

ABSTRACT
Wide-spread cancer-related immunosuppression often curtails immune-mediated antitumoral responses. Immune-checkpoint inhibitors (ICIs) have become a state-of-the-art treatment modality for mismatch repair-deficient (dMMR) tumors. Still, the impact of ICI-treatment on bone marrow perturbations is largely unknown. Using anti-PD1 and anti-LAG-3 ICI treatments, we here investigated the effect of bone marrow hematopoiesis in tumor-bearing Msh2loxP/loxP;TgTg(Vil1-cre) mice. The OS under anti-PD1 antibody treatment was 7.0 weeks (vs. 3.3 weeks and 5.0 weeks, control and isotype, respectively). In the anti-LAG-3 antibody group, OS was 13.3 weeks and thus even longer than in the anti-PD1 group (p = 0.13). Both ICIs induced a stable disease and reduced circulating and splenic regulatory T cells. In the bone marrow, a perturbed hematopoiesis was identified in tumor-bearing control mice, which was partially rescued by ICI treatment. In particular, B cell precursors and innate lymphoid progenitors were significantly increased upon anti-LAG-3 therapy to levels seen in tumor-free control mice. Additional normalizing effects of ICI treatment were observed for lin−c-Kit+IRF8+ hematopoietic stem cells, which function as a “master” negative regulator of the formation of polymorphonuclear-myeloid-derived suppressor cell generation. Accompanying immunofluorescence on the TME revealed significantly reduced numbers of CD206+F4/80+ and CD163+ tumor-associated M2 macrophages and CD11b+Gr1+ myeloid-derived suppressor cells especially upon anti-LAG-3 treatment. This study confirms the perturbed hematopoiesis in solid cancer. Anti-LAG-3 treatment partially restores normal hematopoiesis. The interference of anti-LAG-3 with suppressor cell populations in otherwise inaccessible niches renders this ICI very promising for subsequent clinical application.
KEYWORDS: Hematopoietic progenitor cell, hypermutated tumor, immunotherapy, tumor microenvironment
GRAPHICAL ABSTRACT
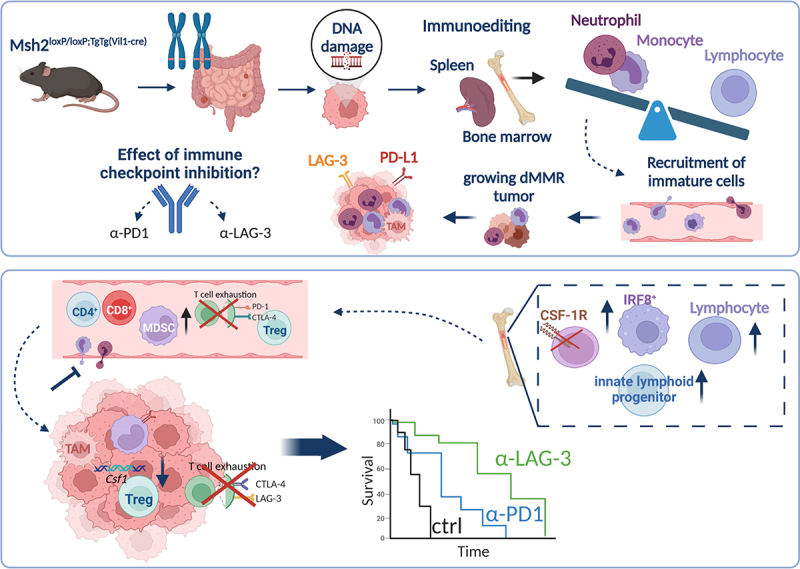
Msh2loxP/loxP;TgTg(Vil1-cre) mice develop tumors in the gastrointestinal tract because of a constitutional Msh2 knock out. Tumor growth is accompanied by global immunological changes in the peripheral blood, along with perturbations in the bone marrow and eventually spleen. These perturbations include a shifted hematopoiesis toward and unbalanced myelopoiesis. These immature cells are recruited to the tumor and polarized toward an immunosuppressive M2-like phenotype. This finally leads to tumor progression and global immunosuppression or exhaustion. Immune-checkpoint inhibitors may have the capacity to break this vicious circle via re-activation of the host’s immunity. Here, we could show that immune-checkpoint inhibition with anti-PD1 or anti-LAG-3 partially rebalances bone marrow perturbations via targeting of CSF-1R on hematopoietic progenitor cells. This suppressed recruitment of myeloid cells to the tumor and thus reshaped the tumor microenvironment. Finally, the outcome of tumor-bearing mice was significantly improved after short-term immune-checkpoint blockade.
Background
Mismatch-repair-deficient (dMMR) tumors are highly immunogenic and predestined to be treated with immunotherapy, such as immune checkpoint inhibitors (ICI)1,2. In dMMR, neoadjuvant and adjuvant ICIs settings are clinically effective and sufficiently safe3,4. Consequently, the anti-PD1 antibody Pembrolizumab received approval as first-line therapy for metastatic dMMR/high-grade microsatellite instability colorectal cancer5. DMMR has become a biomarker for tumor-agnostic anticancer therapy approval6,7. Adding to this, we previously reported prolonged overall survival of dMMR tumor-bearing mice under ICI therapy8,9. Despite the prevailing success of ICIs in treating dMMR-related tumors, developing resistance, mainly attributable to the widespread immunosuppression, is clinically challenging. Finding combination partners for ICIs is another task. Reports for beneficial effects of combined Cyclin-dependent kinase (CDK)/immune-checkpoint inhibition10,11 could only partially withstand clinical testing12,13. Using two different mouse models of spontaneous dMMR tumorigenesis, we additionally reported abrogation of the beneficial immune-modulatory effects of the CDK4/6 inhibitor abemaciclib upon the combination with an anti-PD-L1 antibody. Mechanistically, this was due to the activation of epithelial–mesenchymal transition14.
Hence, targeting alternative immune checkpoints constitutes a novel treatment approach. An increasingly recognized target is the lymphocyte-activation gene 3 (LAG-3). LAG-3 is expressed on activated immune cells15. It blocks cellular proliferation and is involved in dendritic cell signaling16. In addition, LAG-3 is responsible for CD8+ T cell homeostasis. In tumor cells, binding of LAG-3 to MHC II works as a tumor escape mechanism by calming T and NK cells due to its structural homology to CD4 that LAG-3 outcompetes for binding to MHC II17. A current study described reduced anti-apoptotic signaling in tumor cells and restored T and NK cell responses upon inhibition of LAG-318. The safety of anti-LAG-3 antibody treatment either alone or in combination with anti-PD1 was additionally proven in phase I/II studies on patients with advanced/metastatic solid tumors19,20. Still, the activity against dMMR gastrointestinal tumors is unknown.
The tumor microenvironment (TME) significantly affects the response to ICI treatment. In solid tumors, the TME consists of highly immunosuppressive myeloid cells, i.e. tumor-associated macrophages (TAM), myeloid-derived suppressor cells (MDSC), and tumor-associated neutrophils (TANs)21–23. These usually immature, bone marrow-derived cells are heterogeneous and have high plasticity, i.e. their activation, differentiation, and maturation status varies within tumors reviewed in 24,25. Recent elegant work even identified erythroid precursor-differentiated myeloid cells as causes of reduced ICI treatment efficacy26. Hence, the bone marrow is an important reservoir to maintain supply with immunosuppressive precursors into the tumor. A better understanding of the tumor-driven hematopoietic changes and the impact of ICI treatment on bone marrow may help improve diagnosis and ultimately treatment.
In this preclinical study, we examined the efficacy of anti-LAG-3 monotherapy compared to the approved anti-PD-1 antibody in a model of spontaneous dMMR-driven tumorigenesis. Additionally to the clinical outcome, we studied the bone marrow compartment in tumor-bearing mice to decipher factors that might contribute to the dysregulation of cancer-driven hematopoiesis. We found that Msh2loxP/loxP;TgTg(Vil1-cre) mice with clinically proven gastrointestinal tumors experienced prolonged overall survival upon LAG-3 blockade, which was even better than under anti-PD1 treatment. Notably, therapeutic effects were partially traceable in the bone marrow, characterized by a trend toward rebalanced hematopoiesis, including reduced the numbers of CSF-1 R+ precursors and mature monocytic cells. Therefore, we propose anti-LAG-3 as a good alternative for treating dMMR-related tumors.
Methods
In vivo experiments
Ethical statement
The German local authority approved all animal experiments: Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischerei Mecklenburg‐Vorpommern (7221.3‐1‐062/19), under the German animal protection law and the EU Guideline 2010/63/EU. Mice were bred in the animal facility of the University Medical Center in Rostock under specific pathogen‐free conditions. Msh2 genotyping was done according to27,28. During their whole life-time, all animals received enrichment in the form of mouse-igloos (ANT Tierhaltungsbedarf, Buxtehude, Germany), nesting material (shredded tissue paper, Verbandmittel GmbH, Frankenberg, Deutschland), paper roles (75 × 38 mm, H 0528–151, ssniff‐Spezialdiäten GmbH), and wooden sticks (40 × 16 × 10 mm, Abedd, Vienna, Austria). During the experiment, mice were kept in type III cages (Zoonlab GmbH, Castrop‐Rauxel, Germany) at 12‐h dark:light cycle, the temperature of 21 ± 2°C, and relative humidity of 60 ± 20% with food (pellets, 10 mm, ssniff‐Spezialdiäten GmbH, Soest, Germany) and tap water ad libitum. When mice were subjected to treatment (= time of tumor development or prophylactic interventions), they were given daily-prepared soaked pellets to ensure proper food intake.
Experimental protocol
Mice with suspected gastrointestinal tumors were subjected to 18F-FDG PET/CT screening and then treated with the following therapies (Figure 1a): (I) -PD-1 (2.5 mg/kg bw, i.p., q2Wx3, n = 9 mice) or (II) -LAG-3 antibody (2.5 mg/kg bw, i.p., q2Wx3 n = 8 mice). Control mice were given the isotype (2.5 mg/kg bw, q2Wx3, i.p., n = 9 mice) or were left untreated (n = 10 mice). The health status of the mice was monitored daily. Mice were euthanized according to predefined human endpoints (weight loss > 15%, modified social behavior). Blood, spleen, tumors, and bone marrow were removed for further analysis.
Figure 1.
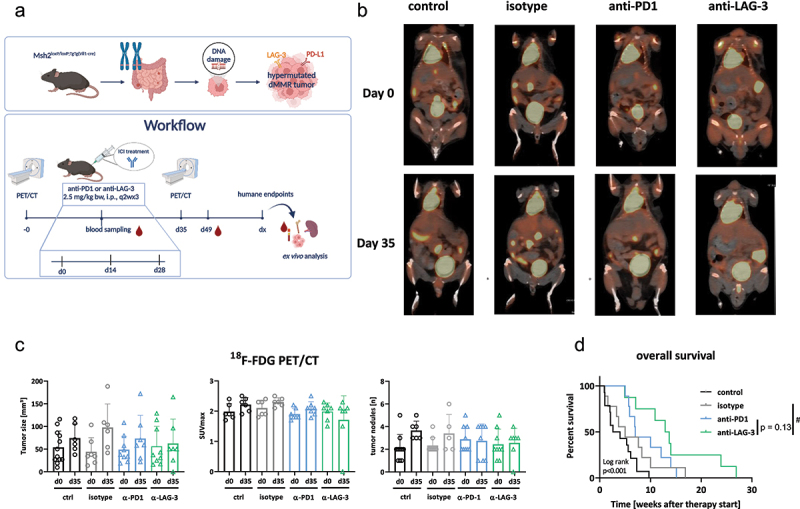
Workflow, 18F-FDG PET/CT imaging in vivo, and overall survival of Msh2loxP/loxP;TgTg(Vil1-cre) mice treated with ICIs.
Notes: (a) Preclinical treatment schedule. (b, c) Tumor volume and SUVmax were measured via18F-FDG PET/CT imaging at start of therapy and after five weeks. (b) representative PET/CT images showing the impact of ICIs on tumor growth. (c) Longitudinal assessment of tumor volumes. Quantification was done at start of treatment and at day 35. Mean + SD. Each dot is representative for one mouse/timepoint. (d) Kaplan–Meier curve representing overall survival of mice. Control n = 10; isotype n = 10; -PD1n = 9; -LAG-3 n = 8, # p < 0.05 vs. isotype; Log-rank analysis (Mantel–Cox test).
PET/CT imaging
A small animal PET/CT scanner (Inveon PET/CT, Siemens Medical Solutions, Knoxville, TN, USA) was used to measure the tumor size following a standard protocol as described29. Mice were anesthetized and intravenously injected with 18F-FDG. Images were evaluated as described8.
Immune phenotyping
Blood was taken routinely from the retrobulbar venous plexus of anesthetized mice. At the endpoint, spleen and tumor were removed and homogenized. A panel of monoclonal antibodies was used to stain certain extra- and intracellular biomarkers14. First, staining with the Zombie NIR™ Fixable Viability Kit (30 min, RT, 1:5000, Biolegend, San Diego, United States) was performed following the manufacturers’ protocol. Afterward, extracellular markers were stained using the BD Horizon Brilliant Stain Buffer (BD Bioscience, Heidelberg, Germany). Subsequently, fixation and permeabilization were conducted and intracellular markers were stained employing the protocol of True-Nuclear™ Transcription Factor Buffer Set by Biolegend. For extracellular stainings, the following monoclonal anti-mouse antibodies were used (20 min, RT, doses were titrated before): Alexa Fluor™700 Gr-1 (clone RB6-8C5), FITC CD8 (clone 53–6.7), APC/Fire™750 CD4 (clone GK1.5), BV570 CD11b (clone M1/70), BV421 PD-L1 (clone 10F.9G2), BV605 NK1.1 (clone PK136), Spark Blue™ 550 CD19 (clone 6D5), (all from BioLegend), PerCP-eFluor™ 710 CD25 (clone PC61.5) (Thermofisher, Waltham, Massachusetts, USA), BV750 CD83 (clone Michel-19), BV650 PD-1 (clone RMP1–30, BD Bioscience). Intracellular staining was done with the following monoclonal anti-mouse antibodies: PE/Cy7 CTLA-4 (clone UC10-4B9), PerCP CD3 (clone 145-2C11), and Alexa Fluor™ 647 Foxp3 (clone MF-14), (BioLegend). Flow cytometry measurement was executed using a spectral flow cytometer (Cytek™ Aurora) with 100,000 recorded events/sample. Analysis was done using SpectroFlow™ Version 2.2.0.3. and FlowJo™ Version 10.6.1. For data analysis, specific T cell subpopulations were characterized, i.e. naïve T cells (CD3+CD4+CD8− CCR7+CD62L+CD44−), effector memory CD3+CD4+ or CD3+CD8+ T cells (TEM, CCR7lowCD62LlowCD44+), and central memory CD3+CD4+ or CD3+CD8+ T cells (TCM, CCR7+CD62L+CD44−).
Bone marrow isolation and further processing
The bone marrow was obtained from the femur and tibia of both hind legs, followed by extensive washing in PBS. Resulting cell suspensions were counted and parts were taken for pappenheim staining. Therefore, 100,000 cells were centrifuged on a slide at 700 rpm for 10 min via cytospin, air-dried, fixed, and stained for 6 min with May-Grünwald solution (MERCK, Darmstadt, Germany). Afterward, the slides were washed three times with buffer at pH 7.2 (MERCK) followed by staining with 10% Giemsa (MERCK) (in buffer at pH 7.2) for 20 min and subsequent washing steps. Air-dried slides were analyzed under the microscope. From each slide, 200 cells were counted and differentiated into the following cell types: myeloblasts, neutrophil pro-/myelocytes, metamyelocytes/neutrophils, eosinophilic pro-/myelocytes, metamyelocytes/eosinophils, lymphocytes, plasma cells, monoblasts, and monocytes. Quantification was done blinded by an experienced researcher.
Remaining bone marrow cells were conducted to flow cytometry following staining with the appropriate monoclonal fluorochrome-labeled antibodies. Firstly, staining with the Zombie NIR™ Fixable Viability Kit (Biolegend) was done, followed by blocking of Fc receptors using the TruStain FcX™ PLUS (anti-mouse CD16/32) incubated for 10 min on ice) and True-Stain Monocyte Blocker™ (Biolegend). Extracellular staining was done using the following monoclonal anti-mouse antibodies (20 min, RT, doses were titrated before): Pacific Blue™ Lineage Cocktail, BV421 CD34, BV510™ CD127 (IL-7 Rα), BV605™ CD90.2 (Thy-1.2), BV711™ Ly-6A/E (Sca-1), BV785™ CD117 (c-Kit), PE CD123, PE/-Dazzle™ 594 CD71, PE/Fire™ 640 CD38, PE/Cyanine5 CD150 (SLAM), PE/Cy7 CD41, PE/Fire™ 810 CD279, Alexa Fluor® 647 CD115 (CSF-1 R), APC/Cy7 CD63, APC/Fire™ 810 CX3Cr1, Vio® Bright B515 REAfinity™ CD48 (Miltenyi Biotec, Bergisch Gladbach, Germany), Alexa Fluor™ 700 CD170 (Siglec F). Intracellular staining was done after fixation and permeabilization (True Nuclear Transcription Factor Buffer Set, BioLegend) using the following monoclonal antibodies (45 min, RT): APC MCP-1 (BioLegend), FITC CXCL12 (Thermo Fisher Scientific, Waltham, Massachusetts, United States), and PerCP-eFluor™ 710 IRF8 (ebioscience, Thermo Fisher Scientific). Flow cytometry measurement was executed using a spectral flow cytometer (Cytek™ Aurora) with 100,000 events/sample. Analysis was done using SpectroFlow™ Version 2.2.0.3. and FlowJo™ Version 10.6.1. Specific subpopulations were defined as outlined in the results part.
Additionally, a classical colony formation was performed with bone marrow cells for isotype-controlled and ICI-treated mice. Therefore, 100.000 cells were seeded in 25 cm2 flasks and cultured for 14 days in defined medium. Thereafter, colonies were stained with crystal violet (0.2%) and colonies were counted manually.
RNA isolation, cDNA synthesis & quantitative real-time PCR of spleen cells and GIT
RNA isolation, cDNA synthesis, and real-time PCR were done as described14. Reactions were performed in triplicate wells. The expression level of each sample was considered by calculating 2−ΔCT (ΔCt = Cttarget – CtHousekeeping gene), followed by 2−ΔΔCT quantification, taking values of controls as calibrator.
Immunofluorescence
Cryostat sections of 4 µm were processed as described9,14. Staining was done using the extracellular surface marker via Alexa Fluor 488, Alexa Fluor 594, and Alexa Fluor 647 labeled antibodies CD206, F4/80, CD11b, Gr1, CD163, and PD-L1 (Biolegend). After staining, sections were washed with PBS to remove leftover antibodies and were embedded in Roti Mount Fluor Care DAPI (Roth, Karlsruhe). Visualization was performed on a confocal laser scanning microscope (ZEISS Elyra 7 Confocal Laser Microscope, Zeiss, Jena, Germany). Regulatory granulocytes and tumor-associated macrophages (TAM) were semi-quantitatively analyzed using a validated scoring system: 0 = no; 1 = mild (1–20 cells/HPF); 2 = moderate (21–40 cells/HPF); 3 = strong (>40 cells/HPF)14.
Functional assays evaluating immunosuppression mediated by myeloid-derived suppressor cells
Splenocytes were first stained with predefined amounts of Alexa Fluor® 488 CD11b (clone M1/70) and Alexa Fluor® 700 Ly-6 G/Ly-6C (Gr-1) antibodies (30 min, RT). Prior sorting of the positive fraction, splenocytes were stained with DAPI for dead cell exclusion. The top CD11b/Gr1-positive cells were bulk sorted on a BD FACSAria™IIIu. Sorting was performed using a 100 µm nozzle, with max. 2000 evts/sec and an efficiency of > 90%. Data acquisition was done with the BD FACSDiva™-software v9.4. Thereafter, sorted cells (1 × 105 cells/ml) were incubated with LPS from E. coli (100 ng/ml) overnight. ROS production from total splenocytes and sorted MDSCs was determined using ROS Brite™ 670 (100 µM, AAT Bioquest). Fluorescence was measured on a preheated Tecan plate reader (ROS Brite™ 670 Ex/Em = 640/680 nm).
Statistics
Statistical analysis was performed using GraphPad PRISM software, version 8.0.2 (GraphPad Software, San Diego, CA, USA). The value of significance was set to p < 0.05. First normality was tested via the Shapiro–Wilk test. In case of normality, one-way ANOVA (Tukey´s or Sidak’s multiple comparison) was performed, and in the case of non-parametric data, Kruskal-Wallis was executed. Kaplan–Meier survival curves were analyzed using log rank test (Mantel cox). Outliers of flow cytometry data were eliminated when data was not in the range of average plus/minus two times the standard deviation. * day 0 vs. experimental endpoint; # - ICI treatment vs. isotype; $ - anti-PD1 vs. anti-LAG-3; § - tumor-free vs. early vs. late-stage (refers to bone marrow analysis only).
Results
ICI treatment prolongs the overall survival of tumor-bearing mice
Longitudinal quantification of tumor size showed a slight increase with anti-PD1 therapy (Figure 1b, c). In contrast, anti-LAG-3 treatment induced stable disease or partial remission within the observation period. Notably, the standardized uptake value (SUV)max was decreased only in the anti-LAG-3 treatment group, while an increase was observed in the anti-PD1 and control tumors (Figure 1c, middle). Regarding the number of tumor nodules, no progression was seen under either ICI treatment, while new tumor lesions were seen in control mice (Figure 1c, right). The therapeutic effects of both ICIs resulted in a significantly prolonged overall survival (OS, Figure 1d). The OS under anti-PD1 antibody treatment was 7.0 weeks (vs. 3.3 weeks and 5.0 weeks, control and isotype, respectively, Figure 1d). In the anti-LAG-3 antibody group, OS was 13.3 weeks and thus longer than under anti-PD1 treatment (p = 0.13).
ICI treatment induces phenotypic changes in the blood
Focusing on the major cell populations in the peripheral blood, we observed increased numbers of T helper and cytotoxic T cells under anti-PD1 treatment and slightly decreased numbers under anti-LAG-3 treatment (Figure 2a,b and supplementary Figure S1A). Both ICIs led to an increase in myeloid-derived suppressor cells (MDSC), which was, however, similar to controls. NK cells remained virtually unchanged upon treatment (Figure 2b). Looking at exhausted T cells, i.e. PD1+, CTLA-4+, LAG-3+, and TIM-3+ cells, the effects of the two ICIs were different (Figure 2c). Anti-PD1 treatment reduced the number of LAG-3+ T cells. In contrast, the impact of the anti-LAG-3 antibody on exhaustion markers was more global, resulting in a decrease in all T cell populations studied (Figure 2c). Both ICIs reduced the number of circulating regulatory T cells.
Figure 2.
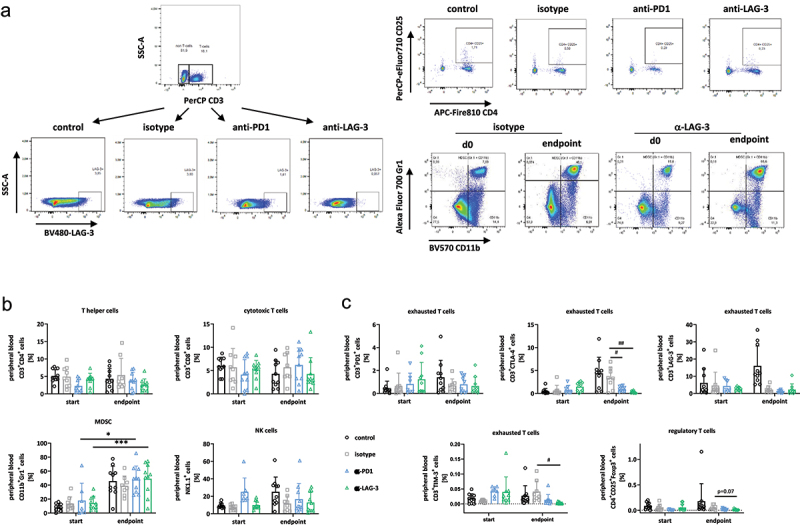
Spectral flow cytometry of the peripheral blood.
Notes: Blood was taken routinely from the retrobulbar venous plexus. (a) Gating strategy for identifying immune cell subtypes in peripheral blood using multi-color spectral flow cytometry. (b, c) Given is the number of % immune cells before treatment (= day 0) and at the experimental endpoint resulting from 100,000 events measured on a flow cytometer. Each symbol represents one case. Control n = 10; isotype n = 10; -PD1n = 9; -LAG-3 n = 8. * p < 0.05 and *** p < 0.001 vs. start (= day 0); # p < 0.05 and ## p < 0.01 vs. isotype. Two-way ANOVA (Tukey’s multiple comparisons test). (b) different immune cell populations; (c) T cell exhaustion marker (PD1, CTLA-4, LAG-3, and TIM-3) and regulatory T cells.
In addition, the immunomodulatory effect of ICI treatment was evaluated on day 49, shortly after the end of treatment (supplementary Figure S1A). The exhaustion markers PD1, LAG-3, and TIM-3 were lower in the ICI-treatment groups compared to the isotype control (where only 4–6 values could be included). Notably, Tregs showed an inverse trend between anti-PD1 and anti-LAG-3 treated mice. LAG-3 led to a continuous reduction, while in anti-PD-1 treated mice, this effect was only seen at the experimental endpoint (supplementary Figure S1A).
Altered immunophenotype and hematopoiesis in the spleen
During tumorigenesis, the spleen becomes an important source for myeloid cells, and tumors frequently reprogram splenic hematopoiesis to a process called extramedullary hematopoiesis. Macroscopically evident is a splenomegaly, as seen here especially in control mice (approximately a twofold increase in size and splenocyte number). Hence, we analyzed the splenic immune subsets in detail (Figure 3a). The number of MDSCs significantly increased in both treatment groups. Likewise, NK cells were higher under therapy, particularly upon PD1 blockade. T helper and cytotoxic T cell level remained unchanged and were comparable to controls. Still, regulatory T cells and the level of exhaustion were lower upon both ICI treatments. Amounts of CD3+CD4+CD25+Foxp3+ cells decreased, and CD3+CTLA-4+ T cells were even significantly lower. CD3+PD1+ and CD3+LAG-3+ T cells showed a comparable trend.
Figure 3.
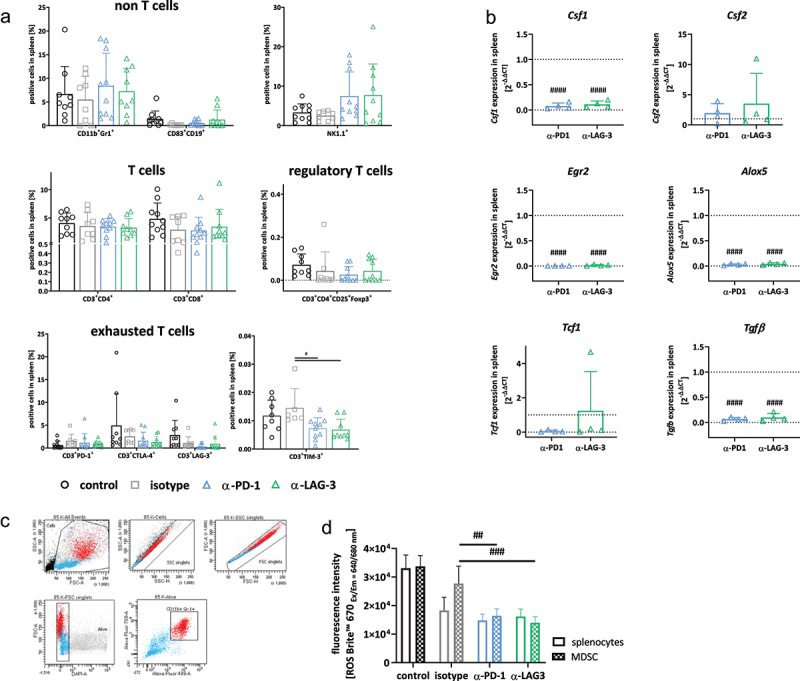
Spectral flow cytometry and gene expression analysis of spleens.
Notes: (a) Given is the number of % immune cells at the experimental endpoint resulting from 100,000 events measured on a flow cytometer. Each symbol represents one case. Control n = 10; isotype n = 8–9; -PD1n = 8–10; -LAG-3 n = 8–10; Two-way ANOVA (Tukey’s multiple comparisons test). (b) Quantitative qPCR of myeloid markers in the spleen. Total RNA from spleens was reverse transcribed into cDNA and qPCR was done as described in material and methods. Analysis was done in triplicates with n = 3–4 mice/group, respectively. All data are given as 2-ΔΔCT values + SD. Each symbol represents one case. #### p < 0.0001 vs. isotype. Two-way ANOVA (Tukey’s multiple comparisons test). (c, d) Sorting approach (CD11b+Gr1+ fraction) and determination of reactive oxygen species for functional myeloid-derived suppressor cell function analysis. (c) Whole spleen cells were labeled with anti-CD11b and anti-Gr1 antibodies and double positive cells were sorted. (d) Thereafter, cells were stimulated overnight with lipopolysaccharide from E. coli (100 ng/ml). ROS production from unsorted and CD11b+Gr1+ sorted splenic MDSCs was determined using ROS NIR™ 670. Fluorescence was measured on a preheated Tecan plate reader. Given is the fluorescence intensity (ROS determination). Mean + SD, n = 3–4 samples/group. # p < 0.05 and ## p < 0.01 vs. isotype; Two-way ANOVA (Tukey’s multiple comparisons test).
To study the effects on myelopoiesis in more detail, we performed gene expression analysis and focused on specific markers involved in macrophage (Csf1/Csf2) and neutrophil polarization (Egr2, Alox5, Figure 3b). Colony stimulating factor 1 (Csf1), a marker for M2 macrophages, was significantly downregulated in the spleens of ICI-treated mice. By contrast, Csf2, which indicates an M1-like phenotype was higher upon anti-LAG-3 treatment compared to controls and anti-PD1 therapy (Figure 3b). Notably, the early growth response gene 2 (Egr2) and Alox5 were significantly lower in the spleens of both treatment groups compared to controls (p < 0.0001). Vice versa, T-cell factor 1 (Tcf1), a transcription factor involved in T-cell lineage specification and thymocyte differentiation30, was slightly elevated upon anti-LAG-3 treatment. The expression of transforming growth factor beta (Tgf) was significantly lower upon ICI treatment. While the latter is a well-established trigger for immunosuppression, we interpret this finding as – at least partial – reconstitution of physiological immunity. To investigate this further, the function of total splenocytes and CD11b+Gr1+-sorted MDSCs (Figure 3c) was analyzed upon stimulation with LPS. We determined ROS production after overnight culture (Figure 3d). Given the fact that sorting may have activated MDSCs in the spleen, a comparative approach was performed with total splenocytes and sorted MDSCs. This experiment nicely confirmed the reduced ROS production of MDSCs from ICI-treated mice (Figure 3d). Notably, both splenocytes and positively sorted CD11b+Gr1+ MDSCs from -LAG-3 treatment showed the lowest level of ROS production. Hence, a reversal of the immunosuppressive function of MDSC by ICI treatment can be expected.
Changes in the tumor microenvironment upon ICI treatment
Spectral flow cytometry was done on residual tumors (Figure 4a). Non-T cells and T cells were only marginally affected by either treatment. Specifically, MDSCs, CD83+CD19+ B cells, and NK1.1.+ NK cells did not change significantly upon PD1 or LAG-3 blockade. Regulatory T cells were lower in ICI-treated tumors. Because of the variation within individual groups, this did not reach statistical significance. Exhausted T cells were significantly reduced upon anti-PD1 and/or anti-LAG-3 application. We found treatment-specific influences on specific exhaustion markers, i.e. PD1+ T cells were massively reduced upon anti-LAG-3, while anti-PD1 significantly suppressed CTLA-4+ T cells. LAG-3+ T cells were lower in both therapy groups.
Figure 4.
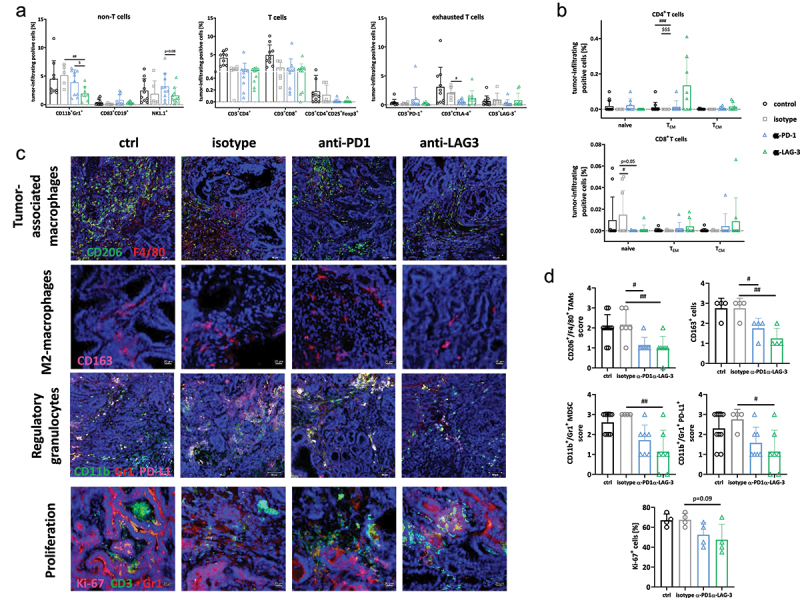
Characterization of the TME using spectral flow cytometry and immunofluorescence.
Notes: (a, b) Spectral flow cytometry. A panel of antibodies was used to examine the infiltration pattern within residual tumors. Given is the number of % immune cells at the experimental endpoint resulting from 100,000 events measured on a flow cytometer. Each symbol represents one case. Control n = 10; isotype n = 6–9; -PD1n = 6; -LAG-3 n = 6. # p < 0.05, ## p < 0.01 and ### p< 0.001 vs. isotype; $$$ p < 0.001 vs. ICI; Two-way ANOVA (Tukey’s multiple comparisons test). (c, d) The infiltration pattern of regulatory and tumor-associated (M2) macrophages, as well as Ki-67 proliferating cells was examined in residual tumor slides. Confocal laser scanning microscopy was done on a Zeiss Elyra 7 microscope. (c) Representative images of tumor slides. (d) Quantitative analysis of tumor-infiltrating immune cells counted in 2–3 HPFs/slide. Each symbol represents one case. Control n = 7–9; isotype n = 4–6; -PD1n = 4–7; -LAG-3 n = 4–7. # p < 0.05 and ## p < 0.01 vs. isotype; One-way ANOVA (Tukey’s multiple comparisons test).
Looking at naive, effector (TEM) and central memory (TCM) T cells, additional differences between the two treatment groups were observed (Figure 4b). The number of CD4+ TEM was significantly higher in the anti-LAG-3 treatment group. In contrast, the number of CD8+ naive T cells decreased significantly (Figure 4b).
Accompanying immunofluorescence deciphered the spatial localization of immune subsets within tumors (Figure 4c,d). We focused on TAMs (including M2-subtype), MDSCs, and regulatory granulocytes as major immunosuppressive cell types. Notably, CD206+F4/80+ TAMs were significantly decreased under ICI treatment (Figure 4c,d). In particular, the CD163+ M2-subtype was significantly reduced. Residual tumors from anti-LAG-3 treated mice tended to harbor fewer tumor-infiltrating M2 macrophages than those following anti-PD1 treatment. A comparable reduction in CD11b+Gr1+ MDSCs and CD11b+Gr1+PD-L1+ was observed, particularly in anti-LAG-3 treated tumors. The Ki-67 proliferation index was then determined. This was done to determine whether the treatment also affects tumor cell proliferation and whether infiltrating cells actually proliferate within tumors (Figure 4c,d). Ki-67 staining identified fewer proliferating tumor cells after ICI treatment. Conversely, some infiltrating T cells were positive for Ki-67, but only after ICI-treatment. Effects were more pronounced after anti-PD1 than after anti-LAG-3 immunotherapy (Figure 4c,d).
Accompanying gene expression analysis for Csf1, Csf2, Egr2, Alox5, Tcf1, and Tgf uncovered additional differences between the two interventions (Figure 5a). Anti-LAG-3-treated tumors exhibited higher levels of Csf1 and Csf2 than tumors from anti-PD1-treated mice, indicative of recruitment of hematopoietic cells into the tumors to polarize macrophages. Levels of Egr2 and Alox5 were comparable between the two treatment arms but remained higher compared to controls. Vice versa, the proto-oncogene Anterior gradient-2 (Agr2) was significantly suppressed by ICI-treatment (Figure 5a). Agr2 is associated with an immunologically “hot” and immunosuppressive phenotype, accompanied by increased level of TGF-β and activation of epithelial–mesenchymal transition (EMT) pathways31,32. Here, Tgf expression remained unaffected by either therapy, but EMT markers vimentin and N-Cadherin tended to be higher after ICI treatment compared to isotype controls, indicating long-term immune-stimulation (Figure 5a).
Figure 5.
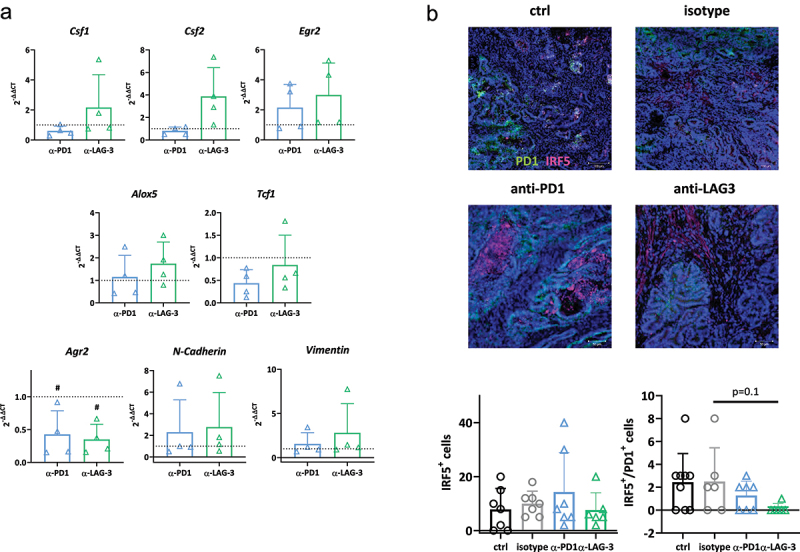
Gene expression analysis in residual tumors and IRF5 detection by tumor immunofluorescence.
Notes: (a) Quantitative qPCR of myeloid and epithelial–mesenchymal transition markers in the tumor. Total RNA from residual tumors was reverse transcribed into cDNA and qPCR was done as described in material and methods. Analysis was done in triplicates with n = 4 mice/group, respectively. All data are given as 2-ΔΔCT values + SD. Each symbol represents one case. # p < 0.05 vs. isotype; One-way ANOVA (Tukey’s multiple comparisons test). (b) The infiltration pattern of IRF5+/PD1+ cells was examined in residual tumor slides. Confocal laser scanning microscopy was done on a Zeiss Elyra 7 microscope. (a) Representative images of tumor slides. (b) Quantitative analysis of tumor-infiltrating immune cells counted in 2–3 HPFs/slide. Control n = 10; isotype n = 4–6/marker; -PD1n = 7; -LAG-3 n = 7. The infiltration pattern was semi-quantitatively analyzed using a scoring system. 0 = no; 1 = mild; 2 = moderate; 3 = strong. Each symbol represents one case. One-way ANOVA (Tukey’s multiple comparisons test).
Then, the amount and distribution of IRF5+, a regulator of host immunity and B cell activation, were studied (Figure 5b). Both ICIs slightly increased IRF5+ cells within tumors. Co-staining for PD1 was done to uncover the functional status of these cells. Notably, tumors from anti-LAG-3 treated mice had no exhausted IRF5+ cells, while few double-positive cells were detectable in the control and anti-PD1 groups (Figure 5b).
DMMR-driven tumorigenesis is characterized by widespread changes in the hematopoiesis
In the hematopoietic compartment, myeloblasts, neutrophil pro-/myelocytes, lymphocytes, and plasma cells were dramatically reduced during tumor development compared to their tumor-free counterparts (Figure 6a). Reticulocytes and erythrocytes were also lower in tumor-bearing mice, due to widespread anemia (not shown). Vice versa, the amounts of metamyelocytes/neutrophils increased significantly with tumor development (Figure 6a). Hence, dMMR-driven tumorigenesis is characterized by perturbed erythropoiesis and myelopoiesis. These changes in hematopoiesis were confirmed by flow cytometry (Figure 6b,c, and supplementary Figure S2). Specific subpopulations (please see Figure 6b for details) were detected in tumor-free, early-, and late-stage tumor-bearing mice. Here, we focused on lin−c-Kit+ progenitor cells. Long-term hematopoietic stem cells (LT-HSC, Sca-1+CD150+CD48−), including their respective quiescent CD63high LT-HSC subtype, and short-term HSC remained comparable between tumor-free and tumor-bearing mice. However, we observed significant differences in the number of multipotent progenitors (MPP, Sca-1+CD150−CD48−), which increased during tumor development. This increase was due to an imbalance between common myeloid precursors (CMP, Sca-1−CD34+) and common lymphoid precursors (CLP, c-kitlowSca-1low-IL7RThy1−) (Figure 6c, lower part). Similarly, the number of CD41+ megakaryocyte progenitors was lower in tumor-bearing mice compared to tumor-free mice. A similar reduction was seen for B cell precursors (IL-7 R+Thy1−CD38+) and innate lymphoid progenitors (IL-7 R+Thy1−PD1+), which were significantly reduced in late-stage tumor-bearing mice. This reduction was counteracted by a significant increase in CD34+ CMPs, phenotypically assignable to the granulocyte-macrophage (GMP) lineage, i.e. positivity for CxCr3+ or CSF-1 R+. IRF8+ cells reduced during tumor development. IRF8 acts in the lineage-committed progenitors to selectively limit neutrophil production by granulocyte progenitors and promote monocyte production by their respective progenitors33. PD1, a marker of T cell exhaustion that is also found on innate lymphoid progenitors, differed between tumor-free and tumor-bearing mice and among specific subpopulations. On innate lymphoid progenitors, PD1 was less frequently detected, whereas on hematopoietic progenitors, defined as lin−c-Kit+Sca-1+PD1+, PD1 increased after tumor development. Hence, we interpret this finding as bone marrow exhaustion due to the perturbed hematopoiesis during dMMR-driven gastrointestinal tumor development.
Figure 6.
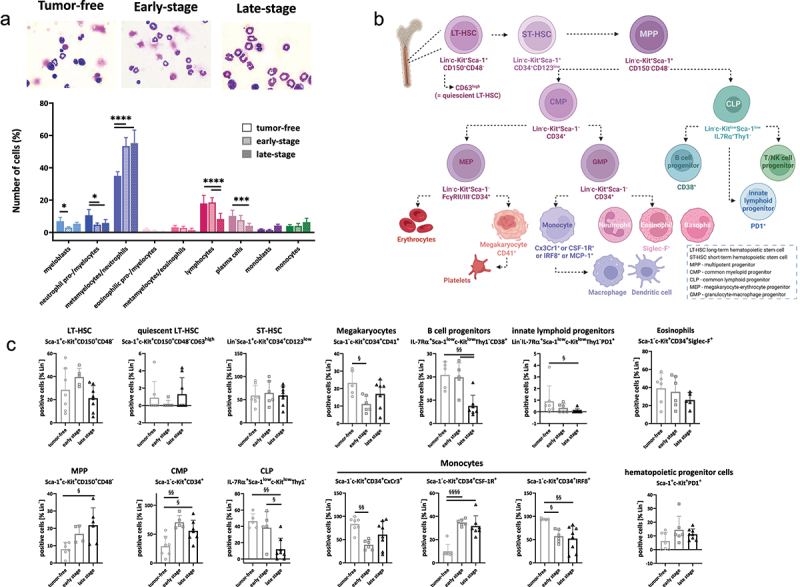
Altered murine haematopoiesis during tumor development.
Notes: (a) Pappenheim staining of bone marrow cells in normal, tumor-free and tumor-bearingMsh2loxP/loxPVillCre mice. From each slide, 200 cells were counted and differentiated according to a pre-designed differentiation scheme into the following cell types: myeloblasts, neutrophil pro-/myelocytes, metamyelocytes/neutrophil, eosinophilic pro-/myelocytes, metamyelocytes/eosinophils, lymphocytes, plasma cells, monoblasts, and monocytes. Upper part: representative images of each treatment. Magnification x1000; lower part: Quantification was done blinded by an experienced researcher. n = 7 mice/group. Mean + SD, * p<0.05; ** p<0.01; **** p<0.0001; One-way ANOVA (Sidak’s multiple comparisons test). (b, c) Bone marrow differentiation using multi-color spectral flow cytometry. (b) Diagram of hematopoiesis based on the markers used in this study. (c) A 20-marker panel was applied as described in material and methods. (c) Quantitative analysis of lin−c-Kit+ precursors according to the diagram shown in Figure 6b. Results show data from 100,000 cells/sample. Mean + SD, each symbol represents one case; § p < 0.05; §§ p < 0.01; §§§§ p < 0.0001; One-way ANOVA (Sidak’s multiple comparisons test).
Short-term ICI treatment partially rebalances hematopoiesis
We then investigated whether short-term ICI rescues the perturbed hematopoiesis in dMMR-driven tumors. First, by assessing cell counts from Pappenheim-stained cells, we identified significantly reduced amounts of metamyelocytes/neutrophils after anti-LAG-3, but not anti-PD1 treatment (p < 0.001, Figure 7a). Vice versa, lymphocyte levels were significantly higher in both treatment groups, approaching levels seen in tumor-free mice (please see Figure 6a for comparison). Plasma cells increased slightly with ICI therapy, while monoblasts decreased slightly.
Figure 7.
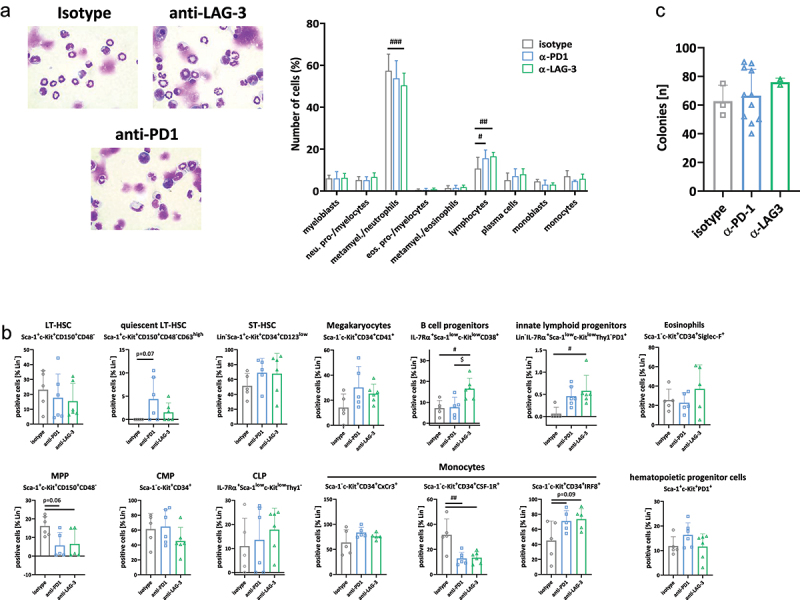
Impact of ICI treatment on murine haematopoiesis.
Notes: (a) Pappenheim staining of bone marrow cells in tumor-bearingMsh2loxP/loxPVillCre mice. From each slide, 200 cells were counted and differentiated according to a pre-designed differentiation scheme into the following cell types: myeloblasts, neutrophil pro-/myelocytes, metamyelocytes/neutrophil, eosinophilic pro-/myelocytes, metamyelocytes/eosinophils, lymphocytes, plasma cells, monoblasts, and monocytes. Upper part: representative images of each treatment. Magnification x1000; lower part: Quantification was done-blinded by an experienced researcher. isotype: n = 7 mice; anti-PD1 and anti-LAG-3: n = 6 mice/group. Mean + SD, # p<0.05; ## p<0.01; ### p<0.001; One-way ANOVA (Sidak’s multiple comparisons test’s multiple comparisons test). (b) Bone marrow differentiation using multi-color spectral flow cytometry. A 20-marker panel was applied as described in material & methods. Quantitative analysis of lin−c-Kit+ precursors. Results show data from 100,000 cells/sample. Mean + SD, each symbol represents one case. Isotype n = 7; -PD1n = 5; -LAG-3 n = 5; # p < 0.05; ## p < 0.01; $ p < 0.05; two-way ANOVA (Tukey’s multiple comparison test). (c) Colony formation assay. The number of viable colony-forming cells was counted after 14 days of in vitro culture. Mean + SD.
To confirm these positive effects of ICI treatment on hematopoiesis, our in-house flow cytometric marker panel was applied (Figure 7b). LT-HSCs were not affected by either treatment. We observed a trend toward higher numbers of the quiescent LT-HSC subtype, ST-HSC, and megakaryocyte progenitors. Conversely, MPPs were reduced. Another interesting finding was the slightly rebalanced CMP:CLP ratio, where CMP decreased and CMP increased after anti-LAG-3 treatment (Figure 7b, lower part). Also, B cell precursors and innate lymphoid progenitors significantly increased upon anti-LAG-3 immunotherapy. Having a look on the CD34+ CMPs identified significantly lower numbers of CSF-1 R+ cells after both ICI and higher numbers of IRF8+. Notably, numbers of IRF8+ progenitors were nearly comparable to tumor-free mice. On hematopoietic progenitors, PD1 was not significantly altered after treatment. Using a functional colony formation assay, the number of viable colony forming cells was slightly higher after anti-LAG-3 treatment (Figure 7c).
To sum up, this analysis identified a partial conversion of the tumor-driven perturbed hematopoiesis, in which lin−c-Kit+Sca-1−CD34+CSF-1 R+ precursors are the main targets of ICI treatment.
Discussion
In this study, we confirm the therapeutic potential of ICI treatment by targeting two different checkpoints: PD1 and LAG-3. While the former has been targeted in different trials, the latter recently entered the clinic, and preclinical data are also scarce. In dMMR cancers, LAG-3+ T cells are detectable at early stages and high LAG-3 expression is associated with a poor prognosis34. Hence, targeting this molecule is reasonable for this molecular subtype.
Low-dose treatment of Msh2loxP/loxP;TgTg(Vil1-cre) mice was well tolerated, resulting in significantly prolonged overall survival after LAG-3 blockade. Notably, this was even longer than after anti-PD1 treatment. Accompanying in vivo PET/CT imaging revealed no significant differences in tumor growth between anti-PD1 and anti-LAG-3. Nevertheless, both ICIs induced a stable disease and prevented the occurrence of novel tumors in virtually all cases during short-term follow-up, explaining the improved outcome compared to control mice. In addition, the standardized uptake value (SUV)max is an emerging imaging biomarker for response. Here, it was found to decrease only in the anti-LAG-3 treatment group, while an increase was observed in the anti-PD1 and control tumors. Furthermore, pseudoprogression, i.e. a transient increase in diameter due to local inflammation during ICI treatment is a common side effect that complicates the interpretation of PET/CT data35–37. By combining three markers: tumor size, number of tumor nodules, and SUVmax, differences were seen only after anti-LAG-3 treatment, most likely leading to the superior survival.
ICI treatment also positively reshaped the tumor microenvironment. The number of TAMs, including the highly suppressive M2-subtype, MDSCs, and regulatory granulocytes decreased – especially after LAG-3 blockade. Conversely, the transcription factor IRF5 was slightly higher, indicating type I interferon signaling via reactivation of the innate immune response38. Previous data showed that IRF5 is a direct target of p53, but is also involved in DNA damage- and IFN-induced apoptosis and cell death39. The finding of higher focal IRF5 levels in the TME of ICI-treated tumors may indicate enhanced (T cell-mediated) cell death upon treatment. In support of this, tumor-infiltrating effector memory T cells (CD4+ subtype) increased after LAG-3 blockade.
The number of circulating T helper and cytotoxic T cells was largely unaffected by either treatment, consistent with our previous findings with anti-PD-L1 ICI treatment8,9,14. However, levels of exhausted T cells, i.e. PD1+, CTLA-4+, LAG-3+, and TIM-3+ cells, and regulatory T cells were reduced after checkpoint blockade and represent a likely immune correlate of treatment. In support of this, a sustained reduction of the exhausted T-cell phenotype was recently reported in clinical samples from Hodgkin lymphoma patients receiving anti-PD1 antibodies40. In melanoma patients, circulating LAG-3+ subsets have also been identified as a blood-based biomarker that significantly correlates with both survival and response outcomes41. Hence, the inhibition of circulating LAG-3 levels is imperative to reconstitute immunity. The beneficial effect of both ICIs on exhausted T cells was not seen on circulating MDSCs, whose numbers were highly elevated after treatment. This can be interpreted as a compensatory mechanism to maintain systemic immunosuppression. Indeed, circulating MDSCs are essential for TAM accumulation within tumors and the finding of elevated MDSC levels in the spleen of ICI-treated mice add further credence to the relevance of these cells in curtailing effective anticancer immunity. By applying functional assays, we evaluated the immunosuppressive potential mediated by CD11b+Gr1+-sorted splenic MDSCs. ROS production was quantified after LPS stimulation. These analyses nicely confirmed the reduced immunosuppressive activity of MDSCs from ICI-treated mice. Hence, immunotherapy, as conducted in this study, reversed the suppressive function of MDSC. Still, the high plasticity of this cell type to differentiate into either macrophages or dendritic cells remains challenging42,43. It is worth mentioning that the spleen, in addition to the bone marrow, is an important “immune reservoir” of suppressive immune cell subsets. Therefore, ICI treatment may put pressure on tumors to recruit suppressor cells to avoid a vigorous T cell-mediated immune attack. Here, both ICIs reduced the number of CD163+ M2-macrophages, and concomitantly led to the downregulation of genes involved in M2-macrophage (Csf1) and neutrophil polarization (Egr2, Alox5). The finding that Csf2, which indicates an M1-like phenotype, was higher after anti-LAG-3 treatment compared to controls and anti-PD1 therapy suggests at least partial immunological counterregulation to mediate tumor growth control in vivo. Furthermore, it can be speculated that PD1 inhibition affects extramedullary hematopoiesis and thus the regulation of macrophage polarization and immune suppression. Consistent with this, the expression level of the transcription factor Tcf1, which is involved in T cell lineage specification and thymocyte differentiation30, was higher after anti-LAG-3 treatment in the spleen. TCF1+PD-1+ tumor-infiltrating lymphocytes were recently identified as positive predictors of response and survival after ICI treatment of non-small-cell lung cancer patients44. Also, dual inhibition of TGFβ signaling and CSF1/CSF1R provides a novel approach to reprogram tumor-infiltrating macrophages and improve chemotherapy response via PD-L1 suppression45. Here, splenic Tgfβ expression levels were lower after ICI treatment, implicating a minor role in tumor suppression at least in the models used here.
A previous study reported dysregulated hematopoiesis in a mammary carcinoma mouse model46. In addition to blood and spleen perturbations, tumor development was associated with FLT3L and GM-CSF, which synergized with tumor-derived G-CSF to promote hematopoietic stem/progenitor cell production46. Together, these tumor-secreted factors resulted in leukocytosis, anemia, and bone marrow defects. In our study, impaired hematopoiesis was also observed in the bone marrow of early- and late-stage tumor-bearing Msh2loxP/loxP;TgTg(Vil1-cre) mice. The number of metamyelocytes/neutrophils increased to an extent corresponding to the reduction of lymphocytes and plasma cells. Notably, this alteration increased with tumor progression, providing further evidence for the global impact of solid tumors on bone marrow hematopoiesis. Besides, these data also show that hematopoietic dysregulation is an early event in Msh2loxP/loxP;TgTg(Vil1-cre)-driven carcinogenesis, highlighting the need for early intervention to rebalance hematopoiesis. Here, the number of common myeloid progenitors increased. CD34+ CMPs, phenotypically belonging to the granulocyte-macrophage (GMP) lineage, i.e. were mostly positive for CxCr3+ or CSF-1 R+. At the same time, the number of common lymphoid progenitors decreased. This decrease was due to a reduction in B cell precursors and innate lymphoid progenitors, especially in late-stage tumor-bearing mice. Conversely, PD1 on hematopoietic progenitors, defined as lin−c-Kit+Sca-1+PD1+, PD1 increased after tumor development. Therefore, one can speculate that the increased level of CMPs is a tumor-driven mechanism for the recruitment of immature neutrophils to distant (tumor) sites. This is fueled by monocytic precursors via CSF-1 R to regulate the production, differentiation, and activation of TAMs and thus drive immunosuppression. Indeed, tumor-infiltrating CSF-1 R+ myeloid cells predict poor survival in cancer patients47–49, and targeting of this molecule with specific inhibitors is ongoing50,51.
Thus, “bone marrow” exhaustion is the result of persistent hematopoietic dysbalance during dMMR-driven gastrointestinal tumor development. This imbalance was partially reversed by ICI treatment, characterized by a slightly rebalanced CMP:CLP ratio, with CMP decreasing and CLP increasing, especially after anti-LAG-3 treatment. The rebalance of CLPs was due to an increase in B cell precursors and innate lymphoid progenitors. Another interesting finding was the reduction of CSF-1 R+ progenitor cells after both ICI and the higher numbers of IRF8+. The latter limits granulocyte development and functions as a “master” negative regulator of the formation of polymorphonuclear-MDSC in vivo52,53.
To sum up our findings, we have identified LAG-3 blockade as a novel promising approach for the treatment of dMMR-driven tumors. Sustained induction of stable disease, accompanied by successful targeting of global immune escape and partial conversion of the tumor-driven perturbed hematopoiesis, involves the underlying effects of LAG-3 targeting. Given these positive effects also seen here on OS, the combination of anti-PD1 and anti-LAG-3 is a very attractive option for the treatment of dMMR-related tumors. In preclinical models, dual blockade has shown synergistic effects54,55. In addition, the FDA recently approved the first anti-PD1/LAG-3-blocking antibody combination for patients with unresectable or metastatic melanoma19. This will be the next experimental approach.
List of abbreviations
- Ctrl
control
- dMMR
mismatch-repair deficiency
- ICI
Immune checkpoint inhibitor
- MDSC
myeloid-derived suppressor cell
- TAM
tumor-associated-macrophages
- Treg
regulatory T cells
Supplementary Material
Acknowledgments
We gratefully thank Mrs. Ilona Klamfuss and Ms. Chantal von Hoersten for breeding mice, Brigitte Vollmar and Bernd Krause for their continuous support in their efforts of chairing the Core Facility of Multimodal Small Animal Imaging. We also gratefully acknowledge the excellent technical assistance of Dr. Anna Schildt. Furthermore, we thank Carina Bergner and Anja Gummesson, radiopharmacy team of the Department of Nuclear Medicine of the University Medical Centre Rostock, for providing 18F-FDG for the small animal PET/CT experiments. Moreover, we thank Prof. Winfried Edelmann and Prof. Christoph Gasche for providing Msh2loxP/loxP;TgTg(Vil1-cre) breeding pairs and giving the permission to breed mice in our facility.
Funding Statement
This work was supported by grants from the German research foundation [DFG grant number MA5799/2-1 and MA5799/2-2] and the Brigitte und Dr. Konstanze Wegener-Stiftung to CM.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Authors’ contributions
I.S. participated in in vivo experiments, isolated tumor RNA, and performed flow cytometry of blood, spleen, and bone marrow. P.K., L.E. J.T., J.M., and J.H. performed in vivo monitoring of health status and ex vivo analysis (including staining of blood and organs for flow cytometry), P.K. analyzed the bone marrow and isolated RNA from bone marrow, A.W. assisted in ex vivo analysis, including immunofluorescence staining and quantification, W.B. assisted in flow cytometry panel design, C.J. critically revised the manuscript, C.M. designed the study, analyzed data, prepared figures, and wrote the manuscript.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Ethics approval and consent to participate
The German local authority approved all animal experiments: Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischerei Mecklenburg‐Vorpommern (7221.3‐1‐062/19), under the German animal protection law and the EU Guideline 2010/63/EU. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2023.2230669
References
- 1.Seth S, Ager A, Arends MJ, Frayling IM.. Lynch syndrome - cancer pathways, heterogeneity and immune escape. J Pathol. 2018;246(2):129–14. doi: 10.1002/PATH.5139. [DOI] [PubMed] [Google Scholar]
- 2.Lim SM, Choi JG, Cho SH, Kang EJ, Hwang IG, Yun T, Choi JW, Lee KW, Chang H, Kim JH, et al. Investigating the feasibility of targeted next-generation sequencing to guide the treatment of head and neck squamous cell carcinoma. Cancer Res Treat. 2019;51(1):300–312. doi: 10.4143/crt.2018.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chalabi M, Fanchi LF, Dijkstra KK, Van den Berg JG, Aalbers AG, Sikorska K, Lopez-Yurda M, Grootscholten C, Beets GL, Snaebjornsson P, et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat Med. 2020;26(4):566–576. 264. 2020. doi: 10.1038/s41591-020-0805-8. [DOI] [PubMed] [Google Scholar]
- 4.Trullas A, Delgado J, Genazzani A, Mueller-Berghaus J, Migali C, Müller-Egert S, Zander H, Enzmann H, Pignatti F.. The EMA assessment of pembrolizumab as monotherapy for the first-line treatment of adult patients with metastatic microsatellite instability-high or mismatch repair deficient colorectal cancer. ESMO Open. 2021;6(3):100145. doi: 10.1016/j.esmoop.2021.100145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.André T, Shiu K-K, Kim TW, Jensen BV, Jensen LH, Punt C, Smith D, Garcia-Carbonero R, Benavides M, Gibbs P, et al. Pembrolizumab in microsatellite-instability–high advanced colorectal cancer. N Engl J Med. 2020;383(23):2207–2218. doi: 10.1056/NEJMoa2017699. [DOI] [PubMed] [Google Scholar]
- 6.Konstantinopoulos PA, Luo W, Liu JF, Gulhan DC, Krasner C, Ishizuka JJ, Gockley AA, Buss M, Growdon WB, Crowe H, et al. Phase II study of avelumab in patients with mismatch repair deficient and mismatch repair proficient recurrent/persistent endometrial cancer. J Clin Oncol. 2019;37(30):2786–2794. doi: 10.1200/JCO.19.01021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salewski I, Kuntoff S, Kuemmel A, Feldtmann R, Felix SB, Henze L, Junghanss C, Maletzki C. Combined vaccine-immune-checkpoint inhibition constitutes a promising strategy for treatment of dMMR tumors. Cancer Immunol Immunother. 2021;70(12):3405–3419. doi: 10.1007/s00262-021-02933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salewski I, Henne J, Engster L, Schneider B, Lemcke H, Skorska A, Berlin P, Henze L, Junghanss C, Maletzki C, et al. Combined gemcitabine and immune-checkpoint inhibition conquers Anti-PD-L1 resistance in low-immunogenic mismatch repair-deficient tumors. Int J Mol Sci. 2021;22(11):5990. doi: 10.3390/IJMS22115990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heckler M, Ali LR, Clancy-Thompson E, Qiang L, Ventre KS, Lenehan P, Roehle K, Luoma A, Boelaars K, Peters V, et al. Inhibition of CDK4/6 promotes CD8 T-cell memory formation. Cancer Discov. 2021;11(10):2564–2581. doi: 10.1158/2159-8290.cd-20-1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schaer DA, Beckmann RP, Dempsey JA, Huber L, Forest A, Amaladas N, Li Y, Wang YC, Rasmussen ER, Chin D, et al. The CDK4/6 inhibitor abemaciclib induces a T cell inflamed tumor microenvironment and enhances the efficacy of PD-L1 checkpoint blockade. Cell Rep. 2018;22(11):2809–2817. doi: 10.1016/j.celrep.2018.02.053. [DOI] [PubMed] [Google Scholar]
- 12.Egelston C, Guo W, Yost S, Lee JS, Rose D, Avalos C, Ye J, Frankel P, Schmolze D, Waisman J, et al. Pre-existing effector T-cell levels and augmented myeloid cell composition denote response to CDK4/6 inhibitor palbociclib and pembrolizumab in hormone receptor-positive metastatic breast cancer. J ImmunoTher Cancer. 2021;9(3):e002084. doi: 10.1136/JITC-2020-002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patnaik A, Yap TA, Chung HC, de Miguel MJ, Bang Y-J, Lin C-C, Su W-C, Italiano A, Chow KH, Szpurka AM, et al. Safety and clinical activity of a New Anti-PD-L1 antibody as monotherapy or combined with targeted therapy in advanced solid tumors: the pact phase Ia/Ib trial. Clin Cancer Res. 2021;27(5):1267–1277. doi: 10.1158/1078-0432.CCR-20-2821. [DOI] [PubMed] [Google Scholar]
- 14.Salewski I, Henne J, Engster L, Krone P, Schneider B, Redwanz C, Lemcke H, Henze L, Junghanss C, Salewski I, et al. CDK4/6 blockade provides an alternative approach for treatment of mismatch-repair deficient tumors. Oncoimmunology. 2022;11(1). doi: 10.1080/2162402X.2022.2094583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phenotypic analysis of the murine CD4-related glycoprotein, CD223 (LAG-3) - PubMed . [DOI] [PubMed]
- 16.Triebel F. LAG-3: a regulator of T-cell and DC responses and its use in therapeutic vaccination. Trends Immunol. 2003;24(12):619–622. doi: 10.1016/J.IT.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 17.Maruhashi T, Sugiura D, Okazaki IM, Okazaki T. LAG-3: from molecular functions to clinical applications. J ImmunoTher Cancer. 2020;8(2):e001014. doi: 10.1136/JITC-2020-001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sordo‐Bahamonde C, Lorenzo‐Herrero S, González‐Rodríguez AP, Payer ÁR, González‐García E, López‐Soto A, Gonzalez S. LAG-3 blockade with relatlimab (BMS-986016) restores anti-leukemic responses in chronic lymphocytic leukemia. Cancers Basel. 2021;13(9):2112. doi: 10.3390/CANCERS13092112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tawbi HA, Schadendorf D, Lipson EJ, Ascierto PA, Matamala L, Castillo Gutiérrez E, Rutkowski P, Gogas HJ, Lao CD, De Menezes JJ, et al. Relatlimab and Nivolumab versus Nivolumab in untreated advanced melanoma. N Engl J Med. 2022;386(1):24–34. doi: 10.1056/NEJMOA2109970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schöffski P, Tan DSW, Martín M, Ochoa-De-Olza M, Sarantopoulos J, Carvajal RD, Kyi C, Esaki T, Prawira A, Akerley W, et al. Phase I/II study of the LAG-3 inhibitor ieramilimab (LAG525) ± anti-PD-1 spartalizumab (PDR001) in patients with advanced malignancies. J ImmunoTher Cancer. 2022;10(2):e003776. doi: 10.1136/JITC-2021-003776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Väyrynen JP, Haruki K, Lau MC, Väyrynen SA, Zhong R, Costa AD, Borowsky J, Zhao M, Fujiyoshi K, Arima K, et al. The prognostic role of macrophage polarization in the colorectal cancer microenvironment. Cancer Immunol Res. 2021;9(1):8–19. doi: 10.1158/2326-6066.CIR-20-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao R, Wan Q, Wang Y, Wu Y, Xiao S, Li Q, Shen X, Zhuang W, Zhou Y, Xia L, et al. M1-like TAMs are required for the efficacy of PD-L1/PD-1 blockades in gastric cancer. Oncoimmunology. 2021;10(1). doi: 10.1080/2162402X.2020.1862520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boudesco C, Verhoeyen E, Martin L, Chassagne-Clement C, Salmi L, Mhaidly R, Pangault C, Fest T, Ramla S, Jardin F, et al. HSP110 sustains chronic NF-κB signaling in activated B-cell diffuse large B-cell lymphoma through MyD88 stabilization. Blood. 2018;132(5):510–520. doi: 10.1182/BLOOD-2017-12-819706. [DOI] [PubMed] [Google Scholar]
- 24.He K, Liu X, Hoffman RD, Shi R, Lv G, Gao J. G-CSF / GM-CSF -induced hematopoietic dysregulation in the progression of solid tumors. FEBS Open Bio. 2022 Jul 9;12(7):1268–1285. doi: 10.1002/2211-5463.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wildes TJ, DiVita Dean B, Flores CT. Myelopoiesis during solid cancers and strategies for immunotherapy. Cells. 2021;10(5):968. doi: 10.3390/cells10050968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Long H, Jia Q, Wang L, Fang W, Wang Z, Jiang T, Zhou F, Jin Z, Huang J, Zhou L, et al. Tumor-induced erythroid precursor-differentiated myeloid cells mediate immunosuppression and curtail anti-PD-1/PD-L1 treatment efficacy. Cancer Cell. 2022;40(6):674–693.e7. doi: 10.1016/j.ccell.2022.04.018. [DOI] [PubMed] [Google Scholar]
- 27.Kucherlapati MH, Lee K, Nguyen AA, Clark AB, Hou HJ, Rosulek A, Li H, Yang K, Fan K, Lipkin M, et al. An Msh2 conditional knockout mouse for studying intestinal cancer and testing anticancer agents. Gastroenterology. 2010;138(3):993–1002.e1. doi: 10.1053/j.gastro.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kortüm B, Campregher C, Lang M, Khare V, Pinter M, Evstatiev R, Schmid G, Mittlböck M, Scharl T, Kucherlapati MH, et al. Mesalazine and thymoquinone attenuate intestinal tumour development in Msh2loxP/loxP Villin-Cre mice. Gut. 2015;64(12):1905–1912. doi: 10.1136/gutjnl-2014-307663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maletzki C, Beyrich F, Hühns M, Klar E, Linnebacher M. The mutational profile and infiltration pattern of murine MLH1-/- tumors: concurrences, disparities and cell line establishment for functional analysis. Oncotarget. 2016;7(33):53583–53598. doi: 10.18632/oncotarget.10677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gounari F, Khazaie K. TCF-1: a maverick in T cell development and function. Nat Immunol. 2022;23(5):671–678. doi: 10.1038/S41590-022-01194-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang S, Liu Q, Wei Y, Xiong Y, Gu Y, Huang Y, Tang F, Ouyang Y. Anterior gradient-2 regulates cell communication by coordinating cytokine–chemokine signaling and immune infiltration in breast cancer. Cancer Sci. 2023 Mar 17;114(6):2238–2253. doi: 10.1111/CAS.15775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martisova A, Sommerova L, Krejci A, Selingerova I, Kolarova T, Zavadil Kokas F, Holanek M, Podhorec J, Kazda T, Hrstka R. Identification of AGR2 gene-specific expression patterns associated with epithelial-mesenchymal transition. Int J Mol Sci. 2022;23(18):10845. doi: 10.3390/ijms231810845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yáñez A, Ng MY, Hassanzadeh-Kiabi N, Goodridge HS. IRF8 acts in lineage-committed rather than oligopotent progenitors to control neutrophil vs monocyte production. Blood. 2015;125(9):1452–1459. doi: 10.1182/BLOOD-2014-09-600833. [DOI] [PubMed] [Google Scholar]
- 34.Al-Mterin MA, Murshed K, Elkord E. Correlations between circulating and tumor-infiltrating CD4+ Treg subsets with immune checkpoints in colorectal cancer patients with early and advanced stages. Vaccines. 2022;10(9):1471. doi: 10.3390/VACCINES10091471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dercle L, Sun S, Seban R-D, Mekki A, Sun R, Tselikas L, Hans S, Bernard-Tessier A, Bouvier FM, Aide N, et al. Emerging and evolving concepts in cancer immunotherapy imaging. Radiology. 2022;306(1):32–46. doi: 10.1148/RADIOL.210518. [DOI] [PubMed] [Google Scholar]
- 36.Guan Y, Feng D, Yin B, Li K, Wang J. Immune-related dissociated response as a specific atypical response pattern in solid tumors with immune checkpoint blockade. Ther Adv Med Oncol. 2022;14:14. doi: 10.1177/17588359221096877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Decazes P, Bohn P. Immunotherapy by immune checkpoint inhibitors and nuclear medicine imaging: current and future applications. Cancers Basel. 2020;12(2):371. doi: 10.3390/CANCERS12020371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santana-de Anda K, Gómez-Martín D, Díaz-Zamudio M, Alcocer-Varela J. Interferon regulatory factors: beyond the antiviral response and their link to the development of autoimmune pathology. Autoimmunity Rev. 2011;11(2):98–103. doi: 10.1016/J.AUTREV.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 39.Hu G, Barnes BJ. Interferon regulatory factor-5-regulated pathways as a target for colorectal cancer therapeutics. Expert Rev Anticancer Ther. 2014;6(5):775–784. doi: 10.1586/14737140.6.5.775. [DOI] [PubMed] [Google Scholar]
- 40.Garcia-Marquez MA, Thelen M, Reinke S, Keller D, Wennhold K, Lehmann J, Veldman J, Borchmann S, Rosenwald A, Sasse S, et al. Reverted exhaustion phenotype of circulating lymphocytes as immune correlate of anti-PD1 first-line treatment in Hodgkin lymphoma. Leukemia. 2022;36(3):760–771. doi: 10.1038/S41375-021-01421-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen R, Postow MA, Adamow M, Arora A, Hannum M, Maher C, Wong P, Curran MA, Hollmann TJ, Jia L, et al. LAG-3 expression on peripheral blood cells identifies patients with poorer outcomes after immune checkpoint blockade. Sci Transl Med. 2021;13(608). doi: 10.1126/SCITRANSLMED.ABF5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Ginderachter JA, Meerschaut S, Liu Y, Brys L, De Groeve K, Ghassabeh GH, Raes G, De Baetselier P. Peroxisome proliferator-activated receptor gamma (PPARgamma) ligands reverse CTL suppression by alternatively activated (M2) macrophages in cancer. Blood. 2006;108(2):525–535. doi: 10.1182/BLOOD-2005-09-3777. [DOI] [PubMed] [Google Scholar]
- 43.Wu Y, Yi M, Niu M, Mei Q, Wu K. Myeloid-derived suppressor cells: an emerging target for anticancer immunotherapy. Mol Cancer. 2022;21(1):184. doi: 10.1186/S12943-022-01657-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koh J, Kim S, Woo YD, Song SG, Yim J, Han B, Lim S, Ahn HK, Mun S, Kim JS, et al. TCF1 + PD-1 + tumour-infiltrating lymphocytes predict a favorable response and prolonged survival after immune checkpoint inhibitor therapy for non-small-cell lung cancer. Eur J Cancer. 2022;174:10–20. doi: 10.1016/J.EJCA.2022.07.004. [DOI] [PubMed] [Google Scholar]
- 45.Chen TW, Hung WZ, Chiang SF, Chen WTL, Ke TW, Liang JA, Huang CY, Yang PC, Huang KCY, Chao KSC. Dual inhibition of TGFβ signaling and CSF1/CSF1R reprograms tumor-infiltrating macrophages and improves response to chemotherapy via suppressing PD-L1. Cancer Lett. 2022;543:543. doi: 10.1016/J.CANLET.2022.215795. [DOI] [PubMed] [Google Scholar]
- 46.Sio A, Chehal MK, Tsai K, Fan X, Roberts ME, Nelson BH, Grembecka J, Cierpicki T, Krebs DL, Harder KW. Dysregulated hematopoiesiscausedbymammary Cancer is associated with epigenetic changes and Hox gene expression in hematopoietic cells. Cancer Res. 2013;73(19):5892–5904. doi: 10.1158/0008-5472.CAN-13-0842. [DOI] [PubMed] [Google Scholar]
- 47.Mao Y, Eissler N, Blanc KL, Johnsen JI, Kogner P, Kiessling R. Targeting Suppressive myeloid cells potentiates checkpoint inhibitors to control spontaneous neuroblastoma. Clin Cancer Res. 2016;22(15):3849–3859. doi: 10.1158/1078-0432.CCR-15-1912. [DOI] [PubMed] [Google Scholar]
- 48.Valero JG, Matas-Céspedes A, Arenas F, Rodriguez V, Carreras J, Serrat N, Guerrero-Hernández M, Yahiaoui A, Balagué O, Martin S, et al. The receptor of the colony-stimulating factor-1 (CSF-1R) is a novel prognostic factor and therapeutic target in follicular lymphoma. Leukemia. 2021;35(9):2635–2649. doi: 10.1038/S41375-021-01201-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Riaz N, Burugu S, Cheng AS, Leung SCY, Gao D, Nielsen TO. Prognostic sgnificance of CSF-1R expression in early invasive breast cancer. Cancers Basel. 2021;13(22):5769. doi: 10.3390/CANCERS13225769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Z, Ding Y, Liu J, Wang J, Mo F, Wang Y, Chen-Mayfield TJ, Sondel PM, Hong S, Hu Q. Depletion of tumor associated macrophages enhances local and systemic platelet-mediated anti-PD-1 delivery for post-surgery tumor recurrence treatment. Nat Commun. 2022;13(1):1–15. 2022 131. doi: 10.1038/s41467-022-29388-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gomez-Roca C, Cassier P, Zamarin D, Machiels JP, Luis Perez Gracia J, Stephen Hodi F, Taus A, Martinez Garcia M, Boni V, Eder JP, et al. Anti-CSF-1R emactuzumab in combination with anti-PD-L1 atezolizumab in advanced solid tumor patients naïve or experienced for immune checkpoint blockade. J ImmunoTher Cancer. 2022;10(5):e004076. doi: 10.1136/JITC-2021-004076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abrams SI, Netherby CS, Twum DYF, Messmer MN. Relevance of interferon regulatory Factor-8 expression in myeloid-tumor interactions. J Interferon Cytokine Res. 2016;36(7):442–453. doi: 10.1089/JIR.2015.0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Netherby CS, Messmer MN, Burkard-Mandel L, Colligan S, Miller A, Cortes Gomez E, Wang J, Nemeth MJ, Abrams SI. The granulocyte progenitor stage is a key target of IRF8-Mediated regulation of myeloid-derived suppressor cell production. J Immunol. 2017;198(10):4129–4139. doi: 10.4049/JIMMUNOL.1601722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang R-Y, Eppolito C, Lele S, Shrikant P, Matsuzaki J, Odunsi K, Huang R-Y, Eppolito C, Lele S, Shrikant P, et al. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8 + T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget. 2015;6(29):27359–27377. doi: 10.18632/ONCOTARGET.4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thudium K, Selby M, Zorn JA, Rak G, Wang X-T, Bunch RT, Hogan JM, Strop P, Korman AJ. Preclinical characterization of relatlimab, a human LAG-3–Blocking antibody, alone or in combination with Nivolumab. Cancer Immunol Res. 2022 Sep 16;10(10):1175–1189. doi: 10.1158/2326-6066.CIR-22-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article.