

Abstract

We have previously identified 5-chloro-2-methyl-2-(3-(4-(pyridin-2-yl)piperazin-1-yl)propyl)-2,3-dihydro-1H-inden-1-one (SYA0340) as a dual 5-HT1A and 5-HT7 receptor ligand, and we posited such ligands might find utility in the treatment of various CNS related illnesses including cognitive and anxiolytic impairments. However, SYA0340 has a chiral center and its enantiomers may confound the readouts for their functional characteristics. Thus, in this study, we resynthesized SYA0340, separated the enantiomers, identified the absolute configurations, and evaluated their binding affinities and functional characteristics at both the 5-HT1A and 5-HT7A receptors. The results of this study show that the (+)-SYA0340-P1 [specific rotation [α] = +18.4 (deg.mL)/(g.dm)] has a binding affinity constant, Ki = 1.73 ± 0.55 nM at 5-HT1AR and Ki = 2.20 ± 0.33 nM at 5-HT7AR and (−)-SYA0340-P2 [specific rotation [α] = −18.2 (deg.mL)/(g.dm)] has Ki = 1.06 ± 0.32 nM (5-HT1AR) and 4.7 ± 1.1 nM (5-HT7AR). Using X-ray crystallographic techniques, the absolute configuration of the P2 isomer was identified as the S-enantiomer and, therefore, the P1 isomer as the R-enantiomer. Functionally, both SYA0340-P1 (EC50 = 1.12 ± 0.41 nM; Emax = 94.6 ± 3.1%) and SYA0340-P2 (EC50 = 2.21 ± 0.59 nM; Emax = 96.8 ± 5.1%) display similar agonist properties at the 5-HT1AR while both enantiomers display antagonist properties at the 5-HT7AR with P1 (IC50 = 32.1 ± 9.2 nM) displaying over 8 times greater potency as P2 (IC50 = 277 ± 46 nM). Thus, based on the functional evaluation results, SYA0340-P1 is considered as the eutomer of the pair of enantiomers of SYA0340. It is expected that these enantiomers will serve as new pharmacological probes for the 5-HT1A and 5-HT7A receptors.
1. Introduction
The G-protein coupled receptor (GPCR), serotonin 5-HT1A receptor (5-HT1AR), exerts its effects through Gi/o proteins to inhibit adenylyl cyclase, and other second messenger cascades such as MAPK pathways and NMDA receptor channels.1,2 These receptors exist as presynaptic autoreceptors on serotonergic cell bodies in the raphe nuclei and as postsynaptic heteroreceptors in the limbic system, including the hippocampus, septum, amygdala and entorhinal cortex, the hypothalamus, cortex, and dorsal horn.1−3 When activated, the 5-HT1A autoreceptors inhibit the firing of 5-HT neurons and may induce anxiolytic activity in rodent behavioral tests,4,5 whereas antidepressant-like responses are seen upon activation of postsynaptic 5-HT1A heteroreceptors.4−6 Additionally, 5-HT1AR knockout mice display increased anxiety and depressive behaviors and have been used as animal models for these disorders, thus revealing the important roles 5-HT1AR plays in CNS disorders.7 In fact, antidepressants such as vilazodone that inhibit 5-HT reuptake and partially activate 5-HT1ARs are more efficacious, fast acting, and tolerable in part because of their partial agonist actions at postsynaptic 5-HT1A receptors.8
The serotonin 5-HT7 receptor (5-HT7R) is another closely related 5-HTR that activates adenylate cyclase and mediates key physiological functions including sleep, mood, learning memory, and cognition.1,9 The receptor was cloned in multiple laboratories in 199310 with the human 5-HT7 receptor being cloned by Bard et al.11 The human 5-HT7R has three splice variants (A, B, and D) with the 5-HT7AR as the most prevalent variant expressed in the CNS.12
A key interest in targeting the 5-HT7AR is its association with depression. Indeed, it has been suggested that the antidepressant effects of amisulpride may be attributed to its antagonism at the 5-HT7 receptor.13 Current knowledge regarding the 5-HT1AR and 5-HT7AR suggests that 5-HT7AR is co-localized with 5-HT1AR and cross talks resulting from heterodimers of these receptors have been implicated in CNS diseases such as depression.14−16 Not only do these receptors share over 40% sequence homology, ligands that activate these receptors tend to exhibit cross activity.15,16 It is our view that dual ligands for 5-HT1A and 5-HT7 receptors may serve as potential treatment options for depression, anxiety, and other diseases characterized by cognitive deficits.
In our previous publications,17−19 we reported studies involving N-heteroaryl piperazines such as SYA16263 (1) (Figure 1), and its modification to obtain 5-chloro-2-methyl-2-(3-(4-(pyridin-2-yl)piperazin-1-yl)propyl)-2,3-dihydro-1H-inden-1-one (SYA0340) (Figure 2) as a high affinity dual 5-HT1A and 5-HT7A receptor ligand which acts as an agonist and an antagonist at the 5-HT1AR and 5-HT7AR, respectively. We then hypothesized that such ligands might find utility in the treatment of various CNS related illnesses including cognitive and anxiolytic impairments. However, SYA0340 has a chiral center and a racemic mixture limited our ability to fully evaluate its potential as a probe agent for evaluating the activities of 5-HT1A and 5-HT7A receptors. To that end, we have now resynthesized SYA0340, separated its enantiomers, identified the absolute configuration of the eutomer through X-ray crystallographic studies, and evaluated each enantiomer for binding and functional activities at 5-HT1AR and 5-HT7AR.
Figure 1.

Structures of some FDA-approved CNS drugs containing N-aryl/heteroaryl piperazine moieties along with SYA16263 (1).
Figure 2.

Structures of SYA0340 and its enantiomers.
2. Results and Discussion
2.1. Synthesis of SYA0340
2.1.1. Synthesis
The synthesis of SYA0340 was carried out using our previously reported method with minor modifications.17 First, the α-methylated 5-chloroindanone 3c was synthesized from β-keto ester, 3b, which was obtained from commercially available 5-chloroindanone, 3a. Deprotonated 5-chloroindanone was subjected to condensation with dimethyl carbonate (DMC) to yield an intermediate which underwent the second deprotonation and then methylation under basic conditions to produce intermediate 3b. Decarboxylation of 3b was accomplished by heating in HCl and AcOH under microwave-assisted condition to produce 3c (Scheme 1). An alkylation step using 1-bromo-3-chloropropane to produce 3d then allowed a final N-alkylation reaction with 1-(pyridine-2-yl)piperazine to afford SYA0340 as a racemic mixture whose NMR spectra matched that which was previously reported.17 For the current manuscript, a microwave-assisted synthesis (MWAS) was carried out by refluxing 1-(pyridine-2-yl)piperazine, with the alkylating agent 3d in toluene, in the presence of K2CO3 as a base and a catalytic amount of KI to obtain racemic SYA0340. The resulting product was purified by flash chromatography, and the pure racemic SYA0340 was subjected to enantiomeric separation to yield SYA0340-P1 and SYA0340-P2 which were subsequently converted to the oxalate salts. The synthetic steps are shown in Scheme 1.
Scheme 1. Synthesis of SYA0340.

Reagents and conditions: (a) NaH, DMC, 0 °C-rt, then 60 °C, 12 h; (b) (i) NaH, DMF, MeI, rt, 24 h; (ii) conc. HCl in AcOH (glacial), MWAS; (c) NaH, DMF, 1-bromo-3-chloropropane, rt, 24 h; (d) 1-(pyridin-2-yl)piperazine, K2CO3/KI, toluene, MWAS. (e) Enantiomeric separation by HPLC using a Chiral-Pak AD-H semi-prep column.
2.2. Enantiomeric Separation
Enantiomeric separation was carried out using a Gilson HPLC system with a 306 pump, 119 UV/VIS detector, and manual injection and collection of fractions. A ChiralPak AD-H semi-prep HPLC column (1 × 25 cm) with particle size 5 μ was used with mobile phase 85:15:0.1 v/v/v Hex:EtOH:Et2NH at a flowrate of 4.7 mL/min and a UV detection wavelength of 254 nm. Injection size was 1.00 mL. Racemate SYA0340 was dissolved in basic ethanol (pH 8–10) and centrifuged for 10 min at 3000 rpm, and the supernatant was used for the chiral separations.
The enantiomeric purity of the separated enantiomers was determined using a Waters 2695 analytical HPLC system with a 2996 PDA detector and autosampler. A ChiralPak AD-H analytical HPLC column (4.6 mm × 25 cm) with particle size 5 μ was used with mobile phase 85:15:0.1 v/v/v Hex:EtOH:Et2NH at a flowrate 1.00 mL/min and a UV detection wavelength of 254 nm. Injection size was 60 μL.
The identity of the collected enantiomers was verified by comparison of retention times and UV spectra for the racemate and the individual collected enantiomers. Retention times of enantiomers matched those in the racemate within experimental error (Table 1 and Figure 3A–C). The enantiomeric purity was 100% for each enantiomer as determined by HPLC peak areas.
Table 1. Retention Time of SYA0340 and the Separated Enantiomers on the Analytical HPLC Column.
| sample | peak 1 retention time, min | peak 2 retention time, min |
|---|---|---|
| SYA0340 (racemate) | 12 | 21 |
| SYA0340-P1 (+)-enantiomer | 14.5 | NDa |
| SYA0340-P2 (−)-enantiomer | NDa | 21.5 |
ND =not detected.
Figure 3.

HPLC chromatograms of SYA0340 (A), SYA0340-P1 (B), and SYA0340-P2 (C).
Ultraviolet spectra were recorded and were identical within experimental error for both enantiomers as expected, and they matched those in the racemate within experimental error with local wavelength maximums of 250.9 and 294.6 nm (Figure 4A,B).
Figure 4.

UV spectra of SYA0340-P1 (A) and SYA0340-P2 (B).
2.3. Polarimetric Determination of Specific Rotations
Specific rotations [α], for the free base of SYA0340-P1 and SYA0340-P2 in EtOH, at an average cell temperature of 23.2 °C, were determined on a JASCO 1020 polarimeter. The mean specific rotations ± SD, n = 3, are SYA0340-P1 [α] = +18.4 ± 0.5 (deg.mL)/(g.dm); and SYA0340-P2 [α] = −18.2 ± 0.5 (deg.mL)/(g.dm).
2.4. X-ray Crystallographic Analyses and Determination of Absolute Configuration of Enantiomers
Results from the X-ray crystallographic study of the oxalate salt of SYA0340-P2 produced the structure shown in Figure 5. The crystal structure data for C26H30ClN3O9 (M = 563.98 g/mol) are monoclinic, space group P21 (no. 4), a = 15.7212(3) Å, b = 5.65480(10) Å, c = 31.4047(5) Å, β = 93.1580(10)°, V = 2787.65(9) Å3, Z = 4, T = 150.00(10) K, μ(Cu Kα) = 1.701 mm–1, Dcalc = 1.344 g/cm3, 50,390 reflections measured (11.274° ≤ 2Θ ≤ 156.956°), and 11,345 unique (Rint = 0.0730, Rsigma = 0.0495). The Cahn Ingold Prelog rule applied to the structure produced an absolute configuration of “S.” This was confirmed and verified by importing the coordinates of the single crystal X-ray diffraction (SCXRD) structure into Chem3D in ChemDraw 19.1. Therefore, the other enantiomer, SYA0340-P1, is assigned as the R-enantiomer.
Figure 5.

X-ray crystallographic structure of the oxalate salt of SYA0340-P2.
2.5. Binding and Functional Evaluation of Enantiomers at 5-HT1A and 5-HT7A Receptors
2.5.1. Evaluation of Binding Affinities
Racemic SYA0340 and enantiomers P1 and P2 were screened for their binding affinities at serotonin 5-HT1AR and 5-HT7AR, and the results are reported in Table 2. SYA0340 shows comparable binding affinity (Ki = 1.76 nM) at 5-HT1AR to 5-HT (Ki = 2.24 nM) which served as the positive standard. The enantiomers, SYA0340-P1 and SYA0340-P2, exhibit similar binding at 5-HT1AR with Ki = 1.73 nM for P1 and Ki = 1.06 nM for P2, respectively (Table 2). Compared to lurasidone, SYA0340 displayed similar binding affinity (Ki = 3.89 nM) versus (Ki = 4.3 nM) at 5-HT7AR. It is also noted that SYA0340-P1 exhibits a two-fold higher binding affinity (Ki = 2.20 nM) than lurasidone at the 5-HT7AR while the other enantiomer SYA0340-P2 binds with a similar Ki of 4.7 ± 1.1 nM (Table 2 and Figure 6). Overall, SYA0340-P1, the dextrorotatory enantiomer, appears to be the eutomer of the two enantiomers.
Table 2. Radioligand Binding Affinities for SYA0340-P1 and SYA0340-P2 at 5-HT1A and 5-HT7A Receptors.
| compound | 5-HT1AR [3H]8-OH-DPAT binding |
5-HT7AR [3H]5-HT binding |
||||
|---|---|---|---|---|---|---|
| Ki (nM) ± SEM | n | hill slope ± SEMb | Ki (nM) ± SEM | n | hill slope ± SEMb | |
| 5-HT | 2.24 ± 0.24 | 4 | –0.96 ± 0.06 | 0.46 ± 0.11 | 6 | –1.07 ± 0.09 |
| SYA0340 | 1.76 ± 0.51 | 10 | –0.66 ± 0.09 | 3.89 ± 0.79 | 6 | –0.59 ± 0.03 |
| SYA0340-P1 | 1.73 ± 0.55 | 4 | –0.58 ± 0.06 | 2.20 ± 0.33 | 6 | –0.59 ± 0.02 |
| SYA0340-P2 | 1.06 ± 0.32 | 3 | –0.53 ± 0.03 | 4.70 ± 1.10 | 6 | –0.59 ± 0.05 |
| lurasidone | NDa | NDa | NDa | 4.3 ± 1.3 | 7 | –0.76 ± 0.11 |
ND = not determined.
A Hill slope of less than −1.0 may suggest that the ligands ([3H]5-HT and (±)-, R- and S-enantiomers) interact with more than one (radioligand) binding site or possibly are involved in negative cooperative binding.
Figure 6.

In vitro 5-HT1AR (left) and 5-HT7AR (right) radioligand binding graphs for SYA0340-P1 and SYA0340-P2. The graph to the left shows the binding affinities at 5-HT1AR, and the graph on the right shows affinities at 5-HT7AR. Serotonin and lurasidone were used as positive controls at 5-HT1AR and 5-HT7AR, respectively.
2.5.2. Functional Assays for SYA0340 and Enantiomers, SYA0340-P1 and SYA0340-P2
Functional assays were also conducted for functional activities at the 5-HT1A and 5-HT7A receptors. SYA0340 and the enantiomers were tested using GTPγS binding and Gs-mediated cAMP production for their agonist and antagonist functional activities toward 5-HT1A and 5-HT7A receptors, respectively. Serotonin and lurasidone were used as positive controls for the evaluation of agonist (at 5-HT1A and 5-HT7A) and antagonist properties (at 5-HT7A) of the compounds, respectively. The results are reported in Tables 3 and 4 and Figures 7, 8 and 9.
Table 3. 5-HT1AR GTPγS and 5-HT7AR Gs cAMP Functional Assay for Agonist Effects.
| compound | 5-HT1AR [35S]GTPγS
assay |
5-HT7AR adenylate
cyclase |
||||
|---|---|---|---|---|---|---|
| EC50 (nM) ± SEM | n | % max ± SEM | EC50 (nM) ± SEM | n | % max ± SEM | |
| 5-HT | 2.60 ± 0.56 | 8 | 101.2 ± 1.1 | 5.3 ± 1.6 | 3 | 95.8 ± 1.6 |
| SYA0340 | 2.66 ± 0.57 | 7 | 101.5 ± 2.8 | >10 μM | N/Aa | N/Aa |
| SYA0340-P1 | 1.12 ± 0.41 | 3 | 94.6 ± 3.1 | >10 μM | N/Aa | N/Aa |
| SYA0340-P2 | 2.21 ± 0.59 | 3 | 96.8 ± 5.1 | >10 μM | N/Aa | N/Aa |
N/A = not applicable.
Table 4. 5-HT7AR Gs cAMP Functional Assay Antagonist Effects of SYA0340, SYA0340-P1, and SYA0340-P2.
| compound | 5-HT1AR [35S]GTPγS
assay |
5-HT7AR adenylate
cyclase |
||||
|---|---|---|---|---|---|---|
| EC50 (nM) ± SEM | n | % max ± SEM | IC50 (nM) ± SEM | n | % max inhib. ± SEM | |
| SYA0340 | agonist | N/Aa | 89 ± 14 | 4 | 94.7 ± 4.1 | |
| SYA0340-P1 | agonist | N/Aa | 32.1 ± 9.2 | 3 | 93.5 ± 4.3 | |
| SYA0340-P2 | agonist | N/Aa | 277 ± 46 | 4 | 94.3 ± 3.7 | |
| lurasidone | ND | NDa | 22.5 ± 2.2 | 5 | 98.0 ± 2.0 | |
ND = not determined; N/A = not applicable.
Figure 7.

Functional assay graph of in vitro 5-HT1AR GTPγS binding showing the [35S]GTPγS agonist activity of SYA0340, SYA0340-P1, and SYA0340-P2 to 5-HT1AR. Serotonin served as the positive control.
Figure 8.

In vitro serotonin receptor 5-HT7AR Gs agonist functional assay graph. Graph showing the Gs-mediated specific cAMP formation for agonist activity of SYA0340, SYA0340-P1, and SYA0340-P2 toward 5-HT7AR. Serotonin served as the positive control.
Figure 9.
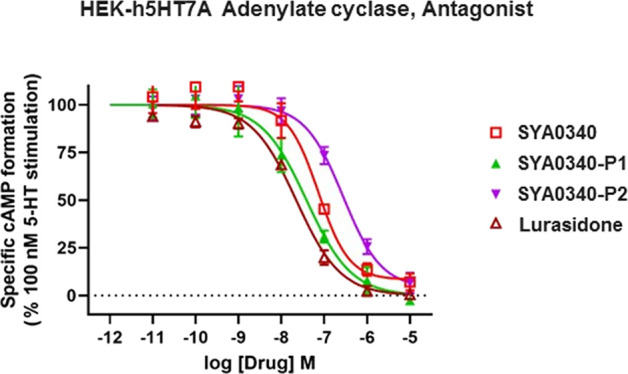
In vitro 5-HT7A receptor adenylate cyclase antagonist functional assay results for compounds SYA0340, SYA0340-P1, and SYA0340-P2 and the graph showing the antagonist activities of the compounds at 5-HT7AR. Lurasidone served as the positive control in this assay.
The results of the functional activities indicated that serotonin (5-HT) has an EC50 of 2.60 nM with Emax of 101.2% showing its full agonist property at 5-HT1AR (Table 3 and Figure 7). Compound SYA0340 exhibits an agonist profile at 5-HT1AR with an EC50 of 2.66 nM and Emax of 101.5% which is similar to 5-HT. Enantiomer SYA0340-P1 has more than two-fold potency (EC50 = 1.12, % max 94.6) at 5-HT1AR as compared to 5-HT (EC50 = 2.60 nM). The other enantiomer, SYA0340-P2, demonstrates agonist activity (EC50 = 2.21 nM, % max 96.8) which is similar to the racemic compound SYA0340 and 5-HT (Table 3 and Figure 7). Thus, SYA0340, SYA0340-P1, and SYA0340-P2 show full agonist characteristics toward 5-HT1AR. SYA0340-P1 has more than two-fold higher potency as compared to 5-HT and SYA0340. Upon evaluating the agonist effects of these compounds at 5-HT7AR, 5-HT alone displays an agonist effect toward 5HT7AR with an EC50 of 5.3 nM and % max stimulation of 95.8%, while SYA0340 and both enantiomers do not show any agonist activity at 5-HT7AR (Table 3 and Figure 8).
Compounds SYA0340, SYA0340-P1, and SYA0340-P2 were evaluated for their antagonism at the 5-HT7A receptor along with the standard drug lurasidone (Table 4 and Figure 9). SYA0340 blocked 94.7% of adenylyl cyclase activity with an IC50 = 89 nM. Likewise, SYA0340-P1 blocked adenylyl cyclase activity (93.5% max inhibition) with an IC50 of 32.1 nM, which is comparable to the standard drug lurasidone (IC50 = 22.5 nM, 98.0% max inhibition), and over 8 times the potency of P2 as an antagonist at 5-HT7AR (IC50 = 277 nM, 94.3% max inhibition) (Table 4 and Figure 9). Overall, SYA0340-P1 and lurasidone exhibit similar adenylyl cyclase inhibition with SYA0340-P1 being a full antagonist at 5-HT7AR and having 8 times the potency of P2.
3. Conclusions
In summary, the synthesis and separation of racemic SYA0340 to obtain enantiomers SYA0340-P1 and SYA0340-P2 and their study show P1 as dextrorotatory with a specific rotation, [α] = +18.4 ± 0.5 (deg.mL)/(g.dm); and SYA0340-P2 as levorotary with a specific rotation, [α] = −18.2 ± 0.5 (deg.mL)/(g.dm). An X-ray crystallographic study indicates SYA0340-P2 has an absolute configuration of S, and thus SYA0340-P1 has the R-configuration. The R-enantiomer binds with a high affinity at both 5-HT1A and 5-HT7A receptors and is a full agonist at the 5-HT1AR and an antagonist at the 5-HT7AR. Indeed, the R-enantiomer is over 8 times as potent as the S-enantiomer as an antagonist at the 5-HT7AR. These functional characteristics warrant an evaluation of the eutomer, SYA0340-P1, which is the R enantiomer, in an in vivo model of depression, and may find application in the development of antidepressant drugs and cognitive impairment treatment.
4. Materials and Methods
4.1. Chemistry
Gallenkamp (U.K.) apparatus was used to determine melting points and is uncorrected. All NMR spectra were obtained either on a Varian 300 MHz Mercury Spectrometer or 600 MHz Bruker Avance Spectrometer, and the free induction decay (FID) data were processed using Mestrelab’s Mnova NMR software (version 8.1) to obtain the reported NMR data. Elemental analyses were carried out by Atlantic Microlab, Inc., Norcross, GA, and are within 0.4% of theory unless otherwise noted. Flash chromatography was performed using a Biotage Isolera flash chromatographic system with Davisil grade 634 silica gel. Starting materials were obtained from Sigma-Aldrich and were used without further purification. All microwave-assisted syntheses (MWASs) were carried out using a Biotage Initiator. 1H and 13C NMR data were obtained for all the intermediates and were consistent with our previously reported NMR spectral data.171H and 13C NMR data were obtained for SYA0340 free base as reported here.
4.2. Synthesis of SYA0340
4.2.1. Ethyl 5-chloro-2-methyl-1-oxo-2,3-dihydro-1H-indene-2-carboxylate (3b)
Synthesis of ethyl 5-chloro-2-methyl-1-oxo-2,3-dihydro-1H-indene-2-carboxylate 3b was achieved in two steps using previously reported methods with minor modification.17 Briefly, NaH (88.2 mmol, 60% in mineral oil) and DMC (126 mmol) in dry THF (50 mL) was stirred at 0 °C. To this stirred suspension of NaH and DMC, 5-chloro indanone 3a (42 mmol) in dry THF (50 mL) was added dropwise. When evolution of gas has ceased, the mixture was stirred at 60 °C for 12 h. At completion, the reaction mixture was diluted with CH2Cl2 and treated with aqueous acetic acid solution at 0 °C with stirring. The aqueous phase was extracted with CH2Cl2, and the combined organic extracts were dried (Na2SO4) and concentrated under reduced pressure to yield the crude 5-substituted β-keto ester as a brown thick oil. Using flash chromatography, the crude extract was purified using a gradient elution (up to 10% EtOAc in hexanes) to afford ethyl 5-chloro-1-oxo-2,3-dihydro-1H-indene-2-carboxylate (not shown in the scheme) as needle-like white crystals in 86% yield.
Next, to the stirred suspension of NaH (44.4 mmol, 60% in mineral oil) in dry DMF was added slowly a solution of ethyl 5-chloro-1-oxo-2,3-dihydro-1H-indene-2-carboxylate (22.2 mmol) in dry DMF under N2, and the mixture was stirred for 2 h at room temperature. Methyl iodide (44.4 mmol) was added dropwise to the stirred mixture and stirring continued for 24 h. The reaction was quenched with slow addition of water (50 mL) and extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were washed with brine and dried over Na2SO4, and the solvent was evaporated under reduced pressure to obtain the crude product. The crude compound was purified by flash chromatography using a gradient of up to 10% EtOAc in hexanes to obtain the intermediate compound, ethyl 5-chloro-2-methyl-1-oxo-2,3-dihydro-1H-indene-2-carboxylate 3b. Yield: 60%.
4.2.2. 5-Chloro-2-methyl-2,3-dihydro-1H-inden-1-one (3c)
Ethyl 5-chloro-2-methyl-1-oxo-2,3-dihydro-1H-indene-2-carboxylate, 3b (33.5 mmol), was hydrolyzed and decarboxylated using a microwave-assisted reaction. Intermediate 3b was dissolved in acetic acid (10 mL) and concentrated HCl (4 mL) and transferred into a 20 mL-microwave vial with a stirrer, and the reaction vial was tightly sealed. The reaction mixture was subjected for microwave heating at 80 °C for 1 h, and then the reaction was allowed to cool to room temperature. After completion of the reaction, reaction mixture was neutralized with slow addition of saturated NaHCO3 solution and extracted with CH2Cl2. The combined organic layers were washed with brine and dried over Na2SO4. The solvent was removed under reduced pressure to yield the crude compound. The crude was then purified by flash chromatography using a gradient of 5% EtOAc in hexanes to afford 5-chloro-2-methyl-2,3-dihydro-1H-inden-1-one 3c in 82% yield.
4.2.3. 5-Chloro-2-(3-chloropropyl)-2-methyl-2,3-dihydro-1H-inden-1-one (3d)
To the stirred suspension of NaH (22 mmol, 60% in mineral oil) in dry DMF was added dropwise a solution of the 5-chloro-2-methyl-2,3-dihydro-1H-inden-1-one 3c (11 mmol) in dry DMF. After the reaction mixture was stirred for an hour, 1-bromo-3-chloropropane (22 mmol) was added dropwise and stirred for 24 h at room temperature. The reaction was quenched at completion with water and extracted with CH2Cl2. The combined organic layers were washed with brine, dried over Na2SO4, and evaporated to dryness under reduced pressure to obtain the crude product which was used for N-alkylation reaction in the next step without further purification.
4.2.4. 5-Chloro-2-methyl-2-(3-(4-(pyridin-2-yl)piperazin-1-yl)propyl)-2,3-dihydro-1H-inden-1-one (SYA0340)
A mixture of alkylating agent, 5-chloro-2-(3-chloropropyl)-2-methyl-2,3-dihydro-1H-inden-1-one 3d (11.7 mmol), 1-(pyridin-2-yl)piperazine (14.04 mmol), K2CO3 (23.4 mmol), and catalytic amount of KI in toluene was placed in a 20 mL microwave vial with a stirrer and tightly sealed. The reaction mixture was subjected for microwave reaction conditions (100 °C, 60 min) and allowed to cool to room temperature, the mixture was filtered, and the solvent evaporated under reduced pressure to obtain the crude compound. The crude was then purified by flash chromatography using EtOAc:MeOH gradient up to 4% to afford compound SYA0340 as its free base. Yield: 65%.
1H NMR (600 MHz, CDCl3) δ 8.23–8.17 (m, 1H), 7.70 (d, J = 8.2 Hz, 1H), 7.50–7.44 (m, 2H), 7.37 (dd, J = 8.2, 1.7 Hz, 1H), 6.67–6.60 (m, 2H), 3.59–3.48 (m, 4H), 3.13 (d, J = 17.4 Hz, 1H), 2.93–2.87 (m, 1H), 2.50 (dd, J = 17.3, 12.2 Hz, 4H), 2.39–2.30 (m, 2H), 1.65 (dtd, J = 25.7, 13.4, 4.5 Hz, 2H), 1.55–1.46 (m, 1H), 1.42–1.33 (m, 1H), 1.26–1.20 (m, 3H).
13C NMR (151 MHz, CDCl3) δ 209.64, 159.55, 154.05, 147.97, 141.39, 137.40, 134.52, 128.31, 126.78, 125.38, 113.26, 107.01, 58.86, 53.07 (2C), 49.20, 45.17 (2C), 39.98, 36.08, 24.09, 22.15.
4.3. Enantiomeric Separation
Fractions of enantiomers were collected as peak 1 (P1) and peak 2 (P2), concentrated by rotary evaporation and nitrogen blow-down, and then dried under high vacuum. The oxalate salt of each enantiomer was prepared by dissolving the free base in EtOAc and adding an ethereal oxalic acid solution. The resulting precipitate was recrystallized from a mixture of Et2O/MeOH solution to produce SYA0340-P1.2OX and SYA0340-P2.2OX as white crystals. Enantiomeric purity was confirmed by analytical HPLC as 100%, and melting points of the oxalate salts were determined as SYA0340-P1.2OX, Mp 176.6–178.0 °C, and SYA0340-P2.2OX, Mp 176.2–177.8 °C. CHN analyses of the enantiomers are consistent with the structure and are within 0.4% of the theoretical value as follows:
Calculated for SYA0340-P1: C26H30ClN3O9; C, 55.37; H, 5.36; N, 7.45; found: C26H30ClN3O9; C, 55.19; H, 5.28; N, 7.35.
Calculated for SYA0340-P2: C26H30ClN3O9; C, 55.37; H, 5.36; N, 7.45; found: C26H30ClN3O9; C, 55.14; H, 5.40; N, 7.29.
4.4. Polarimetric Determination of Specific Rotations
Compounds were dissolved in HPLC grade ethanol to give 0.800 g/100 mL. The solution was transferred to a 3.5 × 100 mm cylindrical cell and allowed to equilibrate to room temperature. A Jasco 1020 polarimeter, with settings of 589 nm wavelength, aperture 3, integration 5 s, interval 120 s, and repeat 9 times was used. Optical rotations were collected for the solvent blank before and after each enantiomer was analyzed. Mean (n = 9 readings of same solution) blank values were subtracted from the mean (n = 9) rotation for each enantiomer, and this value was used to calculate the specific rotation for day 1 for each compound. This was repeated for 2 more days giving a total of n = 3 values for the specific rotation for each enantiomer. The mean (n = 3) specific rotation ± SD of the 3 days is reported.
4.5. X-ray Crystallographic Studies
For the determination of the absolute configuration of the enantiomers, the oxalate salt of SYA0340-P2 was recrystallized for a SCXRD measurement. A crystal (0.557 × 0.051 × 0.033 mm3) was placed onto a nylon loop and mounted on a XtaLAB-Synergy-S diffractometer with a Hypix-6000HE (hybrid photon counting) detector for data collection at 150 K. The full data collection was carried out by the program suite CrysAlisPro using a PhotonJet (Cu) X-ray source in 0.50° oscillations for each frame with the intensity that is more than 10:1 for data-to-parameter ratio. The structure was solved using SHELXT and refined using SHELXL-2014 using the GUI Olex2.20 The space group P21 was determined based on systematic absences for the compound [C22H28ClN3O]2+·2[COO–COOH]. Most of non-hydrogen atoms were assigned from the solution. Full matrix least squares or difference Fourier cycles were performed to locate the remaining non-hydrogen atoms. All non-hydrogen atoms were refined with anisotropic displacement parameters, while all hydrogen atoms were placed in ideal positions and refined as riding atoms with relative isotropic displacement parameters.
4.6. Receptor Binding Affinity Studies
Binding affinities reported in Table 2 were conducted at VA Portland Health Care System, Portland, OR. For affinity studies at the 5-HT7A receptor, the receptor was transiently transfected into the HEK cell line, which do not express serotonin receptors. The cDNA used to transfect the cell line only contains the sequence for the receptor and not the introns; therefore, there are no other splice variants present in the cells. For the preparation of stock solutions, compounds were dissolved in DMSO to make a 10 mM stock solution followed by dilution to 50 μM in assay buffer or water for binding. For binding and functional assays, subsequent dilutions with buffer supplemented with DMSO were made to maintain a final concentration of 0.1% DMSO in all wells. Pipetting was carried out using a Biomek 4000 robotic workstation.
4.6.1. [3H]8-OH-DPAT Binding at the 5-HT1A Receptor
Human embryonic kidney cells expressing the human 5HT1A receptor (HEK-h5HT1A) were used. The methods for cell membrane preparation and [3H]8-OH-DPAT binding assays have been described previously.21 In brief, the assay was performed as follows: 96-well assay tubes containing (in duplicate) serial dilutions of drugs were made using a Biomek 4000 robotics system. The reaction mixture contains drug, cell homogenate (0.05 mg protein/well), [3H]8-OH-DPAT (0.5 nM final concentration, 170 Ci/mmol, Perkin Elmer), and assay buffer (25 mM Tris HCl, pH 7.4, containing 100 μM ascorbic acid and 10 μM pargyline). Non-specific binding was determined with 1.0 μM dihydroergotamine. The plates were incubated at room temperature for 60 min and then filtered on a Tomtec cell harvester. The filters were washed with cold 50 mM Tris buffer (pH 7.4), dried, spotted with scintillation cocktail, and counted for 2 min on a Perkin Elmer microbetaplate counter. IC50 values were calculated with GraphPad Prism, and IC50 values were converted to Ki values using the Cheng–Prusoff correction.
4.6.2. [3H]5-HT Binding at 5HT7A Receptors
Human embryonic kidney cells were transfected with 10 μg h5HT7A cDNA (cDNA Resource Center, Bloomsburg, PA) using PEI in unsupplemented DMEM. After 5–15 h, the medium was changed to DMEM supplemented with 10% Fetal Clone and Pen/strep. Two days later, the media was removed, cells were rinsed with phosphate-buffered saline (PBS), scraped into 10 mL PBS, and centrifuged at 1000 × g for 10 min. The pellet was resuspended in 5 mL of 50 mM Tris (pH 7.4 at 4 °C) and polytronned (5 s at setting 6). The homogenate was centrifuged at 15,000 rpm for 20 min. The resuspension of the pellet followed by centrifugation was repeated 2 more times in order to minimize residual serotonin. Then the pellet was resuspended in 8 mL buffer/plate of transfected cells. The binding assay includes drug, 5-HT or buffer, cell homogenate, [3H]5-HT (∼1–2 nM, final), and buffer (50 mM Tris, pH 7.4 at 37 °C, with 0.1% ascorbic acid, 5 mM CaCl2, and 10 μM pargyline). Specific binding was defined as the difference between total binding and binding in the presence of 10 μM lurasidone. The reaction was incubated for 60 min at 25 °C and terminated by filtration using a Tomtec 96-well harvester. Radioactivity remaining on filters was counted on a Perkin Elmer microbetaplate 1405 liquid scintillation counter.
4.7. Receptor Functional Assays
Functional assay results reported in Tables 3 and 4 were conducted at the VA Portland Health Care System, Portland, OR. Preparation of stock solutions and dilutions were made as described in Section 4.6.
4.7.1. HEK-5HT1A GTPγS Binding Studies
The method from Newman-Tancredi et al.22 was adapted for assessment of the functional status of compounds at 5HT1A receptors, and the assays were conducted as described by Ofori et al.17 Briefly, HEK-h5HT1A cells at 80–90% confluency were used for membrane preparation. The assay buffer consisted of 20 mM HEPES, pH 7.4, 10 mM MgCl2, 100 mM NaCl, and 0.2 mM DTT. The cells were scraped from the plates into buffer and centrifuged at 200 rpm for 15 min, the supernatant was removed, and the pellet was homogenized in 10 mL buffer/plate of cells. The homogenate was centrifuged at 17,500 rpm for 15 min, and the resulting pellet was resuspended. This was repeated a total of 3 times to minimize any serotonin that was present in the medium. Finally, the supernatant was removed, and pellet was resuspended in assay buffer.
For the assay, cell membranes (40–75 μg protein) were preincubated (10 min, room temperature) with the test compound in duplicate in assay buffer. The reaction was initiated by the addition of GDP (3 mM) and [35S] GTPγS (0.1 nM,1350 Ci/mmol, PerkinElmer, ∼150,000 cpm). The reaction was incubated at room temperature for 60 min on a rotating platform. Non-specific binding was defined with 1 μM WAY-100635. Agonist efficacy was expressed relative to that of 100 nM 5-HT (which is a maximally effective concentration) in each experiment. In each experiment, a dose response curve with the agonist serotonin was conducted to help identify full and partial agonist compounds. EC50 values were calculated with GraphPad Prism, and maximal effect for a given drug each day was normalized to the maximal effect of serotonin.
4.7.2. HEK-5HT7A/cAMP Functional Assay
For HEK-5HT7A/cAMP agonist assay, human embryonic kidney cells were transfected as described in Section 4.6.2. The agonist assays were conducted as described by Ofori et al.17 In brief, cells were incubated for 20 min. Next agonists were added followed by a 20 min incubation. The antagonists were added at 10 min followed 10 min later with addition of the agonist, serotonin (100 nM). For both conditions, after another 20 min incubation, the reaction was terminated by aspiration. Finally, 0.1 mL 3% trichloroacetic acid was added, followed by incubation in the dark on a rotator for 2 h. Agonists stimulate adenylate cyclase production, which was measured using a cyclic AMP EIA kit (Cayman). Aliquots from each well were diluted 1:5 in EIA buffer from the kit, and 50 μL of the dilution was added to the EIA plate. The cyclase assay was conducted following the instructions in the EIA kit and as described in ref (17). Maximal stimulation of 5HT7A receptors was defined with serotonin. The maximal drug effect was normalized to maximal serotonin effect in the tables. For antagonists, maximal inhibition of cAMP formation was defined with 10 μM lurasidone.
4.8. Data Analysis
For binding affinity studies, data were normalized to the binding in the absence of drug. Three or more independent competition experiments were conducted with duplicate determinations. GraphPad Prism was used to analyze the ensuing data, with IC50 values converted to Ki values using the Cheng–Prusoff equation (Ki = IC50/(1 + ([drug*]/Kd drug*))), where drug* is the labeled ligand used in the binding assays. The Kd values used in the equations are 5.02 nM for [3H]8-OH-DPAT at 5HT1A receptors and 0.347 nM for [3H]5HT at 5HT7A receptors. For functional assays, GraphPad Prism was used to calculate either EC50 (agonists) or IC50 (antagonists) values using data expressed as pg cAMP for adenylate cyclase activity or % serotonin-stimulation for GTPγS binding.
For functional assays, GraphPad Prism was used to calculate either EC50 (agonists) or IC50 (antagonists) values using data expressed as pg cAMP for adenylate cyclase activity or % serotonin stimulation for GTPγS binding.
Acknowledgments
This work was financially supported by an NIH/NIGMS R16 grant number 1R16GM145581-01 and an NIGMS Endowment grant in support of an Eminent Scholar Chair to S.Y.A. The research was also partly supported with funding from the U.S. Department of Education, Title III Part B, Strengthening Historically Black Graduate Institutions Programs (HBGI), awarded to Florida A&M University and an RCMI grant #NIMHD U54 MD 007582. Ki determinations and receptor binding assays were performed by the Oregon Health and Sciences University. This research made use of a Rigaku Synergy-S single-crystal X-ray diffractometer which was acquired through the NSF MRI program (award CHE-1828362). Funding sources acknowledged had no involvement in the study design, data collection and interpretation, or article preparation and submission of this manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c01283.
NMR data for SYA0340 and its intermediates, and elemental (C, H, and N) analysis data (PDF)
Author Contributions
S.Y.A. conceived the idea and designed the project, experiments, and outline of the manuscript; B.A.B., C.V., E.K.O., U.M.G., X.L., T.L.S., L.B.K., J.L.S., S.H.B., and A.J.J. performed the experiments and analyzed the data. S.Y.A., B.A.B., C.V., L.B.K., and A.J.J. wrote the manuscript. S.Y.A., B.A.B., C.V., X.L., and A.J.J. contributed the critical review of the manuscript; all authors read and approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Barnes N. M.; Ahern G. P.; Becamel C.; Bockaert J.; Camilleri M.; Chaumont-Dubel S.; Claeysen S.; Cunningham K. A.; Fone K. C.; Gershon M.; et al. International Union of Basic and Clinical Pharmacology. CX. Classification of Receptors for 5-hydroxytryptamine; Pharmacology and Function. Pharmacol. Rev. 2021, 73, 310–520. 10.1124/pr.118.015552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drago A.; Ronchi D. D.; Serretti A. 5-HT1A gene variants and psychiatric disorders: a review of current literature and selection of SNPs for future studies. Int. J. Neuropsychopharmacol. 2008, 11, 701–721. 10.1017/S1461145707008218. [DOI] [PubMed] [Google Scholar]
- Newman-Tancredi A. Biased agonism at serotonin 5-HT1A receptors: preferential postsynaptic activity for improved therapy of CNS disorders. Neuropsychiatry 2011, 1, 149–164. 10.2217/Npy.11.12. [DOI] [Google Scholar]
- De Vry J.; Schreiber R.; Melon C.; Dalmus M.; Jentzsch K. R. 5-HT1A receptors are differentially involved in the anxiolytic- and antidepressant-like effects of 8-OH-DPAT and fluoxetine in the rat. Eur. Neuropsychopharmacol. 2004, 14, 487–495. 10.1016/j.euroneuro.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Akimova E.; Lanzenberger R.; Kasper S. The serotonin-1A receptor in anxiety disorders. Biol. Psychiatry 2009, 66, 627–635. 10.1016/j.biopsych.2009.03.012. [DOI] [PubMed] [Google Scholar]
- Altieri S. C.; Garcia-Garcia A. L.; Leonardo E. D.; Andrews A. M. Rethinking 5-HT1A receptors: emerging modes of inhibitory feedback of relevance to emotion-related behavior. ACS Chem. Neurosci. 2013, 4, 72–83. 10.1021/cn3002174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ramboz S.; Oosting R.; Amara D. A.; Kung H. F.; Blier P.; Mendelsohn M.; Mann J. J.; Brunner D.; Hen R. Serotonin receptor 1A knockout: an animal model of anxiety-related disorder. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 14476–14481. 10.1073/pnas.95.24.14476. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Heisler L. K.; Chu H. M.; Brennan T. J.; Danao J. A.; Bajwa P.; Parsons L. H.; Tecott L. H. Elevated anxiety and antidepressant-like responses in serotonin 5-HT1A receptor mutant mice. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 15049–15054. 10.1073/pnas.95.25.15049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wang S.-M.; Han C.; Lee S.-J.; Patkar A. A.; Masand P. S.; Pae C.-U. Vilazodone for the Treatment of Depression: An Update. Chonnam Med. J. 2016, 52, 91–100. 10.4068/cmj.2016.52.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Frampton J. E. Vilazodone: in major depressive disorder. CNS Drugs 2011, 25, 615–627. 10.2165/11207550-000000000-00000. [DOI] [PubMed] [Google Scholar]
- a Modica M. N.; Lacivita E.; Intagliata S.; Salerno L.; Romeo G.; Pittala V.; Leopoldo M. Structure-Activity Relationships and Therapeutic Potentials of 5-HT(7) Receptor Ligands: An Update. J. Med. Chem. 2018, 61, 8475–8503. 10.1021/acs.jmedchem.7b01898. [DOI] [PubMed] [Google Scholar]; b Glass J. D.; Grossman G. H.; Farnbauch L.; DiNardo L. Midbrain raphe modulation of nonphotic circadian clock resetting and 5-HT release in the mammalian suprachiasmatic nucleus. J. Neurosci. 2003, 23, 7451–7460. 10.1523/JNEUROSCI.23-20-07451.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hedlund P. B.; Sutcliffe J. G. Functional, molecular and pharmacological advances in 5-HT7 receptor research. Trends Pharmacol. Sci. 2004, 25, 481–486. 10.1016/j.tips.2004.07.002. [DOI] [PubMed] [Google Scholar]; d Roberts A. J.; Hedlund P. B. The 5-HT(7) receptor in learning and memory. Hippocampus 2012, 22, 762–771. 10.1002/hipo.20938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellynck E.; Heyninck K.; Andressen K. W.; Haegeman G.; Levy F. O.; Vanhoenacker P.; Van Craenenbroeck K. The serotonin 5-HT7 receptors: two decades of research. Exp. Brain Res. 2013, 230, 555–568. 10.1007/s00221-013-3694-y. [DOI] [PubMed] [Google Scholar]
- Bard J. A.; Zgombick J.; Adham N.; Vaysse P.; Branchek T. A.; Weinshank R. L.. Cloning of a novel human serotonin receptor (5-HT7) positively linked to adenylate cyclase. J. Biol. Chem. 1993, 268( (31), ), 23422–23426, PMID: 8226867, 10.1016/S0021-9258(19)49479-9. [DOI] [PubMed] [Google Scholar]
- Heidmann D. E.; Szot P.; Kohen R.; Hamblin M. W. Function and distribution of three rat 5-hydroxytryptamine7 (5-HT7) receptor isoforms produced by alternative splicing. Neuropharmacology 1998, 37, 1621–1632. 10.1016/s0028-3908(98)00070-7. [DOI] [PubMed] [Google Scholar]
- Abbas A. I.; Hedlund P. B.; Huang X. P.; Tran T. B.; Meltzer H. Y.; Roth B. L. Amisulpride is a potent 5-HT7 antagonist: relevance for antidepressant actions in vivo. Psychopharmacology 2009, 205, 119–128. 10.1007/s00213-009-1521-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumenko V. S.; Popova N. K.; Lacivita E.; Leopoldo M.; Ponimaskin E. G. Interplay between serotonin 5-HT1A and 5-HT7 receptors in depressive disorders. CNS Neurosci. Ther. 2014, 20, 582–590. 10.1111/cns.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner U.; Zeug A.; Woehler A.; Niebert M.; Dityatev A.; Dityateva G.; Gorinski N.; Guseva D.; Abdel-Galil D.; Frohlich M.; et al. Heterodimerization of serotonin receptors 5-HT1A and 5-HT7 differentially regulates receptor signalling and trafficking. J. Cell Sci. 2012, 125, 2486–2499. 10.1242/jcs.101337. [DOI] [PubMed] [Google Scholar]
- Hoyer D.; Hannon J. P.; Martin G. R. Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol., Biochem. Behav. 2002, 71, 533–554. 10.1016/s0091-3057(01)00746-8. [DOI] [PubMed] [Google Scholar]
- Ofori E.; Onyameh E. K.; Gonela U. M.; Voshavar C.; Bricker B.; Swanson T. L.; Eshleman A. J.; Schmachtenberg J. L.; Bloom S. H.; Janowsky A. J.; et al. New dual 5-HT1A and 5-HT7 receptor ligands derived from SYA16263. Eur. J. Med. Chem. 2021, 214, 113243 10.1016/j.ejmech.2021.113243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyameh E. K.; Bricker B. A.; Eyunni S. V. K.; Voshavar C.; Gonela U. M.; Ofori E.; Jenkins A.; Ablordeppey S. Y. A study of the structure-affinity relationship in SYA16263; is a D(2) receptor interaction essential for inhibition of apormorphine-induced climbing behavior in mice?. Bioorg. Med. Chem. 2021, 30, 115943 10.1016/j.bmc.2020.115943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyameh E. K.; Ofori E.; Bricker B. A.; Gonela U. M.; Eyunni S. V. K.; Kang H. J.; Voshavar C.; Ablordeppey S. Y. Design and discovery of a high affinity, selective and beta-arrestin biased 5-HT(7) Receptor Agonist. Med. Chem. Res. 2022, 31, 274–283. 10.1007/s00044-021-02797-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Gatch M. B.; Forster M. J.; Janowsky A.; Eshleman A. J. Abuse liability profile of three substituted tryptamines. J. Pharmacol. Exp. Ther. 2011, 338, 280–289. 10.1124/jpet.111.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman-Tancredi A.; Gavaudan S.; Conte C.; Chaput C.; Touzard M.; Verriele L.; Audinot V.; Millan M. J. Agonist and antagonist actions of antipsychotic agents at 5-HT1A receptors: a [35S]GTPgammaS binding study. Eur. J. Pharmacol. 1998, 355, 245–256. 10.1016/s0014-2999(98)00483-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.