

Summary
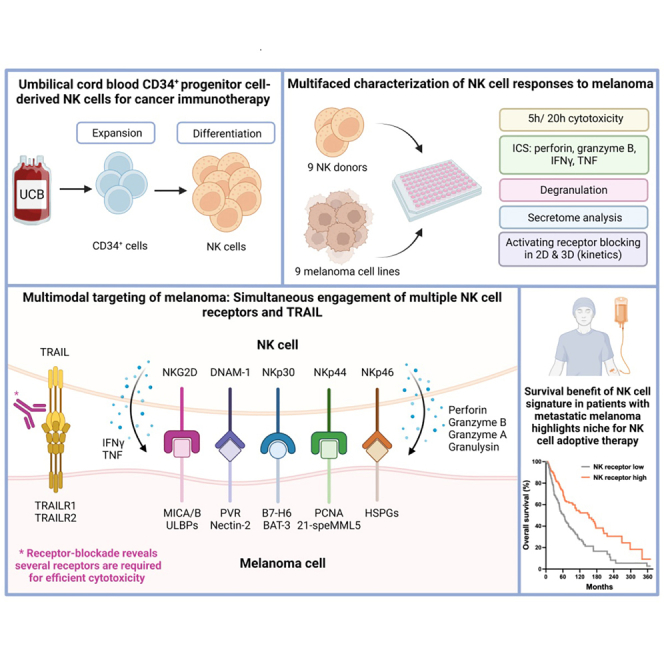
Umbilical cord blood (UCB) CD34+ progenitor cell-derived natural killer (NK) cells exert efficient cytotoxicity against various melanoma cell lines. Of interest, the relative cytotoxic performance of individual UCB donors was consistent throughout the melanoma panel and correlated with IFNγ, TNF, perforin and granzyme B levels. Importantly, intrinsic perforin and Granzyme B load predicts NK cell cytotoxic capacity. Exploring the mode of action revealed involvement of the activating receptors NKG2D, DNAM-1, NKp30, NKp44, NKp46 and most importantly of TRAIL. Strikingly, combinatorial receptor blocking led to more pronounced inhibition of cytotoxicity (up to 95%) than individual receptor blocking, especially in combination with TRAIL-blocking, suggesting synergistic cytotoxic NK cell activity via engagement of multiple receptors which was also confirmed in a spheroid model. Importantly, lack of NK cell-related gene signature in metastatic melanomas correlates with poor survival highlighting the clinical significance of NK cell therapies as a promising treatment for high-risk melanoma patients.
Subject areas: Therapy, Components of the immune system, Cancer
Graphical abstract
Highlights
-
•
NK cells exert potent multimodal cytotoxicity against various melanoma cell lines
-
•
TRAIL is a major contributor to NK cell mediated multimodal targeting of melanoma
-
•
Mechanism of action was confirmed in vitro in a 3D spheroid model of melanoma
-
•
NK cells are a promising cell therapy candidate for the treatment of melanoma
Therapy; Components of the immune system; Cancer
Introduction
Melanoma is a very immunogenic malignancy which can induce potent adaptive immune responses.1 However, in advanced melanoma, the immune system is often impaired leading to tumor immune escape. In particular, immune cells such as T regulatory cells, tumor associated macrophages and myeloid derived suppressor cells influence the tumor microenvironment (TME) of melanoma to become immunosuppressive.2 In addition, immune checkpoints such as PD-1 and TIM-3 play an important role in further suppressing immune responses in melanoma patients.3,4 Because melanoma forms a complex network of interactions with the immune system, it constitutes an excellent candidate for immunotherapy, and it was indeed the first indication for which adoptive cell therapy was tested. Early clinical trials using ex vivo expanded tumor infiltrating lymphocytes (TILs) achieved objective response rates of 49–72% where 22% of the patients obtained complete tumor regression.5 Later, metastatic melanoma was the first indication for which checkpoint inhibitors, such as anti-CTLA-4 antibody ipilimumab and anti-PD1 antibody nivolumab, received FDA approval.6,7
Despite the promising clinical outcomes of adoptive TIL therapy, there are some disadvantages of using T cells. To avoid graft-versus-host disease (GvHD), these therapies are used in autologous settings which limit the chance of successful treatment because of unsuccessful ex vivo expansion cultures.8 Moreover, although HLA class I expression is required to initiate a T cell immune response; it is often downregulated in melanoma, causing resistance to T cell-based immunotherapies.9 On the other hand, T cells with a chimeric antigen receptor (CAR) are independent of HLA-expression and are currently under clinical investigation in melanoma patients.10
Natural killer (NK) cells exert a cytotoxic response on engagement with tumor or virally infected cells when the net signal from activating receptors exceeds that of inhibitory receptors.11 Thus, NK cell activation is not solely dependent on antigen presentation via HLA-expression on tumor cells which, among other reasons, makes NK cells an attractive candidate for immunotherapy, possibly even after development of primary or secondary resistance to immune checkpoint blockade. Upon activation, NK cells release cytotoxic granules and secrete pro-inflammatory cytokines, such as IFNγ or TNF, as part of the antitumor response.12 Crosslinking of activating receptors results in release of perforin and granzyme B, inducing caspase-dependent or -independent apoptosis. In addition, cytotoxicity can also be induced by the death receptors FASL or TRAIL.13
Besides other immune cells, function of NK cells in melanoma patients is also often impaired which is associated with decreased levels of CD161 and NKG2D, and increased TGF-β1 levels.14,15 Therefore, introducing NK cells as adoptive cell therapy might overcome dysfunction of the patient’s own immune system. Clinical investigations of adoptive NK cell therapy have so far shown a safe profile with minimal risk for GvHD and no severe toxicities.16,17,18,19 Although NK cell therapy is more established for hematological malignancies, there is growing interest in investigating its potential for solid tumors, such as melanoma.20 Melanoma cell lines express various ligands for NK cell activating receptors NKG2D, DNAM-1, NKp30, NKp44, NKp46 and TRAIL, indicating that it is a suitable indication for NK cell therapy.21,22,23,24 Potent anti-tumor responses have been observed in vitro with peripheral NK cells against melanoma cell lines and patient samples.25,26,27 In addition, a few clinical trials using NK cells from allogeneic,28 autologous29 or cell line-derived30 sources, showed no GvHD but clinical responses could be further improved. Current NK cell-based adoptive therapy trials for melanoma are limited and still in early phases.31
Glycostem Therapeutics has developed a unique process for the expansion and differentiation of CD34+ hematopoietic umbilical cord blood (UCB)-derived stem cells into a highly potent off-the-shelf NK cell product, called GTA002, which is cryopreserved. GTA002’s non-cryopreserved predecessor has proven to be safe in a phase I clinical trial in acute myeloid leukemia (AML) patients.16 Currently, GTA002 is under clinical investigation for the treatment of AML and is also being preclinically assessed as a potential therapy for other hematological and solid malignancies.
In this study, we perform a comprehensive pre-clinical evaluation of UCB CD34+ progenitor cell-derived NK cells and demonstrate potent anti-tumor activity against a collection of melanoma cell lines. Therefore, we evaluated target cell susceptibility but also assessed donor cytotoxic potency by measuring release of effector molecules granzyme A, granzyme B, perforin and granulysin and cytokines IFNγ and TNF. The cytotoxic capacity of different UCB donors correlates with IFNγ, TNF, perforin and granzyme B levels, whereas the cytolytic activity of NK cells against melanoma is dependent on multimodal activation through different receptors such as NKG2D, DNAM-1, NKp30, NKp44, NKp46 and TRAIL. Of interest, we observed that rapid involvement of TRAIL is central for the mode of action of NK cells against melanoma, accounting for a large proportion of the cytolytic activity. Pronounced cytotoxicity was also seen against melanoma spheroids via the same activation mechanisms. These results highlight NK cells as a promising new adoptive cell therapy candidate for melanoma.
Results
NK cells exert potent anti-tumor activity against melanoma cell lines
A well-established cell culture system was used to promote hematopoietic stem cell expansion and differentiation into highly cytotoxic NK cells (CD56+ > 98%). A collection of 9 UCB donors was selected based on their overnight cytotoxic potencies against target cell line K562, with potent, but various cytotoxicity of 44.1%–84.0% in a 1:1 E:T ratio (Figure S1A). Further increase of the E:T ratio resulted in more cytotoxicity, already reaching a plateau in 5:1 E:T ratio (Figure S1B). Thus, a challenging low E:T ratio was selected in order to assess differences between UCB donors. NK cells derived from UCB CD34+ progenitor cells showed characteristic high expression of NKG2D (79.2% ± 3.3), DNAM-1 (87.0% ± 3.1) and NKp44 (93.1% ± 1.6), high expression of NKG2A (62.9% ± 3.7) and low expression of KIR2D receptors (1.4% ± 0.3) (Figure 1A), as previously published.32 Differences in expression levels between donors were minimal as shown by geometric Mean of Fluorescence Intensity (gMFI) (Figure 1A). In parallel, a set of 9 melanoma cell lines, derived from primary melanoma or lymph node metastasis, was phenotypically characterized for the expression of ligands for certain NK cell receptors (Figure S2). Low expression levels were found for ULBPs and Nectin-2 (CD112), whereas higher expression levels were found for MICA/B and PVR (CD155) with more variability between cell lines. To assess cytotoxic potency of NK cells, the set of 9 different melanoma cell lines was tested in 5- and 20-h flow cytometry-based cytotoxicity assays, measuring 7AAD+ late apoptotic cells, at an E:T of 1:1 (gating strategy in Figure S3). At the 5h timepoint, the highest cytotoxicity was observed against A375 (40.2% ± 5.0) and the lowest against SK-MEL-28 (7.8% ± 1.7) (Figure 1B). Cell lines WM9 and MEL-BRO reached similar levels of cytotoxicity as A375 after 20h of assessment (42.1% ± 5.0, 41.8% ± 3.1 and 40.7% ± 5.6 respectively), whereas for A375, MeWo and G361 a high cytolytic effect was already reached after 5 h (Figures 1B and 1C). SK-MEL-28 still showed lower levels of cytotoxicity (14.0% ± 2.0) after 20h (Figure 1C), suggesting higher resistance toward NK cytotoxicity.
Figure 1.

NK cells exert potent anti-tumor activity against melanoma cell lines
(A) Terminally differentiated NK cells were phenotypically characterized at day 35 for NK cell receptors, and percentages and gMFI are shown.
(B and C) Flowcytometry-based cytotoxicity results against 9 melanoma cell lines of a (B) 5h co-culture and (C) 20h co-culture assay, E:T ratio of 1:1. The order of the cell lines in the graphs is arranged from high to low susceptibility for NK cells in the 5-h co-culture assay.
(D) Percentage of degranulating cells after 5h co-culture with melanoma cells. Each data point represents an individual donor and data are shown as mean ± SEM (n = 9, for C n = 8, divided among 3 independent experiments).
(E) The corresponding heatmap for cytotoxicity 5h and degranulation 5h of the individual donors. The individual donors are arranged in increasing cytotoxic capacity based on the geometric mean (gMean) of % cytotoxicity of each individual donor against all cell lines.
In addition, NK cell degranulation was assessed by measuring CD107a/LAMP-1, which was remarkably high against MEL-BRO, with a percentage of 39.5 ± 3.6 (Figure 1D), in comparison to the rest of the melanoma cell lines. However, the 5h cytotoxicity was only 12.8% ± 4.2 (Figure 1B), indicating that degranulation does not directly correlate with susceptibility of the target cell lines at the same time point. Thus, although differences in average donor cytotoxicity of individual cell lines can be attributed to varying target susceptibility, this is not reflected by degranulation. Comparison of individual donor cytotoxicity and degranulation capacity against the panel of melanoma cell lines showed different response patterns as indicated in the heatmaps (Figure 1E). Correlation tests revealed that degranulation did not correlate with cytotoxicity for 7/9 cell lines, except for MeWo (p = 0.016) and G361 (p = 0.028) (see Figure 3E). Moreover, the range of susceptibility of the melanoma cell lines to NK cytotoxicity implies differential engagement of cytotoxic pathways in a target-dependent manner. The observed differences in cytotoxicity prompted further investigation into other aspects of NK functionality to gauge the range of effector responses against melanoma.
Figure 3.

Intracellular levels of cytokines and cytotoxic effector molecules on co-culture of NK cells with melanoma cell lines
(A–F) NK cells were co-cultured with melanoma cell lines for 5h at the E:T ratio of 1:1 and percentage of (A) IFNγ+ cells, (B) TNF+ cells and gMFI of (C) perforin and (D) granzyme B was determined. Cell lines are arranged from high to low susceptibility. Red dashed line indicates baseline level of no target condition. Each data point represents an individual donor and data are shown as mean ± SEM (n = 9, divided among 3 independent experiments). Statistical analysis was performed comparing no target condition vs. each individual cell line using one-way ANOVA. Below each bar graph, the corresponding heatmaps of the 9 individual UCB donors are shown.
(E) Pearson correlation coefficient (r) of percentage of cytotoxicity vs. % degranulation, % IFNγ+ cells, % TNF+ cells, gMFI of perforin or gMFI of granzyme B of effector + target (NK + tumor) conditions per cell line and data of all cell lines.
(F) Pearson correlation coefficient (r) of % cytotoxicity vs. intrinsic levels of perforin or granzyme B per cell line and gMean of all cell lines. Significance is shown as p < 0.05 ∗, <0.01 ∗∗, 0.001 ∗∗∗, 0.0001 ∗∗∗∗.
NK cells effectively secrete pro-inflammatory cytokines and effector molecules on melanoma engagement
The observed differences in melanoma susceptibility to NK cytotoxicity, and the discrepancy compared to degranulation levels, motivated the investigation of cytokines and effector molecules which are key players of the cytotoxic cascade. We probed the induction of cytokine and cytolytic effector molecule release into the supernatant of 5h-cytotoxicity assays as potential early indicators of response. Significant increases of IFNγ and TNF were found on co-culture of NK cells with A375, MeWo, RPMI79651, WM9, MEL-JUSO and MEL-BRO (Figures 2A and 2B). Remarkably, co-culture with MEL-BRO caused high release of IFNγ (2212 ± 1308 pg/mL) and TNF (912.3 ± 203.9 pg/mL), despite previously observed low cytotoxicity at 5h which increased at 20h. A similar assessment was performed for released levels of cytolytic effector molecules. More specifically, perforin release increased significantly in 6/9 cell lines and granzyme A in 5/9 cell lines, indicating target-specific differences in activation (Figures 2C and 2D). Similarly, Granzyme B and granulysin were significantly increased on co-culture of NK cells with 6/9 cell lines (Figures 2E and 2F).
Figure 2.

Release of cytokines and cytotoxic effector molecules in supernatant on co-culture of NK cells with melanoma cell lines
(A–F) NK cells were co-cultured with melanoma cell lines for 5h, E:T ratio of 1:1 and release of (A) IFNγ, (B) TNF, (C) perforin, (D) granzyme A, (E) granzyme B and (F) granulysin was measured in the supernatant. Cell lines are arranged in order of NK cytotoxic susceptibility. Each data point represents an individual donor and data are shown as mean ± SEM (n = 9, divided among 3 independent experiments). Statistical analysis was performed comparing no target condition vs. each individual cell line using one-way ANOVA. Below each bar graph, the corresponding heatmaps of the 9 individual UCB donors are shown. The individual donors are arranged in increasing cytotoxic capacity based on the geometric mean (gMean) of % cytotoxicity of each individual donor against all cell lines.
(G) Pearson correlation coefficient (r) between percentage of cytotoxicity vs. IFNγ, TNF, perforin, granzyme A, granzyme B or granulysin per cell line. Significance is shown as p < 0.05 ∗, <0.01 ∗∗, 0.001 ∗∗∗, 0.0001 ∗∗∗∗.
Overall, we observed that even cell lines that are less susceptible to NK cytotoxicity were capable of triggering efficient secretion of the aforementioned cytokines and effector molecules. Consequently, we did not observe any correlation between target susceptibility and release of effector molecules, visualized in Figure 2, where cell lines are arranged in high to low susceptibility to NK cytotoxicity.
In contrast, when assessing the cell line-specific cytotoxic capacity of the 9 UCB donors, we do indeed observe that increased secretion of effector molecules correlated with efficient cytotoxicity against some of the cell lines (Figure 2G). IFNγ release correlated with cytotoxicity only for 2/9 cell lines, but granulysin release correlated with cytotoxicity for 6/9 cell lines. Thus, UCB CD34+ progenitor cell-derived NK cells actively secrete cytokines and effector molecules already after a short co-culture period with melanoma cells, and their release in the supernatant correlates with the cytotoxic capacity of the UCB donors.
Intracellular levels of pro-inflammatory cytokines and cytolytic effector molecules predict NK cell cytotoxic capacity
The initial assessment of released cytokines and cytolytic effector molecules into the supernatant on co-culture with melanoma cells suggests that the proportion of responding NK cells may vary between different donors. To investigate this, we assessed the percentage of cells that were positive for IFNγ and TNF after 5h of co-culture with melanoma cell lines by intracellular staining (see gating strategy Figure S4). The percentage of IFNγ+ NK cells significantly increased on co-culture with each individual melanoma cell line compared to no target control, suggesting that the majority of the UCB donors could initiate a functional response to target engagement. However, only a proportion of cells from each UCB donor were IFNγ+ (Figure 3A) after 5h co-culture with melanoma cells. In contrast, a significant increase in the percentages of TNF+ cells was only seen on engagement with A375, MeWo, WM9 and MEL-BRO (Figure 3B). The pre-existing load of intracellular cytotoxic granules seems to be an important determinant of cytotoxic capacity. On melanoma co-culture, a decrease in intracellular perforin and granzyme B levels measured as a reduction in gMFI in the co-culture condition versus the no target control, was interpreted as a functional response (Figures 3C and 3D). The gMFI of granzyme B was significantly decreased after engagement with all cell lines, whereas 7/9 cell lines (except G361 and A375) induced significant depletion of intracellular perforin levels.
It was observed that the induction of pro-inflammatory cytokine production varied between individual donors and specific target cell lines, with UCB donor #1 showing minimal IFNγ production against the melanoma cell lines, whereas donor #9 showed high percentage of IFNγ+ cells (Figure 3A, heatmap). Correlation analyses showed that IFNγ correlated with cytotoxic capacity for 7/9 cell lines (Figure 3E). Because the percentage of TNF+ cells per UCB donor was generally low, a correlation with cytotoxic capacity was observed only for 3/9 cell lines (A375, MeWo and SK-MEL-28) (Figure 3E). Of interest, the average gMFI of intracellular perforin and granzyme B of all UCB donors against the different melanoma cell lines, was very similar. However, it showed a large spread between UCB donors suggesting that perforin and granzyme B levels are associated with cytotoxic capacity rather than melanoma susceptibility. Indeed, cytotoxicity correlated with the gMFI of perforin for 6/9 cell lines and with the gMFI of granzyme B for 5/9 cell lines (Figure 3E). Taking the datapoints of all 9 cell lines vs. all 9 UCB donors together, the cytotoxic capacity still correlated with either IFNγ (p=<0.0001), TNF (p = 0.0089), perforin (p=<0.0001) or granzyme B (p=<0.0001), but not with degranulation (p = 0.068) (Figures 3E and S5). Thus, these 4 factors appear predictive of an individual donor’s capacity to exert potent cytotoxic activity, independent of target cell line. The different intrinsic levels of perforin and granzyme B of the UCB donors before co-culture, suggested that these may be predictive factors of the cytotoxic capacity of NK cells. Correlation tests revealed that cytotoxicity correlated with intrinsic perforin levels for 7/9 (except RPMI7951 and MEL-BRO) and with intrinsic granzyme B levels for 7/9 (except RPMI7951 and SK-MEL-28) melanoma cell lines (Figure 3F). The gMean of the cytotoxicity percentages of all cell lines per donor showed a strong correlation with the intrinsic perforin and granzyme B levels, with p values of 0.002 and 0.0002 respectively (Figure 3F). Therefore, the intrinsic levels of perforin and granzyme B can accurately predict the inherent cytotoxic potential of NK cells.
The phenotype of UCB CD34+progenitor cell-derived NK cells supports anti-melanoma activity
To further explore the observed differences in cytotoxic potency of NK cells against melanoma, a more in-depth analysis of the kinetics of the cytotoxic response was performed on 3 selected target cell lines. A375 and SK-MEL-28 were selected based on high and low susceptibility to NK cytotoxicity in both 5h and 20h cytotoxicity assays, respectively (Figures 1B and 1C). MEL-BRO was selected for its low susceptibility to NK cytotoxicity in the 5h assay and high susceptibility in the 20h assay (Figures 1B and 1C) and for inducing a strong pro-inflammatory response (Figures 2A and 2B). UCB donors with different cytotoxic capacity (donor #1, #5, #7) were selected to evaluate the kinetics of anti-melanoma responses, using an impedance-based cytotoxicity assay for 30h. Similar to the flowcytometry-based cytotoxicity assays, we observed high susceptibility for A375 and low susceptibility for SK-MEL-28, whereas MEL-BRO showed unexpectedly high susceptibility early during the assessment (Figure S6A). More importantly, the selected UCB donors showed similar levels of cytotoxicity as in the flowcytometry-based assays (Figures 1A and 1B). In addition, using higher E:T ratios than 1:1 in the impedance-based cytotoxicity resulted in reaching maximal cytotoxicity against almost all cell lines, and therefore not allowing the observation of differences in target susceptibility (Figure S6B).
A more detailed phenotyping of receptors expressed on NK cells was performed for donors #5 and #7, as they showed high cytotoxicity and therefore provided a good basis to investigate NK cell activation. The donors expressed DNAM-1, NKG2D, NKp44, NKp30, NKp46, TRAIL and very low FASL (Figures 4A and 4B). More specifically, donor #7 showed slightly higher percentages of expression of all receptors compared to donor #5, except for NKp44. Extensive phenotyping of the selected melanoma cell lines for the ligands of the corresponding NK cell receptors showed differences in the expression levels of ULBP3, MICA/B, Nectin-2, B7-H6, FAS and TRAILR1 and R2 (Figure 4C). MEL-BRO expressed very low levels of FAS, whereas A375 and SK-MEL-28 showed moderate expression. TRAILR1 was highly expressed on A375 and MEL-BRO, but very low on SK-MEL-28, whereas TRAILR2 was expressed on all three cell lines but with lower expression on SK-MEL-28. The decoy receptors TRAILR3 and R4 were minimally expressed by all cell lines. Thus, expression of activating receptors on UCB CD34+ progenitor cell-derived NK cells and the presence of their corresponding ligands on melanoma cells, suggest contribution of these interactions to cytotoxicity can be expected.
Figure 4.

Phenotyping of NK cells and melanoma cell lines
(A) Representative gating strategy for characterization of NK cells.
(B) NK cell receptor expression of donor #5 and donor #7. NK cells were pre-gated based on 7AAD−/CD56+ and percentage is shown for each receptor.
(C) Fold MFI compared to unstained for ligand expression of MEL-BRO, SK-MEL-28 and A375 shown as mean ± SEM (n = 3 independent experiments).
TRAIL enhances the multimodal activation of NK cell cytotoxic activity against melanoma
To elucidate the mechanism of activation of NK cytotoxicity against melanoma and confirm receptor involvement, we used blocking mAbs alone or in combinations to interfere with specific NK receptor/ligand interactions in potent donors (#5 and #7). Blocking of NKG2D, DNAM-1, NKp30, NKp44, NKp46, TRAIL and FASL was implemented in an impedance-based assay, previously used to assess NK cell anti-tumor responses,33 to allow for time course detection of differences in cytotoxicity. Because simultaneous engagement of several activating receptors can lead to a cumulative cytolytic response, we explored whether combinatorial receptor blocking would cause more pronounced inhibition of cytotoxicity. Of interest, TRAIL- and FASL-signaling have been reported as later responses compared to granzyme B/perforin release,34 suggesting that combinatorial blocking of activating receptors and TRAIL/FASL would lead to prolonged impairment of cytotoxicity. Early investigations showed that blocking FASL alone did not lead to decreased cytotoxicity and did not contribute to more inhibition of cytotoxicity in combination with NKG2D, DNAM-1 and TRAIL (Figure S7), which could be explained by low FASL expression on UCB CD34+ progenitor cell-derived NK cells (Figure 4B) and was thus not further explored. The most prominent inhibitory effects were observed for donor #7 in co-culture with A375 (Figures 5A–5F), although donor #5 showed very similar results (Figure 5G). Of interest, blocking DNAM-1 or NKG2D alone resulted in a moderate reduction of cytotoxic capacity, whereas blocking TRAIL caused a dramatic reduction implying a more dominant role of death receptor-mediated cytotoxicity (Figure 5A). Moreover, a major decrease in cytotoxicity was seen when both NKG2D and DNAM-1 were blocked (Figure 5B), suggesting an additive effect in stimulating NK cytotoxicity. Of interest, blocking either NKG2D or DNAM-1, in combination with TRAIL led to a dramatic decrease in cytotoxicity, which was more pronounced when all three receptors were blocked (Figure 5B). Further exploration of the involvement of NCRs in NK cytotoxicity revealed that blocking of NKp30 or NKp44 alone did not lead to inhibition of A375 cytotoxicity, whereas NKp46-blocking resulted in minor reduction (Figure 5C). Because TRAIL-blocking alone showed great inhibition of cytotoxic capacity, we next investigated the effect of combinatorial TRAIL- and NCR-blocking. Although no effect was observed for NKp30-blocking alone, cytotoxicity was strongly inhibited when combined with TRAIL-blocking, resulting in more inhibition than TRAIL-blocking alone (Figure 5D). A similar effect was observed with combinatorial blocking of NKp44 and TRAIL, but not of NKp46 and TRAIL (Figure 5D). Of interest, concurrent blocking of NKp30 and NKp44 led to moderate reduction in cytotoxicity, whereas blocking individual NCRs did not result in any decrease (Figures 5E and 5C). Moreover, combinatorial NKp30- and NKp46-blocking resulted in intermediate effect, whereas NKp44- and NKp46-blocking led to a dramatic decrease in cytotoxicity (Figure 5E). Simultaneous blocking of all NCRs almost completely abrogated the cytotoxicity of A375 (Figure 5E). Most importantly, blocking 2 or 3 NCR as well as TRAIL, led to similar levels of very pronounced inhibition of A375 cytotoxicity (Figure 5F). Overall, combinations of TRAIL-blocking and blocking of other receptors consistently resulted in profound inhibition of cytotoxicity, compared to single blockings thus highlighting the major contribution of TRAIL in multimodal cytotoxic function of UCB CD34+ progenitor cell-derived NK cells.
Figure 5.

Effect on cytotoxicity of melanoma cell lines on blocking of various receptors
NK cells were pre-incubated with different combinations of blocking mAbs and seeded in co-culture with A375 in a 1:1 E:T ratio for 24h. Representative graphs of A375 cytotoxicity in co-culture with donor #7 show the mean of technical triplicates after blocking:
(A) DNAM-1, NKG2D, or TRAIL.
(B) Combinations of DNAM-1, NKG2D and TRAIL.
(C) NKp30, NKp44 or NKp46.
(D) Combinations of NKp30, NKp44 or NKp46 with TRAIL.
(E) Combinations of NKp30, NKp44 and NKp46.
(F) Combinations of two out of NKp30, NKp44 or NKp46 with TRAIL or all four combined. All blocking mAbs were used at 10 μg/mL. Concentration of isotype ctrl Ab is matched to the concentration of four added blocking mAbs.
(G) Heatmap of results of blocking studies for 2 donors against 3 melanoma cell lines. The percentage of cytotoxicity relative to isotype ctrl for each blocking combination is shown for MEL-BRO, SK-MEL-28 and A375 for donor #5 and #7. Percentage was calculated as (AUC condition/AUC isotype ctrl)∗100. AUC = area under the curve.
An extensive assessment of the effects of receptor blockings for donor #7 in co-culture with MEL-BRO and SK-MEL-28 is reported in Figures S8 and S9, respectively. A heatmap of the cumulative percentages of cytotoxicity measured as area under the curve (AUC), relative to the maximum cytotoxicity value (isotype ctrl, no blocking mAbs) for each target cell line and UCB donor combination, provides a comprehensive overview of the receptors’ contribution to cytotoxicity (Figure 5G). We observed that cytotoxicity of A375 and SK-MEL-28 was almost completely abrogated with combinatorial blocking of NCRs with or without TRAIL, indicating an essential role of multiple receptor-ligand interactions in targeting these melanoma cell lines. The cytotoxicity of MEL-BRO was very difficult to block, suggesting that other ligand-receptor interactions, which are yet to be determined, may be important for NK activation against MEL-BRO. Moreover, decreases in cytotoxicity of MEL-BRO on blocking of various receptors alone or in combinations were observed later during the co-culture, typically starting at approximately 5 h post co-culture initiation (Figure S8), suggesting that the remaining live target cells in the co-culture proliferate faster than the cytotoxicity rate induced by NK cells after blocking with mAbs.
TRAIL-dependent activation of NK cells leads to efficient lysis of A375 spheroids
To further evaluate and confirm the significance of TRAIL-mediated rapid NK cell anti-melanoma cytotoxicity, we investigated the TRAIL-pathway status in melanoma cell lines. First, we examined the sensitivity of A375, MEL-BRO and SK-MEL-28 to TRAIL-mediated apoptosis by treating the cells with soluble rhTRAIL and monitoring caspase 3/7 activity over time. A375 showed caspases 3/7 activity to a higher extent than SK-MEL-28 (Figure 6A), which could be explained by lower expression levels of TRAILR1 and TRAILR2 on SK-MEL-28 (Figure 4C). Therefore, A375 was selected to further explore caspase-involvement in-depth. On engagement of TRAIL receptors on target cells, caspase 8 is activated followed by caspase 3/7, leading to apoptosis.35 However, TRAIL-engagement could also induce other NK effector functions, such as degranulation and IFNγ release.36 To investigate whether TRAIL-engagement on NK cells leads to downstream signaling of TRAIL-mediated apoptosis in tumor cells, we blocked TRAIL and measured caspases 3/7 and caspase 8 activity simultaneously after 3h of NK co-culture with A375. Both caspases 3/7 as well as caspase 8 activity decreased on TRAIL-blocking, suggesting that TRAIL-engagement indeed led to downstream signaling (Figure 6B). The same was observed when NKp30, NKp44, NKp46, DNAM-1 and NKG2D were simultaneously blocked, suggesting that caspase 8 is also involved in those signaling pathways (Figure 6B). Next, we investigated blocking combinations that had shown significant involvement of TRAIL, in a 3D spheroid model of A375. Here, we observed that TRAIL-blocking alone resulted in inhibition of cytotoxicity to some extent but not significant compared to isotype ctrl. However, as soon as TRAIL-blocking was combined with either NKp30-, NKp30/NKp44-or NKG2D/DNAM-1-blocking, dramatic significant reductions in the cytolytic capacity of NK cells were observed compared to the combination without TRAIL blocking (Figures 6C and 6D), corroborating the extensive contribution of the TRAIL-pathway also in the 3D setting. Exposure of spheroids to NK cells treated with the isotype control mAbs led to complete dissociation and decrease in red fluorescence intensity indicating potent cytotoxic function. In contrast, spheroids that were exposed to NK cells treated with blocking mAbs against NKp30/NKp44/TRAIL had almost completely intact morphology (Figure 6E) confirming the results of the 2D impedance-based cytotoxicity blocking assays (Figure 5).
Figure 6.

TRAIL involvement in NK activation
(A) Caspase 3/7 activity of A375, MEL-BRO and SK-MEL-28 was measured every hour after addition of rhTRAIL in increasing concentrations. Caspase 3/7 activity is shown as total green fluorescence area of the mean ± SEM of technical quadruplicates of a representative experiment.
(B) Caspase 3/7 and caspase 8 activity was measured after 3h of co-culture with A375 using various blocking combinations. Data is shown as mean ± SEM of the caspase activity relative to isotype ctrl, n = 2 donors.
(C–E) NLR-A375 cells were seeded at 1∗103 cells/well and were allowed to form spheroids for 2 days. NK cells were pre-incubated with the various blocking mAbs combinations and added to the spheroid in a final E:T ratio of approximately 25:1.
(C) Representative graph of total integrated intensity of the red calibrated unit (RCU) shown as the mean ± SEM of technical quadruplicates for donor #5.
(D) Percentage of cytotoxicity relative to isotype ctrl. Calculated by first determining % cytotoxicity of each condition using the formula 100-(AUC condition/AUC A375 only∗100) followed by calculating % of cytotoxicity relative to isotype ctrl (=100%). Data is shown as mean ± SEM of n = 3 donors, divided among 3 independent experiments. Statistical analysis was performed using paired t-test between de indicated condition.
(E) Representative images of A375 spheroid after 1h and 10h of co-culture with NK cells at 4× magnification. A375 cells are visible in red and NK cells are surrounding the A375 spheroid. Significance is shown as p < 0.05 ∗, <0.01 ∗∗, 0.001 ∗∗∗, 0.0001 ∗∗∗∗.
NK cell cytotoxic response gene signature in metastatic melanoma patients confers survival benefit
The observed correlation between release of effector molecules and cytokines, and the cytotoxic capacity of NK cells against melanoma cell lines, prompted us to investigate the relevance of the expression of those markers for the survival of melanoma patients. Therefore, we consulted the open access TCGA-SKCM; Cancer Genome Atlas, https://doi.org/10.25504/FAIRsharing.m8wewa (skin cutaneous melanoma) dataset and compared gene expression with survival data.37 Of itnerest, higher expression of granulysin, granzyme A, granzyme B, TNF, IFNγ, or perforin was associated with better overall survival (Figure 7A), suggesting that an NK cell response gene signature contributes to a more favorable clinical outcome and thus supporting our claim for the potential use of adoptive NK cell therapy for metastatic melanoma patients. Moreover, DNAM-1 and the NCRs as their expression in the patient samples was rather low, indicating low numbers of NK cells infiltrating the TME, which further strengthens the relevance for NK cell adoptive therapy. Nevertheless, higher expression of TRAIL, DNAM-1, NKG2D, NKp30 and NKp46 correlated with better survival (Figure 7B). These results underline the clinical relevance and the potential benefit of adoptive NK cell therapy for the treatment of metastatic melanoma.
Figure 7.

Survival data of metastatic melanoma patients subdivided by high and low gene expression of NK cell cytotoxicity related genes
(A) Probability of overall survival of metastatic melanoma patients subdivided by high and low gene expression of granulysin (GNLY), granzyme A (GZMA), granzyme B (GZMB), TNF, IFNγ (IFNG), perforin (PRF1).
(B) Probability of overall survival of metastatic melanoma patients subdivided by high and low gene expression of TRAIL (TNFSF10), DNAM-1 (CD226), NKG2D (KLRK1), NKp46 (NCR1), NKp44 (NCR2), NKp30 (NCR3). Subgroups with high and low expression were generated based on median value, n = 352. Statistical analysis was performed using the Log rank test.
Discussion
Despite recent successes in immunotherapy of metastatic melanoma, development of resistance still poses a challenge, spurring the development of new treatments like adoptive NK cell therapy. Granzyme family genes were previously shown to predict better survival of melanoma patients,38 suggesting an important role for NK cells or cytotoxic T lymphocytes (CTL) in melanoma. Furthermore, melanoma patients have better survival when an NK cell-specific gene signature was detected in the tumor.39 Indeed, we also found a positive correlation between expression of NK cell cytotoxicity-related genes and overall survival, implying that increased infiltration of active NK cells into the tumor could be beneficial for metastatic melanoma patients. Moreover, melanoma cells that develop resistance against BRAF-inhibitors, upregulate ligands important for NK cell recognition and become more susceptible to NK cell mediated cytotoxicity.40 Although this supports the implementation of adoptive NK cell therapy as a treatment option for metastatic melanoma, currently only ∼4% of the allogeneic NK cell-based clinical trials target melanoma as indication.41
This study presents a first systematic in vitro pre-clinical assessment of efficacy of the novel off-the-shelf NK cell therapy against melanoma. Here, we show that UCB CD34+ progenitor cell-derived NK cells exert potent cytotoxic activity against a broad panel of melanoma cell lines, accompanied by the induction of pro-inflammatory responses under challenging E:T conditions. Historically, anti-melanoma activity has been observed for NK cells in vitro at higher E:T, i.e. 20:1,21,27,42 which highlights the superior cytotoxic potency of UCB CD34+ progenitor cell-derived NK cells. One of the initial highlights of this study is that NK cells degranulation and cytotoxicity do not generally correlate (thus, any extrapolation of NK cell potency based on degranulation could potentially be misleading as cytotoxicity would be the most relevant assessment to determine NK potency). Secretion of pro-inflammatory cytokines IFNγ, TNF and cytolytic molecules perforin and granzyme B however strongly correlated with the cytotoxic capacity of individual UCB donors, providing additional parameters for prediction of NK activity. Although differences were observed in release of cytokines and effector molecules on exposure to different melanoma cell lines, these were not predictive for target susceptibility. However, cell lines with high susceptibility like A375 does initiate more release of cytokines such as IFNγ and TNF compared to the low susceptible cell line SK-MEL-28, thus these markers could still be indicative for target susceptibility. Moreover, a major learning from this study is that the intrinsic high levels of perforin and granzyme B before target exposure were predictive of the cytotoxic capacity, suggesting that UCB donors with a higher cytotoxic load are predisposed to be more potent.
An extensive matrix of receptor blockades on NK cells revealed the receptor-ligand interactions that are required to achieve efficient cytotoxicity. Investigations of the contribution of individual receptors and receptor combinations to cytotoxic activity, showed that multimodal activation of NK cells is potent and also target specific. This is in line with other findings where blocking two activating receptors on peripheral blood NK cells resulted in more profound inhibition of cytotoxicity than blocking a single activating receptor.25 Remarkably, NKp30 did not show any involvement in cytotoxicity alone but exhibited a more pronounced deleterious effect on cytotoxicity when blocked simultaneously with other NCRs, suggesting a certain interdependency. Indeed, it has been shown that engagement of one NCR could activate the signaling cascade of another.43 Of interest, although NKp44 can be inhibitory or activating, depending on the specific ligand interaction,44 in the melanoma setting it seems to have an activating function. Furthermore, TRAIL showed strong involvement in NK cytotoxicity, particularly early in the response, which was unexpected as it is usually described as a slower response.13 As mentioned above, TRAIL-engagement to its receptors, including the decoy receptors, can also induce degranulation and IFNγ release.36 Therefore, TRAIL-blockade can negatively affect cytotoxicity both directly because of inactivation of the death receptor apoptosis pathway and indirectly by limiting degranulation. However, the decoy receptors were not expressed by the assessed melanoma cell lines and caspase 8 activity was decreased after TRAIL-blocking, which implied that the apoptosis pathway was activated on TRAIL-engagement. Further investigations are required to fully understand the involvement of TRAIL in activation of UCB CD34+ progenitor cell-derived NK cells as well as NK cells from other sources. In contrast, the death receptor FASL was only poorly expressed on these NK cells and did not play a role in cytotoxicity.
Overall, the NK cells efficiently lysed 8/9 melanoma cell lines after a 20h co-culture at low E:T ratio. MEL-BRO showed low susceptibility to NK cytotoxicity in the short-term cytotoxicity assay, which was, interestingly, accompanied by high cytokine and cytolytic effector molecule response. This secretory response may thus serve as an early indicator of the high susceptibility of MEL-BRO to NK cytotoxicity which was observed at later timepoints. SK-MEL-28 showed very low susceptibility to UCB CD34+ progenitor cell-derived NK cells, which has also been reported for NK-92,45 but still induced significant release of granzyme A, B and granulysin in the supernatant. However, only a slight increase in E:T ratio was still sufficient to achieve more enhanced cytotoxicity. Assessment of NK cell efficacy across a spectrum of melanoma cell lines thus revealed that target susceptibility is not only dependent on efficient recognition by NK cells for potent target elimination, but also on sufficient activation of downstream apoptotic signaling pathways in the target for potent cytotoxicity to be exerted.
Although NK cell therapy is gaining more interest as a treatment of solid tumors, the immunosuppressive TME and migration to the tumor site might be challenges for NK cell infusion therapy. Cellular immunotherapy is more often supported by combination therapy to improve migration, to convert the immunosuppressive TME or to overcome checkpoint inhibition and increase direct cytotoxicity.31 Therefore, increasing the understanding of the mode of action of NK cells is crucial to efficiently screen for potentially advantageous combination therapies. Knowing that NK cytotoxicity is partially dependent on NKG2D for instance, could steer the development of combination therapies preventing shedding of its ligand MICA/B from tumor cells,46 or boosting ligand expression using an inhibitor of heat shock protein 90.47 Moreover, we observed that NKp46 seems to play an important role in NK activation. Therefore, combination with NK cell engagers that allow simultaneous targeting of NKp46 on NK cells and a target antigen on the tumor could be an interesting approach.48 Furthermore, blocking experiments revealed that TRAIL-activation played a major role in NK cytotoxicity, which opens up opportunities to explore combination of NK cell therapy with, for example, the proteasome inhibitor Bortezomib which is known to have a deleterious effect on glioblastoma cells but also to increase susceptibility to NK cell cytotoxicity through TRAIL/TRAILR2-mediated apoptosis.49 The clinical significance of this finding is further supported by the analysis of the TCGA-SKCM metastatic melanoma patient cohort which indicated a positive survival outcome associated with higher TRAIL-expression. In addition, the already very high levels of TRAIL-expression on UCB CD34+ progenitor cell-derived NK cells eliminate the need for genetic modification which is required by other NK cell products, such as the KHYG-1 cell line to overcome immune resistance of multiple myeloma.50
GTA002 NK cells derived from UCB progenitor cells is a novel off-the-shelf cell therapy product with the capacity to exert potent anti-tumor activity against melanoma, using multimodal activation of several NK cell receptors and with TRAIL being a major contributor to the enhancement of early cytotoxicity in both 2D and 3D settings. Together with survival data from a melanoma patient cohort which suggest a beneficial role for NK cells in melanoma, multimodal activation driven by TRAIL is essential for efficient cytotoxicity and could be harnessed to further enhance NK cell therapies. Overall, our results highlight that GTA002 and potentially other therapeutic NK cell products can be a promising efficient new adoptive cell therapy candidate for the treatment of metastatic melanoma.
Limitations of the study
The role of several important activating receptors in NK cell cytotoxicity has been extensively examined, but this assessment does not exhaustively include all activating receptors as there are practical limitations with regards to availability of blocking antibodies against other NK receptors. Moreover, the role of inhibiting receptors is not examined in the specific scope of this study. This is a potentially interesting focus point for a follow-up future study which would focus on abrogation of inhibitory signaling input for further enhancement of NK cytotoxic activity. Again, for such an approach there are certain limitations as to the availability of blocking antibodies against some of the inhibitory NK receptors, but for other inhibitory receptors antibodies are indeed available and could be used to interfere with inhibitory signaling. A similar extensive matrix of receptor blocking antibody combinations would be necessary to properly assess the contribution of each inhibitory receptor to diminishing NK cell cytotoxic activity in a cell line-specific manner.
Ethical statement
Glycostem is working with cells from commercial tissue banks and blood products. This work is in general regulated by the Dutch law. Glycostem has the permission to obtain donor tissue material, such as serum, from Sanquin blood bank in Amsterdam and Leiden, the Netherlands (VWS article 1). Glycostem obtain cord blood from NHS, London UK, and from Anthony Nolan, Nottingham, UK. The ethical procedures regarding the donors are regulated and under the responsibility of the blood banks. All samples are obtained anonymously. Animals are not used in this study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-CD45 KO (J.33) | Beckman Coulter | Cat# B36294; RRID: AB_2833027 |
| anti-CD56 APCA750 (N901) | Beckman Coulter | Cat# B46024 |
| anti-NKG2D PE (ON72) | Beckman Coulter | Cat# A08934; RRID: AB_2801262 |
| anti-NKp44 PE (Z231) | Beckman Coulter | Cat# A66903 |
| anti-NKG2A PE (Z199) | Beckman Coulter | Cat# IM3291U; RRID: AB_10643228 |
| anti- KIR2DL1 KIR2DS1 PE (EB6.B) | Beckman Coulter | Cat# A09778; RRID: AB_2801261 |
| anti-DNAM-1 PE (REA1040) | Miltenyi Biotec B.V. | Cat# 130-117-489; RRID: AB_2727959 |
| anti-NKp30 BV605 (p30-15) | BD Biosciences | Cat# IM3709 |
| anti-NKp46 BV786 (9E2) | BD Biosciences | Cat# 563329; RRID: AB_2738139 |
| anti-FASL BV605 (NOK-1) | BD Biosciences | Cat# 306418 |
| anti-TRAIL BV786 (RIK-2) | BD Biosciences | Cat# 743723; RRID: AB_2741699 |
| anti-MICA/B APC (6D4) | Biolegend | Cat# 320908; RRID: AB_493195 |
| anti-nectin-2 PECy7 (TX31) | Biolegend | Cat# 337414; RRID: AB_2565732 |
| anti-PVR PerCP/Cy5.5 (SKII.4) | Biolegend | Cat# 337612; RRID: AB_2565536 |
| anti-PCNA AF488 (PC10) | Biolegend | Cat# 307909; RRID: AB_2160546 |
| anti-FAS AF488 (DX2), | Biolegend | Cat# 305616; RRID: AB_528891 |
| anti-TRAILR1 APC (DJR1) | Biolegend | Cat# 307208; RRID: AB_2256112 |
| and anti-TRAILR2 PE (DJR2-4) | Biolegend | Cat# 307406; RRID: AB_2204667 |
| anti-ULBP- 2/5/6 BUV661 (165903) | BD Biosciences | Cat# 750011; RRID: AB_2874231 |
| anti-ULBP-1 AF488 (170818) | R&D systems | Cat# FAB1380G |
| anti-ULBP-3 PE (166510) | R&D systems | Cat# FAB1517P; RRID: AB_10719122 |
| anti-B7-H6 AF647 (1A5) | BD Biosciences | Cat# 566733; RRID: AB_2869835 |
| anti-TRAILR3 BV421 (B-D44) | BD Biosciences | Cat# 744764; RRID: AB_2742464 |
| anti-TRAILR4 FITC (TRAIL-R4-01) | Thermofisher | Cat# 12392183 |
| anti-CD107a PE (H4A3) | Biolegend | Cat# 328608; RRID: AB_1186040 |
| anti-IFNγ PE (B27) | Biolegend | Cat# 506507; RRID: AB_315440 |
| anti-TNF AF488 (MAb11) | Biolegend | Cat# 502915; RRID: AB_493121 |
| anti-CD56 BV421 (NCAM16.2) | BD Biosciences | Cat# 562751; RRID: AB_2732054 |
| anti-Perforin PerCP-Cy5.5 (B-D48) | Biolegend | Cat# 353314; RRID: AB_2571971 |
| anti-Granzyme B AF647 (GB11) | Biolegend | Cat# 515406; RRID: AB_2566333 |
| mouse IgG1 isotype ctrl PE (MOPC-21) | Biolegend | Cat# 400114; RRID: AB_2847829 |
| mouse IgG1 isotype ctrl AF488 (MOPC-21) | Biolegend | Cat# 400129; RRID: AB_2890263 |
| mouse IgG1 isotype ctrl PerCP-Cy5.5 (MOPC-21) | Biolegend | Cat# 400149; RRID: AB_893680 |
| mouse IgG1 isotype ctrl AF647 (MOPC-21) | Biolegend | Cat# 400130; RRID: AB_2800436 |
| anti-NKG2D (1D11) | Biolegend | Cat# 320814; RRID: AB_2561488 |
| anti-TRAIL (RIK-2), | Biolegend | Cat# 308214; RRID: AB_2814155 |
| anti-NKp30 (P30-15), | Biolegend | Cat# 325224; RRID: AB_2814183 |
| anti-NKp44 (P44-8), | Biolegend | Cat# 325122; RRID: AB_2819955 |
| anti-NKp46 (9E2) | Biolegend | Cat# 331948; RRID: AB_2814217 |
| anti-DNAM-1 (DX11) | BD Biosciences | Cat# 559786; RRID: AB_397327 |
| Anti-FASL (NOK-1) | Biolegend | Cat# 306416; RRID: AB_2810459 |
| Mouse IgG1, kappa (MOPC-21) | Biolegend | Cat# 400166; RRID: AB_11146992 |
| Bacterial and virus strains | ||
| IncuCyte NucLight Red (NLR) Lentivirus | Essen Biosciences | Cat# 4476 |
| Biological samples | ||
| Fresh umbilical cord blood | Anthony Nolan cord blood bank | NA |
| Fresh umbilical cord blood | NHS Blood and Transplant | Cat# M185 |
| Human serum | Sanquin | Cat#B0618 |
| Chemicals, peptides, and recombinant proteins | ||
| 7-aminoactinomycin D | Sigma | Cat# A9400-1MG |
| Ficoll Paque Plus | GE Healthcare | Cat# GE17-1440-03 |
| pacific blue succinimidyl ester (PBSE) | Thermofisher | Cat#P-10163 |
| PMA | Merck Life Sciences | Cat#P1585-1MG |
| Ionomycin | Merck Life Sciences | Cat#I9657 |
| Live/dead fixable aqua | Thermofisher | Cat#L34957 |
| rhTRAIL | Biolegend | Cat#752906 |
| Critical commercial assays | ||
| CD34+ microbead kit | Miltenyi Biotech B.V. | Cat#130-046-702 |
| cytofix/cytoperm kit | BD Biosciences | Cat# 554715 |
| CD8/NK LEGENDplex™ | Biolegend | Cat# 740267 |
| Incucyte® Caspase-3/7 Dye | Sartorius | Cat# 4440 |
| Cell Meter™ Multiplexing Caspase 3/7, 8 and 9 Activity Assay Kit | Bioconnect | Cat# 22820_100tests |
| Experimental models: Cell lines | ||
| A375 | ATCC | Cat# CRL-1619; RRID: CVCL_0132 |
| COLO829 | ATCC | Cat# CRL-1974; RRID: CVCL_1137 |
| G361 | ATCC | Cat# CRL-1424; RRID: CVCL_1220 |
| RPMI-7951 | ATCC | Cat# HTB-66; RRID: CVCL_1666 |
| MeWo | ATCC | Cat# HTB-65; RRID: CVCL_0445 |
| SK-MEL-28 | CLS | Cat#300337; RRID: CVCL_0526 |
| MEL-JUSO | DSMZ | Cat# ACC 74; RRID: CVCL_1403 |
| MEL-BRO | NA | RRID: CVCL_7036 |
| WM9 | NA | RRID: CVCL_6806 |
| Software and algorithms | ||
| Graphpad Prism software 9.0 | GraphPad Software | RRID: SCR_002798 |
| Kaluza v2.1 | Beckman Coulter | RRID: SCR_016182 |
| Cbioportal for Cancer Genomics | Cbioportal for Cancer Genomics | RRID:SCR_014555 |
| TCGA-SKCM | PanCancer Atlas | https://doi.org/10.25504/FAIRsharing.m8wewa |
| BioRender | BioRender | BioRender.com |
| Other | ||
| DMEM with UltraGlutamine | Lonza/Westburg | Cat# LO BE12-604F/U1 |
| RPMI 1640 with UltraGlutamine | Lonza/Westburg | Cat# LO BE12-702F/U1 |
| EMEM with L-Glutamine | Lonza/Westburg | Cat# LO BE12-611F |
| McCoy5a with L-Glutamine | Lonza/Westburg | Cat# LO BE12-688F |
| FBS | Gibco/Thermofisher | Cat# 10500-064 |
| TryplExpress | Thermofisher | Cat#10718463 |
| Glycostem Basal Growth Medium | Fertipro | Cat# GBGM-500 |
| TPO | Cellgenix | Cat# 1017-050 |
| IL-7 | Cellgenix | Cat# 1010-050 |
| Flt-3L | Cellgenix | Cat# 1015-050 |
| GM-CSF | Cellgenix | Cat# 1012-050 |
| Neupogen (G-CSF) | Amgen B.V. | NA |
| Il-6 | Cellgenix | Cat#1004-050 |
| SCF | Cellgenix | Cat#1018-050 |
| IL-15 | Cellgenix | Cat# 1013-050 |
| Proleukin (IL-2) | Novartis | NA |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Amanda van Vliet (amanda@glycostem.com)
Materials availability
This study did not generate new unique reagents.
Experimental model and study participant
The human melanoma cell lines A375 (ATCC-CRL-1619), COLO829 (ATCC-CRL-1974), G361 (ATCC-CRL-1424), RPMI-7951 (ATCC-HTB-66), MeWo (ATCC-HTB-65), SK-MEL-28 (CLS 300337), MEL-JUSO (DSMZ ACC 74), MEL-BRO (CVCL_7036) and WM9 (CVCL_6806) were purchased from the relevant suppliers and authentication was confirmed by STR profiling. Fresh UCB units were purchased from Anthony Nolan cord blood bank or NHS Blood and Transplant.
Method details
Melanoma cell line culture
A375 and SK-MEL-28 were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Lonza), COLO829, MEL-BRO, MEL-JUSO and WM9 in Roswell Park Memorial Institute 1640 Medium (RPMI; Lonza) and both MeWo and RPMI-7951 were cultured in Eagle’s Minimum Essential Medium (EMEM; Lonza). G361 was cultured in McCoy5a medium (Lonza). All media were supplemented with 10% fetal bovine serum (Gibco). All cell lines were cultured at 37°C and 5% CO2 and regularly tested for mycoplasma.
UCB CD34+ progenitor cell-derived NK cell generation and phenotypic characterization
Mononuclear cells (MNCs) were isolated by Ficoll Paque Plus (1.077 g/ml; GE Healthcare) density gradient centrifugation. Hematopoietic CD34+ stem cells were selected from the MNCs using the CD34+ microbead kit (Miltenyi Biotech B.V.) according to manufacturer’s protocol. CD34+ cells (purity 67.5% ± 16%) were seeded in 6-wells tissue culture treated plates (Corning Incorporated) for expansion culture in Glycostem Basal Growth Medium (GBGM; Fertipro, not commercially available) supplemented with human serum (Sanquin), TPO, IL-7, Flt-3L, GM-CSF, IL-6, SCF (all from Cellgenix) and Neupogen (G-CSF; Amgen BV) as previously described [28]. After 9 days, TPO was replaced by IL-15 (Cellgenix) and after 14 days of expansion, Flt-3L was replaced by Proleukin (IL-2; Novartis) and cultured NK cells were cryopreserved up to 4 years in vapor phase liquid nitrogen (LN) (<-135°C) until further use. To characterize receptor expression profile on NK cells, the following antibodies were used: anti-CD45 KO (J.33), anti-CD56 APCA750 (N901), anti-NKG2D PE (ON72), anti-NKp44 PE (Z231), anti-NKG2A PE (Z199), anti- KIR2DL1 KIR2DS1 PE (EB6.B) (all from Beckman Coulter), anti-DNAM-1 PE (REA1040) (Miltenyi Biotec B.V.), anti-NKp30 BV605 (p30-15), anti-NKp46 BV786 (9E2), anti-FASL BV605 (NOK-1), anti-TRAIL BV786 (RIK-2) (all from BD Biosciences) and 7-aminoactinomycin D (7-AAD; Sigma). All antibodies were used at recommended concentrations; 1∗105 cells were stained for 15 minutes at room temperature followed by washing with FACS buffer and data was acquired with the Cytoflex LX (Beckman Coulter). Flow cytometry data were analyzed using Kaluza v2.1 (Beckman Coulter)
Ligand expression measurement by flow cytometry
Melanoma cell lines were detached using TrypLE Express (Gibco) and stained in FACS buffer for 15 minutes at 4°C followed by washing. The following antibodies were used: anti-MICA/B APC (6D4), anti-nectin-2 PECy7 (TX31), anti-PVR PerCP/Cy5.5 (SKII.4), anti-PCNA AF488 (PC10), anti-FAS AF488 (DX2), anti-TRAILR1 APC (DJR1), and anti-TRAILR2 PE (DJR2-4) from Biolegend; anti-ULBP- 2/5/6 BUV661 (165903; BD Biosciences), anti-ULBP-1 AF488 (170818), and anti-ULBP-3 PE (166510) from R&D systems; anti-B7-H6 AF647 (1A5) and anti-TRAILR3 BV421 (B-D44) from BD Biosciences; and anti-TRAILR4 FITC (TRAIL-R4-01; Thermofisher). All antibodies were used at the recommended dilution.
In vitro 5h cytotoxicity/degranulation assay and 20h cytotoxicity assay
Melanoma cell lines were washed with PBS before labelling with 5 μM pacific blue succinimidyl ester (PBSE; Thermofisher). NK cells were thawed 7 days before the cytotoxicity assay (>92.2% viability after thawing) and cultured in differentiation medium until use. Melanoma cells were co-cultured with NK cells at an effector to target (E:T) ratio of 1:1 in a 96-well plate. As controls, melanoma cells and NK cells were plated alone. To check for maximum degranulation 0.5 μg/ml PMA and 0.5 μg/ml ionomycin (both Merck Life Sciences) were added to NK cells. At the start of the co-culture 1:400 anti-CD107a PE (H4A3, Biolegend) was added. For the 20h set up, melanoma and NK cells were seeded without addition of anti-CD107a. After 5h or 20h incubation at 37°C, cells were detached using TrypLE Express and transferred to a V-bottom plate and stained with 7-AAD (Sigma) and anti-CD56 APC-A750 (N901, Beckman Coulter). Acquisition of the samples from the cytotoxicity assay was done using the Cytoflex LX (Beckman Coulter) and samples were assessed in technical triplicates. Data analysis was done using Kaluza software (Beckman Coulter). Cytotoxicity was determined as follows: Δ %7AAD cytotoxicity = (%7AAD+/PBSE+ melanoma cells in co-culture) – (%7AAD+/PBSE+ melanoma cells in control). Degranulation was calculated as Δ % degranulation = (% CD107a+/CD56+/7AAD− cells in co-culture) – (% CD107a+/CD56+/7AAD− cells in NK cells alone).
Intracellular staining
The same co-culture set up as the 5h cytotoxicity assay was used to measure intracellular cytokines/effector molecules. To measure accumulating levels of IFNγ and TNF, BD GolgiStop™ was added after 1 hour of co-culture. This was not used for perforin and Granzyme B, so that the decrease of intracellular levels upon co-culture could be observed. After 5h co-culture, cells were fixated and permeabilized using the cytofix/cytoperm kit (BD Biosciences) according to the manufacturer’s instructions. The following antibodies were used at recommended concentration: anti-IFNγ PE (B27, Biolegend), anti-TNF AF488 (MAb11, Biolegend), anti-CD56 BV421 (NCAM16.2, BD), anti-Perforin PerCP-Cy5.5 (B-D48, Biolegend), anti-Granzyme B AF647 (GB11, Biolegend), mouse IgG1 isotype ctrls in PE, AF488, PerCP-Cy5.5 and AF647 (MOPC-21, Biolegend), and live/dead fixable aqua (Thermofisher).
Secretome analysis
Supernatants were collected from the 5-hour cytotoxicity co-culture. The CD8/NK LEGENDplex™ (Biolegend) kit was used according to manufacturer’s protocol to determine the following analytes: TNF, IFNγ, Granzyme A, Granzyme B, Perforin, Granulysin. Briefly, supernatant was incubated with beads conjugated to specific antibodies for the different analytes. After washing, biotinylated detection antibodies were added, followed by addition of streptavidin-phycoerythrin resulting in fluorescent signal proportionate to the amount of bound analytes. Stained beads were measured using a Cytoflex LX (Beckman Coulter) and data was analyzed using the LEGENDplex™ data analysis software.
Impedance-based cytotoxicity assay
To explore receptor-ligand interactions by blocking antibodies, the MaestroZ (Axion Biosystems) impedance-based system was used. To correct for background resistance, wells were first measured in 50 μl medium as a baseline. Target cells were seeded at appropriate cell concentrations in a total volume of 100 μl and were allowed to adhere for 5 hours at 37°C. NK cells were pre-incubated with 10 μg/ml blocking antibody for 15 minutes at 37°C. The following blocking monoclonal antibodies (mAbs) were used: NKG2D (1D11), TRAIL (RIK-2), FASL (NOK-1), NKp30 (P30-15), NKp44 (P44-8), NKp46 (9E2) all from Biolegend and DNAM-1 (DX11) from BD Biosciences. Mouse IgG1, kappa (MOPC-21) from Biolegend was used as isotype control antibody at a maximum concentration of 40 μg/ml (corresponding to the concentration of combining the blocking Abs). After incubation, NK cells were added at a 1:1 E:T ratio. The co-culture was followed for 24 hours, and data was analyzed using the Axis Z software (Axion Biosystems).
Spheroid generation and cytotoxicity assay
A375 cells were transduced with IncuCyte NucLight Red (NLR) Lentivirus (Essen Bioscience) to stably express the nucleus-restricted red fluorescent protein mKate2. Transduced cells were banked after selection using 1 μg/ml puromycin (Sigma-Aldrich). To grow spheroids, 1∗103 NLR-A375 cells/well were seeded in an ultra-low attachment U-bottom plate (Corning) and centrifuged for 2 min, 200g. The cells were allowed to form spheroids for 2 days at 37°C and 5% CO2. NK cells were preincubated with 25 μg/ml blocking mAbs at a cell concentration of 1∗106 cells/ml for 15 minutes followed by seeding 100 μl per well to the spheroids, resulting in a 25:1 E:T ratio at time of seeding. The red fluorescent signal was imaged every 1h for 24 h at 4x magnification by the IncuCyte S3 live-cell analysis system (Essen Bioscience).
Caspase activity measurement
To measure sensitivity of TRAIL-mediated cytotoxicity on melanoma, cell lines A375, MEL-BRO and SK-MEL-28 were seeded at 1∗104 cells/well and allowed to adhere for 6h. Recombinant human TRAIL (rhTRAIL; Biolegend) was added at various concentration ranging from 50 ng/ml to 5 μg/ml. The Incucyte® Caspase-3/7 Dye (Sartorius) was added at 0.5 μM. The fluorescent signal was imaged every 1h for 20 h at 20x magnification by the IncuCyte S3 live-cell analysis system (Essen Bioscience). To measure caspase 3/7 and caspase 8 simultaneously, the Cell Meter™ Multiplexing Caspase 3/7, 8 and 9 Activity Assay Kit (Bioconnect) was used according to manufacturer’s instruction. A375 was seeded at 1∗104 cells/well and allowed to adhere overnight. NK cells were preincubated with blocking antibodies and seeded at 2∗104 cells/well to obtain a 1:1 E:T ratio. Fluorescent signal of the caspase activity was measured on a BioTek Synergy H1 plate reader.
Bioinformatic survival analysis
The Cbioportal for Cancer Genomics (https://www.cbioportal.org/)51,52 was used to perform survival analysis for the data set TCGA-SKCM (Skin Cutaneous melanoma) Cancer Genome Atlas, https://doi.org/10.25504/FAIRsharing.m8wewa. The data set contains a total of 448 samples of which 367 samples were taken from metastasis. A total of 352 patients were included in the analysis, excluding samples taken from the same patient and regardless of treatment. Samples from primary lesions were excluded since they are most often excised and as the focus of adoptive NK cell therapy is targeting of the metastatic disease. Using the median mRNA expression of each gene of interest as a cut-off value, the samples were divided into high and low expression groups and Kaplan-Meier plots were generated.
Quantification and statistical analysis
Statistical analysis was performed using GraphPad Prism software 9.0. Normality distribution was checked by using the Shapiro-Wilk test and the analyses were performed accordingly. One-way ANOVA with Tukey multiple comparison analysis was performed for normally distributed data and Friedman test with Dunn multiple comparison analysis was performed for skewed data. Paired t-test was used where indicated. Pearson r test was performed for correlation analysis. Log-rank test was performed for survival data. P-values lower than 0.05 were considered statistically significant. Statistical significance is shown as p value <0.05 ∗, <0.01 ∗∗, 0.001 ∗∗∗, 0.0001 ∗∗∗∗.
Acknowledgments
This research was funded by Eurostars, E∗11275 IMAGINe
Author contributions
A.V. developed methodologies, performed the majority of the experimental work, analyzed results, prepared figures and drafted and wrote the manuscript. E.P. contributed by planning and performing blocking experiments. D.V. contributed with the planning and execution of potency assays and functional experiments. D.S. contributed with planning and execution of generating NK cells. M.R. contributed with planning and execution of generating NK cells and reviewed the manuscript. S.G. contributed access to melanoma cell lines, secured funding and reviewed the manuscript. T.G. contributed access to melanoma cell lines, secured funding and reviewed the manuscript. A.D. contributed with experiment conceptualization and reviewed the manuscript. J.S. conceived the project idea, secured funding, reviewed the manuscript. A.M.G. performed potency experiments and functional assays, developed methodologies, conceived experimental plan, wrote and reviewed the manuscript.
Declaration of interests
Authors A.A.V., D.V., D.S., A.D.D., M.R., A.M.G., and J.S. are employees of Glycostem Therapeutics.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: June 9, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107078.
Supplemental information
Data and code availability
-
•
Raw data files of the results reported in this paper can be shared by the lead contact upon specific request. Survival analysis was performed on publicly available data sets which are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Passarelli A., Mannavola F., Stucci L.S., Tucci M., Silvestris F. Immune system and melanoma biology: a balance between immunosurveillance and immune escape. Oncotarget. 2017;8:106132–106142. doi: 10.18632/oncotarget.22190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray S., Lundqvist A. Targeting the tumor microenvironment to improve natural killer cell-based immunotherapies: on being in the right place at the right time, with resilience. Hum. Vaccines Immunother. 2016;12:607–611. doi: 10.1080/21645515.2015.1096458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmadzadeh M., Johnson L.A., Heemskerk B., Wunderlich J.R., Dudley M.E., White D.E., Rosenberg S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fourcade J., Sun Z., Benallaoua M., Guillaume P., Luescher I.F., Sander C., Kirkwood J.M., Kuchroo V., Zarour H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010;207:2175–2186. doi: 10.1084/jem.20100637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg S.A., Yang J.C., Sherry R.M., Kammula U.S., Hughes M.S., Phan G.Q., Citrin D.E., Restifo N.P., Robbins P.F., Wunderlich J.R., et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011;17:4550–4557. doi: 10.1158/1078-0432.ccr-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lipson E.J., Drake C.G. Ipilimumab: an anti-CTLA-4 antibody for metastatic melanoma. Clin. Cancer Res. 2011;17:6958–6962. doi: 10.1158/1078-0432.Ccr-11-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hazarika M., Chuk M.K., Theoret M.R., Mushti S., He K., Weis S.L., Putman A.H., Helms W.S., Cao X., Li H., et al. U.S. FDA approval summary: nivolumab for treatment of unresectable or metastatic melanoma following progression on ipilimumab. Clin. Cancer Res. 2017;23:3484–3488. doi: 10.1158/1078-0432.ccr-16-0712. [DOI] [PubMed] [Google Scholar]
- 8.Dudley M.E., Wunderlich J.R., Shelton T.E., Even J., Rosenberg S.A. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J. Immunother. 2003;26:332–342. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kageshita T., Ishihara T., Campoli M., Ferrone S. Selective monomorphic and polymorphic HLA class I antigenic determinant loss in surgically removed melanoma lesions. Tissue Antigens. 2005;65:419–428. doi: 10.1111/j.1399-0039.2005.00381.x. [DOI] [PubMed] [Google Scholar]
- 10.Soltantoyeh T., Akbari B., Karimi A., Mahmoodi Chalbatani G., Ghahri-Saremi N., Hadjati J., Hamblin M.R., Mirzaei H.R. Chimeric antigen receptor (CAR) T cell therapy for metastatic melanoma: challenges and road ahead. Cells. 2021;10 doi: 10.3390/cells10061450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vivier E., Tomasello E., Baratin M., Walzer T., Ugolini S. Functions of natural killer cells. Nat. Immunol. 2008;9:503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 12.Wang R., Jaw J.J., Stutzman N.C., Zou Z., Sun P.D. Natural killer cell-produced IFN-γ and TNF-α induce target cell cytolysis through up-regulation of ICAM-1. J. Leukoc. Biol. 2012;91:299–309. doi: 10.1189/jlb.0611308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prager I., Watzl C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019;105:1319–1329. doi: 10.1002/jlb.Mr0718-269r. [DOI] [PubMed] [Google Scholar]
- 14.Konjević G., Mirjacić Martinović K., Vuletić A., Jović V., Jurisić V., Babović N., Spuzić I. Low expression of CD161 and NKG2D activating NK receptor is associated with impaired NK cell cytotoxicity in metastatic melanoma patients. Clin. Exp. Metastasis. 2007;24:1–11. doi: 10.1007/s10585-006-9043-9. [DOI] [PubMed] [Google Scholar]
- 15.Mirjačić Martinović K., Vuletić A., Mališić E., Srdić-Rajić T., Tišma Miletić N., Babović N., Jurišić V. Increased circulating TGF-β1 is associated with impairment in NK cell effector functions in metastatic melanoma patients. Growth Factors. 2022;40:231–239. doi: 10.1080/08977194.2022.2124915. [DOI] [PubMed] [Google Scholar]
- 16.Dolstra H., Roeven M.W.H., Spanholtz J., Hangalapura B.N., Tordoir M., Maas F., Leenders M., Bohme F., Kok N., Trilsbeek C., et al. Successful transfer of umbilical cord blood CD34(+) hematopoietic stem and progenitor-derived NK cells in older acute myeloid leukemia patients. Clin. Cancer Res. 2017;23:4107–4118. doi: 10.1158/1078-0432.Ccr-16-2981. [DOI] [PubMed] [Google Scholar]
- 17.Yang Y., Lim O., Kim T.M., Ahn Y.O., Choi H., Chung H., Min B., Her J.H., Cho S.Y., Keam B., et al. Phase I study of random healthy donor-derived allogeneic natural killer cell therapy in patients with malignant lymphoma or advanced solid tumors. Cancer Immunol. Res. 2016;4:215–224. doi: 10.1158/2326-6066.Cir-15-0118. [DOI] [PubMed] [Google Scholar]
- 18.Romee R., Rosario M., Berrien-Elliott M.M., Wagner J.A., Jewell B.A., Schappe T., Leong J.W., Abdel-Latif S., Schneider S.E., Willey S., et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016;8:357ra123. doi: 10.1126/scitranslmed.aaf2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tonn T., Schwabe D., Klingemann H.G., Becker S., Esser R., Koehl U., Suttorp M., Seifried E., Ottmann O.G., Bug G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy. 2013;15:1563–1570. doi: 10.1016/j.jcyt.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 20.Oh S., Lee J.H., Kwack K., Choi S.W. Natural killer cell therapy: a new treatment paradigm for solid tumors. Cancers. 2019;11 doi: 10.3390/cancers11101534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Casado J.G., Pawelec G., Morgado S., Sanchez-Correa B., Delgado E., Gayoso I., Duran E., Solana R., Tarazona R. Expression of adhesion molecules and ligands for activating and costimulatory receptors involved in cell-mediated cytotoxicity in a large panel of human melanoma cell lines. Cancer immunology. 2009;58:1517–1526. doi: 10.1007/s00262-009-0682-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cagnano E., Hershkovitz O., Zilka A., Bar-Ilan A., Golder A., Sion-Vardy N., Bogdanov-Berezovsky A., Mandelboim O., Benharroch D., Porgador A. Expression of ligands to NKp46 in benign and malignant melanocytes. J. Invest. Dermatol. 2008;128:972–979. doi: 10.1038/sj.jid.5701111. [DOI] [PubMed] [Google Scholar]
- 23.Byrd A., Hoffmann S.C., Jarahian M., Momburg F., Watzl C. Expression analysis of the ligands for the Natural Killer cell receptors NKp30 and NKp44. PLoS One. 2007;2:e1339. doi: 10.1371/journal.pone.0001339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griffith T.S., Chin W.A., Jackson G.C., Lynch D.H., Kubin M.Z. Intracellular regulation of TRAIL-induced apoptosis in human melanoma cells. J. Immunol. 1998;161:2833–2840. [PubMed] [Google Scholar]
- 25.Pietra G., Manzini C., Vitale M., Balsamo M., Ognio E., Boitano M., Queirolo P., Moretta L., Mingari M.C. Natural killer cells kill human melanoma cells with characteristics of cancer stem cells. Int. Immunol. 2009;21:793–801. doi: 10.1093/intimm/dxp047. [DOI] [PubMed] [Google Scholar]
- 26.Carrega P., Pezzino G., Queirolo P., Bonaccorsi I., Falco M., Vita G., Pende D., Misefari A., Moretta A., Mingari M.C., et al. Susceptibility of human melanoma cells to autologous natural killer (NK) cell killing: HLA-related effector mechanisms and role of unlicensed NK cells. PLoS One. 2009;4 doi: 10.1371/journal.pone.0008132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lakshmikanth T., Burke S., Ali T.H., Kimpfler S., Ursini F., Ruggeri L., Capanni M., Umansky V., Paschen A., Sucker A., et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J. Clin. Invest. 2009;119:1251–1263. doi: 10.1172/jci36022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller J.S., Soignier Y., Panoskaltsis-Mortari A., McNearney S.A., Yun G.H., Fautsch S.K., McKenna D., Le C., Defor T.E., Burns L.J., et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051–3057. doi: 10.1182/blood-2004-07-2974. [DOI] [PubMed] [Google Scholar]
- 29.Parkhurst M.R., Riley J.P., Dudley M.E., Rosenberg S.A. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin. Cancer Res. 2011;17:6287–6297. doi: 10.1158/1078-0432.CCR-11-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arai S., Meagher R., Swearingen M., Myint H., Rich E., Martinson J., Klingemann H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy. 2008;10:625–632. doi: 10.1080/14653240802301872. [DOI] [PubMed] [Google Scholar]
- 31.van Vliet A.A., Georgoudaki A.M., Raimo M., de Gruijl T.D., Spanholtz J. Adoptive NK cell therapy: a promising treatment prospect for metastatic melanoma. Cancers. 2021;13 doi: 10.3390/cancers13184722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spanholtz J., Tordoir M., Eissens D., Preijers F., van der Meer A., Joosten I., Schaap N., de Witte T.M., Dolstra H. High log-scale expansion of functional human natural killer cells from umbilical cord blood CD34-positive cells for adoptive cancer immunotherapy. PLoS One. 2010;5 doi: 10.1371/journal.pone.0009221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tóth G., Szöllősi J., Vereb G. Quantitating ADCC against adherent cells: impedance-based detection is superior to release, membrane permeability, or caspase activation assays in resolving antibody dose response. Cytometry A. 2017;91:1021–1029. doi: 10.1002/cyto.a.23247. [DOI] [PubMed] [Google Scholar]
- 34.Li J., Figueira S.K., Vrazo A.C.A., Binkowski B.F., Butler B.L., Tabata Y., Filipovich A., Jordan M.B., Risma K.A. Real-time detection of CTL function reveals distinct patterns of caspase activation mediated by Fas versus granzyme B. J. Immunol. 2014;193:519–528. doi: 10.4049/jimmunol.1301668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Azijli K., Weyhenmeyer B., Peters G.J., de Jong S., Kruyt F.A.E. Non-canonical kinase signaling by the death ligand TRAIL in cancer cells: discord in the death receptor family. Cell Death Differ. 2013;20:858–868. doi: 10.1038/cdd.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Höfle J., Trenkner T., Kleist N., Schwane V., Vollmers S., Barcelona B., Niehrs A., Fittje P., Huynh-Tran V.H., Sauter J., et al. Engagement of TRAIL triggers degranulation and IFNγ production in human natural killer cells. EMBO Rep. 2022:e54133. doi: 10.15252/embr.202154133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cancer Genome Atlas Network Genomic classification of cutaneous melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu X., Wang X., Zhao Y., Li K., Yu B., Zhang J. Granzyme family acts as a predict biomarker in cutaneous melanoma and indicates more benefit from anti-PD-1 immunotherapy. Int. J. Med. Sci. 2021;18:1657–1669. doi: 10.7150/ijms.54747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cursons J., Souza-Fonseca-Guimaraes F., Foroutan M., Anderson A., Hollande F., Hediyeh-Zadeh S., Behren A., Huntington N.D., Davis M.J. A gene signature predicting natural killer cell infiltration and improved survival in melanoma patients. Cancer Immunol. Res. 2019;7:1162–1174. doi: 10.1158/2326-6066.Cir-18-0500. [DOI] [PubMed] [Google Scholar]
- 40.Frazao A., Rethacker L., Jeudy G., Colombo M., Pasmant E., Avril M.F., Toubert A., Moins-Teisserenc H., Roelens M., Dalac S., et al. BRAF inhibitor resistance of melanoma cells triggers increased susceptibility to natural killer cell-mediated lysis. J. Immunother. Cancer. 2020;8 doi: 10.1136/jitc-2019-000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lamers-Kok N., Panella D., Georgoudaki A.M., Liu H., Özkazanc D., Kučerová L., Duru A.D., Spanholtz J., Raimo M. Natural killer cells in clinical development as non-engineered, engineered, and combination therapies. J. Hematol. Oncol. 2022;15:164. doi: 10.1186/s13045-022-01382-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pende D., Rivera P., Marcenaro S., Chang C.C., Biassoni R., Conte R., Kubin M., Cosman D., Ferrone S., Moretta L., Moretta A. Major histocompatibility complex class I-related chain A and UL16-binding protein expression on tumor cell lines of different histotypes: analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res. 2002;62:6178–6186. [PubMed] [Google Scholar]
- 43.Augugliaro R., Parolini S., Castriconi R., Marcenaro E., Cantoni C., Nanni M., Moretta L., Moretta A., Bottino C. Selective cross-talk among natural cytotoxicity receptors in human natural killer cells. Eur. J. Immunol. 2003;33:1235–1241. doi: 10.1002/eji.200323896. [DOI] [PubMed] [Google Scholar]
- 44.Rosental B., Brusilovsky M., Hadad U., Oz D., Appel M.Y., Afergan F., Yossef R., Rosenberg L.A., Aharoni A., Cerwenka A., et al. Proliferating cell nuclear antigen is a novel inhibitory ligand for the natural cytotoxicity receptor NKp44. J. Immunol. 2011;187:5693–5702. doi: 10.4049/jimmunol.1102267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee Y.S., Heo W., Choi H.J., Cho H.R., Nam J.H., Ki Y.G., Lee H.R., Son W.C., Park Y.S., Kang C.D., Bae J. An inhibitor of programmed death ligand 1 enhances natural killer cell-mediated immunity against malignant melanoma cells. PLoS One. 2021;16 doi: 10.1371/journal.pone.0248870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferrari de Andrade L., Tay R.E., Pan D., Luoma A.M., Ito Y., Badrinath S., Tsoucas D., Franz B., May K.F., Jr., Harvey C.J., et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science (New York, N.Y.) 2018;359:1537–1542. doi: 10.1126/science.aao0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saha T., van Vliet A.A., Cui C., Macias J.J., Kulkarni A., Pham L.N., Lawler S., Spanholtz J., Georgoudaki A.M., Duru A.D., Goldman A. Boosting natural killer cell therapies in glioblastoma multiforme using supramolecular cationic inhibitors of heat shock protein 90. Front. Mol. Biosci. 2021;8 doi: 10.3389/fmolb.2021.754443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gauthier L., Morel A., Anceriz N., Rossi B., Blanchard-Alvarez A., Grondin G., Trichard S., Cesari C., Sapet M., Bosco F., et al. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell. 2019;177:1701–1713.e16. doi: 10.1016/j.cell.2019.04.041. [DOI] [PubMed] [Google Scholar]
- 49.Gras Navarro A., Espedal H., Joseph J.V., Trachsel-Moncho L., Bahador M., Gjertsen B.T., Kristoffersen E.K., Simonsen A., Miletic H., Enger P.Ø., et al. Pretreatment of glioblastoma with Bortezomib potentiates natural killer cell cytotoxicity through TRAIL/DR5 mediated apoptosis and prolongs animal survival. Cancers. 2019;11 doi: 10.3390/cancers11070996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holthof L.C., Stikvoort A., van der Horst H.J., Gelderloos A.T., Poels R., Li F., Groen R.W.J., Zweegman S., van de Donk N.W.C.J., O'Dwyer M., Mutis T. Bone marrow mesenchymal stromal cell-mediated resistance in multiple myeloma against NK cells can be overcome by introduction of CD38-CAR or TRAIL-variant. Hemasphere. 2021;5 doi: 10.1097/hs9.0000000000000561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E., et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.Cd-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E., et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Raw data files of the results reported in this paper can be shared by the lead contact upon specific request. Survival analysis was performed on publicly available data sets which are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.