


Abstract
Flux balance analysis (FBA) is an important method for calculating optimal pathways to produce industrially important chemicals in genome-scale metabolic models (GEMs). However, for biologists, the requirement of coding skills poses a significant obstacle to using FBA for pathway analysis and engineering target identification. Additionally, a time-consuming manual drawing process is often needed to illustrate the mass flow in an FBA-calculated pathway, making it challenging to detect errors or discover interesting metabolic features. To solve this problem, we developed CAVE, a cloud-based platform for the integrated calculation, visualization, examination and correction of metabolic pathways. CAVE can analyze and visualize pathways for over 100 published GEMs or user-uploaded GEMs, allowing for quicker examination and identification of special metabolic features in a particular GEM. Additionally, CAVE offers model modification functions, such as gene/reaction removal or addition, making it easy for users to correct errors found in pathway analysis and obtain more reliable pathways. With a focus on the design and analysis of optimal pathways for biochemicals, CAVE complements existing visualization tools based on manually drawn global maps and can be applied to a broader range of organisms for rational metabolic engineering. CAVE is available at https://cave.biodesign.ac.cn/.
Graphical Abstract
Graphical Abstract.

The general user flows for metabolic network quality control and metabolic pathway calculation/visualization using CAVE.
INTRODUCTION
Genome-scale metabolic model (GEM) analysis has become an indispensable tool for guiding the engineering of microorganisms to improve the production of industrially important chemicals. Flux balance analysis (FBA) is one of the most widely used algorithms for GEM analysis, allowing for the prediction of optimal pathways from a substrate to a product in a GEM and subsequent identification of possible metabolic engineering targets (1). Several software tools, including COBRA (2) and CellNetAnalyzer (3), have been developed for GEM analysis using FBA (4). However, the programming skills required (Python/MATLAB) pose a significant challenge for biologists to perform FBA using these tools. Furthermore, the optimal pathway calculated from these tools is often presented as a list of reactions with flux values, making it difficult to discover interesting metabolic properties without a visual representation of the pathway. Manual drawing of the pathway map from the reaction list is often necessary to properly understand the mass flow in the pathway.
Several network visualization tools have been developed to simplify this process. For example, Escher (5), Pathview Web (6), IMFLer (7) and iPath3.0 (8) use pre-drawn metabolic maps to map and visualize the FBA-calculated pathways. However, relying on pre-drawn maps limits the application of these tools to a small number of GEMs with available manually drawn maps. Another drawback of this approach is that the manually drawn maps often display only a small subset of reactions in a GEM to make it visually inspectable. This limitation makes it difficult to obtain a complete picture of a calculated pathway using FBA, which contains not only the backbone conversion pathway but also the pathways to balance co-substrates, co-products, reducing powers and energy. Certain general graph-based visualization tools [e.g. Cytoscape (9), VisANT (10) and ModelExplorer (11)] have also been adopted for metabolic pathway visualization. However, these tools require additional effort to convert the pathway reaction list to a metabolite graph by treating metabolites as nodes and reactions as edges. This conversion process often requires coding skills, and the handling of currency metabolites presents another challenge in generating biologically meaningful pathway graphs (12).
In summary, while several tools exist to help biologists use FBA for metabolic pathway analysis, the requirement for programming skills and the limited functional coverage of existing tools for the entire model-to-discovery workflow remain significant obstacles. Therefore, an integrated, easy-to-use tool is highly desirable to streamline the pathway calculation, analysis and visualization processes. Recently, the web application Fluxer (13) was developed to compute, analyze and visualize GEMs using a spanning tree approach to visualize FBA-calculated pathways. This approach is particularly useful for visualizing complex pathways for biomass growth that involve synthesizing dozens of cellular components through hundreds of reactions. However, Fluxer is not suitable for visualizing pathways from a substrate to a desired product, which is of particular interest to synthetic biologists who aim to create artificial cell factories for producing valuable biochemicals. Additionally, there is currently no way to change the FBA optimization objective from biomass growth to the production of a specific compound in Fluxer.
To overcome these challenges, we have developed CAVE, a cloud-based web tool for the analysis and visualization of metabolic pathways. With CAVE, users can select a GEM from the BiGG database (14) or upload their own model, specify a substrate and a target product, and calculate the optimal pathway using FBA or pFBA (parsimonious enzyme usage FBA) (15). Pathway maps are generated directly from the pathway reactions using d3flux, eliminating the need for pre-drawn maps. This allows for a quick and intuitive examination of the flux distribution in the pathway, facilitating the discovery of interesting metabolic features for metabolic engineering. In addition, CAVE provides model quality checking functions and enables users to add, remove or modify reactions in the network to correct any errors found during pathway analysis. This iterative modification/calculation/examination process results in more reliable pathways. To our knowledge, CAVE is the first tool to offer such an integrated and streamlined function for metabolic network check and modification, FBA-based pathway analysis and interactive pathway visualization in a biologist-friendly manner.
IMPLEMENTATION
CAVE is built on Amazon Web Services (AWS) using an innovative serverless architecture with automatic scaling, allowing for high concurrency performance (Supplementary Figure S1). Every submission from the front end initiates a parallel computing process, with no regard to the website’s current demand. Our platform was built on a three-tier architecture consisting of a data storage tier, logic computation tier and front-end presentation tier.
Data storage
CAVE provides a wide selection of high-quality GEMs for users to choose from, including 108 GEMs from the BiGG database, as well as two additional high-quality GEMs for two model organisms, iCW773R (Corynebacterium glutamicum) (16) and iBsu1147R (Bacillus subtilis) (17). All models are stored on AWS S3 in JSON format. Users can also upload their own models to CAVE, and custom models and the calculated pathways are temporarily stored on S3 for later retrieval. DynamoDB, a fully managed NoSQL database service provided by AWS, is used to store and process information related to models and jobs. The model form contains data related to model ID and names, while the job form stores job-specific information, including substrates, products and the ID of the model used for pathway calculation. Once the pathway calculation is completed, the job form is updated with the S3 address of the calculated pathway.
Logic computation
AWS API Gateway directs HTTP requests from the front end to the serverless Lambda functions in the backend. To execute model parsing/editing and pathway computation using FBA and pFBA, a Lambda layer containing the COBRApy package (18) is created. Additional Lambda functions are utilized to retrieve flux distributions in the optimal pathway, compare pathways and add supplementary information in the pathway file for visualization.
Front end
The CAVE front-end website is built with the React framework (https://github.com/facebook/react/) and the Ant Design UI library (https://github.com/ant-design/ant-design), ensuring secure access to AWS cloud services through the AWS Amplify toolkit. The website’s static files, such as HTML pages and JavaScript code, are stored on AWS S3 and distributed through AWS CloudFront, providing fast content delivery to users worldwide.
Visualization
CAVE uses d3flux (https://github.com/pstjohn/d3flux) to automatically generate and visualize metabolic pathway graphs on the web page. d3flux provides various functions for producing biologically meaningful pathway maps, such as the ability to hide or show cofactors/currency metabolites differently from main metabolites to avoid ambiguous links. This makes the pathway map more biologically understandable, helping biologists quickly identify interesting metabolic features or pathway errors. Users can interact with the pathway map to access detailed information on metabolites and reactions or refine the layout by dragging nodes.
MAIN FUNCTIONALITY OF CAVE
CAVE provides a modular set of tools for metabolic pathway design and analysis, comprising model input/preview, pathway calculation, pathway analysis and visualization, model quality control and model modification (Figure 1). Users can choose to use any combination of these functions, from a single pathway calculation to a comprehensive iterative workflow involving multiple functions. By visually inspecting the pathway, identifying errors and correcting them through model modification, users can achieve more reliable pathway results.
Figure 1.

Main function modules of CAVE and two typical workflows using CAVE. Users can obtain and visualize a pathway for high-quality GEMs without errors using the modules connected through the green lines. However, for GEMs with errors, users may need to iteratively use all the modules to calculate, check and correct until a reliable pathway is obtained, as shown in the red lines.
Model input/preview
CAVE provides a user-friendly interface for model selection and uploading, featuring a sorted select box by species name and a dynamic search function. The platform currently supports most GEM formats for user-uploaded models, including JSON, SBML, YAML and MATLAB. After selecting or uploading a model, users can check its basic information on the preview page, such as the number of genes, reactions and metabolites. The input metabolite list allows for quick identification of transportable metabolites and those with suspectable bounds. Users can modify the bounds of an exchange reaction directly from the preview page to turn off a carbon source or increase its uptake rate (Figure 2A). Carbon source selection can also be performed at the pathway calculation page, which automatically turns off the uptake reaction of all other carbon sources.
Figure 2.

Optimal pathway design for 2-oxoglutarate (AKG) production in iML1515 using CAVE. (A) Choose a model and preview its basic information. (B) Define the simulation conditions and calculate the optimal AKG production pathway and the flux distribution. (C) Summary information of the optimal pathway. (D) Visualization of the pathway shows the threonine bypass, which can improve the product yield.
Pathway calculation
This module empowers users to efficiently calculate metabolic pathways from a given substrate to a desired product (Figure 2B). An auto-fill input box ensures accurate metabolite selection and avoids typos. GEMs typically use biomass growth as the default objective reaction and introduce a non-growth-associated ATP maintenance (NGAM) reaction to represent energy consumption for cellular maintenance. However, the NGAM reaction often has a lower bound that causes unnecessary ATP consumption during pathway calculation. To address this issue, CAVE provides an option to set the lower bound of the maintenance reaction to zero. Users can also specify the oxygen uptake rate to calculate pathways under different conditions (aerobic/microaerobic/anaerobic). Additionally, some organisms can fix CO2 from the environment as a carbon source, and the parameter ‘Allow CO2 assimilation from environment’ has been added to accommodate this. CAVE requires the users to provide a pathway label before submitting their job for easy retrieval later on for analysis, visualization and comparison.
The settings mentioned earlier, namely ‘ATP maintenance’, ‘Oxygen uptake rate’ and ‘Substrate uptake rate’, are utilized to modify the parameters of the model via server-side code. If a user sets the ‘ATP maintenance’ to 0, for example, the code will search for the NGAM reaction from a list of possible reaction IDs (e.g. ATPM: atp + h2o → adp + h + pi) in the model and change its boundary to 0–1000, effectively deactivating NGAM. However, there is a possibility that a user-uploaded model may contain an ID that is not listed in the saved reaction ID list, in which case the code will be unable to locate and modify the NGAM reaction. In such a scenario, a warning window will appear, prompting the user to change the boundary in the ‘Model modification’ module of CAVE. Optimal pathway calculation can be carried out using either FBA or pFBA, depending on the user’s choice of ‘Simulating with’. pFBA is a variant of FBA that seeks to minimize the flux associated with each reaction in the model while maintaining optimal flux through the objective function (15). Most pathways are calculated within seconds, and the results are presented in a searchable and sortable smart table. The table lists the pathway reactions and their fluxes (Figure 2B), with the objective reaction for the target product highlighted and placed at the top. Furthermore, this table grants users the ability to select and filter reactions based on their fluxes, thus facilitating the selection of a list of reactions of interest for visualization or obtaining the backbone reactions in the pathway through flux filtering. A summary page provides important statistical information about the pathway to help the users evaluate its reliability. The product yield and carbon yield (representing the percentage of carbon atoms transferred from the substrate to the product) are displayed on this page. A carbon yield value >1 indicates additional carbon inputs from other metabolites besides the substrate, while a very small value indicates that a significant amount of substrate is converted to other metabolites rather than the target product. Both cases require careful examination of the pathway. Correspondingly, CAVE shows the exchange reactions in the pathway for the users to easily check what other metabolites are consumed or produced in the pathway (Figure 2C). Furthermore, the reactions for consumption/production of ATP and NAD(P)H are summarized to allow the users to quickly check the balance of energy and reducing powers.
Pathway analysis/visualization
This module facilitates the process of obtaining target strains in metabolic engineering, which often involves multiple rounds of transformation. To streamline this process, CAVE enables computational simulation and documentation of each step, presenting all simulations to the user through the ‘Pathway analysis/comparison’ module. With this module, users can easily select, display and visualize previously calculated pathways. Selecting a single pathway and clicking ‘Submit’ displays the reactions in the pathway in a smart table, similar to the ‘Pathway Calculation’ page. Furthermore, CAVE allows users to automatically generate an interactive pathway map using d3flux for visual examination of the pathway. Users have the option to hide cofactors to obtain a clean map showing actual mass flow in the pathway (Figure 2D) or show the cofactors to easily identify where ATP and NAD(P)H are consumed or produced. Both reaction nodes and metabolite nodes can be dragged to refine the layout, and the node coordinates of the manually adjusted pathway map can be exported for future reuse. Nodes and edges are clickable, displaying detailed information about the related metabolites and reactions. Another useful function of CAVE is the comparison of two different pathways obtained from different models or at different parameter configurations. This allows users to identify interesting metabolic features and see their effects on product yield, potentially aiding in the discovery of potential metabolic engineering targets to improve product yield or identifying possible errors in a model by comparing it with a high-quality standard model (19).
Quality control
CAVE offers users the possibility to find the optimal conversion pathway in just a few simple steps on the website, taking only a minute to complete. However, it is important to note that errors in the metabolic models, even in published high-quality GEMs, may result in pathways that are not correct. To ensure the accuracy of the model, tools such as MEMOTE (20) have been proposed for model testing and to ensure the model meets certain criteria (e.g. detailed annotation for genes/metabolites, stoichiometrically consistent) before publication. For pathway calculation, CAVE provides pathway quality control functions, which are especially important for checking energy/reducing power production pathways. An unreasonably high ATP production rate may indicate problems in the model, significantly affecting pathway analysis results. Additionally, CAVE allows users to batch check the pathway for the synthesis of biomass building blocks (e.g. 20 amino acids). This can help users identify gaps in the network or pinpoint metabolites with yields higher than the theoretical maximum yield.
Model modification
CAVE offers users the ability to modify the network by adding, removing or editing reactions to correct errors found during pathway analysis or quality control. Users can update the reaction equation to include a missing compound, correct a wrong annotation in the gene–protein–reaction relationships, adjust reaction bounds to change reversibility or introduce new rate constraints. The ‘New Reaction Addition’ function allows users to fill gaps in the model or add heterologous reactions to expand the biosynthetic capabilities of the model when calculating pathways for a foreign product. CAVE provides two methods for creating new reactions: directly inputting the reaction equation for the new reaction, ensuring that metabolite IDs in the equation match those in the model, or gradually building reactions by selecting substrates and products from a dropdown list of metabolites in the model or a new metabolite from BiGG. The latter method is recommended to ensure that the new reaction is correctly linked to the reactions in the model. CAVE also enables users to simulate the effect of gene knockout on the pathways by removing one or more genes in the model. The modified network can be saved in JSON, SBML and MATLAB formats for future analysis.
CASE STUDIES
To demonstrate the usefulness of CAVE, we present two case studies. The first case illustrates how CAVE was utilized to compute the pathway for 2-oxoglutarate (AKG) and discover a new metabolic engineering strategy. The second case exemplifies how various function modules in CAVE were employed iteratively to identify errors in the pathway, modify the model and obtain a more reliable pathway.
Case 1: computing pathways for AKG production and discovering engineering strategies
AKG is a crucial precursor for synthesizing various amino acids and a central metabolite in the tricarboxylic acid (TCA) cycle pathway (21). To compute the optimal pathway for AKG production, we employed the latest Escherichia coli GEM, iML1515 (22), in CAVE. Upon selecting the model on the home page, CAVE displayed basic information about iML1515, including 2712 reactions, 1877 metabolites and 1516 genes, as well as culture conditions of carbon source, nitrogen source and other elements of the model for boundary modification and viewing (Figure 2A). On the ‘Pathway Calculation’ page, we selected AKG as the objective product and set the glucose uptake rate to 10 mmol/g dry cell weight (DCW)/h (Figure 2B). The oxygen uptake rate was kept at its default value to calculate pathways under aerobic conditions, which can be changed to 0 mmol/g DCW/h for anaerobic conditions. The objective was set to AKG, and the ATP maintenance rate was set to 0 for computing optimal pathways. Upon submission, the pathway flux distribution results were displayed as a smart table that could be sorted and searched (Figure 2B). The first highlighted line showed that the optimal rate for AKG production was 12 mmol/g DCW/h. On the ‘Results Summary’ tab, the carbon yield of this pathway was 1, indicating that all carbon atoms from the substrate glucose are transferred to AKG (Figure 2C).
By clicking the ‘Visualization’ button at the bottom of the result page, users can generate a pathway map for visual examination. The map shown in Figure 2D reveals that in addition to the main flux backbone for AKG synthesis, there is a cycle pathway starting from oxaloacetate to reproduce acetyl-CoA and pyruvate. This pathway is the threonine bypass we reported previously, which can improve PHB production by reutilizing the CO2 (or formate) lost in the pyruvate dehydrogenation step (23). Notably, we found that the threonine bypass was also necessary for the optimal production of AKG. By using CAVE, we discovered this engineering strategy in about a minute through a few steps for pathway calculation and visualization. For comparison, we computed the AKG production pathway in the E. coli core model, ‘e_coli_core’ (24), which contains only the main reactions in the metabolic network. The AKG carbon yield of the pathway was only 0.833, as there was no threonine bypass in the model. Supplementary Figure S2 shows the pathway maps of AKG computed from the e_coli_core model and the comparison maps of the two pathways.
Case 2: error correction to obtain a reliable pathway for serine production
As an important amino acid product, l-serine (SER) has diverse applications in various fields, including food, pharmaceuticals and cosmetics (25). To illustrate CAVE’s ability to identify and correct errors in pathway analysis, we used the iJN746 (26) model of Pseudomonas putida to calculate the optimal pathway for SER production. For this study, we set the glucose and oxygen uptake rates at 10 and 1000 mmol/g DCW/h, respectively (aerobic conditions). Additionally, we changed the ‘Allow CO2 assimilation from environment’ to ‘YES’ to investigate whether this model can use CO2 as a carbon source. Surprisingly, the results show that the pathway is very complex, with 359 reactions and an SER carbon yield of only 0.454, implying a large amount of carbon loss (Figure 3A). From the ‘Result Summary’ tab, we can see that the biomass reaction ‘BIOMASS_KT_TEMP’ is also included in the pathway with a flux of 1 g/g DCW/h. By using the ‘Modification’ module, we discovered that the lower bound of this reaction is set at 1 instead of 0 (Figure 3B), which explains the carbon loss and the complexity of the pathway (as many reactions are required to synthesize biomass building blocks). We changed the boundary to 0, and the resulting pathway has a more reasonable 41 reactions. Additionally, we noticed that the SER carbon yield was increased to 1.2, implying that there is CO2 fixation in the pathway.
Figure 3.

Identification and correction of errors in the SER synthesis pathway of iJN746 model using CAVE. (A) Calculation of the optimal synthesis pathway for SER based on the iJN746 model. (B) Search ‘BIOMASS_KT_TEMP’ reaction on the ‘Modification’ page and update its lower boundary from 1 to 0. (C) Visualization of the SER production pathway calculated from the original iJN746 model. (D) SER production pathway calculated using the updated iJN746 model.
However, the visualized pathway shown in Figure 3C indicates that this carbon fixation is caused by some errors in the model. The first error is that SER is synthesized directly from pyruvate by reacting with ammonium. This reaction should be irreversible and can only happen in the reverse direction; therefore, the upper bound of this reaction was changed to zero. The second error is caused by a net energy generation cycle, as shown in Figure 3C. This cycle was corrected by changing the lower bound of reaction ALDD2x_copy2 (acald_c + h2o_c + nad_c  ac_c + 2.0 h_c + nadh_c) to 0, so that it can only happen in the forward direction (27). The third error is in the carbon fixation cycle, where reaction AKGDa (akg_c + h_c + lpam_c
ac_c + 2.0 h_c + nadh_c) to 0, so that it can only happen in the forward direction (27). The third error is in the carbon fixation cycle, where reaction AKGDa (akg_c + h_c + lpam_c 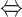 co2_c + sdhlam_c) should also be changed to irreversible. The visualized pathway maps can help users with biochemistry knowledge quickly identify and correct abnormal reactions in the pathway without leaving CAVE. After these corrections, a biologically more reasonable pathway for serine synthesis was obtained (Figure 3D). In this pathway, the SER precursor phosphoglycerate (3PG) is produced by the Entner–Doudoroff (ED) pathway, and the pathway from 3PG to SER agrees with the pathway reported in biochemistry textbooks.
co2_c + sdhlam_c) should also be changed to irreversible. The visualized pathway maps can help users with biochemistry knowledge quickly identify and correct abnormal reactions in the pathway without leaving CAVE. After these corrections, a biologically more reasonable pathway for serine synthesis was obtained (Figure 3D). In this pathway, the SER precursor phosphoglycerate (3PG) is produced by the Entner–Doudoroff (ED) pathway, and the pathway from 3PG to SER agrees with the pathway reported in biochemistry textbooks.
It is important to note that some of the errors mentioned above can be detected prior to pathway analysis by utilizing the ‘Quality control’ module. For instance, by checking the ATP production, users can identify that the model iJN746 is capable of generating 1000 mmol ATP from 10 mmol glucose. Moreover, the net energy generation cycle error, mentioned earlier, can be detected by visually inspecting the ATP production pathway as shown in Supplementary Figure S3. Following the correction, the ATP production rate was enhanced to 217.5 mmol/g DCW/h from 10 mmol/g DCW/h glucose, and the pathways reveal that ATP is predominantly produced through the ED pathway and the TCA cycle (Supplementary Figure S3). Similar energy generation errors not only exist in iJN746, but are also commonly observed in other models, including the E. coli model iECIAI1_1343 (28), the Acinetobacter baumannii AYE model iCN718 (29) and the Homo sapiens model Recon1 (30). With CAVE, it is possible to detect and rectify such errors, thereby obtaining more dependable pathway design outcomes.
DISCUSSION
Cellular metabolism can be engineered to redirect metabolic fluxes toward valuable biochemicals, and synthetic biology aims to achieve this goal. GEM analysis is a useful tool for predicting optimal engineering strategies for flux diversion. CAVE is designed to help experimental biologists, who lack coding skills, to harness the power of GEM analysis in engineering organisms. By integrating COBRApy (18) for pathway calculation at the backend and d3flux for pathway visualization at the front end, CAVE allows users to choose a model, specify the substrate and product, set the conditions (aerobic/anaerobic), calculate and visually check a pathway in about a minute. Currently, no other tools offer such an easy-to-use one-stop pathway design/visualization service. Some tools, such as Escher-FBA (31), Fluxer (13) and MetExplore (32), provide pathway analysis and visualization functions, but changing the optimization objective from biomass to a target product can be challenging. Users have to download the SBML file of the model and manually edit it to add a demand reaction for the product and change the objective to the newly added demand reaction. This is difficult for experimental biologists who may not even understand the SBML file format.
In contrast, CAVE allows users to easily choose any metabolite in the specified GEM as the target product. The auto-fill input box ensures that only metabolites in the GEM can be selected. CAVE also provides functions to add external reactions to a model, making it possible to calculate pathways to a heterologous product. Designing and analyzing optimal product synthesis pathways are at the center of CAVE development. CAVE does not aim to visualize the global network organization like Fluxer or map pathways to the entire network like Escher (5). Although large networks can be impressive, they are difficult for biologists to understand and do not offer much help in identifying possible engineering targets. Instead, CAVE focuses on product synthesis pathways, which typically contain <100 reactions. Therefore, the automatically generated pathway map is more accessible to biologists. Users can generate a clear mass flow pathway by simply dragging and dropping a few nodes. In comparison with the dendrogram maps generated by Fluxer, the pathway maps generated by CAVE (as shown in Figures 2 and 3) are more biologically meaningful. CAVE can be used for pathway design for a wider range of organisms than other tools since there is no requirement for pre-drawn maps.
Another one of the distinctive features of CAVE is its ability to integrate model quality control and modification functions. Despite the existence of over 200 so-called high-quality GEMs, many of them have not been adequately tested for the design of product synthesis pathways, and errors in the calculated pathways are not uncommon. Through visualization, CAVE enables biologists to inspect pathways visually and identify reactions that may be incorrect. The output of the pathway analysis is also designed to facilitate error identification. The ‘Result Summary’ page, for instance, provides key information on carbon, ATP and reducing power balances. After identifying errors, users can directly remove or modify incorrect reactions in the model using the ‘Modification’ function. Additionally, CAVE offers certain pre-analysis quality control measures for frequently occurring errors (such as ATP/NADPH/NADH net production, mass imbalance and network gaps), allowing users to evaluate the model’s accuracy before using it for pathway design.
CAVE is more than just a tool for biologists to calculate and visualize pathways without programming skills. It also enables them to obtain more dependable pathways through an iterative calculation/examination/modification process using the provided functions. This is particularly crucial since incorrect pathways result in incorrect targets and may lead to wasted time and resources on costly genetic engineering to overexpress, insert or delete the gene targets.
DATA AVAILABILITY
CAVE is a web server freely accessible without login requirement at https://cave.biodesign.ac.cn/.
Supplementary Material
Contributor Information
Zhitao Mao, Biodesign Center, Key Laboratory of Engineering Biology for Low-Carbon Manufacturing, Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, China; National Center of Technology Innovation for Synthetic Biology, Tianjin 300308, China.
Qianqian Yuan, Biodesign Center, Key Laboratory of Engineering Biology for Low-Carbon Manufacturing, Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, China; National Center of Technology Innovation for Synthetic Biology, Tianjin 300308, China.
Haoran Li, Biodesign Center, Key Laboratory of Engineering Biology for Low-Carbon Manufacturing, Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, China; National Center of Technology Innovation for Synthetic Biology, Tianjin 300308, China.
Yue Zhang, Biodesign Center, Key Laboratory of Engineering Biology for Low-Carbon Manufacturing, Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, China; National Center of Technology Innovation for Synthetic Biology, Tianjin 300308, China; College of Biotechnology, Tianjin University of Science and Technology, Tianjin 300457, China.
Yuanyuan Huang, Biodesign Center, Key Laboratory of Engineering Biology for Low-Carbon Manufacturing, Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, China; National Center of Technology Innovation for Synthetic Biology, Tianjin 300308, China; College of Biotechnology, Tianjin University of Science and Technology, Tianjin 300457, China.
Chunhe Yang, Biodesign Center, Key Laboratory of Engineering Biology for Low-Carbon Manufacturing, Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, China; National Center of Technology Innovation for Synthetic Biology, Tianjin 300308, China; College of Biotechnology, Tianjin University of Science and Technology, Tianjin 300457, China.
Ruoyu Wang, Biodesign Center, Key Laboratory of Engineering Biology for Low-Carbon Manufacturing, Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, China; National Center of Technology Innovation for Synthetic Biology, Tianjin 300308, China.
Yongfu Yang, State Key Laboratory of Biocatalysis and Enzyme Engineering, Environmental Microbial Technology Center of Hubei Province and School of Life Sciences, Hubei University, Wuhan 430000, China.
Yalun Wu, State Key Laboratory of Biocatalysis and Enzyme Engineering, Environmental Microbial Technology Center of Hubei Province and School of Life Sciences, Hubei University, Wuhan 430000, China.
Shihui Yang, State Key Laboratory of Biocatalysis and Enzyme Engineering, Environmental Microbial Technology Center of Hubei Province and School of Life Sciences, Hubei University, Wuhan 430000, China.
Xiaoping Liao, Biodesign Center, Key Laboratory of Engineering Biology for Low-Carbon Manufacturing, Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, China; National Center of Technology Innovation for Synthetic Biology, Tianjin 300308, China; Haihe Laboratory of Synthetic Biology, Tianjin 300308, China.
Hongwu Ma, Biodesign Center, Key Laboratory of Engineering Biology for Low-Carbon Manufacturing, Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, China; National Center of Technology Innovation for Synthetic Biology, Tianjin 300308, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key Research and Development Program of China [2021YFC2100700]; Strategic Priority Research Program of the Chinese Academy of Sciences [XDB0480000]; Tianjin Synthetic Biotechnology Innovation Capacity Improvement Projects [TSBICIP-PTJS-001, TSBICIP-PTJJ-007]; Innovation Fund of Haihe Laboratory of Synthetic Biology [22HHSWSS00021]; Science and Technology Partnership Program, Ministry of Science of China [KY202001017]; Youth Innovation Promotion Association CAS. Funding for open access charge: National Key Research and Development Program of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Fang X., Lloyd C.J., Palsson B.O.. Reconstructing organisms in silico: genome-scale models and their emerging applications. Nat. Rev. Microbiol. 2020; 18:731–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schellenberger J., Que R., Fleming R.M.T., Thiele I., Orth J.D., Feist A.M., Zielinski D.C., Bordbar A., Lewis N.E., Rahmanian S.et al.. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat. Protoc. 2011; 6:1290–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. von Kamp A., Thiele S., Hädicke O., Klamt S.. Use of CellNetAnalyzer in biotechnology and metabolic engineering. J. Biotechnol. 2017; 261:221–228. [DOI] [PubMed] [Google Scholar]
- 4. Orth J.D., Thiele I., Palsson B.Ø.. What is flux balance analysis. Nat. Biotechnol. 2010; 28:245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. King Z.A., Dräger A., Ebrahim A., Sonnenschein N., Lewis N.E., Palsson B.O.. Escher: a web application for building, sharing, and embedding data-rich visualizations of biological pathways. PLoS Comput. Biol. 2015; 11:e1004321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Luo W., Pant G., Bhavnasi Y.K., Blanchard S.G. Jr, Brouwer C. Pathview Web: user friendly pathway visualization and data integration. Nucleic Acids Res. 2017; 45:W501–W508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Petrovs R., Stalidzans E., Pentjuss A.. IMFLer: a web application for interactive metabolic flux analysis and visualization. J. Comput. Biol. 2021; 28:1021–1032. [DOI] [PubMed] [Google Scholar]
- 8. Darzi Y., Letunic I., Bork P., Yamada T.. iPath3.0: interactive pathways explorer v3. Nucleic Acids Res. 2018; 46:W510–W513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T.. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Granger B.R., Chang Y.-C., Wang Y., DeLisi C., Segrè D., Hu Z.. Visualization of metabolic interaction networks in microbial communities using VisANT 5.0. PLoS Comput. Biol. 2016; 12:e1004875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Martyushenko N., Almaas E.. ModelExplorer—software for visual inspection and inconsistency correction of genome-scale metabolic reconstructions. BMC Bioinformatics. 2019; 20:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ma H., Zeng A.-P.. Reconstruction of metabolic networks from genome data and analysis of their global structure for various organisms. Bioinformatics. 2003; 19:270–277. [DOI] [PubMed] [Google Scholar]
- 13. Hari A., Lobo D. Fluxer: a web application to compute, analyze and visualize genome-scale metabolic flux networks. Nucleic Acids Res. 2020; 48:W427–W435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. King Z.A., Lu J., Dräger A., Miller P., Federowicz S., Lerman J.A., Ebrahim A., Palsson B.O., Lewis N.E.. BiGG Models: a platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2015; 44:D515–D522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lewis N.E., Hixson K.K., Conrad T.M., Lerman J.A., Charusanti P., Polpitiya A.D., Adkins J.N., Schramm G., Purvine S.O., Lopez-Ferrer D.et al.. Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol. Syst. Biol. 2010; 6:390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Niu J., Mao Z., Mao Y., Wu K., Shi Z., Yuan Q., Cai J., Ma H.. Construction and analysis of an enzyme-constrained metabolic model of Corynebacteriumglutamicum. Biomolecules. 2022; 12:1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu K., Mao Z., Mao Y., Niu J., Cai J., Yuan Q., Yun L., Liao X., Wang Z., Ma H.. ecBSU1: a genome-scale enzyme-constrained model of Bacillussubtilis based on the ECMpy workflow. Microorganisms. 2023; 11:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ebrahim A., Lerman J.A., Palsson B.O., Hyduke D.R.. COBRApy: constraints-based reconstruction and analysis for Python. BMC Syst. Biol. 2013; 7:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yuan Q., Huang T., Li P., Hao T., Li F., Ma H., Wang Z., Zhao X., Chen T., Goryanin I.. Pathway-consensus approach to metabolic network reconstruction for Pseudomonasputida KT2440 by systematic comparison of published models. PLoS One. 2017; 12:e0169437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lieven C., Beber M.E., Olivier B.G., Bergmann F.T., Ataman M., Babaei P., Bartell J.A., Blank L.M., Chauhan S., Correia K.et al.. MEMOTE for standardized genome-scale metabolic model testing. Nat. Biotechnol. 2020; 38:272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gyanwali B., Lim Z.X., Soh J., Lim C., Guan S.P., Goh J., Maier A.B., Kennedy B.K.. Alpha-ketoglutarate dietary supplementation to improve health in humans. Trends Endocrinol. Metab. 2022; 33:136–146. [DOI] [PubMed] [Google Scholar]
- 22. Monk J.M., Lloyd C.J., Brunk E., Mih N., Sastry A., King Z., Takeuchi R., Nomura W., Zhang Z., Mori H.et al.. iML1515, a knowledgebase that computes Escherichia coli traits. Nat. Biotechnol. 2017; 35:904–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin Z., Zhang Y., Yuan Q., Liu Q., Li Y., Wang Z., Ma H., Chen T., Zhao X.. Metabolic engineering of Escherichia coli for poly(3-hydroxybutyrate) production via threonine bypass. Microb. Cell Fact. 2015; 14:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Orth J.D., Fleming R.M.T., Palsson B.Ø.. Reconstruction and use of microbial metabolic networks: the core Escherichia coli metabolic model as an educational guide. EcoSal Plus. 2010; 4: 10.1128/ecosalplus.10.2.1. [DOI] [PubMed] [Google Scholar]
- 25. Zhang X., Xu G., Shi J., Koffas M.A.G., Xu Z.. Microbial production of L-serine from renewable feedstocks. Trends Biotechnol. 2018; 36:700–712. [DOI] [PubMed] [Google Scholar]
- 26. Nogales J., Palsson B.Ø., Thiele I.. A genome-scale metabolic reconstruction of Pseudomonasputida KT2440:IJN746 as a cell factory. BMC Syst. Biol. 2008; 2:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fritzemeier C.J., Hartleb D., Szappanos B., Papp B., Lercher M.J.. Erroneous energy-generating cycles in published genome scale metabolic networks: identification and removal. PLoS Comput. Biol. 2017; 13:e1005494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Monk J.M., Charusanti P., Aziz R.K., Lerman J.A., Premyodhin N., Orth J.D., Feist A.M., Palsson B.Ø.. Genome-scale metabolic reconstructions of multiple Escherichia coli strains highlight strain-specific adaptations to nutritional environments. Proc. Natl Acad. Sci. U.S.A. 2013; 110:20338–20343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Norsigian C.J., Kavvas E., Seif Y., Palsson B.O., Monk J.M.. iCN718, an updated and improved genome-scale metabolic network reconstruction of Acinetobacterbaumannii AYE. Front. Genet. 2018; 9:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Duarte N.C., Becker S.A., Jamshidi N., Thiele I., Mo M.L., Vo T.D., Srivas R., Palsson B.Ø.. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc. Natl Acad. Sci. U.S.A. 2007; 104:1777–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rowe E., Palsson B.O., King Z.A.. Escher-FBA: a web application for interactive flux balance analysis. BMC Syst. Biol. 2018; 12:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cottret L., Frainay C., Chazalviel M., Cabanettes F., Gloaguen Y., Camenen E., Merlet B., Heux S., Portais J.-C., Poupin N.et al.. MetExplore: collaborative edition and exploration of metabolic networks. Nucleic Acids Res. 2018; 46:W495–W502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
CAVE is a web server freely accessible without login requirement at https://cave.biodesign.ac.cn/.