

Abstract

The current–voltage characteristics of a single-molecule junction are determined by the electronic coupling Γ between the electronic states of the electrodes and the dominant transport channel(s) of the molecule. Γ is profoundly affected by the choice of the anchoring groups and their binding positions on the tip facets and the tip–tip separation. In this work, mechanically controllable break junction experiments on the N,N′-bis(5-ethynylbenzenethiol-salicylidene)ethylenediamine are presented, in particular, the stretch evolution of Γ with increasing tip–tip separation. The stretch evolution of Γ is characterized by recurring local maxima and can be related to the deformation of the molecule and sliding of the anchoring groups above the tip facets and along the tip edges. A dynamic simulation approach is implemented to model the stretch evolution of Γ, which captures the experimentally observed features remarkably well and establishes a link to the microscopic structure of the single-molecule junction.
Many different extensions of the classical silicon technologies have been proposed to meet the constant demand of miniaturization of electronic devices during the past decades.1−3 Molecular electronics is one of the proposed extensions, which focuses on using single molecules as electronic components, which is the ultimate goal for miniaturization. Elementary electronic components like molecular transistors, switches, and rectifiers4−8 have already been demonstrated using single-molecule junctions. Recently, logic-in-memory operations in single-metallofullerene devices have been reported at room-temperature.9 Characterizing the physical and electronic properties of such single-molecular components requires selectivity and resolution at the nanoscale. This can be achieved using scanning tunneling microscopy (STM),10,11 atomic force microscopy (AFM),12−14 mechanically controllable break junctions (MCBJs),15,16 and electromigrated (EM) break junctions. The electron transport regimes, where these different measurement techniques are employed, have been discussed in detail in the literature.17 Among the above-mentioned techniques, MCBJs enable a more systematic study of the current–voltage (I–V) characteristics and conductance-breaking traces owing to the mechanical stability of the junctions. The stability of such molecule junctions is dictated by the binding strength of the anchoring group, deformation of the molecule, and thermal vibrations.16,18−20 Nevertheless, molecules can bind to the metallic electrodes at different positions and in various orientations depending on the specific chemical nature of the anchoring groups and the local symmetry of binding sites,16,21−31 which makes it a challenging task to elucidate the underlying physical mechanism governing the electronic behavior of single-molecule junctions.
Usually, 1-D conductance histograms and conductance-distance 2-D histograms are constructed from few hundreds or thousands of individual traces. The most probable I–V characteristic for a particular molecule–metal combination is associated with the most prominent conductance peak observed in the conductance histograms.16,18,19,29,32,33 The experimental characterization of single-molecule junctions via MCBJ is complemented with ab initio transport calculations, in order to estimate the relative positions of molecular levels, which act as transport channels, and the chemical potential of the metallic leads.1,26,34−37 However, the theoretical modeling of the broad distribution in the experimental conductance histograms is not feasible because (1) in MCBJ experiments, the adsorption geometries of the molecule between the metallic electrodes are a priori unknown,38−40 (2) many experimental factors remain elusive and are therefore not considered, and (3) the ab initio transport calculations are computationally expensive. Consequently, a limited number of energetically most favorable configurations are selected as representative junction geometries for theoretical investigations.8,35,41,42 However, recent technological advancements allow the simultaneous measurement of mechanical and electronic properties of single-molecule junctions, which enables deeper understanding of the structural information on junction geometries. For example, the most probable junction geometries for hexanedithiol and octanedithiol with gold electrodes in multiple traces can be probed in STM break junctions by applying a high-frequency sinusoidal mechanical signal during junction formation.11 In another study, metallocenes have been shown to form reproducible metal-molecule-metal junctions at low and room temperatures, that correlate with the atomic shape of the metal electrode and ineraction with electron-rich rings giving rise to extended conductance plateaus.33 Yet, to our knowledge, the evolution of junction geometries and associated electronic parameters of single-level model (SLM)1 has not been investigated in individual bridge openings so far.
In this work, we present a combined theoretical and experimental approach to describe the microstates of junction geometries and present the stretch evolution of the electronic coupling extracted from experimentally measured IV curves during individual bridge openings in a MCBJ setup for dithiolated-N,N′-bis(5-ethynylbenzenethiol-salicylidene)ethylenediamine cobalt complex (Co-Salen-S) (see Figure 1(a)).43 The evolution of the electronic coupling Γ with increasing tip–tip separation is accompanied by an initial decrease and a final increase, separated by a flat region of low Γ values with recurrent peaks. We use a novel high-throughput dynamic simulation approach for evaluating the stretch evolution of the SLM parameters by including a considerably large set of junction geometries and evaluating their respective nonequilibrium thermodynamical weights. Our theoretical approach is different from the previous works8,35,42 in that we consider multiple thermodynamically relevant configurations to evaluate the electronic properties of single-molecule junctions. We investigate the effects of the local symmetry of binding sites and of the deformation of the molecule upon anchoring on the I–V characteristics as the gap is widened and the anchoring groups either lock at specific binding sites or slide along the facets of the metallic electrodes toward the tip. The transmission functions T(E) from ballistic transport simulations for thermodynamically favorable junction geometries are used to extract the SLM parameters in the same way as for the measured IV curves, which allows a direct comparison with experimental findings. We relate the recurring peaks in the evolution of Γ with increasing tip–tip separation (Stip–tip) to the deformation of the molecule and steric boundary conditions at certain Stip–tip, which lead to energetically less favorable configurations. Finally, we present first efforts to establish a link between features observed in the evolution of the electronic coupling of stretching curves in MCBJs and the microstates of junction geometries, which are not accessible experimentally.
Figure 1.

Schematic representation of (a) dithiolated-N,N′-bis(5-ethynylbenzenethiol-salicylidene)ethylenediamine cobalt complex (Co-Salen-S) and (b) dithiolated-1,4-bis(phenylethynyl)-2,5-bis(ethoxy)benzene (PEEB-S).
In an MCBJ setup, adjustable atomically sharp gold contacts44,45 are produced by deforming a lithographically manufactured nanoscopic gold constriction on a flexible substrate. The stretching of the nanoscopic gold constriction46 can be controlled to few Å/min. After breaking the direct metallic contact, this allows the formation of a single-molecule junction consisting of a single molecule connected to two metal electrodes in a solution containing the molecules to be measured. The experimental measurements were performed for the molecule Co-Salen-S (see Figure 1(a)). We chose thiol (SH) groups as anchoring groups since they provide stable bonds between molecules and metallic electrodes in charge transport measurements of single molecules. Moreover, sulfur atoms bind to high-symmetry top, hcp-, and fcc-hollow sites on the Au(111) surface (see SI Section S2) leading to a change of the I–V characteristics, which can be monitored in MCBJ experiments. The local binding environment of the SH linkers can be inferred from such measurements. For the MCBJ experiments, the acetyl-protected derivatives of Co-Salen-S were dissolved in toluene. A small amount of 20% ammonia solution in water was added as in situ deprotection agent right before the measurement. The solution was pipetted into the liquid cell on top of the lithographically defined nanogold contact of the MCBJ sample. After detecting the molecular signature16 in the electric signal obtained from the junction by a conductance measurement, the stretching of the junction is halted, and an I–V acquisition is initiated. The I–V measurements were recorded following a butterfly sweep (0 V → 0.8 V → – 0.8 V → 0.8 V → 0 V). Two full-range I–V-curves are obtained for a single bridge position. We wait at least 90 s between the I–V measurements following a butterfly sweep and until the standard deviation of the measured signal is lower than a threshold.16 The opening speed of the tips is of the order of 2 × 10–12 m/s. After completing the data acquisition, the stretching of the bridge is continued as long as the plateau persists and the I–V butterfly sweep is repeated. This procedure is performed until the junction is fully open, and no molecular signal is detected anymore. Before initiating another stretching, the junction is closed until the conductance value of 20 G0(47) is reached, where G0 denotes the quantum of conductance 7.748 × 10–5 S. All measurements were performed in vivo at room temperature. We performed 700 stretching cycles and rejected measurements that failed to fullfil the goodness-of-fit (GOF) criterion.16 We observed partial signatures of the stretch evolution of the electronic coupling (ΓSE) for Co-Salen-S in 5 measurements: 2 measurements for falling trend, 2 measurements for rising trend, and a single measurement for the intermiediate flattened region. We obtained only for a single measurement the complete ΓSE with all the trends, namely the falling and rising trend, and an intermediate flattened region. For further details we refer to SI Sections S1 and S11.
We performed self-consistent density-functional-based tight-binding simulations for the geometry, electronic structure, and ballistic transport properties as implemented in the program package DFTB+ 20.1.48 The parameter set ”auorg-1-1” has been utilized in all calculations,49 which is an extension of the ”mio-1-1”50 parameter set. The ”mio-1-1” set has been developed for organic molecules including O, N, C, H, and S, whereas the ”auorg-1-1” set was designed to describe thiolates on gold nanoclusters.51,52 Dispersion corrections were included using the universal force field.53 The geometry relaxations were performed until the maximum force components were reduced to 0.0001 eV/Å. For calculating the transmission function, periodic boundary conditions in x and y directions and a shifted 2 × 2 Monkhorst scheme54 were used. In order to reduce the computational effort, we chose dithiolated-1,4-bis(phenylethynyl)-2,5-bis(ethoxy)benzene (PEEB-S) over Co-Salen-S, because Co-Salen-S binds to the electrodes via two peripheral PEEB-S moieties. Note that, it is not possible to determine the ΓSE of PEEB-S in our MCBJ setup, because at shorter distances of the order of the dimensions of the PEEB-S, tunneling currents between the tip electrodes dominate over the molecular signature. Nonetheless, the influence of the central metal complex on the stretch evolution of Γ̅ can be neglected due to the length of the peripheral PEEB-S moieties. We evaluate the I–V characteristics by assuming that the electronic transport is dominated by a single-level model1,36,37 and by extracting ϵ0 and Γ, i.e., the energy and the coupling of that molecular orbital (see SI Sections S1 and S8). In addition to standard electronic structure methods such as density functional theory (DFT) or Hartree–Fock (HF), ballistic transport simulations are performed using the equilibrium Green’s function (EGF) formalism.55−57 Since the MCBJ experiments are performed in liquid conditions and the I–V measurements are recorded at the time scale of seconds, it can be assumed that the experimentally obtained SLM parameters do not originate from a single, energetically most favorable junction geometry, but rather represent a mean of many thermodynamically accessible configurations. Usually, the thermodynamically meaningful average is calculated by statistically averaging the quantities of interest for a selected set of snapshots from molecular dynamics trajectories.58 The main disadvantages of this approach are (a) an exhaustive sampling of the configuration space can only be achieved in long trajectories and (b) suitable exclusion algorithms for geometrically nearly identical snapshots are indispensible in order to avoid multiple evaluation of SLM parameters, using computationally expensive transport calculations.
Our dynamic simulation approach samples the configuration space systematically using a dense grid and addresses numerous thermodynamically accessible configurations. It is essential to include the thermodynamically accessible configurations in the case of thiol anchoring groups, because the local minima are separated from the global minimum through high barriers (see SI Section 2 on binding energy landscapes), which are unlikely to be surmounted during the short time scale of the experimental measurement. We employ a stochastic process (random walk) based on Metropolis transition probabilities,59 described by a master equation,60 and evaluate a transition matrix to propagate the probability density by utilizing the Metropolis criterion59 for neighboring configurations and zero elsewhere (see SI Section S3 for more details). Finally, we perform transport calculations for configurations which contribute most to the thermodynamical mean of the SLM parameters (see SI Sections S4 and S5). Our approach is computationally advantageous, because (a) the systematic sampling of the configuration space is trivially parallelized, and (b) the transport calculations are solely performed for thermodynamically relevant and unique geometrical configurations.
The experimentally measured stretch evolution
of the electronic
coupling (ΓE) and the theoretically determined stretch
evolution of the electronic coupling (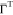 ) are shown in Figure 2. Please note the superscripts E and T to
differentiate between the evolution of the electronic coupling determined
experimentally and theoretically, resepectively.
) are shown in Figure 2. Please note the superscripts E and T to
differentiate between the evolution of the electronic coupling determined
experimentally and theoretically, resepectively.
Figure 2.

Experimentally measured
stretch evolution of the electronic coupling
(ΓE) for a single opening measurement of Co-Salen-S
and theoretically determined stretch evolution of the electronic coupling
(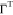 ) evaluated using the SLM applied to the
transmission function T(E), which
are calculated for 1000 thermodynamically most relevant configurations
for individual tip–tip separation (Stip–tip). (a) ΓE - the data points with peaks are marked
using dark blue arrows. (b)
) evaluated using the SLM applied to the
transmission function T(E), which
are calculated for 1000 thermodynamically most relevant configurations
for individual tip–tip separation (Stip–tip). (a) ΓE - the data points with peaks are marked
using dark blue arrows. (b)  ,
, 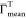 , and
, and  reveal a falling trend for Stip–tip interval (11.51 Å < Stip–tip < 21.72 Å) and a rising trend for Stip–tip > 21.72 Å. Peaks are
visible
for
reveal a falling trend for Stip–tip interval (11.51 Å < Stip–tip < 21.72 Å) and a rising trend for Stip–tip > 21.72 Å. Peaks are
visible
for 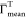 at Stip–tip: 13.08, 17.79, and 21.72 Å, respectively. The four subdivisions
I, II, III, and IV define the regions with the dominant anchoring
position pairs: edge–edge, tip–edge, tip–edge
+ tip–tip, and tip–tip, respectively. Refer to the dominant
anchoring positions in Figure 3(a).
at Stip–tip: 13.08, 17.79, and 21.72 Å, respectively. The four subdivisions
I, II, III, and IV define the regions with the dominant anchoring
position pairs: edge–edge, tip–edge, tip–edge
+ tip–tip, and tip–tip, respectively. Refer to the dominant
anchoring positions in Figure 3(a).
The ΓE values follow a general
trend and reveal
distinct peaks. In the initial opening phase, we notice a falling
trend in ΓE which flattens out for the larger part
of the opening curve, only to be superseded by a surge in the final
phase before break-off. The decrease in ΓE is visible
in the interval (0 < S̃ < 20), the flattening
out in the interval (20 < S̃ < 50) and
the increase thereafter (S̃ > 50) until
break-off.
Please note that the tip–tip separation in experiments cannot
be determined accurately and hence is denoted by S̃. Apart from this general trend in ΓE, we also observe
peaks at various time intervals marked using dark blue arrows in Figure 2(a). Such peaks are
also observed for other opening cycles (see SI section S1). Similarly, 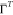 reveals a drop in ΓE for
the Stip–tip interval (11.51 Å
< Stip–tip < 21.72 Å)
and a surge for Stip–tip > 21.72
Å (see Figure 2(b)). Additionally, peaks are observed at Stip–tip values of: 13.08, 17.79, and 21.72 Å.
reveals a drop in ΓE for
the Stip–tip interval (11.51 Å
< Stip–tip < 21.72 Å)
and a surge for Stip–tip > 21.72
Å (see Figure 2(b)). Additionally, peaks are observed at Stip–tip values of: 13.08, 17.79, and 21.72 Å.
In the following, we explore correlations between the stretch evolution
of the electronic coupling and the stretch evolution of three geometrical
descriptors: 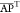 ,
,  , and
, and 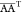 . The geometrical descriptor
. The geometrical descriptor 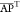 quantifies the fraction of configurations
with dominating anchoring positions of the sulfur atoms on Au(111)
facets on a scale between 0 and 1. The geometrical descriptors
quantifies the fraction of configurations
with dominating anchoring positions of the sulfur atoms on Au(111)
facets on a scale between 0 and 1. The geometrical descriptors 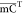 and
and 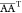 quantify the mean curvature and the mean
anchoring angle, respectively. The anchoring positions of the sulfur
atoms on the Au(111) facets (AP) define the anchoring position doublet
for both end groups on the left and right facets for each configuration.
The mean curvature (mC) is a measure of the overall curvature of the
molecular backbone (MB) of PEEB-S. The anchoring angle (AA) specifies
the angle measured between the S–C-bond and the facet normal.
The geometrical descriptors collectively capture both the deformation
of the molecule and the anchoring position of the sulfur atoms at
the gold surfaces. For more details on AP, mC, and AA, we refer to
the SI Sections S5, S6, and S7, respectively.
The theoretically determined stretch evolution of these geometrical
descriptors is then averaged using the same statistical approach employed
for the SLM parameters (see SI Section S3). The theoretically determined stretch-evolution of AP, mC, and AA are defined as
quantify the mean curvature and the mean
anchoring angle, respectively. The anchoring positions of the sulfur
atoms on the Au(111) facets (AP) define the anchoring position doublet
for both end groups on the left and right facets for each configuration.
The mean curvature (mC) is a measure of the overall curvature of the
molecular backbone (MB) of PEEB-S. The anchoring angle (AA) specifies
the angle measured between the S–C-bond and the facet normal.
The geometrical descriptors collectively capture both the deformation
of the molecule and the anchoring position of the sulfur atoms at
the gold surfaces. For more details on AP, mC, and AA, we refer to
the SI Sections S5, S6, and S7, respectively.
The theoretically determined stretch evolution of these geometrical
descriptors is then averaged using the same statistical approach employed
for the SLM parameters (see SI Section S3). The theoretically determined stretch-evolution of AP, mC, and AA are defined as 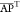 ,
,  , and
, and 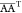 and shown in Figure 3. The trend associated
with
and shown in Figure 3. The trend associated
with 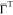 and its peaks can be explained by analyzing
and its peaks can be explained by analyzing 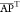 ,
, 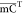 , and
, and 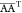 .
.
Figure 3.

(a) Theoretically determined stretch evolution
of the mean anchoring
position on the Au(111) facets (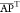 ) - four distinct Stip–tip regions are discernible. For Stip–tip < 13.87 Å, the dominating mean
anchoring positions of the sulfur atoms on the Au(111) facets (
) - four distinct Stip–tip regions are discernible. For Stip–tip < 13.87 Å, the dominating mean
anchoring positions of the sulfur atoms on the Au(111) facets (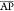 ) are either edge–edge or tip–edge.
For Stip–tip between 14.65 and
18.58 Å, the tip-edge
) are either edge–edge or tip–edge.
For Stip–tip between 14.65 and
18.58 Å, the tip-edge  dominates over edge–edge
dominates over edge–edge 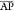 . The contribution of edge–edge
. The contribution of edge–edge 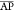 diminishes for Stip–tip > 19.36 Å with the emergence of tip–tip
diminishes for Stip–tip > 19.36 Å with the emergence of tip–tip 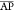 . For Stip–tip between 19.36 and 20.9 Å, the tip-edge
. For Stip–tip between 19.36 and 20.9 Å, the tip-edge 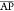 dominate. For Stip–tip > 21.72 Å, the
dominate. For Stip–tip > 21.72 Å, the  are solely tip–tip. (b) Theoretically
determined stretch evolution of the mean curvature (
are solely tip–tip. (b) Theoretically
determined stretch evolution of the mean curvature (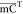 ) - an overall falling trend is discernible.
Trough-like features are visible at Stip–tip: 13.87, 17.79, and 20.9 Å, respectively. A distinct sharp peak
is visible at Stip–tip: 21.72 Å.
(c) Theoretically determined stretch evolution of the mean anchoring
angle (
) - an overall falling trend is discernible.
Trough-like features are visible at Stip–tip: 13.87, 17.79, and 20.9 Å, respectively. A distinct sharp peak
is visible at Stip–tip: 21.72 Å.
(c) Theoretically determined stretch evolution of the mean anchoring
angle (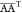 ) - An overall rising trend until saturation
is discernible. Peaks are visible at Stip–tip: 15.44 and 18.58 Å.
) - An overall rising trend until saturation
is discernible. Peaks are visible at Stip–tip: 15.44 and 18.58 Å.
We recall that the stretch evolution of the SLM parameters can be determined theoretically from the transmission function (see SI Sections S4 and S8). Ballistic transport calculations were performed for the 1000 thermodynamically most relevant configurations (see SI Sections S2, S3, S4, and S5).
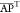 as a function of the tip–tip separation
(Figure 3(a)) reveals
four distinct regions, (I) Stip–tip < 13.9 Å, (II) 13.9 Å < Stip–tip < 14.7 Å, (III) 14.7 Å < Stip–tip < 20.9 Å, and (IV) Stip–tip > 20.9 Å. At short Stip–tip (region I) and large Stip–tip (region IV) the dominant anchoring positions
are edge–edge and tip–tip, respectively. In region I,
the dominant electronic channel can interact with additional surface
states from the adjacent facets which may lead to an enhanced electronic
coupling Γ. In region IV, the molecule is anchored symmetrically
at the tip apexes assuming a planar configuration and can form an
optimal anchoring angle with the facet normal vectors (∠LA - left anchoring angle, ∠RA - right anchoring
angle) (see SI Section S7) that enhances
the electronic coupling Γ significantly.
as a function of the tip–tip separation
(Figure 3(a)) reveals
four distinct regions, (I) Stip–tip < 13.9 Å, (II) 13.9 Å < Stip–tip < 14.7 Å, (III) 14.7 Å < Stip–tip < 20.9 Å, and (IV) Stip–tip > 20.9 Å. At short Stip–tip (region I) and large Stip–tip (region IV) the dominant anchoring positions
are edge–edge and tip–tip, respectively. In region I,
the dominant electronic channel can interact with additional surface
states from the adjacent facets which may lead to an enhanced electronic
coupling Γ. In region IV, the molecule is anchored symmetrically
at the tip apexes assuming a planar configuration and can form an
optimal anchoring angle with the facet normal vectors (∠LA - left anchoring angle, ∠RA - right anchoring
angle) (see SI Section S7) that enhances
the electronic coupling Γ significantly.
In intermediate
regions II and III, mixed tip-edge configurations
become dominant, which lowers the electronic coupling Γ in comparison
to the symmetric cases edge–edge and tip–tip. In particular,
the planar conformations in these regions result in trough-like features
of the 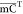 curve (see Figure 3(b)) at Stip–tip values of 13.08, 17.79, and 21.72 Å, coinciding with the peaks
in the theoretically determined stretch evolution of
curve (see Figure 3(b)) at Stip–tip values of 13.08, 17.79, and 21.72 Å, coinciding with the peaks
in the theoretically determined stretch evolution of  (
( ) at Stip–tip: 13.08 Å, and of the theoretically determined stretch evolution
of
) at Stip–tip: 13.08 Å, and of the theoretically determined stretch evolution
of 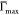 (
(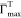 ) at Stip–tip: 17.79 and 21.72 Å, respectively. Furthermore, the minima of
the curvature coincide also with peaks in the evolution of the
) at Stip–tip: 17.79 and 21.72 Å, respectively. Furthermore, the minima of
the curvature coincide also with peaks in the evolution of the 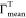 , implying that the electronic coupling
rises at certain distances Stip–tip, where the molecule is predominantly planar. Similarly, the
, implying that the electronic coupling
rises at certain distances Stip–tip, where the molecule is predominantly planar. Similarly, the 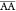 adopts peaks especially at Stip–tip: 15.44 and 18.58 Å, which may also
increase the electronic coupling further. For Stip–tip > 22.5 Å the dominant
adopts peaks especially at Stip–tip: 15.44 and 18.58 Å, which may also
increase the electronic coupling further. For Stip–tip > 22.5 Å the dominant  are tip–tip. When anchored on both
ends at the tip regions, the PEEB-S molecule can assume optimal anchoring
angles due to additional rotational degrees of freedom; there it can
rotate freely about the molecular backbone. The
are tip–tip. When anchored on both
ends at the tip regions, the PEEB-S molecule can assume optimal anchoring
angles due to additional rotational degrees of freedom; there it can
rotate freely about the molecular backbone. The 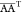 saturates for Stip–tip > 20.15 Å to values between 70° and 90°, suggesting
that most of the relevant configurations have formed optimal anchoring
angles and form a fully planar PEEB-S molecule aligned in transport
direction. Selected configurations are shown in SI Section S12.
saturates for Stip–tip > 20.15 Å to values between 70° and 90°, suggesting
that most of the relevant configurations have formed optimal anchoring
angles and form a fully planar PEEB-S molecule aligned in transport
direction. Selected configurations are shown in SI Section S12.
The effect of the bending plays a more
dominant role than the effect
of forming the optimal anchoring angles. This is further confirmed
by the sharp dip of the 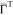 curve at Stip–tip = 18.58 Å followed by a surge up to a
curve at Stip–tip = 18.58 Å followed by a surge up to a 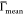 value of 0.06 eV until break-off in theoretically
determined stretch evolution of Γ̅ (
value of 0.06 eV until break-off in theoretically
determined stretch evolution of Γ̅ (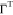 ) (Compared with the steep increase at Stip–tip = 18.58 Å followed by a
drop in
) (Compared with the steep increase at Stip–tip = 18.58 Å followed by a
drop in 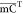 ). A cross-correlation plot between Stip–tip,
). A cross-correlation plot between Stip–tip, 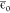 ,
,  ,
, 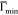 ,
,  ,
, 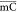 , and
, and 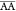 is shown in SI Section
S10 for completeness.
is shown in SI Section
S10 for completeness.
In addition, the electronic coupling of the site-specific anchoring of the molecule on the Au(111) facets can be visualized using the real-space projection of the stretch evolution of the electronic coupling. The real-space projection of Γ of a single configuration is obtained by replacing the clamped molecule between Au(111) electrodes with a pair of colored circles at the anchoring sulfur atoms, where the color and the radius of the circles represent the Γmean (see Figure 4(c) and SI Section S9).
Figure 4.

Real-space projection of Γmean (a) side view and (b) top view (not to scale) of the configuration with strongest coupling for the Stip–tip = 12.33 Å. (c) The value of Γmean is represented by red circles at the sites of the anchoring sulfur atoms on the left and right electrodes in top view. The gold atoms of the electrodes are colored to depict the top, hcp-hollow, and fcc-hollow sites using yellow, brown, and salmon colored circles, respectively. The solid gray and dotted red lines as shown to indicate the anchoring of the sulfur atoms at top and bridge sites along the Au(111) facet edges of both the left and right electrodes.
In Figure 5, the real-space projection of the theoretically determined Γmean is shown for the 50 configurations with the highest random walk weights for each Stip–tip. We analyze three intervals for the tip–tip separation: (1) 13.9 Å < Stip–tip < 14.7 Å, (2) 14.7 Å < Stip–tip < 20.9 Å, and (3) Stip–tip > 20.9 Å separately.
Figure 5.

Real-space projection of the theoretically determined stretch evolution of Γmean for those 50 configurations for each Stip–tip, with the highest random walk weights. The gold electrodes are represented using yellow, brown, and salmon colored circles, representing the top, hcp-hollow, and fcc-hollow sites, respectively. The other colored circles appear in pairs, which reveal the binding sites for the anchoring sulfur atoms on the left and the right gold electrode. Higher and lower Γmean values are represented using a color scale spanning from red to gray, augmented together with the size of circle. Additionally, solid red and dotted gray lines are placed to differentiate between bridge sites and top sites along the Au(111) facet edges, respectively.
In the first interval (see Figure 5(a)–(e)), the anchoring positions
with the strongest
electronic coupling shift from bridge to top sites along the Au(111)
facet edges. The larger red circles situated at bridge sites for Stip–tip 11.5 and 12.3 Å evolve into
smaller red circles closer to the top sites for Stip–tip 13.1, 13.9, and 14.7 Å. For the corresponding Stip–tip interval 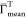 reveals a falling trend, suggesting that
at bridge sites the transport channel couples more strongly to the
metal states than at the top sites (Figure 2(b)).
reveals a falling trend, suggesting that
at bridge sites the transport channel couples more strongly to the
metal states than at the top sites (Figure 2(b)).
Using similar arguments, we dintinguish
two trends in the second
interval. The first trend (see Figure 5 (e) → (f) → (g) → (h) →
(i)) reveals a shift in the dominating anchoring positions from top
sites toward bridge sites along the Au(111) facet edges. For the corresponding Stip–tip interval in Figure 2(b), we notice a rising trend
in  and the maxima at Stip–tip values of 16.2 and 17.8 Å. The second trend
(see Figure 5 (j) →
(k) → (l) → (m) → (n)) reveals a shift in the
dominating anchoring positions from bridge sites toward top sites
along the Au(111) facet edges. For the corresponding Stip–tip interval in Figure 2(b), the
and the maxima at Stip–tip values of 16.2 and 17.8 Å. The second trend
(see Figure 5 (j) →
(k) → (l) → (m) → (n)) reveals a shift in the
dominating anchoring positions from bridge sites toward top sites
along the Au(111) facet edges. For the corresponding Stip–tip interval in Figure 2(b), the  reveals a falling trend to the global minimum
at Stip–tip (21.7 Å), where
the dominating anchoring positions are centered around the top sites.
These trends coincide with the earlier observation that the metallic
state at the bridge site couples more strongly to the molecular transport
channel than that at the top site.
reveals a falling trend to the global minimum
at Stip–tip (21.7 Å), where
the dominating anchoring positions are centered around the top sites.
These trends coincide with the earlier observation that the metallic
state at the bridge site couples more strongly to the molecular transport
channel than that at the top site.
In the third interval for Stip–tip > 21.7 Å, the molecule
predominantely anchors at the tip regions
of both electrodes (see Figure 5(n)–(o)), adopts planar geometry (see Figure 3(b)) and optimal anchoring
angles (see Figure 3(c)). This leads to the enhancement in the electronic coupling, which
is observed as a surge in  (see Figure 2(b)).
(see Figure 2(b)).
In summary, the trend observed in the stretch
evolution of Γmean originates from the complex interplay
of the anchoring
sulfur atoms sliding between bridge and top sites along the Au(111)
facet edges, the molecule ideally adopting a planar geometry and the
optimal anchoring angle at the contact. At small Stip–tip, the anchoring positions with the strongest
electronic coupling are centered around the bridge sites along the
Au(111) facet edges. At large Stip–tip, the molecule anchors to the apex region of the tips, where it experiences
less steric constraints and can more readily assume an optimal geometry
and anchoring angle. This is reflected in the increase of 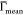 before break-off. In the intermediate interval,
mixed tip-edge configurations become dominant, which exhibit lower
values for the overall transmission than the symmetric edge–edge
and tip–tip configurations. Peaks and minima alternate in this
region as the molecule slides over bridge and top positions along
the edge.
before break-off. In the intermediate interval,
mixed tip-edge configurations become dominant, which exhibit lower
values for the overall transmission than the symmetric edge–edge
and tip–tip configurations. Peaks and minima alternate in this
region as the molecule slides over bridge and top positions along
the edge.
To conclude, we present a combined theoretical and
experimental
approach to describe the microstates of junction geometries in individual
bridge opening curves in MCBJs. We employ a novel, high-throughput
dynamic simulation approach to model the theoretically determined
stretch evolution of the electronic coupling Γ. We perform transport
calculations using the self-consistent density-functional-based tight-binding
approach and the Green’s function formalism for not only just
one representative but many thermodynamically relevant configurations.
We average the obtained single-level model parameters (ϵ0 and Γ) using statistical weights obtained from random
walk Metropolis simulations. The behavior of the theoretically determined
stretch evolution of the electronic coupling ( ) reflects the experimentally measured stretch
evolution of the electronic coupling (ΓE) well. We
associate the recurring maxima of Γ with an overall reduction
of curvature of the molecular backbone, increased anchoring angle,
and symmetric, preferentially edge–edge or tip–tip configurations.
) reflects the experimentally measured stretch
evolution of the electronic coupling (ΓE) well. We
associate the recurring maxima of Γ with an overall reduction
of curvature of the molecular backbone, increased anchoring angle,
and symmetric, preferentially edge–edge or tip–tip configurations.
Our theoretical approach in combination with MCBJ experiments elucidates the dependency of the electronic coupling of thiol anchoring groups on the distance of the gold contacts, which has so far not been reported in the literature. We correlate strong electronic coupling at short and large Stip–tip and weak electronic coupling in the intermediate region to geometrical descriptors: mean curvature, mean anchoring positions of the sulfur atoms on the Au(111) facets, and mean anchoring angle. The comparison of our theoretical analysis with the experimental measurements suggests that a strong electronic coupling is obtained for Stip–tip, where symmetrical contact geometries are statistically more common. The peaks in the intermediate region in the evolution of Γ arise from the contribution of statistically dominant, energetically less favorable molecule-tip-Au(111) structures, which are asymmetric due to steric boundary conditions. Thus, with our novel approach, we establish a link between the evolution of Γ of stretching curves in MCBJ measurements and the microscopic structure of a single molecule anchored between gold electrodes.
Acknowledgments
The authors acknowledge computing time provided through HZDR and the Center for Information Services and HPC (ZIH) at TU Dresden on HRSK-II. L. acknowledges the German Federal Ministry of Education and Research (BMBF, 01/S18026A-F) for funding the competence center for Big Data and AI “ScaDS.AI Dresden/Leipzig” and financial support from the Initiative and Networking Funds of the Helmholtz Association (HGF) through Nanonet Research School Funding (VH-KO-606), the HGF-W2/W3 Programme (W2/W3-026) and the HGF-Excellence Network DCM-MatDNA (ExNet-0028-Phase2-3). F.K. and A.S. acknowledge financial support from the Initiative and Networking Funds of the Helmholtz Association (HGF) through Nanonet Research School Funding (VH-KO-606). F.G. acknowledges the Fundaça õ de Amparo a’Pesquisa do Estado de São Paulo FAPESP for financial support through the project number 2018/15670-5. S.G. acknowledges support by the Deutsche Forschungsgemeinschaft for funding the Central Facility for Materials, Architectures and Integration of Nanomembranes (MAIN) via project INST 270/290-1 FUGB.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.3c00370.
The authors declare no competing financial interest.
Supplementary Material
References
- Cuevas J. C.; Scheer E.. Molecular electronics: an introduction to theory and experiment; World Scientific Series in Nanoscience and Nanotechnology; 1; World Scientific, New Jersey, 2010. [Google Scholar]
- Ratner M. A brief history of molecular electronics. Nat. Nanotechnol. 2013, 8, 378–381. 10.1038/nnano.2013.110. [DOI] [PubMed] [Google Scholar]
- Lörtscher E. Wiring molecules into circuits. Nat. Nanotechnol. 2013, 8, 381–384. 10.1038/nnano.2013.105. [DOI] [PubMed] [Google Scholar]
- Perrin M. L.; Burzurí E.; van der Zant H. S. J. Single-molecule transistors. Chem. Soc. Rev. 2015, 44, 902–919. 10.1039/C4CS00231H. [DOI] [PubMed] [Google Scholar]
- van der Molen S. J.; Liljeroth P. Charge transport through molecular switches. J. Phys.: Condens. Matter 2010, 22, 133001. 10.1088/0953-8984/22/13/133001. [DOI] [PubMed] [Google Scholar]
- Trasobares J.; Vuillaume D.; Théron D.; Clément N. A 17 GHz molecular rectifier. Nat. Commun. 2016, 7, 12850. 10.1038/ncomms12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin M. L.; Doelman M.; Eelkema R.; van der Zant H. S. J. Design of an efficient coherent multi-site single-molecule rectifier. Phys. Chem. Chem. Phys. 2017, 19, 29187–29194. 10.1039/C7CP04456A. [DOI] [PubMed] [Google Scholar]
- Gerhard L.; Edelmann K.; Homberg J.; Valášek M.; Bahoosh S. G.; Lukas M.; Pauly F.; Mayor M.; Wulfhekel W. An electrically actuated molecular toggle switch. Nat. Commun. 2017, 8, 14672. 10.1038/ncomms14672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; et al. Room-temperature logic-in-memory operations in single-metallofullerene devices. Nat. Mater. 2022, 21, 917–923. 10.1038/s41563-022-01309-y. [DOI] [PubMed] [Google Scholar]
- Xu B.; Xiao X.; Tao N. J. Measurements of Single-Molecule Electromechanical Properties. J. Am. Chem. Soc. 2003, 125, 16164–16165. 10.1021/ja038949j. [DOI] [PubMed] [Google Scholar]
- Rascón-Ramos H.; Artés J. M.; Li Y.; Hihath J. Binding Configurations and Intramolecular Strain in Single-Molecule Devices. Nat. Mater. 2015, 14, 517–522. 10.1038/nmat4216. [DOI] [PubMed] [Google Scholar]
- Wold D. J.; Frisbie C. D. Fabrication and Characterization of Metal-Molecule-Metal Junctions by Conducting Probe Atomic Force Microscopy. J. Am. Chem. Soc. 2001, 123, 5549–5556. 10.1021/ja0101532. [DOI] [PubMed] [Google Scholar]
- Morita T.; Lindsay S. Determination of Single Molecule Conductances of Alkanedithiols by Conducting-Atomic Force Microscopy with Large Gold Nanoparticles. J. Am. Chem. Soc. 2007, 129, 7262–7263. 10.1021/ja072040+. [DOI] [PubMed] [Google Scholar]
- Fang S.; Hu Y. H. Open the door to the atomic world by single-molecule atomic force microscopy. Matter 2021, 4, 1189–1223. 10.1016/j.matt.2021.01.013. [DOI] [Google Scholar]
- Van Ruitenbeek J.; Alvarez A.; Pineyro I.; Grahmann C.; Joyez P.; Devoret M.; Esteve D.; Urbina C. Adjustable nanofabricated atomic size contacts. Rev. Sci. Instrum. 1996, 67, 108–111. 10.1063/1.1146558. [DOI] [Google Scholar]
- Kilibarda F.; Strobel A.; Sendler T.; Wieser M.; Mortensen M.; Trads J. B.; Helm M.; Kerbusch J.; Scheer E.; Gemming S.; Gothelf K. V.; Erbe A. Single-Molecule Doping: Conductance Changed By Transition Metal Centers in Salen Molecules. Adv. Electron. Mater. 2021, 7, 2100252. 10.1002/aelm.202100252. [DOI] [Google Scholar]
- Gehring P.; Thijssen J. M.; van der Zant H. S. J. Single-molecule quantum-transport phenomena in break junctions. Nature Reviews Physics 2019, 1, 381–396. 10.1038/s42254-019-0055-1. [DOI] [Google Scholar]
- Scheer E.; Agraït N.; Cuevas J. C.; Yeyati A. L.; Ludoph B.; Martín-Rodero A.; Bollinger G. R.; van Ruitenbeek J. M.; Urbina C. The signature of chemical valence in the electrical conduction through a single-atom contact. Nature 1998, 394, 154–157. 10.1038/28112. [DOI] [Google Scholar]
- Böhler T.; Grebing J.; Mayer-Gindner A.; v Löhneysen H.; Scheer E. Mechanically controllable break-junctions for use as electrodes for molecular electronics. Nanotechnology 2004, 15, S465–S471. 10.1088/0957-4484/15/7/054. [DOI] [Google Scholar]
- Hong W.; Valkenier H.; Mészáros G.; Manrique D. Z.; Mishchenko A.; Putz A.; García P. M.; Lambert C. J.; Hummelen J. C.; Wandlowski T. An MCBJ case study: The influence of π-conjugation on the single-molecule conductance at a solid/liquid interface. Beilstein Journal of Nanotechnology 2011, 2, 699–713. 10.3762/bjnano.2.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer J.; Wadewitz A.; Lokamani; Toher C.; Gresser R.; Leo K.; Riede M.; Moresco F.; Cuniberti G. Molecules for organic electronics studied one by one. Phys. Chem. Chem. Phys. 2011, 13, 14421–14426. 10.1039/c1cp20999j. [DOI] [PubMed] [Google Scholar]
- Lokamani M.; Kelling J.; Ohmann R.; Meyer J.; Kühne T.; Cuniberti G.; Wolf J.; Huhn T.; Zahn P.; Moresco F.; Gemming S. A combined experimental and theoretical study of 1,4-bis(phenylethynyl)-2,5-bis(ethoxy)benzene adsorption on Au(111). Surf. Sci. 2021, 712, 121877. 10.1016/j.susc.2021.121877. [DOI] [Google Scholar]
- Kaliginedi V.; V. Rudnev A.; Moreno-García P.; Baghernejad M.; Huang C.; Hong W.; Wandlowski T. Promising anchoring groups for single-molecule conductance measurements. Phys. Chem. Chem. Phys. 2014, 16, 23529–23539. 10.1039/C4CP03605K. [DOI] [PubMed] [Google Scholar]
- Frei M.; Aradhya S. V.; Hybertsen M. S.; Venkataraman L. Linker Dependent Bond Rupture Force Measurements in Single-Molecule Junctions. J. Am. Chem. Soc. 2012, 134, 4003–4006. 10.1021/ja211590d. [DOI] [PubMed] [Google Scholar]
- Xing Y.; Park T.-H.; Venkatramani R.; Keinan S.; Beratan D. N.; Therien M. J.; Borguet E. Optimizing Single-Molecule Conductivity of Conjugated Organic Oligomers with Carbodithioate Linkers. J. Am. Chem. Soc. 2010, 132, 7946–7956. 10.1021/ja909559m. [DOI] [PubMed] [Google Scholar]
- Zotti L. A.; Kirchner T.; Cuevas J.-C.; Pauly F.; Huhn T.; Scheer E.; Erbe A. Revealing the Role of Anchoring Groups in the Electrical Conduction Through Single-Molecule Junctions. Small 2010, 6, 1529–1535. 10.1002/smll.200902227. [DOI] [PubMed] [Google Scholar]
- Hong W.; Manrique D. Z.; Moreno-García P.; Gulcur M.; Mishchenko A.; Lambert C. J.; Bryce M. R.; Wandlowski T. Single molecular conductance of tolanes: experimental and theoretical study on the junction evolution dependent on the anchoring group. J. Am. Chem. Soc. 2012, 134, 2292–2304. 10.1021/ja209844r. [DOI] [PubMed] [Google Scholar]
- Frisenda R.; Tarkuç S.; Galán E.; Perrin M. L.; Eelkema R.; Grozema F. C.; van der Zant H. S. J. Electrical properties and mechanical stability of anchoring groups for single-molecule electronics. Beilstein Journal of Nanotechnology 2015, 6, 1558–1567. 10.3762/bjnano.6.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishchenko A.; Vonlanthen D.; Meded V.; Bürkle M.; Li C.; Pobelov I. V.; Bagrets A.; Viljas J. K.; Pauly F.; Evers F.; Mayor M.; Wandlowski T. Influence of Conformation on Conductance of Biphenyl-Dithiol Single-Molecule Contacts. Nano Lett. 2010, 10, 156–163. 10.1021/nl903084b. [DOI] [PubMed] [Google Scholar]
- Haiss W.; Martín S.; Leary E.; Zalinge H. v.; Higgins S. J.; Bouffier L.; Nichols R. J. Impact of Junction Formation Method and Surface Roughness on Single Molecule Conductance. J. Phys. Chem. C 2009, 113, 5823–5833. 10.1021/jp811142d. [DOI] [Google Scholar]
- Lokamani; Kelling J.; Ohmann R.; Meyer J.; Kühne T.; Cuniberti G.; Wolf J.; Juckeland G.; Huhn T.; Zahn P.; Moresco F.; Gemming S. Describing chain-like assembly of ethoxygroup-functionalized organic molecules on Au(111) using high-throughput simulations. Sci. Rep. 2021, 11, 14649. 10.1038/s41598-021-93724-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haiss W.; Wang C.; Jitchati R.; Grace I.; Martín S.; Batsanov A. S.; Higgins S. J.; Bryce M. R.; Lambert C. J.; Jensen P. S.; Nichols R. J. Variable contact gap single-molecule conductance determination for a series of conjugated molecular bridges. J. Phys.: Condens. Matter 2008, 20, 374119. 10.1088/0953-8984/20/37/374119. [DOI] [PubMed] [Google Scholar]
- Lawson B.; Zahl P.; Hybertsen M. S.; Kamenetska M. Formation and Evolution of Metallocene Single-Molecule Circuits with Direct Gold-π Links. J. Am. Chem. Soc. 2022, 144, 6504–6515. 10.1021/jacs.2c01322. [DOI] [PubMed] [Google Scholar]
- Lörtscher E.; Weber H. B.; Riel H. Statistical Approach to Investigating Transport through Single Molecules. Phys. Rev. Lett. 2007, 98, 176807. 10.1103/PhysRevLett.98.176807. [DOI] [PubMed] [Google Scholar]
- Hybertsen M. S. Modeling Single Molecule Junction Mechanics as a Probe of Interface Bonding. J. Chem. Phys. 2017, 146, 092323. 10.1063/1.4975769. [DOI] [Google Scholar]
- Kim Y.; Pietsch T.; Erbe A.; Belzig W.; Scheer E. Benzenedithiol: A Broad-Range Single-Channel Molecular Conductor. Nano Lett. 2011, 11, 3734–3738. 10.1021/nl201777m. [DOI] [PubMed] [Google Scholar]
- Komoto Y.; Fujii S.; Nakamura H.; Tada T.; Nishino T.; Kiguchi M. Resolving metal-molecule interfaces at single-molecule junctions. Sci. Rep. 2016, 6, 26606. 10.1038/srep26606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaliginedi V.; Moreno-García P.; Valkenier H.; Hong W.; García-Suárez V. M.; Buiter P.; Otten J. L. H.; Hummelen J. C.; Lambert C. J.; Wandlowski T. Correlations between Molecular Structure and Single-Junction Conductance: A Case Study with Oligo(phenylene-ethynylene)-Type Wires. J. Am. Chem. Soc. 2012, 134, 5262–5275. 10.1021/ja211555x. [DOI] [PubMed] [Google Scholar]
- French W. R.; Iacovella C. R.; Cummings P. T. Large-Scale Atomistic Simulations of Environmental Effects on the Formation and Properties of Molecular Junctions. ACS Nano 2012, 6, 2779–2789. 10.1021/nn300276m. [DOI] [PubMed] [Google Scholar]
- Mejía L.; Renaud N.; Franco I. Signatures of Conformational Dynamics and Electrode-Molecule Interactions in the Conductance Profile During Pulling of Single-Molecule Junctions. J. Phys. Chem. Lett. 2018, 9, 745–750. 10.1021/acs.jpclett.7b03323. [DOI] [PubMed] [Google Scholar]
- Kamenetska M.; Koentopp M.; Whalley A. C.; Park Y. S.; Steigerwald M. L.; Nuckolls C.; Hybertsen M. S.; Venkataraman L. Formation and Evolution of Single-Molecule Junctions. Phys. Rev. Lett. 2009, 102, 126803. 10.1103/PhysRevLett.102.126803. [DOI] [PubMed] [Google Scholar]
- Schosser W. M.; Hsu C.; Zwick P.; Beltako K.; Dulić D.; Mayor M.; van der Zant H. S. J.; Pauly F. Mechanical Conductance Tunability of a Porphyrin–Cyclophane Single-Molecule Junction. Nanoscale 2022, 14, 984–992. 10.1039/D1NR06484C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M.; Gothelf K. V. Synthesis and catalytic properties of p-acylthio(phenylacetylene)n substituted chiral manganese salen complexes. J. Chem. Soc., Perkin Trans. 1 2001, 2440–2444. 10.1039/b104103g. [DOI] [Google Scholar]
- Agrait N.; Yeyati A. L.; van Ruitenbeek J. M. Quantum properties of atomic-sized conductors. Phys. Rep. 2003, 377, 81–279. 10.1016/S0370-1573(02)00633-6. [DOI] [Google Scholar]
- van Ruitenbeek J. M.; Alvarez A.; Piñeyro I.; Grahmann C.; Joyez P.; Devoret M. H.; Esteve D.; Urbina C. Adjustable nanofabricated atomic size contacts. Rev. Sci. Instrum. 1996, 67, 108–111. 10.1063/1.1146558. [DOI] [Google Scholar]
- Zhao Y.; Liu W.; Zhao J.; Wang Y.; Zheng J.; Liu J.; Hong W.; Tian Z.-Q. The fabrication, characterization and functionalization in molecular electronics. International Journal of Extreme Manufacturing 2022, 4, 022003. 10.1088/2631-7990/ac5f78. [DOI] [Google Scholar]
- van Wees B. J.; van Houten H.; Beenakker C. W. J.; Williamson J. G.; Kouwenhoven L. P.; van der Marel D.; Foxon C. T. Quantized conductance of point contacts in a two-dimensional electron gas. Phys. Rev. Lett. 1988, 60, 848–850. 10.1103/PhysRevLett.60.848. [DOI] [PubMed] [Google Scholar]
- Aradi B.; Hourahine B.; Frauenheim T. DFTB+, a Sparse Matrix-Based Implementation of the DFTB Method. J. Phys. Chem. A 2007, 111, 5678–5684. 10.1021/jp070186p. [DOI] [PubMed] [Google Scholar]
- Fihey A.; Hettich C.; Touzeau J.; Maurel F.; Perrier A.; Köhler C.; Aradi B.; Frauenheim T. SCC-DFTB parameters for simulating hybrid gold-thiolates compounds. J. Comput. Chem. 2015, 36, 2075–2087. 10.1002/jcc.24046. [DOI] [PubMed] [Google Scholar]
- Richard R. M.; Herbert J. M. Time-Dependent Density-Functional Description of the 1La State in Polycyclic Aromatic Hydrocarbons: Charge-Transfer Character in Disguise?. J. Chem. Theory Comput. 2011, 7, 1296–1306. 10.1021/ct100607w. [DOI] [PubMed] [Google Scholar]
- Elstner M.; Porezag D.; Jungnickel G.; Elsner J.; Haugk M.; Frauenheim T.; Suhai S.; Seifert G. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 1998, 58, 7260–7268. 10.1103/PhysRevB.58.7260. [DOI] [Google Scholar]
- Niehaus T.; Elstner M.; Frauenheim T.; Suhai S. Application of an approximate density-functional method to sulfur containing compounds. Journal of Molecular Structure: THEOCHEM 2001, 541, 185–194. 10.1016/S0166-1280(00)00762-4. [DOI] [Google Scholar]
- Rappe A. K.; Casewit C. J.; Colwell K. S.; Goddard W. A.; Skiff W. M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. 10.1021/ja00051a040. [DOI] [Google Scholar]
- Monkhorst H. J.; Pack J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. 10.1103/PhysRevB.13.5188. [DOI] [Google Scholar]
- Martin P. C.; Schwinger J. Theory of Many-Particle Systems. I. Phys. Rev. 1959, 115, 1342–1373. 10.1103/PhysRev.115.1342. [DOI] [Google Scholar]
- Danielewicz P. Quantum theory of nonequilibrium processes, I. Annals of Physics 1984, 152, 239–304. 10.1016/0003-4916(84)90092-7. [DOI] [Google Scholar]
- Mahan G. Quantum transport equation for electric and magnetic fields. Phys. Rep. 1987, 145, 251–318. 10.1016/0370-1573(87)90004-4. [DOI] [Google Scholar]
- Stefani D.; Weiland K. J.; Skripnik M.; Hsu C.; Perrin M. L.; Mayor M.; Pauly F.; van der Zant H. S. J. Large Conductance Variations in a Mechanosensitive Single-Molecule Junction. Nano Lett. 2018, 18, 5981–5988. 10.1021/acs.nanolett.8b02810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metropolis N.; Rosenbluth A. W.; Rosenbluth M. N.; Teller A. H.; Teller E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087–1092. 10.1063/1.1699114. [DOI] [Google Scholar]
- Van Kampen N. In Stochastic Processes in Physics and Chemistry, 3rd ed., Van Kampen N., Ed.; North-Holland Personal Library; Elsevier: Amsterdam, 2007; pp 96–133. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.