
Figure 7: EZH2i hinders ACC growth, proliferation, and differentiation in vivo.
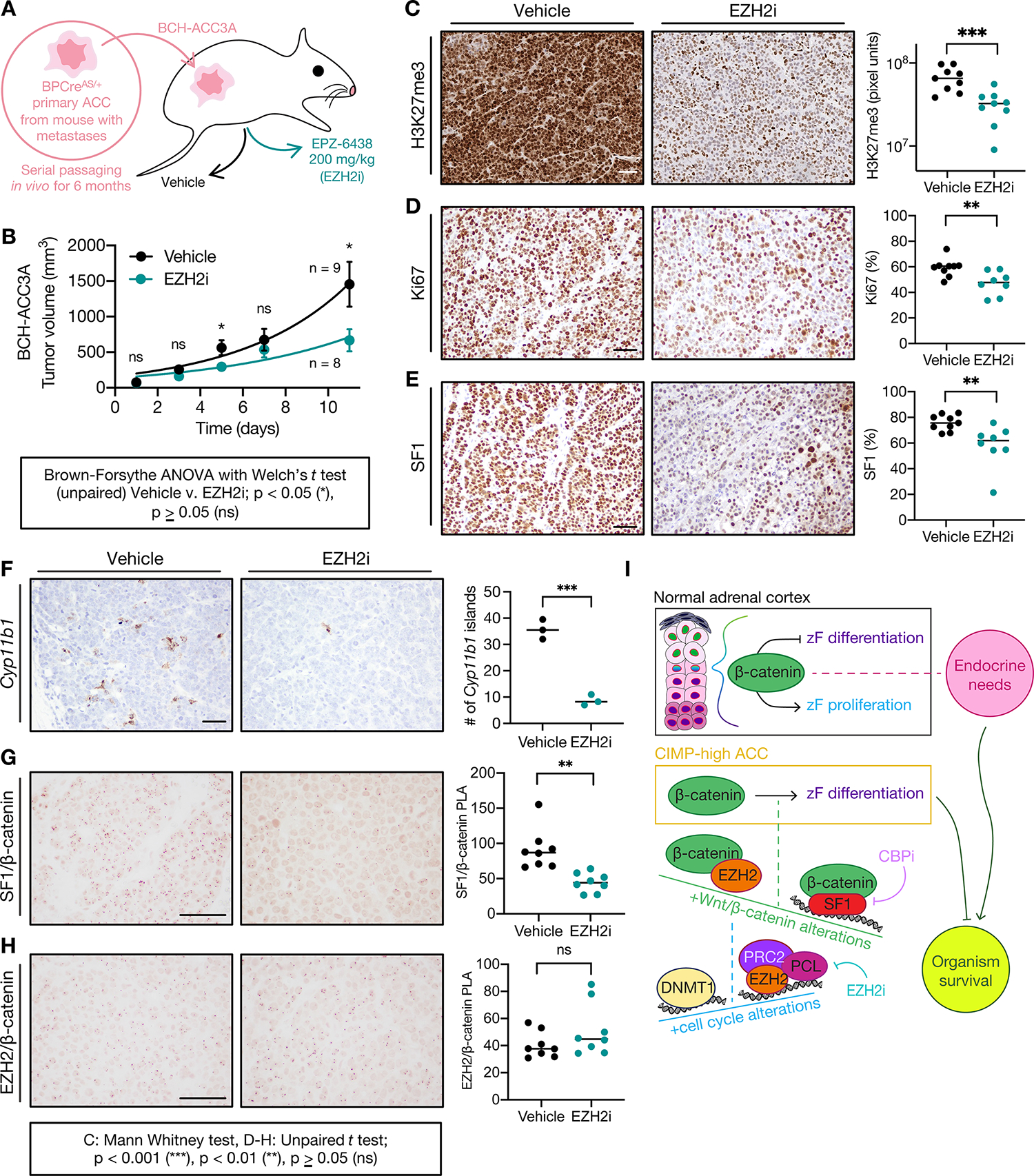
A. Derivation of BCH-ACC3A cell line and subcutaneous NSG mouse allograft model, randomized to vehicle or EZH2i treatment at tumor volume 100 mm3.
B. Tumor growth across treatment groups; data shown as mean with SEM.
C. H3K27me3 IHC across treatment groups. Left, bar=50 μm; right, each sample represented by 3 points, H3K7me3 quantified by MATLAB, line at median.
D-H. Ki67 IHC, SF1 IHC, Cyp11b1 RNA in situ hybridization, SF1/β-catenin PLA or EZH2/β-catenin PLA across treatment groups. Left, bar=100 μm; right, each sample is represented by a point and % nuclear signal, Cyp11b1 islands, or normalized PLA signal quantified by Fiji, line at median.
I. Model: In the upper zF of the normal adrenal cortex, β-catenin restrains zF differentiation or permits zF proliferation depending on endocrine demands (systemic need for glucocorticoids and flux through ACTH). This homeostasis is required for organism survival. In CIMP-high ACC, β-catenin drives zF differentiation through SF1/β-catenin hijacking of genome-wide SE. SF1/β-catenin’s actions on chromatin are limited by EZH2/β-catenin, an off-chromatin complex that completes for β-catenin binding. EZH2/β-catenin abundance is limited by on chromatin EZH2 and PRC2 catalytic activity. PRC2 remains catalytically active in CIMP-high ACC despite displacement by CpGi hypermethylation (written by DNA methyltransferases like DNMT1). Recurrent Wnt pathway and cell cycle alterations in CIMP-high ACC promote the formation of β-catenin-containing and EZH2-containing complexes. Ultimately, β-catenin-dependent zF differentiation is required for sustained ACC proliferation at the cost of organism survival. This program is erased by ACC dedifferentiating agents like EZH2i or CBPi, representing a promising therapeutic avenue.