surface proteins in the Agaricomycetes, with almost all species expressing developmental stage-specific suites of hydrophobin genes.
Lectins
Lectins will be also discussed in the next chapter on defence-related proteins, however, a role in defence has been proven for only a subset of the diverse lectin repertoires of mushroom-forming fungi; the remaining non-defence, or functionally unknown lectins are discussed in this chapter (even though not all lectins may be secreted or their secretion status is unclear at the moment (Boulianne et al. 2000). Lectins are proteins that specifically recognize and bind various carbohydrate moieties, mostly on the cell surface. This ability makes them suitable for multiple biological roles, from bacterial agglutination and cytotoxicity against mushroom grazers (Bleuler-MartÍnez et al. 2011, Künzler 2015) to carbohydrate binding modules that fulfil roles related to the fungal cell wall (e.g., remodelling), adhesion, cell-to-cell communication or other functions in the extracellular matrix. Their carbohydrate binding ability makes them interesting in a wide range of biomedical fields (Hassan et al. 2015), including cancer research, which led to the broader lectin family rapidly expanding. Mushroom fruiting bodies remain one of the richest sources of lectins for applied research. However, regarding their physiological role for the fungi, boundaries between defence-related and other lectins are far from clear at the moment (Bleuler-MartÍnez et al. 2011).
Due to interest in their practical applications, there is often more information on the structure of mushroom lectins than on their roles in the fungus’ life. Fruiting bodies express a diverse set of lectins (reviewed by Varrot et al. 2013, Kobayashi & Kawagishi 2014, Kobayashi et al. 2014) and several of the well-known lectin families were first isolated from mushrooms. For example, the fungal fruit body lectin family was established based on the Xerocomus chrysenteron XCL protein, which was suggested to have insecticidal activity (Brick et al. 2004). Besides interest in their biotechnological applications, lectins have been in the focus of research on fruiting bodies. Early reports identified specific hyphal knot and primordium upregulated galectins (Cgl1 and Cgl2) in C. cinerea (Boulianne et al. 2000, Bertossa et al. 2004), which were initially postulated to be cell-cell adhesive or communication related molecules that may bind basidiolipids (Walser et al. 2005). Simultaneous silencing of the cgl1 and cgl2 genes did, however, not disturb fruiting, arguing against his hypothesis (Wälti et al. 2006). The lectins were rather shown to inhibit the development of insect and nematode larvae, in nematodes via binding to a specific N-glycan epitope, suggesting a function in defence against predators (Butschi et al. 2010, Bleuler-MartÍnez et al. 2011). Both genes were found upregulated in primordia, relative to vegetative mycelium in recent RNA-Seq based studies (cgl1 = C. cinerea 473274, cgl2 = C. cinerea 488611). Later, Wälti et al. (2008) described cgl3 (=C. cinerea 539794), which resembles galectins, but binds chitooligosaccharides and had no effect on larval development of insects and nematodes. Beta-glucan binding lectins of Serepindita indica may be involved in fungal cell wall remodelling at the plant-fungal interface (Wawra et al. 2019). Several other recent reports of individual lectins from a diverse set of medicinal or edible mushrooms is available but their detailed discussion is beyond the scope of this paper. Lectins are also involved in Ascomycota fruiting body (perithecium) formation and interestingly, the gene described in Podospora anserina showed highest similarity to Xerocomus lectins (Nowrousian & Cebula 2005). Several lectin families are overrepresented in mushroom-forming fungi (Krizsán et al. 2019), suggesting key roles in their life cycle.
‘Lectomes’ evolve fast (like other defence genes) both in terms of sequence and gene content. Most lectin families a phylogenetically patchily distributed in mushroom forming fungi, with the probable sole exception of ricin-B lectins, which occur in all examined species. It has been suggested that the fast gene turnover can contribute to the expression of defence gene cocktails by the fungus, which is advantageous and that horizontal gene transfer from bacteria can provide a mechanistic explanation for the phylogenetic patchiness of several lectin families (Moran et al. 2012, Künzler 2015).
We here provide an overview of lectin copy numbers in the examined Agaricomycetes in the context of 8 main families, but note that more recent classifications (Lebreton et al. 2021) significantly reorganized lectin families and may provide a more nuanced view on lectin repertoires. Lectin copy numbers and expression patterns were summarised in the context of 6 Agaricomycetes species by Krizsán et al. (2019) with lectins showing the RicinB fold (which includes protease inhibitors too, see above and Sabotič et al. 2016) being most diverse and present in all species, whereas other families are patchier. Nevertheless, all lectin families had developmentally regulated genes, often showing strong induction in primordium stages.
As noted earlier (e.g., Künzler 2015), lectins are poorly conserved and hence mostly did not form conserved orthogroups in our analysis. Only two CDE orthogroups, all belonging to the RicinB family were detected (C. cinerea protein ID: 495335, 353931) (Supplementary Table S34). Both orthogroups contained secreted proteins. Expression patterns were poorly conserved within these orthogroups, but we note that each had FB-init genes in several species, indicating that these secreted Ricin-B family members are involved in the initiation of fruiting body development.
Other cell surface proteins (HsbA, Con6, PriA, AA7, CFEM, wax synthases, fasciclins, peptidases, intradiol ring cleavage dioxygenase)
The HsbA family comprises poorly known cell wall galactomannoproteins that includes HsbA from Aspergillus oryzae (Ohtaki et al. 2006), a Magnaporthe homolog (Soanes et al. 2012) as well as genes named Mp1 in Talaromyces marneffei (Cao et al. 1998) and in Aspergillus fumigatus (Yuen et al. 2001). Detailed functional information is not available for either of these genes. The family contains GPI-anchored cell surface proteins that can bind to various surfaces and was circumscribed in the context of appressorium attachment and lignocellulose degradation in Ascomycota. The common denominator between these circumstances may be the need for adhesion of hyphae to various surfaces, enabling secreted lytic enzymes to exert their action. In the context of fruiting bodies, the family was first reported in fruiting bodies of A. ostoyae (Sipos et al. 2017). In our current analysis, we detected 0–3 members of the family in Agaricomycetes (missing from L. tigrinus, Ph. chrysosporium, A. ampla. S. commune), all of which were developmentally expressed. The family did not form CDE orthogroups. Expression of members of this family showed significant dynamics (>100-fold), mostly with gill- or cap-specific expression peaks late during development. Functional analysis of this gene family in mushroom-forming fungi would be very interesting. If, as speculated above, the family is related to adhesion, then it may provide a missing link to adhesion in complex multicellularity in mushrooms.
The Con6 protein family include small hydrophilic peptides that were first discovered as conidium-specific proteins in N. crassa and later shown in A. nidulans (ConF and ConJ) to be responsible for desiccation resistance of asexual spores (Suzuki et al. 2013). The family also includes the meiotically expressed Sch. pombe gene cum1. The Con6 family is conserved in Agaricomycetes, with 1–7 copies in the examined genomes, most of which are developmentally regulated. They showed two major expression patterns in all species: (i) strong induction in mature gills or combined gill+cap samples and/or (ii) upregulation in primordia relative to vegetative mycelia (Fig. 12, Supplementary Fig. S8). For some genes both for others only one of these expression patterns were observable. The family seems poorly conserved, nevertheless, they formed three CDE orthogroups (represented by C. cinerea 470234, 186809, S. commune 2624349) (Supplementary Table S35). The gill-specific upregulation, combined with the conidium-specific expression of the gene family in filamentous Ascomycota, suggests that Con6 proteins may be involved in producing a spore-specific protein coating in these species. The orthogroup represented by C. cinerea 186809 showed upregulation in primordia of most species and has low expression in vegetative mycelia and later developmental stages. The strong upregulation of several Con6-encoding genes in primordia relative to vegetative mycelia in virtually all species suggests that it has general roles in fruiting bodies as well, possibly in regulating hydrophobicity/hydrophilicity of the cell surface, although this requires further research.
Fig. 12.

Expression heatmap of Con6 family cell surface protein-encoding genes in C. cinerea. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively. Developmental stages are abbreviated as follows: VM – vegetative mycelium, H – hyphal knot, P1 – stage 1 primordium, P2 – stage 2 primordium, YFB.C – young fruiting body cap, YFB.L – young fruiting body gills, YFB.S – young fruiting body stipe, FB.C – mature fruiting body cap (including gills), FB.S – mature fruiting body stipe.
Wax synthases are long-chain alcohol O-fatty-acyltransferases that synthesize wax esters, a typical constituent of the cuticula of plants or the sebum of the animals (Li et al. 2008). To our best knowledge, only Krizsán et al. (2019) indicated developmental expression for this family previously.
We detected 4–16 wax synthases in the examined Agaricomycetes, the fewest in A. bisporus, but found no CDE orthogroups, suggesting a high duplication rate in the family. However, each species (except Pt. gracilis and A. bisporus) had 1–2 FB-init wax synthase genes, which suggests fruiting body specific roles for these genes. We observed considerable (>100-fold) upregulation compared to vegetative mycelia in many cases (Fig. 13, Supplementary Fig. S9). Given the widespread upregulation of wax synthases in mushroom-forming fungi, it is possible that they produce a wax-like layer on the surface of fruiting body cells. On plants, wax-coating of leaves serves as a defence system that inhibits penetration of pathogenic fungi and water-sealing (Lewandowska et al. 2020). Products of fungal wax synthases, although unknown at the moment, could have similar properties in fruiting bodies, worth investigating in the future.
Fig. 13.

Expression heatmap of putative wax synthase encoding genes in C. cinerea. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively. Developmental stages are abbreviated as follows: VM – vegetative mycelium, H – hyphal knot, P1 – stage 1 primordium, P2 – stage 2 primordium, YFB.C – young fruiting body cap, YFB.L – young fruiting body gills, YFB.S – young fruiting body stipe, FB.C – mature fruiting body cap (including gills), FB.S – mature fruiting body stipe.
PriA family - this family comprises secreted cell surface proteins first described as a primordium-induced protein in L. edodes (hence its name) (Kajiwara et al. 1992). The gene has been quite extensively studied in L. edodes (Ishizaki & Shishido 2000, Hirano et al. 2004), where a link to zinc homeostasis has been suggested. It was also detected as developmentally regulated in F. velutipes (Liu et al. 2020b), P. eryngii (Fu et al. 2017), L. edodes (Kajiwara et al. 1992) and in six species by Krizsán et al. (2019). The family also includes the virulence-related secreted adhesin Cfl1 of C. neoformans, which is highly induced during sexual development and has been thoroughly researched in C. neoformans (Wang et al. 2012, 2013). Cfl1 has been suggested to act as a morphogen that induces neighbouring cells to phenocopy the producing cells’ morphology (Wang et al. 2013). Cfl1 has 4 homologs in C. neoformans, which have stage-specific expression including Dha1, which is primarily expressed in basidia (Gyawali et al. 2017). The family is present in 0–4 copies in the Agaricomycetes and formed two CDE orthogroups. The first of these (C. cinerea protein ID: 359866) (Supplementary Table S35) showed a very conserved, mycelium biased expression; it was highly expressed in vegetative mycelia and the first primordium stage, then virtually unexpressed in all later stages in A. ampla S. commune, M. kentingensis and P. ostreatus. In A. ostoyae expression dropped immediately after the mycelium sample. The second CDE orthogroup (represented by C. cinerea 419688) contains best hits of C. neoformans Dha1 and showed a peak expression in gills and caps of C. cinerea, A. bisporus, C. aegerita, L. edodes and late stages of S. commune and A. ampla. At the same time, these genes were significantly upregulated (up to 50–100x) in primordia relative to vegetative mycelium. In L. edodes, our RNA-Seq based observations are consistent with the first reports of the PriA gene as a primordium-upregulated one (Kajiwara et al. 1992). The third orthogroup (represented by S. commune 2681702), was present in only 8 species, and was FB-init in A. ampla, A. ostoyae, P. ostreatus, M. kentingensis and S. commune (Supplementary Table S35). In summary, the PriA family appears to be a very interesting family from the perspective of fruiting body development. In C. neoformans adhesive and signalling roles have been suggested for the family, although exact mechanisms of function are still unknown.
Aspartic peptidase A1 - This family is a versatile group of endopeptidases that has undergone a characteristic expansion in fungi (Revuelta et al. 2014). They were reported to be involved in pathogenesis, nutrition, cell wall maintenance and a range of other functions. The best researched family of A1 aspartic peptidases are yapsins, which are involved in cell wall assembly and remodelling, although their function is not clear; limited evidence suggests they proteolytically process secreted (cell wall) proteins (Gagnon-Arsenault et al. 2006, Gow et al. 2017, Lozančić et al. 2021). Based on functional knowledge on yapsins, and the abundance of GPI-anchored A1 aspartic peptidases in fungi, we discuss this family in the cell surface section, despite almost certainly not all of them are related to the cell wall. The family has been reported to be upregulated in fruiting bodies by several studies previously (Rahmad et al. 2014, Song et al. 2018, Krizsán et al. 2019). Aspartic peptidase A1 family was found as 15–60 copies in Agaricomycetes genomes. We detected 3 CDE orthogroups and several species-specifically developmentally expressed genes (Supplementary Table S35), though these did not display strong expression conservation in the examined species. Nevertheless, the developmental expression of several members of this family, combined with evidence on yeast yapsins (Gagnon-Arsenault et al. 2006) and the secreted nature of these proteins suggest a cell wall related role for aspartic peptidases which may be worth investigating. Of note, we detected further CDE orthogroups of various peptidase families (M20, M28, M16, S10), which may also be involved in fruiting body development (Supplementary Table S35), but our functional knowledge at the moment is too limited to speculate about their roles. Other peptidase families that were reported in mushroom-forming fungi include the metalloprotease M43 family, which was reported from P. ostreatus (Joh et al. 2004) and has a strong (>700x) induction upon fruiting in the data published by Merényi et al. (2022).
AA7 family - One of the CDE orthogroups (represented by C. cinerea 501135) (Supplementary Table S35) were top hits of Fusarium graminearum ChitO, the only experimentally examined member of the family known to us (Heuts et al. 2007), which oxidizes chitooligosaccharide species. AA7 comprises oligosaccharide oxidizing flavo-enzymes which also contain a Berberine-like domain and produce H2O2 during their action. They have been speculated to be involved in the detoxification of lignocellulose degradation intermediates and in the production of various secondary metabolites (Levasseur et al. 2013). Based on the activity of their homologs and their expression in fruiting bodies, it is likely that these AA7 family members are active on chitin, although their exact roles remain obscure for now.
CFEM domain proteins - We detected 2 CDE orthogroups of CFEM domain containing proteins (Supplementary Table S36). Homologous CFEM domain proteins may be involved in defence, pathogenicity, host-pathogen interactions or iron uptake, as is the rbt5 family of Candida spp. (Bairwa et al. 2017) In C. cinerea certain mycelium expressed CFEM domain proteins were reported to be involved in defence (Plaza et al. 2014). The detected CDE orthogroups comprise cell surface proteins with unknown functions, though they might be involved in defence, however, evidence for that is missing at the moment.
Fasciclins are adhesive cell surface proteins that facilitate the attachment of cells to various surfaces. The few fungal mentions include a report of a role in the adhesion of Magnaporthe oryzae to host surfaces (Liu et al. 2009) as well as a primordium-upregulated gene in L. edodes (Le.flp1) (Miyazaki et al. 2007). Recently, fasciclins were hypothesized to have been co-opted for fruiting body development independently in both the Agarico- and Pezizomycotina (Merényi et al. 2020). We identified 2–8 fasciclin-encoding genes in the examined Agaricomycetes. All examined species have developmentally expressed fascicling genes, although these formed only small CDE orthogroups (Supplementary Table S35). Several genes showed an upregulation in primordia relative to vegetative mycelium (as reported in L. edodes), suggesting that this family has widespread roles in mushroom formation, possibly by facilitating adhesion. We found that the sequence deposited by Miyazaki et al. (2007) (Accession number: AB287432) as well as ascomycete homologs to which they analysed are cupredoxins, not fasciclins, leaving doubts as to the identity of Le.flp1. Nevertheless, this does not undermine the observation that fasciclin encoding genes are widespread developmentally expressed genes in mushroom-forming fungi, probably with roles in fruiting body development.
Intradiol ring cleavage dioxygenases catalyse a ring-opening step in the degradation of aromatic compounds, and were hypothesized to be involved in xenobiotic detoxification or the utilization of aromatic compounds as carbon sources (Semana & Powlowski 2019). However, the physiological role of intradiol ring cleavage dioxygenases in fungi is not known. The only previous mention of a potential relationship to fruiting bodies reported developmental regulation in 4 out of 6 species (Krizsán et al. 2019). We identified 2–7 copies (28 in A. ostoyae) in the examined Agaricomycetes, with 1–3 developmentally expressed genes in most of the species that, however, did not form conserved orthogroups. Members of the family showed a significant upregulation in primordia relative to vegetative mycelium in A. ampla, S. commune, C. cinerea, A. ostoyae as well as a strong upregulation (>100fold) during late development of C. neoformans. Two genes show a very distinct upregulation in young fruiting body caps of C. cinerea. No developmentally expressed genes were found in Ph. chrysosporium and M. kentingensis. In summary, intradiol ring cleavage dioxygenases might be important genes during fruiting body development of several species; they might degrade aromatic compounds during fruiting; however, the identity of those compounds are unknown at the moment.
Defence
Agaricomycete fruiting bodies (mushrooms) are, due to the concentration of biomass, attractive targets of predators, parasites and pathogens. Accordingly, these structures are known to be subject to grazing by slugs, to infestations with larvae of phorid and sciarid flies and to infections by bacteria and molds (Wood et al. 2004, Krivosheina 2008, Lakkireddy et al. 2020, Hamidizade et al. 2020). Since mushrooms are the sexual reproduction organs and a considerable investment in terms of resources and energy, Agaricomycetes have evolved several strategies for the protection and defence of these structures (Künzler 2018). Such strategies include physical barriers (Varga et al. 2021) and chemical defence i.e. the production of molecules (toxins) impairing the growth, development and/or viability of antagonists (Spiteller 2008, Lim & Keller 2014, Stadler & Hoffmeister 2015).
Using saprobic Agaricomycete species, whose sexual cycles can be completed under laboratory conditions, it could be demonstrated that the production of these toxins occurs axenically i.e. in the absence of the antagonist (Plaza et al. 2014, Tayyrov et al. 2019). This axenic toxin production is spatiotemporally regulated in that the toxin-encoding genes are expressed solely during fruiting body formation and in the fruiting body tissue. Some of these genes belong to the most highly upregulated genes of the entire genome during fruiting body formation. This expression pattern results in an efficient, constitutive protection of the fruiting body because at least some toxins are already in place in case the structure is attacked by an antagonist. It is unclear whether the chemical defence of the Agaricomycete fruiting body can be fortified by the induction of toxin production in response to environmental factors e.g., the presence of antagonists. Antagonist-specific induction of antibacterial and nematotoxic proteins upon challenge with bacteria and fungivorous nematodes, respectively, has however, recently been reported for the vegetative mycelium of C. cinerea (Kombrink et al. 2019, Tayyrov et al. 2019).
Toxins of Agaricomycete fruiting bodies include small molecules (secondary metabolites) (Kües et al. 2018), peptides (ribosomally or non-ribosomally synthesized) and proteins, all of which act by binding to and (in case of toxins with enzymatic activity) converting specific target molecules in the antagonists. In the following, we will summarise the current knowledge about the structure, function, biosynthesis, regulation and distribution of Agaricomycete fruiting body toxins. In accordance with the focus of this review on genomics, we restrict our summary to compounds where the biosynthetic genes have been identified. We also make references to available gene expression data, but it must be noted that most toxin-encoding genes evolve fast and thus they do not easily conform to orthology-based categorizations that are central to the rest of this paper.
Small molecules - The structurally related alkaloids ibotenic acid and muscimol of the fly agaric Amanita muscaria are two well-known examples of defensive secondary metabolites of Agaricomycete fruiting bodies (Michelot & Melendez-Howell 2003). The toxicity of these compounds toward flies gave rise to the name of the producer fungus. Their bioactivity is due to their structural similarity to the neurotransmitters glutamate and GABA and their binding to respective receptors in the brains of both invertebrates and vertebrates. The recent discovery of the biosynthetic gene cluster for the compounds revealed that their biosynthesis initiates from glutamate and makes it possible to identify additional Amanita species producing these compounds on a genetic basis (Obermaier & Müller 2020). Another psychotropic alkaloid produced by Agaricomycete fruiting bodies is psilocybin produced by several Psilocybe species (Fricke et al. 2017, Torrens-Spence et al. 2018). This tryptophan derivative is an agonist of serotonergic receptors and is currently tested for treatment of depressions (Tullis 2021). The transcriptional analysis of the recently identified biosynthetic genes confirms its fruiting body-specific production (Demmler et al. 2020). The blueing reaction of psilocybin-producing fruiting bodies upon injury has recently been demonstrated to be due to enzyme-mediated oxidative oligomerization of psilocybin suggesting that at least part of the defence function of this compound may be based on optical deterrence (Lenz et al. 2020). Wound-activation by injury (oxidation) has been reported for other secondary metabolites of Agaricomycete fruiting bodies (Stadler & Sterner 1998, Spiteller 2008) but the genetic basis of these compounds and reactions is often not known.
Defence peptides - The bicyclic octapeptide α-amanitin produced by some Amanita, Galerina, Conocybe and Lepiota species is probably the most famous toxin of Agaricomycete fruiting bodies (Walton 2018). The thermal stability and the very low lethal dose (0.1 mg/kg body weight) of the peptide have led to the common names ‘death cap’ (Amanita phalloides) or ‘destroying angel’ (Amanita bisporigera) of α-amanitin producers. Upon ingestion, the peptide is released from the fruiting body tissue and able to passively enter the intestinal epithelial cells where it binds and inactivates the essential enzyme RNA polymerase II in the nucleus (Liu et al. 2018). The fungus protects itself from the toxin by expressing an insensitive variant of the target enzyme (Horgen et al. 1978). In 2007, Walton et al. (2007) demonstrated that α-amanitin and its relative phallacidin are derived from ribosomally synthesized precursor peptides with common flanking sequences (MSDIN) by the action of a dual function prolyloligopeptidase-macrocyclase (POPB) (Hallen et al. 2007, Luo et al. 2012). Genome surveys of above Agaricomycete species revealed a large family of peptide precursor genes differing in the core peptide sequence and, thus, coding for α-amanitin- and phallacidin-related, but structurally different peptides, referred to as cycloamanides, whose function is unknown (Pulman et al. 2016, Luo et al. 2018. 2018, He et al. 2020, Zhou et al. 2021). Localization of POPB expression confirmed the fruiting body-specific expression of α-amanitin and phallacidin (Luo et al. 2010, Zhang et al. 2018).
Above described secondary metabolites and peptides have received considerable attention, mainly due to their activity towards humans. During the last two decades of research on fungal defence, it has become evident, however, that a large part of the chemical defence of Agaricomycete fruiting bodies, in particular against invertebrate predators and parasites, is based on protein toxins (Tayyrov et al. 2018, Sabotič et al. 2016, Wang et al. 2012)). These toxins have received less attention because their activity is usually lost upon boiling and is specific for a particular group of invertebrates. In addition, based on their fruiting body-specific expression, some of these proteins have been implicated in fruiting body formation rather than defence (see the example of C. cinerea galectins CGL1 and CGL2 mentioned above), which has delayed their recognition and functional analysis as toxins (Berne et al. 2007, Paneske et al. 2019). Most of these protein toxins are stored intracellularly and released upon ingestion of the fruiting body tissue by fungivores (Künzler Künzler 2015, Künzler 2018). Once released, they interact with the digestive tract of the fungivore. Characterised classes of protein toxins from Agaricomycete fruiting bodies include lectins (Goldstein & Winter 2007, Bleuler-Martínez et al. 2011, Varriot et al. 2013), protease inhibitors (Žurga et al. 2015, Sabotič et al. 2019), pore-forming toxins (PFTs) (Anderluh & Maček 2003, Mancheño et al. 2010, Panevska et al. 2020), ribotoxins (Tayyrov et al. 2019b, Ragucci et al. 2021) and biotin-binding proteins (Takakura et al. 2009, Bleuler-Martinez et al. 2012).
Defence related lectins - Fruiting body lectins have long been used as tools in glycobiology without addressing their biological function (Kobayashi et al. 2014). In the meantime, it has been established that these proteins bind to specific glycoepitopes on the epithelial cells of the digestive tract of the fungivore which can ultimately lead to disruption of the structure and function of this tissue (Stutz et al. 2015). The mechanism of lectin-mediated toxicity is not clear yet but, in case of lectins without additional functional domain (hololectins), the bioactivity appears to depend on the crosslinking of protein-bound glycoepitopes on the epithelial cell surface (Bleuler-Martinez et al. 2017), whereas in case of lectins with additional functional domains (chimerolectins), the bioactivity appears to depend on binding to lipid-bound glycoepitopes and endocytosis (Wohlschlager et al. 2021, Juillot et al. 2016). Fruiting body lectins can adopt various folds and carbohydrate specificities amongst which the RicinB-like (β-trefoil) fold and fucoside-binding, respectively, are most common. A prototypic example for such a fruiting body lectin is the hololectin CCL2 (C. cinerea protein ID: 408852) from the model Agaricomycete C. cinerea which is present in other Agaricomycete species, adopts a RicinB-like fold, forms a dimer, binds to a specific fucoside present on nematode N-glycan cores and is toxic towards bacterivorous and fungivorous nematodes (Schubert et al. 2012, Bleuler-Martinez et al. 2017, Tayyrov et al. 2018). A summary of diverse entomo- and nematotoxic lectins from fruiting bodies has been given by Sabotič et al. (2016). Our data confirm previous reports on the upregulation (>150-fold) of CCL1 (C. cinerea protein ID: 456849) and CCL2 (C. cinerea protein ID: 408852) in primordia (Supplementary Table S36).
The RicinB-like (beta-trefoil) fold is also adopted by protease-inhibitors isolated from Agaricomycete fruiting bodies (Sabotič et al. 2016). These proteins were shown to inhibit serine and cysteine proteases of various insect larvae. A prototypic example for this type of fruiting body toxin is Clitocypin, an insecticidal cysteine-protease inhibitor isolated from the Agaricomycete Clitocybe nebularis (Pohleven et al. 2011, Šmid et al. 2015). Of the examined species, the genomes of C. aegerita and L. bicolor encode 1 and 20 putative clitocypins, respectively. Our data identified a putatively defence-related RicinB protein orthogroup (represented by C. cinerea 485770) (Supplementary Table S36). Another example is the six-bladed beta-propeller lectin Tectonin2 from L. bicolor whose production is induced in fruiting bodies and ectomycorrhizae (Martin et al. 2008). This lectin forms tetramers harbouring 24 binding sites for O-methylated glycoepitopes and exhibits toxicity towards nematodes and agglutination of bacteria displaying respective glycoepitopes on their cell surfaces (Wohlschlager et al. 2014, Sommer et al. 2018). We detected tectonin homologs in the genomes of L. bicolor and C. aegerita.
Pore-forming toxins - A common and probably very ancient class of protein toxins of Agaricomycete fruiting bodies are pore-forming toxins (PFTs) Dal Peraro et al. 2016). These toxins are divided into α- and β-PFTs depending on whether the predominant secondary structure motif is an α-helix or β-sheet. Within the producing cell, these proteins are usually monomeric and soluble, but upon release and binding to a target cell membrane, they undergo a dramatic conformational change. This change in conformation leads to oligomerization and insertion of the protein into the membrane. The result of this complex process is a proteinaceous pore that leads to lysis of the target cell. Among PFTs occurring in Agaricomycete fruiting bodies, members of the aerolysin and Membrane Attack Complex PerForin/Cholesterol Dependent Cytolysin (MACPF/CDC) β-PFT superfamilies have been characterised most extensively (Reboul et al. 2016, Cirauqui et al. 2017). Examples for aerolysins are the hemolytic Laetiporus sulphureus lectin (LSL) (Mancheño et al. 2010), a homologous and recently identified nematotoxic protein, CC1G_11805 (C. cinerea protein ID: 546868) and its two paralogs, from C. cinerea (Plaza et al. 2014) and the hemolytic flammutoxin from F. velutipes (Tomita et al. 2004). Pri1, a primordium-expressed gene in C. aegerita (Agrae_CAA7262647) also belongs to pore-forming toxins (Fernandez Espinar et al. 1997). The MACPF/CDC superfamily is represented by the insecticidal pleurotolysin complex, consisting of PlyA and PlyB, from the edible mushroom P. ostreatus (Ota et al. 2014, Novak et al. 2015, Lukoyanova et al. 2015, Panevska et al. 2019). The main representatives of the two β-PFT superfamilies differ in the specificity determinant of the target membrane and the dimensions of the formed membrane pore. Whereas LSL likely binds to lactose-containing glycoepitopes (as part of glycolipids or GPI-anchored glycoproteins) on the target membrane via an N-terminal RicinB-type (β-trefoil) lectin domain and forms homomeric transmembrane β-barrels (pores) of seven or eight protomers with an inner diameter of ~4 nm (Angulo et al. 2011, Szczesny et al. 2011), pleurotolysin PlyA binds to sphingophospholipids (sphingomyelins) of cholesterol-rich membranes and directs PlyB to the formation of transmembrane β-barrels (pores) consisting of 13 heteromeric protomers and an inner diameter of ~ 8 nm (Novak et al. 2020).
Ribotoxins are ribonucleases cleaving a single phosphodiester bond in the universally conserved sarcin/ricin loop (SRL) of 23S or 28S rRNA in intact prokaryotic or eukaryotic ribosomes, respectively (Olombrada et al. 2017b). Upon this action, the ribosome is unable to interact with translation elongation factors and translation is stalled. The SRL is named after α-sarcin, one of the best characterised ribotoxins secreted by the ascomycetous mold Aspergillus giganteus (Olombrada et al. 2017b). Other fungal ribotoxins have been isolated from ascomycetous entomopathogens and are generally highly active against insects (Viegas et al. 2009, Olombrada et al. 2017a). Recently, the first basidiomycetous ribotoxin, ageritin (Agt1), from fruiting bodies of the edible mushroom C. aegerita was isolated (Landi et al. 2017). Heterologous expression of the respective cDNA in Escherichia coli and phylogenetic analysis showed that the protein is widespread but patchy among Agaricomycetes and displays entomotoxic activity similar to its ascomycetous counterparts (Tayyrov et al. 2019b). In contrast to its ascomycetous counterparts, however, ageritin does not contain a canonical signal peptide for secretion suggesting that the protein, as most protein toxins of Agaricomycete fruiting bodies, is stored intracellularly (Baglivo et al. 2020). In order to avoid damage to the ribosomes of the producing fungal cell, the protein might be sequestered into a specific subcellular compartment which might involve N-terminal truncation of the protein (Ragucci et al. 2021). The agt1 gene (C. aegerita CAA7259527) was induced considerably in primordia relative to vegetative mycelium in C. aegerita, based on data produced by Orban et al. (2021)
Tamavadins - Finally, defence proteins of some Agaricomycete fruiting bodies, similar to animals and bacteria, include proteins that are able to avidly bind and thus sequester biotin at equimolar concentrations (Green 1990). The prototype of these proteins, avidin, is thought to protect the eggs of birds from bacterial infections, but the protein and its relatives also exhibit broad and strong insecticidal activity (Christeller et al. 2010). A family of avidin-related biotin-binding proteins, tamavidin (called Tam1 and Tam2), was isolated from the edible mushroom Pleurotus cornucopiae (Tamogitake mushroom) and shown to possess very strong entomotoxic and nematotoxic activity (Takakura et al. 2009, Bleuler-Martinez et al. 2012). Amongst the fruiting body protein toxins tested against two fungivorous nematode species, tamavidin was the most effective one (Tayyrov et al. 2018). It is unclear how the producing cells shield their own biotin against this cytoplasmic protein.
Taken together, this data shows that the chemical armory of Agaricomycete fruiting bodies is very diverse, in terms of compound classes, molecular targets and antagonist specificity of the toxins, but three common themes can be recognized: (1) Toxin-encoding genes are phylogenetically patchy, with each family present in only a subset of species (2) protein toxins provide a large part of the chemical defence of these structures; and (3) intracellularly stored toxins directed against nematodes and arthropods are predominant. Latter finding may reflect the use of Agaricomycete fruiting bodies as habitat by these organisms (Ruess et al. 2005, Krivosheina 2008). It has even been postulated that some mycophagous Drosophila flies exploit fruiting body nematotoxins to purge themselves from insect-parasitic nematodes (Cevallos et al. 2017).
Secondary metabolism and natural product biosynthesis
Secondary metabolites are one of the most important classes of molecules in chemical warfare and developmental signalling in fungi (Spiteller 2015, Keller 2019, Gressler et al. 2021). Based on recent genome surveys it appears that the fungal secondary metabolome is extremely diverse and is a largely untapped resource of natural products and bioactive molecules (Keller 2019). This may be particularly true for the Agaricomycetes, despite several lines of evidence for a large diversity of secondary metabolites produced by Agaricomycetes (Spiteller 2015, Gressler et al. 2021). From the perspective of fruiting body formation, it is important to note that secondary metabolites can serve defence or repellent purposes in fruiting bodies (see chapter “Defense”) but can also influence developmental processes. For example, the deletion of a polyketide synthase gene resulted in malformed perithecia of the ascomycete S. macrospora (Schindler & Nowrousian 2014). The lack of coprinoferrin production in C. cinerea resulted in the loss of fruiting body formation (Tsunematsu et al. 2019).
Due to differences in basic genome organization between Asco- and Basidiomycota, the latter possess fewer secondary metabolism gene clusters, yet, according to a recent study some developmentally regulated genes formed ‘clusters’ in the genomes of 8 species and some of these ‘clusters’ overlapped with predicted biosynthetic gene clusters that might encode secondary metabolism genes (Merényi et al. 2022).
The number of functionally characterised secondary metabolism genes and linked natural products of Agaricomycetes is rapidly increasing. For example, ArmB and orsellinic acid in Armillaria spp. (Lackner et al. 2013), LpaA and laetiporic acid in Laetiporus sp. (Seibold et al. 2020), or St.pks1 and strobilurin in Strobilurus tenacellus (Nofiani et al. 2018) as well as numerous terpene synthases have been circumscribed in the past decade (reviewed in more detail by Gressler et al. 2021). Strobilurin derivatives are available commercially and are the world’s most sold fungicides (Bartlett et al. 2002). Siderophore-type secondary metabolites, coprinoferrin, ferrichrome A and basidioferrin in C. cinerea, Omphalotus olearius and Ceriporiopsis subvermispora, respectively, and their putative synthesizing genes were also identified. In C. cinerea cfp1 (C. cinerea protein ID: 354233) was reported to be responsible for coprinoferrin production (Tsunematsu et al. 2019), whereas in O. olearius a cluster composed of fso1, omo1 and ato1 (Welzel et al. 2005). Coprinoferrin proved necessary for fruiting body development, as the cfp1 mutant did not, only in the presence of exogenously administered coprinoferrin produce fruiting bodies. Contrary to expectations based on Ascomycota knowledge, deleting a homolog (C. cinerea protein ID: 357031) of A. nidulans LaeA did not compromise coprinoferrin production (Tsunematsu et al. 2019). Despite recent advances in understanding Agaricomycetes secondary metabolism, given the number of putative secondary metabolism genes encoded by fungal genomes, we are likely hardly scratching the surface of natural product diversity in mushroom-forming fungi.
Since many secondary metabolites specifically occur in fruiting bodies of Agaricomycetes, it is expected that their biosynthetic gene clusters may be developmentally regulated. However, secondary metabolism related genes evolve fast, which does not make them easily detectable in the orthology-based framework we use here. Nevertheless, we identified several secondary metabolism related genes that showed shared and conserved developmental expression profiles in the examined Agaricomycetes, although the natural products they produce are unknown in most cases.
Polyketide synthases (PKS) are large (often >1 500 amino acid) multi-domain enzymes that produce diverse secondary metabolites. They are widely distributed in fungi and are subject to intense research, mostly in the Ascomycota. Polyketide synthases and the few examples of known natural products of the Basidiomycota have recently been reviewed (Gressler et al. 2021). Basidiomycete PKSs mostly belong to type 1, which is divided into highly-, partially- and non-reducing PKSs. Hybrid nonribosomal peptide synthase-PKS genes have been shown to underlie fungal bioluminescence (Ke et al. 2020, Gressler et al. 2021).
We identified orthologs of previously described siderophore biosynthesis related genes in the examined Agaricomycetes. Members of the biosynthetic cluster described from O. olearius (Welzel et al. 2005) seem to be conserved, with the fso1 protein being orthologous to C. cinerea cfp1 (C. cinerea protein ID: 354233), whereas L-ornithine N (5)-monooxygenase omo1 and the N-acyltransferase ato1 seem single-copy proteins in Agaricomycetes genomes (represented by C. cinerea 492586 and 161175). These genes were not developmentally regulated and had highest expression in vegetative mycelia. We further identified a single CDE orthogroup of polyketide synthases (represented by C. cinerea 446258, Supplementary Table S37), which was developmentally regulated in 11/8 species (FC>2/4). These genes were stipe upregulated (albeit weakly) in most species (C. cinerea, P. ostreatus, M. kentingensis, A. ostoyae), although their expression also increased in mature caps of P. ostreatus, A. ostoyae, M. kentingensis relative to younger cap stages and in mature fruiting bodies of C. aegerita, A. ampla and S. commune relative to earlier stages. The C. cinerea gene shows peak expression in meiotic gill tissues, whereas in P. ostreatus and A. ostoyae, where gills and caps were sampled separately like in C. cinerea, highest expression was observed in caps but not in gills.
Terpene synthases have been a focus of secondary metabolism studies in mushroom-forming fungi. They encode highly promiscuous enzymes that can generate a large diversity of metabolites (Slot & Gluck-Thaler 2019) and thus provide the basis for fast adaptive evolution. Slot & Gluck-Thaler showed that morphologically complex fungi harbour more terpene synthases than simpler counterparts (Slot & Gluck-Thaler 2019). Similarly, terpene synthases were found to be strongly overrepresented in the genomes of Agaricomycetes compared to other fungi (FDR corrected p-value < 10-125) (Krizsán et al. 2019). Twelve sesquiterpene synthases (Fla1-12) were identified in F. velutipes (Tao et al. 2016), eleven in C. aegerita (Agr1-Agr11) (Zhang et al. 2020, 2021a) and in Omphalotus olearius (Omp1-10) (Wawrzyn et al. 2012) and five (Cop1-Cop6) in C. cinerea (Agger et al. 2009, Stöckli et al. 2019), with information on the potential metabolite products in many cases (e.g., viridiflorene/viridiflorol, Δ6-protoilludene, γ-cadinene, germacrene, lagopodin B).
Agger et al. identified six genes, Cop1–Cop6, related to terpene biosynthesis in C. cinerea (Agger et al. 2009). Of the genes reported by Agger et al, orthologs of Cop2 and Cop4 appear interesting from the perspective of mushroom development. Orthologs of Cop1 show an upregulation in primordia relative to vegetative mycelium in C. aegerita, A. ampla (only FC>2), L. bicolor, Ph. chrysosporium and were cap-upregulated in A. ostoyae, P. ostreatus, L. edodes and M. kentingensis. On the other hand, orthologs of Cop4 (C. cinerea protein ID: 30510) were FB-init in C. cinerea, C. aegerita, A. ampla, L. bicolor, L. edodes and P. ostreatus (Supplementary Table S37). Cop2 (C. cinerea protein ID: 443805) is only present in C. cinerea, A. bisporus and P. ostreatus and is upregulated in primordia relative to vegetative mycelium in all species, suggesting it functions during fruiting body initiation. Products of these genes were reported to as δ-cadinene and germacrene A, but the functions of these natural products remain unknown.
We found 13–28 (63 in A. ostoyae) putative terpene synthases in the examined Agaricomycetes genomes. Of these, several were developmentally regulated, showing one of two most typical expression profiles: (i) highest expression in vegetative mycelium or (ii) a strong induction in primordia and other fruiting stages relative to vegetative mycelium. The latter expression profile is reminiscent of that of typical defence genes (e.g., pore-forming toxins, see above), but it is compatible with several other putative functions as well. Terpene synthases formed three CDE orthogroups (Supplementary Table S37), two of these correspond to C. cinerea Cop1 (C. cinerea protein ID: 469738) and Cop4 (C. cinerea protein ID: 30510, contains C. aegerita Agr4 also), discussed above. The third CDE orthogroup (represented by C. cinerea 488324) is developmentally regulated in 8/6 species, but shows mixed expression profiles.
Taken together, terpene synthases might participate in important fruiting body functions, including events around the initiation of primordium development, yet their exact functions, whether the production of defence/repellent metabolites and/or compounds for communication, remain unknown for the moment.
Phenylalanine ammonia-lyases - We detected two highly conserved CDE orthogroups of phenylalanine ammonia-lyases (PAL), one of which (C. cinerea protein ID: 386671, ortholog of pal1 of P. ostreatus) is developmentally regulated in 11/9 species, the other (C. cinerea protein ID: 361541, ortholog of pal2 of P. ostreatus, Hou et al. 2019) is in 8/5 (out of 9 species in which present) (Supplementary Table S37). PAL enzymes are best known from plants, where they are part of the phenylpropanoid pathway, which is involved in the production of diverse secondary metabolites, associated with defence, lignin and flavonoid biosynthesis (Dixon et al. 2002). In fungi, phenylalanine ammonia-lyases are discussed mostly in the context of catabolism of phenylalanine to cinnamic acid (Hyun et al. 2011), although some reports of the production of diverse compounds have been published (Hyun et al. 2011). Recently, an association with fruiting body formation has been reported in F. velutipes (Yun et al. 2015) and P. ostreatus (Hou et al. 2019).
Our analyses revealed two PAL-encoding genes in most of the examined Agaricomycetes (up to 4 in A. ostoyae). Expression profiles of genes within the two CDE orthogroups were not conserved, although several genes showed an upregulation in primordia (e.g., both genes in C. cinerea). At the moment the physiological role of PALs is unknown, to our best knowledge, in any fungus. They could be involved in the production of defence-related metabolites, as in plants, or other fruiting body related functionalities.
Cytoskeleton
A plastic cytoskeleton is a key factor for Eukaryotic Cell shape regulation and has been named one of the prerequisites for diversification of size, form and function in multicellular eukaryotes (Knoll 2011). It is involved in the regulation of diverse morphogenetic processes in fungi, including hyphal growth, conidiation, biofilm formation, dimorphic switching, to name a few (Riquelme et al. 2018). In line with this, although at a somewhat limited scale, evidence is available for the role of cytoskeleton rearrangement in fruiting body development. Evidence from Gene Ontology analyses pointed at differential cytoskeletal gene expression in P. microcarpus (Pereira et al. 2017). Microscopy observations provided evidence for a key role of cytoskeletal elements during the development of mushroom-forming fungi. For example, longitudinally oriented microtubules were observed in the sterigmata during spore formation in C. cinerea, along which Golgi vesicles carry carbohydrates to the developing spore and spore wall (Kües & Liu 2000).
In terms of expression levels, we found only limited dynamics in cytoskeletal proteins, likely reflecting the need for continuous supply of its components for the cell. Cheng et al. reported an upregulation of tubulin and tubulin-specific chaperone genes in primordium stages of C. cinerea (Cheng et al. 2013) using 5’-SAGE technology. Although newer data did not confirm these observations (Muraguchi et al. 2015, Krizsán et al. 2019), both recent RNA-Seq studies reported upregulation of these genes in stipes of C. cinerea.
Following these lines of evidence, we annotated the main cytoskeletal protein groups in C. cinerea based on homology and domain composition and checked their orthologs’ expression patterns across mushroom-forming fungi. We grouped 67 cytoskeletal and associated protein coding genes into the following categories (Supplementary Table S38): actin, actin nucleation, actin filament organization, actin-based motor protein, microtubule, microtubule heterodimerization, microtubule-based motor protein, regulation of microtubule-based motor proteins. Copy numbers in these categories agreed with previous annotations, including 2 copies of formins per species and alpha-tubulin gene duplication in the common ancestor of Basidiomycota (Zhao et al. 2014). Of cytoskeletal genes, only kinesin genes and the Dynactin complex showed consistent expression dynamics. In both cases, expression peaks were observed in young fruiting body gills and stipes in C. cinerea (Fig. 14, Supplementary Fig. S10).
Fig. 14.

Expression heatmap of kinesin genes and members of the Dynactin complex in C. cinerea. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively. Developmental stages are abbreviated as follows: VM – vegetative mycelium, H – hyphal knot, P1 – stage 1 primordium, P2 – stage 2 primordium, YFB.C – young fruiting body cap, YFB.L – young fruiting body gills, YFB.S – young fruiting body stipe, FB.C – mature fruiting body cap (including gills), FB.S – mature fruiting body stipe.
Tubulin gene expression (alpha1-, alpha2-, beta-tubulin and tubulin chaperone) was previously reported to be regulated during early fruiting events of C. cinerea, with upregulation in stage 1 primordia based on 5’-SAGE data (Cheng et al. 2013). Our data on the corresponding genes revealed that the three of these, alpha1- (C. cinerea protein ID: 176636), beta-tubulin (C. cinerea protein ID: 393528) and tubulin chaperone (C. cinerea protein ID: 413977) had constant expression in primordia, but show expression peak in young fruiting body stipes, whereas alpha2-tubulin (C. cinerea protein ID: 359665) was upregulated in young fruiting body caps. These expression patterns were not consistent across species (Supplementary Table S38) and possibly reflect the fast stipe elongation of C. cinerea.
Septins - Septins form a highly conserved family of proteins involved in morphogenesis, development and in generating cell asymmetries in fungi and in eukaryotes in general (Gladfelter 2006, Khan et al. 2015). They have been shown to be responsible for morphogenesis in a number of model fungi, such as bud emergence in S. cerevisiae, septum formation in hyphae or appressorium formation in Magnaporthe oryzae, among others (Gladfelter 2006). Their broad involvement in morphogenesis and limited evidence from fruiting body forming fungi suggest that septins have important, although to date underappreciated, roles in fruiting body development. For example, they have been reported in stipe elongation in C. cinerea (Shioya et al. 2013).
We annotated septins based on their domain composition: the combination of two IPR domain terms (IPR030379, IPR016491) are diagnostic for fungal septins. Most mushroom-forming fungi have seven septin genes (C. cinerea protein ID: 367587, 416864 [cdc3, = A. nidulans AspE, =AspB], 103773, 415573 [top hit of A. nidulans AspE], 103809, 455217, 481878), with some having only 6 while a few having 8, which is similar to the septin repertoires seen in filamentous fungi and yeasts (Douglas et al. 2005).
Septins are highly variable genes, and did not form conserved orthogroups, but nevertheless maintain some functional conservation in mushroom-forming fungi. Two orthogroups of septins showed consistent upregulation at the initiation of fruiting body development, in the very first primordium stages in all species in which they are present (Krizsán et al. 2019, Merényi et al. 2022). One of these is the top hit of AspE of A. nidulans, which is involved in conidiation and spore production (Hernández-Rodríguez et al. 2014). Septin expression seems to be highly induced in stipes in C. cinerea, M. kentingensis, A. ostoyae, but following a different expression tendency in P. ostreatus (mostly in primordia) and A. bisporus (mostly in young fruiting bodies). The stipe induced septins included Cc.cdc3 of C. cinerea, which has been shown to be involved in stipe elongation (Shioya et al. 2013). The ortholog of C. cinerea cdc3 was also mostly stipe-induced in A. bisporus (Lu et al. 2019). Muraguchi et al. reported a downregulation of septin expression from the 4_0hrPri primordia (approximately coinciding with stage 2 primordia as defined by Kües 2000, Krizsán et al. 2019, to the 5_12hrPri stage of C. cinerea by Muraguchi et al. 2015).
The limited diversity of septins raises the question how they can fulfil the diverse roles they have in fungal morphogenesis. Although most septins had peak expression in stipes of mushroom-forming fungi (except in certain species) their expression level was dynamically changing in other tissues and from one developmental stage to the other as well, suggesting that there is still a long way to uncover their roles in fruiting body development. Understanding their protein-protein interaction networks is a promising future avenue of research.
Ubiquitin-proteasome system
Cop9 signalosome
The Cop9 signalosome is a protein complex conserved across all eukaryotes, which catalyses the deactivation of cullin-RING ubiquitin ligase (CRL) complexes by removing the Nedd8 protein from the cullin subunit of the complex (deneddylation reaction). As such, it is an important component of various signal transduction pathways and is involved primarily in multicellular developmental processes. In fungi, it was found dispensable for vegetative growth, but necessary for sexual fruiting body formation (Busch et al. 2003, Braus et al. 2010), secondary metabolism and it was reported to interact with the Velvet complex (von Zeska Kress et al. 2012, Meister et al. 2019). Recognition of the COP9 signalosome (CSN) as an important player in fruiting body formation started with genetic screens that identified CsnE and CsnD, which displayed identical defects in fruiting body initiation in A. nidulans (Busch et al. 2003, 2007). The A. nidulans Cop9 complex represents an 8-subunit, stereotypical complex, whereas that of other model fungi (N. crassa, S. cerevisiae or S. pombe) possess fewer subunits and are not typical (Braus et al. 2010). Agaricomycetes genomes appear to encode a typical 8-subunit CSN (Supplementary Table S39), with an additional gene carrying a CSN8/PSMD8/EIF3K domain (IPR033464), making it putatively related to the Cop9 signalosome. Neither we nor previous studies found significant expression dynamics of any subunit of the CSN in mushroom-forming fungi. It is not known whether the complex is involved in fruiting body development as it is in Ascomycota, nevertheless, it will be an interesting target for future experiments, especially because it is involved in light regulation in N. crassa and can interact with the Velvet complex, two key factors that regulate Basidiomycota development also.
Protein ubiquitination
Covalent protein modification by the ubiquitin-proteasome system is a widely conserved regulatory machinery that functions in several essential processes in eukaryotes and was recently detected as a potentially very interesting function during fruiting body development (Krizsán et al. 2019). The regulatory potential of the ubiquitin-proteasome system stems from its ability to attach the 76-amino acid ubiquitin protein to lysines of target proteins, which either destines them for degradation by the proteasome or modifies their activity, cellular localization or DNA-binding ability (Trotman et al. 2007, Liu & Xue 2011, Finley et al. 2012). Ubiquitination can modify the activity of transcription factors, which provides an additional layer of transcriptional regulation (Freiman & Tjian 2003).
The protein ubiquitination machinery is composed of E1 ubiquitin-activating, E2 ubiquitin-conjugating enzymes and E3 ubiquitin ligases, which attach the ubiquitin to target proteins. While E1 and E2 enzymes are generic components of the system and occur in limited numbers in eukaryotic genomes (Supplementary Table S40), ubiquitin ligases provide the substrate specificity and come in many subtypes and exhibit considerable diversity due to interchangeable protein components, of which hundreds may exist in the genomes of some species (Hutchins et al. 2013, Zheng & Shabek 2017). The most diverse such parts are F-box and BTB/POZ domain proteins, both of which can confer substrate-specificity to the E3 complexes (Geyer et al. 2003) and RING-type zinc finger proteins, which are scaffold proteins linking target proteins with E2 enzymes (Metzger et al. 2012). These three gene families were found to be strongly expanded in mushroom-forming fungi, with frequent tissue-specific developmental regulation seen in RNA-Seq data (Almási et al. 2019, Krizsán et al. 2019) (see also Supplementary Table S41). The expansion stems from Agaricomycetes-specific duplication events and a correlation between fruiting body complexity and F-Box, BTB/POZ and RING-type zinc finger copy number and expression has been suggested (Krizsán et al. 2019), which could explain the evolution of increasingly complex mushroom morphologies in the class.
The widespread cellular roles of ubiquitination suggests that it should be instrumental during fruiting body morphogenesis too. In line with this, the protein ubiquitination system was found differentially expressed in F. velutipes fruiting bodies (Yamadat et al. 2006, Liu et al. 2020) and in caps of the same species subjected to differential CO2 treatments (Yan et al. 2019). Recent comparative transcriptomics analyses provided evidence for the developmental regulation of several components, mostly those that confer substrate specificity to E3 ligase complexes, in multiple Agaricomycetes species, including C. cinerea, A. ostoyae, A. ampla, S. commune or P. ostreatus, among others (Almási et al. 2019, Krizsán et al. 2019, Merényi et al. 2022). BTB domain-containing proteins, a family of E3 proteins, were reported to be upregulated in fruiting bodies in Antrodia cinnamomea (Lu et al. 2014). The combination of a remarkable gene family expansion (reported by Krizsán et al. 2019) with developmental regulation suggests that the ubiquitin-proteasome system could play a pivotal role in fruiting body development, although mechanistic details of the process remain to be understood. Ubiquitylation is key in developmental processes of the Ascomycota also, including fruiting body formation, and in some cases more mechanistic details have been uncovered (reviewed in Riquelme et al. 2018). However, a similar expansion of F-Box domains, as seen in Agaricomycetes, has not been observed in the Ascomycota.
Hereafter, we present an annotation of components of the protein ubiquitination machinery in selected mushroom-forming fungi, with particular emphasis on C. cinerea genes and comparisons across species. In this presentation, we follow the classification of E3 ubiquitin ligases used by Hutchins et al. (2013) and identify protein diversity of each protein family based on Interpro domain HMM-s. Results of the annotation along with orthologous genes and conservation levels are shown in Supplementary Table S40. A limitation of this approach is that the presence of a defining domain in the protein does not in all the cases, indicate a function in the ubiquitination machinery. For example, BTB/POZ domains, which contribute to expanding the diversity of E3 ligase complexes (Geyer et al. 2003) can, in rare cases, also occur in proteins not related to ubiquitination (e.g., cytoskeleton or transcription regulating proteins, Stogios et al. 2005).
Several components of the ubiquitination machinery are very conserved in Agaricomycetes genomes, including E1 (on average 3 genes) and E2 enzymes (19–24 genes), Cullin genes (4–10 genes) and deubiquitinating enzyme-encoding genes (23–27 genes) (Supplementary Table S40). Most of these genes show no or modest expression dynamics, which is not surprising given that the ubiquitination machinery is needed constantly in the cell. Of the E1 enzymes, only S. cerevisiae Uba3 orthologs (C. cinerea protein ID: 485679), which belong to the neddylation pathway, are developmentally regulated to some extent (8/3 species at FC>2/4). In C. cinerea 14 of the 23 E2 enzymes and half (13 of 26) of deubiquitinating enzymes are developmentally regulated, potentially indicating functional diversification of E2 enzymes and deubiquitinating enzymes during development (Supplementary Table S42). We found that none of the cullin genes in C. cinerea are developmentally regulated. Significantly larger dynamics can be observed in E3 ligase components, which are discussed in detail below.
RING-type Zinc finger proteins - RING-domain containing proteins, named after RING (really interesting new gene), are E3 ubiquitin ligases that confer interchangeability to the ligase complexes (Deshaies & Joazeiro 2009). Eukaryotic genomes contain large numbers of RING-domain proteins, which, along with other E3 ligases, enable the ubiquitin system to selectively ubiquitinate proteins. RING-domain proteins are generally poorly known in filamentous fungi, though it was recently noticed that they are strongly overrepresented in Agaricomycetes (Krizsán et al. 2019). The mfbAc (=Le.MFB1) gene of L. edodes (Lenedo1_1172639) described as a protein with adhesive properties by Kondoh et al. (1995) also belongs to this gene family.
The examined Agaricomycetes possess 81–148 RING-domain containing proteins (Supplementary Table S41), consistent with a previous broader sampling across 200 species. On average, 30–50 RING-domain proteins were developmentally regulated in the examined species (Almási et al. 2019, Krizsán et al. 2019), representing a significant pool of potentially important regulators. RING-domain proteins are highly variable, with most of them not showing clear orthology across species. Nevertheless, we found seven CDE orthogroups which displayed conserved developmental expression, mostly at FC>2 (Supplementary Table S41). For example, one of these (represented by C. cinerea 378120) showed a very distinct peak in gills of C. cinerea, A. ostoyae, P. ostreatus and caps (containing gills) of M. kentingensis, L. edodes as well as mature fruiting bodies of S. commune and A. ampla. Another CDE orthogroup (represented by C. cinerea 440772) was upregulated in gill tissues of mature fruiting bodies in P. ostreatus, A. ostoyae, M. kentingensis, L. bicolor and mature fruiting bodies of S. commune and A. ampla, but not in A. bisporus, L. edodes or C. cinerea. These two orthogroups coincide (partly) with meiosis and sporulation, so they could be involved in these, or other gill-specific processes. Genes in another CDE orthogroup (C. cinerea protein ID: 543850) showed an abrupt, although modest upregulation at the initiation of fruiting body development and expression peak in mature fruiting bodies or gills therein in C. cinerea, A. ostoyae, C. aegerita, A. ampla, L. bicolor, L. tigrinus, Ph. chrysosporium, P. ostreatus and S. commune (not in L. edodes).
F-box proteins - F-box proteins were recently identified as a potentially key group of proteins in fruiting body development (Krizsán et al. 2019). They are generally responsible for providing target specificity to the ubiquitination machinery, by binding and docking substrate proteins to Cullin E3 ubiquitin ligase complexes. Some F-box proteins interact with multiple target proteins while others only with a single protein (Jonkers & Rep 2009). Similarly, some F-Box proteins function independently of the SCF complex (Jonkers & Rep 2009), allowing for additional, yet poorly known regulatory functions to emerge. For example, F-box proteins can act as transcriptional co-regulators in plants (Chae et al. 2008) and are present in large numbers in plant genomes (up to 700–1000 genes, Xu et al. 2009), providing the possibility to assemble an enormous diversity of alternative ubiquitin ligase complexes.
The F-box family is highly expanded in mushroom-forming fungi and is composed of fast-evolving proteins with little similarity to each other. Agaricomycetes possess 69 to ~1 200 F-box proteins (on average ~200) in their genomes, whereas other filamentous fungi and yeasts possess up to 90 (on average ~40) and 20 such genes (Krizsán et al. 2019), respectively. This represents a strong enrichment in Agaricomycetes genomes (P<10-140), considering that the number of other proteins is not proportionately higher in this group than in other fungi. F-box protein encoding genes often showed tissue-specific expression and their numbers showed a positive correlation with the complexity level of the fruiting bodies (Krizsán et al. 2019).
F-Box proteins are highly variable and form few conserved orthogroups, compared to their diversity in fungal genomes. However, we found eleven orthogroups that showed some level of conservation and consistent developmental regulation across species (Supplementary Table S41). For example, orthologs of A. nidulans GrrA (C. cinerea protein ID: 358351), which is needed for meiosis and ascospore maturation (Krappmann et al. 2006, Riquelme et al. 2018), were conserved across Agaricomycetes and developmentally regulated in five species (FC>2). This indicates that some F-Box proteins are conserved despite the general volatility of the family. Due to this high variability, F-box proteins are hard to examine systematically using a comparative transcriptomics approach. Rather, they likely often fulfill species- or clade-specific roles, which could be studied either with more densely sampled transcriptomic data, or reverse genetics approaches.
BTB/POZ domain proteins - Proteins that contain the BTB/POZ motif, a protein-protein interaction (homodimerization) domain, can possess diverse cellular functions, the most frequent of which are regulation of chromatin structure (thus, transcription) and protein ubiquitination, as members of E3 ubiquitin ligase complexes (Zollman et al. 1994, Geyer et al. 2003). They are poorly known in fungi.
In animals, plants as well as fungi outside the Agaricomycetes, BTB domains are mostly fused to other conserved protein domains (Stogios et al. 2005) (in animals, mostly to zinc fingers (Zollman et al. 1994). In contrast, we observed that in the Agaricomycetes, less than 10 % of BTB/POZ domain proteins associate with any other domains and virtually never with DNA binding transcription factor domains, indicating that the gene family expansion in mushroom-forming fungi is concerned with proteins that only contain the BTB/POZ domain. This domain composition suggests that the BTB/POZ family is primarily involved in E3 ligases in Agaricomycetes, therefore we discuss this family under the chapter on protein ubiquitination.
We found ten CDE orthogroups, which showed some level of conserved developmental expression in Agaricomycetes (Supplementary Table S41). Members of two (C. cinerea protein ID: 379603, 449824) of these orthogroups are co-expressed in stages or tissues in which meiosis and sporulation occurs: gills of A. ostoyae and P. ostreatus, mature fruiting bodies of Ph. chrysosporium, A. ampla and S. commune, mature caps of M. kentingensis, L. bicolor, L. edodes (but not developmentally regulated in C. cinerea). Another CDE orthogroup (C. cinerea protein ID: 459680) was upregulated in stipes of C. cinerea, A. ostoyae, M. kentingensis, L. edodes (but not in P. ostreatus and A. bisporus), primordium stages of A. ampla and S. commune and during early development (primordia + young fruiting bodies) of L. bicolor. A fourth CDE orthogroup, which contains reciprocal best hits to the putative E3 ubiquitin ligase protein btb1 of Sch. pombe (C. cinerea protein ID: 545078), but contains also domains that suggest it regulates chromatin condensation and thus gene expression. Members of this orthogroup were upregulated typically late during development.
Other gene groups
Transporters
Among the conserved developmentally expressed genes (CDE orthogroups) we detected 49 putatively transport-related gene families. Many of these genes belong to the Major Facilitator Superfamily or ABC transporters, fast-evolving gene families that are represented by hundreds of genes in fungal genomes. Transporters have been reported in transcriptomic studies of several mushroom-forming species, both in fruiting body (Song et al. 2018b, Krizsán et al. 2019) and ectomycorrhiza formation (Xu et al. 2015). Due to the fast evolution of these families, orthology to genes of Ascomycota model systems and even within the Agaricomycetes is lacking in most cases, making it hard to infer their functions. Nevertheless, some conserved transporter genes were detected, including orthologs of S. cerevisiae Pns1 (choline transporter, C. cinerea 355745), A. nidulans MepA (ammonium permease, C. cinerea 538158), Sch. pombe Kha1 (plasma membrane potassium ion/proton symporter, C. cinerea 449705), S. cerevisiae Emp70 (endosomal transport protein, C. cinerea 495380), S. cerevisiae Opt1 (plasma membrane oligopeptide transporter, C. cinerea 496395), among others (see Supplementary Table S43). In the absence of functional annotations for the vast majority of these genes, we list the genes in Supplementary Table S43, but do not go into detailed analyses, because it is unlikely that we can arrive at any reasonable speculation about their function.
Notable CDE orthogroups include an MFS transporter C. cinerea 465489, which was developmentally regulated in 9/5 species (FC>2/4) and FB-init in C. cinerea, A. ostoyae, P. ostreatus, S. commune (in the latter at FC>2 only). A phosphate transporter CDE orthogroup (C. cinerea protein ID: 30849) was FB-init in C. cinerea, C. aegerita, A. ostoyae, A. ampla, S. commune, M. kentingensis and, at FC>2 in L. bicolor (not in L. edodes) (Supplementary Table S43). A group of zinc-regulated transceptors (transporter + receptor, C. cinerea protein ID: 455616) orthologous to S. cerevisiae Zrt1 was also detected. In summary, transporters can be expected to fulfil key functions in fruiting bodies, in moving around goods, signal molecules, modulating osmolarity to name a few, however, information about these in fruiting bodies is still limited.
Aquaporins are water and solute-transporting membrane channels that are integral to several different aspects of the fungal lifestyle. They were reported to be overexpressed in primordia and stipes of F. filiformis, relative to other tissues and developmental stages (Liu et al. 2020). They were also reported to be upregulated in L. bicolor fruiting bodies and ectomycorrhizae (Xu et al. 2015).
We found 2–11 aquaporin genes in the examined Agaricomycetes and observed conserved aquaporin expression in two aquaporin orthogroups. One of these (C. cinerea protein ID: 373141) contains putative orthologs of yeast Fps1, an aquaglyceroporin that has suggested roles in acetate uptake or glycerol transport (Nehls & Dietz 2014), not water uptake (see under chapter “Acetyl-CoA production by central carbon metabolism”). It is worth noting that aquaglyceroporins were also differentially regulated in L. bicolor fruiting bodies (Xu et al. 2015) and that these proteins were shown to have a capacity to transport various solutes, including NH3 and glycerol. This led Xu et al. to hypothesize that these genes could be involved in osmosis regulation or signalling (Xu et al. 2015), which is different from the conclusion we make above on acetate transport.
The other orthogroup (represented by S. commune 2501677) comprises stipe-specific aquaporin genes, which show similar expression trajectories in A. bisporus, A. ostoyae, M kentingensis and P. ostreatus, as well as in an external F. filiformis dataset (Liu et al. 2020). In all of these species, the genes showed expression peaks in stipes, mostly in young and mature fruiting bodies. It is notable that this pattern is seen in pileate-stipitate species and has been noted in a previous 3-species comparison of transcriptomes (Plaza et al. 2014). Species without separate cap and stipe tissues either do not have this gene (Pt. gracilis) or they do, but the gene is not developmentally regulated (S. commune, Ph. chrysosporium). The current genome assembly of C. cinerea does not contain an orthologue in this group, which might be because it became dispensable for the unique developmental trajectory of this species or because the genome assembly is incomplete at this genomic region. The marked stipe expression peak suggests a widespread role of water transport during mushroom development. Further, the increasing expression parallel with developmental progression could indicate a role in water supply needed for cellular expansion. On a related note, C. neoformans also shows an upregulation of an aquaporin gene (Cryne_JEC21_1_3081) late in its development.
A consequence of growth by cell expansion is that growth rate is limited by the speed of water transport, not cell division and CW material deposition (Roper & Seminara 2019). Therefore, aquaporin-facilitated water transport might be a selected trait in many mushroom-forming fungi that undergo fast growth. Although unknown at this moment, if fruiting body expansion is mediated by locally produced osmolytes (e.g., by breaking down glycogen), then that could produce a gradient that draws water towards the expanding tissues. This may be further facilitated by water channels formed by aquaporins. Finally, it is possible that other physical solutions, e.g., inter-hyphal water passageways also help transporting (e.g., via surface tension) water in the fruiting body.
The shared stipe upregulation of aquaporins can shed light on the origins of the typical mushroom (pileate-stipitate) morphology. All examined stipitate Agaricales species showed a stipe-specific aquaporin expression, which supports a single origin of pileate-stipitate fruiting bodies in the Agaricales. On the other hand, L. tigrinus, which evolved pileate-stipitate fruiting bodies independently in the Polyporales, does not show stipe-specific aquaporin expression.
Stress response genes
Diverse stress-related genes were detected among CDE orthogroups. Based on functional annotations and orthology relationships, many of these seem to be related to protection from oxidative stress, either through direct protection by their protein products (peroxiredoxins) or via the produced metabolites (ergothioneine) that protect from oxidative stress. We found evidence for all three pathways of reactive oxygen detoxification, the peroxiredoxin pathway, cytoplasmic catalases and glutathione peroxidases (Breitenbach et al. 2015), being active in fruiting bodies. The dynamic expression of these genes may point to the existence of an oxidative environment in fruiting bodies. However, it is currently unknown in which tissues/stages and in what cellular contexts this may arise. One possibility is that the action of cell wall modifying enzymes (e.g., peroxidases), which require an oxidative environment (H2O2), results in excess reactive oxygen species, though this has not been confirmed yet and remains a hypothesis for now. Below we discuss the main stress-related CDE orthogroups which we detected in the developmental transcriptomes of Agaricomycetes.
Peroxiredoxins - Peroxiredoxins are antioxidant proteins that can reduce diverse reactive oxygen species (ROS), including H2O2, and are one of the three cellular pathways of ROS detoxification in fungi. They have also been reported to be involved in signal transduction. The function of peroxiredoxins is not known in fruiting bodies and we identified a single report of peroxiredoxin upregulation in fruiting bodies, in Termitomyces (Rahmad et al. 2014). Another gene from Antrodia camphorata was cloned and biochemically characterised (Wen et al. 2007). We found orthologs of peroxiredoxin Tpx1 of Sch. pombe and Tsa1 of S. cerevisiae (C. cinerea protein ID: 468305) to be developmentally regulated in 12/8 species (FC>2/4) (Supplementary Table S44). Tpx1 has been characterised as the H2O2 sensor that relays the redox signal to the transcription factor Pap1 by inducing the formation of intramolecular disulfide bonds in Pap1 (Jara et al. 2007). Orthologs of Tpx1 are upregulated mostly in gill and cap tissues of mature fruiting bodies in the examined species. Orthologs of the Pap1 transcription factor (represented by C. cinerea 378471) are expressed constantly in mushroom-forming fungi. Orthologs of yeast Ahp1 peroxiredoxin (C. cinerea protein ID: 444300) were also developmentally regulated in the majority of species (10/4 species at FC>2/4, Supplementary Table S44). This gene preferentially eliminates organic peroxides, rather than H2O2 and relays signal to the Cad1 transcription factor in S. cerevisiae (Iwai et al. 2010). In C. cinerea this gene shows a very strong induction in young fruiting body gills, whereas in other species no evidence for a transcript enrichment in gill tissues was found. Finally, we detected orthologs of S. cerevisiae Dot5 (C. cinerea protein ID: 379600) (Supplementary Table S44), a thioredoxin (thiol-specific peroxidase) that is involved in the detoxification of peroxides (Cha et al. 2003, Izawa et al. 2004), but was also reported to be involved in the regulation of telomere length. Thioredoxins, together with peroxiredoxins (thioredoxin peroxidase) make up the thioredoxin system, which acts similarly to the glutaredoxin system in ROS scavenging. This orthogroup showed a characteristic upregulation in primordia relative to vegetative mycelium, then lower expression in late developmental stages in C. cinerea, L. edodes, C. aegerita, A. ostoyae and A. ampla, but not in other species.
Ergothionine biosynthesis - We also detected orthologs of the ergothioneine biosynthesis protein Egt1 of S. pombe. Ergothioneine is a histidine-derived antioxidant metabolite known to confer beneficial health effects to mushroom fruiting bodies (Weigand-Heller et al. 2012, Kalaras et al. 2017). For eukaryotic cells, it might be needed for growth and survival under oxidative stress conditions, for example during conidial survival and germination (Bello et al. 2012, Sheridan et al. 2016). Ergothioneine is produced via a two-step reaction pathway in S. pombe (Pluskal et al. 2014), comprising Egt1 then Egt2. Both are conserved in C. cinerea (C. cinerea protein ID: 356399 and 258252, respectively, Supplementary Table S44), however, only the first gene is developmentally regulated in the majority of the species, the other is not. Expression patterns of the Egt1 orthologs varied, in certain species they are FB-init, while in others it showed stipe-upregulation (A. ostoyae, L. bicolor, L. edodes, L. tigrinus, M. kentingensis, but not C. cinerea). How this can be explained is not clear, nevertheless, our data provide some evidence for the upregulation of ergothioneine biosynthesis in fruiting bodies. Orthologs of Sch. pombe Egt2, which catalyses the second step of ergothioneine production (C. cinerea protein ID: 258252) were developmentally regulated in a more restricted set of species.
Dehydro-D-arabinono-1;4-lactone - Two orthogroups that make up the biosynthesis pathway of dehydro-D-arabinono-1;4-lactone were developmentally expressed in several species. Dehydro-D-arabinono-1;4-lactone (previously known as D-erythroascorbate) is a protective metabolite against oxidative stress and is produced by a pathway comprising Ara1, Ara2 and Alo1 in S. cerevisiae (Amako et al.2006). Of these, Ara2 and Alo1 have clear orthologs in Agaricomycetes (represented by C. cinerea 365550 and 432313, respectively) (Supplementary Table S44), whereas Ara1 does not. Both Ara2 and Alo1 orthologs are strongly upregulated in fruiting bodies. ALO1 orthologs showed a clear upregulation in stipe tissues of C. cinerea, A. ostoyae and P. ostreatus (but not in L. bicolor or L. edodes), whereas in S. commune, A. ampla and L. edodes it is upregulated in primordia and stays high throughout fruiting body development. In C. aegerita, the gene is strongly upregulated in the first primordium stage, then expression drops to low levels.
Catalases - Two catalase orthogroups (represented by C. cinerea 472224 and 420157) (Supplementary Table S44) were developmentally regulated in the majority of species. Catalases are involved in H2O2 scavenging and thus protection against reactive oxygen species, but also in other pathways (such as beta-oxidation, see above), so their assignment to oxidative stress response is less certain than that of the above-mentioned genes.
Glutathione system - The glutathione system is composed of (i) glutathione, a 3-amino acid redox-active metabolite (Pócsi et al. 2004, Sato et al. 2009); (ii) glutathione peroxidases, which reduce H2O2; (iii) glutathione reductases, which convert oxidized glutathione back to its non-oxidized form; as well as (iv) glutaredoxins, which repair oxidatively damaged proteins. In S. cerevisiae glutathione is produced by a two-step reaction from L-cysteine and L-glutamate by the enzymes GSH1 and GSH2 (CM 2001). We annotated members of this pipeline in C. cinerea based on orthology information to components of the glutathione system in S. cerevisiae (Pócsi et al. 2004, Sato et al. 2009) (Supplementary Table S44). We identified single-copy orthologs of two proteins in the glutathione synthesis pathway, glutathione reductase, glutathione peroxidase. Glutaredoxins are present, on average, as 5–6 distinct genes in Agaricomycetes genomes, except for Pt. gracilis and M. kentingensis, which harbour 9 and 11 glutaredoxin genes in their genomes, respectively. Unlike the other antioxidant systems, the expression patterns in the glutathione system (peroxiredoxins, glutaredoxins, glutathione reductases and synthetases) show only moderate conservation across species, being developmentally regulated in 3–6 species (Supplementary Table S44).
Glutathione-S-transferases are also involved in defence against oxidative damage. They represent a diverse gene family also involved in other cellular detoxification tasks, such as the neutralization of xenobiotics, drugs or pesticides (Morel et al. 2009). Leon-Ramirez et al. hypothesized that in U. maydis ‘basidiocarps’ glutathione-S-transferases are involved in the protection against oxidizing intracellular conditions generated by some enzymes, such as NAPDH oxidases (León-Ramírez et al. 2017). Therefore, we only marginally treat them under this chapter, noting that any glutathione-S-transferase orthogroup, also by virtue of the lack of clear orthology to functionally characterised genes from model species, could be related to any of the possible functions of this family, not necessarily protection from oxidative damage. Glutathione S-transferases are a diverse gene family in fungi, with 20–49 genes (on average 32) in the genomes of Agaricomycetes. In each of the species, ~50 % of glutathione S-transferase encoding genes (up to 70 % in Coprinopsis) were developmentally regulated, indicating that these play important roles in fruiting body development, however, due to the general lack of conservation of these genes, these did not form CDE orthogroups. Interestingly, each species had genes that showed a marked upregulation (FC>4) at the initiation of fruiting body development.
Unannotated genes
Genes that encode proteins with no known conserved domain (Pfam or InterPro) signatures, referred to as unannotated genes here, are abundant in fungal genomes and represent some of the most exciting gene families. At the same time, they represent a challenge to link to fruiting body development, due to the complete lack of functional clues. In general, few literature reports are available on unannotated genes, possibly due to the difficulty of tackling their functions. An exception from this is a report of the enrichment of unannotated genes in mature peridioles of Pisolithus microcarpus fruiting bodies (de Freitas Pereira et al. 2017). An example of unannotated genes that turn out to be developmentally relevant is the LPMO-like X325 gene family (Labourel et al. 2020), which was circumscribed just recently and appears to be related to the cell wall (see chapter “Chitin-active enzyme families”).
We identified 176 CDE orthogroups of unannotated genes (Supplementary Table S45). These included, for example, Spc14 and Spc33 of S. commune, which are involved in the formation of the septal pore cap (Van Peer et al. 2010), are unannotated proteins but form conserved, single-copy gene families in the Agaricomycetes (represented by C. cinerea 419716 and 473658) that was developmentally regulated in 10/6 and 12/5 species, respectively. In C. cinerea alone, we identified 32 FB-init unannotated genes, several of which showed very significant fold changes (up to 150–200x).
This category of genes included Ctg1 of L. edodes and Cc.Ctg1 of C. cinerea. Ctg1 was originally identified in L. edodes as a target of the pre-mRNA splicing factor Cdc5 and was later shown to influence stipe elongation in C. cinerea (Nakazawa et al. 2008). In the latter study knockdown experiments were unsuccessful, whereas overexpression C. cinerea mutants showed accelerated stipe elongation (Nakazawa et al. 2009). In our data Ctg1 orthologs formed a CDE orthogroup (represented by C. cinerea 528942) which was developmentally regulated in 10/8 species (FC>2/4) (Supplementary Table S45). Despite the conservation of developmental regulation, expression profiles were only partially conserved across species. For example, the gene was upregulated in mature fruiting bodies in C. aegerita, A. ampla, C. cinerea, L. edodes, P. ostreatus, whereas in other species (e.g., A. ostoyae), it showed upregulation in primordia relative to vegetative mycelium. Overall, CTG1 orthologs seem to be a very exciting group of developmental genes, with no conserved domains and no mechanistic information on how they might contribute to fruiting body formation. Its role in stipe elongation needs to be conserved in other species, if expression profile conservation correlates with phenotype, we may expect other phenotypic effects in other species.
In summary, unannotated genes may contain very interesting, novel gene families involved in fruiting body development, yet circumscribing their functions remains challenging at the moment and will require extensive genetic manipulation techniques.
Functionally poorly known genes
In this group of genes we placed ones that had some (mostly automatic) annotations, but we could not clearly link them to cellular or fruiting-related processes. Thus, this category differs from unannotated genes in that unannotated genes are completely devoid of all (even automated) annotations. Altogether, we assigned 182 CDE orthogroups to this category (Supplementary Table S46). Some of these genes are discussed briefly below, but we note that many more interesting, fruiting-related genes are potentially hidden among the 182 orthogroups, however, clarifying their roles is left for future studies.
Cytochrome p450 family - We discuss cytochrome p450s in this category, because they participate in diverse functions, from secondary metabolite biosynthesis (as part of gene clusters, e.g., Nofiani et al. 2018) to unknown cellular pathways mediating stipe elongation (Muraguchi & Kamada 2000). We detected 8 CDE orthogroups of cytochrome p450s, most of which are functionally unknown. Among the cytochrome p450s, C. cinerea Eln2 should be mentioned. This gene was identified associated with a dominant mutation that confers an elongation less stipe phenotype (Muraguchi & Kamada 2000). Eln2 was reported to be a constitutively expressed cytochrome p450. In accordance with this, we did not find considerable expression dynamics on the orthogroup containing eln2 (represented by C. cinerea 381558). However, the developmentally regulated orthogroups may be worthy targets of functional analyses in the future.
GT25 family - This family includes enzymes with diverse activities (lipopolysaccharide β-1,4-galactosyltransferase, β-1,3- or β-1,2-glucosyltransferase). In bacteria, GT25 are responsible for the biosynthesis of the membrane component lipopolysaccharides (Bertani & Ruiz 2018) (‘endotoxins’). Most known GT25 enzymes are of bacterial origin, we found no functional study of fungal genes to date. The examined Agaricomycetes had mostly a single gene encoding GT25, which showed sporulation-associated expression peaks in most species: young fruiting body gills in C. cinerea, fruiting body gills in P. ostreatus, fruiting body cap in L. edodes and C. aegerita, mature fruiting bodies in A. ampla and S. commune, etc. GT25 encoding genes showed very low expression in general, in many cases only detectable in the peak expressing stage/tissue. The broader orthogroup (C. cinerea protein ID: 501720) that contains the detected GT25 genes is specific to the Basidiomycota (result not shown), which could indicate bacterial origin into the phylum. The function of these proteins in fruiting bodies is currently unknown, however, expression peaks coincident with sporulation suggest they are related to late developmental events.
UstYa-like mycotoxin biosynthesis proteins - this gene family was first reported to be involved in the synthesis of the ribosomally synthesised peptide toxin ustiloxin in the ascomycete Ustilaginoidea virens (Tsukui et al. 2015). This family of genes was found to participate in the production of other toxic cyclic peptides, collectively named ‘dikaritins’, in plant pathogenic fungi (Ding et al. 2016). Biochemically, they are oxidases of ribosomally produced peptides, including ustiloxins but also alpha-pheromones of S. cerevisiae (collectively Kex2-processed repeat proteins, KEPs) (Umemura 2020). Using a simple domain-based search, we detected 2–16 (28 in A. ostoyae) genes in the examined Agaricomycetes. Of these, each species had one to multiple genes that showed an induction (>4-fold) in primordia relative to vegetative mycelia (except C. cinerea, in which two genes were induced in young fruiting body caps), and high expression in all subsequent fruiting body stages, suggesting that their protein product has an increased abundance in fruiting bodies. Although the role of these genes in the Basidiomycota remains unknown for now, we speculate they may be responsible for the production of toxic compounds, possibly with a role in defence.
Carbonic anhydrases (CA) - convert CO2 to HCO3- which is then used in various ways by eukaryotic cells. CAs have been shown to be involved in fruiting body formation: CA knockout strains of the ascomycete S. macrospora showed slower vegetative growth and delayed fruiting body formation or defects in ascospore germination (Elleuche & Pöggeler 2009, 2010). The delay in fruiting body formation suggests a lower rate of HCO3- build-up necessary for some as yet unknown downstream processes, that, however, can be made up in longer time. Similar roles of CAs have not yet been reported in Basidiomycota, however, they would be worth investigating as many CAs appeared developmentally regulated in fruiting bodies. We found 2–9 CA encoding genes in the examined Agaricomycetes and that each species had at least one developmentally regulated carbonic anhydrase gene, although clear patterns of expression conservation were not noticeable. We found one CDE orthogroup (C. cinerea protein ID: 450047) that was widely developmentally regulated in mushroom-forming fungi (8/5 species at FC>2/4), showing diverse expression peaks in different species. It should be noted that CA expression levels potentially react also to ambient CO2 concentrations, so some of the expression variation we encountered may be due to the growth conditions used during fruiting.
CipB gene family - This category of genes included the cipB gene reported from C. cinerea (Nakazawa et al. 2009). The cipB gene was first identified as an interacting partner of the splicing factor cdc5 in L. edodes (Nakazawa et al. 2008). We found that the orthogroup that contained cipB orthologs (represented by C. cinerea 401749) was developmentally regulated in 10/2 species (FC>2/4), indicating that these genes have widespread although moderate expression dynamics (Supplementary Table S46). Based on conserved domain annotations, cipB genes are transmembrane ATP synthases located in the mitochondria; its contribution to fruiting body formation is not known at the moment.
Ferric reductase superfamily proteins and iron metabolism
The Ferric reductase superfamily comprises a conserved group of proteins that include ferric reductases and NAPDH oxidases, both of which are located in the plasma membrane (Zhang et al. 2013). Ferric reductases are part of a reductive iron uptake system which, in many model fungi, are involved in maintaining iron and potentially also copper homeostasis (Bairwa et al. 2017). These genes have mostly been studied in the context of iron acquisition of pathogenic fungi from their hosts. Agaricomycetes don’t share clear orthology with experimentally characterised ferric reductases of the Ascomycota (reviewed in Bairwa et al. 2017). Nevertheless, we identified six ferric reductase orthogroups, of which two were developmentally regulated in multiple species (Supplementary Table S47), suggesting that ferric reductases have conserved roles in fruiting body development, although their function in the process is unknown. One of these was developmentally expressed in 9/5 species (represented by C. cinerea 354442, FC>2/4) contains genes orthologous to S. cerevisiae FRE7, which might be involved in copper homeostasis in S. cerevisiae (Gross et al. 2000). Another orthogroup, which contains orthologs of A. nidulans NoxA and N. crassa nox-1 (represented by C. cinerea 260659), was developmentally regulated in 5/1 species. This gene is a NADPH oxidase, which has been shown to be involved in developmental signalling and tissue differentiation (Nguyen et al. 2017).
The Ferric reductase superfamily also includes NADPH oxidases (NOXs) (Zhang et al. 2013), which are reactive oxygen-generating cell membrane proteins involved in differentiation-related signalling processes and tissue patterning (Blackstone 2000). NOX genes and their regulator NoxR were reported to have been lost repeatedly in yeast lineages, underscoring their role in multicellularity (Nguyen et al. 2017). There is evidence for NOXs regulating sexual, but not asexual or vegetative development in A. nidulans (Lara-Ortíz et al. 2003) and G. lucidum and there is expression evidence in H. marmoreus (Zhang et al. 2015b). We identified two putative NOX orthogroups in Agaricomycetes (C. cinerea protein ID: 461481, 260659). A third (represented by C. cinerea 152927) was interpreted as a NOX by León-Ramírez et al. (2017), but appears to be a ferric reductase. Despite clear reports of NOXs being induced in sexual structures of Ascomycota (Lara-Ortíz et al. 2003), we did not find evidence for consistent induction of these genes in fruiting bodies, rather, expression patterns vary among fruiting body forming fungi. NOX genes show modest expression dynamics in mushroom forming fungi (2<FC<4, Supplementary Table S47). C. cinerea 448679 appears to be a single-copy homolog of noxR that is conserved among mushroom-forming fungi. Members of this orthogroup are not developmentally expressed.
A SYNOPTIC MODEL OF FRUITING BODY DEVELOPMENT
This study aimed to provide a catalogue of conserved fruiting body development genes based on a literature review and a meta-analysis of developmental transcriptomes of 12 mushroom-forming fungi. Based on sequence analyses of genes showing developmentally expression during fruiting body morphogenesis in these 12 species, we identified 921 conserved developmentally expressed orthogroups and several gene families. To more broadly explore pathways linked to these genes, as well as genes that are known to be involved in fruiting but do not show significant expression dynamics, we analysed further 558 gene families. As a result, altogether 1 479 genes of C. cinerea, and the orthologs of these in 11 other species are treated in this paper. These allowed us to identify developmental expression patterns that are shared across species. For most of the conserved developmental orthogroups, we provided broad functional classifications and speculations on their probable role in fruiting body development, although elucidating their exact function and molecular mechanisms of their action remains for future studies. In this chapter we synthesize these data into an updated, synoptic model of fruiting body development in the Agaricomycetes (Fig. 15). This model strongly builds on work by previous scholars, who attempted to provide overviews of developmental and/or molecular mechanisms of mushroom formation. In a series of papers Moore provided detailed descriptions of developmental events based on morphological and biochemical analyses (Moore 2005). Later, Kües (2000) and Kües & Liu (2000) summarised our knowledge on the life history of C. cinerea and details of its developmental biology. More recently, Ohm et al. (2011), Plaza et al. (2014) and Muraguchi et al. (2015) provided examples of transcription networks active during development and a molecular overview of C. cinerea development based on gene ontology, respectively. This paper further elaborates these models with more detail, and a selective presentation of only those processes that, based on gene expression patterns are conserved across twelve Agaricomycetes species. We hereafter discuss developmental events in three phases: early events (primordium formation and cell proliferation), growth by cell expansion and late events (sexual processes, sporogenesis).
Fig. 15.

The most important, conserved genes belonging to each phase are shown - genes with limited expression conservation or species-specific expression are not depicted.
[footnote: *pigment patterns vary by species] TP: trehalose phosphorylase, PKS: pyloketide synthase; OSH: Oxysterol Binding Protein Homologue; AQP: aquaporins; EXP: expansins; MCO: multicopper oxidase; RRM: RNA recognition motif family proteins; PAP2: diacylglycerol pyrophosphate phosphatases; FAD: fatty acid desaturase; GD-PDE: Glycerophosphodiester phosphodiesterases; S-C9-M: sphingolipid C9-methyltransferases; IRC: intradiol ring cleavage dioxygenase; PAL: phenylalanine ammonia-lyase; SM: secondary metabolism; HTP: heme-thiolate peroxidase; CP: cerato-platanin. genes associated with cap autolysis are not shown on this Figure, because it is a species-specific trait of C. cinerea. Note that light induction schemes are not shown on this Figure, as this varies from species to species.
Early events: primordium formation and cell proliferation
Expression patterns of developmentally regulated gene groups outline several distinct processes and a temporal sequence of cellular events in fruiting bodies. The first in this sequence and probably the most interesting is the initiation of fruiting body development, which involves the rearrangement of hyphal branching patterns, and the light-dependent formation of a primary, then a compact, secondary hyphal knot (Kües 2000, Liu et al. 2006, Sakamoto et al. 2018). Hyphal knot formation represents a transition from loosely arranged hyphae with fractal dimensions to true complex multicellular structures (Nagy et al. 2018). Fruiting body initiation has been a subject of intense research. Several gene expression changes have been described in this transition, related to widely known genes and functionalities of fruiting body morphogenesis. These include light-regulated (Sakamoto et al. 2018), defence (e.g., galectins, pore-forming toxins, Plaza et al. 2014); lectins, Boulianne et al. 2000, Walser et al. 2005); fatty acid biosynthesis, Liu et al. 2006; transcriptional regulation (TFs, velvet proteins, Ohm et al. 2011, Plaza et al. 2014), cell surface (hydrophobins, PriA, fasciclins (Kajiwara et al. 1992, Miyazaki et al. 2007), cell wall (Kre9/Knh1, laccases, Kües et al. 2011) as well as other genes (Liu et al. 2006, Hou et al. 2019). We update these observations by several functionalities in this work.
The largest gene group that shows an upregulation in primordium samples is related to mitotic cell division (See Fig. 3 in chapter “Meiotic and mitotic gene expression” above), which coincides with tissue differentiation in the primordium and produces the final number of cells that makes up the fruiting body. Ribosomal and chromatin- remodelling related gene (e.g., histones) expression show very similar expression profiles in primordia (see Fig. 4 & 10 in chapter/session “Ribosomal genes” and “Histone gene expression”), suggesting that novel protein synthesis and chromatin remodelling are required cell proliferation/division in primordia. Chromatin remodelling might be required for mitotic chromosome condensation and/or for rearranging chromatin accessibility for fruiting-specific trans regulatory factors. We term this the proliferative phase of development, which, in C. cinerea, encompasses hyphal knots to stage 2 primordia (Fig. 15). This pattern of mitotic, histone and ribosomal gene expression has been observed in most species, suggesting that fruiting body development can, in general, be divided into proliferative phase and growth by cell division. However, some species (e.g., A. bisporus (Craig et al. 1977), see chapter “Growth by cell expansion”) might not fully conform to this pattern and we expect non-Agaricales species (e.g., Polyporales, bracket fungi) to not follow this rule.
While the vegetative mycelium is entangled in the substrate and sensitive to bacterial and fungal competitors, fruiting bodies grow above ground and are, thus, more exposed to more extreme environmental conditions and biotic stress (animals grazers or infections). Accordingly, it has been reported that mycelial- and fruiting body-expressed defence gene repertoires show little overlap, indicating a dramatic switch in defence strategy upon fruiting (Plaza et al. 2014). defence genes generally show little conservation and high phylogenetic patchiness and did not form conserved orthogroups in our analyses. We, therefore, discussed them in the context of broader gene families (for detailed reviews see Künzler 2015, 2018). Putative secondary metabolism and oxylipin-related genes might also serve defence and/or antimicrobial purposes.
Probably as a response to differences in environment compared to the substrate, such as higher temperature or humidity fluctuations, several cell surface protein-encoding and fatty acid metabolism genes showed induction in primordia. These included hydrophobins, cerato-platanins, wax synthases and the Con6 family, among others. Hydrophobins probably help retaining water or provide protection from excess water above-ground (Lugones et al. 1999b). Wax synthases and Con6 proteins, although haven’t been examined to date, might act in a similar way. We infer changes in cell membrane composition in primordia from expression changes in genes influencing membrane composition. For example, fatty acid desaturases related to linoleic acid biosynthesis are upregulated in fruiting bodies, probably contributing to the higher linoleic acid contents of fruiting bodies reported before (Shaw 1967; Song et al. 1989), with potentially to oxylipin synthesis too (Orban et al. 2020). Putative stress-related gene expression changes (peroxiredoxins and ergothioneine-biosynthesis genes) might also correspond to genetically encoded mechanisms for coping with adverse above-ground conditions.
Cell wall remodelling emerges as a potentially important process in fruiting body initiation both in this study and previous ones (e.g., Liu et al. 2021). Fruiting body-specific cell wall architectures exist and are probably produced by a diverse suite of lytic and cell wall remodelling enzymes that are induced in primordia (Fig. 15). From the array of conserved orthogroups, it appears that glucan remodelling is more important than chitin modification. Additionally, oxidative enzyme genes (HTPs, DyP, MCOs, AA5_1) seem to be important in primordium development, although their functions have not been elucidated, yet. Cell wall related gene families show conservation at the gene family level, not always at the level of strict CDE orthogroups. Therefore, the complete array of cell wall remodelling enzymes induced in primordia is much broader in any species than shown on Fig. 15. Several cell wall remodelling related gene families described from the Ascomycota were developmentally regulated in the Agaricomycetes as well (e.g., GH16/17, Kre9/Knh1), whereas some, that are central to Ascomycota cell wall remodelling showed constant expression (e.g., GH72) or are missing in the Agaricomycetes (e.g., GH76). Further, it appears that dynamic expression is mostly observed in cell wall-active enzyme genes, not in upstream (e.g., ER-localized) components of the cell wall assembly line.
Although the light-dependency of fruiting body initiation differs from species to species (Sakamoto 2018), we detected some orthogroups that probably correspond to light reception. These include phytochromes, the C. cinerea Dst2 family and potentially PAS domain containing proteins. The latter is particularly interesting, given that homologs in N. crassa regulate the white collar complex and mediate adaptation to changing light intensities during the day (Fuller et al. 2015).
The initiation of fruiting body development represents a transition from fractal-like to complex multicellularity. Accordingly, several complex multicellular processes, such as cell-to-cell communication, adhesion and the regulation of differentiation (Knoll 2011, Nagy et al. 2018) should turn on in primordia. Like for previous studies, decoding these aspects proved difficult for us too. We expect regulatory factors, such as transcription factors, kinases, phosphatases, potentially F-box proteins or non-proteinogenic factors (e.g., small RNA) to participate in cellular differentiation. We and previous work detected some transcription factor, kinase, NOX, pheromone, F-box orthogroups upregulated in primordia. However, these generally did not show significant conservation of expression, so the main regulatory mechanisms of cellular differentiation in fruiting bodies remain an enigma. It further remains to be established if differentiation mechanisms described from plants and animals, such as morphogens, hormone-like substances (e.g., oxylipins) or highly conserved gene regulatory circuits exist in fungi. Similarly, adhesion-related proteins are mostly unknown at the moment. Fasciclins and cell wall modification (e.g., by laccases) have been speculated to be related to adhesion (Nagy et al. 2018, Merényi et al. 2020), though glucan remodelling enzymes, wax synthases, the carbohydrate-binding properties of lectins or species-specific mechanisms could also be key and more research is needed to elucidate what genes glue hyphae together in 3-dimension fruiting bodies.
Transition to growth by expansion
At the stage 2 primordium stage (following nomenclature in C. cinerea, Fig. 15), cell and tissue differentiation, cell proliferation has largely completed. This is reflected in the downregulation of mitotic, ribosomal and chromatin-remodelling genes. At this point, the fruiting body transitions to a different developmental mechanism, which we term ‘growth by cell expansion’. Growth by cell expansion is a well-known fungal trait and was already noted by de Bary in 1887 (De Bary et al. 1887), however, the molecular details have remained unknown. The phenomenon is marginally similar to fruit ripening in plants, but the mechanisms are probably completely different, though both involve cell wall loosening, remodelling and cell expansion.
Based on analyses of developmental transcriptomes, we detected what we believe is a plausible molecular mechanism for growth by expansion. This involves the upregulation of membrane biosynthesis genes and the rewiring of basic metabolic pathways for the production of excess Acetyl-CoA (see chapter “Acetyl-CoA production by central carbon metabolism”), which is the starting point of the biosynthesis of membrane components (e.g., ergosterol). Upregulation of genes involved in sterol transport, ergosterol biosynthesis correlates with the rewiring of basic metabolism. At the same time, aquaporin gene expression increases in stipes, which probably supports water transport driving the expansion of cells via turgor. Turgor manipulation requires an osmolyte, however, despite speculations of trehalose or mannitol serving as osmolytes, its identity remain unresolved; the expression data analysed here did not provide unequivocal evidence for any given osmotically active molecule in fruiting bodies. Trehalose, glycogen and mannitol metabolism genes are upregulated at this stage, but many of them in gills, which suggest an involvement in spore storage material production, rather than turgor manipulation.
For enabling cell volume increases, the fungus deploys a diverse and mostly species-specific array of cell wall remodelling enzymes. Cell expansion also probably follows different mechanisms in each tissue. For example, according to the model of stipe elongation in C. cinerea proposed by Liu et al. (2021), CE4 and GH30 genes may contribute to the rigidification of the cell wall in the stipe base, causing it to be more resistant to loosening by lytic enzymes than the stipe apex, thereby reducing its elongation potential. It is reasonable to assume that similar mechanisms underlie the differential expansion potential of different tissues within the fruiting body. We detected a large suite of conserved CAZyme orthogroups upregulated in elongating stipes (Fig. 15), whereas only one conserved orthogroup (GH55) was found in expanding caps. Since caps are composed of multiple tissues, it is possible that RNA-Seq techniques with higher resolution (e.g., single-cell RNA-Seq) are needed to uncover conserved tissue-specific cell wall remodelling gene expression.
What regulatory genes underlie growth by cell expansion will need to be determined in future studies. We detected conserved orthogroups of several developmentally expressed regulators (transcription factors, RING-type zinc fingers, F-box, BTB and RNA-binding proteins) but most showed tissue-specific expression patterns and no expression profile clearly suggested involvement in regulating cell expansion in general. Interestingly, the Exp1 transcription factor reported to be responsible for cap expansion in C. cinerea (Muraguchi et al. 2008b) was more frequently upregulated in stipes than in caps across the examined species. We detected a red light-sensing phytochrome orthogroup upregulated in stipes, which we hypothesize is responsible directing stipe growth away from closed spaces towards the open air.
Late events: meiosis and spore production
The young and mature fruiting body (in C. cinerea nomenclature) stages are the scene for most sexual events, including meiosis and spore production. Accordingly, meiotic genes as well as several genes involved in DNA replication/repair, chromatin remodelling, ribosome assembly, which showed a peak in the proliferative phase in primordia, show a very sharp expression peak associated with meiosis (see chapter “Cell division, proliferation and growth” and “Chromatin regulators”). We found several conserved orthogroups of BTB/POZ proteins and RING-type zinc fingers with expression peaks in gills; these might regulate key events associated with meiosis. A conserved transcriptional co-repressor, Cag1 of C. cinerea should be mentioned here. Although this gene and its orthologs did not show significant expression dynamics, its deletion caused abnormalities in gill development (Masuda et al. 2016).
Genes associated with storage carbohydrate metabolism were upregulated in gills in most species, which is consistent with a rich body of literature reporting glycogen fluxes in fruiting bodies (Ji & Moore 1993, Kües 2000). These genes included ones related to glycogen mobilization, trehalose and mannitol metabolism. We found evidence for glycogen synthesis (Gsy1 gene orthologs), suggesting that this carbohydrate is packaged into spores.
OPEN QUESTIONS RELATED TO FRUITING BODY MORPHOGENESIS
Despite the advances in understanding cellular events of fruiting body morphogenesis, several significant questions remain open and await further research. We below outline eight key questions that we think are instrumental to develop a better understanding of fungal fruiting body formation and complex multicellularity. This represents a subjective assessment of significant challenges of fruiting body research, there may be other perspectives on remaining key challenges to be solved.
1. Although new information on fruiting body morphogenesis accumulates at a steady pace, several questions related to multicellular organization remain open. Fruiting bodies are complex multicellular structures and thus display complex developmental programs involving tissue differentiation and cell type specification. Besides a developmental program, adhesion and cell-to-cell communication are two key traits for complex multicellularity (Knoll 2011). While we are starting to understand transcriptional networks governing tissue differentiation (see chapter “Transcription factors”), we still know very little about the genes/proteins mediating hypha-hypha adhesion, communication or whether an extracellular matrix (other than the cell wall) exists in mushroom-forming fungi. Hypotheses have been put forth on adhesion (via laccases or fasciclins), however, these have not yet been tested experimentally. Tissue differentiation results in distinct cell types within the fruiting body, however, our current understanding of cell type diversity in mushroom-forming fungi is limited. All current estimates for cell type diversity are based on morphology, which probably represent underestimates; technologies like single-cell RNA-Seq might provide more insights into cell type classification.
2. Regarding regulatory genes, it is an open question whether ‘master’ regulators of fruiting body development or tissue differentiation exists, similarly to animal and plant developmental regulator systems (Meyerowitz 2002). Developmental processes are often organized modularly and hierarchically so that the resulting modules have a high level of autonomy. Such autonomy of mushroom development, to our best knowledge, has rarely been documented. An exception from this is monokaryotic fruiting, the process of forming basidiocarps on monokaryotic cultures without mating in certain species (e.g., Herzog et al. 2016). The fact that fruiting body initiation can bypass mating suggests that the process has a certain degree of autonomy, which might be reflected as modularity or hierarchy in the underlying regulatory networks. Moore et al. characterised fruiting body morphogenesis as being made up of developmental subroutines that are somewhat independent of each other and that makes the whole process resilient to a certain degree of developmental imprecision (Loftus et al. 2020).
3. Transcriptional changes during the initiation of fruiting are significant. Most studies with a sufficient number of time points sampled (e.g., Plaza et al. 2014, Muraguchi et al. 2015, Sipos et al. 2017, Almási et al. 2019, Krizsán et al. 2019, Merényi et al. 2022, Ruytinx et al. 2021, Zhang et al. 2021d) found the largest number of differentially expressed genes at the transition from vegetative mycelium to the hyphal knot/primordium stage. However, whether this transcriptional rewiring event is purely a result of transcription factor activity or whether there are more general changes, for example, in chromatin accessibility or epigenetics, is unknown at the moment. Assays such as ATAC-Seq (Buenrostro et al. 2015) or ChIP-Seq (see e.g., Vonk & Ohm 2021) could provide insight into the magnitude of transcriptional versus chromatin-related rearrangement during fruiting body initiation. Similarly, posttranslational modification of proteins may be a significant regulatory mechanism in fruiting body morphogenesis (Pelkmans et al. 2017). Gene methylation, an important developmental mechanism in animals, seems to be missing in fungi, on the other hand (Bewick et al. 2019).
4. Understanding tissue specificity of genes and encoded proteins is key to uncovering mechanisms of tissue differentiation and predicting gene function. The bulk expression studies and a few studies involving fluorescent protein tagging revealed numerous examples of tissue-specifically expressed genes; however, more systematic studies will be needed to understand tissue-specific expression and the mechanisms that generate it.
5. We identified altogether 176 conserved developmentally expressed orthogroups of unannotated genes. These genes encode proteins that contain no known protein domains, therefore, they lack even automated functional annotations. Unannotated genes are common in fungal genomes and represent an enigma for mycology. Their conservation implies that they have important functions these are, however, unknown in the vast majority of cases (see Spc genes of S. commune for an exception, chapter “Unannotated genes”). Therefore, unannotated genes might represent a future treasure trove for developmental biology.
6. Growth by cell expansion is a widely conserved trait of mushroom-forming fungi. Above we provided evidence for this process involving excess Ac-CoA production, the upregulation of membrane biosynthetic processes and cell wall remodelling genes (chapter “Acetyl-CoA production by central carbon metabolism” and “Lipid metabolism”). However, the identity of the osmolyte that drives turgor increase in fruiting bodies remains unknown and its identification represents a significant challenge for future research.
7. Although not strictly morphogenetic, how the fruiting body is supported by the mycelium is a question with important practical implications in the mushroom industry. It is known that the directionality of the transport of goods in the mycelium switches as fruiting body development is initiated (Herman et al. 2020). Also, it has recently been suggested that secondary metabolite biosynthesis during fruiting body development depends also on the mycelium (Orban et al. 2021). Thus, transcriptional and metabolic reprogramming has to affect the entire fungal colony and not only the part where primordia emerge. Autophagy has been implicated in fruiting body development in the Ascomycota (Voigt & Poggeler 2013), however, to the best of our knowledge, it has not yet been reported in Basidiomycota fruiting bodies. The feeding and nutrient transport mechanisms as well as regulation of fruiting in relation to nutrient availability are exciting avenues of research, but beyond the primarily morphogenetic scope of the present paper.
8. While useful for discovering shared aspects of fruiting body morphogenesis, the comparative approach used by us, ignores species-specific aspects of morphology, ecological adaptation, gene repertoires and expression dynamics. Non-conserved aspects of development are certainly important, particularly for understanding the biology of individual cultivated species and will deserve further study in the future. However, given the available information, making functional inferences for taxonomically restricted or species-specific genes is even harder than for conserved genes, therefore, we here restrict ourselves to the discussion of the latter.
CONCLUSIONS
The development of mushroom fruiting bodies is one of the most complex morphogenetic processes in fungi. A significant body of previous research has focused on understanding the underlying genetics and has made significant progress in cataloguing genes related to the initiation, morphogenesis, defence, pigmentation of fruiting bodies, among others. However, available knowledge is still patchy and lacks key details in multiple aspects, such as conserved events in fruiting body morphogenesis and corresponding genes or key regulators of major differentiation steps.
This study aimed at providing a catalogue of conserved fruiting body development genes based on a literature review and a meta-analysis of developmental transcriptomes of 12 mushroom-forming fungi. We identified gene orthogroups that show conserved developmental expression across multiple species. Despite some limitations of the genomic and transcriptomic approach used here, this study provided novel insights (such as gene families, orthogroups, conserved expression profiles) into the morphogenesis of mushroom fruiting bodies. We hope these data reveal some aspects of the core genetic program of agaricomycete fruiting body morphogenesis and will provide a roadmap for functional analyses of fruiting body development. We are aware that the summary provided here is still an incomplete survey of developmental genes in fruiting body forming fungi. Comparisons at finer scales, such as within economically relevant genera, denser sampling of time points during development or at better spatial resolution (e.g., single-cell RNA-Seq) could yield additional insights. Joint analyses of gene expression patterns with gene phylogenies or with gene family copy number dynamics, functional genomics assays, the inference of gene co-expression networks or population genomics (e.g., GWAS analyses) among others, could all provide more detailed views on the synergy between evolutionary changes in gene content, expression regulation and morphogenesis. We anticipate that with advances in functional genomics of Agaricomycetes, it will become easier to conduct such studies and shed light on conserved and species-specific aspects of fruiting body morphogenesis.
Acknowledgments
We are grateful to all friends and colleagues who, over the years, inspired our work on fruiting body development through collaborations or discussions. We appreciate the help of Levente Karaffa in making sense of Acetyl-CoA production and basic metabolism. We appreciate the permission of Francis Martin and Jason Tsai to use photographs of L. bicolor and M. kentingensis in Fig. 1, respectively. We are grateful to Annegret Kohler for providing pre-publication access to the transcriptome of L. bicolor. This work was supported by the European Research Council (grant no. 758161 to L.G.N.), the “Momentum” program of the Hungarian Academy of Sciences (contract No. LP2019-13/2019 to L.G.N.), the Hungarian National Research, Development, and Innovation Office (contract No. GINOP-2.3.2-15-2016-00052), the Szeged Scientists Academy under the sponsorship of the Hungarian Ministry of Innovation and Technology (FEIF/646-4/2021-ITM_SZERZ, to B.Sz., A. Cs. And L.G.N.) and with the professional support of the Doctoral Student Scholarship Program of the Co-operative Doctoral Program of the Ministry of Innovation and Technology financed from the National Research, Development and Innovation Fund (KDP-17-4/PALY-2021, to Cs.F.). T.V. was supported by the National Talent Programme (NTP-NFTÖ-21-B-0074) of the Hungarian Government. FH gratefully acknowledges funding from the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) under grant HE 7849/3-1.
DECLARATION ON CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
1We deviated from this rule where justified.
Supplementary Material: https://studiesinmycology.org/
Expression heatmap of DNA replication, repair, mitosis and meiosis related genes in A. ostoyae, M. kentingensis, P. ostreatus and the simple S. commune. Well-delimited complexes mentioned in the paper are shown separately. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively.
{kind=link}
{kind=link}
{kind=link}
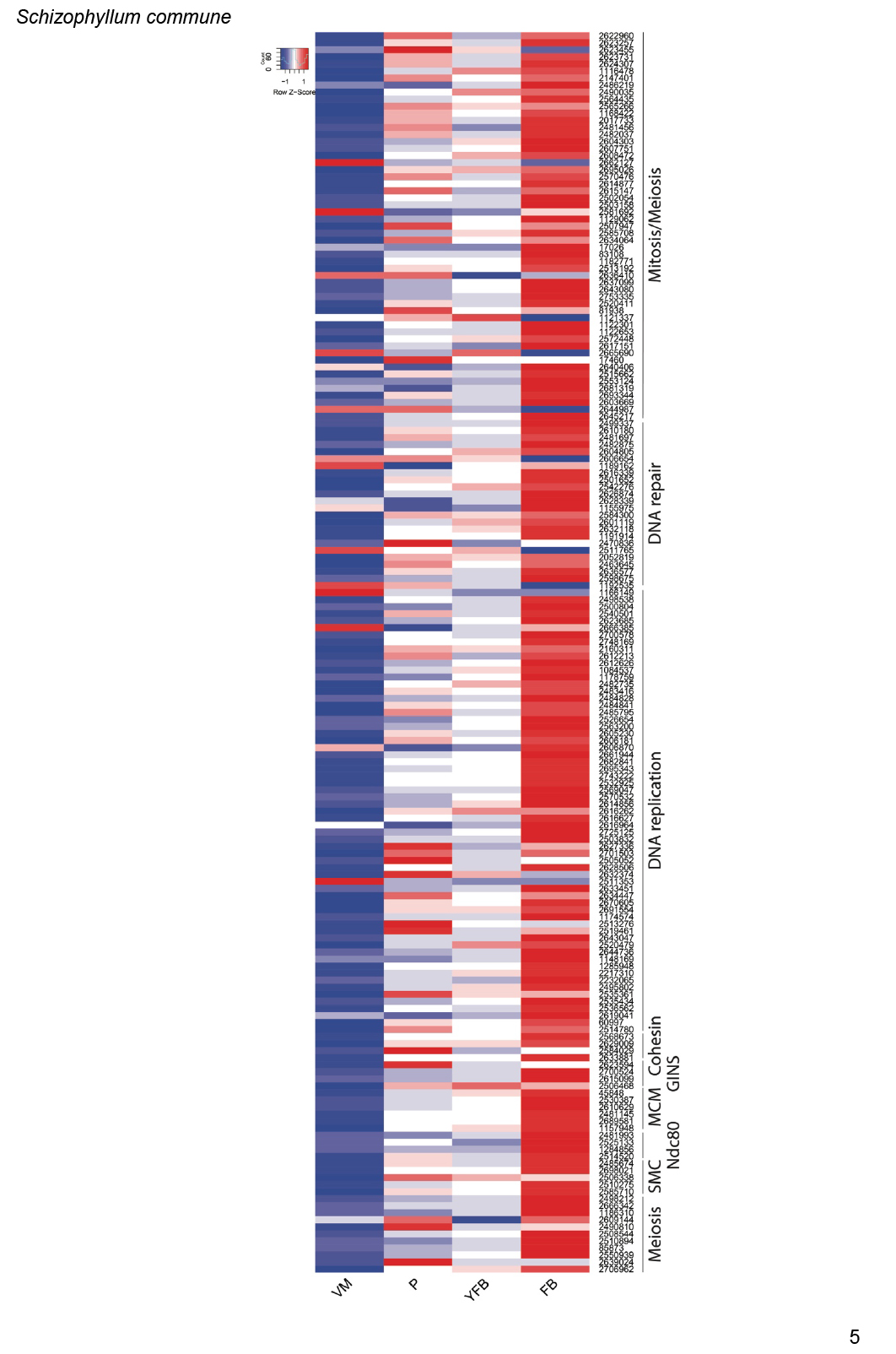{kind=link}
Expression heatmap of ribosomal protein encoding genes in A. bisporus, A. ostoyae and P. ostreatus. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively.
{kind=link}
{kind=link}
{kind=link}
Annotated glycolysis pathway of C. cinerea. Genes in the pathway are denoted by the S. cerevisiae gene name, followed by the protein ID of the C. cinerea ortholog and, in parentheses, the number of species in which the gene was developmentally regulated at fold change >2/>4.
{kind=link}
Expression heatmap of ergosterol and sphingolipid biosynthesis related genes in A. ostoyae and P. ostreatus. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively.
{kind=link}
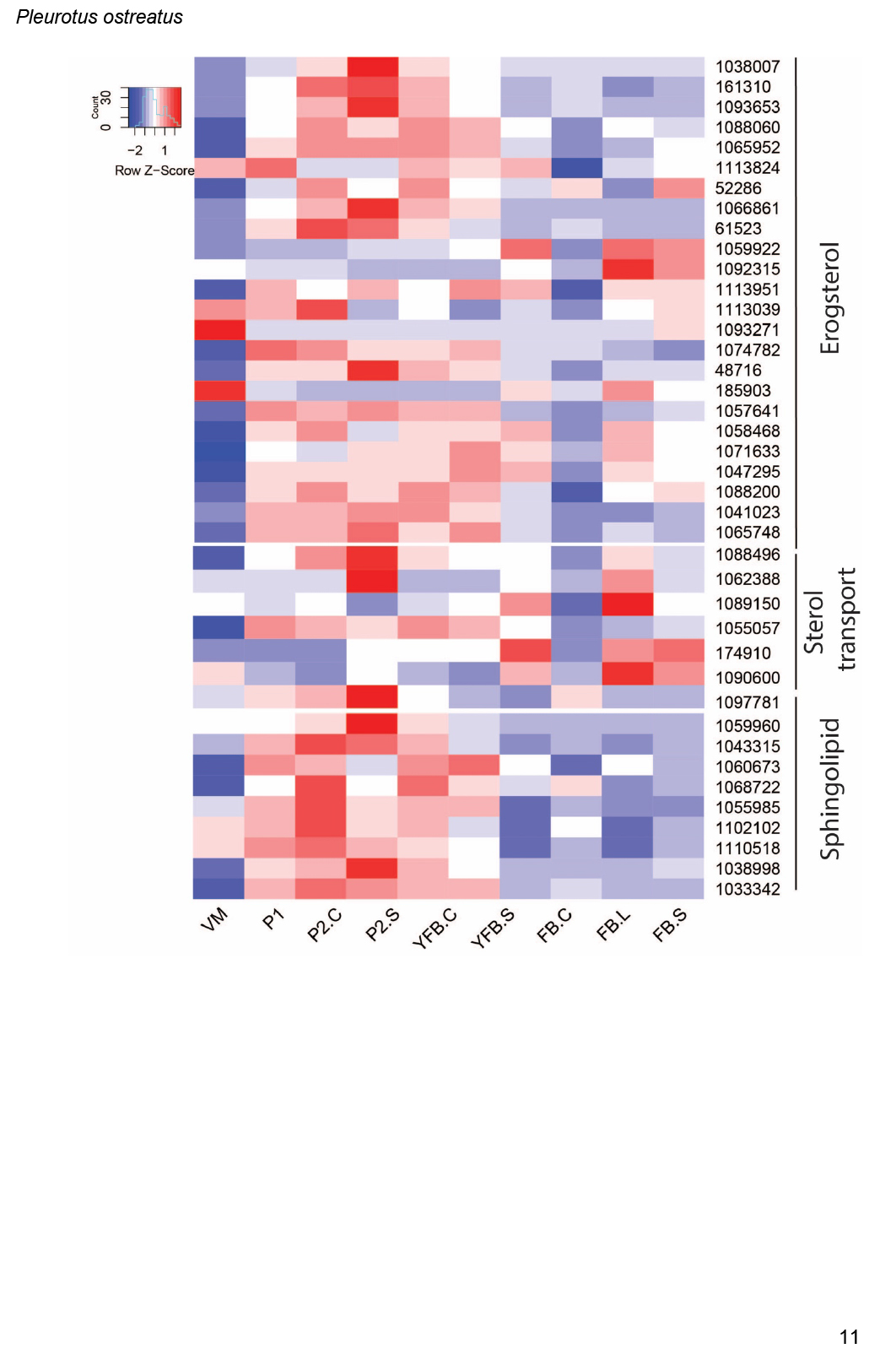{kind=link}
Expression heatmap of membrane phospholipid and fatty acid biosynthetic genes in A. bisporus, A. ostoyae, M. kentingensis and P. ostreatus. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively.
{kind=link}
{kind=link}
{kind=link}
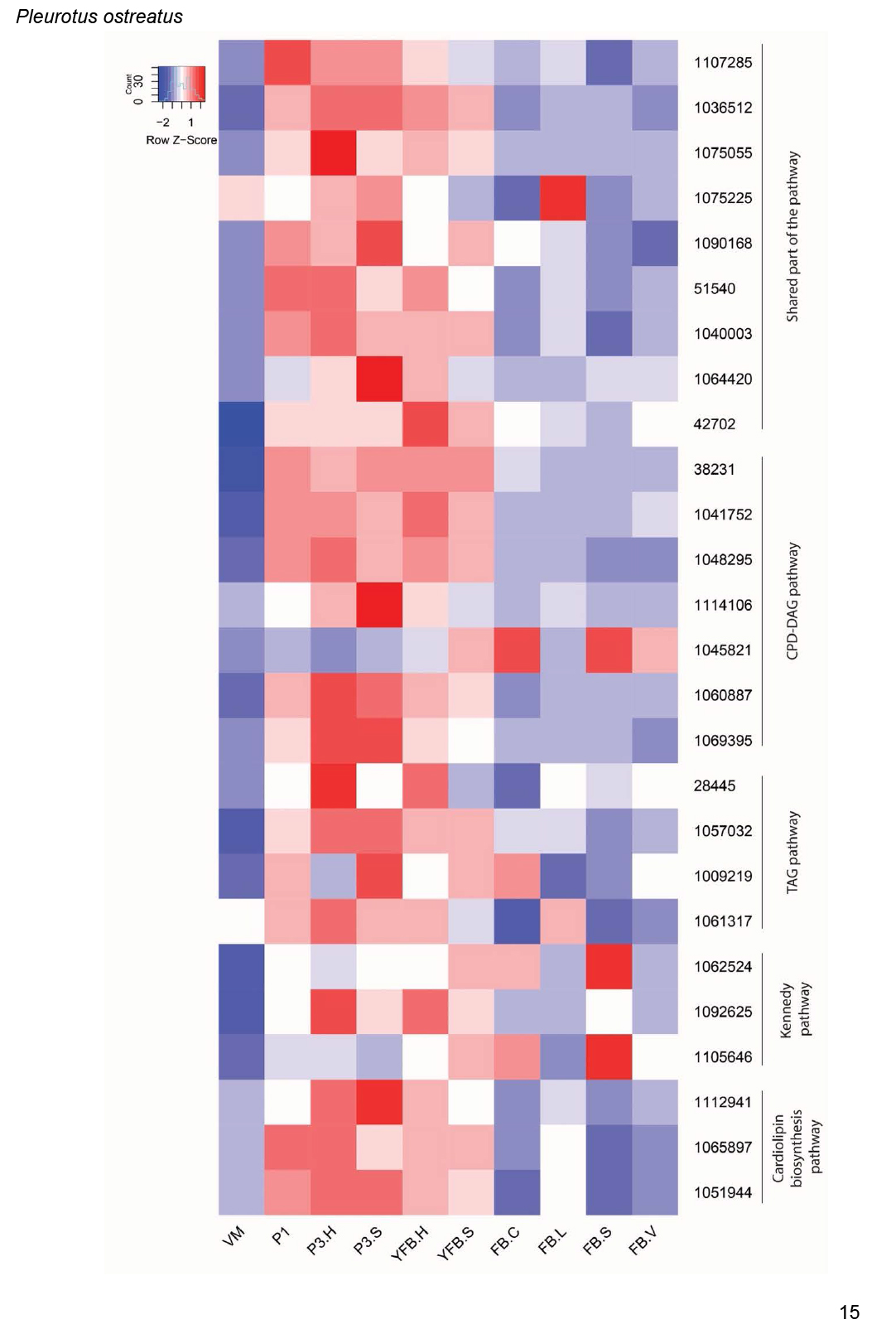{kind=link}
Expression heatmap of storage carbohydrate metabolism genes in A. ostoyae, L. bicolor and P. ostreatus. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively.
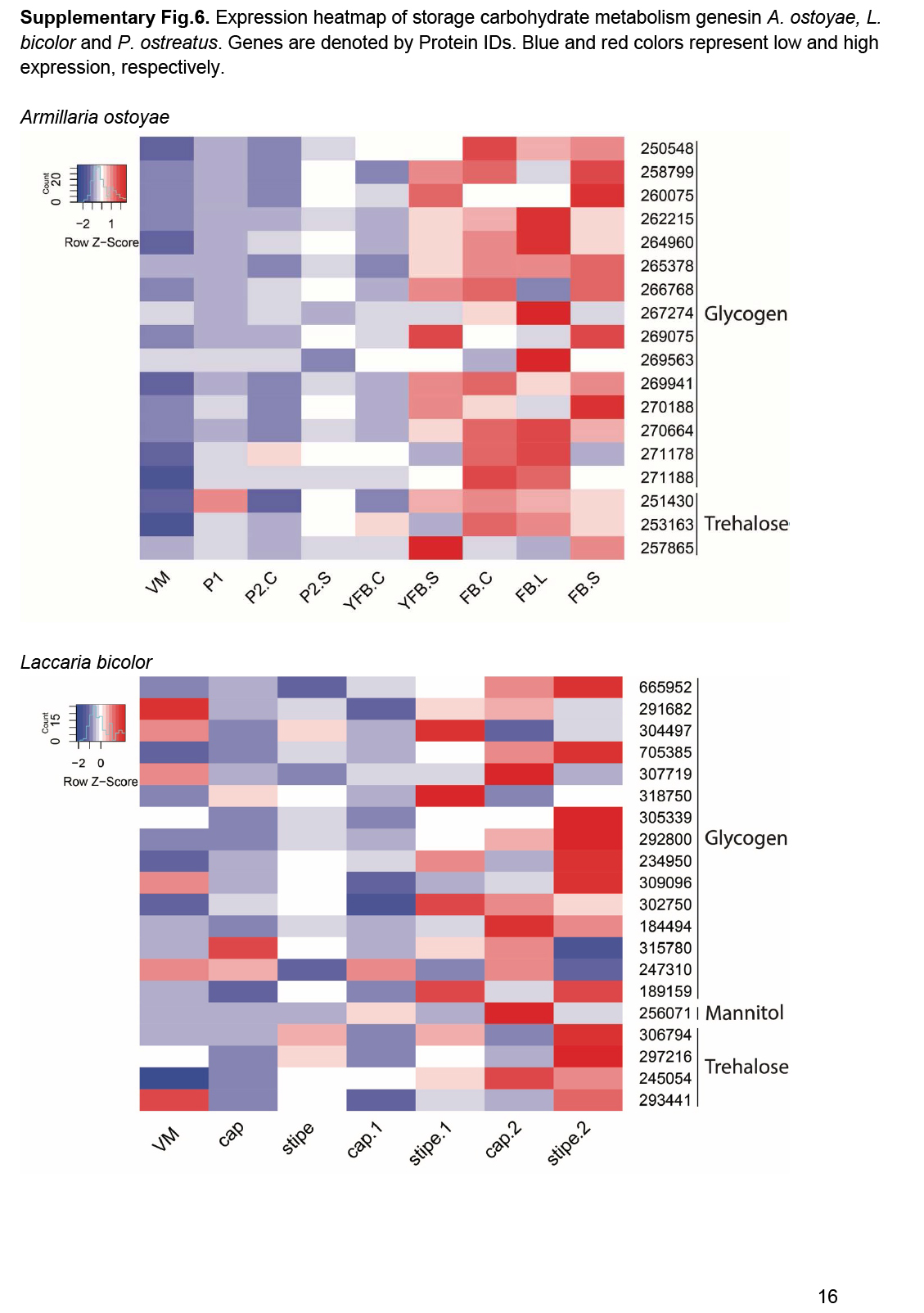{kind=link}
{kind=link}
Expression heatmap of chromatin remodeling related genes (histones, histone chaperones, histone deacethylases) in A. ampla, A. ostoyae, L. tigrinus, M. kentingensis and P. ostreatus. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively.
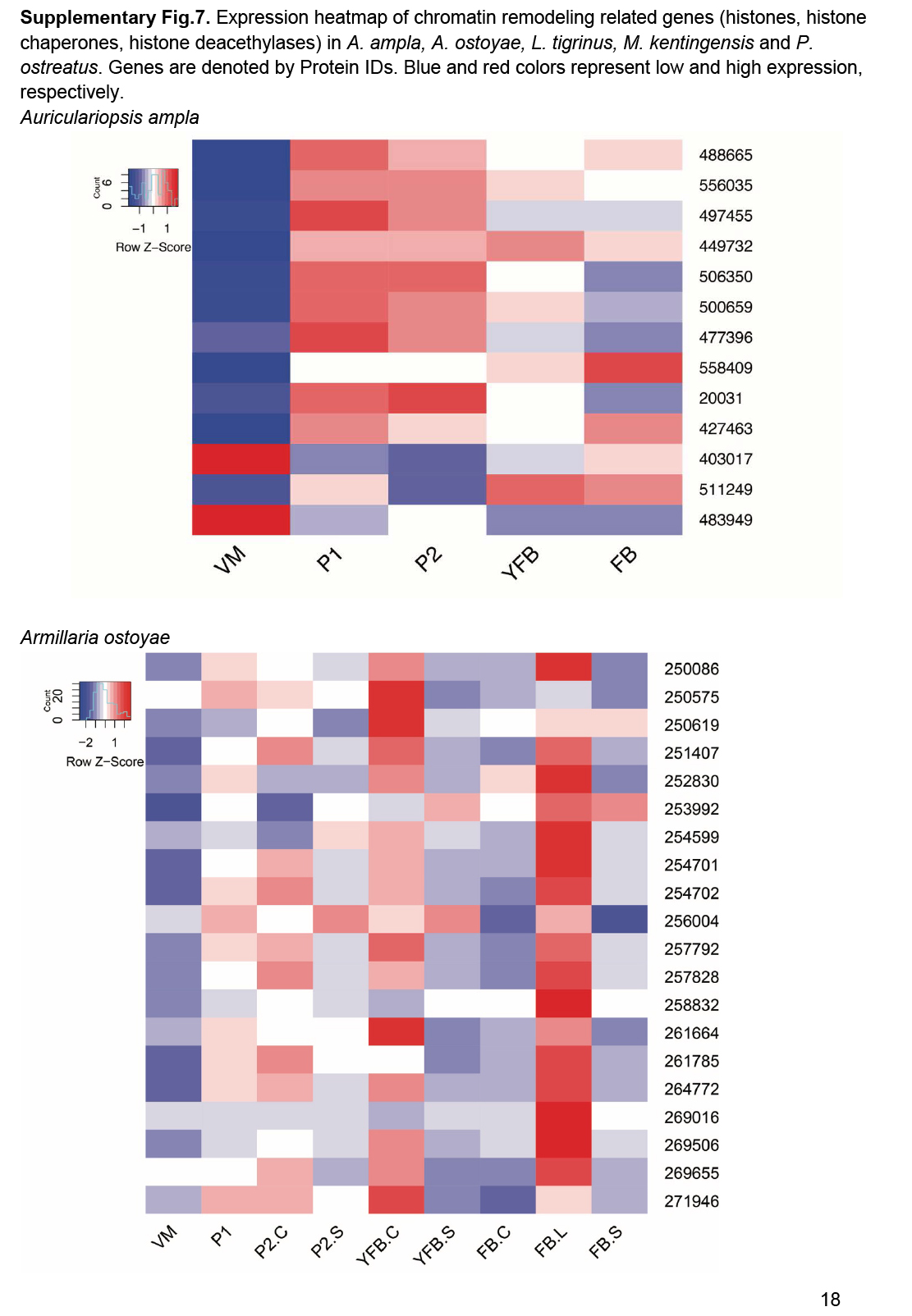{kind=link}
{kind=link}
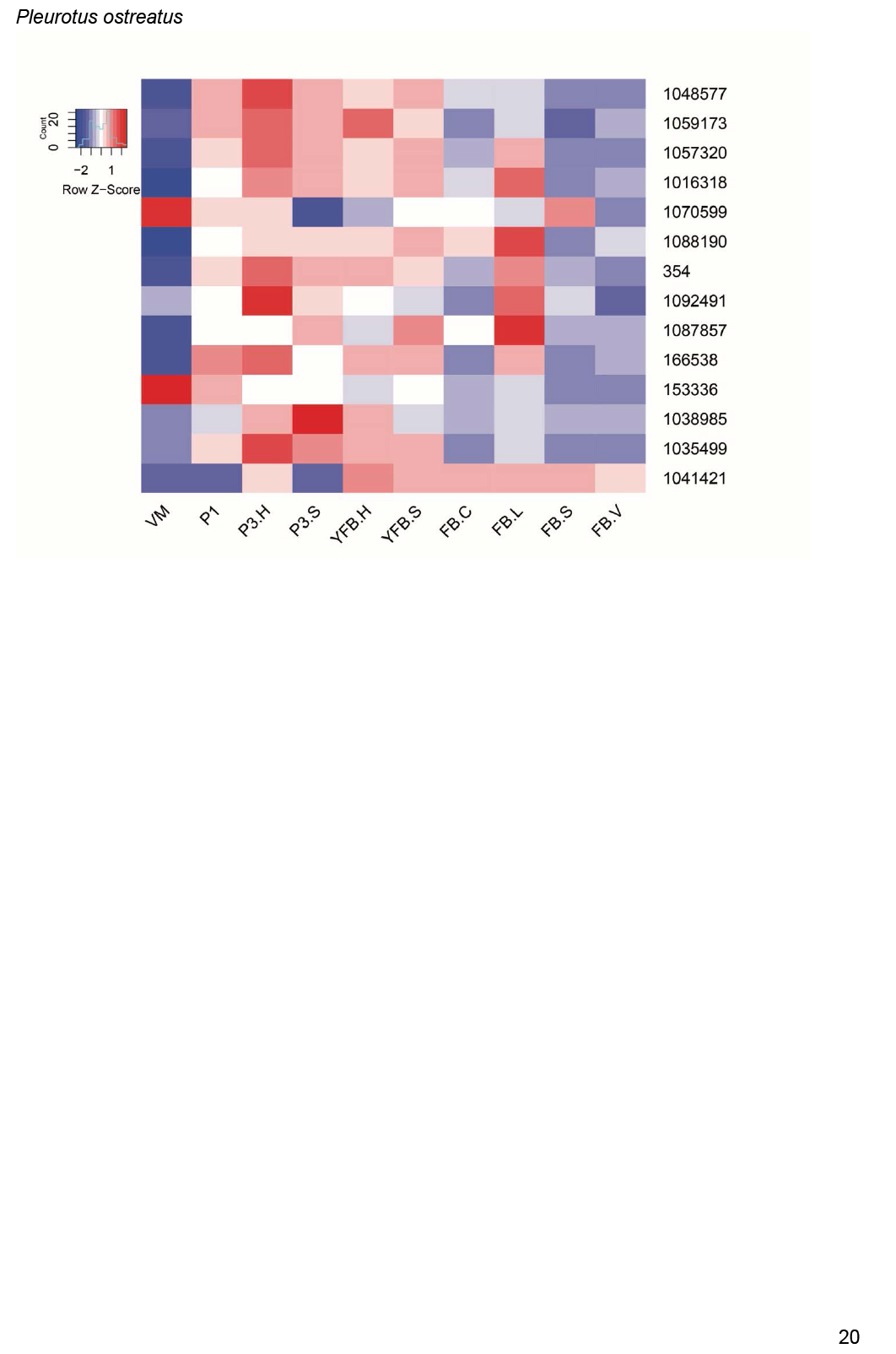{kind=link}
Expression heatmap of Con6 family cell surface protein-encoding genes in A. ostoyae, A. ampla, L. tigrinus, M. kentingensis and P. ostreatus. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively.
{kind=link}
{kind=link}
Expression heatmap of putative wax synthase encoding genes in A. ostoyae and S. commune. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively.
{kind=link}
Expression heatmap of kinesin genes and members of the Dynactin complex in A. ostoyae and P. ostreatus. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively.
{kind=link}
Table summarizing ortholog protein ID for all discussed orthogroups in twelve Agaricomycetes.
Summary of read numbers and alignments in the analysed fruiting body transcriptomes.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of cell division-related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
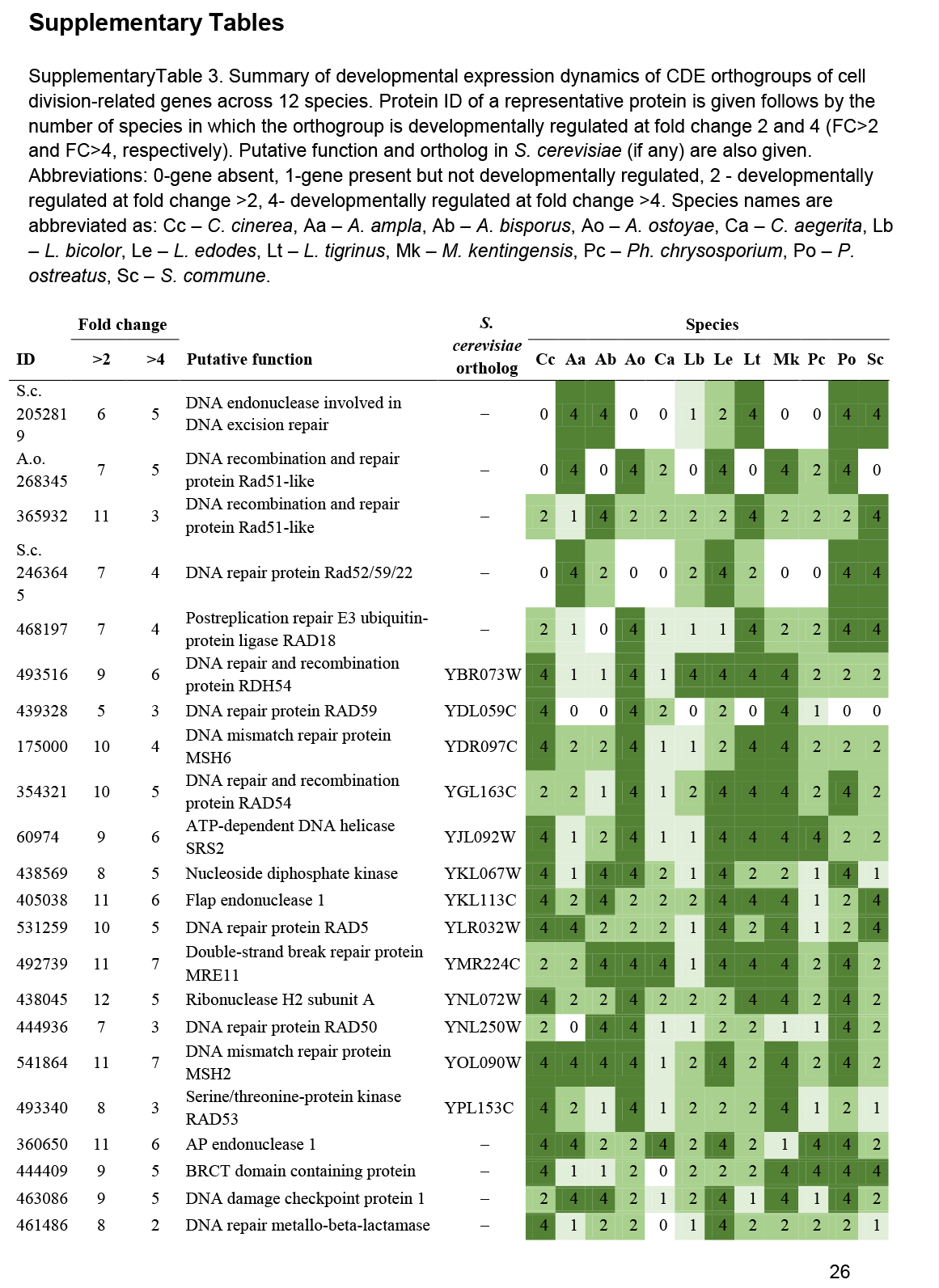{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of ribosomal genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
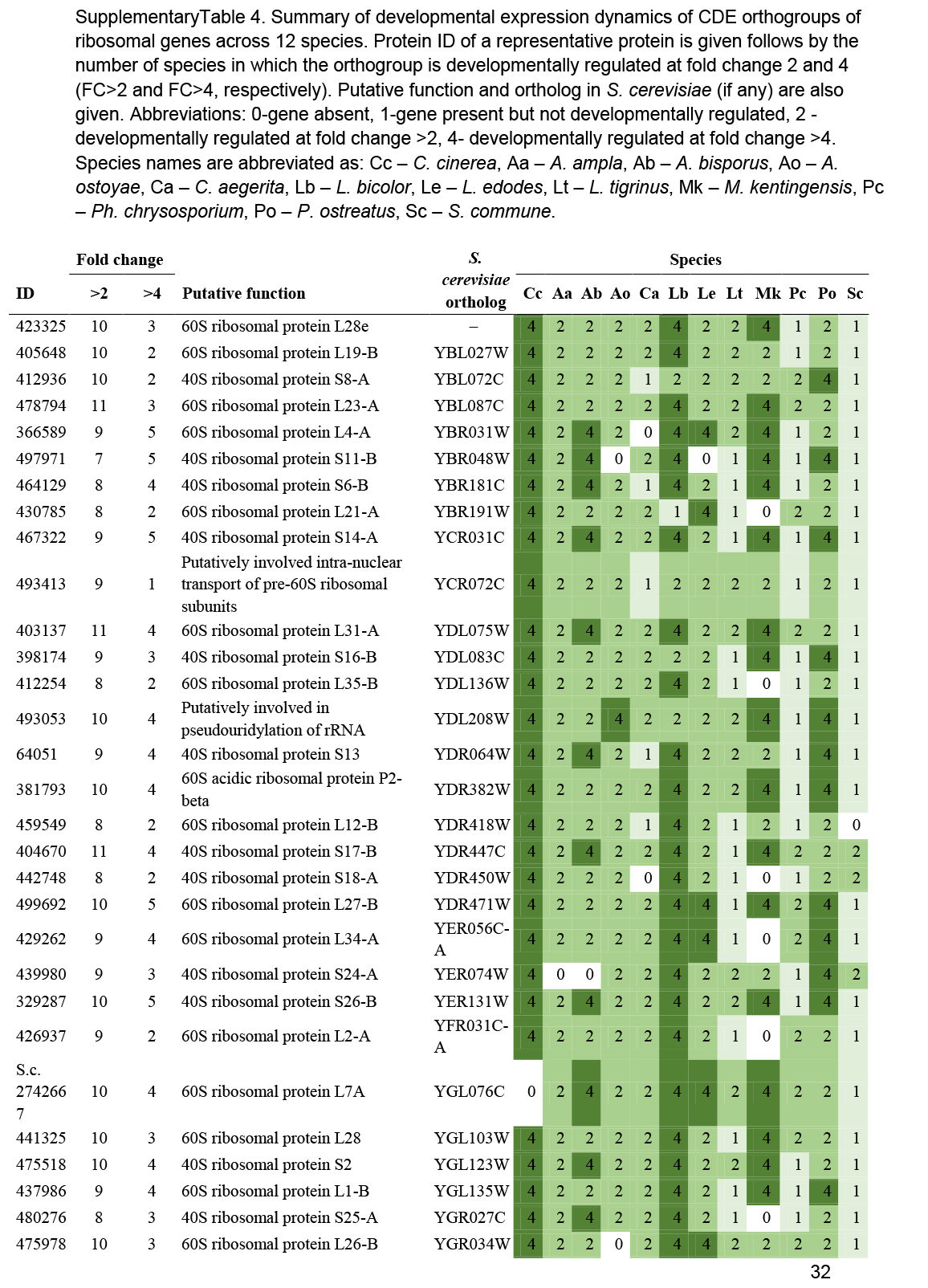{kind=link}
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of genes related to Acetyl-CoA production and basic metabolism across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
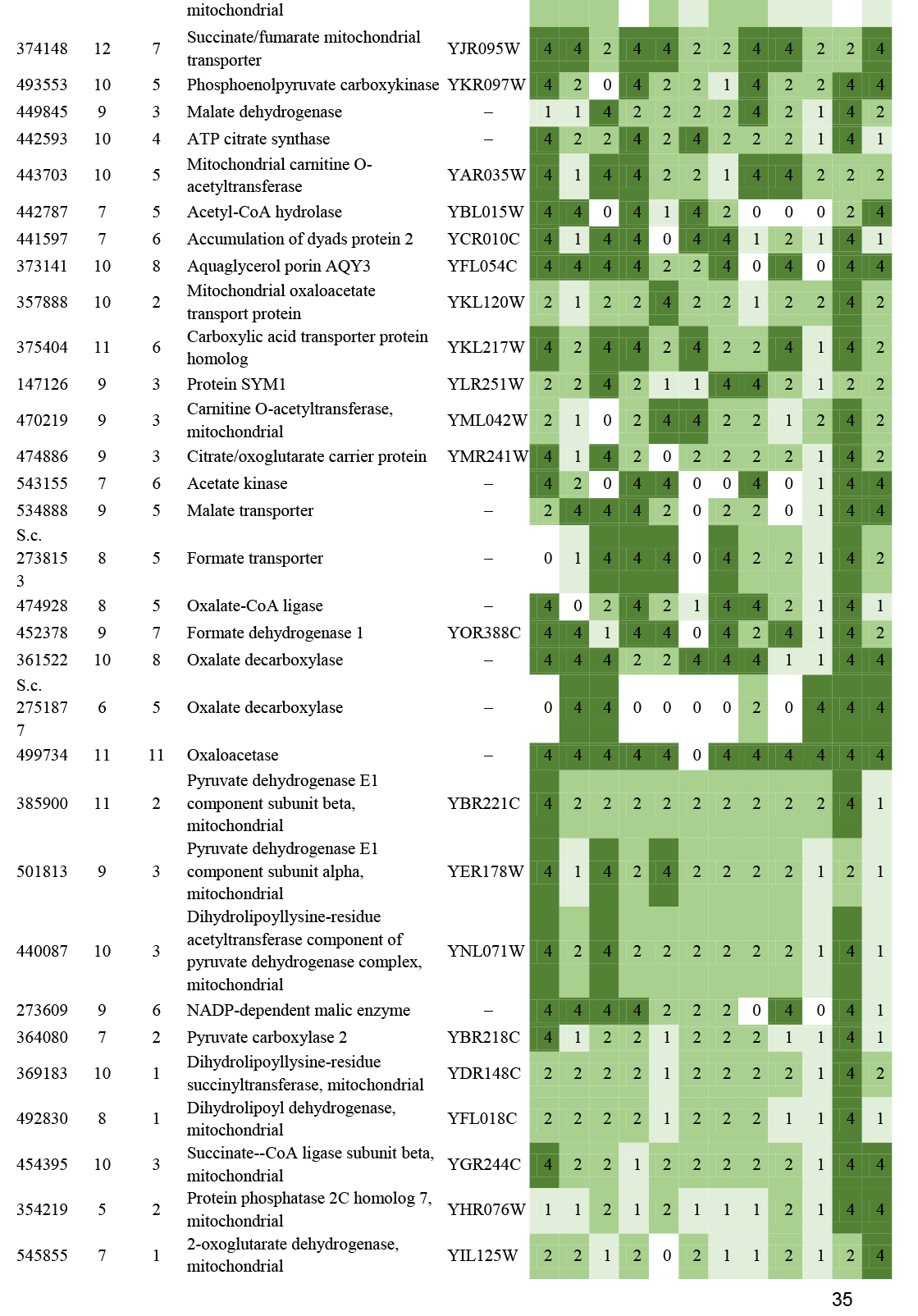{kind=link}
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of fatty acid beta-oxidation related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of ergosterol and sphingolipid biosynthesis related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of sterol transport related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
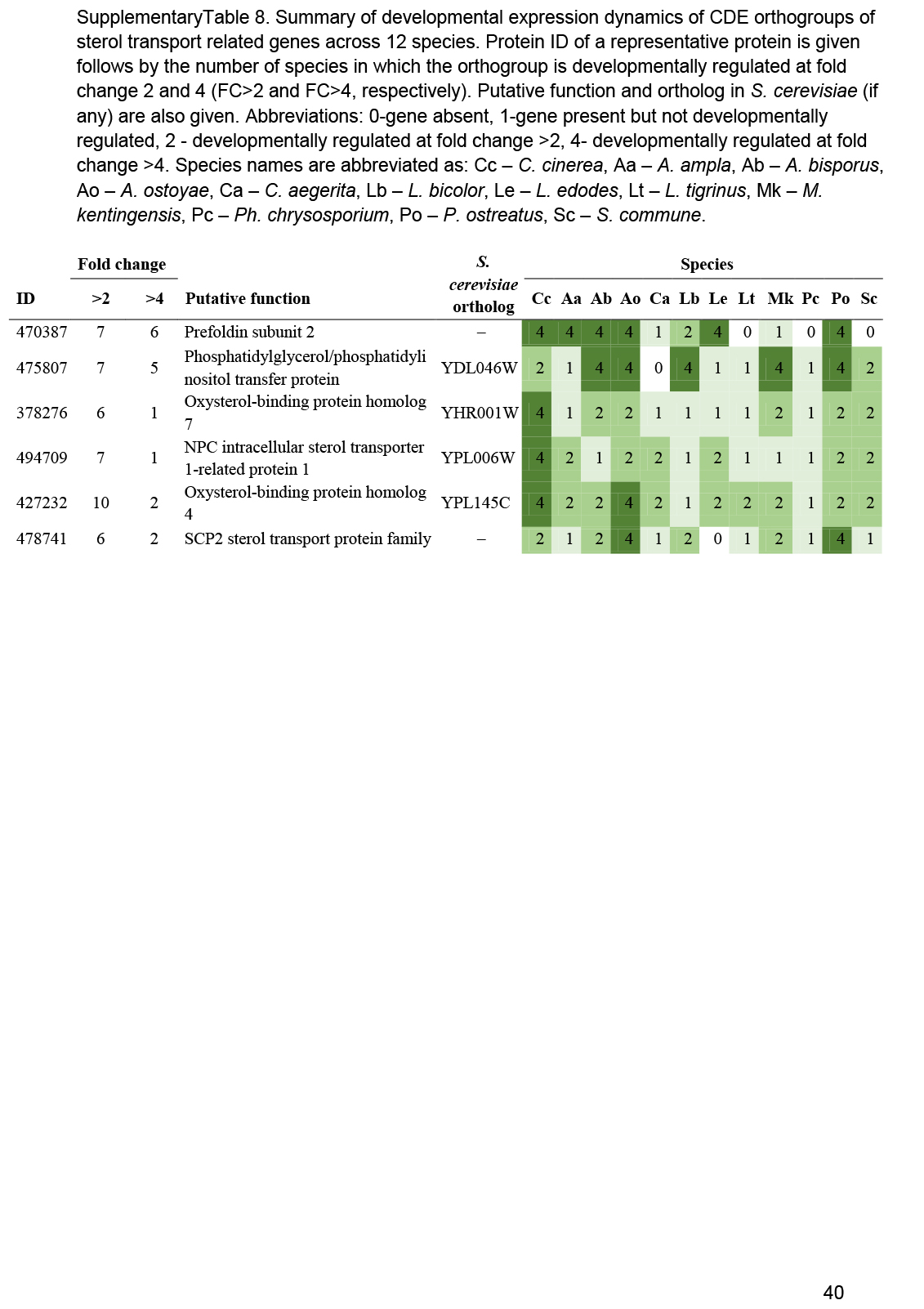{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of membrane phoshpolipid and fatty acid biosynthesis genes genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of linoleic acid biosynthesis related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of other lipid metabolism related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of storage carbohydrate metabolism genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of transcriptional regulator genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
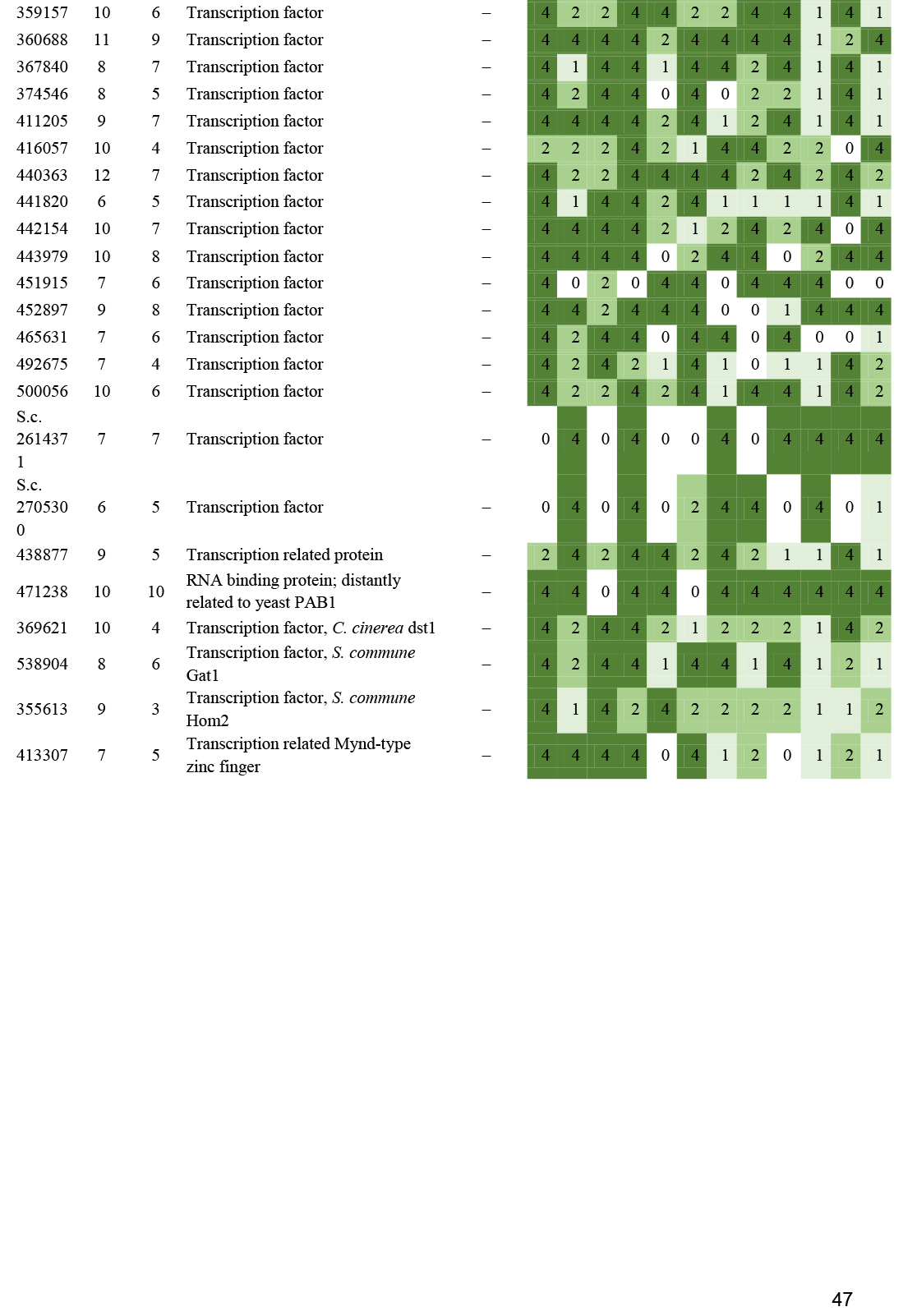{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of RNA-binding protein encoding genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of RNA interference related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
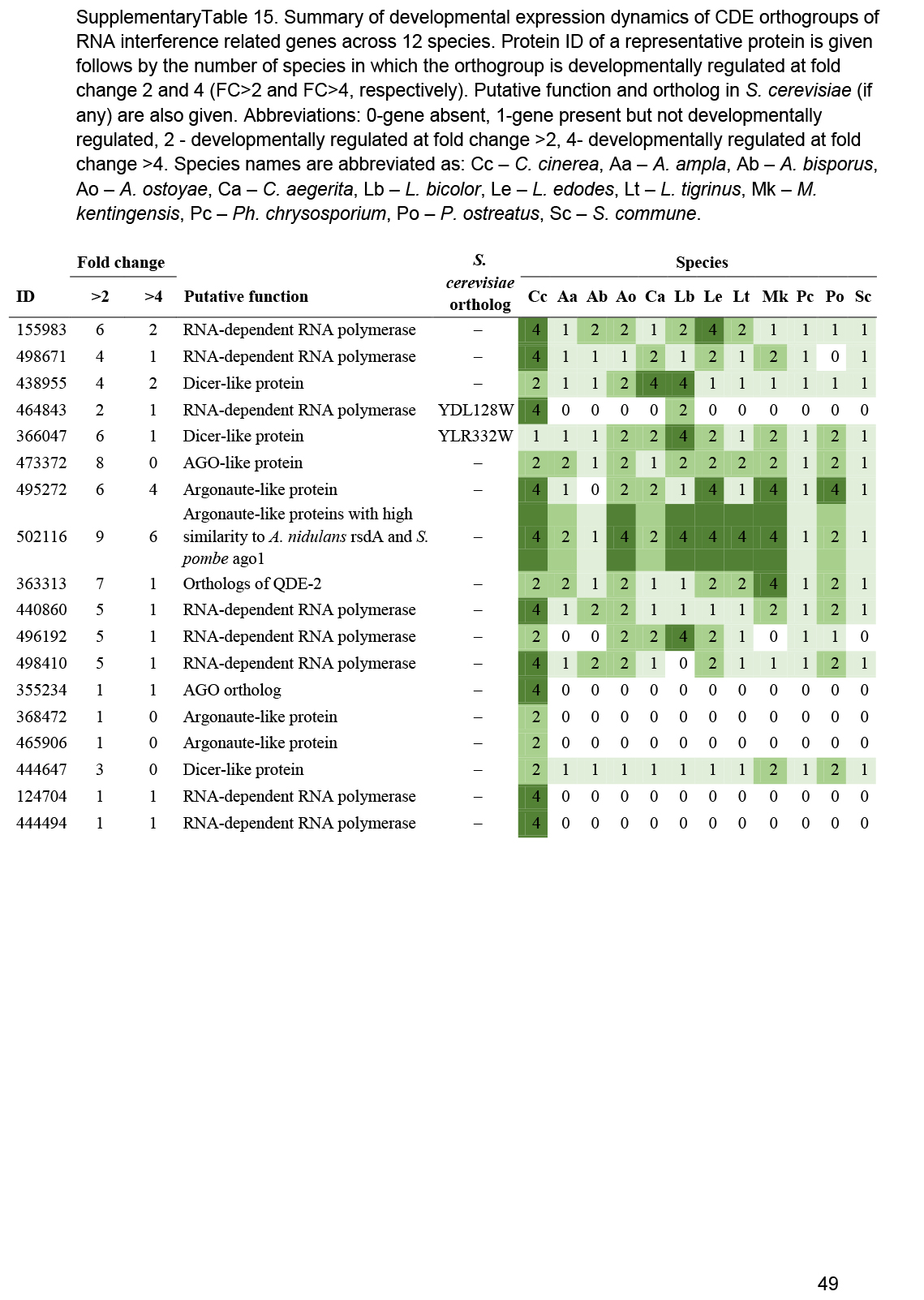{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of chromatin-related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
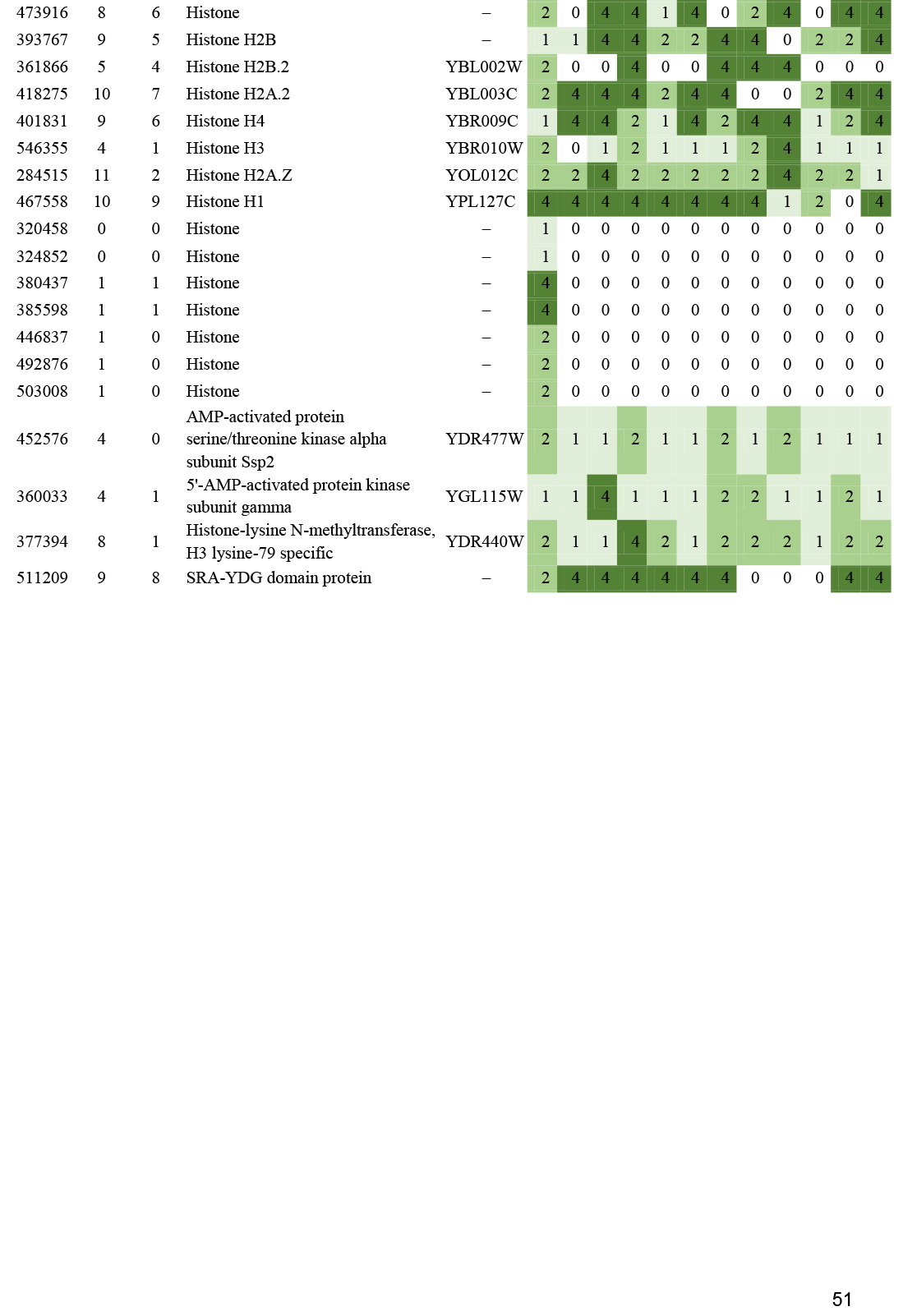{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of genes related to alternative splicing across 12 species. Protein ID of a representative protein is given follows by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4.
{kind=link}
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of signal transduction related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of pheromone precursor genes across 12 species. Protein ID (by default for <I>C.</I> cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of conserved signal transduction pathway genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
{kind=link}
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of G-protein coupled receptor genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of velvet protein encoding genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of STRIPAK complex protein genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of light sensing related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of chitin biosynthesis genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of glucan biosynthesis related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of chitin remodeling genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of glucan remodeling related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of miscellaneous cell wall remodeling genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of multicopper oxidase genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of expansin genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of miscellaneous cell wall related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of hydrophobin genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of genes belonging to conserved lectin orthogroups across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of cell surface protein encoding genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
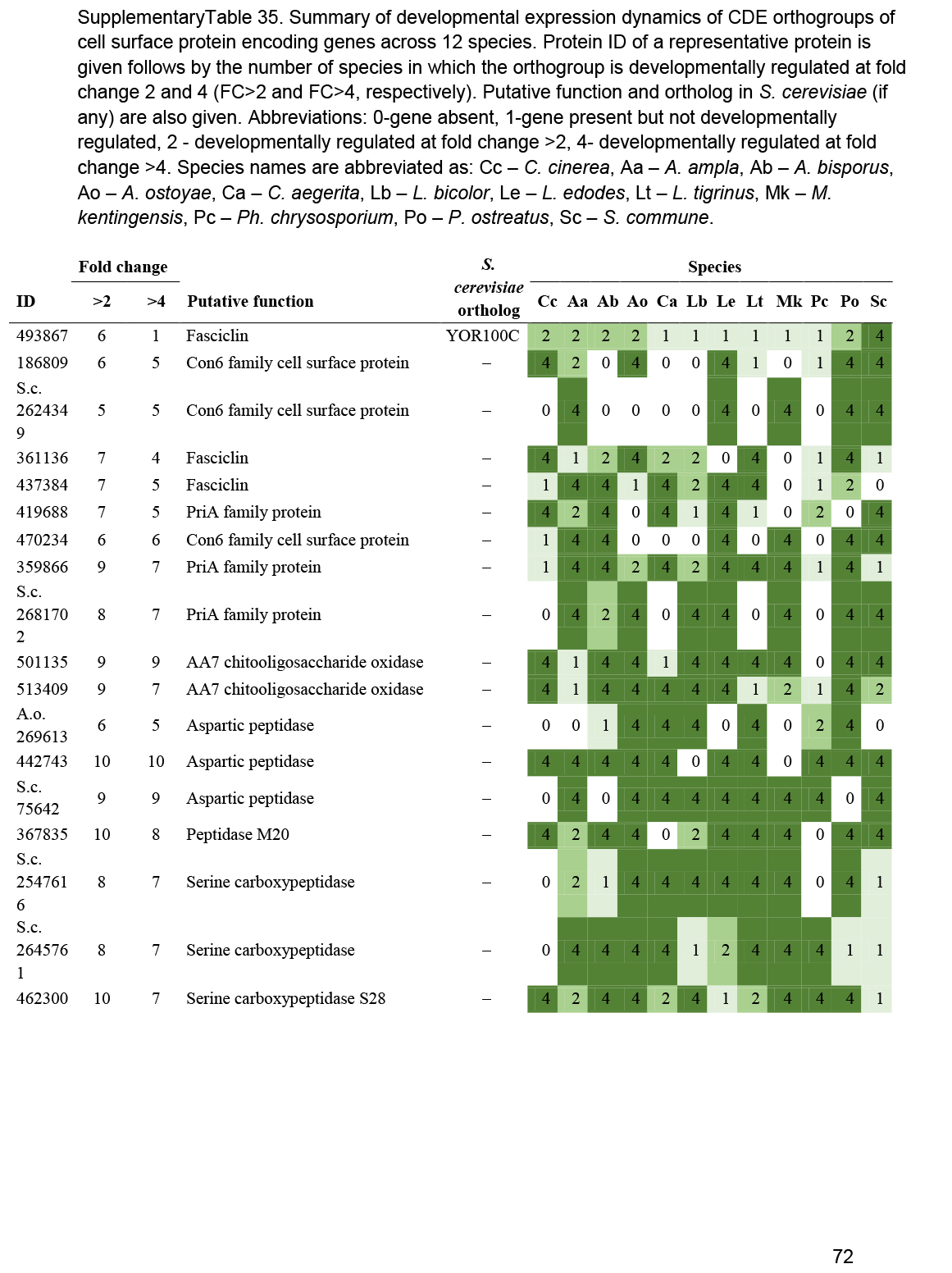{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of defense-related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of terpene synthase genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of cytoskeleton-related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of Cop9 signalosome genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Copy number distributions of genes related to protein ubiquitination and the proteasome in twelve examined Agaricomycetes.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of protein ubiquitination and the proteasome related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
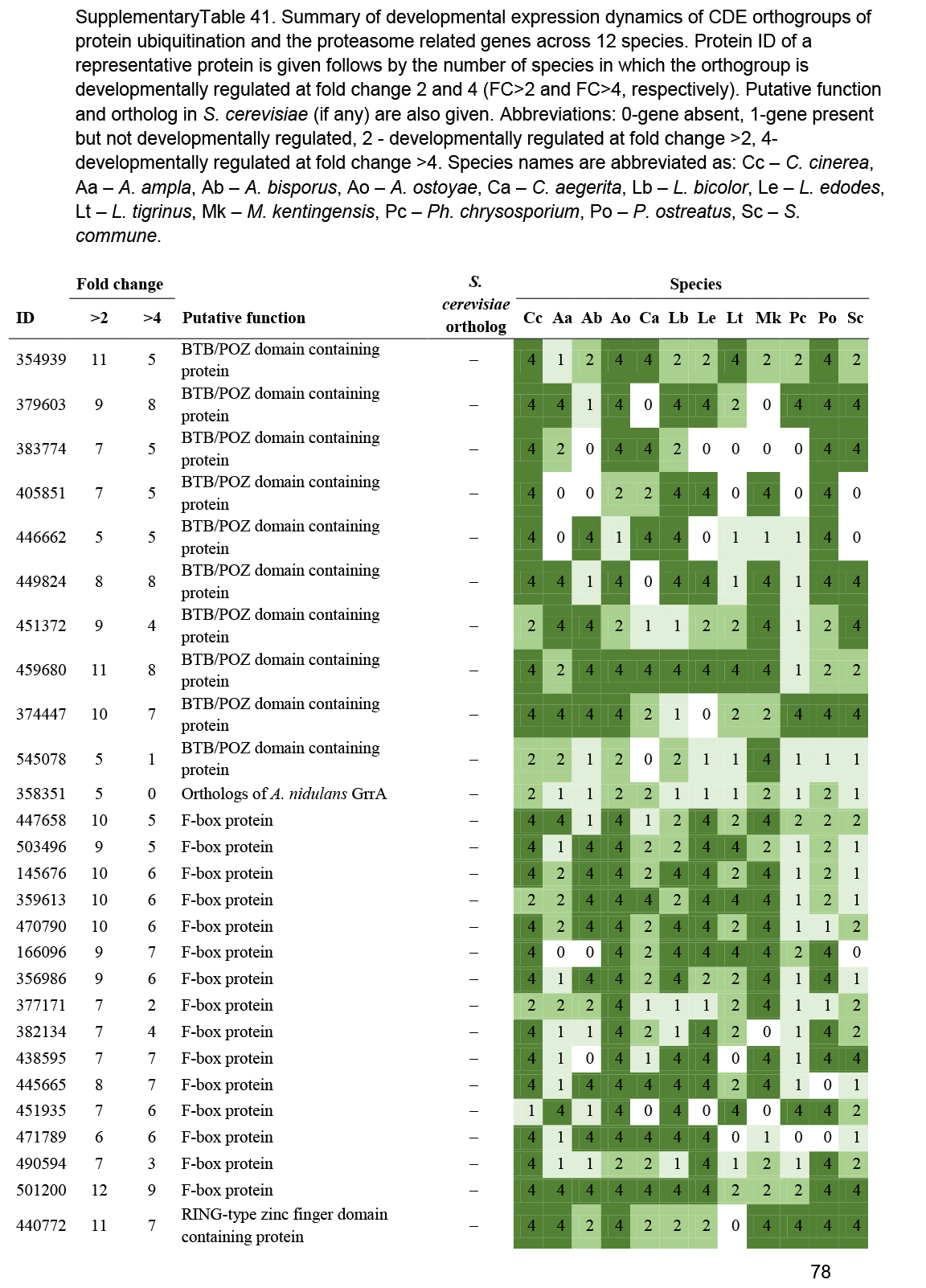{kind=link}
{kind=link}
Genes related to protein ubiquitination and the proteasome in C. cinerea.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of transporter encoding genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
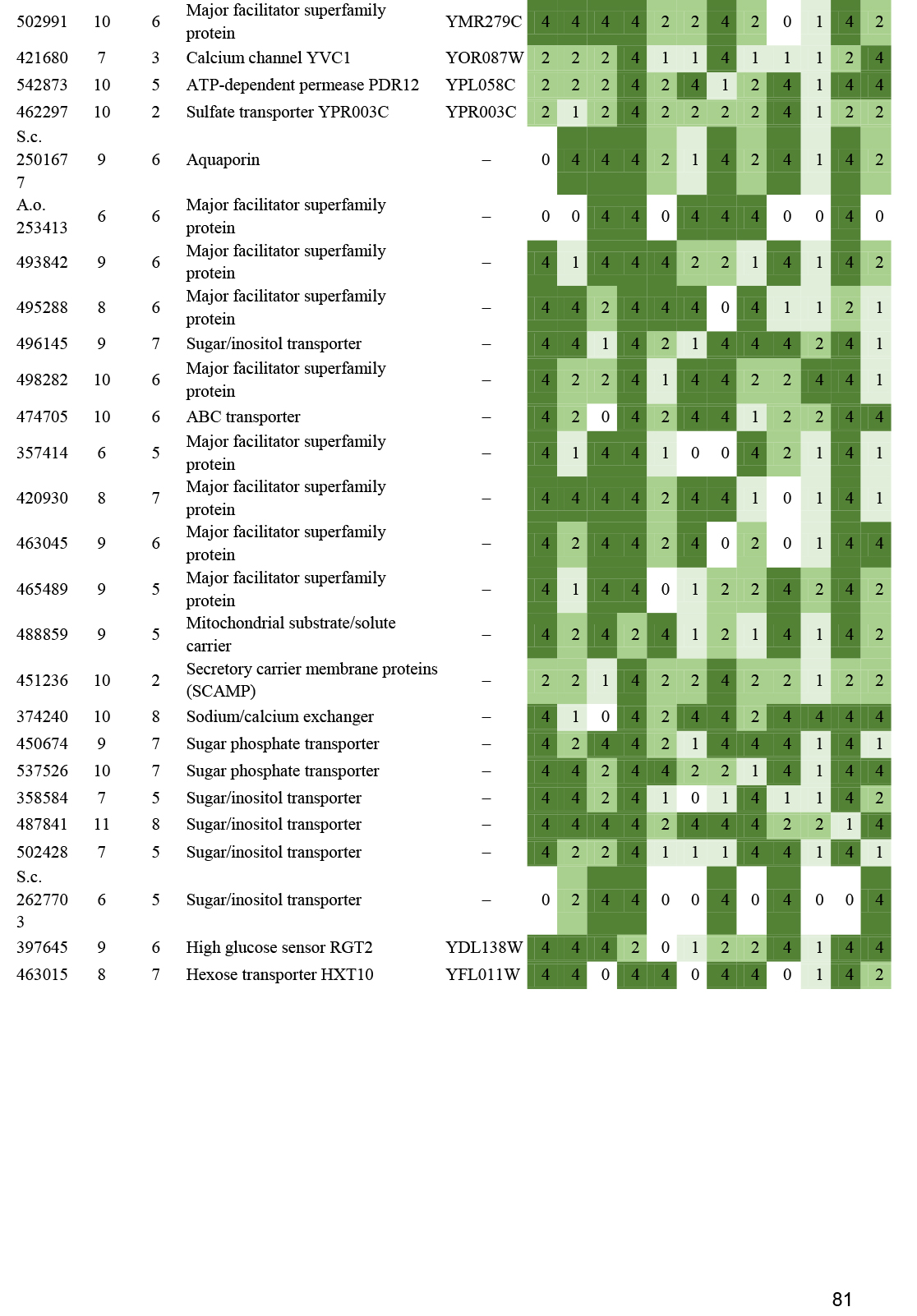{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of stress response genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of unannotated genes across 12 species. Protein ID of a representative protein is given follows by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4 - developmentally regulated at fold change >4.
{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of functionally poorly known genes across 12 species. Protein ID of a representative protein is given follows by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4.
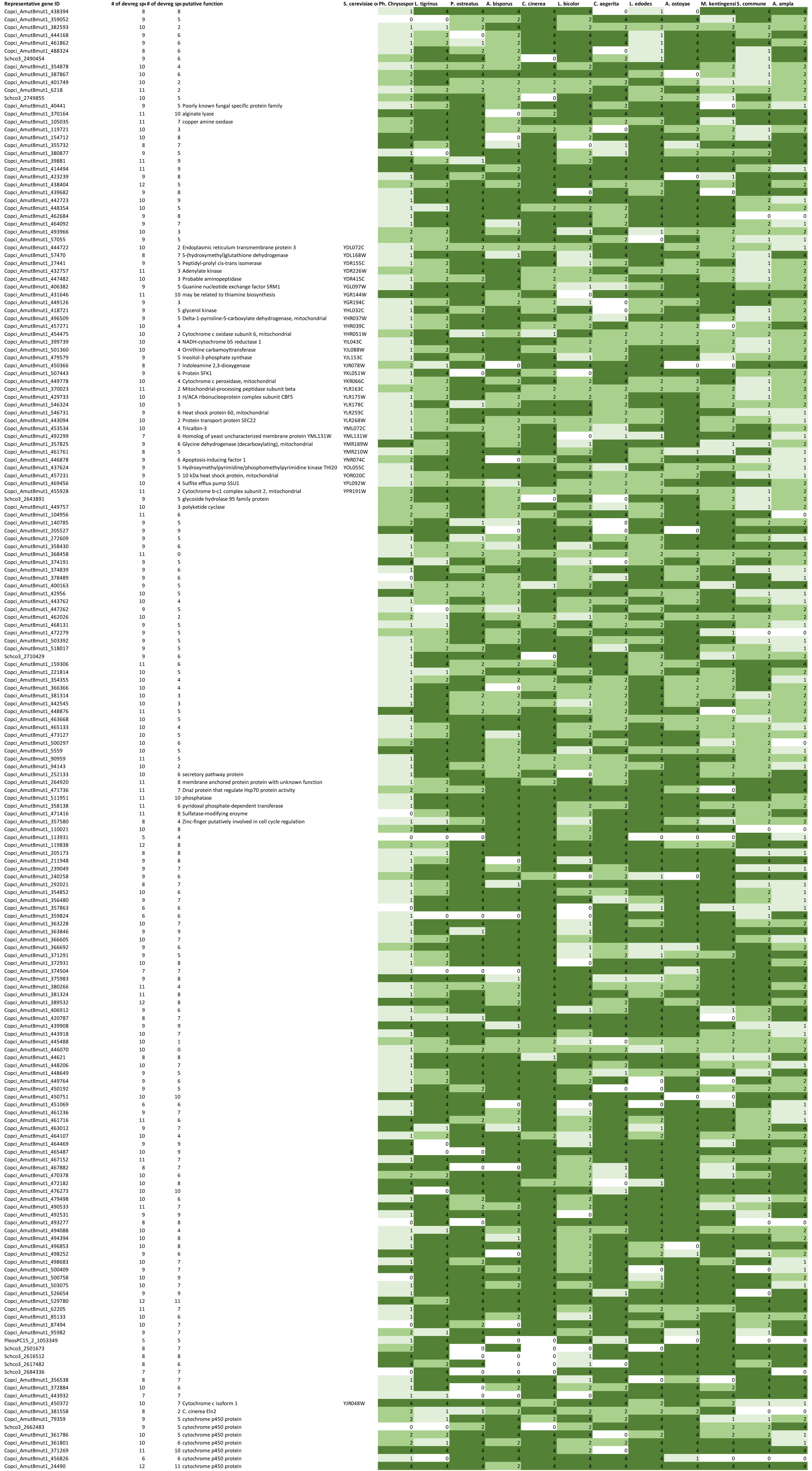{kind=link}
Summary of developmental expression dynamics of CDE orthogroups of ferric reductase genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
{kind=link}
REFERENCES
- Agger S, Lopez-Gallego F, Schmidt-Dannert C. (2009). Diversity of sesquiterpene synthases in the basidiomycete Coprinus cinereus. Molecular Microbiology 72: 1181–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed YL, Gerke J, Park HS, et al. (2013). The velvet family of fungal regulators contains a DNA-binding domain structurally similar to NF-κB. PLoS Biology 11: e1001750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akcapinar GB, Kappel L, Sezerman OU, et al. (2015). Molecular diversity of LysM carbohydrate-binding motifs in fungi. Current Genetics 61: 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcazar-Fuoli L, Clavaud C, Lamarre C, et al. (2011). Functional analysis of the fungal/plant class chitinase family in Aspergillus fumigatus. Fungal Genetics and Biology 48: 418–429. [DOI] [PubMed] [Google Scholar]
- Almási É, Sahu N, Krizsán K, et al. (2019). Comparative genomics reveals unique wood-decay strategies and fruiting body development in the Schizophyllaceae. New Phytologist 224: 902-915. [DOI] [PubMed] [Google Scholar]
- Alvarez FJ, Konopka JB. (2007). Identification of an N-acetylglucosamine transporter that mediates hyphal induction in Candida albicans. Molecular Biology of the Cell 18: 965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amako K, Fujita K, Shimohata TA, et al. (2006). NAD+-specific D-arabinose dehydrogenase and its contribution to erythroascorbic acid production in Saccharomyces cerevisiae. FEBS letters 580: 6428–6434. [DOI] [PubMed] [Google Scholar]
- Anderluh G, Maèek P. (2003) Dissecting the actinoporin pore-forming mechanism. Structure 11: 1312–1313. [DOI] [PubMed] [Google Scholar]
- Ando Y, Nakazawa T, Oka K, et al. (2013). Cc. snf5, a gene encoding a putative component of the SWI/SNF chromatin remodelling complex, is essential for sexual development in the agaricomycete Coprinopsis cinerea. Fungal Genetics and Biology 50: 82–89. [DOI] [PubMed] [Google Scholar]
- Angulo I, Acebrón I, de Las Rivas B, et al. (2011). High-resolution structural insights on the sugar-recognition and fusion tag properties of a versatile β-trefoil lectin domain from the mushroom Laetiporus sulphureus. Glycobiology 21: 1349–1361. [DOI] [PubMed] [Google Scholar]
- Arima T, Yamamoto M, Hirata A, et al. (2004). The eln3 gene involved in fruiting body morphogenesis of Coprinus cinereus encodes a putative membrane protein with a general glycosyltransferase domain. Fungal Genetics and Biology 41: 805–812. [DOI] [PubMed] [Google Scholar]
- Arroyo J, Farkaš V, Sanz AB, et al. (2016). Strengthening the fungal cell wall through chitin–glucan cross-links: effects on morphogenesis and cell integrity. Cellular Microbiology 18: 1239–1250. [DOI] [PubMed] [Google Scholar]
- Arsuffi G, Braybrook SA. (2018). Acid growth: an ongoing trip. Journal of Experimental Botany 69: 137–146. [DOI] [PubMed] [Google Scholar]
- Ashkari T, Nakamura N, Tanaka Y, et al. (1986). Rhizopus raw-starch-degrading glucoamylase: its cloning and expression in yeast. Agricultural and Biological Chemistry 50: 957–964. [Google Scholar]
- Baccelli I. (2015). Cerato-platanin family proteins: one function for multiple biological roles? Frontiers in Plant Science 5: 769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baglivo I, Ragucci S, D’Incecco P, et al. (2020). Gene organization, expression, and localization of ribotoxin-like protein Ageritin in fruiting body and mycelium of Agrocybe aegerita. International Journal of Molecular Sciences 21: 7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Wang Y, Liu X, et al. (2020). Heterologous expression and characterization of a novel chitin deacetylase, CDA3, from the mushroom Coprinopsis cinerea. International Journal of Biological Macromolecules 150: 536–545. [DOI] [PubMed] [Google Scholar]
- Bairwa G, Jung WH, Kronstad JW. (2017). Iron acquisition in fungal pathogens of humans. Metallomics 9: 215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker LG, Specht CA, Donlin MJ, et al. (2007). Chitosan, the deacetylated form of chitin, is necessary for cell wall integrity in Cryptococcus neoformans. Eukaryotic Cell 6: 855–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldrian P. (2006). Fungal laccases–occurrence and properties. FEMS Microbiology Reviews 30: 215–242. [DOI] [PubMed] [Google Scholar]
- Banks IR, Specht CA, Donlin MJ, et al. (2005). A chitin synthase and its regulator protein are critical for chitosan production and growth of the fungal pathogen Cryptococcus neoformans. Eukaryotic Cell 4: 1902–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett DW, Clough JM, Godwin JR, et al. (2002). The strobilurin fungicides. Pest Management Science: formerly Pesticide Science 58: 649–662. [DOI] [PubMed] [Google Scholar]
- Bartoš O, Röslein J, Kotusz J, et al. (2019). The legacy of sexual ancestors in phenotypic variability, gene expression, and homoeolog regulation of asexual hybrids and polyploids. Molecular Biology and Evolution 36: 1902–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayram O, Krappmann S, Ni M, et al. (2008). VelB/VeA/LaeA complex coordinates light signal with fungal development and secondary metabolism. Science 320: 1504–1506. [DOI] [PubMed] [Google Scholar]
- Bayry J, Aimanianda V, Guijarro JI, et al. (2012). Hydrophobins—unique fungal proteins. PLoS Pathogens 8: e1002700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello MH, Barrera-Perez V, Morin D, et al. (2012). The Neurospora crassa mutant NcΔEgt-1 identifies an ergothioneine biosynthetic gene and demonstrates that ergothioneine enhances conidial survival and protects against peroxide toxicity during conidial germination. Fungal Genetics and Biology 49: 160–172. [DOI] [PubMed] [Google Scholar]
- Berends E, Ohm RA, de Jong JF, et al. (2009). Genomic and biochemical analysis of N glycosylation in the mushroom-forming basidiomycete Schizophyllum commune. Applied and Environmental Microbiology 75: 4648–4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen RL, Kalkhove SI, Lugones LG, et al. (2013). Effects of the mushroom-volatile 1-octen-3-ol on dry bubble disease. Applied Microbiology and Biotechnology 97: 5535–5543. [DOI] [PubMed] [Google Scholar]
- Berne S, Pohleven J, Vidic I, et al. (2007). Ostreolysin enhances fruiting initiation in the oyster mushroom (Pleurotus ostreatus). Mycological Research 111: 1431–1436. [DOI] [PubMed] [Google Scholar]
- Bertani B, Ruiz N. (2018). Function and biogenesis of lipopolysaccharides. EcoSal Plus 8: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertossa RC, Kües U, Aebi M, et al. (2004). Promoter analysis of cgl2, a galectin encoding gene transcribed during fruiting body formation in Coprinopsis cinerea (Coprinus cinereus). Fungal Genetics and Biology 41: 1120–1131. [DOI] [PubMed] [Google Scholar]
- Bewick AJ, Hofmeister BT, Powers RA, et al. (2019). Diversity of cytosine methylation across the fungal tree of life. Nature Ecology & Evolution 3: 479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bien CM, Espenshade PJ. (2010). Sterol regulatory element binding proteins in fungi: hypoxic transcription factors linked to pathogenesis. Eukaryotic Cell 9: 352–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieszke JA, Braun EL, Bean LE, et al. (1999). The nop-1 gene of Neurospora crassa encodes a seven transmembrane helix retinal-binding protein homologous to archaeal rhodopsins. Proceedings of the National Academy of Sciences 96: 8034–8039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birck C, Damian L, Marty-Detraves C, et al. (2004). A new lectin family with structure similarity to actinoporins revealed by the crystal structure of Xerocomus chrysenteron lectin XCL. Journal of Molecular Biology 344: 1409–1420. [DOI] [PubMed] [Google Scholar]
- Blackstone NW. (2000). Redox control and the evolution of multicellularity. Bioessays 22: 947–953. [DOI] [PubMed] [Google Scholar]
- Bleuler-Martínez S, Butschi A, Garbani M, et al. (2011). A lectin-mediated resistance of higher fungi against predators and parasites. Molecular Ecology 20: 3056–3070. [DOI] [PubMed] [Google Scholar]
- Bleuler-Martinez S, Schmieder S, Aebi M, et al. (2012). Biotin-binding proteins in the defence of mushrooms against predators and parasites. Applied and Environmental Microbiology 78: 8485–8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleuler-Martinez S, Stutz K, Sieber R, et al. (2017). Dimerization of the fungal defence lectin CCL2 is essential for its toxicity against nematodes. Glycobiology 27: 486–500. [DOI] [PubMed] [Google Scholar]
- Bloemendal S, Kück U. (2013.) Cell-to-cell communication in plants, animals, and fungi: a comparative review. Naturwissenschaften 100: 3–19. [DOI] [PubMed] [Google Scholar]
- Blumenstein A, Vienken K, Tasler R, et al. (2005). The Aspergillus nidulans phytochrome FphA represses sexual development in red light. Current Biology 15: 1833–1838. [DOI] [PubMed] [Google Scholar]
- Bojunga N, Kötter P, Entian KD. (1998). The succinate/fumarate transporter Acr1p of Saccharomyces cerevisiae is part of the gluconeogenic pathway and its expression is regulated by Cat8p. Molecular and General Genetics MGG 260: 453–461. [DOI] [PubMed] [Google Scholar]
- Boontawon T, Nakazawa T, Horii M, et al. (2021). Functional analyses of Pleurotus ostreatus pcc1 and clp1 using CRISPR/Cas9. Fungal Genetics and Biology 154: 103599. [DOI] [PubMed] [Google Scholar]
- Borgognone A, Castanera R, Morselli M, et al. (2018). Transposon-associated epigenetic silencing during Pleurotus ostreatus life cycle. DNA Research 25: 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkovich KA, Alex LA, Yarden O, et al. (2004). Lessons from the genome sequence of Neurospora crassa: tracing the path from genomic blueprint to multicellular organism. Microbiology and Molecular Biology Reviews 68: 1–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulianne RP, Liu Y, Aebi M, et al. (2000). Fruiting body development in Coprinus cinereus: regulated expression of two galectins secreted by a non-classical. Microbiology 146: 1841–1853. [DOI] [PubMed] [Google Scholar]
- Braus GH, Irniger S, Bayram Ö. (2010). Fungal development and the COP9 signalosome. Current opinion in microbiology 13: 672–676. [DOI] [PubMed] [Google Scholar]
- Breitenbach M, Weber M, Rinnerthaler M, et al. (2015). Oxidative stress in fungi: its function in signal transduction, interaction with plant hosts, and lignocellulose degradation. Biomolecules 5: 318–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodhun F, Feussner I. (2011). Oxylipins in fungi. The FEBS Journal 278: 1047–1063. [DOI] [PubMed] [Google Scholar]
- Brown JL, Bussey H. (1993). The yeast KRE9 gene encodes an O glycoprotein involved in cell surface beta-glucan assembly. Molecular and Cellular Biology 13: 6346–6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NA, Schrevens S, Van Dijck P, et al. (2018). Fungal G-protein-coupled receptors: Mediators of pathogenesis and targets for disease control. Nature Microbiology 3: 402–414. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, et al. (2015). ATAC-seq: A method for assaying chromatin accessibility genome-wide. Current Protocols in Molecular Biology 109: 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhian WP, Bensmihen S. (2018). Mini-Review: Nod Factor Regulation of Phytohormone signalling and Homeostasis During Rhizobia-Legume Symbiosis. Frontiers in Plant Science 9: 1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns C, Stajich JE, Rechtsteiner A, et al. (2010). Analysis of the Basidiomycete Coprinopsis cinerea Reveals Conservation of the Core Meiotic Expression Program over Half a Billion Years of Evolution. PLoS Genetics 6: e1001135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch S, Eckert SE, Krappmann S, et al. (2003). The COP9 signalosome is an essential regulator of development in the filamentous fungus Aspergillus nidulans. Molecular Microbiology 49: 717–730. [DOI] [PubMed] [Google Scholar]
- Busch S, Schwier EU, Nahlik K, et al. (2007). An eight-subunit COP9 signalosome with an intact JAMM motif is required for fungal fruit body formation. Proceedings of the National Academy of Sciences 104: 8089–8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buser R, Lazar Z, Käser S, et al. (2010). Identification, characterization, and biosynthesis of a novel N-glycan modification in the fruiting body of the basidiomycete Coprinopsis cinerea. Journal of Biological Chemistry 285: 10715–10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butschi A, Titz A, Wälti MA, et al. (2010). Caenorhabditis elegans N-glycan core β-galactoside confers sensitivity towards nematotoxic fungal galectin CGL2. PLoS Pathogens 6: e1000717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell GA, Naider F, Becker J. M. (1995). Fungal lipopeptide mating pheromones: a model system for the study of protein prenylation. Microbiological Reviews 59: 406–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caligiuri M, Connolly T, Beach D. (1997). Ran1 functions to control the Cdc10/Sct1 complex through Puc1. Molecular Biology of the Cell 8: 1117–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo S, Nicolás FE, Vila A, et al. (2012). Two distinct RNA-dependent RNA polymerases are required for initiation and amplification of RNA silencing in the basal fungus Mucor circinelloides. Molecular Microbiology 83: 379–394. [DOI] [PubMed] [Google Scholar]
- Campbell LT, Padula MP, Harry E, et al. (2015). You are what you secrete: extracellular proteins and virulence in Cryptococcus. Microbiology Australia 36: 93–95. [Google Scholar]
- Cao, Chan C, Lee C, Wong SS, et al. (1998). MP1 encodes an abundant and highly antigenic cell wall mannoprotein in the pathogenic fungus Penicillium marneffei. Infection and Immunity 66: 966–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman GM, Han GS. (2011). Regulation of phospholipid synthesis in the yeast Saccharomyces cerevisiae. Annual Review of Biochemistry 80: 859–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael JB, Provost P, Ekwall K, et al. (2004). ago1 and dcr1, two core components of the RNA interference pathway, functionally diverge from rdp1 in regulating cell cycle events in Schizosaccharomyces pombe. Molecular Biology of the Cell 15: 1425–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casal M, Paiva S, Andrade RP, et al. (1999). The lactate-proton symport of Saccharomyces cerevisiae is encoded by JEN1. Journal of Bacteriology 181: 2620–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cevallos JA, Okubo RP, Perlman SJ, et al. (2017). Olfactory preferences of the parasitic nematode Howardula aoronymphium and its insect host Drosophila falleni. Journal of Chemical Ecology 43: 362–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha MK, Choi YS, Hong SK, et al. (2003). Nuclear thiol peroxidase as a functional alkyl-hydroperoxide reductase necessary for stationary phase growth of Saccharomyces cerevisiae. Journal of Biological Chemistry 278: 24636–24643. [DOI] [PubMed] [Google Scholar]
- Chae E, Tan QKG, Hill TA, et al. (2008). An Arabidopsis F-box protein acts as a transcriptional co-factor to regulate floral development. Development 135: 1235–45. [DOI] [PubMed] [Google Scholar]
- Chakraborty TK, Basu D, Das N, et al. (2004). The mannitol cycle in Pleurotus ostreatus (Florida). FEMS Microbiology Letters 236: 307–311. [DOI] [PubMed] [Google Scholar]
- Chang J, Chan PL, Xie Y, et al. (2019a). Modified recipe to inhibit fruiting body formation for living fungal biomaterial manufacture. PLoS One 14: e0209812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Desirò A, Na H, et al. (2019b). Phylogenomics of Endogonaceae and evolution of mycorrhizas within Mucoromycota. New Phytologist 222: 511–525. [DOI] [PubMed] [Google Scholar]
- Chen DD, Shi L, Yue SN, et al. (2019). The Slt2-MAPK pathway is involved in the mechanism by which target of rapamycin regulates cell wall components in Ganoderma lucidum. Fungal Genetics and Biology 123: 70–77. [DOI] [PubMed] [Google Scholar]
- Chen H, Kovalchuk A, Keriö S, et al. (2017). Distribution and bioinformatic analysis of the cerato-platanin protein family in Dikarya. Mycologia 105: 1479–1488. [DOI] [PubMed] [Google Scholar]
- Chen J, Li JM, Tang YJ, et al. (2020). Genome-wide analysis and prediction of genes involved in the biosynthesis of polysaccharides and bioactive secondary metabolites in high-temperature-tolerant wild Flammulina filiformis. BMC Genomics 21: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Gong Y, Cai Y, et al. (2016). Genome sequence of the edible cultivated mushroom Lentinula edodes (Shiitake) reveals insights into lignocellulose degradation. PLoS One 11: e0160336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Ma D, GeW, et al. (2003). Induction of laccase activity in the edible straw mushroom, Volvariella volvacea. FEMS Microbiology Letters 218: 143–148. [DOI] [PubMed] [Google Scholar]
- Chen S, Xu J, Liu C, et al. (2012). Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nature Communications 3: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhang Y, Siewers V, et al. (2015). Ach1 is involved in shuttling mitochondrial acetyl units for cytosolic C2 provision in Saccharomyces cerevisiae lacking pyruvate decarboxylase. FEMS Yeast Research 15: fov015. [DOI] [PubMed] [Google Scholar]
- Cheng CK, Au CH, Wilke SK, et al. (2013). 5’-Serial Analysis of Gene Expression studies reveal a transcriptomic switch during fruiting body development in Coprinopsis cinerea. BMC Genomics 14: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Hui JHL, Le YY, et al. (2015). A “developmental hourglass” in fungi. Molecular Biology and Evolution 32: 1556–1566. [DOI] [PubMed] [Google Scholar]
- Cherry JM, Hong EL, Amundsen C, et al. (2012). Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Research 40: D700–D705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitarra GS, Abee T, Rombouts FM, et al. (2004). Germination of Penicillium paneum conidia is regulated by 1-octen-3-ol, a volatile self-inhibitor. Applied and Environmental Microbiology 70: 2823–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu EYH, Lin YH, Wu W, et al. (2010). Alternative splicing and genetic diversity of the white collar-1 (wc-1) gene in cereal Phaeosphaeria pathogens. European Journal of Plant Pathology 127: 351–363. [Google Scholar]
- Christeller JT, Markwick NP, Burgess EPJ, et al. (2010). The use of biotin-binding proteins for insect control. Journal of Economic Entomology 103: 497–508. [DOI] [PubMed] [Google Scholar]
- Chu S, DeRisi J, Eisen M, et al. (1998). The transcriptional program of sporulation in budding yeast. Science 282: 699–705. [DOI] [PubMed] [Google Scholar]
- Chum WWY, Ng KTP, Shih RSM, et al. (2008). Gene expression studies of the dikaryotic mycelium and primordium of Lentinula edodes by serial analysis of gene expression. Mycological Research 112: 950–964. [DOI] [PubMed] [Google Scholar]
- Cirauqui N, Abriata LA, Van Der Goot FG, et al. (2017). Structural, physicochemical and dynamic features conserved within the aerolysin pore-forming toxin family. Scientific Reports 7: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Kupiec R, Broglie KE, Friesem D, et al. (1999). Molecular characterization of a novel β-1, 3-exoglucanase related to mycoparasitism of Trichoderma harzianum. Gene 226: 147–154. [DOI] [PubMed] [Google Scholar]
- Combet E, Eastwood DC, Burton KS, et al. (2006). Eight-carbon volatiles in mushrooms and fungi: properties, analysis, and biosynthesis. Mycoscience 47: 317–326. [Google Scholar]
- Combet E, Henderson J, Eastwood DC, et al. (2009). Influence of sporophore development, damage, storage, and tissue specificity on the enzymic formation of volatiles in mushrooms (Agaricus bisporus). Journal of Agricultural and Food Chemistry 57: 3709–3717. [DOI] [PubMed] [Google Scholar]
- Corrochano LM. (2011). Fungal photobiology: a synopsis. IMA fungus 2: 25–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrochano LM. (2019). Light in the fungal world: from photoreception to gene transcription and beyond. Annual Review of Genetics 53: 149–170. [DOI] [PubMed] [Google Scholar]
- Costa ASMB, Thomas DJI., Eastwood D, et al. (2009). Quantifiable downregulation of endogenous genes in Agaricus bisporus mediated by expression of RNA hairpins. Journal of Microbiology and Biotechnology 19: 271–276. [PubMed] [Google Scholar]
- Costachel C, Coddeville B, Latgé JP, et al. (2005). Glycosylphosphatidylinositol-anchored fungal polysaccharide in Aspergillus fumigatus. Journal of Biological Chemistry 280: 39835–39842. [DOI] [PubMed] [Google Scholar]
- Courty PE, Hoegger PJ, Kilaru S, et al. (2009). Phylogenetic analysis, genomic organization, and expression analysis of multi-copper oxidases in the ectomycorrhizal basidiomycete Laccaria bicolor. New Phytologist 182: 736–750. [DOI] [PubMed] [Google Scholar]
- Couturier M, Ladevèze S, Sulzenbacher G, et al. (2018). Lytic xylan oxidases from wood-decay fungi unlock biomass degradation. Nature Chemical Biology 14: 306–310. [DOI] [PubMed] [Google Scholar]
- Craig GD, Gull K, Wood DA. (1977). Stipe elongation in Agaricus bisporus. Microbiology 102: 337–347. [Google Scholar]
- Crestini C, Kovac B, Giovannozzi-Sermanni G. (1996). Production and isolation of chitosan by submerged and solid-state fermentation from Lentinus edodes. Biotechnology and Bioengineering 50: 207–210. [DOI] [PubMed] [Google Scholar]
- Cummings WJ, Merino ST, Young KG, et al. (2002). The Coprinus cinereus adherin Rad9 functions in Mre11-dependent DNA repair, meiotic sister-chromatid cohesion, and meiotic homolog pairing. Proceedings of the National Academy of Sciences 99:14958–14963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza CA, Heitman J. (2001). Conserved cAMP signalling cascades regulate fungal development and virulence. FEMS Microbiology Reviews 25: 349–364. [DOI] [PubMed] [Google Scholar]
- da Rocha Campos AN, Costa MD. (2010). Basidiosporogenesis, meiosis, and post-meiotic mitosis in the ectomycorrhizal fungus Pisolithus microcarpus. Fungal Genetics and Biology 47: 477–483. [DOI] [PubMed] [Google Scholar]
- Dai Y, Sun L, Yin X, et al. (2019). Pleurotus eryngii genomes reveal evolution and adaptation to the Gobi desert environment. Frontiers in Microbiology 10: 2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Peraro M, Van Der Goot FG. (2016). Pore-forming toxins: ancient, but never really out of fashion. Nature Reviews Microbiology 14: 77–92. [DOI] [PubMed] [Google Scholar]
- Dang Y, Yang Q, Xue Z, et al. (2011). RNA Interference in Fungi: Pathways, Functions, and Applications. Eukaryotic Cell 10: 1148–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwiche R, Mène-Saffrané L, Gfeller D, et al. (2017). The pathogen-related yeast protein Pry1, a member of the CAP protein superfamily, is a fatty acid-binding protein. Journal of Biological Chemistry 292: 8304–8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das N, Sengupta S, Mukherjee M. (1997). Importance of laccase in vegetative growth of Pleurotus florida. Applied and Environmental Microbiology 63: 4120–4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bary A, Balfour IB, Garnsey HEF. (1887) Comparative morphology and biology of the Fungi, Mycetozoa and bacteria. Oxford: Clarendon press [Google Scholar]
- de Freitas Pereira M, Narvaes da Rocha Campos A, Anastacio TC, et al. (2017). The transcriptional landscape of basidiosporogenesis in mature Pisolithus microcarpus basidiocarp. BMC Genomics 18: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong JF, Deelstra HJ, Wösten HAB, et al. (2006). RNA-mediated gene silencing in monokaryons and dikaryons of Schizophyllum commune. Applied and Environmental Microbiology 72: 1267–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jong JF, Ohm RA, De Bekker C, et al. (2010). Inactivation of ku80 in the mushroom-forming fungus Schizophyllum commune increases the relative incidence of homologous recombination. FEMS Microbiology Letters 310: 91–95. [DOI] [PubMed] [Google Scholar]
- de Mendoza A, Sebé-Pedrós A, Ruiz-Trillo I. (2014). The Evolution of the GPCR signalling System in Eukaryotes: Modularity, Conservation, and the Transition to Metazoan Multicellularity. Genome biology and evolution 6: 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de O Scale barsottini MR, de Oliveira JF, Adamoski D, et al. (2013). Functional diversification of cerato-platanins in Moniliophthora perniciosa as seen by differential expression and protein function specialization. Molecular Plant-Microbe Interactions 26: 1281–1293. [DOI] [PubMed] [Google Scholar]
- de Oliveira ES, Junges Â, Sbaraini N, et al. (2018). Molecular evolution and transcriptional profile of GH3 and GH20 β-n-acetylglucosaminidases in the entomopathogenic fungus Metarhizium anisopliae. Genetics and Molecular Biology 41: 843–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker N, Speijer D, Grün CH, et al. (2004). Role of the α-glucanase Agn1p in fission-yeast cell separation. Molecular Biology of the Cell 15: 3903–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Poeta M, Nimrichter L, Rodrigues ML, et al. (2014). Synthesis and biological properties of fungal glucosylceramide. PLoS pathogens 10: e1003832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demmler R, Fricke J, Dörner S, et al. (2020). S-Adenosyl-l-Methionine Salvage Impacts Psilocybin Formation in “Magic” Mushrooms. Chembiochem 21: 1364–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, Joazeiro CA. (2009). RING domain E3 ubiquitin ligases. Annual Review of Biochemistry 78: 399–434. [DOI] [PubMed] [Google Scholar]
- Deveau A, Kohler A, Frey-Klett P, et al. (2008). The major pathways of carbohydrate metabolism in the ectomycorrhizal basidiomycete Laccaria bicolor S238N. New Phytologist 180: 379–390. [DOI] [PubMed] [Google Scholar]
- Díaz-Valderrama JR, Aime MC. (2016). The cacao pathogen Moniliophthora roreri (Marasmiaceae) possesses biallelic A and B mating loci but reproduces clonally. Heredity 116: 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkgraaf GJ, Brown JL, Bussey H. (1996). The KNH1 gene of Saccharomyces cerevisiae is a functional homolog of KRE9. Yeast 12: 683–692. [DOI] [PubMed] [Google Scholar]
- Ding W, Liu WQ, Jia Y, et al. (2016). Biosynthetic investigation of phomopsins reveals a widespread pathway for ribosomal natural products in Ascomycetes. Proceedings of the National Academy of Sciences 113: 3521–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon RA, Achnine L, Kota P, et al. (2002). The phenylpropanoid pathway and plant defence - A genomics perspective. Molecular Plant Pathology 3: 371–390. [DOI] [PubMed] [Google Scholar]
- Douglas LM, Alvarez FJ, McCreary C, et al. (2005). Septin function in yeast model systems and pathogenic fungi. Eukaryotic Cell 4: 1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs JA, Lowndes NF, Jackson SP. (2000). A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 408: 1001–1004. [DOI] [PubMed] [Google Scholar]
- Dressaire E, Yamada L, Song B, et al. (2016). Mushrooms use convectively created airflows to disperse their spores. Proceedings of the National Academy of Sciences 113: 2833–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan M, Bao H, Bau T. (2021). Analyses of transcriptomes and the first complete genome of Leucocalocybe mongolica provide new insights into phylogenetic relationships and conservation. Scientific Reports 11: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dueñas-Santero E, Martín-Cuadrado AB, Fontaine, et al. (2010). Characterization of glycoside hydrolase family 5 proteins in Schizosaccharomyces pombe. Eukaryotic Cell 9: 1650–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastwood DC, Herman B, Noble R, et al. (2013). Environmental regulation of reproductive phase change in Agaricus bisporus by 1-octen-3-ol, temperature and CO2. Fungal Genetics and Biology 55: 54–66. [DOI] [PubMed] [Google Scholar]
- Eis C, Nidetzky B. (1999). Characterization of trehalose phosphorylase from Schizophyllum commune. Biochemical Journal 341: 385–393. [PMC free article] [PubMed] [Google Scholar]
- Elleuche S, Pöggeler S. (2009). β-carbonic anhydrases play a role in fruiting body development and ascospore germination in the filamentous fungus Sordaria macrospora. PLoS One 4: e5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elleuche S, Pöggeler S. (2010). Carbonic anhydrases in fungi. Microbiology 156: 23–29. [DOI] [PubMed] [Google Scholar]
- Emms DM, Kelly S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biology 16: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo H, Kajiwara S, Tsunoka O, et al. (1994). A novel cDNA, priBc, encoding a protein with a Zn (II) 2Cys6 zinc cluster DNA-binding motif, derived from the basidiomycete Lentinus edodes. Gene 139: 117–121. [DOI] [PubMed] [Google Scholar]
- Essen LO, Vogt MS, Mösch HU. (2020). Diversity of GPI-anchored fungal adhesins. Biological Chemistry 401: 1389–1405. [DOI] [PubMed] [Google Scholar]
- Fakas S. (2017) Lipid biosynthesis in yeasts: A comparison of the lipid biosynthetic pathway between the model nonoleaginous yeast Saccharomyces cerevisiae and the model oleaginous yeast Yarrowia lipolytica. Engineering in Life Sciences 17: 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fäldt J, Jonsell M, Nordlander G, et al. (1999). Volatiles of bracket fungi Fomitopsis pinicola and Fomes fomentarius and their functions as insect attractants. Journal of Chemical Ecology 25: 567–590. [Google Scholar]
- Fan Y, Ortiz-Urquiza A, Garrett T, et al. (2015). Involvement of a caleosin in lipid storage, spore dispersal, and virulence in the entomopathogenic filamentous fungus, Beauveria bassiana. Environmental Microbiology 17: 4600–4614. [DOI] [PubMed] [Google Scholar]
- Fang H, Zhang W, Niu X, et al. (2014). Stipe wall extension of Flammulina velutipes could be induced by an expansin-like protein from Helix aspersa, Fungal Biology 118: 1–11. [DOI] [PubMed] [Google Scholar]
- Felenbok B, Flipphi M, Nikolaev I. (2001). Ethanol catabolism in Aspergillus nidulans: a model system for studying gene regulation. Progress in Nucleic Acid Research and Molecular Biology 69: 149–204. [DOI] [PubMed] [Google Scholar]
- Fernandez Espinar MT, Labarère J. (1997). Cloning and sequencing of the Aa-Pri1 gene specifically expressed during fruiting initiation in the edible mushroom Agrocybe aegerita, and analysis of the predicted amino-acid sequence. Current Genetics 32: 420–424. [DOI] [PubMed] [Google Scholar]
- Finegold AA, Johnson DI, Farnsworth CC, et al. (1991). Protein geranylgeranyltransferase of Saccharomyces cerevisiae is specific for Cys-Xaa-Xaa-Leu motif proteins and requires the CDC43 gene product but not the DPR1 gene product. Proceedings of the National Academy of Sciences 88: 4448–4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley D, Ulrich HD, Sommer T, et al. (2012). The Ubiquitin–Proteasome System of Saccharomyces cerevisiae. Genetics 192: 319–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Cox J, Davis DJ, et al. (2004). New information on the mechanism of forcible ascospore discharge from Ascobolus immersus. Fungal Genetics and Biology 41: 698–707. [DOI] [PubMed] [Google Scholar]
- Fischer MS, Glass NL. (2019). Communicate and fuse: How filamentous fungi establish and maintain an interconnected mycelial network. Frontiers in Microbiology 10: 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flipphi M, Kocialkowska J, Felenbok B. (2003). Relationships between the ethanol utilization (alc) pathway and unrelated catabolic pathways in Aspergillus nidulans. European Journal of Biochemistry 270: 3555–3564. [DOI] [PubMed] [Google Scholar]
- Floudas D, Binder M, Riley R, et al. (2012). The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 336: 1715–1719. [DOI] [PubMed] [Google Scholar]
- Foster J, Nakata PA. (2014). An oxalyl-CoA synthetase is important for oxalate metabolism in Saccharomyces cerevisiae. FEBS Letters 588: 160–166. [DOI] [PubMed] [Google Scholar]
- Foulongne-Oriol M, Taskent O, Kües U, et al. (2021). Mating-type locus organization and mating-type chromosome differentiation in the bipolar edible button mushroom Agaricus bisporus. Genes 12: 1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- François J, Parrou JL. (2001). Reserve carbohydrates metabolism in the yeast Saccharomyces cerevisiae. FEMS Microbiology Reviews 25: 125–1 [DOI] [PubMed] [Google Scholar]
- Fraser JA, Hsueh YP, Findley KM, et al. (2007). Evolution of the mating-type locus: the basidiomycetes. Sex in fungi: Molecular Determination and Evolutionary Implications, 19–34. [Google Scholar]
- Frawley D, Bayram Ö. (2020). The pheromone response module, a mitogen-activated protein kinase pathway implicated in the regulation of fungal development, secondary metabolism and pathogenicity. Fungal Genetics and Biology 144: 103469. [DOI] [PubMed] [Google Scholar]
- Freihorst D, Fowler TJ, Bartholomew K, et al. (2016). 13 The mating-type genes of the basidiomycetes. In Growth, differentiation and sexuality (pp. 329–349). Springer, Cham. [Google Scholar]
- Freiman RN, Tjian R. (2003). Regulating the regulators: lysine modifications make their mark. Cell 112: 11–17. [DOI] [PubMed] [Google Scholar]
- Freitag M. (2017). Histone methylation by SET domain proteins in fungi. Annual Review of Microbiology 71: 413–439. [DOI] [PubMed] [Google Scholar]
- Fricke J, Blei F, Hoffmeister D. (2017). Enzymatic synthesis of psilocybin. Angewandte Chemie International Edition 56: 12352–12355. [DOI] [PubMed] [Google Scholar]
- Froehlich AC, Chen CH, Belden WJ, et al. (2010). Genetic and molecular characterization of a cryptochrome from the filamentous fungus Neurospora crassa. Eukaryotic Cell 9: 738–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Dai Y, Yang C, et al. (2017). Comparative Transcriptome Analysis Identified Candidate Genes Related to Bailinggu Mushroom Formation and Genetic Markers for Genetic Analyses and Breeding. Scientific Reports 7: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller KK, Loros JJ, Dunlap JC. (2015). Fungal photobiology: visible light as a signal for stress, space and time. Current Genetics 61: 275–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaderer R, Bonazza K, Seidl-Seiboth V. (2014). Cerato-platanins: A fungal protein family with intriguing properties and application potential. Applied Microbiology and Biotechnology 98: 4795–4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon-Arsenault I, Tremblay J, Bourbonnais Y. (2006). Fungal yapsins and cell wall: a unique family of aspartic peptidases for a distinctive cellular function. FEMS Yeast Research 6: 966–978. [DOI] [PubMed] [Google Scholar]
- Gao W, Qu J, Zhang J, et al. (2018.) A genetic linkage map of Pleurotus tuoliensis integrated with physical mapping of the de novo sequenced genome and the mating type loci. BMC Genomics 19: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Rubio R, de Oliveira HC, Rivera J, et al. (2020). The fungal cell wall: Candida, Cryptococcus, and Aspergillus species. Frontiers in Microbiology 2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Santamarina S, Probst C, Festa RA, et al. (2020). A lytic polysaccharide monooxygenase-like protein functions in fungal copper import and meningitis. Nature Chemical Biology 16: 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrmann T, Pelkmans JF, Lugones LG, et al. (2016). Schizophyllum commune has an extensive and functional alternative splicing repertoire. Scientific Reports 6: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrmann T, Pelkmans JF, Ohm RA, et al. (2018). Nucleus-specific expression in the multinuclear mushroom-forming fungus Agaricus bisporus reveals different nuclear regulatory programs. Proceedings of the National Academy of Sciences 115: 4429–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerecke EE, Zolan ME. (2000). An mre11 mutant of Coprinus cinereus has defects in meiotic chromosome pairing, condensation and synapsis. Genetics 154: 1125–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessler NN, Filippovich SY, Bachurina GP, et al.(2017). Oxylipins and oxylipin synthesis pathways in fungi. Applied Biochemistry and Microbiology 53: 628–639. [Google Scholar]
- Geyer R, Wee S, Anderson S, et al. (2003). BTB/POZ domain proteins are putative substrate adaptors for cullin 3 ubiquitin ligases. Molecular Cell 12: 783–790. [DOI] [PubMed] [Google Scholar]
- Gibbs GM, Roelants K, O’bryan MK. (2008). The CAP superfamily: cysteine-rich secretory proteins, antigen 5, and pathogenesis-related 1 proteins—roles in reproduction, cancer, and immune defence. Endocrine Reviews 29: 865–897. [DOI] [PubMed] [Google Scholar]
- Gilberti M, Baruffini E, Donnini C, et al. (2018). Pathological alleles of MPV17 modeled in the yeast Saccharomyces cerevisiae orthologous gene SYM1 reveal their inability to take part in a high molecular weight complex. PLoS One 13: e0205014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladfelter AS. (2006). Control of filamentous fungal cell shape by septins and formins. Nature Reviews Microbiology 4: 223–229. [DOI] [PubMed] [Google Scholar]
- Gold MH, Cheng TM. (1979). Conditions for fruit body formation in the white rot basidiomycete Phanerochaete chrysosporium. Archives of Microbiology 121: 37–41. [Google Scholar]
- Goldstein IJ, Winter HC. (2007). Mushroom Lectins. Comprehensive Glycoscience: From Chemistry to Systems Biology 3–4: 601–621. [Google Scholar]
- Gong W, Wang Y, Xie C, et al. (2020). Whole genome sequence of an edible and medicinal mushroom, Hericium erinaceus (Basidiomycota, Fungi). Genomics 112: 2393–2399. [DOI] [PubMed] [Google Scholar]
- Gong W, Xie C, Zhou Y, et al. (2020). A resequencing-based ultradense genetic map of Hericium erinaceus for anchoring genome sequences and identifying genetic loci associated with monokaryon growth. Frontiers in Microbiology 10: 3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Hilarion S, Paulet D, Lee K, et al. (2016). Intron retention-dependent gene regulation in Cryptococcus neoformans. Scientific Reports 6: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich-Tanrikulu M, Howe K, Stafford A, et al. (1998). Changes in fatty acid composition of Neurospora crassa accompany sexual development and ascospore germination. Microbiology 144: 1713–1720. [DOI] [PubMed] [Google Scholar]
- Gow NAR, Latge JP, Munro CA. (2017). The Fungal Cell Wall: Structure, Biosynthesis, and Function. Microbiology Spectrum 5: 5–3. [DOI] [PubMed] [Google Scholar]
- Grant CM. (2001). Role of the glutathione/glutaredoxin and thioredoxin systems in yeast growth and response to stress conditions. Molecular Microbiology 39: 533–541. [DOI] [PubMed] [Google Scholar]
- Grau-Bové X, Ruiz-Trillo I, Irimia M. (2018). Origin of exon skipping-rich transcriptomes in animals driven by evolution of gene architecture. Genome Biology 19: 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grąz M, Jarosz-Wilkołazka A, Janusz G, et al. (2017). Transcriptome-based analysis of the saprophytic fungus Abortiporus biennis–response to oxalic acid. Microbiological Research 199: 79–88. [DOI] [PubMed] [Google Scholar]
- Green NM. (1990). [5] Avidin and streptavidin. In Methods in Enzymology (Vol. 184, pp. 51–67). Academic Press. [DOI] [PubMed] [Google Scholar]
- Gressler M, Löhr NA, Schäfer T, et al. (2021). Mind the mushroom: natural product biosynthetic genes and enzymes of Basidiomycota. Natural Product Reports 38: 702–722. [DOI] [PubMed] [Google Scholar]
- Gross C, Kelleher M, Iyer VR, et al. (2000). Identification of the copper regulon in Saccharomyces cerevisiae by DNA microarrays. Journal of Biological Chemistry 275: 32310–32316. [DOI] [PubMed] [Google Scholar]
- Grützmann K, Szafranski K, Pohl M, et al. (2014). Fungal alternative splicing is associated with multicellular complexity and virulence: a genome-wide multi-species study. DNA Research 21: 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta DK, Rühl M, Mishra B, et al. (2018). The genome sequence of the commercially cultivated mushroom Agrocybe aegerita reveals a conserved repertoire of fruiting-related genes and a versatile suite of biopolymer-degrading enzymes. BMC Genomics 19: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyawali R, Upadhyay S, Way J, Lin X. (2017). A family of secretory proteins is associated with different morphotypes in Cryptococcus neoformans. Applied and Environmental Microbiology 83: e02967–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha B, Kim S, Kim M, et al. (2018). Activation of the mating pheromone response pathway of Lentinula edodes by synthetic pheromones. Mycobiology 46: 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber JE. (2012). Mating-type genes and MAT switching in Saccharomyces cerevisiae. Genetics 191: 33–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallen HE, Luo H, Scott-Craig JS, et al. (2007). Gene family encoding the major toxins of lethal Amanita mushrooms. Proceedings of the National Academy of Sciences 104: 19097–19101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamidizade M, Taghavi SM, Martins SJ, et al. (2020). Bacterial brown pit, a new disease of edible mushrooms caused by Mycetocola sp. Plant Disease 104: 1445–1454. [DOI] [PubMed] [Google Scholar]
- Hammond TM, Bok JW, Andrewski MD, et al. (2008). RNA silencing gene truncation in the filamentous fungus Aspergillus nidulans. Eukaryotic Cell 7: 339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Adams T. (2001). Complex control of the developmental regulatory locus brlA in Aspergillus nidulans. Molecular Genetics and Genomics 266: 260–270. [DOI] [PubMed] [Google Scholar]
- Han SE, Kwon HB, Lee SB, et al. (2003). Cloning and characterization of a gene encoding trehalose phosphorylase (TP) from Pleurotus sajor-caju. Protein Expression and Purification 30: 194–202. [DOI] [PubMed] [Google Scholar]
- Hartl L, Zach S, Seidl-Seiboth V. (2012). Fungal chitinases: diversity, mechanistic properties and biotechnological potential. Applied Microbiology and Biotechnology 93: 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan MAA, Rouf R, Tiralongo E, et al. (2015). Mushroom lectins: specificity, structure and bioactivity relevant to human disease. International Journal of Molecular Sciences 16: 7802–7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastrup ACS, Green IIIF, Lebow PK, et al. (2012). Enzymatic oxalic acid regulation correlated with wood degradation in four brown-rot fungi. International Biodeterioration & Biodegradation 75: 109–114. [Google Scholar]
- He Z, Long P, Fang F, et al. (2020). Diversity of MSDIN family members in amanitin-producing mushrooms and the phylogeny of the MSDIN and prolyl oligopeptidase genes. BMC Genomics 21: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneghan MN, Costa AMSB, Challen MP, et al. (2007). A comparison of methods for successful triggering of gene silencing in Coprinus cinereus. Molecular Biotechnology 35: 283–296. [DOI] [PubMed] [Google Scholar]
- Herman KC, Wösten HAB, Fricker MD, et al. (2020). Growth induced translocation effectively directs an amino acid analogue to developing zones in Agaricus bisporus. Fungal Biology 124: 1013–1023. [DOI] [PubMed] [Google Scholar]
- Hernández-Rodríguez Y, Masuo S, Johnson D, et al. (2014). Distinct septin heteropolymers co-exist during multicellular development in the filamentous fungus Aspergillus nidulans. PLoS One 9: e92819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz S, Rodríguez JM, Bussink HJ, et al. (2005). Arrestin-related proteins mediate pH signalling in fungi. Proceedings of the National Academy of Sciences 102: 12141–12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog R, Solovyeva I, Rühl M, et al. (2016). Dikaryotic fruiting body development in a single dikaryon of Agrocybe aegerita and the spectrum of monokaryotic fruiting types in its monokaryotic progeny. Mycological Progress 15: 947–957. [Google Scholar]
- Heuts DPHM, Janssen DB, Fraaije MW. (2007). Changing the substrate specificity of a chitooligosaccharide oxidase from Fusarium graminearum by model-inspired site-directed mutagenesis. FEBS Letters 581: 4905–4909. [DOI] [PubMed] [Google Scholar]
- Hibbett DS. (2004). Trends in morphological evolution in homobasidiomycetes inferred using maximum likelihood: a comparison of binary and multistate approaches. Systematic Biology 53: 889–903. [DOI] [PubMed] [Google Scholar]
- Hirano T, Sato T, Enei H. (2004). Isolation of genes specifically expressed in the fruit body of the edible basidiomycete Lentinula edodes. Bioscience, Biotechnology, and Biochemistry 68: 468–472. [DOI] [PubMed] [Google Scholar]
- Hofrichter M, Kellner H, Pecyna MJ, et al. (2015). Fungal unspecific peroxygenases: heme-thiolate proteins that combine peroxidase and cytochrome P450 properties. Monooxygenase, peroxidase and peroxygenase properties and mechanisms of cytochrome P450, pp. 341–368. [DOI] [PubMed] [Google Scholar]
- Holighaus G, Rohlfs M. (2019). Volatile and non-volatile fungal oxylipins in fungus-invertebrate interactions. Fungal Ecology 38: 28–36. [Google Scholar]
- Hood HM, Neafsey DE, Galagan J, et al. (2009). Evolutionary roles of upstream open reading frames in mediating gene regulation in fungi. Annual Review of Microbiology 63: 385–409. [DOI] [PubMed] [Google Scholar]
- Hörer S, Stoop J, Mooibroek H, et al. (2001). The crystallographic structure of the mannitol 2-dehydrogenase NADP+ binary complex from Agaricus bisporus. Journal of Biological Chemistry 276: 27555–27561. [DOI] [PubMed] [Google Scholar]
- Horgen PA, Vaisius AC, Ammirati JF. (1978). The insensitivity of mushroom nuclear RNA polymerase activity to inhibiton by amatoxins. Archives of Microbiology 118: 317–319. [DOI] [PubMed] [Google Scholar]
- Hou L, Wang L, Wu X, et al. (2019). Expression patterns of two pal genes of Pleurotus ostreatus across developmental stages and under heat stress. BMC Microbiology 19: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Z, Chen Q, Zhao M, et al. (2020). Genome-wide characterization of the Zn (II) 2Cys6 zinc cluster-encoding gene family in Pleurotus ostreatus and expression analyses of this family during developmental stages and under heat stress. PeerJ 8: e9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houbraken J, Dyer PS. (2015). Induction of the sexual cycle in filamentous ascomycetes. In Genetic Transformation Systems in Fungi, Volume 2 (pp. 23–46). Springer, Cham. [Google Scholar]
- Huang Q, Han X, Mukhtar I, et al. (2018). Identification and expression patterns of fvexpl1, an expansin-like protein-encoding gene, suggest an auxiliary role in the stipe morphogenesis of Flammulina velutipes. Journal of Microbiology and Biotechnology 28: 622–629. [DOI] [PubMed] [Google Scholar]
- Huang X, Zhang R, Qiu Y, et al. (2020). RNA-seq profiling showed divergent carbohydrate-active enzymes (CAZymes) expression patterns in Lentinula edodes at brown film formation stage under blue light induction. Frontiers in Microbiology 11: 1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber SMFE, Lottspeich F, Kämper J. (2002). A gene that encodes a product with similarity to dioxygenases is highly expressed in teliospores of Ustilago maydis. Molecular Genetics and Genomics 267: 757–771. [DOI] [PubMed] [Google Scholar]
- Hung R, Lee S, Bennett JW. (2014). The effects of low concentrations of the enantiomers of mushroom alcohol (1-octen-3-ol) on Arabidopsis thaliana. Mycology 5: 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins AP, Liu S, Diez D, et al. (2013). The repertoires of ubiquitinating and deubiquitinating enzymes in eukaryotic genomes. Molecular Biology and Evolution 30: 1172–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun MW, Yun YH, Kim JY, et al. (2011). Fungal and plant phenylalanine ammonia-lyase. Mycobiology 39: 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idnurm A, Heitman J. (2005). Photosensing fungi: phytochrome in the spotlight. Current Biology 15: R829–R832. [DOI] [PubMed] [Google Scholar]
- Inoue SB, Qadota H, Arisawa M, et al. (1999). Prenylation of Rho1p is required for activation of yeast 1, 3-β-glucan synthase. Journal of Biological Chemistry 274: 38119–38124. [DOI] [PubMed] [Google Scholar]
- Ishizaki T, Shishido K. (2000). Decreased zinc ion accumulation by the basidiomycete Lentinus edodes over-expressing L. edodes priA gene. FEMS Microbiology Letters 193: 111–115. [DOI] [PubMed] [Google Scholar]
- Ismaya WT, Tjandrawinata RR, Rachmawati H. (2020). Lectins from the edible mushroom Agaricus bisporus and their therapeutic potentials. Molecules 25: 2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai K, Naganuma A, Kuge S. (2010). Peroxiredoxin Ahp1 acts as a receptor for alkylhydroperoxides to induce disulfide bond formation in the Cad1 transcription factor. Journal of Biological Chemistry 285: 10597–10604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa S, Kuroki N, Inoue Y. (2004). Nuclear thioredoxin peroxidase Dot5 in Saccharomyces cerevisiae: roles in oxidative stress response and disruption of telomeric silencing. Applied Microbiology and Biotechnology 64: 120–124. [DOI] [PubMed] [Google Scholar]
- Jai-Sik H, Tae-Young K. (1988). Contents of free-sugars & free-sugaralcohols in Pleurotus ostreatus, Lentinus edodes & Agaricus bisporus. Korean Journal of Food Science and Technology 20: 459–462. [Google Scholar]
- James TY. (2015). Why mushrooms have evolved to be so promiscuous: Insights from evolutionary and ecological patterns. Fungal Biology Reviews 29: 167–178. [Google Scholar]
- James TY, Lee M, van Diepen LTA. (2011). A single mating-type locus composed of homeodomain genes promotes nuclear migration and heterokaryosis in the white-rot fungus Phanerochaete chrysosporium. Eukaryotic Cell 10: 249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jara M, Vivancos AP, Calvo IA, et al. (2007). The peroxiredoxin Tpx1 is essential as a H2O2 scavenger during aerobic growth in fission yeast. Molecular Biology of the Cell 18: 2288–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J, Moore D. (1993). Glycogen metabolism in relation to fruit body maturation in Coprinus cinereus. Mycological Research 97: 283–289. [Google Scholar]
- Jia D, Wang B, Li X, et al. (2017). Proteomic analysis revealed the fruiting-body protein profile of Auricularia polytricha. Current Microbiology 74: 943–951. [DOI] [PubMed] [Google Scholar]
- Jiao X, Li G, Wang Y, et al. (2018). Systematic analysis of the Pleurotus ostreatus laccase gene (PoLac) family and functional characterization of PoLac2 involved in the degradation of cotton-straw lignin. Molecules 23: 880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Li G, Yu D, et al. (2017). Transcriptome analysis reveals the complexity of alternative splicing regulation in the fungus Verticillium dahliae. BMC Genomics 18: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joh JH, Kim BG, Kong WS, et al. (2004). Cloning and developmental expression of a metzincin family metalloprotease cDNA from oyster mushroom Pleurotus ostreatus. FEMS Microbiology Letters 239: 57–62. [DOI] [PubMed] [Google Scholar]
- Jones SK, Jr, Bennett RJ. (2011). Fungal mating pheromones: choreographing the dating game. Fungal Genetics and Biology 48: 668–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers W, Rep M. (2009). Lessons from fungal F-box proteins. Eukaryotic Cell 8: 677–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen P, Nishikawa JL, Breitkreutz BJ, et al. (2002). Systematic identification of pathways that couple cell growth and division in yeast. Science 297: 395–400. [DOI] [PubMed] [Google Scholar]
- Juillot S, Cott C, Madl J, et al. (2016). Uptake of Marasmius oreades agglutinin disrupts integrin-dependent cell adhesion. Biochimica et Biophysica Acta (BBA)-General Subjects 1860: 392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabel MA, Jurak E, Mäkelä MR, et al. (2017). Occurrence and function of enzymes for lignocellulose degradation in commercial Agaricus bisporus cultivation. Applied Microbiology and Biotechnology 101: 4363–4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiwara S, Yamaoka K, Hori K, et al. (1992). Isolation and sequence of a developmentally regulated putative novel gene, priA, from the basidiomycete Lentinus edodes. Gene 114: 173–178. [DOI] [PubMed] [Google Scholar]
- Kalaras MD, Richie JP, Calcagnotto A, et al. (2017). Mushrooms: A rich source of the antioxidants ergothioneine and glutathione. Food Chemistry 233: 429–433. [DOI] [PubMed] [Google Scholar]
- Káldi K, González BH, Brunner M. (2006). Transcriptional regulation of the Neurospora circadian clock gene wc1 affects the phase of circadian output. EMBO Reports 7: 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada T, Sano H, Nakazawa T, et al. (2010). Regulation of fruiting body photomorphogenesis in Coprinopsis cinerea. Fungal Genetics and Biology 47: 917–921. [DOI] [PubMed] [Google Scholar]
- Kamada T, Takemaru T, Prosser JI, et al. (1991). Right and left handed helicity of chitin microfibrils in stipe cells in Coprinus cinereus. Protoplasma 165: 64–70. [Google Scholar]
- Kar B, Patel P, Ao J, et al. (2019). Neurospora crassa family GH72 glucanosyltransferases function to crosslink cell wall glycoprotein N-linked galactomannan to cell wall lichenin. Fungal Genetics and Biology 123: 60–69. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM. (2013). MAFFT multiple sequence alignment software version 7: Improvements in Performance and Usability. Molecular Biology and Evolution 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsimpouras C, Dedes G, Thomaidis NS, et al. (2019). A novel fungal GH30 xylanase with xylobiohydrolase auxiliary activity. Biotechnology for Biofuels 12: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käufer NF, Potashkin J. (2000). Analysis of the splicing machinery in fission yeast: a comparison with budding yeast and mammals. Nucleic Acids Research 28: 3003–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto S, Sasaki T, Itahashi S, et al. (1993). A Mutant Allele skt5 Affecting Protoplast Regeneration and Killer Toxin Resistance. Has Double Mutations in Its Wild-type Structural Gene in Saccharomyces cerevisiae. Bioscience, Biotechnology, and Biochemistry 57: 1391–1393. [DOI] [PubMed] [Google Scholar]
- Ke HM, Lee HH, Lin CYI, et al. (2020). Mycena genomes resolve the evolution of fungal bioluminescence. Proceedings of the National Academy of Sciences 117: 31267–31277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller NP. (2019). Fungal secondary metabolism: regulation, function and drug discovery. Nature Reviews Microbiology 17: 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemppainen M, Duplessis S, Martin F, et al. (2009). RNA silencing in the model mycorrhizal fungus Laccaria bicolor: gene knock-down of nitrate reductase results in inhibition of symbiosis with Populus. Environmental Microbiology 11: 1878–1896. [DOI] [PubMed] [Google Scholar]
- Khan A, McQuilken M, Gladfelter AS. (2015). Septins and generation of asymmetries in fungal cells. Annual Review of Microbiology 69: 487–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HI, Lee CS, Park YJ. (2016). Further characterization of hydrophobin genes in genome of Flammulina velutipes. Mycoscience 57: 320–325. [Google Scholar]
- Kim JY, Kim DY, Park YJ, et al. (2020). Transcriptome analysis of the edible mushroom Lentinula edodes in response to blue light. PLoS ONE 15: e0230680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Kang YM, I CH, et al. (2014). Identification and functional analysis of pheromone and receptor genes in the B3 mating locus of Pleurotus eryngii. PLoS ONE 9: e104693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipreos ET, Pagano M. (2000). The F-box protein family. Genome Biology 1: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamoto Y, Kobayashi A, Mori N, et al. (2001). Metabolic function of glycogen phosphorylase and trehalose phosphorylase in fruit-body formation of Flammulina velutipes. Mycoscience 42: 143–147. [Google Scholar]
- Kiyama R, Furutani Y, Kawaguchi K, et al. (2018). Genome sequence of the cauliflower mushroom Sparassis crispa (Hanabiratake) and its association with beneficial usage. Scientific Reports 8: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klis FM, Boorsma A, De Groot PWJ. (2006). Cell wall construction in Saccharomyces cerevisiae. Yeast 23: 185–202. [DOI] [PubMed] [Google Scholar]
- Knabe N, Jung EM, Freihorst D, et al. (2013). A central role for Ras1 in morphogenesis of the basidiomycete Schizophyllum commune. Eukaryotic Cell 12: 941–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll AH. (2011). The multiple origins of complex multicellularity. Annual Review of Earth and Planetary Sciences 39: 217–239. [Google Scholar]
- Kobayashi Y, Kawagishi H. (2014). Fungal lectins: a growing family. Methods in Molecular Biology 1200: 15–38. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Tateno H, Ogawa H, et al. (2014). Comprehensive list of lectins: origins, natures, and carbohydrate specificities. Methods in Molecular Biology 1200: 555–577. [DOI] [PubMed] [Google Scholar]
- Koch RA, Yoon GM, Aryal UK, et al. (2021). Symbiotic nitrogen fixation in the reproductive structures of a basidiomycete fungus. Current Biology 31: 3905–3914. [DOI] [PubMed] [Google Scholar]
- Kohler A, Kuo A, Nagy LG, et al. (2015). Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nature Genetics 47: 410–415. [DOI] [PubMed] [Google Scholar]
- Kombrink A, Tayyrov A, Essig A, et al. (2019). Induction of antibacterial proteins and peptides in the coprophilous mushroom Coprinopsis cinerea in response to bacteria. The ISME Journal 13: 588–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konada L, Aricthota S, Vadla R, et al. (2018). Fission yeast sirtuin hst4 functions in preserving genomic integrity by regulating replisome component mcl1. Scientific Reports 8: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondoh O, Muto A, Kajiwara S, et al. (1995). A fruiting body-specific cDNA, mfbAc, from the mushroom Lentinus edodes encodes a high-molecular-weight cell-adhesion protein containing an Arg-Gly-Asp motif. Gene 154: 31–37. [DOI] [PubMed] [Google Scholar]
- Kozlov AM, Darriba D, Flouri T, et al. (2019). RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35: 4453–4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraakman LS, Griffioen G, Zerp S, et al. (1993). Growth-related expression of ribosomal protein genes in Saccharomyces cerevisiae. Molecular and General Genetics 239: 196–204. [DOI] [PubMed] [Google Scholar]
- Krappmann S, Jung N, Medic B, et al. (2006). The Aspergillus nidulans F-box protein GrrA links SCF activity to meiosis. Molecular Microbiology 61: 76–88. [DOI] [PubMed] [Google Scholar]
- Krijgsheld P, Bleichrodt RV, Van Veluw GJ, et al. (2008). Development in Aspergillus. Studies in Mycology 60: 1–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivosheina NP. (2008). Macromycete fruit bodies as a habitat for dipterans (Insecta, Diptera). Entomological Review 88: 778–792. [Google Scholar]
- Krizsán K, Almási É, Merényi Z, et al. (2019). Transcriptomic atlas of mushroom development reveals conserved genes behind complex multicellularity in fungi. Proceedings of the National Academy of Sciences 116: 7409–7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kück U, Radchenko D, Teichert I. (2019). STRIPAK, a highly conserved signalling complex, controls multiple eukaryotic cellular and developmental processes and is linked with human diseases. Biological Chemistry 400: 1005–1022. [DOI] [PubMed] [Google Scholar]
- Kües U. (2000). Life history and developmental processes in the basidiomycete Coprinus cinereus. Microbiology and Molecular Biology Reviews 64: 316–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kües U. (2015). From two to many: multiple mating types in Basidiomycetes. Fungal Biology Reviews 29: 126–166. [Google Scholar]
- Kües U, Granado JD, Hermann R, et al. (1998). The A mating type and blue light regulate all known differentiation processes in the basidiomycete Coprinus cinereus. Molecular and General Genetics MGG 260: 81–91. [DOI] [PubMed] [Google Scholar]
- Kües U, Khonsuntia W, Subba S, et al. (2018). Volatiles in communication of Agaricomycetes. In Physiology and Genetics (pp. 149–212). Springer, Cham. [Google Scholar]
- Kües U, Liu Y. (2000). Fruiting body production in basidiomycetes. Applied Microbiology and Biotechnology 54: 141–152. [DOI] [PubMed] [Google Scholar]
- Kües U, Navarro-Gonzalez M. (2015). How do Agaricomycetes shape their fruiting bodies? 1. Morphological aspects of development. Fungal Biology Reviews 29: 63–97. [Google Scholar]
- Kües U, Ruhl M. (2011). Multiple multi-copper oxidase gene families in basidiomycetes-what for? Current Genomics 12: 72–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kües U, Walser PJ, Klaus MJ, et al. (2002). Influence of activated A and B mating-type pathways on developmental processes in the basidiomycete Coprinus cinereus. Molecular Genetics and Genomics 268: 262–271. [DOI] [PubMed] [Google Scholar]
- Kuhad RC, Rosin IV, Moore D. (1987). A possible relation between cyclic-AMP levels and glycogen mobilization in Coprinus cinereus. Transactions of the British Mycological Society 88: 229–236. [Google Scholar]
- Kumar MJ, Jamaluddin MS, Natarajan K, et al. (2000). The inducible N-acetylglucosamine catabolic pathway gene cluster in Candida albicans: discrete N-acetylglucosamine-inducible factors interact at the promoter of NAG1. Proceedings of the National Academy of Sciences 97: 14218–14223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Künzler M. (2015). Hitting the sweet spot: glycans as targets of fungal defence effector proteins. Molecules 20: 8144–8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Künzler M. (2018). How fungi defend themselves against microbial competitors and animal predators. PLoS Pathogens 14: e1007184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuratani M, Tanaka K, Terashima K, et al. (2010). The dst2 gene essential for photomorphogenesis of Coprinopsis cinerea encodes a protein with a putative FAD-binding-4 domain. Fungal Genetics and Biology 47: 152–158. [DOI] [PubMed] [Google Scholar]
- Labourel A, Frandsen KEH, Zhang F, et al. (2020). A fungal family of lytic polysaccharide monooxygenase-like copper proteins. Nature Chemical Biology 16: 345–350. [DOI] [PubMed] [Google Scholar]
- Lackner G, Bohnert M, Wick J, et al. (2013). Assembly of melleolide antibiotics involves a polyketide synthase with cross-coupling activity. Chemistry & Biology 20: 1101–1106. [DOI] [PubMed] [Google Scholar]
- Lakkireddy K, Khonsuntia W, Kües U. (2020). Mycoparasite Hypomyces odoratus infests Agaricus xanthodermus fruiting bodies in nature. AMB Express 10: 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landi N, Pacifico S, Ragucci S, et al. (2017). Purification, characterization and cytotoxicity assessment of Ageritin: The first ribotoxin from the basidiomycete mushroom Agrocybe aegerita. Biochimica et Biophysica Acta (BBA)-General Subjects 1861: 1113–1121. [DOI] [PubMed] [Google Scholar]
- Langner T, Göhre V. (2015). Fungal chitinases: function, regulation, and potential roles in plant/pathogen interactions. Current Genetics 62: 243–254. [DOI] [PubMed] [Google Scholar]
- Lara-Ortíz T, Riveros-Rosas H, Aguirre J. (2003). Reactive oxygen species generated by microbial NADPH oxidase NoxA regulate sexual development in Aspergillus nidulans. Molecular Microbiology 50: 1241–1255. [DOI] [PubMed] [Google Scholar]
- Larrondo LF, Salas L, Melo F, et al. (2003). A novel extracellular multicopper oxidase from Phanerochaete chrysosporium with ferroxidase activity. Applied and Environmental Microbiology 69: 6257–6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau AYT, Cheng X, Cheng CK, et al. (2018). Discovery of microRNA-like RNAs during early fruiting body development in the model mushroom Coprinopsis cinerea. PLoS One 13: e0198234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavrijssen B, Baars JP, Lugones LG, et al. (2020). Interruption of an MSH4 homolog blocks meiosis in metaphase I and eliminates spore formation in Pleurotus ostreatus. PLoS One 15: e0241749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leatham GF, Stahmann MA. (1981). Studies on the laccase of Lentinus edodes: specificity, localization and association with the development of fruiting bodies. Microbiology 125: 147–157. [Google Scholar]
- Lebreton A, Bonnardel F, Dai YC, et al. (2021). A comprehensive phylogenetic and bioinformatics survey of lectins in the fungal kingdom. Journal of Fungi 7: 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KT, et al. Systematic functional analysis of kinases in the fungal pathogen Cryptococcus neoformans. Nature Communications 7: 12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Jang EH, Lee M, et al. (2019). Unraveling melanin biosynthesis and signalling networks in Cryptococcus neoformans. MBio 10: e02267–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YY, Vidal-Diez de Ulzurrun G, Schwarz EM, et al. (2021). Genome sequence of the oyster mushroom Pleurotus ostreatus strain PC9. G3 11: jkaa008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz C, Wick J, Braga D, et al. (2020). Injury-triggered blueing reactions of Psilocybe “magic” mushrooms. Angewandte Chemie 132: 1466–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- León-Ramírez CG, Cabrera-Ponce JL, Martínez-Soto D, et al. (2017). Transcriptomic analysis of basidiocarp development in Ustilago maydis (DC) Cda. Fungal Genetics and Biology 101: 34–45. [DOI] [PubMed] [Google Scholar]
- Lesage G, Bussey H. (2006). Cell wall assembly in Saccharomyces cerevisiae. Microbiology and Molecular Biology Reviews 70: 317–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levasseur A, Drula E, Lombard V, et al. (2013). Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnology for Biofuels 6: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levasseur A, Lomascolo A, Chabrol O, et al. (2014). The genome of the white-rot fungus Pycnoporus cinnabarinus: a basidiomycete model with a versatile arsenal for lignocellulosic biomass breakdown. BMC Genomics 15: 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin DE. (2005). Cell wall integrity signalling in Saccharomyces cerevisiae. Microbiology and Molecular Biology Reviews 69: 262–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowska M, Keyl A, Feussner I. (2020). Wax biosynthesis in response to danger: its regulation upon abiotic and biotic stress. New Phytologist 227: 698–713. [DOI] [PubMed] [Google Scholar]
- Li F, Wu X, Lam P, et al. (2008). Identification of the wax ester synthase/acyl-coenzyme A: diacylglycerol acyltransferase WSD1 required for stem wax ester biosynthesis in Arabidopsis. Plant Physiology 148: 97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Gerecke EE, Zolan ME. (1999). Homolog pairing and meiotic progression in Coprinus cinereus. Chromosoma 108: 384–392. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. [DOI] [PubMed] [Google Scholar]
- Lim FY, Keller NP. (2014). Spatial and temporal control of fungal natural product synthesis. Natural Product Reports 31: 1277–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H, Choi HT. (2009). Enhanced expression of chitinase during the autolysis of mushroom in Coprinellus congregatus. The Journal of Microbiology 47: 225–228. [DOI] [PubMed] [Google Scholar]
- Linder MB, Szilvay GR, Nakari–Setälä T, et al. (2005). Hydrophobins: the protein-amphiphiles of filamentous fungi. FEMS Microbiology Reviews 29: 877–896. [DOI] [PubMed] [Google Scholar]
- Liu C, Bi J, Kang L, et al. (2021). The molecular mechanism of stipe cell wall extension for mushroom stipe elongation growth. Fungal Biology Reviews 35: 14–26. [Google Scholar]
- Liu H, Li Y, Chen D, et al. (2017). A-to-I RNA editing is developmentally regulated and generally adaptive for sexual reproduction in Neurospora crassa. Proceedings of the National Academy of Sciences 114: E7756–E7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Wang Q, He Y, et al. (2016). Genome-wide A-to-I RNA editing in fungi independent of ADAR enzymes. Genome Research 26: 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JH, Shan XD, Liu JY, et al. (2016). Changes in trehalose content, enzyme activity and gene expression related to trehalose metabolism in Flammulina velutipes under heat shock. Microbiology 162: 1274–1285. [DOI] [PubMed] [Google Scholar]
- Liu L, He GJ, Chen L, et al. (2018). Genetic basis for coordination of meiosis and sexual structure maturation in Cryptococcus neoformans. Elife 7: e38683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TB, Chen GQ, Min H, et al. (2009). MoFLP1, encoding a novel fungal fasciclin-like protein, is involved in conidiation and pathogenicity in Magnaporthe oryzae. Journal of Zhejiang University Science B 10: 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TB, Xue C. (2011). The ubiquitin-proteasome system and F-box proteins in pathogenic fungi. Mycobiology 39: 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ding S. (2009). Molecular characterization of a new acetyl xylan esterase (AXEII) from edible straw mushroom Volvariella volvacea with both de-O-acetylation and de-N-acetylation activity. FEMS Microbiology Letters 295: 50–56. [DOI] [PubMed] [Google Scholar]
- Liu X, Farnung L, Wigge C, et al. (2018). Cryo-EM structure of a mammalian RNA polymerase II elongation complex inhibited by α-amanitin. Journal of Biological Chemistry 293: 7189–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XB, Xia EH, Li M, et al. (2020). Transcriptome data reveal conserved patterns of fruiting body development and response to heat stress in the mushroom-forming fungus Flammulina filiformis. PLoS One 15: e0239890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Srivilai P, Loos S, et al. (2006). An essential gene for fruiting body initiation in the basidiomycete Coprinopsis cinerea is homologous to bacterial cyclopropane fatty acid synthase genes. Genetics 172: 873–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodder AL, Lee TK, Ballester R. (1999). Characterization of the Wsc1 protein, a putative receptor in the stress response of Saccharomyces cerevisiae. Genetics 152: 1487–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftus MG, Sánchez C, Moore D, et al. (2020). A 21st century miniguide to sporophore morphogenesis and development in Agaricomycetes and their biotechnological potential. Mexican Journal of Biotechnology 5: 1–50. [Google Scholar]
- Loibl M, Grossmann G, Stradalova V, et al. (2010). C terminus of Nce102 determines the structure and function of microdomains in the Saccharomyces cerevisiae plasma membrane. Eukaryotic Cell 9: 1184–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Mondéjar R, Catalano V, Kubicek CP, et al. (2009). The β-N-acetylglucosaminidases NAG1 and NAG2 are essential for growth of Trichoderma atroviride on chitin. The FEBS Journal 276: 5137–5148. [DOI] [PubMed] [Google Scholar]
- Lozančiæ M, Žunar B, Hrestak D, et al. (2021). Systematic comparison of cell wall-related proteins of different yeasts. Journal of Fungi 7: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu BC, Gallo N, Kües U. (2003). White-cap mutants and meiotic apoptosis in the basidiomycete Coprinus cinereus. Fungal Genetics and Biology 39: 82–93. [DOI] [PubMed] [Google Scholar]
- Lu MYJ, Fan WL, Wang WF, et al. (2014). Genomic and transcriptomic analyses of the medicinal fungus Antrodia cinnamomea for its metabolite biosynthesis and sexual development. Proceedings of the National Academy of Sciences 111: E4743–E4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Guo Z, Cai Z, et al. (2018) Cloning and expression analyses of the septin gene Ab.Cdc3 in Agaricus bisporus. Mycosystema 37: 1635–1642. [Google Scholar]
- Lu YP, Liao JH, Guo ZJ, et al. (2020). Genome survey and transcriptome analysis on mycelia and primordia of Agaricus blazei. BioMed Research International Article ID 1824183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugones LG, Scholtmeijer K, Klootwijk R, et al. (1999a). Introns are necessary for mRNA accumulation in Schizophyllum commune. Molecular Microbiology 32: 681–689. [DOI] [PubMed] [Google Scholar]
- Lugones LG, Wösten HAB, Birkenkamp KU, et al. (1999b). Hydrophobins line air channels in fruiting bodies of Schizophyllum commune and Agaricus bisporus. Mycological Research 103: 635–640. [Google Scholar]
- Lukoyanova N, Kondos SC, Farabella I, et al. (2015). Conformational changes during pore formation by the perforin-related protein pleurotolysin. PLoS Biology 13: e1002049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Cai Q, Lüli Y, et al. (2018). The MSDIN family in amanitin-producing mushrooms and evolution of the prolyl oligopeptidase genes. IMA Fungus 9: 225–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Hallen-Adams HE, Scott-Craig JS, et al. (2010). Colocalization of amanitin and a candidate toxin-processing prolyl oligopeptidase in Amanita basidiocarps. Eukaryotic Cell 9: 1891–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Hallen-Adams HE, Scott-Craig JS, et al. (2012). Ribosomal biosynthesis of α-amanitin in Galerina marginata. Fungal Genetics and Biology 49: 123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luti S, Sella L, Quarantin A, et al. (2020). Twenty years of research on cerato-platanin family proteins: clues, conclusions, and unsolved issues. Fungal Biology Reviews 34: 13–24. [Google Scholar]
- Luyten K, Albertyn J, Skibbe WF, et al. (1995). Fps1, a yeast member of the MIP family of channel proteins, is a facilitator for glycerol uptake and efflux and is inactive under osmotic stress. The EMBO Journal 14: 1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A, Shan L, Wang N, et al. (2007). Characterization of a Pleurotus ostreatus fruiting body-specific hydrophobin gene, Po. hyd. Journal of Basic Microbiology 47: 317–324. [DOI] [PubMed] [Google Scholar]
- Ma G, Yang W, Zhao L, et al. (2018). A critical review on the health promoting effects of mushrooms nutraceuticals. Food Science and Human Wellness 7: 125–133. [Google Scholar]
- Ma X, Fan X, Wang G, et al. (2021). Enhanced Expression of Thaumatin-like Protein Gene (LeTLP1) Endows Resistance to Trichoderma atroviride in Lentinula edodes. Life 11: 863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavan S, Krause K, Jung EM, et al. (2014). Differential regulation of multi-copper oxidases in Schizophyllum commune during sexual development. Mycological Progress 13: 1199–1206. [Google Scholar]
- Mäkelä MR, Sietiö OM, de Vries RP, et al. (2014). Oxalate-metabolising genes of the white-rot fungus Dichomitus squalens are differentially induced on wood and at high proton concentration. PLoS One 9: e87959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkinen M, Kuuskeri J, Laine P, et al. (2019). Genome description of Phlebia radiata 79 with comparative genomics analysis on lignocellulose decomposition machinery of phlebioid fungi. BMC Genomics 20: 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancheño JM, Tateno H, Sher D, et al. (2010). Laetiporus sulphureus lectin and aerolysin protein family. Proteins Membrane Binding and Pore Formation 677: 67–80. [DOI] [PubMed] [Google Scholar]
- Many AM, Melki CS, Savytskyy OP, et al. (2009). Meiotic localization of Mre11 and Rad50 in wild type, spo11-1, and MRN complex mutants of Coprinus cinereus. Chromosoma 118: 471–486. [DOI] [PubMed] [Google Scholar]
- Martin F, Aerts A, Ahrén D, et al. (2008). The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature 452: 88–92. [DOI] [PubMed] [Google Scholar]
- Martinez D, Challacombe J, Morgenstern I, et al. (2009). Genome, transcriptome, and secretome analysis of wood decay fungus Postia placenta supports unique mechanisms of lignocellulose conversion. Proceedings of National Academy of Sciences 106: 1954–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez D, Larrondo LF, Putnam N, et al. (2004). Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nature Biotechnology 22: 695–700. [DOI] [PubMed] [Google Scholar]
- Masuda R, Iguchi N, Tukuta K, et al. (2016). The Coprinopsis cinerea Tup1 homologue Cag1 is required for gill formation during fruiting body morphogenesis. Biology Open 5: 1844–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata J, Lyne R, Burns G, Bähler J. (2002). The transcriptional program of meiosis and sporulation in fission yeast. Nature Genetics 32: 143–147. [DOI] [PubMed] [Google Scholar]
- Matityahu A, Hadar Y, Dosoretz CG, et al. (2008). Gene silencing by RNA interference in the white rot fungus Phanerochaete chrysosporium. Applied and Environmental Microbiology 74: 5359–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister C, Thieme KG, Thieme S, et al. (2019). COP9 signalosome interaction with UspA/Usp15 deubiquitinase controls VeA-mediated fungal multicellular development. Biomolecules 9: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mélida H, Sain D, Stajich JE, et al. (2015). Deciphering the uniqueness of Mucoromycotina cell walls by combining biochemical and phylogenomic approaches. Environmental Microbiology 17: 1649–1662. [DOI] [PubMed] [Google Scholar]
- Meng L, Chou T, Jiang S, et al. (2020). Characterization and expression pattern analysis of pheromone receptor-like genes in Winter Mushroom Flammulina filiformis. Archives of Microbiology 202: 2671–2678. [DOI] [PubMed] [Google Scholar]
- Meng L, Lyu X, Shi L, et al. (2021). The transcription factor FvHmg1 negatively regulates fruiting body development in Winter Mushroom Flammulina velutipes. Gene 785: 145618. [DOI] [PubMed] [Google Scholar]
- Merényi Z, Prasanna AN, Wang Z, et al. (2020). Unmatched level of molecular convergence among deeply divergent complex multicellular fungi. Molecular Biology and Evolution 37: 2228–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merényi Z, Virágh M, Gluck-Thaler E, et al. (2022). Gene age shapes the transcriptional landscape of sexual morphogenesis in mushroom-forming fungi (Agaricomycetes). Elife 11: e71348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger MB, Hristova VA, Weissman AM. (2012). HECT and RING finger families of E3 ubiquitin ligases at a glance. Journal of Cell Science 125: 531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerowitz EM. (2002). Comparative genomics. Plants compared to animals: The broadest comparative study of development. Science 295: 1482–1485. [DOI] [PubMed] [Google Scholar]
- Mgbeahuruike AC, Kovalchuk A, Asiegbu FO. (2013). Comparative genomics and evolutionary analysis of hydrophobins from three species of wood-degrading fungi. Mycologia 105: 1471–1478. [DOI] [PubMed] [Google Scholar]
- Michaelis S. (1993). STE6, the yeast a-factor transporter. In Seminars in Cell Biology (Vol. 4, No. 1, pp. 17–27). Academic Press. [DOI] [PubMed] [Google Scholar]
- Michelot D, Melendez-Howell LM. (2003). Amanita muscaria: chemistry, biology, toxicology, and ethnomycology. Mycological Research 107: 131–146. [DOI] [PubMed] [Google Scholar]
- Middleton R, Gao D, Thomas A, et al. (2017). IRFinder: assessing the impact of intron retention on mammalian gene expression. Genome Biology 18: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min B, Kim S, Oh YL, et al. (2018). Genomic discovery of the hypsin gene and biosynthetic pathways for terpenoids in Hypsizygus marmoreus. BMC Genomics 19: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min B, Yoon H, Park J, et al. (2020). Unusual genome expansion and transcription suppression in ectomycorrhizal Tricholoma matsutake by insertions of transposable elements. PLoS One 15: e0227923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyauchi S, Kiss E, Kuo A, et al. (2020). Large-scale genome sequencing of mycorrhizal fungi provides insights into the early evolution of symbiotic traits. Nature Communications 11: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki Y, Jojima T, Ono T, et al. (2004). A cDNA homologue of Schizosaccharomyces pombe cdc5 + from the mushroom Lentinula edodes: Characterization of the cDNA and its expressed product. Biochimica et Biophysica Acta (BBA)-Gene Structure and Expression 1680: 93–102. [DOI] [PubMed] [Google Scholar]
- Miyazaki Y, Kaneko S, Sunagawa M, et al. (2007). The fruiting-specific Le. flp1 gene, encoding a novel fungal fasciclin-like protein, of the basidiomycetous mushroom Lentinula edodes. Current Genetics 51: 367–375. [DOI] [PubMed] [Google Scholar]
- Mol PC, Vermeulen CA, Wessels JGH. (1990). Diffuse extension of hyphae in stipes of Agaricus bisporus may be based on a unique wall structure. Mycological Research 94: 480–488. [Google Scholar]
- Mol PC, Wessels JGH. (1990). Differences in wall structure between substrate hyphae and hyphae of fruit-body stipes in Agaricus bisporus. Mycological Research 94: 472–479. [Google Scholar]
- Mollapour M, Piper PW. (2007). Hog1 Mitogen-Activated Protein Kinase Phosphorylation Targets the Yeast Fps1 Aquaglyceroporin for Endocytosis, Thereby Rendering Cells Resistant to Acetic Acid. Molecular and Cellular Biology 27: 6446–6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore D. (2005). Principles of Mushroom Developmental Biology. International Journal of Medicinal Mushrooms 7: 79–101. [Google Scholar]
- Moore D. (2013). Coprinopsis: an autobiography. Leipzig, Germany: CreateSpace Independent Publishing Platform. [Google Scholar]
- Moore D, Elhiti MM, Butler RD. (1979). Morphogenensis of the carpophore of Coprinus cinereus. New Phytologist 83: 695–722. [Google Scholar]
- Moran Y, Fredman D, Szczesny P, et al. (2012). Recurrent horizontal transfer of bacterial toxin genes to eukaryotes. Molecular Biology and Evolution 29: 2223–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel M, Ngadin AA, Droux M, et al. (2009). The fungal glutathione S-transferase system. Evidence of new classes in the wood-degrading basidiomycete Phanerochaete chrysosporium. Cellular and Molecular Life Sciences 66: 3711–3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin E, Kohler A, Baker AR, et al. (2012). Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proceedings of the National Academy of Sciences 109: 17501–17506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin-Crini N, Lichtfouse E, Torri G, Crini G. (2019). Applications of chitosan in food, pharmaceuticals, medicine, cosmetics, agriculture, textiles, pulp and paper, biotechnology, and environmental chemistry. Environmental Chemistry Letters 17: 1667–1692. [Google Scholar]
- Morton N, Hammond JBW, Dickerson AG. (1985). The Agaricus bisporus mannitol pathway during sporophore growth. Transactions of the British Mycological Society 85: 671–675. [Google Scholar]
- Munir E, Yoon JJ, Tokimatsu T, et al. (2001). A physiological role for oxalic acid biosynthesis in the wood-rotting basidiomycete Fomitopsis palustris. Proceedings of the National Academy of Sciences 98: 11126–11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraguchi H, Abe K, Nakagawa M, et al. (2008a). Identification and characterisation of structural maintenance of chromosome 1 (smc1) mutants of Coprinopsis cinerea. Molecular Genetics and Genomics 280: 223–232. [DOI] [PubMed] [Google Scholar]
- Muraguchi H, Fujita T, Kishibe Y, et al. (2008b). The exp1 gene essential for pileus expansion and autolysis of the inky cap mushroom Coprinopsis cinerea (Coprinus cinereus) encodes an HMG protein. Fungal Genetics and Biology 45: 890–896. [DOI] [PubMed] [Google Scholar]
- Muraguchi H, Kamada T. (1998). The ich1 gene of the mushroom Coprinus cinereus is essential for pileus formation in fruiting. Development 125: 3133–3141. [DOI] [PubMed] [Google Scholar]
- Muraguchi H, Kamada T. (2000). A mutation in the eln2 gene encoding a cytochrome P450 of Coprinus cinereus affects mushroom morphogenesis. Fungal Genetics and Biology 29: 49–59. [DOI] [PubMed] [Google Scholar]
- Muraguchi H, Umezawa K, Niikura M, et al. (2015). Strand-specific RNA-seq analyses of fruiting body development in Coprinopsis cinerea. PLoS One 10: e0141586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata Y, Fujii M, Zolan ME, et al. (1998). Molecular analysis of pcc1, a gene that leads to A-regulated sexual morphogenesis in Coprinus cinereus. Genetics 149: 1753–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahashi S, Lussier M, Bussey H. (1998). Isolation of Candida glabrata homologs of the Saccharomyces cerevisiae KRE9 and KNH1 genes and their involvement in cell wall β-1, 6-glucan synthesis. Journal of Bacteriology 180: 5020–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy LG. (2017). Evolution: Complex Multicellular Life with 5,500 Genes. Current Biology 27: R609–R612. [DOI] [PubMed] [Google Scholar]
- Nagy LG, Kovács GM, Krizsán K. (2018). Complex multicellularity in fungi: evolutionary convergence, single origin, or both? Biological Reviews 93: 1778–1794. [DOI] [PubMed] [Google Scholar]
- Nagy LG, Ohm RA, Kovács GM, et al. (2014). Latent homology and convergent regulatory evolution underlies the repeated emergence of yeasts. Nature Communications 5: 1–8. [DOI] [PubMed] [Google Scholar]
- Nakade K, Watanabe H, Sakamoto Y, et al. (2011). Gene silencing of the Lentinula edodes lcc1 gene by expression of a homologous inverted repeat sequence. Microbiological Research 166: 484–493. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Ando Y, Hata T, et al. (2016). A mutation in the Cc.arp9 gene encoding a putative actin-related protein causes defects in fruiting initiation and asexual development in the agaricomycete Coprinopsis cinerea. Current Genetics 62: 565–574. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Ando Y, Kitaaki K, et al. (2011). Efficient gene targeting in δCc.ku70 or δCc.lig4 mutants of the agaricomycete Coprinopsis cinerea. Fungal Genetics and Biology 48: 939–946. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Kaneko S, Miyazaki Y, et al. (2008). Basidiomycete Lentinula edodes CDC5 and a novel interacting protein CIPB bind to a newly isolated target gene in an unusual manner. Fungal Genetics and Biology 45: 818–828. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Kaneko S, Murata H, et al. (2009). The homologue of Lentinula edodes ctg1, a target for CDC5 and its interacting partner CIPB, from Coprinopsis cinerea is involved in fruitingbody morphogenesis of C. cinerea. Mycoscience 50: 331–342. [Google Scholar]
- Nakazawa T, Morimoto R, Wu H, et al. (2019). Dominant effects of gat1 mutations on the ligninolytic activity of the white-rot fungus Pleurotus ostreatus. Fungal Biology 123: 209–217. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Tatsuta Y, Fujita T, et al. (2010). Mutations in the Cc.rmt1 gene encoding a putative protein arginine methyltransferase alter developmental programs in the basidiomycete Coprinopsis cinerea. Current Genetics 56: 361–367. [DOI] [PubMed] [Google Scholar]
- Namekawa SH, Hamada FN, Sakaguchi K. (2004). Latest frontiers of meiosis research in Coprinus cinereus. Seikagaku. The Journal of Japanese Biochemical Society 76: 1450–1454. [PubMed] [Google Scholar]
- Namekawa SH, Iwabata K, Sugawara H, et al. (2005). Knockdown of LIM15/DMC1 in the mushroom Coprinus cinereus by double-stranded RNA-mediated gene silencing. Microbiology 151: 3669–3678. [DOI] [PubMed] [Google Scholar]
- Nara T, Hamada F, Namekawa S, et al. (2001). Strand Exchange Reaction in Vitro and DNA-Dependent ATPase Activity of Recombinant LIM15/DMC1 and RAD51 Proteins from Coprinus cinereus. Biochemical and Biophysical Research Communications 285: 92–97. [DOI] [PubMed] [Google Scholar]
- Nehls U, Dietz S. (2014). Fungal aquaporins: cellular functions and ecophysiological perspectives. Applied Microbiology and Biotechnology 98: 8835–8851. [DOI] [PubMed] [Google Scholar]
- Nguyen DV, Roret T, Fernandez-Gonzalez A, et al. (2020). Target of rapamycin pathway in the white-rot fungus Phanerochaete chrysosporium. PLoS One 15: e0224776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TA, Cissé OH, Wong JY, et al. (2017). Innovation and constraint leading to complex multicellularity in the Ascomycota. Nature Communications 8: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson KW, Atkin AL, Hornby JM. (2006). Quorum sensing in dimorphic fungi: farnesol and beyond. Applied and Environmental Microbiology 72: 3805–3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu X, Liu CC, Xiong YJ, et al. (2016). The Modes of Action of ChiIII, a Chitinase from Mushroom Coprinopsis cinerea, Shift with Changes in the Length of GlcNAc Oligomers. Journal of Agricultural and Food Chemistry 64: 6958–6968. [DOI] [PubMed] [Google Scholar]
- Niu X, Liu Z, Zhou Y, et al. (2015). Stipe cell wall architecture varies with the stipe elongation of the mushroom Coprinopsis cinerea. Fungal Biology 119: 946–956. [DOI] [PubMed] [Google Scholar]
- Noble R, Dobrovin-Pennington A, Hobbs PJ, et al. (2009). Volatile C8 compounds and pseudomonads influence primordium formation of Agaricus bisporus. Mycologia 101: 583–591. [DOI] [PubMed] [Google Scholar]
- Nofiani R, de Mattos-Shipley K, Lebe KE, et al. (2018). Strobilurin biosynthesis in Basidiomycete fungi. Nature Communications 9: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novaes-Ledieu M, Garcia Mendoza C. (1981). The cell walls of Agaricus bisporus and Agaricus campestris fruiting body hyphae. Canadian Journal of Microbiology 27: 779–787. [DOI] [PubMed] [Google Scholar]
- Novak M, Kraševec N, Skoèaj M, et al. (2015). Fungal aegerolysin-like proteins: distribution, activities, and applications. Applied Microbiology and Biotechnology 99: 601–610. [DOI] [PubMed] [Google Scholar]
- Novak M, Krpan T, Panevska A, et al. (2020). Binding specificity of ostreolysin A6 towards Sf9 insect cell lipids. Biochimica et Biophysica Acta (BBA)-Biomembranes 1862: 183307. [DOI] [PubMed] [Google Scholar]
- Nowrousian M, Cebula P. (2005). The gene for a lectin-like protein is transcriptionally activated during sexual development, but is not essential for fruiting body formation in the filamentous fungus Sordaria macrospora. BMC Microbiology 5: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor E, Doyle S, Amini A, et al. (2021). Transmission of mushroom virus X and the impact of virus infection on the transcriptomes and proteomes of different strains of Agaricus bisporus. Fungal Biology 125: 704–717. [DOI] [PubMed] [Google Scholar]
- O’Connor E, McGowan J, McCarthy CGP, et al. (2019). Whole genome sequence of the commercially relevant mushroom strain Agaricus bisporus var. bisporus ARP23. G3 9: 3057–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermaier S, Müller M. (2020). Ibotenic Acid Biosynthesis in the Fly Agaric Is Initiated by Glutamate Hydroxylation. Angewandte Chemie 132: 12532–12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohga S, Cho NS, Thurston CF, Wood DA. (2000). Transcriptional regulation of laccase and cellulase in relation to fruit body formation in the mycelium of Lentinula edodes on a sawdust-based substrate. Mycoscience 41: 149–153. [Google Scholar]
- Ohm RA, Aerts D, Wösten HAB, et al. (2013). The blue light receptor complex WC-1/2 of Schizophyllum commune is involved in mushroom formation and protection against phototoxicity. Environmental Microbiology 15: 943–955. [DOI] [PubMed] [Google Scholar]
- Ohm RA, de Jong JF, de Bekker C, et al. (2011). Transcription factor genes of Schizophyllum commune involved in regulation of mushroom formation. Molecular Microbiology 81: 1433–1445. [DOI] [PubMed] [Google Scholar]
- Ohm RA, De Jong JF, Lugones LG, et al. (2010). Genome sequence of the model mushroom Schizophyllum commune. Nature Biotechnology 28: 957–963. [DOI] [PubMed] [Google Scholar]
- Ohm RA, Riley R, Salamov A, et al. (2014). Genomics of wood-degrading fungi. Fungal Genetics and Biology 72: 82–90. [DOI] [PubMed] [Google Scholar]
- Ohtaki S, Maeda H, Takahashi T, et al. (2006). Novel hydrophobic surface binding protein, HsbA, produced by Aspergillus oryzae. Applied and Environmental Microbiology 72: 2407–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oide S, Tanaka Y, Watanabe A, et al. (2019). Carbohydrate-binding property of a cell wall integrity and stress response component (WSC) domain of an alcohol oxidase from the rice blast pathogen Pyricularia oryzae. Enzyme and Microbial Technology 125: 13–20. [DOI] [PubMed] [Google Scholar]
- Ojeda-López M, Chen W, Eagle CE, et al. (2018). Evolution of asexual and sexual reproduction in the aspergilli. Studies in Mycology 91: 37–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olombrada M, Lázaro-Gorines R, López-Rodríguez JC, et al. (2017b). Fungal ribotoxins: a review of potential biotechnological applications. Toxins 9: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olombrada M, Medina P, Budia F, et al. (2017a). Characterization of a new toxin from the entomopathogenic fungus Metarhizium anisopliae: the ribotoxin anisoplin. Biological Chemistry 398: 135–142. [DOI] [PubMed] [Google Scholar]
- Orban A, Hennicke F, Rühl M. (2020). Volatilomes of Cyclocybe aegerita during different stages of monokaryotic and dikaryotic fruiting. Biological Chemistry 401: 995–1004. [DOI] [PubMed] [Google Scholar]
- Orban A, Weber A, Herzog R, et al. (2021). Transcriptome of different fruiting stages in the cultivated mushroom Cyclocybe aegerita suggests a complex regulation of fruiting and reveals enzymes putatively involved in fungal oxylipin biosynthesis. BMC Genomics 22: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlean P. (2012). Architecture and biosynthesis of the Saccharomyces cerevisiae cell wall. Genetics 192: 775–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota K, Butala M, Viero G, et al. (2014). Fungal MACPF-like proteins and aegerolysins: bi-component pore-forming proteins?. MACPF/CDC Proteins-Agents of Defence, Attack and Invasion. Sub-cellular Biochemistry 80: 271–291. [DOI] [PubMed] [Google Scholar]
- Paiva S, Devaux F, Barbosa S, et al. (2004). Ady2p is essential for the acetate permease activity in the yeast Saccharomyces cerevisiae. Yeast 21(3): 201–210. [DOI] [PubMed] [Google Scholar]
- Palmer GE, Horton JS. (2006). Mushrooms by magic: making connections between signal transduction and fruiting body development in the basidiomycete fungus Schizophyllum commune. FEMS Microbiology Letters 262: 1–8. [DOI] [PubMed] [Google Scholar]
- Palmieri F, Estoppey A, House GL, et al. (2019). Oxalic acid, a molecule at the crossroads of bacterial-fungal interactions. Advances in Applied Microbiology 106: 49–77. [DOI] [PubMed] [Google Scholar]
- Panevska A, Hodnik V, Skoèaj M, et al. (2019). Pore-forming protein complexes from Pleurotus mushrooms kill western corn rootworm and Colorado potato beetle through targeting membrane ceramide phosphoethanolamine. Scientific Reports 9: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panevska A, Skoèaj M, Modic Š, et al. (2021). Aegerolysins from the fungal genus Pleurotus–Bioinsecticidal proteins with multiple potential applications. Journal of Invertebrate Pathology 186: 107474. [DOI] [PubMed] [Google Scholar]
- Park M, Kim M, Kim S, et al. (2015). Differential expression of laccase genes in Pleurotus ostreatus and biochemical characterization of laccase isozymes produced in Pichia pastoris. Mycobiology 43: 280–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SG, il Yoo S, Ryu DS, et al. (2017). Long-read transcriptome data for improved gene prediction in Lentinula edodes. Data in Brief 15: 454–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YJ, Baek JH, Lee S, et al. (2014). Whole genome and global gene expression analyses of the model mushroom Flammulina velutipes reveal a high capacity for lignocellulose degradation. PLoS One 9: e93560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YJ, Lee CS, Kong WS. (2019). Genomic insights into the fungal lignocellulolytic machinery of Flammulina rossica. Microorganisms 7: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel PK, Free SJ. (2019). The genetics and biochemistry of cell wall structure and synthesis in Neurospora crassa, a model filamentous fungus. Frontiers in Microbiology 10: 2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patyshakuliyeva A, Jurak E, Kohler A, et al. (2013). Carbohydrate utilization and metabolism is highly differentiated in Agaricus bisporus. BMC Genomics 14: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peisker K, Chiabudini M, Rospert S. (2010). The ribosome-bound Hsp70 homolog Ssb of Saccharomyces cerevisiae. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1803: 662–672. [DOI] [PubMed] [Google Scholar]
- Pelkmans JF. (2016a). Environmental signalling and regulation of mushroom formation. (Doctoral dissertation, University Utrecht). [Google Scholar]
- Pelkmans JF, Patil MB, Gehrmann T, et al. (2017). Transcription factors of Schizophyllum commune involved in mushroom formation and modulation of vegetative growth. Scientific Reports 7: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkmans JF, Vos AM, Scholtmeijer K, et al. (2016b). The transcriptional regulator c2h2 accelerates mushroom formation in Agaricus bisporus. Applied Microbiology and Biotechnology 100: 7151–7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrin C, Morin E, Martin FM, et al. (2015). Comparative analysis of secretomes from ectomycorrhizal fungi with an emphasis on small-secreted proteins. Frontiers in Microbiology 6: 1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peñalva MA, Tilburn J, Bignell E, et al. (2008). Ambient pH gene regulation in fungi: making connections. Trends in Microbiology 16: 291–300. [DOI] [PubMed] [Google Scholar]
- Plagemann I, Zelena K, Arendt P, et al. (2013). LOXPsa1, the first recombinant lipoxygenase from a basidiomycete fungus. Journal of Molecular Catalysis B: Enzymatic 87: 99–104. [Google Scholar]
- Plaza DF, Lin CW, van der Velden NSJ, et al. (2014). Comparative transcriptomics of the model mushroom Coprinopsis cinerea reveals tissue-specific armories and a conserved circuitry for sexual development. BMC Genomics 15: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluskal T, Ueno M, Yanagida M. (2014). Genetic and metabolomic dissection of the ergothioneine and selenoneine biosynthetic pathway in the fission yeast, S. pombe, and construction of an overproduction system. PLoS One 9: e97774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pócsi I, Prade RA, Penninckx MJ. (2004). Glutathione, altruistic metabolite in fungi. Advances in Microbial Physiology 49: 1–76. [DOI] [PubMed] [Google Scholar]
- Pöggeler S, Kück U. (2004). A WD40 repeat protein regulates fungal cell differentiation and can be replaced functionally by the mammalian homologue striatin. Eukaryotic Cell 3: 232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pöggeler S, Nowrousian M, Kück U. (2006). Fruiting-body development in ascomycetes. In Growth, Differentiation and sexuality (pp. 325–355). Springer, Berlin, Heidelberg. [Google Scholar]
- Pohleven J, Brzin J, Vrabec L, et al. (2011). Basidiomycete Clitocybe nebularis is rich in lectins with insecticidal activities. Applied Microbiology and Biotechnology 91: 1141–1148. [DOI] [PubMed] [Google Scholar]
- Polonio Á, Fernández-Ortuño D, de Vicente A, et al. (2021). A haustorial expressed lytic polysaccharide monooxygenase from the cucurbit powdery mildew pathogen Podosphaera xanthii contributes to the suppression of chitin-triggered immunity. Molecular Plant Pathology 22: 580–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanna AN, Gerber D, Kijpornyongpan T, et al. (2020). Model choice, missing data, and taxon sampling impact phylogenomic inference of deep Basidiomycota relationships. Systematic Biology 69: 17–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primig M, Williams RM, Winzeler EA, et al. (2000). The core meiotic transcriptome in budding yeasts. Nature Genetics 26: 415–423. [DOI] [PubMed] [Google Scholar]
- Pulman JA, Childs KL, Sgambelluri RM, et al. (2016). Expansion and diversification of the MSDIN family of cyclic peptide genes in the poisonous agarics Amanita phalloides and A. bisporigera. BMC Genomics 17: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Chen H, Zhang M, et al. (2019). Identification and expression analysis of Pofst3 suggests a role during Pleurotus ostreatus primordia formation. Fungal Biology 123: 200–208. [DOI] [PubMed] [Google Scholar]
- Qi Y, Sun X, Ma L, et al. (2020). Identification of two Pleurotus ostreatus blue light receptor genes (PoWC-1 and PoWC-2) and in vivo confirmation of complex PoWC-12 formation through yeast two hybrid system. Fungal Biology 124: 8–14. [DOI] [PubMed] [Google Scholar]
- Quiroz-Castañeda RE, Martínez-Anaya C, Cuervo-Soto LI, et al. (2011) Loosenin, a novel protein with cellulose-disrupting activity from Bjerkandera adusta. Microbial Cell Factories 10: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragucci S, Landi N, Russo R, et al. (2021). Ageritin from pioppino mushroom: the prototype of ribotoxin-like proteins, a novel family of specific ribonucleases in edible mushrooms. Toxins 13: 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmad N, Al-Obaidi JR, Rashid NMN, et al. (2014). Comparative proteomic analysis of different developmental stages of the edible mushroom Termitomyces heimii. Biological Research 47: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman F, Hassan M, Hanano A, et al. (2018). Evolutionary, structural and functional analysis of the caleosin/peroxygenase gene family in the Fungi. BMC Genomics 19: 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudaskoski M, Kothe E. (2010). Basidiomycete mating type genes and pheromone signalling. Eukaryotic Cell 9: 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayle DL, Cleland RE. (1992). The Acid Growth Theory of auxin-induced cell elongation is alive and well. Plant Physiology 99: 1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboul CF, Whisstock JC, Dunstone MA. (2016). Giant MACPF/CDC pore forming toxins: A class of their own. Biochimica et Biophysica Acta (BBA)-Biomembranes 1858: 475–486. [DOI] [PubMed] [Google Scholar]
- Reinhold R, Krüger V, Meinecke M, et al. (2012). The Channel-Forming Sym1 Protein Is Transported by the TIM23 Complex in a Presequence-Independent Manner. Molecular and Cellular Biology 32: 5009–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennemeier C, Schwab M, Lermann U, et al. (2011). Seminal plasma protects human spermatozoa and pathogenic yeasts from capture by dendritic cells. Human Reproduction 26: 987–999. [DOI] [PubMed] [Google Scholar]
- Reschka EJ, Nordzieke S, Valerius O, et al. (2018). A novel STRIPAK complex component mediates hyphal fusion and fruiting-body development in filamentous fungi. Molecular Microbiology 110: 513–532. [DOI] [PubMed] [Google Scholar]
- Revuelta MV, van Kan JA, Kay J, et al. (2014). Extensive expansion of A1 family aspartic proteinases in fungi revealed by evolutionary analyses of 107 complete eukaryotic proteomes. Genome Biology and Evolution 6: 1480–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley R, Salamov AA, Brown DW, et al. (2014). Extensive sampling of basidiomycete genomes demonstrates inadequacy of the white-rot/brown-rot paradigm for wood decay fungi. Proceedings of the National Academy of Sciences 111: 9923–9928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquelme M, Aguirre J, Bartnicki-García S, et al. (2018). Fungal morphogenesis, from the polarized growth of hyphae to complex reproduction and infection structures. Microbiology and Molecular Biology Reviews 82: e00068–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquelme M, Challen MP, Casselton LA, et al. (2005). The origin of multiple B mating specificities in Coprinus cinereus. Genetics 170: 1105–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenburg SY, Terhem RB, Veloso J, et al. (2018). Functional analysis of mating type genes and transcriptome analysis during fruiting body development of Botrytis cinerea. MBio 9: e01939–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Romero J, Hedtke M, Kastner C, et al. (2010). Fungi, hidden in soil or up in the air: light makes a difference. Annual Review of Microbiology 64: 585–610. [DOI] [PubMed] [Google Scholar]
- Román E, Arana DM, Nombela C, et al. (2007). MAP kinase pathways as regulators of fungal virulence. Trends in Microbiology 15: 181–190. [DOI] [PubMed] [Google Scholar]
- Romano JD, Michaelis S. (2001). Topological and mutational analysis of Saccharomyces cerevisiae Ste14p, founding member of the isoprenylcysteine carboxyl methyltransferase family. Molecular Biology of the Cell 12: 1957–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper M, Seminara A. (2019). Mycofluidics: the fluid mechanics of fungal adaptation. Annual Review of Fluid Mechanics 51: 511–538. [Google Scholar]
- Royse DJ, Baars J, Tan Q. (2017). Current overview of mushroom production in the world. Edible and Medicinal Mushrooms: Technology and Applications 5–13. [Google Scholar]
- Ruess L, Lussenhop J. (2005). Trophic interactions of fungi and animals. Mycology Series 23: 581. [Google Scholar]
- Ruytinx J, Miyauchi S, Hartmann-Wittulsky S, et al. (2021). A Transcriptomic Atlas of the Ectomycorrhizal Fungus Laccaria bicolor. Microorganisms 9: 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabotič J, Ohm RA, Künzler M. (2016). Entomotoxic and nematotoxic lectins and protease inhibitors from fungal fruiting bodies. Applied Microbiology and Biotechnology 100: 91–111. [DOI] [PubMed] [Google Scholar]
- Sabotič J, Renko M, Kos J. (2019). β-trefoil protease inhibitors unique to higher fungi. Acta Chimica Slovenica 66: 28–36. [PubMed] [Google Scholar]
- Saito K, Yamazaki H, Ohnishi Y, et al. (1998). Production of trehalose synthase from a basidiomycete, Grifola frondosa, in Escherichia coli. Applied Microbiology and Biotechnology 50: 193–198. [DOI] [PubMed] [Google Scholar]
- Sakai H, Kajiwara S. (2003). A stearoyl-CoA-specific Δ9 fatty acid desaturase from the basidiomycete Lentinula edodes. Bioscience, Biotechnology, and Biochemistry 67: 2431–2437. [DOI] [PubMed] [Google Scholar]
- Sakai H, Kajiwara S. (2005). Cloning and functional characterization of a Δ12 fatty acid desaturase gene from the basidiomycete Lentinula edodes. Molecular Genetics and Genomics 273: 336–341. [DOI] [PubMed] [Google Scholar]
- Sakamoto Y. (2018). Influences of environmental factors on fruiting body induction, development and maturation in mushroom-forming fungi. Fungal Biology Reviews 32: 236–248. [Google Scholar]
- Sakamoto Y, Irie T, Sato T. (2005). Isolation and characterization of a fruiting body-specific exo-β-1, 3-glucanase-encoding gene, exg1, from Lentinula edodes. Current Genetics 47: 244–252. [DOI] [PubMed] [Google Scholar]
- Sakamoto Y, Minato KI, Nagai M, et al. (2005). Characterization of the Lentinula edodes exg2 gene encoding a lentinan-degrading exo-β-1, 3-glucanase. Current Genetics 48: 195–203. [DOI] [PubMed] [Google Scholar]
- Sakamoto Y, Nakade K, Konno N. (2011). Endo-β-1, 3-glucanase GLU1, from the fruiting body of Lentinula edodes, belongs to a new glycoside hydrolase family. Applied and Environmental Microbiology 77: 8350–8354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto Y, Nakade K, Sato S, et al. (2017). Lentinula edodes genome survey and postharvest transcriptome analysis. Applied and Environmental Microbiology 83: e02990–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto Y, Nakade K, Sato T. (2009). Characterization of the post-harvest changes in gene transcription in the gill of the Lentinula edodes fruiting body. Current Genetics 55: 409–423. [DOI] [PubMed] [Google Scholar]
- Sakamoto Y, Nakade K, Yoshida K, et al. (2015). Grouping of multicopper oxidases in Lentinula edodes by sequence similarities and expression patterns. AMB Express 5: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto Y, Sato S, Ito M, et al. (2018). Blue light exposure and nutrient conditions influence the expression of genes involved in simultaneous hyphal knot formation in Coprinopsis cinerea. Microbiological Research 217: 81–90. [DOI] [PubMed] [Google Scholar]
- Sakamoto Y, Watanabe H, Nagai M, et al. (2006). Lentinula edodes tlg1 encodes a thaumatin-like protein that is involved in lentinan degradation and fruiting body senescence. Plant Physiology 141: 793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saloheimo M, Paloheimo M, Hakola S, et al. (2002). Swollenin, a Trichoderma reesei protein with sequence similarity to the plant expansins, exhibits disruption activity on cellulosic materials. European Journal of Biochemistry 269: 4202–4211. [DOI] [PubMed] [Google Scholar]
- Samalova M, Carr P, Bromley M, et al. (2020). GPI anchored proteins in Aspergillus fumigatus and cell wall morphogenesis. The Fungal Cell Wall 167–186. [DOI] [PubMed] [Google Scholar]
- Sammer D, Krause K, Gube M, et al. (2016). Hydrophobins in the Life Cycle of the Ectomycorrhizal Basidiomycete Tricholoma vaccinum. PLoS One 11: e0167773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampedro J, Cosgrove DJ. (2005). The expansin superfamily. Genome Biology 6: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Arreguin JA, Cabrera-Ponce JL, León-Ramírez CG, et al. (2020). Analysis of the photoreceptors involved in the light-depending basidiocarp formation in Ustilago maydis. Archives of Microbiology 202: 93–103. [DOI] [PubMed] [Google Scholar]
- Sánchez-García M, Ryberg M, Khan FK, et al. (2020). Fruiting body form, not nutritional mode, is the major driver of diversification in mushroom-forming fungi. Proceedings of the National Academy of Sciences 117: 32528–32534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Pulido L, Martýn-Belmonte F, Valencia A, et al. (2002). MARVEL: a conserved domain involved in membrane apposition events. Trends in Biochemical Sciences 27: 599–601. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vallet A, Mesters JR, Thomma BP. (2015). The battle for chitin recognition in plant-microbe interactions. FEMS Microbiology Reviews 39: 171–183. [DOI] [PubMed] [Google Scholar]
- Sano H, Kaneko S, Sakamoto Y, et al. (2009). The basidiomycetous mushroom Lentinula edodes white collar-2 homolog PHRB, a partner of putative blue-light photoreceptor PHRA, binds to a specific site in the promoter region of the L. edodes tyrosinase gene. Fungal Genetics and Biology 46: 333–341. [DOI] [PubMed] [Google Scholar]
- Sato I, Shimizu M, Hoshino T, et al. (2009). The glutathione system of Aspergillus nidulans involves a fungus-specific glutathione S-transferase. Journal of Biological Chemistry 284: 8042–8053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Kurahashi A, Nishibori K, Fujimori F. (2015). Development of a transformation system for the edible mushroom Grifola frondosa: Demonstrating heterologous gene expression and RNAi-mediated gene silencing. Mycoscience 56: 364–372. [Google Scholar]
- Schindler D, Nowrousian M. (2014). The polyketide synthase gene pks4 is essential for sexual development and regulates fruiting body morphology in Sordaria macrospora. Fungal Genetics and Biology 68: 48–59. [DOI] [PubMed] [Google Scholar]
- Schneiter R, Di Pietro A. (2013). The CAP protein superfamily: function in sterol export and fungal virulence. Biomolecular Concepts 4: 519–525. [DOI] [PubMed] [Google Scholar]
- Schubert D, Raudaskoski M, Knabe N, et al. (2006). Ras GTPase-activating protein Gap1 of the homobasidiomycete Schizophyllum commune regulates hyphal growth orientation and sexual development. Eukaryotic Cell 5: 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert M, Bleuler-Martinez S, Butschi A, et al. (2012). Plasticity of the β-trefoil protein fold in the recognition and control of invertebrate predators and parasites by a fungal defence system. PLoS Pathogens 8: e1002706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TA, Prinz WA. (2007). Sterol transport in yeast and the oxysterol binding protein homologue (OSH) family. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids 1771: 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalb MN. (1974). Effect of adenosine 3′, 5′-cyclic monophosphate on the morphogenesis of fruit bodies of Schizophyllum commune. Archives of Microbiology 96: 17–20. [DOI] [PubMed] [Google Scholar]
- Sebastian J, Rouissi T, Brar SK. (2020). Fungal chitosan: prospects and challenges. In Handbook of Chitin and Chitosan (pp. 419–452). Elsevier. [Google Scholar]
- Seibold PS, Lenz C, Gressler M, et al. (2020). The Laetiporus polyketide synthase LpaA produces a series of antifungal polyenes. The Journal of Antibiotics 73: 711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz LC, Tang K, Cummings WJ, et al. (1996). The rad9 gene of Coprinus cinereus encodes a proline-rich protein required for meiotic chromosome condensation and synapsis. Genetics 142: 1105–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semana P, Powlowski J. (2019). Four aromatic intradiol ring cleavage dioxygenases from Aspergillus niger. Applied and Environmental Microbiology 85: e01786–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahinian S, Bussey H. (2000). β-1, 6-Glucan synthesis in Saccharomyces cerevisiae. Molecular Microbiology 35: 477–489. [DOI] [PubMed] [Google Scholar]
- Shaw R. (1967). Fatty acids of fruiting bodies of basidiomycetes. Nature 213: 86–87. [Google Scholar]
- Sheridan KJ, Lechner BE, Keeffe GO, et al. (2016). Ergothioneine biosynthesis and functionality in the opportunistic fungal pathogen, Aspergillus fumigatus. Scientific Reports 6: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim D, Park SG, Kim K, et al. (2016). Whole genome de novo sequencing and genome annotation of the world popular cultivated edible mushroom, Lentinula edodes. Journal of Biotechnology 223: 24–25. [DOI] [PubMed] [Google Scholar]
- Shioya T, Nakamura H, Ishii N, et al. (2013). The Coprinopsis cinerea septin Cc. Cdc3 is involved in stipe cell elongation. Fungal Genetics and Biology 58: 80–90. [DOI] [PubMed] [Google Scholar]
- Simockova M, Holic R, Tahotná D, et al. (2008). Yeast Pgc1p (YPL206c) controls the amount of phosphatidylglycerol via a phospholipase C-type degradation mechanism. Journal of Biological Chemistry 283: 17107–17115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipos G, Prasanna AN, Walter MC, et al. (2017). Genome expansion and lineage-specific genetic innovations in the forest pathogenic fungi Armillaria. Nature Ecology & Evolution 1: 1931–1941. [DOI] [PubMed] [Google Scholar]
- Slot JC, Gluck-Thaler E. (2019). Metabolic gene clusters, fungal diversity, and the generation of accessory functions. Current Opinion in Genetics & Development 58: 17–24. [DOI] [PubMed] [Google Scholar]
- Šmid I, Rotter A, Gruden K, et al. (2015). Clitocypin, a fungal cysteine protease inhibitor, exerts its insecticidal effect on Colorado potato beetle larvae by inhibiting their digestive cysteine proteases. Pesticide Biochemistry and Physiology 122: 59–66. [DOI] [PubMed] [Google Scholar]
- Soanes DM, Chakrabarti A, Paszkiewicz KH, et al. (2012). Genome-wide transcriptional profiling of appressorium development by the rice blast fungus Magnaporthe oryzae. PLoS Pathogens 8: e1002514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer R, Makshakova ON, Wohlschlager T, et al. (2018). Crystal structures of fungal tectonin in complex with O-methylated glycans suggest key role in innate immune defence. Structure 26: 391–402. [DOI] [PubMed] [Google Scholar]
- Song CH, Cho KY, Nair NG, et al. (1989). Growth stimulation and lipid synthesis in Lentinus edodes. Mycologia 81: 514–522. [Google Scholar]
- Song HY, Kim DH, Kim JM. (2018). Comparative transcriptome analysis of dikaryotic mycelia and mature fruiting bodies in the edible mushroom Lentinula edodes. Scientific Reports 8: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg AS, Sedaghat-Telgerd N, Lavrijssen B, et al. (2020). Telomere-to-telomere assembled and centromere annotated genomes of the two main subspecies of the button mushroom Agaricus bisporus reveal especially polymorphic chromosome ends. Scientific Reports 10: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht CA. (1995). Isolation of the Bα and Bβ mating-type loci of Schizophyllum commune. Current Genetics 28: 374–379. [DOI] [PubMed] [Google Scholar]
- Spiteller P. (2008). Chemical defence strategies of higher fungi. Chemistry–A European Journal 14: 9100–9110. [DOI] [PubMed] [Google Scholar]
- Spiteller P. (2015). Chemical ecology of fungi. Natural Product Reports 32: 971–993. [DOI] [PubMed] [Google Scholar]
- Stadler M, Hoffmeister D. (2015). Fungal natural products - the mushroom perspective. Frontiers in Microbiology 6: 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler M, Sterner O. (1998). Production of bioactive secondary metabolites in the fruit bodies of macrofungi as a response to injury. Phytochemistry 49: 1013–1019. [Google Scholar]
- Stajich JE, Berbee ML, Blackwell M, et al. (2009). Primer - the fungi. Current Biology: CB 19: R840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajich JE, Wilke SK, Ahrén D, et al. (2010). Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus). Proceedings of the National Academy of Sciences 107: 11889–11894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stassen NY, Logsdon JM, Jr, Vora GJ, et al. (1997). Isolation and characterization of rad51 orthologs from Coprinus cinereus and Lycopersicon esculentum, and phylogenetic analysis of eukaryotic recA homologs. Current Genetics 31: 144–157. [DOI] [PubMed] [Google Scholar]
- Steinegger M, Söding J. (2017). MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nature Biotechnology 35: 1026–1028. [DOI] [PubMed] [Google Scholar]
- Stogios PJ, Downs GS, Jauhal JJ, et al. (2005). Sequence and structural analysis of BTB domain proteins. Genome Biology 6: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoop JM, Mooibroek H. (1998). Cloning and characterization of NADP-mannitol dehydrogenase cDNA from the button mushroom, Agaricus bisporus, and its expression in response to NaCl stress. Applied and Environmental Microbiology 64: 4689–4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöckli M, Morinaka BI, Lackner G, Kombrink A, Sieber R, Margot C, Stanley CE, deMello AJ, Piel J, Künzler M. (2019). Bacteria-induced production of the antibacterial sesquiterpene lagopodin B in Coprinopsis cinerea. Molecular Microbiology 112: 605–619. [DOI] [PubMed] [Google Scholar]
- Straatsma G, Sonnenberg ASM, van Griensven LJ. (2013). Development and growth of fruit bodies and crops of the button mushroom, Agaricus bisporus. Fungal Biology 117: 697–707. [DOI] [PubMed] [Google Scholar]
- Strijbis K, Distel B. (2010). Intracellular acetyl unit transport in fungal carbon metabolism. Eukaryotic Cell 9: 1809–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutz K, Kaech A, Aebi M, et al. (2015). Disruption of the C. elegans intestinal brush border by the fungal lectin CCL2 phenocopies dietary lectin toxicity in mammals. PLoS One 10: e0129381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Ma Y, Xu X, et al. (2020). Molecular cloning and bioinformatics analyses of a GH3 beta-glucosidase from Auricularia heimuer. Biotechnology & Biotechnological Equipment 34: 850–859. [Google Scholar]
- Sunagawa M, Magae Y. (2005). Isolation of genes differentially expressed during the fruit body development of Pleurotus ostreatus by differential display of RAPD. FEMS Microbiology Letters 246: 279–284. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Bayram OS, Bayram Ö, et al. (2013). conF and conJ contribute to conidia germination and stress response in the filamentous fungus Aspergillus nidulans. Fungal Genetics and Biology 56: 42–53. [DOI] [PubMed] [Google Scholar]
- Swamy S, Uno I, Ishikawa T. (1985). Regulation of cyclic AMP metabolism by the incompatibility factors in Coprinus cinereus. Microbiology 131: 3211–3217. [Google Scholar]
- Szczesny P, Iacovache I, Muszewska A, et al. (2011). Extending the aerolysin family: from bacteria to vertebrates. PLoS One 6: e20349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto CY, Leung GS, Kwan HS. (2007). Le. MAPK and its interacting partner, Le. DRMIP, in fruiting body development in Lentinula edodes. Gene 393: 87–93. [DOI] [PubMed] [Google Scholar]
- Takakura Y, Tsunashima M, Suzuki J, et al. (2009). Tamavidins–novel avidin-like biotin-binding proteins from the Tamogitake mushroom. The FEBS Journal 276: 1383–1397. [DOI] [PubMed] [Google Scholar]
- Takeshita N, Yamashita S, Ohta A, et al. (2006). Aspergillus nidulans class V and VI chitin synthases CsmA and CsmB, each with a myosin motor-like domain, perform compensatory functions that are essential for hyphal tip growth. Molecular Microbiology 59: 1380–1394. [DOI] [PubMed] [Google Scholar]
- Tamaki H. (2007). Glucose-stimulated cAMP-protein kinase A pathway in yeast Saccharomyces cerevisiae. Journal of Bioscience and Bioengineering 104: 245–250. [DOI] [PubMed] [Google Scholar]
- Tan YH, Moore D. (1994). High concentrations of mannitol in the shiitake mushroom Lentinula edodes. Microbios 79: 31–35. [PubMed] [Google Scholar]
- Tang LH, Jian HH, Song CY, et al. (2013). Transcriptome analysis of candidate genes and signalling pathways associated with light-induced brown film formation in Lentinula edodes. Applied Microbiology and Biotechnology 97: 4977–4989. [DOI] [PubMed] [Google Scholar]
- Tang X, Ding X, Hou YL. (2020). Comparative analysis of transcriptomes revealed the molecular mechanism of development of Tricholoma matsutake at different stages of fruiting bodies. Food Science and Biotechnology 29: 939–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Q, Ma K, Yang Y, et al. (2016). Bioactive sesquiterpenes from the edible mushroom Flammulina velutipes and their biosynthetic pathway confirmed by genome analysis and chemical evidence. The Journal of Organic Chemistry 81: 9867–9877. [DOI] [PubMed] [Google Scholar]
- Tao Y, Chen R, Yan J, et al. (2019). A hydrophobin gene, Hyd9, plays an important role in the formation of aerial hyphae and primordia in Flammulina filiformis. Gene 706: 84–90. [DOI] [PubMed] [Google Scholar]
- Tao Y, Xie B, Yang Z, et al. (2013). Identification and expression analysis of a new glycoside hydrolase family 55 exo-beta-1,3-glucanase-encoding gene in Volvariella volvacea suggests a role in fruiting body development. Gene 527: 154–160. [DOI] [PubMed] [Google Scholar]
- Tasaki Y, Toyama S, Kuribayashi T, et al. (2013). Molecular characterization of a lipoxygenase from the basidiomycete mushroom Pleurotus ostreatus. Bioscience, Biotechnology, and Biochemistry 77: 38–45. [DOI] [PubMed] [Google Scholar]
- Tatebe H, Shiozaki K. (2017). Evolutionary conservation of the components in the TOR signalling pathways. Biomolecules 7: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JW, Ellison CE. (2010). Mushrooms: morphological complexity in the fungi. Proceedings of the National Academy of Sciences 107: 11655–11656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayyrov A, Azevedo S, Herzog R, et al. (2019a). Heterologous production and functional characterization of ageritin, a novel type of ribotoxin highly expressed during fruiting of the edible mushroom Agrocybe aegerita. Applied and Environmental Microbiology 85: e01549–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayyrov A, Schmieder SS, Bleuler-Martinez S, et al. (2018). Toxicity of potential fungal defence proteins towards the fungivorous nematodes Aphelenchus avenae and Bursaphelenchus okinawaensis. Applied and Environmental Microbiology 84: e02051–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayyrov A, Stanley CE, Azevedo S, et al. (2019b). Combining microfluidics and RNA-sequencing to assess the inducible defensome of a mushroom against nematodes. BMC Genomics 20: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichert I, Dahlmann TA, Kuck U, et al. (2017). RNA Editing During Sexual Development Occurs in Distantly Related Filamentous Ascomycetes. Genome Biology and Evolution 9: 855–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichert I, Steffens EK, Schnaß N, et al. (2014). PRO40 Is a Scaffold Protein of the Cell Wall Integrity Pathway, Linking the MAP Kinase Module to the Upstream Activator Protein Kinase C. PLoS Genetics 10: e1004582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telzrow CL, Nichols CB, Castro-Lopez N, et al. (2019). A fungal arrestin protein contributes to cell cycle progression and pathogenesis. MBio 10: e02682–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temp U, Eggert C. (1999). Novel interaction between laccase and cellobiose dehydrogenase during pigment synthesis in the white rot fungus Pycnoporus cinnabarinus. Applied and Environmental Microbiology 65: 389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashima K, Yuki K, Muraguchi H, et al. (2005). The dst1 gene involved in mushroom photomorphogenesis of Coprinus cinereus encodes a putative photoreceptor for blue light. Genetics 171: 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thammahong A, Puttikamonkul S, Perfect JR, et al. (2017). Central role of the trehalose biosynthesis pathway in the pathogenesis of human fungal infections: opportunities and challenges for therapeutic development. Microbiology and Molecular Biology Reviews 81: e00053–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita T, Mizumachi Y, Chong K, et al. (2004). Protein sequence analysis, cloning, and expression of flammutoxin, a pore-forming cytolysin from Flammulina velutipes: maturation of dimeric precursor to monomeric active form by carboxyl-terminal truncation. Journal of Biological Chemistry 279: 54161–54172. [DOI] [PubMed] [Google Scholar]
- Tong X, Zhang H, Wang F, et al. (2020). Comparative transcriptome analysis revealed genes involved in the fruiting body development of Ophiocordyceps sinensis. PeerJ 8:e8379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrens-Spence MP, Liu CT, Pluskal T, et al. (2018). Monoamine biosynthesis via a noncanonical calcium-activatable aromatic amino acid decarboxylase in psilocybin mushroom. ACS Chemical Biology 13: 3343–3353. [DOI] [PubMed] [Google Scholar]
- Tovar-Herrera OE, Batista-García RA, Sánchez-Carbente MDR, et al. (2015). A novel expansin protein from the white-rot fungus Schizophyllum commune. PLoS One 10: e0122296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trail F, Gaffoor I, Vogel S. (2005). Ejection mechanics and trajectory of the ascospores of Gibberella zeae (anamorph Fusarium graminearum). Fungal Genetics and Biology 42: 528–533. [DOI] [PubMed] [Google Scholar]
- Trotman LC, Wang X, Alimonti a, et al. (2007). Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128: 141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsitsigiannis DI, Keller NP. (2007). Oxylipins as developmental and host–fungal communication signals. Trends in Microbiology 15: 109–118. [DOI] [PubMed] [Google Scholar]
- Tsukui T, Nagano N, Umemura M, et al. (2015). Ustiloxins, fungal cyclic peptides, are ribosomally synthesized in Ustilaginoidea virens. Bioinformatics 31: 981–985. [DOI] [PubMed] [Google Scholar]
- Tsunematsu Y, Takanishi J, Asai S, et al. (2019). Genomic mushroom hunting decrypts coprinoferrin, a siderophore secondary metabolite vital to fungal cell development. Organic Letters 21: 7582–7586. [DOI] [PubMed] [Google Scholar]
- Tullis P. (2021). How ecstasy and psilocybin are shaking up psychiatry. Nature 589: 506–510. [DOI] [PubMed] [Google Scholar]
- Ullrich R, Nüske J, Scheibner K, et al. (2004). Novel haloperoxidase from the agaric basidiomycete Agrocybe aegerita oxidizes aryl alcohols and aldehydes. Applied and Environmental Microbiology 70: 4575–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemura M. (2020). Peptides derived from Kex2-processed repeat proteins are widely distributed and highly diverse in the Fungi kingdom. Fungal Biology and Biotechnology 7: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno I, Ishikawa T. (1973). Purification and identification of the fruiting-inducing substances in Coprinus macrorhizus. Journal of Bacteriology 113: 1240–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaaje-Kolstad G, Westereng B, Horn SJ, et al. (2010). An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330: 219–222 [DOI] [PubMed] [Google Scholar]
- van den Brink J, de Vries RP. (2011). Fungal enzyme sets for plant polysaccharide degradation. Applied Microbiology and Biotechnology 91: 1477–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Peer AF, Wang F, Van Driel KG, et al. (2010). The septal pore cap is an organelle that functions in vegetative growth and mushroom formation of the wood-rot fungus Schizophyllum commune. Environmental Microbiology 12: 833–844. [DOI] [PubMed] [Google Scholar]
- van Roermund CWT, Waterham HR, Ijlst L, et al. (2003). Fatty acid metabolism in Saccharomyces cerevisiae. Cellular and Molecular Life Sciences CMLS 60: 1838–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Wymelenberg A, Gaskell J, Mozuch M, et al. (2009). Transcriptome and secretome analyses of Phanerochaete chrysosporium reveal complex patterns of gene expression. Applied and Environmental Microbiology 75: 4058–4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga T, Földi C, Bense V, et al. (2021). Developmental innovations promote species diversification in mushroom-forming fungi. BioRxiv. 10.1101/2021.03.10.434564 [DOI] [Google Scholar]
- Varga T, Krizsán K, Földi C, et al. (2019). Megaphylogeny resolves global patterns of mushroom evolution. Nature Ecology & Evolution 3: 668–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varrot A, Basheer SM, Imberty A. (2013). Fungal lectins: structure, function and potential applications. Current Opinion in Structural Biology 23: 678–685. [DOI] [PubMed] [Google Scholar]
- Veeneman BA, Shukla S, Dhanasekaran SM, et al. (2016). Two-pass alignment improves novel splice junction quantification. Bioinformatics 32: 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veneault-Fourrey C, Commun C, Kohler A, et al. (2014). Genomic and transcriptomic analysis of Laccaria bicolor CAZome reveals insights into polysaccharides remodelling during symbiosis establishment. Fungal Genetics and Biology 72: 168–181. [DOI] [PubMed] [Google Scholar]
- Venter P, Kock JLF, Kumar GS, et al. (1997). Production of 3R-hydroxy-polyenoic fatty acids by the yeast Dipodascopsis uninucleata. Lipids 32: 1277–1283. [DOI] [PubMed] [Google Scholar]
- Verdín J, Sánchez-León E, Rico-Ramírez AM, et al. (2019). Off the wall: the rhyme and reason of Neurospora crassa hyphal morphogenesis. The Cell Surface 5: 100020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viegas A, Herrero-Galá E, Oñaderra M, et al. (2009). Solution structure of hirsutellin A–new insights into the active site and interacting interfaces of ribotoxins. The FEBS Journal 276: 2381–2390. [DOI] [PubMed] [Google Scholar]
- Virágh M, Merényi Z, Csernetics Á, et al. (2022). Evolutionary Morphogenesis of Sexual Fruiting Bodies in Basidiomycota: Toward a New Evo-Devo Synthesis. Microbiology and Molecular Biology Reviews 86: e00019–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Zeska Kress MR, Harting R, Bayram Ö, et al. (2012). The COP9 signalosome counteracts the accumulation of cullin SCF ubiquitin E3 RING ligases during fungal development. Molecular Microbiology 83: 1162–1177. [DOI] [PubMed] [Google Scholar]
- Voigt O, Poggeler S. (2013). Autophagy genes Smatg8 and Smatg4 are required for fruiting-body development, vegetative growth and ascospore germination in the filamentous ascomycete Sordaria macrospora. Autophagy 9: 33–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonk PJ, Ohm RA. (2018). The role of homeodomain transcription factors in fungal development. Fungal Biology Reviews 32: 219–230. [Google Scholar]
- Vonk PJ, Ohm RA. (2021). H3K4me2 ChIP-Seq reveals the epigenetic landscape during mushroom formation and novel developmental regulators of Schizophyllum commune. Scientific Reports 11: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walser PJ, Kües U, Aebi M, et al. (2005). Ligand interactions of the Coprinopsis cinerea galectins. Fungal Genetics and Biology 42: 293–305. [DOI] [PubMed] [Google Scholar]
- Wälti MA, Villalba C, Buser RM, et al. (2006). Targeted gene silencing in the model mushroom Coprinopsis cinerea (Coprinus cinereus) by expression of homologous hairpin RNAs. Eukaryotic Cell 5: 732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wälti MA, Walser PJ, Thore S, et al. (2008). Structural basis for chitotetraose coordination by CGL3, a novel galectin-related protein from Coprinopsis cinerea. Journal of Molecular Biology 379: 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton J. (2018). Chemistry of the Amanita peptide toxins. In: The cyclic peptide toxins of Amanita and other poisonous mushrooms (pp. 19–57). Springer, Cham. [Google Scholar]
- Wang F, Song X, Dong X, et al. (2017). DASH-type cryptochromes regulate fruiting body development and secondary metabolism differently than CmWC-1 in the fungus Cordyceps militaris. Applied Microbiology and Biotechnology 101: 4645–4657. [DOI] [PubMed] [Google Scholar]
- Wang HX, Ng TB. (2006). Purification of a laccase from fruiting bodies of the mushroom Pleurotus eryngii. Applied Microbiology and Biotechnology 69: 521–525. [DOI] [PubMed] [Google Scholar]
- Wang L, Gao W, Wu X, et al. (2018). Genome-wide characterization and expression analyses of Pleurotus ostreatus MYB transcription factors during developmental stages and under heat stress based on de novo sequenced genome. International Journal of Molecular Sciences 19: 2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Tian X, Gyawali R, et al. (2013). Fungal adhesion protein guides community behaviors and autoinduction in a paracrine manner. Proceedings of the National Academy of Sciences 110: 11571–11576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhai B, Lin X. (2012). The link between morphotype transition and virulence in Cryptococcus neoformans. PLoS Pathogens 8: e1002765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Triguéros V, Paquereau L, et al. (2002). Proteins as active compounds involved in insecticidal activity of mushroom fruitbodies. Journal of Economic Entomology 95: 603–607. [DOI] [PubMed] [Google Scholar]
- Wang R, Ma P, Li C, et al. (2019). Combining transcriptomics and metabolomics to reveal the underlying molecular mechanism of ergosterol biosynthesis during the fruiting process of Flammulina velutipes. BMC Genomics 20: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Niu X, Guo X, et al. (2018). Heterologous expression, characterization and possible functions of the chitin deacetylases, Cda1 and Cda2, from mushroom Coprinopsis cinerea. Glycobiology 28: 318–332. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zeng X, Liu W. (2018). De novo transcriptomic analysis during Lentinula edodes fruiting body growth. Gene 641: 326–334. [DOI] [PubMed] [Google Scholar]
- Wang Z, Li N, Li J, et al. (2016). The fast-evolving phy-2 gene modulates sexual development in response to light in the model fungus Neurospora crassa. MBio 7: e02148–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wannet WJ, Aben EM, van der Drift C, et al. (1999). Trehalose phosphorylase activity and carbohydrate levels during axenic fruiting in three Agaricus bisporus strains. Current Microbiology 39: 205–210. [DOI] [PubMed] [Google Scholar]
- Watcharawipas A, Watanabe D, Takagi H. (2018). Sodium acetate responses in Saccharomyces cerevisiae and the ubiquitin ligase Rsp5. Frontiers in Microbiology 9: 2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawra S, Fesel P, Widmer H, et al. (2019). FGB1 and WSC3 are in planta-induced β-glucan-binding fungal lectins with different functions. New Phytologist 222: 1493–1506. [DOI] [PubMed] [Google Scholar]
- Wawrzyn GT, Quin MB, Choudhary S, et al. (2012). Draft genome of Omphalotus olearius provides a predictive framework for sesquiterpenoid natural product biosynthesis in Basidiomycota. Chemistry & Biology 19: 772–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiberg A, Wang M, Lin FM, et al. (2013). Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science 342: 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigand-Heller ABJ, Kris-Etherton PM, et al. (2012). The bioavailability of ergothioneine from mushrooms (Agaricus bisporus) and the acute effects on antioxidant capacity and biomarkers of inflammation. Preventive Medicine 54: S75–S78. [DOI] [PubMed] [Google Scholar]
- Wells TK, Hammond JB, Dickerson AG. (1987). Variations in activities of glycogen phosphorylase and trehalase during the periodic fruiting of the edible mushroom Agaricus bisporus (Lange) Imbach. New Phytologist 105: 273–280. [Google Scholar]
- Welzel K, Eisfeld K, Antelo L, et al. (2005). Characterization of the ferrichrome A biosynthetic gene cluster in the homobasidiomycete Omphalotus olearius. FEMS Microbiology Letters 249: 157–163. [DOI] [PubMed] [Google Scholar]
- Wen J, Zhang Z, Gong L, et al. (2019). Transcriptome changes during major developmental transitions accompanied with little alteration of DNA methylome in two Pleurotus species. Genes 10: 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L, Huang HM, Juang RH, et al. (2007). Biochemical characterization of 1-Cys peroxiredoxin from Antrodia camphorata. Applied Microbiology and Biotechnology 73: 1314–1322. [DOI] [PubMed] [Google Scholar]
- Whiteford JR, Thurston CF. (2000). The molecular genetics of cultivated mushrooms. Advances in Microbiological Physiology 42: 1–23. [DOI] [PubMed] [Google Scholar]
- Wilinski D, Buter N, Klocko AD, et al. (2017). Recurrent rewiring and emergence of RNA regulatory networks. Proceedings of the National Academy of Sciences 114: E2816–E2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth S, Freihorst D, Krause K, et al. (2021). What role might non-mating receptors play in Schizophyllum commune? Journal of Fungi 7: 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlschlager T, Butschi A, Grassi P, et al. (2014). Methylated glycans as conserved targets of animal and fungal innate defence. Proceedings of the National Academy of Sciences 111: E2787–E2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlschlager T, Butschi A, Zurfluh K, et al. (2011). Nematotoxicity of Marasmius oreades agglutinin (MOA) depends on glycolipid binding and cysteine protease activity. Journal of Biological Chemistry 286: 30337–30343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood WF, Clark TJ, Bradshaw DE, et al. (2004). Clitolactone: A Banana Slug Antifeedant from Clitocybe flaccida. Mycologia 96: 23–25. [PubMed] [Google Scholar]
- Wösten HA. (2001). Hydrophobins: multipurpose proteins. Annual Reviews in Microbiology, 55: 625–646. [DOI] [PubMed] [Google Scholar]
- Wösten HA, Scholtmeijer K. (2015). Applications of hydrophobins: current state and perspectives. Applied Microbiology and Biotechnology 99: 1587–1597. [DOI] [PubMed] [Google Scholar]
- Wu B, Xu Z, Knudson A, et al. (2018). Genomics and development of Lentinus tigrinus: a white-rot wood-decaying mushroom with dimorphic fruiting bodies. Genome Biology and Evolution 10: 3250–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu FL, Zhang G, Ren A, et al. (2016). The pH-responsive transcription factor PacC regulates mycelial growth, fruiting body development, and ganoderic acid biosynthesis in Ganoderma lucidum. Mycologia 108: 1104–1113. [DOI] [PubMed] [Google Scholar]
- Wu L, van Peer A, Song W, et al. (2013). Cloning of the Lentinula edodes B mating-type locus and identification of the genetic structure controlling B mating. Gene 531: 270–278. [DOI] [PubMed] [Google Scholar]
- Wu T, Hu C, Xie B, et al. (2020a). A putative transcription factor LFC1 negatively regulates development and yield of winter mushroom. Applied Microbiology and Biotechnology 104: 5827–5844. [DOI] [PubMed] [Google Scholar]
- Wu T, Zhang Z, Hu C, et al. (2020b). A WD40 protein encoding gene Fvcpc2 positively regulates mushroom development and yield in Flammulina velutipes. Frontiers in Microbiology 11: 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Gong W, Zhu Z, et al. (2018). Comparative transcriptomics of Pleurotus eryngii reveals blue-light regulation of carbohydrate-active enzymes (CAZymes) expression at primordium differentiated into fruiting body stage. Genomics 110: 201–209. [DOI] [PubMed] [Google Scholar]
- Xie Y, Chang J, Kwan HS. (2020). Carbon metabolism and transcriptome in developmental paths differentiation of a homokaryotic Coprinopsis cinerea strain. Fungal Genetics and Biology 143: 103432. [DOI] [PubMed] [Google Scholar]
- Xu G, Ma H, Nei M, Kong H. (2009). Evolution of F-box genes in plants: Different modes of sequence divergence and their relationships with functional diversification. Proceedings of the National Academy of Sciences 106: 835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Navarro-Ródenas A, Cooke JEK, et al. (2015). Transcript profiling of aquaporins during basidiocarp development in Laccaria bicolor ectomycorrhizal with Picea glauca. Mycorrhiza 26: 19–31. [DOI] [PubMed] [Google Scholar]
- Xue C, Hsueh YP, Heitman J. (2008). Magnificent seven: Roles of G protein-coupled receptors in extracellular sensing in fungi. FEMS Microbiology Reviews 32: 1010–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Kurano M, Inatomi S, et al. (2008). Isolation and characterization of a gene coding for chitin deacetylase specifically expressed during fruiting body development in the basidiomycete Flammulina velutipes and its expression in the yeast Pichia pastoris. FEMS Microbiology Letters 289: 130–137. [DOI] [PubMed] [Google Scholar]
- Yamada M, Sakuraba S, Shibata K, et al. (2006). Isolation and analysis of genes specifically expressed during fruiting body development in the basidiomycete Flammulina velutipes by fluorescence differential display. FEMS Microbiology Letters 254: 165–172. [DOI] [PubMed] [Google Scholar]
- Yamada-Okabe T, Sakamori Y, Mio T, et al. (2001). Identification and characterization of the genes for N-acetylglucosamine kinase and N-acetylglucosamine-phosphate deacetylase in the pathogenic fungus Candida albicans. European Journal of Biochemistry 268: 2498–2505. [DOI] [PubMed] [Google Scholar]
- Yamagishi K, Kimura T, Suzuki M, et al. (2004). Elevation of intracellular cAMP levels by dominant active heterotrimeric G protein alpha subunits ScGP-A and ScGP-C in homobasidiomycete, Schizophyllum commune. Bioscience, Biotechnology, and Biochemistry 68: 1017–1026 [DOI] [PubMed] [Google Scholar]
- Yamagishi K, Kimura T, Suzuki M, et al. (2002). Suppression of fruit-body formation by constitutively active G-protein α-subunits ScGP-A and ScGP-C in the homobasidiomycete Schizophyllum commune. Microbiology 148: 2797–2809. [DOI] [PubMed] [Google Scholar]
- Yan JJ, Tong ZJ, Liu YY, et al. (2019). Comparative Transcriptomics of Flammulina filiformis suggests a high CO2 concentration inhibits early pileus expansion by decreasing cell division control pathways. International Journal of Molecular Sciences 20: 5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Song S, Yan Q, et al. (2014). Biochemical characterization of the first fungal glycoside hydrolyase family 3 β-N-acetylglucosaminidase from Rhizomucor miehei. Journal of Agricultural and Food Chemistry 62: 5181–5190. [DOI] [PubMed] [Google Scholar]
- Ye SQ, Zou Y, Zheng QW, et al. (2021). TMT-MS/MS proteomic analysis of the carbohydrate-active enzymes in the fruiting body of Pleurotus tuoliensis during storage. Journal of the Science of Food and Agriculture 101: 1879–1891. [DOI] [PubMed] [Google Scholar]
- Yin G, Padhi S, Lee S, et al. (2015). Effects of Three Volatile Oxylipins on Colony Development in Two Species of Fungi and on Drosophila Larval Metamorphosis. Current Microbiology 71: 347–356. [DOI] [PubMed] [Google Scholar]
- Yoo S, Lee HY, Markkandan K, et al. (2019). Comparative transcriptome analysis identified candidate genes involved in mycelium browning in Lentinula edodes. BMC Genomics 20: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Armant O, Fischer R. (2016). Fungi use the SakA (HogA) pathway for phytochrome-dependent light signalling. Nature Microbiology 1: 1–7. [DOI] [PubMed] [Google Scholar]
- Yu Z, Fischer R. (2018). Light sensing and responses in fungi. Nature Reviews Microbiology 17: 25–36. [DOI] [PubMed] [Google Scholar]
- Yuen KY, Chan CM, Chan KM, et al. (2001). Characterization of AFMP1: a novel target for serodiagnosis of aspergillosis. Journal of Clinical Microbiology 39: 3830–3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun YH, Koo JS, Kim SH, et al. (2015). Cloning and expression analysis of phenylalanine ammonia-lyase gene in the mycelium and fruit body of the edible mushroom Flammulina velutipes. Mycobiology 43: 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawirska-Wojtasiak R. (2004). Optical purity of (R)-(-)-1-octen-3-ol in the aroma of various species of edible mushrooms. Food Chemistry 86: 113–118. [Google Scholar]
- Zeng X, Ling H, Yang J, et al. (2018). Proteome analysis provides insight into the regulation of bioactive metabolites in Hericium erinaceus. Gene 666: 108–115. [DOI] [PubMed] [Google Scholar]
- Zhang CH, Zou JP, Deng WQ, et al. (2018). Molecular cloning and the expression pattern of AePOPB involved in the α-amanitin biosynthesis in Amanita exitialis fruiting bodies. Gene 662: 123–130. [DOI] [PubMed] [Google Scholar]
- Zhang J, Chen H, Chen M, et al. (2015a). Cloning and functional analysis of a laccase gene during fruiting body formation in Hypsizygus marmoreus. Microbiological Research 179:54–63. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ren A, Chen H, et al. (2015b). Transcriptome Analysis and Its Application in Identifying Genes Associated with Fruiting Body Development in Basidiomycete Hypsizygus marmoreus. PLoS One 10: e0123025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Chen X, Orban A, et al. (2020). Agrocybe aegerita Serves As a Gateway for Identifying Sesquiterpene Biosynthetic Enzymes in Higher Fungi. ACS Chemical Biology 15: 1268–1277. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Xu L, Yuan S, et al. (2020). NGT1 Is Essential for N-Acetylglucosamine-Mediated Filamentous Growth Inhibition and HXK1 Functions as a Positive Regulator of Filamentous Growth in Candida tropicalis. International Journal of Molecular Sciences 21: 4036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Shen N, Li C, et al. (2021c). Population genomics provides insights into the genetic basis of adaptive evolution in the mushroom-forming fungus Lentinula edodes. Journal of Advanced Research 38: 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Gong W, Li C, et al. (2021d). RNA-Seq-based high-resolution linkage map reveals the genetic architecture of fruiting body development in shiitake mushroom, Lentinula edodes. Computational and Structural Biotechnology Journal 19: 1641–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Chen X, Lee RTC, et al. (2021). Bioinformatics–aided identification, characterization and applications of mushroom linalool synthases. Communications Biology 4: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Hao H, Liu H, et al. (2021). Genetic and functional analysis of the Zn(II)2Cys6 transcription factor HADA-1 in Hypsizygus marmoreus. Applied Microbiology and Biotechnology 105: 2815–2829. [DOI] [PubMed] [Google Scholar]
- Zhang S, Sakuradani E, Ito K, et al. (2007). Identification of a novel bifunctional delta12/delta15 fatty acid desaturase from a basidiomycete, Coprinus cinereus TD#822-2. FEBS Letters 581: 315–319. [DOI] [PubMed] [Google Scholar]
- Zhang W, Wu X, Zhou Y, et al. (2014). Characterization of stipe elongation of the mushroom Coprinopsis cinerea. Microbiology 160: 1893–1902. [DOI] [PubMed] [Google Scholar]
- Zhang X, Krause K-H, Xenarios I, et al. (2013). Evolution of the ferric reductase domain (FRD) superfamily: modularity, functional diversification, and signature motifs. PLoS One 8: e58126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZM, Wu WW, Li GK. (2008). A GC--MS study of the volatile organic composition of straw and oyster mushrooms during maturity and its relation to antioxidant activity. Journal of Chromatographic Science 46: 690–696. [DOI] [PubMed] [Google Scholar]
- Zhao C, Waalwijk C, de Wit PJGM, et al. (2013). RNA-Seq analysis reveals new gene models and alternative splicing in the fungal pathogen Fusarium graminearum. BMC Genomics 14: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Kwan HS. (1999). Characterization, molecular cloning, and differential expression analysis of laccase genes from the edible mushroom Lentinula edodes. Applied and Environmental Microbiology 65: 4908–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Liu H, Luo Y, et al. (2014). Molecular evolution and functional divergence of tubulin superfamily in the fungal tree of life. Scientific Reports 4: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, Shabek N. (2017). Ubiquitin ligases: structure, function, and regulation. Annual Review of Biochemistry 86: 129–157. [DOI] [PubMed] [Google Scholar]
- Zhou J, Kang L, Liu C, et al. (2019). Chitinases play a key role in stipe cell wall extension in the mushroom Coprinopsis cinerea. Applied and Environmental Microbiology 85: e00532–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Li X, Lüli Y, et al. (2021). Novel cyclic peptides from lethal Amanita mushrooms through a genome-guided approach. Journal of Fungi 7: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Ma F, Zhang X, et al. (2016a). Carbohydrate changes during growth and fruiting in Pleurotus ostreatus. Fungal Biology 120: 852–861 [DOI] [PubMed] [Google Scholar]
- Zhou S, Zhang J, Ma F, et al. (2018). Investigation of lignocellulolytic enzymes during different growth phases of Ganoderma lucidum strain G0119 using genomic, transcriptomic and secretomic analyses. PLoS One 13: e0198404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Chen L, Fan X, et al. (2014). De novo assembly of Auricularia polytricha transcriptome using Illumina sequencing for gene discovery and SSR marker identification. PLoS One 9: e91740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Kang L, Niu X, et al. (2016b). Purification, characterization and physiological significance of a chitinase from the pilei of Coprinopsis cinerea fruiting bodies. FEMS Microbiology Letters 363: fnv120. [DOI] [PubMed] [Google Scholar]
- Zhu W, Hu J, Li Y, et al. (2019). Comparative proteomic analysis of Pleurotus ostreatus reveals great metabolic differences in the cap and stipe development and the potential Role of Ca2+ in the primordium differentiation. International Journal of Molecular Sciences 20: 6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Luo H, Zhang X, et al. (2014). Abundant and selective RNA-editing events in the medicinal mushroom Ganoderma lucidum. Genetics 196: 1047–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Zhuang L, Goodell B, et al. (2016). Iron sequestration in brown-rot fungi by oxalate and the production of reactive oxygen species (ROS). International Biodeterioration & Biodegradation 109: 185–190. [Google Scholar]
- Zollman S, Godt D, Prive GG, et al. (1994). The BTB domain, found primarily in zinc finger proteins, defines an evolutionarily conserved family that includes several developmentally regulated genes in Drosophila. Proceedings of the National Academy of Sciences 91: 10717–10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Žurga S, Pohleven J, Kos J, et al. (2015). β-Trefoil structure enables interactions between lectins and protease inhibitors that regulate their biological functions. The Journal of Biochemistry 158: 83–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expression heatmap of DNA replication, repair, mitosis and meiosis related genes in A. ostoyae, M. kentingensis, P. ostreatus and the simple S. commune. Well-delimited complexes mentioned in the paper are shown separately. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively.
Expression heatmap of ribosomal protein encoding genes in A. bisporus, A. ostoyae and P. ostreatus. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively.
Annotated glycolysis pathway of C. cinerea. Genes in the pathway are denoted by the S. cerevisiae gene name, followed by the protein ID of the C. cinerea ortholog and, in parentheses, the number of species in which the gene was developmentally regulated at fold change >2/>4.
Expression heatmap of ergosterol and sphingolipid biosynthesis related genes in A. ostoyae and P. ostreatus. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively.
Expression heatmap of membrane phospholipid and fatty acid biosynthetic genes in A. bisporus, A. ostoyae, M. kentingensis and P. ostreatus. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively.
Expression heatmap of storage carbohydrate metabolism genes in A. ostoyae, L. bicolor and P. ostreatus. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively.
Expression heatmap of chromatin remodeling related genes (histones, histone chaperones, histone deacethylases) in A. ampla, A. ostoyae, L. tigrinus, M. kentingensis and P. ostreatus. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively.
Expression heatmap of Con6 family cell surface protein-encoding genes in A. ostoyae, A. ampla, L. tigrinus, M. kentingensis and P. ostreatus. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively.
Expression heatmap of putative wax synthase encoding genes in A. ostoyae and S. commune. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively.
Expression heatmap of kinesin genes and members of the Dynactin complex in A. ostoyae and P. ostreatus. Genes are denoted by Protein IDs. Blue and red colors represent low and high expression, respectively.
Table summarizing ortholog protein ID for all discussed orthogroups in twelve Agaricomycetes.
Summary of read numbers and alignments in the analysed fruiting body transcriptomes.
Summary of developmental expression dynamics of CDE orthogroups of cell division-related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of ribosomal genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of genes related to Acetyl-CoA production and basic metabolism across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of fatty acid beta-oxidation related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of ergosterol and sphingolipid biosynthesis related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of sterol transport related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of membrane phoshpolipid and fatty acid biosynthesis genes genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of linoleic acid biosynthesis related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of other lipid metabolism related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of storage carbohydrate metabolism genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of transcriptional regulator genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of RNA-binding protein encoding genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of RNA interference related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of chromatin-related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of genes related to alternative splicing across 12 species. Protein ID of a representative protein is given follows by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4.
Summary of developmental expression dynamics of CDE orthogroups of signal transduction related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of pheromone precursor genes across 12 species. Protein ID (by default for <I>C.</I> cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of conserved signal transduction pathway genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of G-protein coupled receptor genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of velvet protein encoding genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of STRIPAK complex protein genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of light sensing related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of chitin biosynthesis genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of glucan biosynthesis related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of chitin remodeling genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of glucan remodeling related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of miscellaneous cell wall remodeling genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of multicopper oxidase genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of expansin genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of miscellaneous cell wall related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of hydrophobin genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of genes belonging to conserved lectin orthogroups across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of cell surface protein encoding genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of defense-related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of terpene synthase genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of cytoskeleton-related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of Cop9 signalosome genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Copy number distributions of genes related to protein ubiquitination and the proteasome in twelve examined Agaricomycetes.
Summary of developmental expression dynamics of CDE orthogroups of protein ubiquitination and the proteasome related genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Genes related to protein ubiquitination and the proteasome in C. cinerea.
Summary of developmental expression dynamics of CDE orthogroups of transporter encoding genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of stress response genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.
Summary of developmental expression dynamics of CDE orthogroups of unannotated genes across 12 species. Protein ID of a representative protein is given follows by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4 - developmentally regulated at fold change >4.
Summary of developmental expression dynamics of CDE orthogroups of functionally poorly known genes across 12 species. Protein ID of a representative protein is given follows by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4.
Summary of developmental expression dynamics of CDE orthogroups of ferric reductase genes across 12 species. Protein ID (by default for C. cinerea, unless abbreviation indicates otherwise) of a representative protein is given followed by the number of species in which the orthogroup is developmentally regulated at fold change 2 and 4 (FC>2 and FC>4, respectively). Putative function and ortholog in S. cerevisiae (if any) are also given. Abbreviations: 0-gene absent, 1-gene present but not developmentally regulated, 2 - developmentally regulated at fold change >2, 4- developmentally regulated at fold change >4. Species names are abbreviated as: Cc – C. cinerea, Aa – A. ampla, Ab – A. bisporus, Ao – A. ostoyae, Ca – C. aegerita, Lb – L. bicolor, Le – L. edodes, Lt – L. tigrinus, Mk – M. kentingensis, Pc – Ph. chrysosporium, Po – P. ostreatus, Sc – S. commune.