

Abstract
INTRODUCTION
Donanemab is an amyloid‐targeting therapy that specifically targets brain amyloid plaques. The objective of these analyses was to characterize the relationship of donanemab exposure with plasma biomarkers and clinical efficacy through modeling.
METHODS
Data for the analyses were from participants with Alzheimer's disease from the phase 1 and TRAILBLAZER‐ALZ studies. Indirect‐response models were used to fit plasma phosphorylated tau 217 (p‐tau217) and plasma glial fibrillated acidic protein (GFAP) data over time. Disease‐progression models were developed using pharmacokinetic/pharmacodynamic modeling.
RESULTS
The plasma p‐tau217 and plasma GFAP models adequately predicted the change over time, with donanemab resulting in decreased plasma p‐tau217 and plasma GFAP concentrations. The disease‐progression models confirmed that donanemab significantly reduced the rate of clinical decline. Simulations revealed that donanemab slowed disease progression irrespective of baseline tau positron emission tomography (PET) level within the evaluated population.
DISCUSSION
The disease‐progression models show a clear treatment effect of donanemab on clinical efficacy regardless of baseline disease severity.
Keywords: Alzheimer's disease, amyloid plaques, Clinical Dementia Rating‐Sum of Boxes, donanemab, integrated Alzheimer’s Disease Rating Scale, modeling, pharmacokinetics/pharmacodynamics, plasma glial fibrillary acidic protein, plasma phosphorylated tau 217, tau
1. INTRODUCTION
Donanemab was developed to treat Alzheimer's disease (AD) based on the amyloid cascade hypothesis. It is an antibody therapy that targets amyloid present solely in brain plaques. 1 The phase 2 clinical trial, TRAILBLAZER‐ALZ, showed that donanemab can robustly decrease amyloid plaque levels in participants with early symptomatic AD. 2 It is hypothesized that buildup of amyloid plaques facilitates an increase in (1) tau as measured by plasma phosphorylated tau 217 (p‐tau217) and neurofibrillary tangles in the brain 3 , 4 and (2) neuroinflammation as measured by plasma glial fibrillary acidic protein (GFAP) as a marker for astrocytic activation or proliferation. 5 Exploratory post hoc analyses showed that donanemab treatment significantly reduced the concentration of plasma p‐tau217 and plasma GFAP compared to placebo. 6 Amyloid plaque removal and these subsequent downstream changes that result from donanemab treatment are presumably responsible for the clinical benefits, supporting the amyloid cascade hypothesis. In the TRAILBLAZER‐ALZ trial, donanemab resulted in a 32% slowing of clinical decline on the Integrated Alzheimer's Disease Rating Scale (iADRS) after 76 weeks compared to placebo. 2
Donanemab dosing decisions for the phase 2 TRAILBLAZER‐ALZ trial and ongoing phase 3 TRAILBLAZER‐ALZ 2 trial were based on the phase 1b trial and pharmacokinetic/pharmacodynamic (PK/PD) modeling. 7 In the population PK analysis, donanemab serum concentration–time profiles were best described using a two‐compartment model with first‐order elimination. In the exposure‐response (amyloid plaque) model, the donanemab serum concentration associated with amyloid plaque reduction was found to be 4.43 μg/mL, and at least 80% of participants maintained serum concentrations above this threshold. Simulations showed that at least 75% of participants reached amyloid plaque clearance (<24.1 Centiloids) by 76 weeks of treatment. 8
Here, we further explore the PK/PD of donanemab by characterizing the relationships between (1) donanemab exposure and amyloid plaque with plasma p‐tau217 and plasma GFAP, as well as (2) donanemab exposure and clinical efficacy based on iADRS and Clinical Dementia Rating Scale‐Sum of Boxes (CDR‐SB), and (3) donanemab exposure‐amyloid plaque reduction and clinical efficacy (on iADRS).
2. METHODS
2.1. Participants and study design
The PK/PD models in these analyses are based on data from the donanemab phase 1b study (NCT02624778) and the phase 2 TRAILBLAZER‐ALZ study (NCT03367403). 2 , 7 Both were randomized, double‐blind, placebo‐controlled clinical trials. The phase 1b study enrolled participants with mild cognitive impairment due to AD or mild to moderate dementia due to AD. TRAILBLAZER‐ALZ enrolled participants with early symptomatic AD (mild cognitive impairment or mild dementia due to AD).
Key inclusion criteria include a gradual and progressive change in memory for at least 6 months, a Mini‐Mental State Examination (MMSE) score of 16–30 for the phase 1 study or 20–28 for TRAILBLAZER‐ALZ, and elevated amyloid level as measured using florbetapir F 18 scan. 2 , 7 TRAILBLAZER‐ALZ additionally required participants to meet intermediate tau positron emission tomography (PET)–based criteria (standardized uptake value ratio [SUVR] of 1.10 to 1.46; or less than 1.10 SUVR with topographic deposition pattern consistent with advanced AD). 2
Donanemab or placebo was administered via IV injections. In the phase 1 study, participants received single (10, 20, or 40 mg/kg) or multiple (10 or 20 mg/kg) doses of donanemab for up to 72 weeks. 7 In TRAILBLAZER‐ALZ, participants received donanemab every 4 weeks for up to 76 weeks (700 mg × 3 followed by 1400 mg doses), with blind dose reductions based on amyloid levels on florbetapir PET. 2
All studies were conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. All protocols were approved by the appropriate institutional review board or ethics committee.
RESEARCH IN CONTEXT
Systematic Review: Traditional sources were used to review the literature (e.g., PubMed). This article builds upon previously published donanemab studies and the Alzheimer's natural disease progression model to characterize the clinical efficacy of donanemab using pharmacokinetic/pharmacodynamic modeling.
Interpretation: The models in this article indicate that donanemab decreases levels of the plasma biomarker p‐tau217 and reduces the rate of clinical decline as measured by cognitive scales. Simulations using the models and identified covariates indicate that donanemab benefits patients, regardless of baseline tau levels.
Future Directions: These models can be used for further analyses to simulate the utility of donanemab for the treatment of Alzheimer's disease.
2.2. Consent statement
Written informed consent to participate was obtained from all participants or their legally authorized representatives or caregivers.
2.3. Biomarker analysis
Amyloid and tau PET scans, plasma p‐tau217, and plasma GFAP concentrations were collected as described previously. 6 , 9 Amyloid and tau levels were measured using florbetapir F 18 and flortaucipir F 18 PET scans, respectively. Florbetapir scans were collected at baseline and weeks 12, 24, 36, 48, and 72 in the phase 1b study or baseline and weeks 24, 52, and 76 in TRAILBLAZER‐ALZ. Flortaucipir scans were collected at baseline and at week 76 in TRAILBLAZER‐ALZ. To determine the baseline tau level, a previously published AD signature–weighted neocortical SUVR with respect to a reference signal intensity in white matter (Parametric Estimation of Reference Signal Intensity [PERSI] 10 ) was used. Plasma p‐tau217 and GFAP levels were assessed at baseline and at weeks 12, 24, 36, 52, 64, and 76 using a custom Simoa‐HD‐X assay (Quanterix) and the Simoa Neurology 4‐Plex E Advantage Kit (Quanterix), respectively. 6
2.4. Clinical assessment
In TRAILBLAZER‐ALZ, change in clinical symptoms (cognition and function) was measured using iADRS and CDR‐SB. 2 The iADRS is an integrated assessment of cognition and daily function comprising items from the Alzheimer's Disease Assessment Scale‐Cognitive subscale (ADAS‐Cog13) and the Alzheimer's Disease Cooperative Study—instrumental Activities of Daily Living (ADCS‐iADL). The total score ranges from 0 to 144, with lower scores reflecting greater impairment. 11 CDR‐SB scores range from 0 to 18, with higher scores reflecting greater disease severity. 12
2.5. Plasma p‐tau217 and GFAP model development
Individual participant–observed longitudinal donanemab exposure was used in the population PK/amyloid plaque model, 8 where it was found that when donanemab exposure is maintained above a certain threshold, it resulted in an indirect response (i.e., the effect can be time‐lagged and/or persist over time even in the absence of serum exposure) of a sustained reduction in amyloid plaque over time, which then further induced downstream reduction in plasma p‐tau217 and GFAP. Individual participant parameters from the previously reported final population PK model and the exposure‐response amyloid plaque model 8 , 9 were added to the plasma p‐tau217 and plasma GFAP data sets. Two, separate indirect‐response models were used to fit the plasma p‐tau217 and plasma GFAP data over time using mixed‐effects non‐linear regression with individual participant data from TRAILBLAZER‐ALZ. Individual participant baseline plasma p‐tau217 concentration and estimated rate of plasma p‐tau217 formation were parameters in the model. Two models were tested to predict the plasma p‐tau217 reduction: a treatment‐response model based on donanemab dosing and a model including the impact of change in amyloid levels.
A basic indirect‐response model where donanemab treatment alters the production of plasma GFAP was used to fit the plasma GFAP data over time using the FOCEI method. The model was parameterized in terms of individual participant baseline GFAP concentration and estimated rate of GFAP formation. Individual post hoc participant parameters from the final population PK and the amyloid plaque models were added to the GFAP data set to obtain predicted drug concentrations and amyloid levels for individual participants. A treatment‐effect model driven by dosing information of donanemab and the impact of change in amyloid PET (a relative change from baseline) was evaluated as a predictor, reducing the rate of GFAP formation.
Final model development included covariate selection using a stepwise forward‐inclusion, backward‐deletion process. Linear, power, and exponential covariate relationships were evaluated. The forward‐inclusion and backward‐deletion criteria were p < 0.01 and p < 0.001, respectively. Covariates tested on estimated baseline plasma p‐tau217 concentration and treatment effect parameters include entry age, entry weight, apolipoprotein E (APOE)ε4 carrier status, gender, race, treatment‐emergent anti‐drug antibody status, time since onset of AD symptoms, time since AD diagnosis, and baseline tau PET SUVR. Covariates tested on the baseline GFAP concentration and treatment‐effect parameters include entry age, gender, entry weight, estimated glomerular filtration rate (eGFR), baseline tau PET SUVR, and baseline plasma p‐tau217.
The models were evaluated using standard goodness‐of‐fit plots and visual predictive checks.
2.6. Disease progression model development
Two disease‐progression models were developed with TRAILBLZAER‐ALZ data for CDR‐SB and iADRS using an identical approach. Richard's logistic model was used to describe the non‐linear disease progression. 13 Beta regression was used to account for decreasing variance in residual error as data approached the boundaries (0–144 for iADRS and 0–18 for CDR‐SB). 13 , 14 , 15 A treatment‐effect model using the donanemab dosing information was tested. Another model including the impact of change in amyloid levels was also tested as described previously. 9
In the final model development, covariates were tested on baseline score, disease progression, and drug‐effect parameters. Covariates tested include APOE ε4 carrier status, baseline tau level, age, gender, time since onset of AD symptoms, time since AD diagnosis, and baseline C‐reactive protein level. In addition, the influence of anti‐drug antibodies (ADAs) was tested on the treatment‐effect term of the model but was not significant. Subsequently, a power calculation was conducted to explore the degree of effect that could be detected with the current data set, utilizing all available titer data, assuming the impact on treatment effect decreased with the log of the titer in a linear fashion. The parameter associated with the change in treatment effect with log(titer) was simulated as either 0.05 or 0.15. This analysis was conducted with only the iADRS model, as this was the primary endpoint for the study.
A stepwise forward‐inclusion, backward‐deletion process was used to select covariates. Linear, power, and exponential covariate relationships were evaluated. Forward‐inclusion and backward‐deletion criteria were both p < 0.01.
The model was evaluated using standard goodness‐of‐fit plots and visual predictive checks.
3. RESULTS
3.1. Participants
The data sets used in the population pharmacokinetic analyses included participants from the phase 1 or the phase 2 (TRAILBLAZER‐ALZ) clinical trials assessing donanemab. 2 , 7 Participants with mild cognitive impairment or mild to moderate dementia due to AD were included in these trials. Summaries of participant baseline characteristics for each model are presented in Table 1.
TABLE 1.
Baseline participant characteristics.
| Characteristic | PK | Plasma biomarkers | iADRS and CDR‐SB |
|---|---|---|---|
| Treatment, n (%) | |||
| Donanemab | 177 | 111 (50.5) | 131 (51.2) |
| Placebo | 0 | 109 (49.5) | 125 (48.8) |
| Mean age, years (range) | 74.4 (54, 88) | 75.2 (61, 86) | 75.1 (61, 86) |
| Mean weight, kg (range) | 73.2 (40.3, 123) | 73.9 (42.7, 123.1) | 73.6 (42.7, 123.1) |
| Mean body mass index, kg/m2 (range) | 26.3 (15.9, 44.2) | 26.4 (15.9, 44.2) | 26.4 (15.9, 44.2) |
| Gender, n (%) | |||
| Men | 86 (48.6) | 109 (49.5) | 124 (48.4) |
| Women | 91 (51.4) | 111 (50.5) | 132 (51.6) |
| Race, n (%) | |||
| White | 153 (86.4) | 206 (93.6) | 242 (94.5) |
| Black | 7 (4.0) | 8 (3.6) | 8 (3.1) |
| Asian | 14 (7.9) | 3 (1.4) | 3 (1.2) |
| American Indian or Alaska Native | 2 (1.1) | 2 (0.9) | 2 (0.8) |
| Other | 1 (0.6) | 1 (0.5) | 1 (0.4) |
| Hispanic Origin, n (%) | 7 (4.0) | 7 (3.2) | 8 (3.1) |
| APOE ε4 Status | |||
| Carrier | 132 (74.6) | 153 (69.5) | 187 (73.0) |
| Noncarrier | 45 (25.6) | 66 (30.0) | 68 (26.6) |
| Unknown | 0 | 1 (0.5) | 1 (0.4) |
Abbreviations: APOE ε4, apolipoprotein E ε4 allele; CDR‐SB, Clinical Dementia Rating‐Sum of Boxes; iADRS, Integrated Alzheimer's Disease Rating Scale; n, number of participants; PK, pharmacokinetics.
3.2. Plasma p‐tau217 reduction model
A treatment‐effect model and a model including the change in amyloid plaque level were developed to evaluate the change in plasma p‐tau217 concentration following donanemab treatment. Data from participants in TRAILBLAZER‐ALZ were included in the data set. The 23.4‐point difference in objective function value indicated that the change in amyloid plaque concentrations better predicted the change in plasma p‐tau217 concentration compared to the treatment‐effect model. The parameters of the final model are presented in Table 2.
TABLE 2.
Parameter estimates for plasma biomarker models.
| Plasma p‐tau 217 model | Plasma GFAP model | |||
|---|---|---|---|---|
| Parameter |
Base model Population mean (95% CI) a |
Final model Population mean (95% CI) a |
Base model Population mean (95% CI) a |
Final model Population mean (95% CI) a |
| Kin (U/mL/h) |
0.000372 (0.000247, 0.000613) |
0.000355 (0.000237, 0.000584) |
0.0017 (0.00103, 0.00259) |
0.00179 (0.00099, 0.00250) |
| Baseline p‐tau217 concentration (U/mL) |
0.389 (0.376, 0.403) |
0.384 (0.372, 0.396) |
‐ | ‐ |
| Baseline GFAP concentration (U/mL) | ‐ | ‐ |
201 (188.4, 212.2) |
201 (193.3, 212.0) |
| Effect of amyloid reduction on Kin |
0.273 (0.236, 0.315) |
0.274 (0.231, 0.319) |
–1.17 (–2.113, –0.673) |
–1.17 (–2.31, –0.75) |
| Covariate effect on baseline plasma concentration | ||||
|---|---|---|---|---|
| Effect of baseline tau PET SUVR b | ‐ |
0.966 (0.671, 1.27) |
‐ | ‐ |
| Effect of baseline age c | ‐ | ‐ | ‐ |
0.0180 (0.0106, 0.0251) |
| Effect of baseline weight d | ‐ | ‐ | ‐ |
–0.50 (–0.70, –0.294) |
| Interindividual variability CV% (95% CI) e | ||||
|---|---|---|---|---|
| Baseline p‐tau217 concentration (U/mL) |
26.0% (23.2, 28.6) |
23.4% (20.6, 25.6) |
‐ | ‐ |
| Baseline GFAP concentration (pg/mL) | ‐ | ‐ |
36.9% (33.4‐40.2) |
33.7% (30.1‐38.1) |
| Treatment effect |
39.5% (23.3, 51.2) |
38.3% (21.4, 50.2) |
91.9% (44.0‐151.5) |
91.1% (33.3‐154.6) |
| Residual unexplained variability | ||||
|---|---|---|---|---|
| Proportional |
18.6% (16.8, 20.5) |
18.6% (16.9, 20.7) |
17.9 % (16.9‐19.4) |
17.9% (16.7‐19.3) |
Abbreviations: BTAUSUVR, baseline tau PET SUVR; CI, confidence interval; CV, coefficient of variation; Kin, p‐tau217 synthesis input rate; PET, positron emission tomography; SUVR, standardized uptake value ratio.
95% CI from bootstrap.
Typical logit for baseline p‐tau217 concentration = 0.384* (1 + 0.966*[BTAUSUVR‐1.2]), where BTAUSUVR is baseline tau PET SUVR (mean, CV%: 1.22, 9.68%).
Effect of age on baseline is (1+0.0183*[age‐75]).
Effect of weight on entry on baseline is (WT/72.2)–0.50.
Inter‐individual variability was calculated using the following equation for log‐normal distributions of the random effects , or normal distribution of random effects of , where OMEGAN is the variance of the parameter.
As reported previously, there was a statistically significant association between baseline tau PET SUVR and baseline plasma p‐tau217 concentration (p < 0.001). 6 Higher baseline plasma p‐tau217 concentrations were associated with higher baseline tau PET SUVR. Therefore, baseline tau PET was included as a covariate in the plasma p‐tau217 reduction model. None of the other covariates tested met the inclusion criteria.
A comparison of the observed plasma p‐tau217 concentrations with the modeled data suggests the model adequately describes the data (Figure S1). There was a small number of samples with high concentrations, which widens the 95th percentiles for both placebo and treatment groups. The treatment effect is observed in all quartiles of baseline plasma p‐tau217 (analyzed as a continuous variable), supported by the observation that the estimates of between‐participant variability between baseline plasma p‐tau217 and treatment effect are not correlated.
3.3. Plasma GFAP reduction model
An indirect‐response model was found to adequately describe the time course of plasma GFAP concentrations in placebo‐ and donanemab‐treated participants. The final model parameters are found in Table 2. A treatment‐effect model driven by donanemab‐dosing information as well as a model where donanemab treatment decreased amyloid load (a relative change from baseline) and reduced the rate of GFAP formation described the data well. Although there was no statistically significant decrease in objective function with the latter model, it was selected as final due to the known positive correlation between amyloid load and plasma GFAP. 16 Statistically significant associations between age and body weight with baseline GFAP were identified (p < 0.001). No other covariates tested met the inclusion criteria for the final model.
A visual predictive check suggests that the model adequately describes the data (Figure S1). A bootstrap analysis suggests that the parameters are well estimated (Table 2).
3.4. Disease‐progression model
Disease‐progression models were developed to describe the relationship between donanemab treatment and disease progression as measured by iADRS and CDR‐SB. The data set used for model development came from participants enrolled in TRAILBLAZER‐ALZ with intermediate tau levels. The Richard's logistic model adequately described iADRS and CDR‐SB data (Figures S2 and S3). The parameters of the final model are presented in Table 3. The shape parameter in the iADRS model could not be well estimated so it was fixed to a value of 7.25. This estimate was based on a previously developed disease‐progression model using placebo data from more than 2400 participants. 17
TABLE 3.
Parameter estimates for final disease progression models.
| iADRS | CDR‐SB | |||
|---|---|---|---|---|
| Parameter | Estimate (%SEE) | Bootstrap (95% CI) | Estimate (%SEE) | Bootstrap (95% CI) |
| Baseline score a | 108 (2.64) | (107, 110) | 3.13 (2.73) | (2.90, 3.35) |
| Disease‐progression rate (week–1) | 0.00346 (4.48) | (0.00287, 0.00399) | 0.00517 (6.44) | (0.00452, 0.00636) |
| Shape factor | 7.25 (Fixed) | ‐ | 3.51 (42.2) | (1.50, 13.3) |
| Residual error b | 144 (4.02) | (130, 162) | 65.6 (4.01) | (59.6, 74.7) |
| Reduction in rate of disease progression for APOE ε4 carriers (%) | 41.8 (17.4) | (19.5, 62.2) | 28.6 (39.9) | (9.33, 50.1) |
| Effect of age on baseline score c | 0.0255 (19.6) | (0.0156, 0.0350) | ‐ | ‐ |
| Effect of age on disease progression d | ‐ | ‐ | 0.0246 (36.1) | (0.00484, 0.0492) |
| Effect of baseline tau on baseline score c | 1.17 (19.9) | (0.716, 1.63) | ‐ | ‐ |
| Effect of baseline tau on disease progression d | ‐ | ‐ | 1.33 (32.2) | (0.377, 2.44) |
| Effect of time from diagnosis of Alzheimer's disease on baseline score e | ‐ | ‐ | 0.112 (29.1) | (0.0634, 0.176) |
| Population variability in the baseline score (%CV) | 44.3 (9.22) | (39.0, 49.0) | 61.7 (9.55) | (56.1, 67.7) |
Abbreviations: AADIAG, time from diagnosis of Alzheimer's disease; APOE ε4, apolipoprotein E ε4 allele; BTau, baseline tau; CDR‐SB, Clinical Dementia Rating‐Sum of Boxes; CI, confidence interval; CV, coefficient of variance; iADRS, Integrated Alzheimer's Disease Rating Scale; SEE, standard error of the estimate.
Estimate as back‐transformed from the logit scale.
Tau parameter for beta distribution.
Typical logit for baseline iADRS = –1.06 + 1.17*(BTau–1.2) + 0.0255*(Age–75.5).
Typical disease progression for CDR‐SB = 0.00517* (1 + 1.33 *[BTau–1.2]) *(1 + 0.0246*[Age–75.5]).
Typical logit for baseline CDR‐SB = –1.56 + 0.112*(AADIAG–0.53).
Several covariates impact baseline disease status and disease progression. Regardless of whether the iADRS or CDR‐SB scale were used, APOE ε4 carrier status was identified as a significant covariate on disease‐progression rate. Covariates identified on the baseline score were age and baseline tau for the iADRS scale and the effect of time from diagnosis on the CDR‐SB. Covariates identified in disease‐progression rate were age and baseline tau SUVR on CDR‐SB. Men had significantly higher baseline iADRS scores. Although the effect was statistically significant, it was not clinically relevant, with a small difference in gender covariance (men vs women). Therefore, it was not included in the final model. There was no significant effect between gender and baseline CDR‐SB scores or disease‐progression rates. In the power calculation conducted to determine the degree of ADA effect, at the 0.05 change in treatment effect, the power to detect the change was 19% at α = 0.05 and 10% at α = 0.01. At the 0.15 change in treatment effect, the power to detect the change was 92% at α = 0.05 and 81% at α = 0.01. For a titer value of 20, this results in a 15% decrease in treatment effect using the 0.05 parameter, and 45% decrease at the 0.15 parameter. Thus the model can rule out a substantial effect of ADA on slowing of disease progression associated with donanemab treatment, but a more subtle effect cannot be excluded. However, the fact that ADA was significant on PK, 8 but that PK was not significant on iADRS given the high dose level, suggests that ADA is unlikely to have a clinically meaningful impact on the treatment effect of donanemab.
In these models, donanemab treatment had a significant effect on iADRS (p < 0.001) and CDR‐SB (p < 0.05) endpoints. Covariate analyses for the treatment‐effect model of donanemab on disease progression showed that there was a significant treatment effect modeled for iADRS (p < 0.001, 42% reduction in disease progression rate) and CDR‐SB (p < 0.01, 28% reduction in disease progression rate) in APOE ε4 carriers. In the data set used to develop this model, 187 participants (73%) were APOE ε4 carriers. Within the APOE ε4 carrier group, 53 participants (28%) were homozygous carriers and 134 participants (72%) were heterozygous carriers and there was no statistically significant difference in treatment effect between these groups.
Instead of a treatment‐effect model driven by the administration of donanemab doses, a disease‐progression model where the impact of change in amyloid plaque (absolute change, as well as percent change from baseline) was evaluated as a predictor of disease progression on iADRS. 9 Parameter estimates of this model are presented in Table S1. Percent change from baseline amyloid level was a significant predictor of disease progression in iADRS. The inclusion of percent change in amyloid in addition to a treatment effect did not further improve model fit. Therefore, slowing of disease progression as a result of donanemab administration could be described by either a simple treatment effect or via change in amyloid level (which is driven by donanemab exposure).
3.5. Simulations
Change in plasma biomarker concentration over time with placebo or donanemab treatment was simulated using the plasma biomarker models based on the change in amyloid (Figure 1). The simulations indicate that plasma GFAP concentrations continue to increase over time with placebo. However, with donanemab treatment, both plasma p‐tau217 and plasma GFAP concentrations decrease.
FIGURE 1.
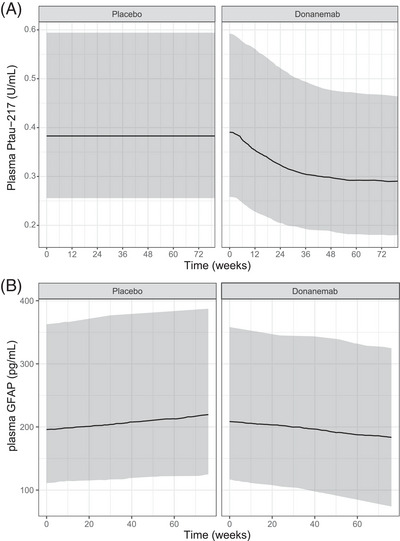
Model‐predicted change in plasma biomarker concentration. Change from baseline simulated for plasma p‐tau217 (A) and plasma GFAP (B) levels with placebo and donanemab treatment. The shaded areas represent a 95% prediction interval. The lines represent the median prediction. Participants were simulated to follow the dosing treatment regimen as described in the Methods section. Actual participant titer time courses, weight, age, and baseline tau PET SUVR were sampled from the NONMEM dataset. GFAP, glial fibrillary acidic protein; NONMEM, nonlinear mixed effects modeling; PET, positron emission tomography; p–tau217, phosphorylated tau 217; SUVR, standarized uptake value ratio.
Simulations in APOE ε4 carriers were conducted using the disease‐progression model based on treatment effect to determine the impact of baseline tau SUVR on disease progression on iADRS and CDR‐SB (Figure 2). The baseline tau range used in this simulation was based on participants within the intermediate tau range included in TRAILBLAZER‐ALZ (0.99 to 1.46 SUVR). The simulation indicates that donanemab slowed disease progression regardless of tau level at baseline. However, participants with higher baseline tau had worse iADRS and CDR‐SB scores at baseline and faster disease progression.
FIGURE 2.
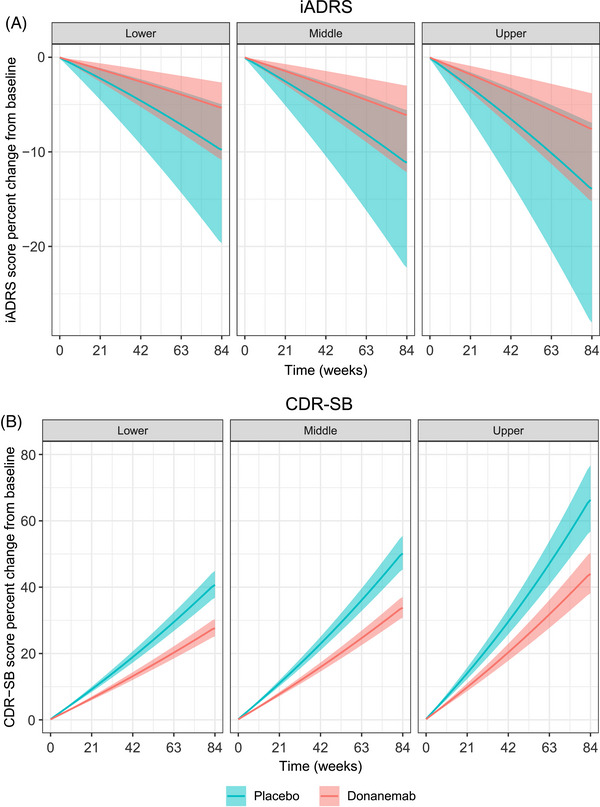
Model‐predicted impact of baseline tau on disease progression in APOE ε4 carriers within the intermediate tau range. Percent change from baseline in iADRS (A) or CDR‐SB (B) score based on tertiles of baseline tau (within the intermediate tau range [0.99–1.46]) simulated using the disease‐progression model based on treatment effect. The shaded areas represent a 95% prediction interval. The lines represent the median prediction. APOE ε4, apolipoprotein E ε4 allele ; CDR‐SB, Clinical Dementia Rating‐Sum of Boxes; iADRS, Integrated Alzheimer's Disease Rating Scale.
Simulations were conducted using the disease‐progression model based on change in amyloid to examine the rate of disease progression in APOE ε4 carriers upon cessation of donanemab (Figure 3). Simulations of the percent change from baseline in iADRS and CDR‐SB scores show that slowing of disease‐progression rate compared to placebo is maintained after stopping donanemab treatment (Figure 3A,B). Using the disease‐progression model, simulations were carried out on the absolute iADRS scores for placebo and 76‐week donanemab treatment (Figure 3C), which show that slowing of the disease progression rate with donanemab treatment results in increasing difference compared to placebo over time (simulation for 264 weeks) (Figure 3D).
FIGURE 3.
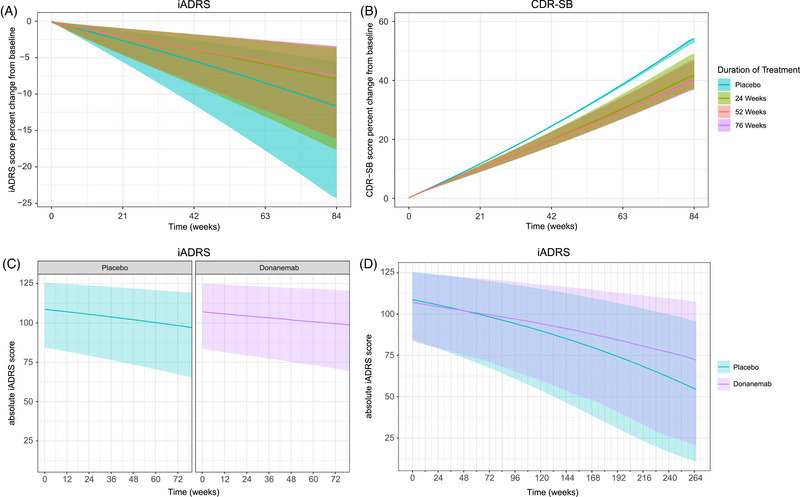
Model‐predicted impact of duration of treatment on disease progression using amyloid level as a predictor of disease progression in APOE ε4 carriers. (A,B) Percent change from baseline in iADRS (A) or CDR‐SB (B) score based on duration of treatment using the disease‐progression model based on amyloid level. (C,D) Absolute iADRS score based on duration of treatment (76 weeks) (C) and simulation to 264 weeks using the disease‐progression model based on amyloid level (D). The y‐axis is plotted across a different range in panels A and B. The shaded areas represent a 95% prediction interval. The lines represent the median prediction. APOE ε4, apolipoprotein E ε4 allele; CDR‐SB, Clinical Dementia Rating‐Sum of Boxes; iADRS, Integrated Alzheimer's Disease Rating Scale.
4. DISCUSSION
The models used in these post hoc exploratory analyses of phase 2 data were built upon the framework of the previously described population PK and donanemab exposure‐response (amyloid reduction model) models. 8 , 9 Here, we have shown how modeling can be used to describe the relationship between donanemab exposure, AD biomarkers, and clinical scales measuring cognition and function.
Plasma p‐tau217 is elevated in patients with AD. 18 We reported previously that a decrease in plasma p‐tau217 was observed following donanemab treatment in TRAILBLAZER‐ALZ. 6 Here, two models were evaluated to examine the relationship between donanemab treatment and plasma p‐tau217 concentration. Between a model based on a simple treatment effect and one based on change in amyloid plaque levels, the latter better predicted the change in plasma p‐tau217. The simple treatment‐effect model, although describing the data, did not offer understanding on the mechanism behind the observed reduction in plasma p‐tau217. Using the indirect‐response model, amyloid plaque reduction, which can be time‐lagged and/or persist over time, affects the synthesis rate of plasma p‐tau217. This supports the hypothesis that plasma p‐tau217 reduction is driven by the reduction in amyloid plaque level. This finding is consistent with donanemab's known mechanism of action, suggesting that donanemab‐induced amyloid plaque reduction results in further downstream changes and disease modification. There were no covariates identified that influenced the relationship between amyloid PET and plasma p‐tau217 over time. However, a statistically significant relationship was identified between baseline plasma p‐tau217 and baseline tau PET SUVR (based on PERSI reference region), with higher plasma p‐tau217 concentrations associated with higher values of tau SUVR, as published previously. 6 These results indicate that plasma p‐tau217 concentrations are higher in more advanced disease states, suggesting that the decrease in plasma p‐tau217 following donanemab treatment may represent slowing of disease progression.
Plasma GFAP is associated with AD pathology and has been shown to predict the progression of cognitive decline. 5 , 19 , 20 , 21 It has also been reported that plasma GFAP levels decrease as eGFR levels and body weight increase, whereas plasma GFAP levels increase with age. 22 Here we show that an indirect‐response model, where donanemab treatment decreased amyloid load (a relative change from baseline) and reduced the rate of GFAP formation, described the data well. A statistically significant relationship was identified between baseline plasma GFAP and age and weight at entry, with higher plasma GFAP concentrations associated with older age and lower weight. On forward search there was also a significant relationship between baseline plasma GFAP and eGFR and baseline p‐tau217, but these were not retained in the final model due to these covariates not meeting the pre‐specified criteria for reducing the between‐participant estimate of baseline plasma GFAP. The baseline disease state was not evaluated as a covariate and is a limitation of this model, as plasma GFAP levels are altered early in the disease process. 23 These results indicate that baseline plasma GFAP concentrations are higher in older participants and are lower in participants with higher weight. Results suggest that the decrease in amyloid plaque load following donanemab treatment leads to a reduction in plasma GFAP.
In the TRAILBLAZER‐ALZ trial, change in iADRS was the primary outcome measure and change in CDR‐SB was a secondary clinical outcome. 2 The iADRS and CDR‐SB disease‐progression modeling approaches tested here compared a model based solely on treatment effect with another previously reported disease‐progression model based on change in amyloid plaque level. 9 Both models indicate that donanemab slows the rate of disease progression, and a simulation up to 264 weeks predicts that donanemab‐treated participants will decline more slowly than placebo‐treated participants, with an increasing difference from placebo after completion of 76 weeks of treatment. Using the iADRS disease‐progression model based on amyloid plaque reduction, we reported previously that simulating the maximum percent decrease in amyloid plaque level would result in a significant reduction in the disease‐progression rate. 9 Here we further use this model to show that slowing of the disease‐progression rate compared to placebo is maintained after stopping donanemab treatment. This is possibly due to the slow re‐accumulation of amyloid once it has been removed by donanemab. 9 The findings from these disease‐progression models support the amyloid cascade hypothesis. If the buildup of amyloid plaques in the brain initiates a series of downstream changes that result in worsening cognition, then removal of amyloid plaques should result in slowing of disease progression, as predicted here.
None of the investigated covariates, including APOE ε4 carrier status, were identified as significant factors in the amyloid PET model, 8 where the individual‐participant predictions of amyloid PET baseline and overall treatment effect were similar between APOE ε4 carriers and non‐carriers. In the plasma p‐tau217 model reported here, APOE ε4 carrier status was also not identified as a significant factor on baseline and treatment‐effect parameters for the plasma p‐tau217 model. This led to the hypotheses that the lack of clinical effect for APOE ε4 noncarriers observed may be attributable to several factors, such as pharmacogenomic differences in treatment response or the relatively few noncarriers (68 noncarriers) in TRAILBLAZER‐ALZ. In fact, in the larger phase 3 lecanemab trial, the amyloid‐targeting therapy was reported to have a greater clinical benefit to APOE ε4 noncarriers. 24 This finding will be evaluated further with data from TRAILBLAZER‐ALZ 2, where the number of noncarriers is expected to be larger compared with TRAILBLAZER‐ALZ study.
There were limitations to the development of these models. First, these results are from post hoc exploratory analyses 6 , 9 and from a comparatively small number of participants; therefore, results will need to be confirmed with data from the larger phase 3 trial. In additionally, the disease‐progression models were built only with data from the TRAILBLAZER‐ALZ study, which used a single dosing regimen. This narrow range of exposures may have prevented characterization of an informative exposure–response relationship. The model could not identify a treatment effect in participants who were not APOE ε4 carriers. APOE ε4 carriers comprised ≈73% of the participant population. The inability to detect a treatment effect in APOE ε4 noncarriers may reflect the difficulty of finding a modest effect in this relatively small population. Further evaluation of the effect of donanemab in a larger population of APOE ε4 noncarriers is needed. Furthermore, baseline tau levels and change in tau as assessed with tau PET was tested as a covariate in these models and used for stimulations. The tau‐inclusion criteria for TRAILBLAZER‐ALZ focuses on the participants with an intermediate tau level, thereby limiting the dynamic range of the data set.
The findings from the disease‐progression models presented here show a clear effect of donanemab on clinical efficacy. Simulations indicate that donanemab slows disease progression regardless of baseline tau and that slowing of disease progression is maintained after participants discontinue donanemab treatment.
CONFLICT OF INTEREST STATEMENT
Ivelina Gueorguieva, Laiyi Chua, Kay Chow, C. Steven Ernest, Jian Wang, Sergey Shcherbinin, John R. Sims, and Emmanuel Chigutsa are all full‐time employees and minor shareholders of Eli Lilly and Company. Brian A. Willis is a former full‐time employee and minor shareholder of Eli Lilly and Company. Author disclosures are available in the supporting information.
CONSENT STATEMENT
Written informed consent to participate was obtained from all participants or their legally authorized representatives or caregivers.
Supporting information
Figure S1. Visual predictive check of final plasma biomarker models. Visual predictive check of final plasma phosphorylated tau 217 (P‐tau217) model for placebo (A) and donanemab (B) and of the final plasma glial fibrillary acidic protein (GFAP) model for placebo (C) and donanemab (D). The points are the observed data. The lines are the 5th, 50th, and 95th percentiles of the observed data. The shaded areas are the model‐predicted 95% confidence interval of the corresponding percentiles.
Figure S2. Goodness of fit for iADRS disease‐progression model. LOWESS fit, a smoothed value given by a weighted linear least‐squares regression over the span of observations, for data are presented (black line) in addition to a line of identity (gray line on top panel). Abbreviations: iADRS, Integrated Alzheimer's Disease Rating Scale; LOWESS, locally weighted scatterplot smoothing.
Figure S3. Goodness of fit for CDR‐SB disease progression model. LOWESS fit, a smoothed value given by a weighted linear least‐squares regression over the span of observations, for data are presented (black line) in addition to a line of identity (gray line on top panel). Abbreviations: CDR‐SB, Clinical Dementia Rating‐Sum of Boxes; LOWESS, locally weighted scatterplot smoothing.
Table S1. Parameter estimates for the model describing impact of exposure and amyloid plaque reduction on iADRS disease progression.
Icmje Disclosure Forms
ACKNOWLEDGMENTS
The authors thank the participants, caregivers, and investigators. Staci E. Engle, PhD, Eli Lilly and Company, provided writing and editorial assistance. The data from this study were presented in part at the International Congress on Alzheimer's and Parkinson's Diseases (AD/PD) in Barcelona, Spain (March 15‐20, 2022) and at the Alzheimer's Association International Conference in San Diego, CA, USA and Online (July 31–August 4, 2022). This study was sponsored/funded/supported by Eli Lilly and Company.
Gueorguieva I, Willis BA, Chua L, et al. Donanemab exposure and efficacy relationship using modeling in Alzheimer's disease. Alzheimer's Dement. 2023;9:e12404. 10.1002/trc2.12404
REFERENCES
- 1. Demattos RB, Lu J, Tang Y, et al. A plaque‐specific antibody clears existing β‐amyloid plaques in Alzheimer's disease mice. Neuron. 2012;76(5):908‐920. [DOI] [PubMed] [Google Scholar]
- 2. Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer's disease. N Engl J Med. 2021;384(18):1691‐1704. [DOI] [PubMed] [Google Scholar]
- 3. Wennström M, Janelidze S, Nilsson KPR, et al. Cellular localization of p‐tau217 in brain and its association with p‐tau217 plasma levels. Acta Neuropathol Commun. 2022;10(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nature Rev Drug Discov. 2011;10(9):698‐712. [DOI] [PubMed] [Google Scholar]
- 5. Benedet AL, Milà‐Alomà M, Vrillon A, et al. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. 2021;78(12):1471‐1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pontecorvo MJ, Lu M, Burnham SC, et al. Association of Donanemab treatment with exploratory plasma biomarkers in early symptomatic alzheimer disease: a secondary analysis of the TRAILBLAZER‐ALZ randomized clinical trial. JAMA Neurol. 2022;79(12):1250‐1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lowe SL, Duggan Evans C, Shcherbinin S, et al. Donanemab (LY3002813) phase 1b study in Alzheimer's disease: rapid and sustained reduction of brain amyloid measured by florbetapir F18 imaging. J Prev Alzheimers Dis. 2021;8(4):414‐424. [DOI] [PubMed] [Google Scholar]
- 8. Gueorguieva I, Willis BA, Chua L, et al. Donanemab population pharmacokinetics, amyloid plaque reduction, and safety in participants with Alzheimer's disease. Clin Pharmacol and Ther. 2023;113(6):1258‐1267. [DOI] [PubMed] [Google Scholar]
- 9. Shcherbinin S, Evans CD, Lu M, et al. Association of amyloid reduction after Donanemab treatment with tau pathology and clinical outcomes: the TRAILBLAZER‐ALZ randomized clinical trial. JAMA Neurol. 2022;79(10):1015‐1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Devous MD, Sr. , Joshi AD, Navitsky M, et al. Test‐retest reproducibility for the tau PET imaging agent flortaucipir F 18. J Nucl Med. 2018;59(6):937‐943. [DOI] [PubMed] [Google Scholar]
- 11. Wessels AM, Siemers ER, Yu P, et al. A combined measure of cognition and function for clinical trials: the integrated Alzheimer's disease rating scale (iADRS). J Prev Alzheimers Dis. 2015;2(4):227‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morris JC. The clinical dementia rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412‐2414. [DOI] [PubMed] [Google Scholar]
- 13. Gueorguieva I, Chua L, Willis BA, Sims JR, Wessels AM. Disease progression model using the integrated Alzheimer's Disease Rating Scale. Alzheimer's Dement. 2023;19(6):2253‐2264. [DOI] [PubMed] [Google Scholar]
- 14. Xu XS, Samtani MN, Dunne A, Nandy P, Vermeulen A, De Ridder F. Mixed‐effects beta regression for modeling continuous bounded outcome scores using NONMEM when data are not on the boundaries. J Pharmacokinet Pharmacodyn. 2013;40(4):537‐544. [DOI] [PubMed] [Google Scholar]
- 15. Conrado DJ, Denney WS, Chen D, Ito K. An updated Alzheimer's disease progression model: incorporating non‐linearity, beta regression, and a third‐level random effect in NONMEM. J Pharmacokinet Pharmacodyn. 2014;41(6):581‐598. [DOI] [PubMed] [Google Scholar]
- 16. Pereira JB, Janelidze S, Smith R, et al. Plasma GFAP is an early marker of amyloid‐β but not tau pathology in Alzheimer's disease. Brain. 2021;144(11):3505‐3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Willis B, Wessels A, Chua L, et al. Natural disease progression model of Alzheimer's disease using the integrated Alzheimer's disease rating scale. J Prev Alzheimers Dis. 2021;8(S1):S77‐S78. [Google Scholar]
- 18. Janelidze S, Berron D, Smith R, et al. Associations of plasma phospho‐Tau217 levels with tau positron emission tomography in early Alzheimer disease. JAMA Neurol. 2021;78(2):149‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jack CR, Wiste HJ, Algeciras‐Schimnich A, et al. Predicting amyloid PET and tau PET stages with plasma biomarkers. Brain. 2023;146(5):2029‐2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cicognola C, Janelidze S, Hertze J, et al. Plasma glial fibrillary acidic protein detects Alzheimer pathology and predicts future conversion to Alzheimer dementia in patients with mild cognitive impairment. Alzheimer's Res Ther. 2021;13(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Salvadó G, Ossenkoppele R, Ashton NJ, et al. Specific associations between plasma biomarkers and postmortem amyloid plaque and tau tangle loads. EMBO Mol Med. 2023;15(5):e17123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rebelos E, Rissanen E, Bucci M, et al. Circulating neurofilament is linked with morbid obesity, renal function, and brain density. Sci Rep. 2022;12(1):7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chatterjee P, Pedrini S, Stoops E, et al. Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer's disease. Transl Psychiatry. 2021;11(1):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer's disease. N Engl J Med. 2022;388(1):9‐21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Visual predictive check of final plasma biomarker models. Visual predictive check of final plasma phosphorylated tau 217 (P‐tau217) model for placebo (A) and donanemab (B) and of the final plasma glial fibrillary acidic protein (GFAP) model for placebo (C) and donanemab (D). The points are the observed data. The lines are the 5th, 50th, and 95th percentiles of the observed data. The shaded areas are the model‐predicted 95% confidence interval of the corresponding percentiles.
Figure S2. Goodness of fit for iADRS disease‐progression model. LOWESS fit, a smoothed value given by a weighted linear least‐squares regression over the span of observations, for data are presented (black line) in addition to a line of identity (gray line on top panel). Abbreviations: iADRS, Integrated Alzheimer's Disease Rating Scale; LOWESS, locally weighted scatterplot smoothing.
Figure S3. Goodness of fit for CDR‐SB disease progression model. LOWESS fit, a smoothed value given by a weighted linear least‐squares regression over the span of observations, for data are presented (black line) in addition to a line of identity (gray line on top panel). Abbreviations: CDR‐SB, Clinical Dementia Rating‐Sum of Boxes; LOWESS, locally weighted scatterplot smoothing.
Table S1. Parameter estimates for the model describing impact of exposure and amyloid plaque reduction on iADRS disease progression.
Icmje Disclosure Forms