

Abstract
Purpose of review
Observational data provide compelling evidence for elevated fibroblast growth factor-23 (FGF23) as a risk factor for heart failure (HF), particularly heart failure with preserved ejection fraction (HFpEF). Given the limitations of observational studies, uncertainties persist regarding the causal role of FGF23 in the pathogenesis of HF and HFpEF. Recently, Mendelian randomization (MR) studies have been performed to examine causal associations between FGF23 and HF phenotypes.
Recent findings
The current review describes the methodological basis of the MR techniques used to examine the causal role of FGF23 on HF phenotypes, highlighting the importance of large-scale multi-omics data. The findings from most of the MR studies indicate an absence of evidence of a causal effect of FGF23 on the risk of HF in general population settings. However, analysis using individual-level data showed a strong association between genetically-predicted FGF23 and HFpEF in individuals with a genetic predisposition to low eGFR.
Summary
Evidence from MR analysis suggests a causal role of FGF23 in the pathogenesis of HFpEF in low eGFR settings – a finding supported by experimental, clinical, and epidemiological data. While future MR studies of FGF23 and HFpEF could provide further evidence, randomized trials of FGF23-lowering agents could provide the most definitive answers on the association in chronic kidney disease (CKD) populations.
Keywords: mendelian randomization, fibroblast growth factor-23, heart failure with preserved ejection fraction, causality, chronic kidney disease
Introduction
Consistent associations across multiple cohort studies provide compelling evidence of the role of mineral metabolism markers, including phosphate, parathyroid hormone, and fibroblast growth factor-23 (FGF23) in the pathophysiology of cardiovascular disease[1–5]. Over the last decade, observational studies have implicated elevated levels of circulating FGF23 as a risk factor for heart failure (HF) in patients with chronic kidney disease (CKD) and individuals in the general population [6–10]. Independent associations were reported between serum FGF23 concentration and multiple HF-related phenotypes, including HF with preserved ejection fraction (HFpEF) and left ventricular hypertrophy (LVH) [11–13]. Observational epidemiological studies suffer from inherent biases such as confounding and reverse causation, limiting their ability to robustly identify causal associations. In addition, the lack of specificity of associations and an unclear dose-response relationship raised the usual questions on whether the observed correlations signify cause and effect.[14]
Recently, several research teams have examined the potential causal role of FGF23 on HF and HF subtypes using mendelian randomization (MR) experiments, designed to mitigate some of the pitfalls of observational studies[15–18]. This review discusses the MR studies that utilize large-scale multi-omics data to bring clarity to the debate regarding the causal nature of these associations. The review also highlights other experimental, animal model and mechanistic data that clarify the potential role of FGF23 on the pathophysiology of HF and HFpEF.
Mendelian randomization methods for the FGF23-heart failure association
MR techniques leverage genetic variants associated with circulating biomarker levels as instruments to investigate the causal effect of the candidate biomarker on the risk of disease [19–22]. Akin to a randomized trial, because of the random assortment and segregation of alleles at meiosis, genetic variants associated with a risk factor through genome-wide association studies (GWAS) can be used as instruments to investigate the relationship between the risk factor and disease in an un-confounded manner (Figure 1). Since genetic variants are assigned at conception and largely stable over the course of an individual’s life, MR studies also have the correct temporal ordering to overcome the possibility of reverse causation.
Figure 1.
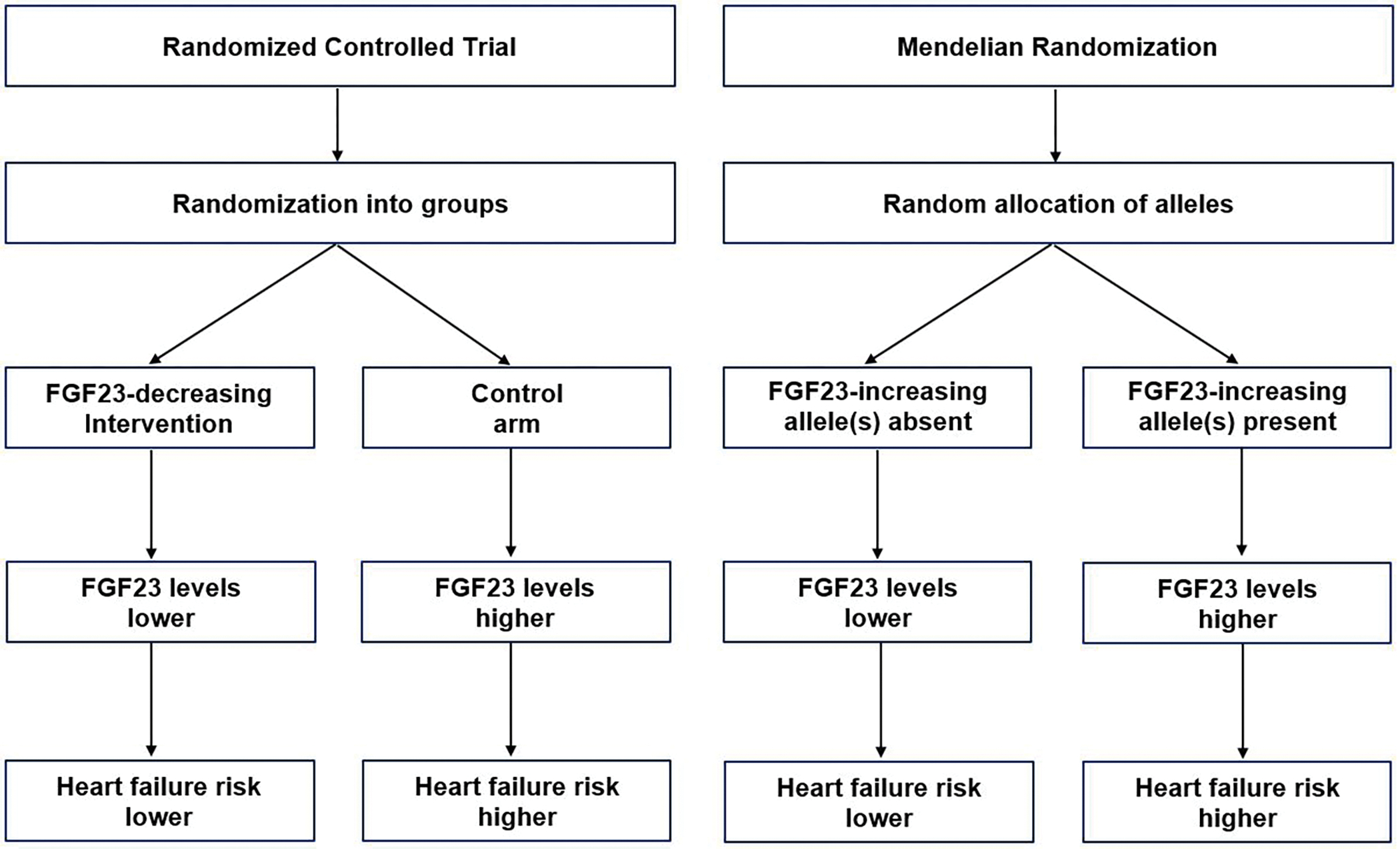
Random allocation of FGF23 alleles in Mendelian randomization versus randomized control trials. Legend: This figure demonstrates the analogy between Mendelian Randomization (MR) designs and randomized controlled trial (RCT) designs. In MR designs, genetic variants are randomly allocated at conception, while in RCT designs, participants are randomly allocated to intervention or control groups.
GWAS can identify genetic variants correlated with interindividual differences in protein levels that may indirectly or directly influence the protein-coding gene [23, 24]. A GWAS of 16,624 individuals of European ancestry in the CHARGE consortium identified five FGF23-associated genetic loci, some of which were located near genes involved in vitamin D metabolism and renal phosphate transport [25]. A second FGF23 GWAS from the SCALLOP consortium using a proteomic assay (multiplex immunoassay) on 19,195 individuals replicated most of the signals from the prior meta-GWAS and additionally identified one genome-wide-significant pQTL (protein quantitative trait locus) near the FGF23 gene itself [26]. Mendelian randomization studies investigating the FGF23-HF association have primarily used a two-sample MR design [27] were the single nucleotide polymorphism (SNP)-exposure (FGF23) and the SNP-outcome (HF) data come from two different studies with similar ancestry or population structure.
MR studies of serum biomarkers often utilize data from GWAS to identify independent genetic variants which together predict a non-negligible proportion of the variance of the circulating biomarker levels [28, 29]. These genetic variants must meet the three assumptions of instrumental variables to be considered valid instruments [30]. For the FGF23-HF association, these are (1) the genetic markers are strongly associated with circulating FGF23 levels (relevance assumption); (2) the variants are independent of any confounders of the association between circulating FGF23 and HF (independence assumption); and (3) the variants affect HF only through their effect on FGF23 (no horizontal pleiotropy) (Figure 2). Even if only one of the genetic variants is not a valid instrumental variable, the causal estimate based on all the variants from a conventional Mendelian randomization analysis will be biased, with inflated false positive error rates. [31, 32] For example, a genetic variant in the ABO gene identified as an independent signal in the GWAS of FGF23 has known pleiotropic effects and was not included in the primary analysis of the MR studies included in the current review.
Figure 2.
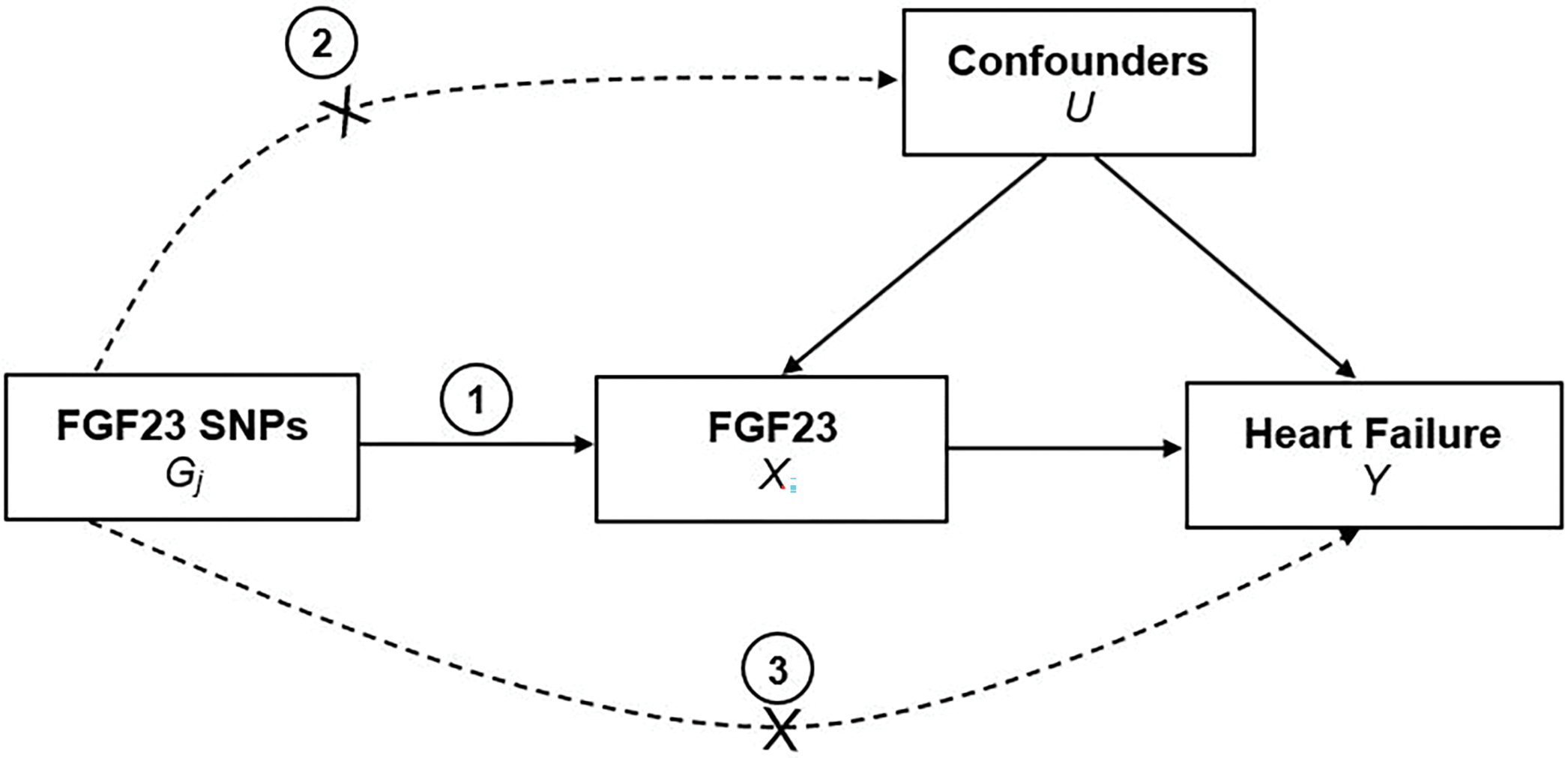
Instrumental variable assumptions for the Mendelian randomization studies of FGF23 and heart failure. Legend: The three assumptions are: (1) the genetic variant must be robustly associated with the exposure (FGF23); (2) the genetic variant should not be associated with confounders of the exposure-outcome (heart failure) association; and (3) the genetic variant must influence the outcome through the exposure only and not through any direct or alternative pathways. The dashed lines represent pathways that violate the assumptions.
When all variants satisfy the assumptions, a Wald ratio estimate is obtained by dividing the beta coefficient of the variant-HF association by the beta coefficient of the variant-FGF23 association. An overall inverse-variance weighted (IVW) estimate is obtained by combining the variants’ ratio estimates using standard meta-analysis [33]. Alternative MR approaches allow consideration or inclusion of pleiotropic variants and are included as sensitivity analyses in MR efforts. The weighted median estimate is more robust to the presence of pleiotropic effects, allowing up to 50% of variants to violate MR assumptions[34]. MR-Egger regression, in contrast to conventional MR, allows for pleiotropy and fits the weighted linear regression model with an intercept term that represents the average pleiotropic effect [33]. Assuming that the distribution of pleiotropic effects is independent from the genetic associations with exposure, (i.e. Instrument Strength Independent of Direct Effect assumption), the MR-Egger approach provides a consistent and valid estimate of the causal effect.[35] In MR studies of FGF23 and HF, additional sensitivity analyses have been performed, including leave-one-out analyses, where variants are removed one at a time to investigate their impact on the primary analysis. Other alternatives include using only cis-PQTLs or utilizing a larger set of genetic instruments meeting a lower threshold for significance (e.g. p<1×10−6) and combining their effects in a genetic risk score to increase the proportion of variance of FGF23 explained by the instruments [16].
Current evidence from Mendelian randomization studies of FGF23 and overall heart failure
Four recent studies have examined the causal nature of the association between FGF23 and overall HF using MR techniques [15–18]. Liang et al performed a two-sample MR using genome-wide association data of individuals of European ancestry from large consortia [18, 36]. Genetic instruments were defined as independent variants (r2 <0.01) that were genome-wide significant signals (p<5 × 10−8)[25] for log-FGF23 in models adjusted for age, sex, and 10 principal components of ancestry and did not have known pleiotropic effects. These variants were rs17216707 upstream of CYP24A1, rs11741640 within RGS14, rs17479566 near LINC01506, rs9925837 near LINC01229. The pleiotropic rs2769071 variant in ABO gene was excluded. There was no weak instrument bias as the F-statistics for these variants ranged from 34.0 to 117.5 [18]. A meta-analysis of 26 GWAS from the HERMES consortium, including 47,309 cases of HF and 930,014 controls [37], was used to capture the association of the genetic instruments with overall HF. Cases of HF were all patients with a clinical diagnosis of HF regardless of left ventricular ejection fraction (LVEF) while controls were individuals without HF. In the primary analysis using an IVW approach, no evidence of a causal effect of genetically-predicted FGF23 on HF was detected (OR: 0.99, 95%CI: 0.75–1.31) (Table 1). A similar finding was obtained using the weighted median approach (OR: 1.01, 95%CI: 0.73–1.38) and there was no evidence of heterogeneity between the SNPs (p-value for Cochran’s Q = 0.24) or directional pleiotropy (p-value for MR-Egger intercept = 0.20) [18].
Table 1.
Methods and summary estimates from mendelian randomization studies of FGF23 and heart failure phenotypes
| Study (First Author, Journal, Publication date) | FGF23 GWAS | FGF23 Instruments | Source of Outcome data (n = number of HF cases) | MR IVW OR (95%CI) |
|---|---|---|---|---|
|
| ||||
| Liang Y et al. Frontiers in Genetics 07/23/2021 | CHARGE Consortium, FGF23 GWAS meta-analysis, n =16,624 | 4 SNPs with p<5×10−8 (rs17216707 near CYP24A1, rs11741640 near RGS14, rs17479566 near LINC01229, rs9925837 near LINC01506) | HERMES (n = 47,309) | 0.99 (0.75, 1.31) |
|
| ||||
| Akwo E et al CJASN 08/01/2022 | CHARGE Consortium, FGF23 GWAS meta-analysis, n=16,624 | 4 SNPs with p<5×10−8 (rs17216707 near CYP24A1, rs11741640 near RGS14, rs17479566 near LINC01229, rs9925837 near LINC01506) | Overall HF | |
| HERMES (n = 47,309) | 1.13 (0.89, 1.42) | |||
| BioVU, Overall (n = 18,415) | 1.32 (0.95, 1.84) | |||
| BioVU, low eGFR PRSa (n=3086) | 3.09 (1.38, 6.91) | |||
| BioVU, low eGFR PRSa (n=15,329) | 1.11 (0.77, 1.54) | |||
| HFpEF | ||||
| BioVU, Overall (n = 13,141) | 1.47 (1.01, 2.14) | |||
| BioVU, low eGFR PRSa (n=2223) | 7.20 (2.80, 18.5) | |||
| BioVU, low eGFR PRSa (n=10,918) | 1.03 (0.67, 1.57) | |||
| HFrEF | ||||
| BioVU, Overall (n = 3394) | 0.86 (0.38, 1.96) | |||
| BioVU, low eGFR PRSa (n=554) | 0.24 (0.03, 1.72) | |||
| BioVU, low eGFR PRSa (n=2840) | 1.12 (0.45, 2.77) | |||
|
| ||||
| Donovan et al CJASN 01/01/2023 | SCALLOP Consortium, FGF23 GWAS meta-analysis, n = 19,195 | Genetic score of 34 SNPs with p<5×10−6 | HERMES (n = 47,309) | 1.00 (0.94, 1.05) |
|
| ||||
| UK Biobank (n=10,177) | 1.01 (0.94, 1.10) | |||
|
| ||||
| Henry et al Circulation 04/19/2022 | SCALLOP Consortium, FGF23 GWAS meta-analysis, n = 19,195 | 1 Cis-variant (rs6489536) near the FGF23 gene with p < 5×10−8 | HERMES (n = 47,309) | 0.79 (0.62, 1.02) |
|
| ||||
| 2 Cis-variants (rs6489536 and rs7955866) near the FGF23 gene with p < 1×10−6 | HERMES (n = 47,309) | 0.80 (0.71–0.89) | ||
BioVU, Vanderbilt University Medical Center DNA Biobank; CHARGE, Cohorts for Heart and Aging Research in Genetic Epidemiology; eGFR, estimated Glomerular Filtration Rate; FGF23, Fibroblast Growth Factor-23; GWAS, Genome-Wide Association Study; HERMES, Heart Failure Molecular Epidemiology Therapeutic Targets; HF, heart failure; HFpEF heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; IVW, Inverse Variance Weighted; MR, mendelian randomization; OR, Odds Ratio; PRS, Polygenic Risk Score; SNP, single nucleotide polymorphism
A subsequent MR study used the same FGF23 GWAS to identify genetic instruments[25], alongside individual-level data from BioVU, the Vanderbilt University Medical Center DNA biobank linked to de-identified electronic medical records to identify heart failure cases (n=18,415), with differentiation for preserved (HFpEF) and reduced ejection fraction (HFrEF) and controls (n=13,903), who had no HF-related ICD codes or text mentions in their record, and never had an LVEF ≤50%.[15, 38]. After obtaining beta coefficients for the association between the FGF23 genetic instruments and overall HF in BioVU, IVW MR analysis showed no evidence of a causal effect of genetically-predicted FGF23 on HF (OR: 1.32, 95%CI: 0.95–1.84) [15] (Table 1). MR analyses were then repeated within strata of estimated glomerular filtration (eGFR) polygenic risk score (PRS) based on eGFR GWAS data from the CKDGen consortium.[15] (Figure 3) In the low eGFR PRS stratum, genetically-predicted FGF23 was associated with 3-fold higher odds of overall HF (OR: 3.09, 95%CI: 1.38–6.91) [15] (Table 1). This suggests that individuals with a genetic liability for low eGFR or CKD may have greater susceptibility to the adverse effects of FGF23 on the risk of heart failure. No evidence of a causal effect of genetically-predicted FGF23 was observed in the high eGFR PRS stratum suggesting perhaps that genetic liability to elevated FGF23 may be benign in settings of preserved kidney function [15].
Figure 3.
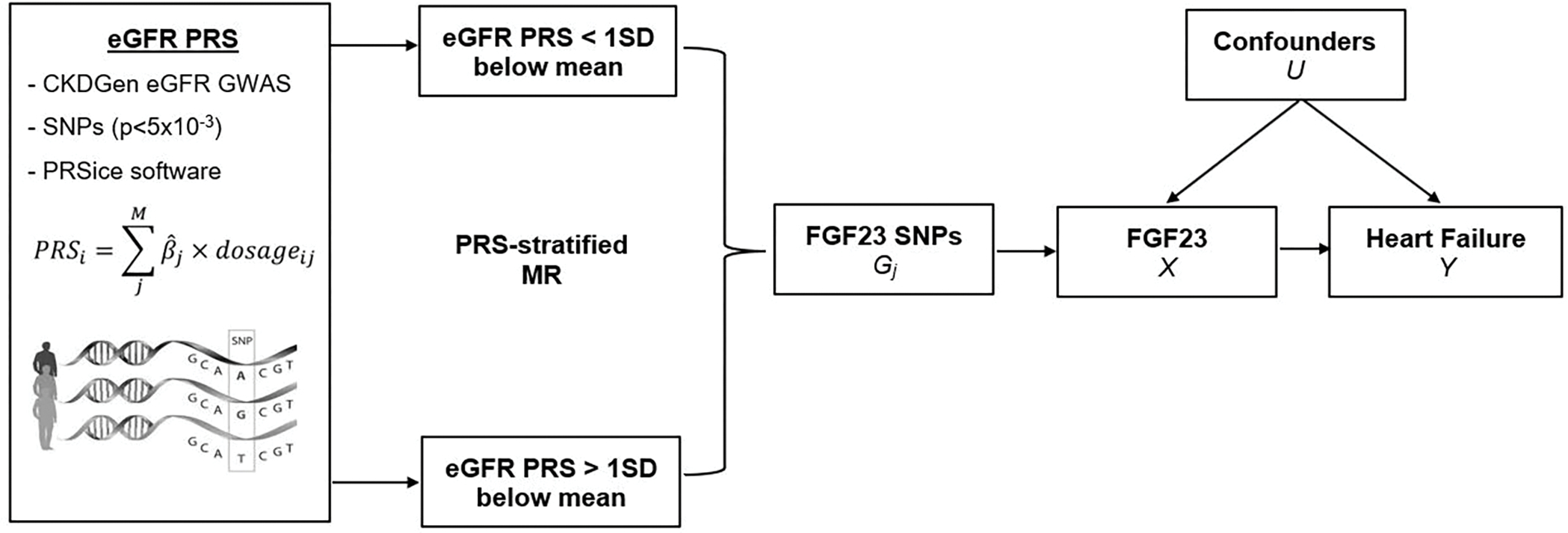
Polygenic risk score-stratified analysis for Mendelian randomization study of FGF23 and heart failure. Figure: An eGFR polygenic risk score was developed using summary statistics from the Chronic Kidney Disease Genetics Consortium (CKDGen) genome-wide association study of eGFR in >1 million individuals and stratified MR analyses of FGF23 and heart failure were performed across eGFR polygenic risk score strata.
More recently, Donovan and collaborators used a different approach to instrument selection, involving building a genetic risk score for FGF23,[39] based on a GWAS of FGF23 in 19,195 individuals of European ancestry from the Systematic and Combined Analysis of Olink Proteins (SCALLOP) consortium, a collaboration of 14 cohorts with proteomic assays for 90 cardiovascular proteins. Genetic instruments included in the GRS were 34 independent (r2<0.1) variants associated with FGF23 at p<5×10−6, explaining up to 6.4% of FGF23 variance. The GRS was validated in the ORIGIN consortium and included two cis-variants (variants within 100 kb of the FGF23 transcription start site, less likely to have unidentified pleiotropic effects, rs6489536 and rs7955866). Outcome data were from the UK Biobank (n=337,448) with 10,177 cases of HF hospitalizations and the HERMES consortium.[37] Both sets of analyses found no evidence of a causal association between genetically-predicted FGF23 and HF, OR = 1.01 (95%CI: 0.94–1.10) in the UK Biobank and 1.00 (95%CI: 0.95–1.05) in the HERMES consortium (Table 1). As these cohorts were both general population cohorts, these data re-emphasize the absence of a causal effect of FGF23 on HF risk in such settings.
Henry et al used the cis-variants in the FGF23 meta-GWAS in the SCALLOP consortium to perform a 2-sample cis-MR analysis of the FGF23-HF association. In the analysis using the only genome-wide significant cis-variant (rs6489536) the effect estimate was not different from the null (OR: 0.79, 95%CI: 0.62–1.02) (Table 1). When including a second cis-variant with a less stringent FGF23 GWAS p-value (p=2.7×10−7) there was unexpectedly evidence of a protective effect of FGF23 on HF risk (OR: 0.80, 95%CI: 0.71–0.89). Since cis-MR studies use variants that are near or within the cognate gene, if their effects are indeed causal and replicated, the findings are often used to inform drug target discovery and validation. However, both cis-variants explained only 0.4% of the variance of FGF23 and the directionality of the MR estimates obtained using these cis-variants was inconsistent with the bulk of the data from other MR studies using instruments explaining a greater proportion of the variance of FGF23. [15, 25, 39] Given that previous studies have established the significant role of trans-acting variants in the regulation of gene expression and consequently the circulating levels of the encoded proteins, it is difficult to predict how omitting trans-regulatory variants in the cis-MR analysis may have impacted the MR estimate. Importantly, analyses within subgroups of eGFR PRS and according to HF-subtype suggest that there is significant heterogeneity of effects. As such, MR analyses that do not account for these differences in associations may produce estimates that obfuscate specific pathophysiological pathways.
Current evidence from mendelian randomization studies of FGF23 and HFpEF
To our knowledge, only one MR study has examined the association of FGF23 with HF subtypes defined by LVEF cut-points. Using individual-level data in BioVU, the algorithm used to ascertain HF allowed for differentiation of cases by LVEF determined using transthoracic echocardiographic measurements [15] [38]. A case-control design was used to estimate the effects of the FGF23 genetic instruments (4 SNPs) on HFpEF (all LVEF measures ≥50%) and HFrEF (any left ventricular ejection fraction ≤40%). In the primary IVW analysis, genetically-predicted FGF23 was significantly associated with HFpEF (OR: 1.47, 95%CI: 1.01–2.14) but not HFrEF (OR: 0.86, 95%CI: 0.38–1.96) [15]. The findings from the eGFR PRS-stratified analysis were particularly striking for HFpEF. In the low eGFR PRS stratum, a one-unit higher genetically predicted logFGF23 was associated with an over 7-fold higher odds of HFpEF (OR:7.20, 95%CI: 2.80–18.5) while the association was not different from the null (OR:1.03, 95%CI: 0.67–1.57) in the high eGFR PRS stratum [15]. There was no evidence of a significant FGF23-HF association for HFrEF in either PRS strata.
Potential causal role of FGF23 on the risk of HFpEF
The contemporary debate on the potential causal role of FGF23 in the pathogenesis of HFpEF must be weighed against the totality of the evidence from observational studies, animal models, and MR experiments leveraging proteogenomic data.
Initial epidemiologic evidence described an association between elevated FGF23 levels and incident HF in general population [6, 8–10] and CKD settings [9] and subsequent data emphasized the association with HFpEF [10] which is thought to be mediated in part via the direct effect of FGF23 on cardiomyocyte hypertrophy [11–13]. Data from observational studies have highlighted the prospective independent association between elevated FGF23 at baseline and incident left ventricular hypertrophy (LVH) that was preponderantly concentric [11–13] – a typical feature of HFpEF.
In animal models, rat ventricular cardiomyocytes treated with FGF23 experienced activation of pro-hypertrophic genes and underwent pathological hypertrophy that was mediated via FGF receptor (FGFR)-dependent activation of the calcineurin-NFAT signaling pathway [11]. Additional experimental data showed that FGF23 induced pathological LVH [40] via activation of FGFR4, [41, 42] and blockade of FGFR4 attenuated this response [43] further emphasizing a direct likely causal effect of FGF23 in the development of LVH and the pathogenesis of HFpEF. Other mechanisms have been suggested to further explain the role of FGF23 on the heart. FGF23 potentially exacerbates diastolic dysfunction by promoting myocardial fibrosis via upregulation of TGF-β and β-catenin [44]. Elevated FGF23 may lead to LVH in a calcium-dependent pathway similar to angiotensin II and this pathway can be inhibited with angiotensin receptor blockers [45] and/or by directly regulating sodium chloride cotransporter expression in the distal convoluted tubule via a signaling mechanism involving the FGFR-klotho complex [46].
Further evidence of a potential causal role of FGF23 on HFpEF in CKD is provided by the striking results of the eGFR PRS-stratified MR analysis of the FGF23-HFpEF association in the BioVU biobank [15] highlighting the importance of large-scale EMR-linked genomics data for investigating causal effects. The observed 3-fold higher odds of overall HF compared to the more than 7-fold higher odds of HFpEF per log-unit higher genetically-predicted FGF23 among patients with a genetic liability for lower eGFR, implies that the association with overall HF was likely driven by the more specific association with HFpEF, which is supported by the biological and experimental data linking elevated FGF23 to concentric LVH that underlies HFpEF [1]. Secreted by osteocytes in the incipient stages of kidney dysfunction, FGF23 is well known for its role in suppressing type 2a sodium sodium-phosphate cotransporters to facilitate renal phosphate excretion. As circulating FGF23 levels rise sharply from 2 to 5-fold above normal in early CKD to reach 100 to 1000-fold in advanced kidney failure [47, 48], this potentially explains the stronger effect of FGF23 observed in low eGFR settings in the stratified MR analysis in the BioVU biobank.[15]
A recent study demonstrated a significant longitudinal association between reduction in serum FGF23 after kidney transplantation and improvements in cardiovascular functional capacity (VO2 max), suggesting a potential surrogate endpoint for randomized trials of FGF23-lowering interventions in advanced CKD [49, 50]. While there was no significant improvement in LV mass index with decreasing FGF23 in the study, the accompanying editorial underscored the fact that VO2 max is a measure of cardiopulmonary fitness for which diastolic dysfunction (which is a feature of HFpEF) is one of the components [49, 50]. Targeting improvements in diastolic dysfunction and consequently VO2 max in randomized trials for FGF23-lowering agents would provide compelling evidence that could influence clinical practice.
Conclusion
The aggregate evidence from epidemiologic, experimental, and genetic data suggests that while the association of FGF23 with overall HF in the general population may be driven in part by residual confounding, there appears to be a causal role for FGF23 in the pathogenesis of HFpEF in CKD patients that is likely mediated via the effect of pronounced chronically-elevated levels of FGF23 on concentric LVH and enhanced by the biological synergism between elevated FGF23 and CKD.
Taken together, these data suggest it may be time to consider designing randomized trials to test the safety and efficacy of FGF23 lowering agents in CKD patients. Meanwhile additional mendelian randomization studies could provide more critical evidence. Nonlinear MR for HFpEF using larger datasets with individual-level data could demonstrate dose-response revealing any threshold effects and multivariable mendelian randomization of several mineral metabolism markers with shared genetic architecture with HFpEF as the outcome could provide critical evidence further deconstructing the mechanisms by which FGF23 is implicated in the pathogenesis of HFpEF.
Summary
Elevated FGF23 has been associated with heart failure in observational and experimental studies, yet it is unknown whether FGF23 levels themselves represent causal processes for complications, how kidney function impacts these processes, and if it may be a promising interventional target.
Mendelian randomization represents an inexpensive alternative or adjunct to randomized controlled trials and incorporates genetic information into traditional epidemiological methods to address the causality of a risk factor for important clinical outcomes.
Several Mendelian randomization studies of FGF23 and heart failure have been conducted and the consensus is that while the association of FGF23 with overall heart failure may be driven by residual confounding, there appears to be a causal role for FGF23 in the pathogenesis of HFpEF in CKD patients.
Results from the MR analyses have the potential to suggest new therapies for mineral metabolism disturbances and to identify pathways/targets amenable to pharmaceutical or biological intervention.
Financial Support and Sponsorship
Dr. Robinson-Cohen and Dr. Akwo’s work is supported in part by the National Institutes of Diabetes and Digestive and Kidney Diseases (NIDDK: R01-DK122075 and R01-DK132155).
Footnotes
Conflicts of Interest
There are no conflicts of interest.
References
- [1].Go AS, Chertow GM, Fan D et al. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. The New England journal of medicine 2004; 351:1296–1305. [DOI] [PubMed] [Google Scholar]
- [2].Herzog CA, Asinger RW, Berger AK et al. Cardiovascular disease in chronic kidney disease. A clinical update from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney international 2011; 80:572–586. [DOI] [PubMed] [Google Scholar]
- [3].London GM, Guérin AP, Marchais SJ et al. Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 2003; 18:1731–1740. [DOI] [PubMed] [Google Scholar]
- [4].Palmer SC, Hayen A, Macaskill P et al. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: a systematic review and meta-analysis. Jama 2011; 305:1119–1127. [DOI] [PubMed] [Google Scholar]
- [5].Covic A, Vervloet M, Massy ZA et al. Bone and mineral disorders in chronic kidney disease: implications for cardiovascular health and ageing in the general population. The lancet. Diabetes & endocrinology 2018; 6:319–331. [DOI] [PubMed] [Google Scholar]
- [6].Ix JH, Katz R, Kestenbaum BR et al. Fibroblast growth factor-23 and death, heart failure, and cardiovascular events in community-living individuals: CHS (Cardiovascular Health Study). J Am Coll Cardiol 2012; 60:200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kestenbaum B, Sachs MC, Hoofnagle AN et al. Fibroblast growth factor-23 and cardiovascular disease in the general population: the Multi-Ethnic Study of Atherosclerosis. Circ Heart Fail 2014; 7:409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lutsey PL, Alonso A, Selvin E et al. Fibroblast growth factor-23 and incident coronary heart disease, heart failure, and cardiovascular mortality: the Atherosclerosis Risk in Communities study. J Am Heart Assoc 2014; 3:e000936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Scialla JJ, Xie H, Rahman M et al. Fibroblast growth factor-23 and cardiovascular events in CKD. J Am Soc Nephrol 2014; 25:349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Almahmoud MF, Soliman EZ, Bertoni AG et al. Fibroblast Growth Factor-23 and Heart Failure With Reduced Versus Preserved Ejection Fraction: MESA. J Am Heart Assoc 2018; 7:e008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Faul C, Amaral AP, Oskouei B et al. FGF23 induces left ventricular hypertrophy. The Journal of clinical investigation 2011; 121:4393–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gutiérrez OM, Januzzi JL, Isakova T et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation 2009; 119:2545–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mirza MA, Larsson A, Melhus H et al. Serum intact FGF23 associate with left ventricular mass, hypertrophy and geometry in an elderly population. Atherosclerosis 2009; 207:546–551. [DOI] [PubMed] [Google Scholar]
- [14].Marthi A, Donovan K, Haynes R et al. Fibroblast Growth Factor-23 and Risks of Cardiovascular and Noncardiovascular Diseases: A Meta-Analysis. J Am Soc Nephrol 2018; 29:2015–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15]. Akwo E, Pike MM, Ertuglu LA et al. Association of Genetically Predicted Fibroblast Growth Factor-23 with Heart Failure: A Mendelian Randomization Study. Clinical journal of the American Society of Nephrology : CJASN 2022; 17:1183–1193. ** This two-sample mendelian randomization study investigates the causal nature of the association of FGF23 with several heart failure phenotypes and specifically examines the heterogeneity in the association between FGF23 and HFpEF across low and high polygenic risk scores strata for estimated glomerular filtration rate using individual-level data from the Vanderbilt DNA Biobank (BioVU).
- [16]. Donovan K, Herrington WG, Paré G et al. Fibroblast Growth Factor-23 and Risk of Cardiovascular Diseases: A Mendelian Randomization Study. Clinical journal of the American Society of Nephrology : CJASN 2023; 18:17–27. ** This mendelian randomization study uses a 34-SNP genetic risk score based on weights from the SCALLOP consortium to investigate the association of FGF23 with heart failure, other nonatherosclerotic cardiovascular outcomes and atherosclerotic cardiovascular disease in the UK Biobank and HERMES consortia.
- [17]. Henry A, Gordillo-Marañón M, Finan C et al. Therapeutic Targets for Heart Failure Identified Using Proteomics and Mendelian Randomization. Circulation 2022; 145:1205–1217. * This study performs cis-MR analyses for 88 cardiovascular proteins including FGF23 measured using proteomic assays and identified via meta-analysis of observational studies as potential therapeutic targets for heart failure.
- [18]. Liang Y, Luo S, Schooling CM, Au Yeung SL. Genetically Predicted Fibroblast Growth Factor 23 and Major Cardiovascular Diseases, Their Risk Factors, Kidney Function, and Longevity: A Two-Sample Mendelian Randomization Study. Frontiers in Genetics 2021; 12. * This study performs two-sample MR analyses of FGF23 with heart failure and several other cardiometabolic outcomes including CAD, diabetes, glycemic and lipid traits using summary-level GWAS data from several large consortia.
- [19].Lawlor DA, Harbord RM, Sterne JA et al. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Statistics in medicine 2008; 27:1133–1163. [DOI] [PubMed] [Google Scholar]
- [20].Smith GD, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 2003; 32:1–22. [DOI] [PubMed] [Google Scholar]
- [21].Davey Smith G, Holmes MV, Davies NM, Ebrahim S. Mendel’s laws, Mendelian randomization and causal inference in observational data: substantive and nomenclatural issues. European journal of epidemiology 2020; 35:99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Matías-García PR, Wilson R, Guo Q et al. Plasma Proteomics of Renal Function: A Transethnic Meta-Analysis and Mendelian Randomization Study. J Am Soc Nephrol 2021; 32:1747–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sun BB, Maranville JC, Peters JE et al. Genomic atlas of the human plasma proteome. Nature 2018; 558:73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fauman EB, Hyde C. An optimal variant to gene distance window derived from an empirical definition of cis and trans protein QTLs. BMC Bioinformatics 2022; 23:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Robinson-Cohen C, Bartz TM, Lai D et al. Genetic Variants Associated with Circulating Fibroblast Growth Factor 23. JASN 2018; 29:2583–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Folkersen L, Gustafsson S, Wang Q et al. Genomic and drug target evaluation of 90 cardiovascular proteins in 30,931 individuals. Nature metabolism 2020; 2:1135–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zheng J, Baird D, Borges MC et al. Recent Developments in Mendelian Randomization Studies. Current epidemiology reports 2017; 4:330–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Burgess S, Thompson SG. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol 2011; 40:755–764. [DOI] [PubMed] [Google Scholar]
- [29].Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genetic epidemiology 2013; 37:658–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Burgess S, Small DS, Thompson SG. A review of instrumental variable estimators for Mendelian randomization. Statistical methods in medical research 2017; 26:2333–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Burgess S, Thompson SG. Use of allele scores as instrumental variables for Mendelian randomization. Int J Epidemiol 2013; 42:1134–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. European journal of epidemiology 2017; 32:377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genetic epidemiology 2016; 40:304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015; 44:512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Psaty BM, O’Donnell CJ, Gudnason V et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet 2009; 2:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shah S, Henry A, Roselli C et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat Commun 2020; 11:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bielinski SJ, Pathak J, Carrell DS et al. A Robust e-Epidemiology Tool in Phenotyping Heart Failure with Differentiation for Preserved and Reduced Ejection Fraction: the Electronic Medical Records and Genomics (eMERGE) Network. Journal of cardiovascular translational research 2015; 8:475–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Donovan K, Herrington WG, Pare G et al. Fibroblast Growth Factor-23 and Risk of Cardiovascular Diseases: A Mendelian Randomization Study. Clin J Am Soc Nephrol 2023; 18:17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nature reviews. Molecular cell biology 2006; 7:589–600. [DOI] [PubMed] [Google Scholar]
- [41].Grabner A, Schramm K, Silswal N et al. FGF23/FGFR4-mediated left ventricular hypertrophy is reversible. Scientific reports 2017; 7:1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Leifheit-Nestler M, Haffner D. Paracrine Effects of FGF23 on the Heart. Frontiers in endocrinology 2018; 9:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Di Marco GS, Reuter S, Kentrup D et al. Treatment of established left ventricular hypertrophy with fibroblast growth factor receptor blockade in an animal model of CKD. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 2014; 29:2028–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hao H, Li X, Li Q et al. FGF23 promotes myocardial fibrosis in mice through activation of β-catenin. Oncotarget 2016; 7:64649–64664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mhatre KN, Wakula P, Klein O et al. Crosstalk between FGF23- and angiotensin II-mediated Ca(2+) signaling in pathological cardiac hypertrophy. Cellular and molecular life sciences : CMLS 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Andrukhova O, Slavic S, Smorodchenko A et al. FGF23 regulates renal sodium handling and blood pressure. EMBO molecular medicine 2014; 6:744–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Isakova T, Wahl P, Vargas GS et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney international 2011; 79:1370–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Shimada T, Urakawa I, Isakova T et al. Circulating fibroblast growth factor 23 in patients with end-stage renal disease treated by peritoneal dialysis is intact and biologically active. The Journal of clinical endocrinology and metabolism 2010; 95:578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Halim A, Burney HN, Li X et al. FGF23 and Cardiovascular Structure and Function in Advanced Chronic Kidney Disease. Kidney360 2022; 3:1529–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Murray SL, Wolf M. Exercising the FGF23-Cardiac Axis. Kidney360 2022; 3:1471–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]