

Summary
Genetic alterations help predict the clinical behavior of diffuse gliomas, but some variability remains uncorrelated. Here, we demonstrate that haploinsufficient deletions of chromatin-bound tumor suppressor NFKB inhibitor alpha (NFKBIA) display distinct patterns of occurrence in relation to other genetic markers and are disproportionately present at recurrence. NFKBIA haploinsufficiency is associated with unfavorable patient outcomes, independent of genetic and clinicopathologic predictors. NFKBIA deletions reshape the DNA and histone methylome antipodal to the IDH mutation and induce a transcriptome landscape partly reminiscent of H3K27M mutant pediatric gliomas. In IDH mutant gliomas, NFKBIA deletions are common in tumors with a clinical course similar to that of IDH wild-type tumors. An externally validated nomogram model for estimating individual patient survival in IDH mutant gliomas confirms that NFKBIA deletions predict comparatively brief survival. Thus, NFKBIA haploinsufficiency aligns with distinct epigenome changes, portends a poor prognosis, and should be incorporated into models predicting the disease fate of diffuse gliomas.
Keywords: glioma, NFKBIA deletion, haploinsufficiency, H3K27M mutation, IDH mutation, nomogram, tumor suppressor, methylome
Graphical abstract
Highlights
-
•
NFKBIA deletions exhibit a distinct pattern relative to other key genetic markers
-
•
NFKBIA deletions reshape the DNA and histone methylome antipodal to the IDH mutation
-
•
NFKBIA deletions engender a transcriptome landscape reminiscent of H3K27M mutant glioma
-
•
NFKBIA deletions independently portend poor patient prognosis
Bredel et al. characterize multiple adult diffuse glioma populations and reveal that haploinsufficient deletions of NFKBIA portend poor patient prognosis and align with epigenome and transcriptome landscapes that are antithetical to those induced by the IDH mutation and partly reminiscent of those induced by the H3K27M mutation in pediatric gliomas.
Introduction
Gliomas are the most frequently occurring primary brain tumors. Comprehensive genome-wide characterization has illuminated their molecular complexity.1,2,3,4,5,6,7 Among their key genetic alterations identified, mutations of isocitrate dehydrogenase 1 (IDH1) and 2 (IDH2) genes (collectively referred to as IDH),8 mutations in the promoter of telomerase reverse transcriptase (TERT) and ATRX chromatin remodeler (ATRX) genes,9,10 and codeletion of chromosome arms 1p and 19q (1p19q codeletion)11 are associated with glioma subsets possessing distinct clinical trajectories.1 Moreover, homozygous deletion of cyclin-dependent kinase inhibitor 2A/B (CDKN2A/B) is a biomarker of grade and prognosis, especially in IDH mutant gliomas.12 These alterations are currently the most potent genetic markers predicting glioma aggressiveness.1 IDH mutations manifest the epigenetic glioma cytosine-phosphate-guanine (CpG) island methylator phenotype (G-CIMP),13 which portends a favorable prognosis,14,15 as does high vs. low G-CIMP.4,16 Mutations (lysine-to-methionine substitution) in histone H3 (H3K27M)—involving the H3F3A locus of histone variant H3.3 and the HIST1H3B locus in the H3.1 or H3.2 variants17—characterize a subgroup of aggressive diffuse midline gliomas in children and young adults.18,19 H3K27M mutant oncohistone induces a dose-dependent inhibition of the H3K27me3 methyltransferase complex, polycomb repressive complex 2 (PRC2).20,21 PRC2 orchestrates genome architecture and mediates silencing of developmental genes during lineage specification and commitment.22
The NFKB inhibitor alpha (NFKBIA) gene at 14q13.2 encodes the alpha subunit of the inhibitors of κB (IκBα), proteins that regulate the activity of transcription factor nuclear factor κB (NF-κB) in the cytoplasm.23 Evidence that, in chromatin, nuclear NFKBIA dynamically interacts with histones H2A and H4 to regulate polycomb-dependent transcriptional repression and, thus, stem cell maturation, lineage specification, and cancer24,25,26 and our characterization of NFKBIA as a tumor suppressor in glioblastoma27 prompted our investigation of the relationship of NFKBIA deletions with other genetic markers, alterations in the methylome, and the clinical course of gliomas. We analyzed the genetic profiles of gliomas of multiple well-characterized patient populations to determine whether incorporation of NFKBIA deletions could enhance the prognostic value of current molecular descriptions.1,4
Results
Overview of patient population characteristics
We analyzed 2,255 patients and their 2,343 diffuse gliomas. The demographic and disease characteristics of patients of seven glioma populations are summarized in Table S1, and their clinicopathologic-genetic details are listed in Table S2. We used population 1 to test the relationship between NFKBIA deletions and key genetic alterations and survival in lower-grade (World Health Organization [WHO] grade 2–3) gliomas and populations 2 and 3 for validation; population 4 to test the relationship of NFKBIA with key genetic alterations and survival in WHO grade 2–4 gliomas and population 5 for validation; population 6 to assess the relationship between NFKBIA deletion and tumor recurrence; and population 7 to study the intersection of gene signatures associated with NFKBIA deleted gliomas and H3K27M mutant diffuse intrinsic pontine gliomas (DIPGs).
Deletion of NFKBIA in lower-grade gliomas
We observed a common hemizygous deletion encompassing NFKBIA in 68 of 513 (13.3%) lower-grade gliomas in population 1 (Figure 1A). NFKBIA deletions ranged from small (12.3 kbp) losses involving only the NFKBIA locus (chr14: 35,401,079–35,404,749) to larger chromosomal regions encompassing NFKBIA (median: 36.6 Mbp, interquartile range: 43.5 Mbp), to those extending to the 14q terminus. In the three main genetic groups of adult lower-grade gliomas—IDH mutant/1p19q codeleted/TERT mutant, IDH mutant/1p19q non-codeleted/ATRX mutant, and IDH wild-type/TERT mutant—we found no predilection for either IDH mutant group (9.3% and 6.7%, respectively) but did find higher presence in the IDH wild-type group (30.6%) (Figure 1A). NFKBIA deletions occurred in 4 of 87 (4.6%) WHO grade 2 gliomas of population 2 and in 60 of 694 (8.6%) lower-grade gliomas of population 3.
Figure 1.

NFKBIA deletions and expression in lower-grade gliomas
(A) Heatmap and two-way contingency tables showing the relationship between NFKBIA deletions and genetic alterations—IDH, TERT, ATRX, and TP53 mutations, 1p19q codeletions, and CDKN2A/B deletions—and clinicopathologic variables in 516 lower-grade gliomas of population 1. OR, odds ratio; CI, confidence interval.
(B) Expression of the six NFKBIA exons—in log2-transformed RPKM (reads per kilobase of exon model per million mapped reads)—in 513 NFKBIA deleted vs. wild-type lower-grade gliomas of population 1. Five rare missense or truncating mutations are mapped in relation to the exons and various NFKBIA protein motifs (DM, destruction motif; NES, nuclear export signal; NIS, nuclear import signal) and ankyrin (Ank) repeats. Error bars represent ±standard deviation. Wilcoxon rank-sum test.
(C) NFKBIA protein expression—by immunofluorescence (IF)—in two glioma samples with (+/–) and without (+/+) hemizygous NFKBIA deletion—by fluorescence in situ hybridization (FISH), indicating a haploinsufficiency effect. Red dots (arrowheads) represent one NFKBIA allele. Scale bars, 20 μm.
Sequencing analysis showed that NFKBIA is rarely mutated in lower-grade gliomas. Only 3 of 508 (0.6%) tumors of population 1 harbored a total of four missense mutations and one truncating mutation in the coding region of NFKBIA (Figure 1B). Expression of all six NFKBIA exons in tumors in which NFKBIA was deleted was significantly lower than in tumors with two intact copies of NFKBIA (Figures 1B and 1C).
NFKBIA and key driver mutations in lower-grade gliomas
We sought to define the relationship between NFKBIA deletion and IDH, TERT, ATRX, and tumor protein P53 (TP53) mutations, 1p19q codeletions, and CDKN2A/B deletions, major early molecular alterations strongly associated with overall survival in lower-grade gliomas.1,12 We observed distinct patterns of occurrence between NFKBIA deletion and the IDH, TERT, and ATRX mutations and between NFKBIA and CDKN2A/B deletions but no relationship with TP53 mutations or 1p19q codeletions in population 1 (Figure 1A). We observed similar patterns between NFKBIA deletions and IDH mutations in population 2 and between NFKBIA deletions and IDH and TERT mutations in population 3 (Figure S1). We also noted an association of NFKBIA deletions with patient age in population 1 and population 3—patients older than 40 had a higher deletion frequency—but not in population 2 (Figures 1A and S1).
NFKBIA deletions and survival in patients with lower-grade gliomas
Association between NFKBIA deletions and survival was established in three groups of patients with newly diagnosed lower-grade glioma. Patients in population 1 with NFKBIA deleted tumors had shorter median survival (4.3 vs. 8.0 years) than those with non-deleted tumors (Figure 2A). Cox proportional-hazards regression confirmed that patients with NFKBIA deleted tumors had briefer survival (Figure S2A). An adjusted Cox model suggested that this association was independent of prognostic covariates, including IDH, TERT, ATRX, and TP53 mutations, 1p19q codeletions, CDKN2A/B deletions, WHO grade, tumor histology, and patient age (Figure S2A). NFKBIA deletion also portended shorter recurrence-free survival (median 3.2 vs. 6.0 years) in population 1 (Figure 2B).
Figure 2.

NFKBIA deletions and survival in patients with lower-grade gliomas
(A−B) Kaplan-Meier estimates of overall survival (A) and recurrence-free survival (B) for 508 and 425 patients in population 1, respectively, with patients stratified according to the presence (del) or absence (wild-type, wt) of the NFKBIA deletion. CI denotes confidence interval. Small vertical lines indicate patients alive at last follow-up assessment. p values were calculated using the Cox model likelihood-ratio test. Patients at risk (No. at Risk) correspond to the x axis timescale.
(C and D) Kaplan-Meier estimates for the relationship between NFKBIA deletions and patient overall survival (C) and recurrence-free survival (D) in 87 low-grade gliomas patients of NRG/RTOG consortium trial 9802 (population 2).
(E) Kaplan-Meier estimates for the relationship between NFKBIA deletions and overall survival in population 3.
Similarly, mRNA expression data of population 1 showed association between NFKBIA expression and duration of overall survival when expression was dichotomized at the median. Association between NFKBIA expression and survival held in an adjusted Cox model that included the IDH mutation and other genetic and clinicopathologic variables (Figure S3).
We validated the relationship between NFKBIA deletions in low-grade (WHO grade 2) glioma patients of NRG Oncology/RTOG 9802 (population 2) and both overall survival and recurrence-free survival (Figures 2C and 2D). Median overall and recurrence-free survival for deleted vs. non-deleted tumors was 1.0 vs. 11.4 years and 0.7 vs. 5.1 years, respectively. Lest the smaller overall sample size and low variant frequency for NFKBIA deletion in population 2 inflate variance estimates, we included only the most relevant pathogenetic variables in an adjusted model. NFKBIA deletion remained independently associated with overall survival after adjusting for IDH mutation, 1p19q codeletion, and patient age (Figure S2B). As reported, patients who received both chemotherapy and radiation therapy survived longer than those who received radiation therapy alone.28 In an adjusted Cox model, NFKBIA was associated with survival independent of treatment randomization (Figure S2B).
We also observed a significant relationship between NFKBIA and survival in the lower-grade gliomas of population 3 (Figure 2E). Because low variant frequency and some collinearity (e.g., between IDH and ATRX mutations and patient age) precluded modeling all available genetic and clinicopathologic predictors for population 3 in one statistical model, we developed separate clinicopathologic and genetic models, both of which showed that NFKBIA deletions were independently associated with overall survival duration (Figure S2C).
Deletion of NFKBIA in WHO grade 2–4 diffuse gliomas
Molecular genetic alterations are used to predict the clinical behavior, response to therapy, and outcome of diffuse gliomas irrespective of glioma grade and histology.1,2,4,12 We therefore examined the occurrence of NFKBIA deletions in WHO grade 2–4 gliomas relative to these alterations (Figure 3A). Among diffuse gliomas of population 4, we observed a patterned occurrence between NFKBIA deletions and IDH, ATRX, TERT, and PTEN mutations, 1p19q codeletions, CDKN2A/B deletions, and chromosome 7 gain/10 loss, but not with TP53 mutations or EGFR amplifications (Figure 3A).
Figure 3.

NFKBIA deletions in WHO grade 2–4 gliomas
(A) NFKBIA deletions in relation to driver genetic alterations—the IDH, TERT, ATRX, TP53, and PTEN mutations, 1p19 codeletions, CDKN2A/B deletions, EGFR amplifications, and combined gains of chromosome 7 and monosomy 10 (7+/10−)—in 1,122 WHO grade 2–4 gliomas of population 4. OR, odds ratio; CI, confidence interval.
(B and C) Kaplan-Meier estimates of overall survival for 1,027 patients in population 4 (B) and 473 patients in population 5 (C), with patients stratified according to the presence (del) or absence (wild-type, wt) of the NFKBIA deletion. Small vertical lines indicate patients alive at last follow-up assessment. p values were calculated using the Cox model likelihood-ratio test. Patients at risk (No. at Risk) correspond to the x axis timescale. Bottom panels show corresponding adjusted Cox proportional-hazards models, with NFKBIA and other genetic (as available) and clinicopathologic factors as the primary predictor. Hazard ratios for death are displayed for presence vs. absence of the genetic alterations.
In two-class models of population 4 and population 5 patients stratified by NFKBIA deletion status, those with NFKBIA deleted tumors had briefer overall survivals (Figures 3B and 3C). Median survival times for tumors with and without NFKBIA deletions were 1.3 and 2.2 years in population 4 and 1.5 and 7.7 years in population 5, respectively. Adjusted Cox models revealed that the association between NFKBIA status and duration of overall survival was independent of covariates IDH, TERT, ATRX, TP53, and PTEN mutations, 1p19q codeletion, CDKN2A/B deletion, chromosome 7 gain/10 loss, EGFR amplification, WHO grade, tumor histology, patient age, and patient gender in population 4 and of available covariates IDH, TERT, and ATRX mutations, 1p19q codeletion, WHO grade, tumor histology, patient age, and patient gender in population 5 (Figures 3B and 3C). On subgroup analysis, the survival association of NFKBIA deletions in populations 4 and 5 was primarily evident in IDH mutant tumors (Figures 7A and 7B). Also, NFKBIA deletion was associated with shorter patient survival for the subgroups of tumors with either TERT mutation or WHO grade 3 class (Figure S4). Median overall survival durations of TERT mutant tumors with or without NFKBIA deletions were 1.9 vs. 4.3 years in population 4 and 1.4 vs. 3.2 years in population 5, respectively (Figures S4A and S4B). Median overall survival durations of WHO grade 3 gliomas with or without NFKBIA deletions were 2.2 vs. 4.6 years in population 4 and 1.7 vs. 14.5 years in population 5, respectively (Figures S4C and S4D).
Figure 7.

NFKBIA deletions and survival in IDH mutant WHO grade 2–4 gliomas
(A and B) Kaplan-Meier estimates of overall survival for 398 IDH mutant glioma patients in population 4 (A) and 291 IDH mutant glioma patients in population 5 (B), with patients stratified according to the presence (del) or absence (wild-type, wt) of the NFKBIA deletion. Small vertical lines indicate patients alive at last follow-up assessment. p values were calculated using the Cox model likelihood-ratio test. Patients at risk (No. at Risk) correspond to the x axis timescale.
(C and D) Corresponding nomograms for predicting the probability of survival at 5 years in IDH mutant gliomas. Nomograms built based on high-dimensional penalized Cox models using the adaptive least absolute shrinkage and selection operator (adaLASSO) function. Models include the covariates NFKBIA deletion, 1p19q codeletion, TERT, and ATRX mutations, WHO grade, and patient age and were internally validated and calibrated by repeated cross-validation in population 4, then externally validated and calibrated in population 5. Displayed variables are those retained as significant. Model performance by time-dependent area under the receiver-operating curve (ROC) (t-AUC) over 180 months (15 years). Curves reflect average performance for each cross-validation event. The solid line represents the mean of the area under the t-AUC and the dashed line the median of the AUC. The darker interval shows 25% and 75% quantiles of the t-AUC, and the lighter interval shows the minimum and maximum of the t-AUC.
Since the NFKBIA deletion is enriched in recurrent diffuse gliomas (Figure 4A), we examined the association between the deletion and recurrence-free survival. In population 4, NFKBIA deletion denoted shorter recurrence-free survival in a two-class model and an adjusted Cox model that included covariates IDH, TERT, and ATRX mutations, 1p19q codeletion, WHO grade, tumor histology, patient age, and patient gender (Figure S5).
Figure 4.

The epigenome landscape of NFKBIA deleted gliomas
(A) Deletion (del) frequencies of NFKBIA in matched primary tumors vs. first and second recurrence in 180 glioma samples belonging to 87 WHO grade 2–4 gliomas in population 6. †Tumor lost a second NFKBIA allele during progression. CI denotes confidence interval.
(B) Contingency table of relationships between NFKBIA deletions and seven methylation subtypes of glioma in 1,122 WHO grade 2–4 gliomas of population 4. Cramér’s V indicates effect size. Panel on the right shows frequencies of these methylation subtypes according to NFKBIA status.
(C) Circular genome (CIRCOS) visualization of DNA regions (or CpG sites) hypomethylated in NFKBIA deleted vs. wild-type gliomas (false discovery rate [FDR] < 0.001). Data were generated in 609 WHO grade 2–4 gliomas of population 4 and validated in 87 WHO grade 2 gliomas of population 2. Orange dots in ring “a” denote genome-wide CpGs validated with an FDR of <0.1 (93.0%), blue dots denote non-validated CpGs. Orange dots in rings “b” and “c” indicate overlap of these hypomethylated CpGs with CpG signatures found to denote glioma methylation subtypes (“pan-glioma-specific”) or to distinguish G-CIMP-low from G-CIMP-high IDH mutant tumors. The height of each ring represents the beta-value—the ratio of methylated probe intensity and the sum of methylated and unmethylated probe intensities—difference range of [0.15, 0.3] between NFKBIA wild-type and deleted tumors. Dots (CpG sites) with an absolute beta difference of ≥0.2 are encapsulated by a black line. Corresponding two-way contingency tables show cosegregation between CpGs hypomethylated in NFKBIA deleted tumors and these CpG signatures. Also shown are two-way contingency tables for the cosegregation of CpGs hypomethylated in NFKBIA deleted tumors in general or in the subpopulation of IDH mutant tumors with NFKBIA deletion and CpGs that map into repressive trimethylated histone 3 lysine 27 (H3K27me3) marks. CI denotes confidence interval.
(D) NFKBIA protein expression in primary cultures of human astrocytes carrying the IDH1-R132H mutation after siRNA-mediated near-complete (si-NFKBIA) knockdown or CRISPR-mediated (crispr [cr]-NFKBIA) knockout, compared with scrambled control (Ctrl) versions. β-Actin as a loading control. Panel on the right shows global H3K27me3 levels in si-NFKBIA knockdown and crispr-NFKBIA knockout cells compared with control cells. Wilcoxon rank-sum test. Error bars represent standard error of the mean (±SEM) from three biological replicates.
(E) Bar graphs show mRNA expression—by real-time PCR average (avg) fold relative quantification (RQ)—of the SLC16A3 glioma CpG island methylator phenotype (G-CIMP) gene, its promoter binding to NFKBIA or the PRC2 catalytic subunit EZH2, and the promoter’s 5-methylcytosine (5-mC) and repressive H3K27me3 and H3K9me3 marks—all based on chromatin immunoprecipitation (ChIP) and expressed as percent of input sample representing the amount of chromatin used in ChIP—following si-NFKBIA knockdown in IDH mutant primary human astrocytes compared with their control counterparts. Wilcoxon rank-sum test. Error bars represent ±SEM from three biological replicates.
Deletions of NFKBIA during glioma progression
Of 180 diffuse—WHO grade 2–4—gliomas from population 6, 42 (23.3%) carried NFKBIA deletions. Among 87 matched pairs of primary and recurrent tumors, 30 (34.5%) pairs had NFKBIA deletions in the primary tumor, the recurrent tumor, or both (Figure 4A). Among the 30 patients with NFKBIA deletions, 27 (90%) retained the deletion or acquired a new deletion during tumor progression (Figure 4A). Deletions were more frequent in recurrent tumors (31.0%) than in primary tumors (16.1%) (Figure 4A). Among 74 multiply recurrent tumors, the deletion frequency was even higher in the second recurrence (45.5%) (Figures 4A and S6A). These data imply that diffuse gliomas acquire, or select for, NFKBIA deletions during disease progression.
Deletion of NFKBIA and molecular glioma subtype
Gliomas have seven cohesive methylation subtypes: three IDH mutant subtypes (G-CIMP-low, G-CIMP-high, and Codel) and four IDH wild-type subtypes (classic-like, mesenchymal-like, LGm6-GBM, and pilocytic astrocytoma-like).4 In population 4, NFKBIA deletions were particularly enriched in three glioma subtypes with low epigenomic DNA methylation2,29: IDH mutant G-CIMP-low (32.0%), IDH-wild-type mesenchymal-like (42.1%), and LGm6 (42.5%) gliomas (Figure 4B). Among IDH mutant gliomas, the G-CIMP-low subtype carries a particularly dismal prognosis.4,16 Despite their cosegregation with this unfavorable subtype (Figure S6B), NFKBIA deletions remained independently associated with survival in an adjusted Cox model that included as covariates the methylation subtypes and IDH, TERT, ATRX, TP53, and PTEN mutations, 1p19q codeletion, CDKN2A/B deletion, chromosome 7 gain/10 loss, EGFR amplification, WHO grade, tumor histology, and patient age and gender (Figure S6C).
NFKBIA deletion is associated with epigenome hypomethylation
We identified genome-wide methylation targets of the NFKBIA deletion in the 609 gliomas of population 4 from genome methylation data. Tumors with NFKBIA deletions had a distinct epigenome footprint of widespread DNA hypomethylation (Figure 4C and Table S3); it encompassed 26.3% of all genes with NFKBIA regulatory regions (Figure S6D and Table S3)24 and was highly enriched for pan-glioma CpGs, by whose prevalence gliomas can be classified into distinct DNA methylation subtypes (Figure 4C and Table S3).4 This was particularly prominent for CpGs whose prevalence robustly distinguishes the two discrete subtypes of 1p19q non-codeleted IDH mutant gliomas, the G-CIMP-high vs. G-CIMP-low subtype (Figure 4C and Table S3).4
In IDH mutant gliomas in population 4, CpGs hypomethylated in NFKBIA deleted tumors (Table S3) overlapped significantly with those hypomethylated in all NFKBIA deleted gliomas (73.7%) (Figure S6E and Table S3), and the association of those CpGs, whose presence denotes the G-CIMP-low subtype, remained highly significant (Figure S7A).
We validated the epigenomic hypomethylation signature associated with NFKBIA deleted gliomas in 87 low-grade gliomas of population 2 for whom whole-genome DNA methylome data were generated. Almost all CpGs (93.0%) were similarly demethylated in tumors with the NFKBIA deletion relative to those without the deletion (Figure 4C and Table S3).
Examination of methylome changes of NFKBIA deleted gliomas for functionally related sets of genes in population 4 found that genes hypomethylated in the NFKBIA deleted tumors relative to wild-type tumors function in nervous system development, in gliomas in general, and IDH mutant gliomas in particular (Figure S7B and Table S3). These genes were highly enriched for a gene cluster in the brain with high-CpG-density promoters bearing trimethylated histone 3 marks (Figure S7C and Table S3). Given this enrichment, the link between DNA and histone methylation30,31 and the importance of chromatin modifications in the regulation of gene transcription,32,33 we further examined the epigenomic location of hypomethylated CpG sites relative to distinct histone marks. Most of these CpGs mapped to defined histone modification regions, and their locations notably overlapped with that of the repressive histone 3 lysine 27 (H3K27me3) mark in all gliomas (56%) and in IDH mutant gliomas (49%) (Figure 4C and Table S3).
NFKBIA deletion reshapes the DNA and histone methylome antithetically to the IDH mutation
Introducing IDH mutations into primary human astrocytes establishes the G-CIMP phenotype.13 The methylation targets of mutant IDH in this astrocyte system significantly overlapped with the epigenomic gene signatures of NFKBIA deleted and IDH mutated/NFKBIA deleted gliomas of population 4 (Figure S8A and Table S3). We therefore tested the effects of near-complete small interfering RNA (siRNA)-mediated NFKBIA knockdown on epigenome reprogramming in this IDH1 mutant system. The NFKBIA knockdown was associated with reduction in global histone H3K27 trimethylation—a hallmark of promoter activation—in the IDH1 mutant astrocytes, but less so in parental wild-type astrocytes (Figures 4D and S8B), indicating that the histone hypermethylation landscape engendered by the IDH mutation13,34 is particularly sensitive to NFKBIA depletion. Clustered regularly interspaced short palindromic repeats (CRISPR)-mediated NFKBIA knockout reduced the global increase in H3K27me3 caused by the IDH mutation to an extent similar to that of siRNA-mediated NFKBIA knockdown (Figure 4D).
We then assessed the effects of NFKBIA knockdown on a representative G-CIMP gene—the monocarboxylate transporter gene SLC16A313,35—in the same IDH1 mutant astrocytes. Although the SLC16A3 promoter is hypermethylated and enriched for repressive heterochromatin H3K27 and H3K9 marks in these IDH1 mutant cells,13 the promoter was hypomethylated in the NFKBIA deleted tumors compared with wild-type tumors of population 4 (Figure S8C). NFKBIA can repress gene expression by binding chromatin in regions containing such histone methylation marks.24 We found that NFKBIA protein directly interacts with the promoter of SLC16A3 and that NFKBIA depletion in these astrocytes reversed the repressive DNA and histone trimethylation marks induced by the IDH1 mutation, thereby switching the promoter into a transcriptionally active state (Figure 4E). The transcriptional derepression in NFKBIA-depleted cells involved reduced binding of NFKBIA to the catalytic PRC2 subunit Enhancer of Zeste 2 (EZH2) (Figure 4E).
To characterize the antipodal epigenome effects of NFKBIA deletion and IDH mutation, we performed chromatin immunoprecipitation sequencing (ChIP-seq) for H3K27me3 in the primary human astrocyte system with and without NFKBIA depletion. This cell system contains a nuclear, chromatin-bound form of phosphorylated-sumoylated (ps-)NFKBIA—which interacts with histones 2A and 4, thereby modulating polycomb recruitment24—that is lost upon NFKBIA depletion (Figure 5A). Consistent with the effect in the global H3K27me3 assay (Figure S8B), a discrete set of 678 of 13,494 (5.0%) sequenced genes lost H3K27me3 peaks upon NFKBIA depletion (Table S4). These genes greatly overlapped with genes CpG hypermethylated upon introducing the IDH1 mutation (36.6%) and those CpG hypermethylated in the G-CIMP phenotype (36.3%) (Figure 5B and Table S4), suggesting that these genetic events have opposed epigenome effects. Over-representation analysis revealed highly significant enrichment of these genes for functions in development and synaptic plasticity and transmission (Figure 5C).
Figure 5.

NFKBIA deletion reshapes the methylome antithetically to the IDH mutation
(A) Immunoblot showing expression of NFKBIA in nuclear (N) and cytoplasmic (C) fractions of control (ΔCtrl) and NFKBIA-depleted (ΔNFKBIA) immortalized primary human astrocytes. Presence of the canonical 37 kDa band in the C fraction captured with anti-phospho-NFKBIA and the ∼48 kDa band in the N fraction of ΔCtrl cells but not ΔNFKBIA cells; the latter is compatible with chromatin-bound, phosporylated-sumoylated NFKBIA (ps-NFKBIA) (red asterisks). α-Tubulin as a cytoplasmic and histone H3 as a nuclear loading control. Protein phosphatase 1 (PP1) as a loading control for both soluble and chromatin fractions.
(B) Euler diagrams showing the proportion of 678 genes with loss of repressive H3K27me3 marks by ChIP-seq upon NFKBIA depletion in primary cultures of human astrocytes and intersection with genes CpG hypermethylated upon introducing the IDH1 mutation into the same astrocyte system or with genes CpG hypermethylated in the G-CIMP phenotype.
(C) Functional enrichment analysis of the 248 genes in (A) that lose H3K27me3 marks upon NFKBIA depletion and are hypermethylated by the IDH1 mutation.
(D) Euler diagrams showing the proportions of the same 678 genes that map into H3K27me3 silencer regions or super-silencers (H3K27me3-rich regions, MRRs), and those that are EZH2-responsive PRC2 targets.
(E) Transcription factor binding site analysis for the 85 MRR-associated genes in (D) that overlap with the H3K27me3-depleted gene set. Scatterplot of terms in the ChIP enrichment analysis (ChEA) gene set library. Terms are plotted based on the first two uniform manifold approximation and projection (UMAP) dimensions. Terms with more similar gene sets are closer together. Terms are colored by automatically identified clusters computed with the Leiden algorithm applied to the term frequency-inverse document frequency (TF-IDF) values. Darker and larger points signify more substantial enrichment. Significant enrichment (points encapsulated by a black line) for components of the PRC2 core complex (SUZ12 and EZH2) and accessory proteins that define PRC2 subcomplexes PRC2.1 (MTF2) and PRC2.2 (JARID2), and other polycomb group (PcG) genes (BMI1 and RNF2). q values calculated by the Benjamini-Hochberg method.
(F) Levels of D-2-hydroxyglutarate (D-2HG) assessed by colorimetric enzymatic assay in primary cultures of IDH wild-type (wt) or IDH1-R132H mutant (mut) human astrocytes with CRISPR-mediated (cr-NFKBIA) or without (cr-Ctrl) NFKBIA depletion. Student’s t test. Error bars represent ±SEM from three biological replicates.
(G) DNA-binding activity of NF-κB proteins p65, p50, RelB, and c-Rel assessed by DNA-binding ELISAs in IDH wt vs. IDH1-R132H mut primary human astrocytes transfected with siRNA (si)-NFKBIA or cr-NFKBIA vs. Ctrl. Multiple-comparison one-way ANOVA with post hoc Tukey’s test. Error bars represent ±SEM from three biological replicates. n.s., non-significant.
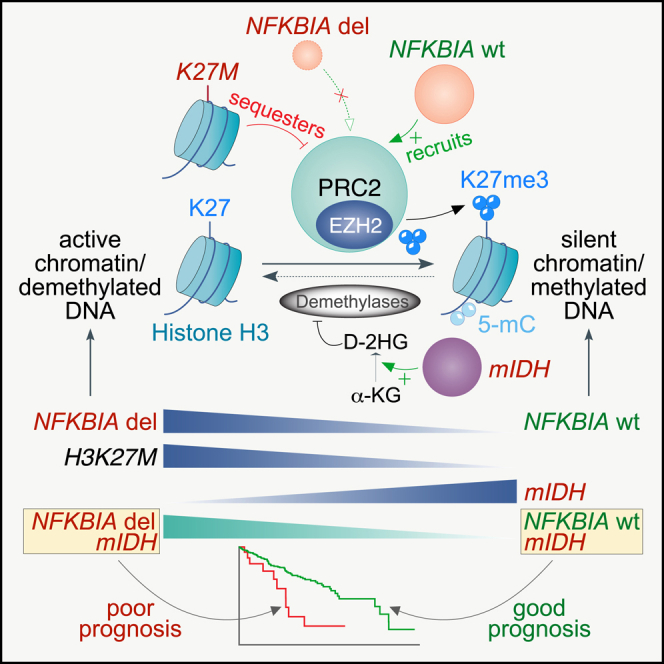
(H) Graphical model of the relationship between the NFKBIA deletion and IDH and H3K27M mutations and polycomb repressive complex 2 (PRC2). Chromatin-bound NFKBIA (“NFKBIA wt”) regulates differentiation-related genes by recruiting PRC2, which promotes the trimethylation of H3K27 (H3K27me3) through its catalytic subunit EZH2, a histone methyltransferase. Genomic sequences occupied by NFKBIA overlap with those regions containing high H3K27me3 levels. Deletion of NFKBIA (NFKBIA del) results in PRC2 release and, thus, loss of H3K27me3. Similarly, H3K27M mutations in diffuse midline gliomas sequester PRC2, thereby reducing global H3K27me3 levels, especially in large unmethylated CpG islands. In turn, the IDH mutation (mIDH) hypermethylates DNA (5-mC, 5-methylcytosine) and increases global H3K27me3 levels—particularly at PRC2-targeted loci—by metabolizing α-ketoglutarate (α-KG) to D-2-hydroxyglutarate (D-2HG) and competitively inhibiting DNA and histone demethylases. Tumors with both the NFKBIA deletion and the IDH mutation demonstrate methylome changes that are more similar to those with an isolated NFKBIA deletion than to those with an isolated IDH mutation.
(I) Area-proportional Euler diagrams depicting the intersection of genes overexpressed in NFKBIA deleted gliomas (NFKBIA deletion signature) in population 4 and those identified as H3K27me3 or PRC2 targets in human embryonic stem (hES) cells. OR, odds ratio.
Much of the H3K27me3-depleted gene set (33.8%) mapped into known H3K27me3 silencer regions (Figure 5D and Table S4). Eighty-five genes (12.5%) mapped to super-silencer regions (Figure 5D and Table S4).36 These H3K27me3-rich genomic regions (MRRs), defined from clusters of H3K27me3 peaks, silence gene expression via proximity or looping.36 This gene set included 113 (16.7%) genes responsive to EZH2 inhibition (Figure 5D and Table S4).36
PRC2 subcomplexes defined by distinct accessory proteins, PRC2.1 (MTF2) and PRC2.2 (JARID2), synergistically orchestrate H3K27me3 methylation.37 Their loss primes lineage choice during exit of pluripotency.38 While MTF2-containing PRC2.1 broadly balances poised lineage-specific gene activation, JARID2-containing PRC2.2 is more selective.38 In analyzing transcription factor DNA-binding sites for the overlapping set of 85 MRR-associated genes, we found binding sites for the PRC2 core complex (SUZ12 and EZH2) and significant enrichment for binding sites targeted by accessory proteins MTF2 (PRC2.1) and JARID2 (PRC2.2), suggesting that loss of both PRC2 subcomplexes is required for mislocalization of PRC2 and thus loss of H3K27me3 upon NFKBIA depletion (Figure 5E and Table S4).37 Moreover, consistent with data indicating that JARID2-PRC2.2 chromatin association depends on PRC1,37 we found significant enrichment for polycomb group (PcG) protein BMI1-binding sites, which regulates PRC1 architecture,39,40 and for PcG protein RNF2-binding sites, the enzymatic component of PRC1 that plays an important role in the migration and differentiation of neural progenitor cells (Figure 5E and Table S4).40,41
NFKBIA deletion induces D-2-hydroxyglutarate but not NF-κB in IDH mutant cells
The IDH1 mutation imparts the ability to produce the oncometabolite D-2-hydroxyglutarate (D-2HG).42 D-2HG levels generated from α-ketoglutarate (α-KG) inhibit demethylases, thus favoring an epigenetic state of high H3K27me3.43 We measured D-2HG levels in the parental wild-type astrocyte and IDH1 mutant astrocyte system with and without NFKBIA depletion. As expected, D-2HG levels increased more in IDH1 mutant cells than in wild-type cells without NFKBIA depletion (Figure 5F). NFKBIA depletion and consequent H3K27me3 loss raised D-2HG production in IDH1 mutant astrocytes but not in the parental system (Figure 5F). Thus, NFKBIA depletion moves the IDH1 mutant hypermethylated system toward a compensatory loop that inhibits demethylation. NFKBIA deletion and IDH mutation appear to affect a shared methylation pathway in opposing ways, each with its preferred epigenetic state.
Given NFKBIA’s regulation of NF-κB23 and NF-κB’s role in glioma propagation and resistance,44,45 we analyzed, with DNA-binding ELISAs, the activation state of NF-κB family proteins p65, p50, RelB, and c-Rel in the same astrocyte system with and without NFKBIA depletion. We found no biologically meaningful difference in the DNA binding of individual NF-κB proteins upon NFKBIA depletion (Figure 5G), indicating that activation of NF-κB is not a primary feature of NFKBIA loss in this cell system.
NFKBIA deletion induces a transcriptome landscape reminiscent of that of H3K27M mutant gliomas
Unmethylated CpG islands are high-affinity sites for PRC2.46,47 PRC2 modifies chromatin structure as its catalytic EZH2 subunit deposits H3K27me3 (Figure 5H).48 Chromatin-bound NFKBIA regulates differentiation-related genes by recruiting PRC2 (Figure 5H), and genomic sequences occupied by NFKBIA strongly overlap with those having high levels of H3K27me3.24 Genes hypermethylated in IDH mutant gliomas are greatly enriched for PRC2-targeted loci,13 and the mutation increases global H3K27me3 levels by competitively inhibiting demethylases (Figure 5H).49 H3K27M mutant diffuse midline gliomas demonstrate defective H3K27me3 deposition due to PRC2 sequestration, especially from large unmethylated CpG islands (Figure 5H).46,50,51 Given our observed enrichment for demethylated CpGs mapping to genes bearing repressive H3K27me3 marks in NFKBIA deleted gliomas, we examined whether the transcriptome landscape in these tumors shares features associated with the H3K27M mutation in diffuse midline gliomas.17,46,52,53,54
We analyzed 2,629 genes significantly overexpressed in NFKBIA deleted vs. NFKBIA wild-type gliomas (NFKBIA deletion signature) in population 4 (Table S4) by comparing them with the presence of H3K27me3 or PRC2 target genes in undifferentiated human embryonic stem cells.55 We found a highly significant intersection of the NFKBIA deletion signature with both H3K27me3- and PRC2-target genes (Figure 5I and Table S4). The NFKBIA deletion signature overlapped significantly with the presence of genes upregulated in H3K27M mutant DIPGs compared with H3K27 wild-type DIPGs in population 7 (comprising 11 patients with H3K27M mutations and 5 with wild-type H3K27), in four additional independent datasets of H3K27M mutant diffuse midline gliomas, and in an inducible H3K27M DIPG model (Figures 6A, 6B, and S9A; Table S4).
Figure 6.

NFKBIA deletion gene signature intersects with a gene signature of H3K27M mutant diffuse midline glioma
(A) Left upper panel shows an area-proportional Euler diagram depicting the intersection of genes overexpressed in NFKBIA deleted gliomas (NFKBIA deletion signature) and those overexpressed in a panel of 11 diffuse intrinsic pontine glioma (DIPG) tumor samples from population 7 carrying H3K27M mutations compared with five DIPGs that are wild-type for H3K27 (H3K27WT). Intersection between NFKBIA deletion signature and genes overexpressed in an inducible pontine model of H3K27M mutant DIPG is also shown. Right upper and lower panels show area-proportional Euler diagrams depicting intersection of the NFKBIA deletion signature and genes overexpressed in three additional independent validation populations or models of pediatric midline glioma (pHGG, pediatric high-grade glioma; DMG, diffuse midline glioma; DIPG, diffuse intrinsic pontine glioma) carrying H3K27M mutations from Harvard University, McGill University, and Memorial Sloan Kettering Cancer Center (MSKCC). Cosegregation of the NFKBIA deletion signature with genes delineating cell subpopulations resembling various differentiation states, cell lineages (AC-like, astrocytic differentiation; OC-like, oligodendrocytic differentiation; OPC-like, oligodendrocyte precursor cells-like) and oncogenic programs (cell cycle) are shown for the Harvard population. In the human embryonic stem (hES)/neural progenitor cell (NPC) DIPG model from MSKCC, the NFKBIA deletion signature more closely resembled H3K27me3 target genes specific to mutant H3K27M than those of wild-type H3K27.
(B) Area-proportional Euler diagram depicting the intersection of the NFKBIA deletion signature with an H3K27M gene expression signature in 119 pediatric high-grade gliomas from Gustave Roussy Paris. Functional enrichment analysis of the intersecting signature of 869 genes between NFKBIA deleted gliomas and H3K27M diffuse midline gliomas. The relationship between the significant gene ontology functions is visualized in a directed acyclic graph. FDR, false discovery rate. p values from Fisher’s exact test.
(C) Euler diagram showing intersection of genes derepressed in H3K27M mutant diffuse pediatric midline gliomas (above five populations) and those that lose repressive H3K27me3 marks by ChIP-seq upon NFKBIA depletion in primary cultures of human astrocytes (a), those that lose repressive H3K27me3 marks upon NFKBIA depletion and map into known silencer regions (b), and those that lose repressive H3K27me3 marks upon NFKBIA depletion and are EZH2-responsive PRC2 targets (c).
(D) Intersection of a cluster of HOX genes—paradigmatic PRC2 targets—that lose H3K27me3 marks upon NFKBIA depletion in primary cultures of human astrocytes with genes overexpressed in the H3K27M mutant DIPGs of population 7, those with NFKBIA regulatory regions, those described as H3K27me3 and PRC2 targets in human embryonic stem cells, those that map into typical H3K27me3 silencer and super-silencer regions (MRRs), and those CpG hypermethylated in the IDH1 mutant condition and the G-CIMP phenotype.
H3K27me3 marks undergo genomic redistribution in the H3K27M mutant condition.46,52,56 Strikingly, the NFKBIA deletion signature closely resembled the pattern of H3K27 receptive genes specific to H3K27M but less so those specific to H3K27WT tumors (Figure 6A and Table S4). Functional enrichment analysis of gene signature interaction between NFKBIA deleted gliomas and H3K27M mutant diffuse midline gliomas revealed—similar to that between NFKBIA deletion and IDH1 mutation (Figure 5B)—significant over-representation of functions in neurogenesis and synaptic plasticity (Figure 6B and Table S4). To exclude the possibility that the enriched overlap of genes reflected IDH mutation status, we assessed the intersection and functional enrichment in IDH mutant tumors. We again noted significant intersection between genes hypomethylated in IDH mutant/NFKBIA deleted (vs. IDH mutant/NFKBIA wild-type) gliomas and genes derepressed in H3K27M mutant diffuse midline gliomas (Figure S9B and Table S5). These overlapping genes demonstrated significant over-representation of functions in neurogenesis and synaptic plasticity, such as regulation of trans-synaptic signaling and modulation of chemical synaptic transmission (Figure S9B).
To examine the biological similarity of the molecular consequences of H3K27M mutation and NFKBIA deletion, we tested the intersection of genes that lose repressive H3K27me3 upon NFKBIA knockdown in the immortalized astrocyte system with genes derepressed in H3K27M mutant pediatric midline gliomas: 21.5% of H3K27me3-depleted genes overlapped with genes overexpressed in the H3K27M mutant condition and 27.9% of H3K27me3-depleted genes mapped into genomic regions that can silence gene expression via chromatin interactions (Figure 6C and Table S5).36 Strikingly, about one-third (32.7%) of H3K27me3-depleted genes that are EZH2 responsive PRC2 target genes intersected with genes derepressed in H3K27M mutant tumors (Figure 6C and Table S5).
This enriched gene set included a cluster of HOX genes (Figure 6D). HOX genes are paradigmatic polycomb targets,57,58 and ps-NFKBIA interacts with histones 2A and 4 at their regulatory region.24 Several of these HOX genes were overexpressed in the H3K27M mutant tumors of population 7, possess NFKBIA regulatory regions, constitute H3K27me3 and PRC2 targets in human embryonic stem cells, map into typical H3K27me3 silencer or super-silencer regions (MRRs), and are CpG hypermethylated in the IDH mutant condition and the G-CIMP phenotype (Figure 6D). These data indicate that both NFKBIA deletion and H3K27M mutation can seize a common trimethylation pathway that leads to a de facto PRC2 loss-of-function epigenetic state.
NFKBIA deletion and outcome in IDH mutant diffuse gliomas
Although generally more favorable, outcomes of patients with IDH mutant gliomas can vary substantially and are difficult to predict. The WHO classification highlights the importance of CDKN2A/B deletions and TP53 mutations in IDH mutant gliomas.12 We found no pattern of occurrence between NFKBIA deletions and either of these two alterations (Figure S10A). A two-class model of the relationship between NFKBIA and survival duration in patients of population 4 with IDH mutant tumors revealed remarkably briefer survival of patients with NFKBIA deleted tumors compared with non-deleted tumors, with median survival estimates of 3.8 and 7.6 years, respectively (Figure 7A). This survival association held in an adjusted Cox model that included IDH, ATRX, TERT, and TP53 mutations, 1p19 codeletions, CDKN2A/B deletions, WHO grade, histology, patient age, and patient gender (Figure S10B). A similar association was found in IDH mutant gliomas of population 5: median survival of 6.8 vs. 14.3 years in the NFKBIA deleted vs. NFKBIA wild-type tumors (Figure 7B); this relationship held in an adjusted Cox model (Figure S10C).
NFKBIA deletion and a nomogram predicting individual patient survival in IDH mutant gliomas
Nomograms are graphical representations of statistical models that allow individualized predictions based on the characteristics of a single patient.59 We generated a nomogram model that uses NFKBIA deletion and the best-established clinicopathologic and molecular genetic markers to estimate the probability of 5-year survival of individual patients with IDH mutant glioma. In choosing this method, we balanced model sparsity (minimal number of needed variables), interpretability, and predictive accuracy. We used the 398 IDH mutant gliomas of population 4 to build, internally validate, and calibrate the model and assess performance over 15 years. This model retained the NFKBIA deletion but not the 1p19q codeletion as a significant variable (Figure 7C). We then used the 291 IDH mutant gliomas of population 5 to validate the model externally. NFKBIA deletion remained an independent survival variable with an effect on survival greater than that of all other variables, including 1p19q codeletion, TERT mutation, ATRX mutation, WHO grade, and patient age (Figure 7D). Similar models incorporating CDKN2A/B deletions and TP53 mutations in the IDH mutant gliomas of population 4 (Figure S11A) or using the 619 IDH mutant lower-grade gliomas of populations 1 and 5 (Figure S11B) confirmed that the NFKBIA deletion is a robust variable in predicting the duration of survival of patients with IDH mutant gliomas.
Discussion
Here, we report chromatin-bound NFKBIA to be a haploinsufficient tumor suppressor whose deletion portends an unfavorable clinical course of diffuse gliomas. The patterned presence of the NFKBIA deletion with the driver mutations IDH, TERT, ATRX, and PTEN, the 1p19q codeletion, the CDKN2A/B deletion, and its enriched occurrence in recurrent tumors support the notion that this deletion is a critical genetic event during glioma progression. Glioma progression is associated with specific demethylation shifts,16,60 some of which we found resemble methylome changes related to the NFKBIA deletion.
NFKBIA is a critical regulator of NF-κB,23 which is involved in glioma propagation and resistance.44,45 Nuclear NFKBIA participates in chromatin-associated epigenetic regulation.24,25 It does so by dynamically associating with histones H2A and H4 to control a subset of polycomb-regulated differentiation genes, thus governing stem cell maturation and lineage specification.24,25 Our observations show that NFKBIA deletions manifest epigenetically in gliomas in two ways: demethylation of glioma-associated CpG sites and loss of repressive histone H3K27 trimethylation marks. Both effects appear to be antithetical to the DNA and histone methylome changes caused by the IDH mutation. Consistently, we found that IDH mutant tumors with the NFKBIA deletion behave similarly to IDH wild-type tumors.
Mutant IDH reshapes the epigenome through extensive DNA hypermethylation and increasing repressive histone methylation.13,49,61 The opposed effects of NFKBIA deletion and IDH mutation on the glioma epigenome may reflect overlapping but antipodal regulatory roles in chromatin remodeling.24,62 DNA methylation and histone modifications participate in chromatin organization.13,30,31 Genes undergoing hypermethylation in response to the IDH mutation are highly enriched for PRC2-targeted loci,13 as DNA methylation and the PRC2 system are intimately linked.63 Similarly, histone-bound NFKBIA interacts with PRC2 and regulates its association with a subset of target genes.24,25,64 As such, NFKBIA deletion and IDH mutation may drive a PRC2-regulated methylome mechanism toward diametrically different epigenetic states.
We also noted that the epigenetic state facilitated through NFKBIA deletion partially mimics the altered chromatin landscape of diffuse midline gliomas carrying H3K27M oncohistone mutations. H3K27M mutations lead to defective spread and significantly lower overall amounts of trimethylated H3K27 by sequestering PRC2 to poised enhancers, wherein PRC2 becomes trapped at H3K27M-H3K27me3 boundaries46,51,52,56; this engenders discrete transcriptome changes that preferentially involve derepression of genes regulating neurogenesis.46,54 We found that the methylome changes of NFKBIA deleted gliomas are highly enriched for neural stem cell and neurogenesis genes bearing repressive H3K27 marks.24,30 Moreover, we observed significant overlap between the transcriptomic landscapes of NFKBIA deleted and H3K27M mutant gliomas, with an over-representation of functions of stemness, neurogenesis, and regulation of synaptic plasticity. This mutual functional enrichment is consistent with compelling evidence indicating that gliomas can manipulate normal elements of neuronal development and synaptic plasticity,65,66,67 a process that may be partly driven through PRC2: neuronal activity can derepress PRC2,68 and PRC2 is a key regulator of epigenetic plasticity in glioblastoma.69 Consistently, our findings imply that the NFKBIA deletion in adult gliomas and the H3K27M mutation in pediatric diffuse midline gliomas define aggressive disease subtypes that may share an epigenetic state reflecting a common PRC2 loss-of-function phenotype antipodal to the quasi PRC2 gain-of-function phenotype of IDH mutant gliomas and their favorable clinical course. This PRC2 loss-of-function phenotype might become a functionally defined therapeutic target in gliomas as histone demethylase inhibitors successfully enter clinical trials.70,71,72,73
NFKBIA maps to chromosome 14q13.2. NFKBIA may be one of multiple yet-to-be-identified targets that drive 14q losses in gliomas via cumulative haploinsufficiency.74 While our results contribute to bridging several important gaps in our understanding of how NFKBIA haploinsufficiency impacts the biology and clinical behavior of gliomas, wider-spanning losses on the long arm of chromosome 14 have been identified in gliomas.75,76,77 These genomic loci map more distally on 14q within commonly deleted regions spanning 14q21.2–14q24.3, 14q22.3–q32.1, 14q31.3–q32.1, and 14q32.1–14qter.75,76,77 No other glioma-associated tumor-suppressor genes have been identified within the region encompassing our NFKBIA deletion and these more distal regions. Chromosome 14q losses correlate with increased complexity of chromosome copy number. We consistently noted greater copy-number complexity in NFKBIA deleted tumors than in non-deleted tumors across every WHO grade of glioma but found that the survival association of NFKBIA remained independent of aneuploidy.
Our data show that lower-grade gliomas harboring NFKBIA deletions behave much like high-grade gliomas. NFKBIA deletions result in a clinical course for IDH mutant gliomas that resembles that of IDH wild-type tumors. Despite the comparatively favorable prognosis of most IDH mutant gliomas, individual outcomes vary greatly. NFKBIA status may be a robust marker in identifying IDH mutant tumors with potential for more aggressive behavior and thus the need for earlier, more substantial therapy. Accordingly, we generated a clinically applicable and externally validated nomogram model, including NFKBIA, to predict the survival duration of patients with IDH mutant gliomas. Estimation of survival duration of an individual patient from such a nomogram is highly preferable to estimation based on membership in a subpopulation defined by multiple disease characteristics.59 Risk grouping forces patients into distinct classes, ignores the individuality of each glioma case, and becomes impractical as more driver genetic events are identified. We developed a linear model that balances the number of selected variables and predictive performance by focusing on NFKBIA deletion and current best-established genetic and clinicopathologic factors. It reflects a reasonable compromise between model sparsity—and thus ease of clinical use and interpretability—and accuracy of outcome prediction. While our model consistently and independently associates the NFKBIA deletion with survival in multiple well-characterized glioma populations, further validation—ideally in a prospective trial—is needed to ascertain its role in clinical prognostication.
Limitations of the study
Despite the inclusion of seven well-characterized glioma populations, including a national randomized consortium trial, the retrospective nature of our analysis of NFKBIA deletions and their correlation with patient outcomes highlights the need for a prospectively conducted cohort study to further affirm the survival significance of NFKBIA. To address the issue of model complexity and predictive ability, we developed a linear nomogram model that focused on the NFKBIA deletion, along with the most well-established genetic and clinicopathologic factors, achieving a good balance between the number of selected variables and predictive performance. However, there is a trade-off between model sparsity and precision of outcome predictions, and any future optimizations of this nomogram model will have to consider a balance between model simplicity, interpretability, and predictive accuracy. We used different molecular platforms to characterize NFKBIA deletions. A simple yet robust assay to screen for NFKBIA deletions would enable wide clinical applicability. Our findings suggest that NFKBIA deletions hold significant promise as a biological and clinical target in diffuse gliomas. However, currently, there is no molecular therapeutic intervention that would allow for the specific restitution of NFKBIA expression, except for first-generation and second-generation proteasome inhibitors—clinically used in multiple myeloma, mantle cell lymphoma, and some types of non-Hodgkin’s lymphoma—that suffer from broad off-target effects. Further research is needed to develop effective and targeted molecular therapies for NFKBIA deletions in gliomas.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal IκBα | Cell Signaling Technology | Cat# 4814S; RRID: AB_390781 |
| Mouse monoclonal β-Actin | Cell Signaling Technology | Cat# 3700; RRID: AB_2242334 |
| Rabbit monoclonal Tri-Methyl-Histone H3(Lys27) | Cell Signaling Technology | Cat# 9733S; RRID: AB_2616029 |
| Mouse monoclonal Di/Tri-Methyl-histone H3 (Lys9) | Cell Signaling Technology | Cat# 5327S; RRID: AB_10695295 |
| Mouse monoclonal Anti-5-methylcytosine (5-mC) | Abcam | Cat# ab10805; RRID: AB_442823 |
| Rabbit monoclonal Ezh2 | Cell Signaling Technology | Cat# 5246S; RRID: AB_10694683 |
| Mouse monoclonal Anti-IDH1 R132H | Dianova manufacture | Cat# DIA-H09; RRID: AB_2335716 |
| Mouse monoclonal PP1 (E−9) | Santa Cruz | Cat# 7482; RRID: AB_628177 |
| Mouse monoclonal Phospho IκBα | Cell signaling | Cat# 9246; RRID: AB_22267145 |
| Mouse monoclonal Anti-α-Tubulin | Sigma Aldrich | Cat# T6074; RRID:AB_477582 |
| Rabbit monoclonal Anti-Histone H3 | Abcam | Cat# ab1791; RRID: AB_302613 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling | Cat# 7076; RRID:AB_330924 |
| Anti-rabbit IgG, HRP-Linked Antibody | Cell Signaling | Cat# 7074; RRID:AB_2099233 |
| Donkey anti-mouse IgG (H + L) Alexa Fluor™ 594 | Thermo Fisher/Invitrogen | Cat# R37115; RRID:AB_2556543 |
| Bacterial and virus strains | ||
| Retroviral vector pLNCX2 | Turcan et al.13 | N/A |
| Biological samples | ||
| Diffuse glioma samples (Populations 1–7) | This study | Table S1 |
| Chemicals, peptides, and recombinant proteins | ||
| Plasmid Transfection Medium | Santa Cruz | Cat# sc-108062 |
| Ultra-Cruz Transfection Reagent | Santa Cruz | Cat# sc-395739 |
| Gibco Geneticin Selective Antibiotic (G418 Sulfate) (50 mg/mL) | Gibco | Cat# 10131-027 |
| TRIzol Reagent | Invitrogen | Cat# 15-596-018 |
| Gibco Opti-MEM I Reduced Serum Medium, no phenol red | Gibco | Cat# 11-058-021 |
| Lipofectamine 2000 Transfection Reagent | Invitrogen | Cat# 11668019 |
| GenomicDNA MiniPrep Kit | Sigma Aldrich | Cat# G1N70-1KT |
| Critical commercial assays | ||
| TransAM NF-κB Activation Assays | Colorimetric Kits | Active Motif | Cat# 43296 |
| Nuclear Extract Kit | Active Motif | Cat# 40010 |
| EpiQuik Global Histone H3K27 Methylation Assay Kit | Epigentek | Cat# P-3020-96 |
| D-2-Hydroxyglutarate Assay Kit (Colorimetric) | Abcam | Cat# ab211070 |
| GenElute Genomic DNA miniprep kit | Sigma Aldrich | Cat# GN170 |
| Deposited data | ||
| Clinical, genetic marker data (Populations 1–7) | This study | Table S2 |
| ChIP-sequencing data | This study | GEO accession # GSE222571 |
| RNA-sequencing data | This study | portal.gdc.cancer.gov |
| RNA-sequencing data | This study | GEO accession #GSE223712 |
| RNA-sequencing data | Nagaraja et al.17 | GEO accession #GSE126319 |
| RNA-sequencing data | Grasso et al.70 | dbGaP study accession phs000900.v1.p1 |
| DNA methylation data | This study | portal.gdc.cancer.gov |
| DNA methylation signature raw data – RTOG 9802 | This study | Table S6 |
| NFKBIA methylation raw data – RTOG 9802 | This study | Table S6 |
| DNA methylation data | De Souza et al.16 | https://data.mendeley.com/datasets/hx566mwxnm/ |
| DNA methylation data | Mazor et al.78 | EGAS00001001255 |
| DNA methylation data | Mazor et al.79 | EGAS00001001854 |
| DNA methylation data | Bai et al.80 | EGAS00001001588 |
| DNA copy number variation data | This study | portal.gdc.cancer.gov |
| NFKBIA DNA CNV raw data – RTOG 9802 | This study | Table S6 |
| NFKBIA DNA CNV raw data – Mayo | This study | Table S6 |
| DNA copy number variation data | Suzuki et al.3 | EGAS00001001044 |
| Sequencing data | This study | portal.gdc.cancer.gov |
| NFKBIA sequencing raw data | This study | https://github.com/SharedExomes/Prim_Recur_Glioma_NFKBIA_exomes |
| Experimental models: Cell lines | ||
| NHA (Normal human immortalized astrocytes) Parental | Sonoda et al.81 | N/A |
| NHA IDH1 WT | Turcan et al.13 | N/A |
| NHA IDH1 R132H | Turcan et al.13 | N/A |
| NHA IDH1 WT/KO IkBa | This study | N/A |
| NHA IDH1 R132H Mut/KO IkBa | This study | N/A |
| Oligonucleotides | ||
| Accell Human NFKBIA siRNA | Dharmacon (Horizon) | Cat# A-004765-13-0020 |
| Accell Non-targeting Control siRNA | Dharmacon (Horizon) | Cat# D-001910-01-20 |
| Primer: SLC16A3 Forward: ACGAAAAGTGGGTTGGTCAG |
Invitrogen | Cat# custome |
| Primer: SLC16A3 Reverse: CCCCAACAGACACAAGACCT |
Invitrogen | Cat# custome |
| Recombinant DNA | ||
| IκB-α Double Nickase Plasmid (h) | Santa Cruz | Cat# sc-400034-NIC |
| NFKBIA FISH probe | Empire Genomics | Cat# NFKBIA-20-OR |
| Software and algorithms | ||
| R | The R project for Statistical Computing | https://www.r-project.org/ |
| R package DNAcopy 1.72.3 | Olshen et al.82 | https://bioconductor.org/packages/release/bioc/html/DNAcopy.html |
| R package conumee 1.9.0 | N/A | https://bioconductor.org/packages/release/bioc/html/conumee.html |
| R package RCircos 1.2.2 | Zhang et al.83 | https://cran.r-project.org/web/packages/RCircos/index.html |
| R package minifi 1.44.0 | Aryee et al.84 | https://www.bioconductor.org/packages/release/bioc/html/minfi.html |
| R package TCGAbiolinksGUI 1.23.0 | https://www.biorxiv.org/content/10.1101/147496v2.full | https://www.bioconductor.org/packages/release/bioc/html/TCGAbiolinksGUI.html |
| R package MissMethyl 1.32.0 | Phipson et al.85 | https://bioconductor.org/packages/release/bioc/html/missMethyl.html |
| R package recount2 1.24.1 | Collado-Torres et al.86 | https://bioconductor.org/packages/release/bioc/html/recount.html |
| R package limma-voom 3.54.2 | Ritchie et al.87 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| R package WebGestaltR 0.4.5 | Zhang et al.88 | https://cran.r-project.org/web/packages/WebGestaltR/index.html |
| R package BioVenn 1.13 | Hulsen et al.89 | https://cran.r-project.org/web/packages/BioVenn/index.html |
| R package hdnom 6.0.1 | https://www.biorxiv.org/content/10.1101/065524v1.full.pdf | https://cran.r-project.org/web/packages/hdnom/index.html |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Markus Bredel (mbredel@uab.edu).
Materials availability
Gene-modified primary human astrocytes used in this study are available upon request; additionally, the reagents used in this study are available from the lead contact with a completed Materials Transfer Agreement.
Experimental model and subject details
Tumor samples and patient populations
Our study utilized seven study populations of patients with diffuse glioma encompassing 2,343 tumors and 2,255 patients. The demographic and disease characteristics of these populations are shown in Table S1, and their clinico-pathologic-genetic details are listed in Table S2. Where appropriate, institutional review board ethics committee approval was obtained. 530 lower-grade—WHO grade 2 and 3—glioma samples profiled as part of The Cancer Genome Atlas (TCGA) Pilot Project (cancergenome.nih.gov/index.asp) constituted Population 1. Gene copy number, gene expression, sequencing, and clinical data for 513, 530, 513, and 525 tumors, respectively, were obtained from the Open Access and Controlled Access Tier of the NIH Genomic Data Commons (GDC) portal (portal.gdc.cancer.gov) of TCGA upon National Human Genome Research Institute (NHGRI) approval. At the time of data retrieval, there was incomplete overlap in the data types available for each sample. Alignment of sample identifiers yielded 516 samples with mutational sequence and gene dosage data, 508 samples with gene dosage and overall survival data, 515 samples with exon expression data and overall survival data, 425 samples with gene dosage and recurrence-free survival data, and 269 samples with complete mutational, gene dosage and overall survival data. Eighty-seven low-grade (WHO grade 2) glioma patients treated as part of NRG Oncology/Radiation Therapy Oncology Group (RTOG) consortium trials 9802 (A Phase III Trial of Radiation Versus Radiation Plus Procarbazine, Lomustine (CCNU), and Vincristine in High-Risk Low-Grade Glioma)28 constituted Population 2. A unified sample population of 688 lower-grade gliomas from the Mayo Clinic Genome Consortium, a multi-institutional Japanese study consortium,3 and the German Glioma Network90 constituted Population 3. A panel of 1,122 WHO grade 2 to 4 glioma samples profiled as part of TCGA and retrieved from the NIH GDC portal comprised Population 4. Gene copy numbers, sequencing, and clinical data were obtained from the NIH GDC portal. At the time of data retrieval, there was incomplete overlap in the data types available for each sample. Alignment of sample identifiers yielded 1,027 samples with gene dosage and overall survival data and 753 samples with gene dosage and recurrence-free survival data. A panel of 473 WHO grade 2 to 4 glioma specimens treated and profiled at Mayo Clinic (Mayo Clinic Genome Consortium) constituted Population 5. A sample population of 180 primary and recurrent tumors from 154 diffuse glioma cases belonging to 74 patients profiled at Henry Ford Hospital (n = 154 samples) and 13 patients profiled at Case Western University (n = 26 samples) constituted Population 6. All specimens were collected under institutional review board-approved guidelines and classified according to standards set by the World Health Organization (WHO).91 A central path review was done for RTOG 9802. Written informed consent was obtained from all patients. Specimens were analyzed by a neuropathologist to confirm the histological diagnosis and the presence of vital tumor tissue without excessive contamination by normal brain and tumor necrosis.
Cell lines
Cultures of immortalized primary human astrocytes81 constitutively expressing wildtype IDH1 or mutant IDH1 (R132H), and corresponding parental cells were obtained from Dr. T.A. Chan (Cleveland Clinic) and prepared as previously described.13 Stable expression of wildtype or mutant IDH1 was achieved via transduction with retroviral vector pLNCX2 and selection with G418 at 600 μg/mL (Gibco).13 Parental cells were grown in Dulbecco’s Modified Eagle Medium (MT-10-090-CV, Gibco) supplemented with 10% fetal bovine serum (Sigma) and 5% Pen-strep (Corning); IDH wildtype and mutant cells were cultured with Geneticin (G418) at 600 μg/mL (Gibco) as the expression of mutant IDH1 presented as negative selection.
Method details
Mutational analyses
Pan-Cancer Analysis of Whole Genomes (PCAWG) sequences of the International Cancer Genome Consortium (icgc.org) for diffuse gliomas (Population 1) were analyzed for non-synonymous somatic NFKBIA mutations and mapped using GenomeViewer and Homo sapiens Genome Reference Consortium Human Build 37 (GRCh37). IDH mutations and TERT and ATRX promoter mutations in Population 1 were obtained from the somatic RNA sequencing-based data of the NIH GDC. In Population 2, immunohistochemistry with the mutation-specific monoclonal antibody IDH1-R132H (Dianova) was used to assess for the canonical IDH1-R132H mutation as previously reported.92 To evaluate for noncanonical IDH1/2 mutations and mutations in ATRX, a customized Ion AmpliSeq (Thermo Fisher Scientific) DNA panel was designed and used. Sequence alignment and variant calling were performed using the Ion Suite and Reporter software. TERT promoter mutations were assessed by Sanger sequencing based on methods previously described.1,92 ATRX mutations in Population 5 were assessed by immunohistochemistry (denoted by loss of expression; HPA001906, Sigma Aldrich) or by somatic exon sequencing.
Copy number analyses
Estimates of NFKBIA deletion and 1p19q codeletion status in 454 patients of Population 1 and 1,090 patients of Population 4 were generated from data from the Open Access and Controlled Access Tier of the NIH Genomic Data Commons (GDC) portal (portal.gdc.cancer.gov) and were based on Affymetrix SNP 6.0 array data using R package DNAcopy to perform a circular binary segmentation (CBS) analysis82 followed by numeric focal-level CNV calls using GISTIC2.93 Masked copy number segments, including those containing the NFKBIA locus, were based on CBS, except that a filtering step removed the Y chromosome and probe sets that were previously indicated to be associated with frequent germline copy-number variation. The Infinium Human Methylation 450K BeadChip array and R package conumee, available from The Comprehensive R Network (cran.r-project.org), were used to determine copy number variation (CNV) for NFKBIA in 87 patients of Population 2 and 154 tumor samples belonging to 74 patients in Population 6. CNV was assessed in 26 samples in Population 6 via whole exome sequencing on an Illumina platform.16,78,79,80 CNV calls on the 450K BeadChip array were made using probes for a region spanning NFKBIA ± one megabase pair based on log R ratios (LRRs) calculated by dividing the probe intensity of the query sample by a linear combination of reference samples consisting of material from 52 healthy patients with nominally no copy number aberrations. The threshold for calling NFKBIA deletions was determined during chromosome 1p19q codeletion calls based on the distribution of segment LRRs and by eye evaluation of the segment scatterplots. The Affymetrix OncoScan Array was used for NFKBIA CNV analysis in 40 patients of Population 2 and 227 patients of Population 5. Probe-level LRRs for the NFKBIA region of interest (gene ±1 Mbp) were generated using Nexus Copy Number Software (BioDiscovery) and GRCh37. NFKBIA CNV for 261 tumors of Population 3 was generated using Affymetrix GeneChip Human Mapping 250K NspI or CytoScan HD arrays. NFKBIA CNV was assessed via array-CGH on a spotted genomic DNA microarray with 10,000 large insert clones allowing for an average resolution of better than 0.5 Mb in 136 patients of Population 3 and on an Agilent custom 8 × 60K aCGH array in 305 patients of Population 5 as described.1,90 Chromosome 1p19q deletions were also determined based on these arrays, except for Population 5, where codeletions were also determined using a clinical fluorescence in situ hybridization (FISH) assay.1,90
Methylome analyses
Methylation data for the samples of Population 1 were downloaded from the Broad GDAC Firehose data portal (http://gdac.broadinstitute.org/). Differentially represented CpG probes were identified with the t.test function in R and an FDR of <0.001, and those with differences of beta values—the ratio of the methylated probe intensity and the sum of the methylated and unmethylated probe intensities—of >0.15 between NFKBIA wildtype and deleted tumors were selected for further analysis. R package RCircos83 was used to map differentially methylated CpG probes according to genome position. In Populations 2 and 4, the Infinium Human Methylation 450K BeadChip was used for DNA methylome profiling. In Population 2, data were processed using the R package minifi.84 Genome annotation was per Genome Reference Consortium Human Build 37 (GRCh37, hg19). Data were Noob-normalized and M-value transformed.94 Noob was used so newly profiled samples did not need to be processed with previously processed samples. Probes failing in >10% (by Illumina’s detection p value) of samples were removed. Samples failing across >10% of probes were removed. SNP-containing probes were removed. Associations between methylation and NFKBIA status were tested using the Wilcoxon rank-sum test on methylation data adjusted by study (residuals from the linear model). FDR-adjusted q-values were computed within the probe list, and an FDR of <0.1 and effect in the same direction was considered validated.95 In Population 4, Infinium Human Methylation 450K BeadChip data from TCGA project were downloaded from the GDC and used for differential DNA methylome analysis in NFKBIA deleted vs. non-deleted gliomas. The Beta-value (β) method was used to estimate epigenome-wide methylation levels using the ratio of intensities between methylated and unmethylated alleles.96 Methylation changes with a mean β (NFKBIA deleted tumors) – mean β (NFKBIA-wildtype) of >0.15 and an FDR-adjusted p value of <0.001 were deemed significant.
G-CIMP-low vs. G-CIMP-high calls in 87 tumor samples of Population 2 were made using R package TCGAbiolinksGUI.4,16 163 methylation probes were used to establish a random forest model with 10-fold cross-validation and >97% accuracy. Analysis of enrichment of cytosine-phosphate-guanine (CpG) sites that are hypomethylated in NFKBIA deleted tumors of Population 4 for genes with repressive H3K27me3 marks compared to mid-frontal lobe of normal brain tissue was done using the ENCODE Uniform Processing Pipeline that allows for making uniform peak calls in histone ChiP-sequencing data.97 MACS2 peak caller was used to identify broader regions of H3K27me3 enrichment (broadPeaks) that pass a Poisson p value threshold of 0.1 (using MACS2’s broad peak mode) and gapped/chained regions of enrichment (gappedPeaks) defined as broadPeaks that contain at least one strong narrowPeak (the latter defined as contiguous enrichment that passes a Poisson p value threshold of 0.01). H3K27me3 marks were intersected with a 3-base pair (bp) region to locate the CpGs. The R package missMethyl85 was used to identify CpG sites in gene promoters across the genome that showed differential DNA methylation and to define functionally related sets of genes.
RNA sequencing analyses
Raw read counts of RNA-seq data for the diffuse gliomas of Population 1 and Population 4 profiled as part of TCGA were downloaded using R package recount2.86 Very low expressed genes across samples were filtered out. More specifically, genes were kept when their count-per-million (CPM) values were larger than 1.0 for at least three samples among all samples, which resulted in 27,319 genes. The filtered count matrix was normalized by the trimmed mean of M-values (TMM) method.98 Differentially expressed genes between two groups (i.e., NFKBIA wildtype vs. NFKBIA deleted samples) were identified by R package limma-voom87 with an FDR of 0.05.
Primary RNA-seq data from eleven H3K27M mutant diffuse intrinsic pontine gliomas (DIPGs), five H3K27 wildtype DIPGs, and three normal pontine tissue samples in Population 7 were obtained from Nagaraja et al.,17 Grasso et al.,70 or generated for this study. For the newly generated data, RNA was extracted from approximately 50 mg of tissue frozen at autopsy. RNA was extracted using the RNeasy Mini Plus Kit (Qiagen), and RNA quality was confirmed using an Agilent Bioanalyzer 2100. Following RNA extraction, sequencing libraries were prepared as described by Nagaraja et al.17 and sequenced on a NextSeq 500 by the Stanford Functional Genomics Facility. RNA-seq data from two biological replicates (3k3 and 3k16) of human pluripotent stem cell (hPSC) derived oligodendrocyte precursor cells (OPCs) with inducible H3K27M expression was obtained from Nagaraja et al.17 For all RNA-seq analyses, sequencing adaptors were first trimmed using cutadapt (version 1.11), and reads were aligned to the hg19 reference genome using tophat2 version 2.11.99 Raw counts were obtained by assigning mapped reads to RefSeq genes using Rsubread featureCounts.100 Using the gene-level counts from the primary data, differential expression analysis was performed using DESeq2 with default settings and a Benjamini-Hochberg FDR threshold of 0.1.101 For the hPSC-derived oligodencrocyte progentior cell (OPC) dataset, gene-level counts were first converted to transcript per million (TPM). Genes upregulated in cells with induced H3K27M expression were defined as features with a TPM log2 fold-change greater than one relative to their H3K27 wildtype OPC counterparts across both biological replicates.
Functional enrichment analysis of differentially expressed genes was done using the Over-Representation Analysis (ORA) module via R package WebGestaltR.88 Enriched gene sets were post-processed for redundancy reduction by the weighted set cover function. R package BioVenn was used to graph area-proportional Euler diagrams for the intersection of various gene sets.
Fluorescence in situ hybridization (FISH)
FISH was done as described by Renfrow et al.102 and an NFKBIA-specific probe (NFKBIA-20-OR) from Empire Genomics. Briefly, paraffin-embedded slides were deparaffinized starting in xylene, followed by 100% ethanol. Slides were placed in a 10 mM citrate acid solution and microwaved for 5 min. Samples were digested in a pepsin solution at 37°C for 40 min, followed by washing in 70% ethanol for 1 min, 85% ethanol for 1 min, and 100% ethanol for 10 min. Slides were dried on a slide warmer. FISH probes targeting NFKBIA (Empire Genomics) were applied per manufacturer instructions, and coverslips were applied and sealed with rubber cement. Probes were denatured at 72°C for 3 min and left to hybridize overnight at 37°C in a dark humidity chamber. The next day slides were washed in 2X SSC (saline sodium citrate) 0.5% tween at 42°C for 2 min, followed by 2X SSC at room temperature for 1 min. After air drying in the dark, VECTASHIELD with DAPI (Vector Laboratories) mounting media and a coverslip prepared the specimen for viewing. Pictures were taken using appropriate fluorescent filters on a Zeiss AXIO Observer D1 microscope. There was no centromere control available as chromosome 14 is acrocentric. We used the criteria that at least 80% of the cells needed to be missing one NFKBIA signal to be classified as a hemizygous loss.
Immunofluorescence
Paraffin-embedded slides were placed in two washes of xylene followed by decreasing alcohol content washes and then rehydrated in water. Slides were then placed in an antigen retrieval citrate pH 6.0 solution and microwaved for 5 min. After cooling, slides were washed in TBS and permeabilized in TBS with 0.1% Triton X-100 for 10 min. Slides were then washed in TBS twice for 5 min each. A 1% BSA in TBS with 0.1% Tween 20 blocking buffer was applied for 1 h at 37°C. Primary antibody for NFKBIA (Cat 4814, Cell Signaling) at 1:100 dilution was added and incubated at 4°C overnight. Slides were washed in TBS-T twice for 5 min. A donkey anti-mouse594 secondary antibody (Cat R37115, Thermo Fisher) was then added at a 1:1,000 dilution for 1 h at 37°C in dark conditions. Slides were repeatedly washed with TBS-T twice for 5 min each. VECTASHIELD with DAPI (Vector Laboratories) mounting media and a coverslip prepared the specimen for viewing. Pictures were taken using appropriate fluorescent filters on a Zeiss AXIO Observer D1 microscope.
Knockdown and knockout of NFKBIA
For near-complete knockdown of NFKBIA, G418-resistant primary human astrocytes transduced to stably express wildtype IDH1 or mutant IDH1-(R132H) were transfected with Accell human NFKBIA small interfering (si)RNA or non-targeting control siRNA (Dharmacon), at 20nM concentration using lipofectamine 2000 reagent (Invitrogen) at 1:1 ratio for 48 h. For complete clustered regularly interspaced short palindromic repeats (CRISPR) knockout of NFKBIA, astrocytes were transfected with IκBα Double Nickase Plasmid (h) (sc-400034-NIC, Santa Cruz)—consisting of a pair of plasmids each encoding a D10A mutated Cas9 nuclease and a target-specific 20-nucleotide (nt) guide RNA (gRNA)—using plasmid transfection medium (sc-108062) and Ultra-Cruz transfection reagent (sc-395739) at 1:1.5 ratio and were incubated for 72 h. After incubation, cells were screened for GFP-positivity to select for successfully transfected cells. Cells were then sorted and selected into single cells in a 96-well plate with standard growth medium containing G418 at 0.5 μg/mL as a selection agent to complete screening for successfully transfected cells. Clonally selected cells were expanded into bigger plates. Knockdown and knockout efficiency were assayed by immunoblotting.
mRNA extraction and quantitative polymerase chain reaction (PCR)
Total RNA was extracted from immortalized astrocytes expressing various combinations of wildtype and mutant IDH1 with or without NFKBIA knockdown using TRIzol (Thermo Fisher). To assess mRNA expression levels, 1 μg of total RNA was reverse-transcribed and analyzed by quantitative polymerase chain reaction (qPCR). Reactions for each sample were performed in triplicate using a PCR protocol (95°C activation for 10 min followed by 40 cycles of 95°C for 15 s and 60°C for 1 min) in an ABI StepOnePlus Detection System (Applied Biosystems).
Protein extraction and immunoblotting
Primary human astrocytes were grown to 70% confluency overnight. Whole cell lysate was collected in RIPA buffer containing protease inhibitor, and protein estimation was done using the BCA Assay. Primary antibodies used were NFKBIA/IκBα (ab 4814, Cell Signaling), IDH1-R132H (ab DIA-H09, Dianova), β-Actin (ab 3700, Cell Signaling). Secondary IgG-conjugated horseradish peroxidase (Cell Signaling) was followed by enhanced chemiluminescent (ECL) detection (Thermo Fisher). For the assessment of nuclear phosphorylated-sumoylated NFKBIA (ps-NFKBIA), for cell fractionation, cells were lysed with buffer A (10mM HEPES, 1.5mM MgCl2, 10mM KCl, 0.5mM DTT, 0.05% NP40 (or 0.05% Igepal or Tergitol)) pH 7.9, protease inhibitor cocktail, 4mM N-Ethylmaleimide (NEM) (Sigma, Ref. E3876) (sumoylases inhibitor), in PBS for 20 min on ice and centrifuged at 4°C at 1,000 rcf for 10 min. Supernatants were recovered as the cytoplasmic fraction. 6X Laemmli buffer (SDS–PAGE buffer plus β-mercaptoethanol (Sigma, Ref. M−3148])) was added to the supernatant and sonicated. Pellets were washed with buffer A, centrifuged at 4°C at 1,000 rcf for 10 min, and resuspended with RIPA buffer on ice (181mL H2O, 2mL DOC 10%, 2mL 1M Tris-HCl pH 8.0, 5.6mL 5M NaCl, 2mL Triton X-100 100% from Merck, 2mL SDS 10%, 400μL 500mM EDTA pH 8.0, 250μL 200mM EGTA, 4mL of 1M β-Glycerol-phosphate, 200μL of 100mM NaOrtovanadate, 500μL 4M NaButyrate), and then sonicated and centrifuged at 13,000 rcf for 10 min at 4°C. Supernatants were recovered as the nuclear fraction, and 1X Laemmli buffer was added. Lysates were analyzed by western blotting using standard SDS–polyacrylamide gel electrophoresis (SDS–PAGE) techniques. In brief, protein samples were boiled in Laemmli buffer for 10 min, run in polyacrylamide gels, and transferred onto polyvinylidene–difluoride (PVDF) membranes (Millipore Ref. IPVH00010). Membranes were incubated overnight at 4°C with the appropriate primary antibodies (anti-Phospho-IkBa (Ser32/36) (5A5) (Cell Signaling, ref. 9246); anti-α-Tubulin (clone B-5-1-2) (Sigma-Aldrich, Ref. T6074); anti-Histone H3 (Abcam, Ref. ab1791); anti-PP1 E−9 (Santa Cruz, Ref. sc-7482), extensively washed, and then incubated with specific secondary horseradish peroxidase-linked antibodies from Dako (Ref. P0260 and P0448). Peroxidase activity was visualized using the enhanced chemiluminescence reagent (Biological Industries ref. 20-500-120) and autoradiography films (GE Healthcare ref. 28906835).
Genomic DNA preparation
Genomic DNA from primary human astrocytes was isolated using the GenElute Mammalian Genomic DNA Miniprep Kit (Sigma Aldrich) and quantified via spectrophotometry.
Global histone methylation analysis
The EpiQuik Global H3-K27 Methylation Assay Kit (EpiGentek) was used per manufacturer’s instructions to measure global histone H3-K27 methylation across cultured primary human astrocytes expressing wildtype IDH1 or mutant IDH1 with or without knockdown/knockout of NFKBIA.
Chromatin immunoprecipitation (ChIP)
After transfection with siRNA targeting NFKBIA or a scrambled control, primary human astrocytes were fixed with 37% formaldehyde, and nuclei were prepared. Samples were sonicated, and DNA was measured using a NanoDrop (Thermo Scientific). Two units of DNA were immunoprecipitated with 5-methylcytosine (10805, Abcam), H3K27me3 (9733S, Cell Signaling), H3K9me3 (5327S, Cell Signaling), IκBα (4814S), or EZH2 (5246S, all Cell Signaling). After DNA recovery, results were evaluated by quantitative PCR using Fast SYBR Green master mix (Applied Biosystem) of the histone mark for the G-CIMP gene SLC16A3 (ACGAAAAGTGGGTTGGTCAG/CCCCAACAGACACAAGACCT).
ChIP sequencing
Control or NFKBIA-depleted primary human astrocytes were incubated and crosslinked with 37% formaldehyde and processed according to the above ChIP protocol. For immunoprecipitation, a Tri-Methyl-Histone H3 (Lys27) H3K27me3 antibody (9733S, Cell Signaling) was used at 1:50 dilution overnight at 4°C to elute the ChIP DNA samples. DNA samples were sequenced using an Illumina HiSeq 4000 platform. Raw paired-end sequences were filtered by quality (Q > 30) and length (length >20 bp) with Trim Galore.103 Total filtered sequences were aligned against the reference genome (hg38) with Bowtie2.104 MACS2 software105 was run for each replicate considering unique alignments (q-value <0.1). Peak annotation was performed using the ChIPker package.106
Transcription factor DNA-binding site analysis
Transcription factor binding site analysis for genes mapping into regions of super-silencers (H3K27me3-rich genomic regions, MRRs) in NFKBIA depleted primary immortalized astrocytes was done in EnrichR107 based on ChIP-X data curated in ChIP Enrichment Analysis (ChEA).108 A scatterplot of all terms in the ChEA_2022 gene set library was visualized via Appyter.109 Term frequency-inverse document frequency (TF-IDF) values were computed for the gene set corresponding to each term, and Uniform Mani-fold Approximation and Projection (UMAP) was applied to the resulting values for dimension reduction. Terms were plotted based on the first two UMAP dimensions.
D-2-hydroxyglutarate assay
IDH1 mutant or wildtype primary human astrocytes transfected with siRNA or CRISPR targeting NFKBIA or scrambled control were seeded in 150 mm dishes at a 1 × 107 cells/well density. A colorimetric enzymatic assay (ab211070, Abcam) was used to assess D-2-Hydroxyglutarate (D-2HG) levels according to the manufacturer’s instructions. Samples were spin-filtered using a 10kD spin column for deproteinization to reduce high background. Briefly, 20 μL of each cell lysate were transferred onto 96-well plates in triplicate, and 5 μL of 1 mM D-2HG internal standards were added to each sample well. A freshly prepared reaction mix containing D-2HG assay buffer, D-2HG enzyme, and D-2HG substrate mix was added to each well. Reaction mixes with the cell lysates were incubated for 60 min at 37°C. The samples' optical density (OD) was analyzed at 450 nm using a plate reader. D-2HG concentrations were converted to nmol/μL. Experiments were performed in triplicate.
NF-κB family protein activation assay
Nuclear extracts from IDH1 mutant or wildtype primary human astrocytes transfected with siRNA or CRISPR targeting NFKBIA or scrambled control were prepared using an Active Motif Nuclear Extract Kit (40010, Active Motif) according to the manufacturer’s protocol. Nucleotide-binding of nuclear NF-κB proteins p65, p50, c-Rel, and RelB was quantified via DNA-binding ELISAs (TransAM NF-κB Family Kit, Active Motif) according to the manufacturer’s protocol. 10 μg of nuclear extracts were added to a 96-well plate pre-coated with complete binding buffer. For positive control, 5 μg Raji nuclear extract was used. Activated p65, p50, c-Rel, and RelB nucleotide binding was detected by secondary antibodies conjugated to HRP. Sample absorbance was read at 450 nm using a plate reader. Experiments were performed in triplicate.
Quantification and statistical analysis
Unless otherwise specified, all statistical calculations were performed using R software and packages from Bioconductor (www.bioconductor.org/). Median duration of follow-up was calculated with the reverse Kaplan-Meier method.110 Survival curves were estimated with the Kaplan-Meier method, and survival distributions were compared across groups with the log rank test. We also used the Cox proportional-hazards model likelihood-ratio test to test the significance of Kaplan-Meier survival estimate separations and to avoid overestimating significance when the number of samples in each group is not balanced (low variant frequency of the NFKBIA deletion).111 The assumption of proportional hazards was tested using interactions of the predictor variables with time. Estimation of the hazard ratios (HRs) and 95%-confidence (CI) intervals used the Cox regression method. A Cox regression model with likelihood-ratio tests was computed in NRG Oncology/RTOG 9802 with age, NFKBIA deletion status, adjuvant therapy randomization (combined chemoradiation vs. radiation alone), and the interaction between NFKBIA and adjuvant treatment as covariates. ANOVA was then applied to this model to obtain the overall p value for effects with more than two levels (e.g., the interaction effect). We performed unadjusted and multivariate Cox proportional-hazards regression analyses for overall survival and recurrence-free survival (time from initial diagnosis to death and tumor recurrence, respectively), adjusting for NFKBIA and other genetic and clinicopathologic factors. All categorical variables were treated as nominal categorical variables. During the model-building process, we used alterations of gene dosage (wildtype vs. deleted NFKBIA or CDKN2A/B, wildtype vs. codeleted chromosome 1p19q, amplified or non-amplified EGFR, and presence or absence of chromosome 7 gain/10 loss) or IDH1/2, TERT, ATRX, TP53, and PTEN status (wildtype vs. mutant) as binary predictors to allow for results that are easier to interpret.
Wilcoxon rank-sum test, t-tests, and multiple-comparison one-way ANOVA with post hoc Tukey’s test were used to analyze between-group differences for continuous variables and rxc contingency-table analysis based on McNemar’s test with continuity correction on paired nominal data and Fisher’s exact test otherwise. We computed odds ratios (ORs) in contingency-table analyses according to the equation (a/b)/(c/d) using Woolf’s method for variance estimation,112 and CIs for estimated parameters were computed for ‘constant chi-square boundaries.’ Cramér’s V was used as a post-test to estimate effect size in contingency tables with more than 1° of freedom. Standardized residuals for contingency table cells were calculated as the ratio of the difference between the observed count and the expected count to the standard deviation of the expected count. In association studies between NFKBIA deletions and patient age, we dichotomized age at 40 (≤40 vs. >40) as this cutoff has been used to define low vs. high-risk disease in low(er)-grade gliomas.28 The Benjamini-Hochberg procedure was used in multiple testing to control the false-discover rate (FDR).95
Nomograms for penalized Cox models were developed based on the R package hdnom. Sparse high-dimensional models were built using an adaptive least absolute shrinkage and selection operator (adaLASSO) with a two-step estimation procedure for automatic variable selection113 among the covariates NFKBIA deletion, TERT and ATRX mutations, 1p19q codeletion, WHO grade, and patient age (and CDKN2A/B deletion and TP53 mutation in a select model). Built models were internally validated and calibrated using repeated 5-fold cross-validation and performance assessed by time-dependent area under the receiver operating curves (ROC) (t-AUC) proposed by Chambless & Diao.114 For each round of cross-validation, the data were randomly separated into five groups of equal size, according to package defaults in hdnom. Each of the five groups was used for model validation after parameter estimation based on the other four groups within each distinct round of cross-validation. Models generated in the IDH mutant WHO grade 2 to 4 tumors of Population 4 were externally validated and calibrated in the IDH mutant WHO grade 2 to 4 tumors of Population 5 as an independent dataset with the same variables, as well as in the pooled lower-grade gliomas of Populations 1 and 5.
Acknowledgments
This project was supported by grants UG1CA189867 (NCORP), U10CA180822 (NRG Oncology SDMC), U10CA180868 (NRG Oncology Operations), and U24CA196067 (NRG Specimen Bank) from the National Cancer Institute; R01NS117641 (NIH/NINDS), PI22/00069 (Instituto de Salud Carlos III); POSTD21975LOBO (Asociación Española Contra el Cáncer); and 70-3163-Wi 3 (German Glioma Network) from German Cancer Aid.
Author contributions
Conceptualization, M.B.; methodology, M.B., L.E., H.K., D.M.S., J.P.M., and S.L.P.; validation, M.B., D.M.S., and J.P.M.; formal analysis, M.B., L.E., H.K., D.M.S., J.P.M., W.M., T.M.K., T.M.M., M.A.Q., A.W., R.G.W., B.H., J.S.B.-S., H.N., and R.B.J.; investigation, M.B., L.E., H.K., R.R., W.M., T.M.K., T.M.M., M.A.Q., T.L.-J., A.M., L.S., S. Nagaraja, S. Nair, J.J.W., G.R.H., N.K.T., J.L.F., A.N., S.O., J.B., S.J.H., A.-L.G., M.W., J.S.B.-S., M.M., H.N., R.B.J., and A.C.; resources, T.A.C., C.L.R., and D.R.M.; data curation, M.B., H.K., J.P.M., A.W., and S.L.P.; writing – original draft, M.B.; writing – review & editing, all authors; visualization, M.B., H.K., and J.P.M.; supervision, M.B.; project administration, M.B., S.L.P., and A.C.; funding acquisition: M.B., L.E., T.L.-J., M.W., and A.C. All authors discussed the results and commented on the manuscript.
Declaration of interests
M.B.: NIH/NINDS grant R01NS117641, advisory board membership and grant (Varian Medical Systems), consulting (MRIMath). L.E.: Instituto de Salud Carlos III grant PI22/00069. T.L.-J.: an Asociación Española Contra el Cáncer grant. G.R.H.: NIH/NIBIB grant (P41-EB032840-01). J.S.B.-S.: NIH/NCI grants to Case Western Reserve University School of Medicine. J.P.M.: MOU between OSU Radiation Oncology and OSU Center for Biostatistics. R.B.J.: NIH grants. A.W.: MOU between OSU Radiation Oncology and OSU Biomedical Informatics. T.A.C.: grants (PGDX, Pfizer, AstraZeneca, Illumina, NysnoBio); royalties (PGDX); consulting (Illumina, NysnoBio, Pfizer, BMS, Merck, LG Chem); patents (TMB as predictor of immunotherapy response); stock (Griststone Bio, Nysno Bio). A.N.: consulting (Daiichi-Sankyo, NGK, SparkPlug) to his institution; $2,000 as honoraria. M.W.: grants (Apogenix, Merck, Sharp & Dohme, Merck [EMD], Philogen, Quercis); consulting (Adastra, Medac, Merck [EMD], Nerviano Medical Sciences, Novartis); honoraria (Bristol Meyer Squibb, Medac, Merck, Sharp & Dohme); Data Safety Monitoring Board or Advisory Board (Orbus and Philogen); leadership (EORTC). R.G.W.: grant 70-3163-Wi 3 (German Cancer Aid). S.O.: grant (Cordia Therapeutics, Dainippon-Sumitomo, Pharmaceuticals, Otsuka Assay Inc.); royalties (ASAHI Genomics, Qiagen); consulting (Cordia Therapeutics, Kan Research Institute, Novartis Pharmaceuticals); honoraria (MSD Japan, Kyowa Hakko Kirin, Pfizer); stocks (ASAHI Genomics, Cordia Therapeutics, Rebirthell). M.M.: NIH grants (R01NS092597, DP1NS111132, P50CA165962), grants from Kleberg Foundation Cancer Research, Ludwig Fund for Cancer Research; consulting (Cygnal Therapeutics); honoraria for lectures. C.R.M. and F.B.F.: NIH grant (R01CA258248). F.B.F.: NIH grant (R01NS080939). D.R.M.: clinical trials support (INTELLANCE-1 study [M13-813], CCTG CE.8 [EORTC-1709-BTG], INDIGO study [AG-881], Abbvie, Celgene, Agios [now Servier]).
Inclusion and diversity
We worked to ensure gender balance in the recruitment of human subjects, ethnic or other types of diversity in the recruitment of human subjects, that the study questionnaires were prepared in an inclusive way, sex balance in the selection of non-human subjects, diversity in experimental samples through the selection of the cell lines, diversity in experimental samples through the selection of the genomic datasets. Multiple authors self-identify as an underrepresented ethnic minority in their field of research or within their geographical location or a gender minority in their field of research. One or more of the authors of this paper received support from a program designed to increase minority representation in their field of research. While citing references scientifically relevant to this work, we also actively worked to promote gender balance in our reference list.
Published: June 20, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101082.
Supplemental information
Data and code availability
-
•
RNA-sequencing and ChIP-sequencing data have been deposited into the NIH Gene Expression Omnibus (GEO) repository. The accession numbers for the datasets are listed in the key resources table. This manuscript also analyzes existing, publicly available data. The sources and accession numbers for these datasets are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in the work paper is available from the lead contact upon request.
References
- 1.Eckel-Passow J.E., Lachance D.H., Molinaro A.M., Walsh K.M., Decker P.A., Sicotte H., Pekmezci M., Rice T., Kosel M.L., Smirnov I.V., et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N. Engl. J. Med. 2015;372:2499–2508. doi: 10.1056/NEJMoa1407279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research Network. Brat D.J., Verhaak R.G.W., Aldape K.D., Yung W.K.A., Salama S.R., Cooper L.A.D., Rheinbay E., Miller C.R., Vitucci M., et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N. Engl. J. Med. 2015;372:2481–2498. doi: 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suzuki H., Aoki K., Chiba K., Sato Y., Shiozawa Y., Shiraishi Y., Shimamura T., Niida A., Motomura K., Ohka F., et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 2015;47:458–468. doi: 10.1038/ng.3273. [DOI] [PubMed] [Google Scholar]
- 4.Ceccarelli M., Barthel F.P., Malta T.M., Sabedot T.S., Salama S.R., Murray B.A., Morozova O., Newton Y., Radenbaugh A., Pagnotta S.M., et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164:550–563. doi: 10.1016/j.cell.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson K.C., Anderson K.J., Courtois E.T., Gujar A.D., Barthel F.P., Varn F.S., Luo D., Seignon M., Yi E., Kim H., et al. Single-cell multimodal glioma analyses identify epigenetic regulators of cellular plasticity and environmental stress response. Nat. Genet. 2021;53:1456–1468. doi: 10.1038/s41588-021-00926-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verhaak R.G.W., Hoadley K.A., Purdom E., Wang V., Qi Y., Wilkerson M.D., Miller C.R., Ding L., Golub T., Mesirov J.P., et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan H., Parsons D.W., Jin G., McLendon R., Rasheed B.A., Yuan W., Kos I., Batinic-Haberle I., Jones S., Riggins G.J., et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Killela P.J., Reitman Z.J., Jiao Y., Bettegowda C., Agrawal N., Diaz L.A., Jr., Friedman A.H., Friedman H., Gallia G.L., Giovanella B.C., et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA. 2013;110:6021–6026. doi: 10.1073/pnas.1303607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heaphy C.M., de Wilde R.F., Jiao Y., Klein A.P., Edil B.H., Shi C., Bettegowda C., Rodriguez F.J., Eberhart C.G., Hebbar S., et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425. doi: 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jenkins R.B., Blair H., Ballman K.V., Giannini C., Arusell R.M., Law M., Flynn H., Passe S., Felten S., Brown P.D., et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006;66:9852–9861. doi: 10.1158/0008-5472.CAN-06-1796. [DOI] [PubMed] [Google Scholar]
- 12.Louis D.N., Perry A., Wesseling P., Brat D.J., Cree I.A., Figarella-Branger D., Hawkins C., Ng H.K., Pfister S.M., Reifenberger G., et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23:1231–1251. doi: 10.1093/neuonc/noab106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turcan S., Rohle D., Goenka A., Walsh L.A., Fang F., Yilmaz E., Campos C., Fabius A.W.M., Lu C., Ward P.S., et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malta T.M., de Souza C.F., Sabedot T.S., Silva T.C., Mosella M.S., Kalkanis S.N., Snyder J., Castro A.V.B., Noushmehr H. Glioma CpG island methylator phenotype (G-CIMP): biological and clinical implications. Neuro Oncol. 2018;20:608–620. doi: 10.1093/neuonc/nox183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noushmehr H., Weisenberger D.J., Diefes K., Phillips H.S., Pujara K., Berman B.P., Pan F., Pelloski C.E., Sulman E.P., Bhat K.P., et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Souza C.F., Sabedot T.S., Malta T.M., Stetson L., Morozova O., Sokolov A., Laird P.W., Wiznerowicz M., Iavarone A., Snyder J., et al. A distinct DNA methylation shift in a subset of glioma CpG island methylator phenotypes during tumor recurrence. Cell Rep. 2018;23:637–651. doi: 10.1016/j.celrep.2018.03.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagaraja S., Quezada M.A., Gillespie S.M., Arzt M., Lennon J.J., Woo P.J., Hovestadt V., Kambhampati M., Filbin M.G., Suva M.L., et al. Histone variant and cell context determine H3K27M reprogramming of the enhancer landscape and oncogenic state. Mol. Cell. 2019;76:965–980.e12. doi: 10.1016/j.molcel.2019.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwartzentruber J., Korshunov A., Liu X.Y., Jones D.T.W., Pfaff E., Jacob K., Sturm D., Fontebasso A.M., Quang D.A.K., Tönjes M., et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 19.Wu G., Diaz A.K., Paugh B.S., Rankin S.L., Ju B., Li Y., Zhu X., Qu C., Chen X., Zhang J., et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014;46:444–450. doi: 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis P.W., Müller M.M., Koletsky M.S., Cordero F., Lin S., Banaszynski L.A., Garcia B.A., Muir T.W., Becher O.J., Allis C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857–861. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bender S., Tang Y., Lindroth A.M., Hovestadt V., Jones D.T.W., Kool M., Zapatka M., Northcott P.A., Sturm D., Wang W., et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. 2013;24:660–672. doi: 10.1016/j.ccr.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 22.Müller J. Transcriptional silencing by the Polycomb protein in Drosophila embryos. EMBO J. 1995;14:1209–1220. doi: 10.1002/j.1460-2075.1995.tb07104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beg A.A., Ruben S.M., Scheinman R.I., Haskill S., Rosen C.A., Baldwin A.S. I kappa B interacts with the nuclear localization sequences of the subunits of NF-kappa B: a mechanism for cytoplasmic retention. Genes Dev. 1992;6:1899–1913. doi: 10.1101/gad.6.10.1899. [DOI] [PubMed] [Google Scholar]
- 24.Mulero M.C., Ferres-Marco D., Islam A., Margalef P., Pecoraro M., Toll A., Drechsel N., Charneco C., Davis S., Bellora N., et al. Chromatin-bound IκBα regulates a subset of polycomb target genes in differentiation and cancer. Cancer Cell. 2013;24:151–166. doi: 10.1016/j.ccr.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marruecos L., Bertran J., Álvarez-Villanueva D., Mulero M.C., Guillén Y., Palma L.G., Floor M., Vert A., Arce-Gallego S., Pecharroman I., et al. Dynamic chromatin association of IκBα is regulated by acetylation and cleavage of histone H4. EMBO Rep. 2021;22:e52649. doi: 10.15252/embr.202152649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Espinosa L., Marruecos L. NF-κB-Dependent and -independent (moonlighting) IκBα functions in differentiation and cancer. Biomedicines. 2021;9:1278. doi: 10.3390/biomedicines9091278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bredel M., Scholtens D.M., Yadav A.K., Alvarez A.A., Renfrow J.J., Chandler J.P., Yu I.L.Y., Carro M.S., Dai F., Tagge M.J., et al. NFKBIA deletion in glioblastomas. N. Engl. J. Med. 2011;364:627–637. doi: 10.1056/NEJMoa1006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buckner J.C., Shaw E.G., Pugh S.L., Chakravarti A., Gilbert M.R., Barger G.R., Coons S., Ricci P., Bullard D., Brown P.D., et al. Radiation plus procarbazine, CCNU, and vincristine in low-grade glioma. N. Engl. J. Med. 2016;374:1344–1355. doi: 10.1056/NEJMoa1500925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D'Angelo F., Ceccarelli M., Tala, Garofano L., Zhang J., Frattini V., Caruso F.P., Lewis G., Alfaro K.D., Bauchet L., et al. The molecular landscape of glioma in patients with Neurofibromatosis 1. Nat. Med. 2019;25:176–187. doi: 10.1038/s41591-018-0263-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meissner A., Mikkelsen T.S., Gu H., Wernig M., Hanna J., Sivachenko A., Zhang X., Bernstein B.E., Nusbaum C., Jaffe D.B., et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Estève P.O., Chin H.G., Smallwood A., Feehery G.R., Gangisetty O., Karpf A.R., Carey M.F., Pradhan S. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 2006;20:3089–3103. doi: 10.1101/gad.1463706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allis C.D., Jenuwein T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016;17:487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- 33.Dixit D., Prager B.C., Gimple R.C., Miller T.E., Wu Q., Yomtoubian S., Kidwell R.L., Lv D., Zhao L., Qiu Z., et al. Glioblastoma stem cells reprogram chromatin in vivo to generate selective therapeutic dependencies on DPY30 and phosphodiesterases. Sci. Transl. Med. 2022;14:eabf3917. doi: 10.1126/scitranslmed.abf3917. [DOI] [PubMed] [Google Scholar]
- 34.Xu W., Yang H., Liu Y., Yang Y., Wang P., Kim S.H., Ito S., Yang C., Wang P., Xiao M.T., et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Viswanath P., Najac C., Izquierdo-Garcia J.L., Pankov A., Hong C., Eriksson P., Costello J.F., Pieper R.O., Ronen S.M. Mutant IDH1 expression is associated with down-regulation of monocarboxylate transporters. Oncotarget. 2016;7:34942–34955. doi: 10.18632/oncotarget.9006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cai Y., Zhang Y., Loh Y.P., Tng J.Q., Lim M.C., Cao Z., Raju A., Lieberman Aiden E., Li S., Manikandan L., et al. H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions. Nat. Commun. 2021;12:719. doi: 10.1038/s41467-021-20940-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Healy E., Mucha M., Glancy E., Fitzpatrick D.J., Conway E., Neikes H.K., Monger C., Van Mierlo G., Baltissen M.P., Koseki Y., et al. PRC2.1 and PRC2.2 synergize to coordinate H3K27 trimethylation. Mol. Cell. 2019;76:437–452.e6. doi: 10.1016/j.molcel.2019.08.012. [DOI] [PubMed] [Google Scholar]
- 38.Loh C.H., van Genesen S., Perino M., Bark M.R., Veenstra G.J.C. Loss of PRC2 subunits primes lineage choice during exit of pluripotency. Nat. Commun. 2021;12:6985. doi: 10.1038/s41467-021-27314-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gray F., Cho H.J., Shukla S., He S., Harris A., Boytsov B., Jaremko Ł., Jaremko M., Demeler B., Lawlor E.R., et al. BMI1 regulates PRC1 architecture and activity through homo- and hetero-oligomerization. Nat. Commun. 2016;7:13343. doi: 10.1038/ncomms13343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang H., Wang L., Erdjument-Bromage H., Vidal M., Tempst P., Jones R.S., Zhang Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431:873–878. doi: 10.1038/nature02985. [DOI] [PubMed] [Google Scholar]
- 41.Feng G., Sun Y. The Polycomb group gene. Front. Neurosci. 2022;16:960149. doi: 10.3389/fnins.2022.960149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dang L., White D.W., Gross S., Bennett B.D., Bittinger M.A., Driggers E.M., Fantin V.R., Jang H.G., Jin S., Keenan M.C., et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2010;465:966. doi: 10.1038/nature09132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chung C., Sweha S.R., Pratt D., Tamrazi B., Panwalkar P., Banda A., Bayliss J., Hawes D., Yang F., Lee H.J., et al. Integrated metabolic and epigenomic reprograming by H3K27M mutations in diffuse intrinsic pontine gliomas. Cancer Cell. 2020;38:334–349.e9. doi: 10.1016/j.ccell.2020.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhat K.P.L., Balasubramaniyan V., Vaillant B., Ezhilarasan R., Hummelink K., Hollingsworth F., Wani K., Heathcock L., James J.D., Goodman L.D., et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell. 2013;24:331–346. doi: 10.1016/j.ccr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soubannier V., Stifani S. NF-κB signalling in glioblastoma. Biomedicines. 2017;5:29. doi: 10.3390/biomedicines5020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harutyunyan A.S., Krug B., Chen H., Papillon-Cavanagh S., Zeinieh M., De Jay N., Deshmukh S., Chen C.C.L., Belle J., Mikael L.G., et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun. 2019;10:1262. doi: 10.1038/s41467-019-09140-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Højfeldt J.W., Laugesen A., Willumsen B.M., Damhofer H., Hedehus L., Tvardovskiy A., Mohammad F., Jensen O.N., Helin K. Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat. Struct. Mol. Biol. 2018;25:225–232. doi: 10.1038/s41594-018-0036-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R.S., Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 49.Lu C., Ward P.S., Kapoor G.S., Rohle D., Turcan S., Abdel-Wahab O., Edwards C.R., Khanin R., Figueroa M.E., Melnick A., et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harutyunyan A.S., Chen H., Lu T., Horth C., Nikbakht H., Krug B., Russo C., Bareke E., Marchione D.M., Coradin M., et al. H3K27M in gliomas causes a one-step decrease in H3K27 methylation and reduced spreading within the constraints of H3K36 methylation. Cell Rep. 2020;33:108390. doi: 10.1016/j.celrep.2020.108390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Diehl K.L., Ge E.J., Weinberg D.N., Jani K.S., Allis C.D., Muir T.W. PRC2 engages a bivalent H3K27M-H3K27me3 dinucleosome inhibitor. Proc. Natl. Acad. Sci. USA. 2019;116:22152–22157. doi: 10.1073/pnas.1911775116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Funato K., Major T., Lewis P.W., Allis C.D., Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science. 2014;346:1529–1533. doi: 10.1126/science.1253799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Filbin M.G., Tirosh I., Hovestadt V., Shaw M.L., Escalante L.E., Mathewson N.D., Neftel C., Frank N., Pelton K., Hebert C.M., et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science. 2018;360:331–335. doi: 10.1126/science.aao4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Castel D., Philippe C., Kergrohen T., Sill M., Merlevede J., Barret E., Puget S., Sainte-Rose C., Kramm C.M., Jones C., et al. Transcriptomic and epigenetic profiling of ‘diffuse midline gliomas, H3 K27M-mutant' discriminate two subgroups based on the type of histone H3 mutated and not supratentorial or infratentorial location. Acta Neuropathol. Commun. 2018;6:117. doi: 10.1186/s40478-018-0614-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ben-Porath I., Thomson M.W., Carey V.J., Ge R., Bell G.W., Regev A., Weinberg R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fang D., Gan H., Cheng L., Lee J.H., Zhou H., Sarkaria J.N., Daniels D.J., Zhang Z. H3.3K27M mutant proteins reprogram epigenome by sequestering the PRC2 complex to poised enhancers. Elife. 2018;7:e36696. doi: 10.7554/eLife.36696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lewis E.B. A gene complex controlling segmentation in Drosophila. Nature. 1978;276:565–570. doi: 10.1038/276565a0. [DOI] [PubMed] [Google Scholar]
- 58.Gentile C., Kmita M. Polycomb repressive complexes in hox gene regulation: silencing and beyond: the functional dynamics of polycomb repressive complexes in hox gene regulation. Bioessays. 2020;42:e1900249. doi: 10.1002/bies.201900249. [DOI] [PubMed] [Google Scholar]
- 59.Gorlia T., van den Bent M.J., Hegi M.E., Mirimanoff R.O., Weller M., Cairncross J.G., Eisenhauer E., Belanger K., Brandes A.A., Allgeier A., et al. Nomograms for predicting survival of patients with newly diagnosed glioblastoma: prognostic factor analysis of EORTC and NCIC trial 26981-22981/CE.3. Lancet Oncol. 2008;9:29–38. doi: 10.1016/S1470-2045(07)70384-4. [DOI] [PubMed] [Google Scholar]
- 60.Nomura M., Saito K., Aihara K., Nagae G., Yamamoto S., Tatsuno K., Ueda H., Fukuda S., Umeda T., Tanaka S., et al. DNA demethylation is associated with malignant progression of lower-grade gliomas. Sci. Rep. 2019;9:1903. doi: 10.1038/s41598-019-38510-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chaligne R., Gaiti F., Silverbush D., Schiffman J.S., Weisman H.R., Kluegel L., Gritsch S., Deochand S.D., Gonzalez Castro L.N., Richman A.R., et al. Epigenetic encoding, heritability and plasticity of glioma transcriptional cell states. Nat. Genet. 2021;53:1469–1479. doi: 10.1038/s41588-021-00927-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Raineri S., Mellor J. Linking metabolism and epigenetics. Front. Genet. 2018;9:493. doi: 10.3389/fgene.2018.00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reddington J.P., Perricone S.M., Nestor C.E., Reichmann J., Youngson N.A., Suzuki M., Reinhardt D., Dunican D.S., Prendergast J.G., Mjoseng H., et al. Redistribution of H3K27me3 upon DNA hypomethylation results in de-repression of Polycomb target genes. Genome Biol. 2013;14:R25. doi: 10.1186/gb-2013-14-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brena D., Bertran J., Porta-de-la-Riva M., Guillén Y., Cornes E., Kukhtar D., Campos-Vicens L., Fernández L., Pecharroman I., García-López A., et al. Ancestral function of Inhibitors-of-kappaB regulates Caenorhabditis elegans development. Sci. Rep. 2020;10:16153. doi: 10.1038/s41598-020-73146-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Venkatesh H.S., Morishita W., Geraghty A.C., Silverbush D., Gillespie S.M., Arzt M., Tam L.T., Espenel C., Ponnuswami A., Ni L., et al. Electrical and synaptic integration of glioma into neural circuits. Nature. 2019;573:539–545. doi: 10.1038/s41586-019-1563-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Venkatesh H.S., Johung T.B., Caretti V., Noll A., Tang Y., Nagaraja S., Gibson E.M., Mount C.W., Polepalli J., Mitra S.S., et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell. 2015;161:803–816. doi: 10.1016/j.cell.2015.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Venkataramani V., Tanev D.I., Strahle C., Studier-Fischer A., Fankhauser L., Kessler T., Körber C., Kardorff M., Ratliff M., Xie R., et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature. 2019;573:532–538. doi: 10.1038/s41586-019-1564-x. [DOI] [PubMed] [Google Scholar]
- 68.Palomer E., Carretero J., Benvegnù S., Dotti C.G., Martin M.G. Neuronal activity controls Bdnf expression via Polycomb de-repression and CREB/CBP/JMJD3 activation in mature neurons. Nat. Commun. 2016;7:11081. doi: 10.1038/ncomms11081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Natsume A., Ito M., Katsushima K., Ohka F., Hatanaka A., Shinjo K., Sato S., Takahashi S., Ishikawa Y., Takeuchi I., et al. Chromatin regulator PRC2 is a key regulator of epigenetic plasticity in glioblastoma. Cancer Res. 2013;73:4559–4570. doi: 10.1158/0008-5472.CAN-13-0109. [DOI] [PubMed] [Google Scholar]
- 70.Grasso C.S., Tang Y., Truffaux N., Berlow N.E., Liu L., Debily M.A., Quist M.J., Davis L.E., Huang E.C., Woo P.J., et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 2015;21:555–559. doi: 10.1038/nm.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Katagi H., Louis N., Unruh D., Sasaki T., He X., Zhang A., Ma Q., Piunti A., Shimazu Y., Lamano J.B., et al. Radiosensitization by histone H3 demethylase inhibition in diffuse intrinsic pontine glioma. Clin. Cancer Res. 2019;25:5572–5583. doi: 10.1158/1078-0432.CCR-18-3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fang Y., Liao G., Yu B. LSD1/KDM1A inhibitors in clinical trials: advances and prospects. J. Hematol. Oncol. 2019;12:129. doi: 10.1186/s13045-019-0811-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hashizume R., Andor N., Ihara Y., Lerner R., Gan H., Chen X., Fang D., Huang X., Tom M.W., Ngo V., et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 2014;20:1394–1396. doi: 10.1038/nm.3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Davoli T., Xu A.W., Mengwasser K.E., Sack L.M., Yoon J.C., Park P.J., Elledge S.J. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013;155:948–962. doi: 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dichamp C., Taillibert S., Aguirre-Cruz L., Lejeune J., Marie Y., Kujas M., Delattre J.Y., Hoang-Xuan K., Sanson M. Loss of 14q chromosome in oligodendroglial and astrocytic tumors. J. Neuro Oncol. 2004;67:281–285. doi: 10.1023/b:neon.0000024218.68120.8d. [DOI] [PubMed] [Google Scholar]
- 76.Felsberg J., Yan P.S., Huang T.H.M., Milde U., Schramm J., Wiestler O.D., Reifenberger G., Pietsch T., Waha A. DNA methylation and allelic losses on chromosome arm 14q in oligodendroglial tumours. Neuropathol. Appl. Neurobiol. 2006;32:517–524. doi: 10.1111/j.1365-2990.2006.00759.x. [DOI] [PubMed] [Google Scholar]
- 77.Hu J., Pang J.C.S., Tong C.Y.K., Lau B., Yin X.L., Poon W.S., Jiang C.C., Zhou L.F., Ng H.K. High-resolution genome-wide allelotype analysis identifies loss of chromosome 14q as a recurrent genetic alteration in astrocytic tumours. Br. J. Cancer. 2002;87:218–224. doi: 10.1038/sj.bjc.6600430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mazor T., Pankov A., Johnson B.E., Hong C., Hamilton E.G., Bell R.J.A., Smirnov I.V., Reis G.F., Phillips J.J., Barnes M.J., et al. DNA methylation and somatic mutations converge on the cell cycle and define similar evolutionary histories in brain tumors. Cancer Cell. 2015;28:307–317. doi: 10.1016/j.ccell.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mazor T., Chesnelong C., Pankov A., Jalbert L.E., Hong C., Hayes J., Smirnov I.V., Marshall R., Souza C.F., Shen Y., et al. Clonal expansion and epigenetic reprogramming following deletion or amplification of mutant. Proc. Natl. Acad. Sci. USA. 2017;114:10743–10748. doi: 10.1073/pnas.1708914114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bai H., Harmancı A.S., Erson-Omay E.Z., Li J., Coşkun S., Simon M., Krischek B., Özduman K., Omay S.B., Sorensen E.A., et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat. Genet. 2016;48:59–66. doi: 10.1038/ng.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sonoda Y., Ozawa T., Hirose Y., Aldape K.D., McMahon M., Berger M.S., Pieper R.O. Formation of intracranial tumors by genetically modified human astrocytes defines four pathways critical in the development of human anaplastic astrocytoma. Cancer Res. 2001;61:4956–4960. [PubMed] [Google Scholar]
- 82.Olshen A.B., Venkatraman E.S., Lucito R., Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 83.Zhang H., Meltzer P., Davis S. RCircos: an R package for Circos 2D track plots. BMC Bioinformatics. 2013;14:244. doi: 10.1186/1471-2105-14-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Aryee M.J., Jaffe A.E., Corrada-Bravo H., Ladd-Acosta C., Feinberg A.P., Hansen K.D., Irizarry R.A. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Phipson B., Maksimovic J., Oshlack A. missMethyl: an R package for analyzing data from Illumina's HumanMethylation450 platform. Bioinformatics. 2016;32:286–288. doi: 10.1093/bioinformatics/btv560. [DOI] [PubMed] [Google Scholar]
- 86.Collado-Torres L., Nellore A., Kammers K., Ellis S.E., Taub M.A., Hansen K.D., Jaffe A.E., Langmead B., Leek J.T. Reproducible RNA-seq analysis using recount2. Nat. Biotechnol. 2017;35:319–321. doi: 10.1038/nbt.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang B., Kirov S., Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33:W741–W748. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hulsen T., de Vlieg J., Alkema W. BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008;9:488. doi: 10.1186/1471-2164-9-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Weller M., Weber R.G., Willscher E., Riehmer V., Hentschel B., Kreuz M., Felsberg J., Beyer U., Löffler-Wirth H., Kaulich K., et al. Molecular classification of diffuse cerebral WHO grade II/III gliomas using genome- and transcriptome-wide profiling improves stratification of prognostically distinct patient groups. Acta Neuropathol. 2015;129:679–693. doi: 10.1007/s00401-015-1409-0. [DOI] [PubMed] [Google Scholar]
- 91.Louis D.N., Perry A., Reifenberger G., von Deimling A., Figarella-Branger D., Cavenee W.K., Ohgaki H., Wiestler O.D., Kleihues P., Ellison D.W. The 2016 World Health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 92.Bell E.H., Zhang P., Shaw E.G., Buckner J.C., Barger G.R., Bullard D.E., Mehta M.P., Gilbert M.R., Brown P.D., Stelzer K.J., et al. Comprehensive genomic analysis in NRG oncology/RTOG 9802: a Phase III trial of radiation versus radiation plus procarbazine, lomustine (CCNU), and vincristine in high-risk low-grade glioma. J. Clin. Oncol. 2020;38:3407–3417. doi: 10.1200/JCO.19.02983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mermel C.H., Schumacher S.E., Hill B., Meyerson M.L., Beroukhim R., Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Du P., Zhang X., Huang C.C., Jafari N., Kibbe W.A., Hou L., Lin S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. B. 1995;57:289–300. [Google Scholar]
- 96.Bibikova M., Lin Z., Zhou L., Chudin E., Garcia E.W., Wu B., Doucet D., Thomas N.J., Wang Y., Vollmer E., et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006;16:383–393. doi: 10.1101/gr.4410706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kellis M., Wold B., Snyder M.P., Bernstein B.E., Kundaje A., Marinov G.K., Ward L.D., Birney E., Crawford G.E., Dekker J., et al. Defining functional DNA elements in the human genome. Proc. Natl. Acad. Sci. USA. 2014;111:6131–6138. doi: 10.1073/pnas.1318948111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Robinson M.D., Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 101.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Renfrow J.J., Scheck A.C., Dhawan N.S., Lukac P.J., Vogel H., Chandler J.P., Raizer J.J., Harsh G.R., Chakravarti A., Bredel M. Gene-protein correlation in single cells. Neuro Oncol. 2011;13:880–885. doi: 10.1093/neuonc/nor071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Krueger B., Friedrich T., Förster F., Bernhardt J., Gross R., Dandekar T. Different evolutionary modifications as a guide to rewire two-component systems. Bioinform. Biol. Insights. 2012;6:97–128. doi: 10.4137/BBI.S9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yu G., Wang L.G., He Q.Y. ChIPker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–2383. doi: 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- 107.Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lachmann A., Xu H., Krishnan J., Berger S.I., Mazloom A.R., Ma'ayan A. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics. 2010;26:2438–2444. doi: 10.1093/bioinformatics/btq466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Clarke D.J.B., Jeon M., Stein D.J., Moiseyev N., Kropiwnicki E., Dai C., Xie Z., Wojciechowicz M.L., Litz S., Hom J., et al. Appyters: turning jupyter notebooks into data-driven web apps. Patterns. 2021;2:100213. doi: 10.1016/j.patter.2021.100213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schemper M., Smith T.L. A note on quantifying follow-up in studies of failure time. Control. Clin. Trials. 1996;17:343–346. doi: 10.1016/0197-2456(96)00075-x. [DOI] [PubMed] [Google Scholar]
- 111.Heinze G., Gnant M., Schemper M. Exact log-rank tests for unequal follow-up. Biometrics. 2003;59:1151–1157. doi: 10.1111/j.0006-341x.2003.00132.x. [DOI] [PubMed] [Google Scholar]
- 112.WOOLF B. On estimating the relation between blood group and disease. Ann. Hum. Genet. 1955;19:251–253. doi: 10.1111/j.1469-1809.1955.tb01348.x. [DOI] [PubMed] [Google Scholar]
- 113.Wahid A., Khan D.M., Hussain I. Robust Adaptive Lasso method for parameter's estimation and variable selection in high-dimensional sparse models. PLoS One. 2017;12:e0183518. doi: 10.1371/journal.pone.0183518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chambless L.E., Diao G. Estimation of time-dependent area under the ROC curve for long-term risk prediction. Stat. Med. 2006;25:3474–3486. doi: 10.1002/sim.2299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNA-sequencing and ChIP-sequencing data have been deposited into the NIH Gene Expression Omnibus (GEO) repository. The accession numbers for the datasets are listed in the key resources table. This manuscript also analyzes existing, publicly available data. The sources and accession numbers for these datasets are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in the work paper is available from the lead contact upon request.