

Abstract
LCK is a novel therapeutic target in ~40% of T-cell acute lymphoblastic leukemia (T-ALL), and dasatinib and ponatinib can act as LCK inhibitors with therapeutic effects. We herein report a comprehensive preclinical pharmacokinetic and pharmacodynamic evaluation of dasatinib and ponatinib in LCK-activated T-ALL. In 51 human T-ALL cases, these two drugs showed similar patterns of cytotoxic activity, with ponatinib being slightly more potent. Given orally in mice, ponatinib was associated with slower clearance with a longer Tmax and higher AUC0–24hrs, although maximum pLCK inhibition was comparable between the two drugs. After establishing the exposure-to-response models, we simulated the steady-state pLCK inhibitory effects of each drug at currently-approved dosages in humans: dasatinib at 140 mg and ponatinib at 45 mg once daily are both sufficient to achieve >50% pLCK inhibition for 13.0 and 13.9 hours per day, respectively, comparable to pharmacodynamic profiles of these agents in BCR::ABL1 leukemias. Moreover, we developed a dasatinib-resistant T-ALL cell line model with LCK T316I mutation, in which ponatinib retained partial activity against LCK. In conclusion, we described the pharmacokinetic and pharmacodynamic profiles of dasatinib and ponatinib as LCK inhibitors in T-ALL, providing critical data for the development of human trials of these agents.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common cancer in children and can arise in both B- and T-cell lineages. With the advent of risk-directed combination chemotherapy, 5-year overall survival of B-ALL has improved significantly in the past decades(1–6). However, cure rates of T-ALL persistently lag by 5–10%, and the prognosis is particularly dismal with relapsed disease(7, 8). This can be attributed to a number of factors: T-ALL blasts are inherently more resistant to certain cytotoxic agents that are highly effective in B-ALL(9); T-ALL is also more common in older children who are less tolerant to conventional chemotherapy(10). Genomic profiling of T-ALL provides important insights into the biology of this disease but this has not directly led to opportunities for novel targeted therapies, unlike those seen in B-ALL(11–15). Therefore, the treatment options for T-ALL remain limited and novel strategies are needed to improve the cure of this type of leukemia.
We and others recently discovered that ~40% of T-ALLs are exquisitely sensitive to dasatinib ex vivo(16–18). Using network-based systems pharmacology, we identified that preTCR-LCK signaling caused by differentiation arrest at the DN3/DN4 stages is the driver of this drug response phenotype in T-ALL(16). Dasatinib monotherapy exhibited significant anti-leukemic efficacy in vivo in patient-derived xenograft (PDX) models of T-ALL(16), and dasatinib-based PROTACs showed further improvement in leukemia-free survival in these models(19). Dasatinib can be safely combined with ALL chemotherapy as shown in patients with BCR::ABL1 B-ALL(20–23). Therefore, extending this tyrosine kinase inhibitor to T-ALL treatment regimens is feasible and highly attractive. In fact, there is emerging evidence that LCK-activated T-ALL cases fare poorly with conventional chemotherapy(24, 25), providing a clinical rationale for implementing LCK inhibitor therapy in this population. However, it remains unknown what exact dose and dosing schedule of dasatinib are needed to inhibit LCK in patients with T-ALL, e.g., whether it is sufficient to simply adopt the regimen currently used for BCR::ABL1 ALL.
Like dasatinib, ponatinib is also a multi-target tyrosine kinase inhibitor originally developed as an ABL inhibitor, especially for patients with BCR::ABL1 leukemia who acquired resistance to imatinib(13, 26). Both dasatinib and ponatinib are more potent against ABL1 than imatinib, with overlapping but not identical other targets spectrum(27). For example, dasatinib can efficiently inhibit LCK, LYN, CSK as well as EGFR and TGFBR1(28). By contrast, ponatinib targets LCK, FLT3, LYN, and CSK, but does not target EGFR and TGFBR1(29). There are appreciable differences in pharmacokinetics and toxicity profile between these two drugs in BCR::ABL1 leukemia patients(20, 30–34). Therefore, although dasatinib and ponatinib are both potent LCK inhibitors, pharmacology evaluation of each agent is needed to determine their clinical potential in patients with T-ALL.
To this end, we sought to perform preclinical pharmacokinetic (PK) and pharmacodynamic (PD) characterization of dasatinib and ponatinib, and to establish their exposure-to-response relationships in T-ALL PDX models. Using these preclinical data, we simulated the steady-state PD effect of each drug on LCK with the equivalent of the clinically approved dosages in humans in a 7-day course of treatment, to infer effective dose and dosing schedule in future trials in T-ALL. Moreover, we explore the possible utility of ponatinib as an LCK inhibitor in dasatinib-resistant T-ALL.
Material and methods
Patients and samples
T-ALL cases evaluated for dasatinib and/or ponatinib sensitivity consisted of 22 primary patient samples and 32 patient-derived xenograft (PDX) samples. The pediatric cases were obtained from St. Jude Children’s Research Hospital (St. Jude), the Children’s Oncology Group, and Children’s Hospital of Philadelphia (CHOP); and the adult cases were from MD Anderson Cancer Center, University of Chicago, and the Alliance for Clinical Trials in Oncology. For primary patient samples, T-ALL blasts were collected from bone marrow or peripheral blood after Ficoll gradient centrifugation and were subjected to further enrichment by magnetic-activated cell sorting if blast was <85%. Whenever necessary, human T-ALL blasts were expanded using the NOD.CgPrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice. This study was approved by the respective institutional review boards at St. Jude, CHOP, Children’s Oncology Group, MD Anderson Cancer Center, University of Chicago, and the Alliance for Clinical Trials in Oncology. Informed consent was obtained from parents, guardians and/or patients, as appropriate.
To establish xenografts, human T-ALL cells were injected into female NSG mice aged 8 to 12 weeks through the tail vein (1–2 million cells per mouse). All NSG mice were housed in sterilized conditions at 20–23 °C and 40–60% humidity, a 12 hrs light–12 hrs dark cycle was applied. Health statuses of all injected mice were monitored daily. All animal studies were approved by the Institutional Animal Care and Use Committee of St. Jude. Starting from two weeks after injection, peripheral blood was collected every other week for monitoring the level of human leukemia by flow cytometry: cells were stained with mTER119 (BD Pharmingen, #560512), mCD45 (BD Pharmingen, #557659), hCD45 (BD Pharmingen, #555482) and hCD7 (BD Pharmingen, #561604), all diluted 1:100; hCD45 and hCD7 double-positive percentage was determined using a BD FACS LSR II machine (BD FACS Diva Software v.8.0.1). Mice were euthanized when leukemia cells reached 80% in peripheral blood, or they became moribund. Human leukemia cells were collected from spleen and bone marrow and enriched using immunomagnetic isolation kit (Stemcell Technologies, #19849) for further in vivo studies.
In vivo evaluation of ponatinib efficacy in T-ALL PDXs
Ponatinib efficacy was evaluated in vivo in three T-ALL PDX models. T-ALL blasts were reinjected into 12 or 16 NSG mice (1–2 million cells per mouse). After seven days, the mice were randomly grouped (N=6 or 8 per group) and the drug treatment was initiated. Ponatinib (Takeda Pharmaceutical) was dissolved in citric acid (Jena Bioscience, #CSS-508) and was administered at 30 mg/kg once daily through oral gavage. Control mice received DMSO (Fisher BioReagents, #BP231–100) dissolved in citric acid. Leukemia burden was monitored in peripheral blood weekly with the same procedure and end points as described above. The sample size was considered sufficient to detect the ponatinib efficacy at lease 20% increase in leukemia survival compared to vehicle-treated mice and was estimated using Hmisc package in R (version 4.1.0) using log-rank test. Investigators were not blinded to the group allocation during treatment.
Dasatinib and ponatinib PK studies
Sample collection
Plasma samples from NSG mice were collected at different time points following a single oral dose of dasatinib (LC Laboratories, #D-3307) 20 or 40 mg/kg, or ponatinib 15 or 30 mg/kg in citric acid so that at least three mice were evaluated at each time point. For dasatinib, a total of 27 mice were bled at 0.5, 1, 2, 3, 5, 7 and 8 hrs after dosing (3–12 mice for each time point). For ponatinib, 24 mice were bled at 0.5, 1, 2, 3, 5, 8, 12 and 24 hrs after dosing (3–9 mice for each time point). Plasma samples were collected by centrifugation and frozen at −80°C until measurement. Details of dasatinib and ponatinib quantification are provided in Supplemental Methods.
PK modeling
The PK was evaluated using non-linear mixed effects modeling analysis via Monolix (version 5.1.0) using the Stochastic Approximation Expectation-Maximization (SAEM) method. A one-compartment PK model with first-order absorption and linear elimination was used to model both dasatinib and ponatinib. The PK parameters estimated included: ka (1/hrs), the absorption rate constant; ke (1/hrs), the elimination rate constant; and V/f (ml/kg), apparent volume. The parameter “f” is the unknown bioavailability. The inter-individual variability of the parameters was assumed to be log-normally distributed. A proportional residual error model was used with assumed normal distribution of the residuals. The median and 90th percent prediction interval for the estimated AUC0–24hrs, AUC0-infinity, Cmax, Tmax, t1/2, and apparent clearance were determined using N=100 sets of PK parameters randomly sampled from the population PK parameter distribution.
Dasatinib and ponatinib PD studies
Sample collection and phosphorylated LCK (pLCK) measurement
Two LCK-activated T-ALL PDX models (PDX #2 and #3 in Fig. 1E,1F) were selected for PD evaluations. 84 NSG mice bearing leukemia (i.e., the blast % in peripheral blood reaching 70%) were randomly regrouped in four groups (N=21), and a single dose of dasatinib at 20 or 40 mg/kg or ponatinib at 15 or 30 mg/kg was orally given to each group. Bone marrow cells from three mice were harvested at each time point: 0, 3, 8, 12, 18, 24, and 48 hrs after dosing. After red blood cells were lysed using RBC Lysis Solution (QIAGEN, #158904), proteins were immediately extracted and denatured, followed by Western blotting where the levels of LCK, pLCK at Y394 and GAPDH were quantified as described below.
Figure 1. Ponatinib shows antileukemic activity in LCK-activated T-ALL ex vivo and in vivo.
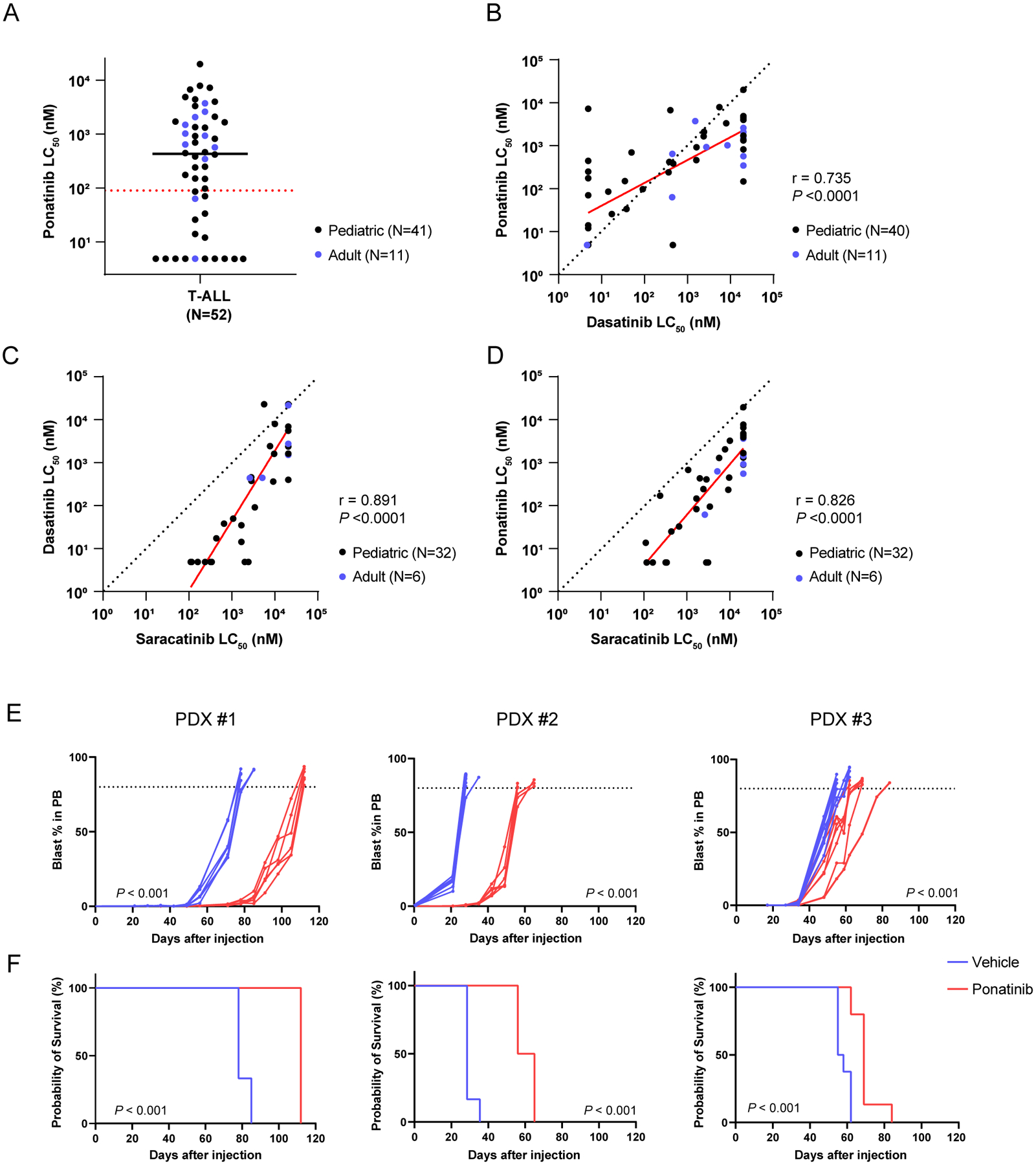
A, Ponatinib LC50 distribution in human T-ALL (N=52). The black solid line and red dash line indicate the median LC50 and the cut-off (90nM) for ponatinib sensitivity, respectively. B-D, Comparison of LC50 between dasatinib and ponatinib (B, N=51), saracatinib and dasatinib (C, N=38), and saracatinib and ponatinib (D, N=38) in T-ALL samples evaluated by Pearson tests. In each panel, the red line indicates the regression line (R2=0.540, P <0.0001 in B; R2=0.793, P <0.0001 in C; R2=0.682, P <0.0001 in D) and the black dashed line represents the line of identity. E, F, In vivo efficacy of ponatinib therapy in three LCK-activated T-ALL PDX models. Leukemic burden of the mice treated with either ponatinib 30 mg/kg or vehicle were monitored by blast % in peripheral blood. Each curve represents an individual mouse and P-value was estimated using a Wilcoxon matched-pairs signed rank test (E). Survival after the injection was estimated for each T-ALL PDX model. P-value was calculated using a log-rank test (F). The ponatinib treatment arms are shown in red, while the vehicle treatment arms are in blue. Each treatment arm included six mice for PDXs #1 and #2, and eight mice per arm for PDX #3 (left, middle, and right panels, respectively). PB, peripheral blood.
PD modeling
The dynamics of pLCK in the presence of dasatinib or ponatinib are modeled with an indirect response model where the drug de-phosphorylates LCK(19). Specifically, the rate constant describing de-phosphorylation in the following model (koff) is an increasing function of the drug concentration. The model is defined as follows:
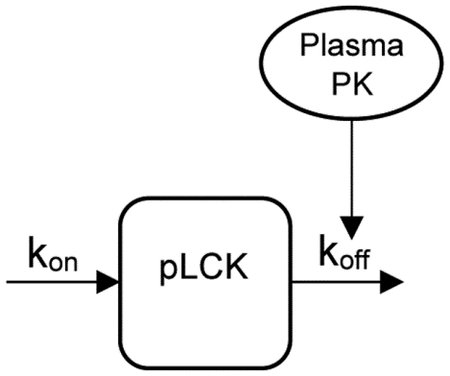 |
Where:
In the above equation kon (1/hr) is the rate constant describing phosphorylation of LCK, koff (1/hr) is the rate constant describing de-phoshorylation of pLCK (% relative to baseline). The equation relating kon to koff maintains steady-state concentrations of pLCK (pLCKbaseline) in the absence of drug. The effect of dasatinib or ponatinib concentration (C: determined using the above PK model) was described by the saturable Hill function where Emax is the maximum effect, EC50 (ng/mL) is the concentration that causes a 50% of maximum effect, and n is the Hill coefficient that quantifies the steepness of the Hill function(35). The PD model parameters were estimated with maximum likelihood estimation using a naive pooled data approach by first estimating the PK model parameters, next fixing these PK parameters and then estimating the PD parameters.
PK/PD simulations for steady-state drug exposure and pLCK inhibition
Simulations were performed based on mice and human PK/PD data. Specifically, the results of PK and PD modeling from mice treated with dasatinib 20 or 40 mg/kg or ponatinib 15 or 30 mg/kg in this study were used for the mouse simulations. For humans, the PK simulations were based on previously reported clinical results(30, 32), dasatinib doses of 100 and 140 mg and ponatinib doses of 40 and 45 mg. The PD model parameters were assumed to be the same as in the mouse studies described in this study. All simulations performed were based on various doses given daily for seven days. For each case the time pLCK was depleted below 50% was determined at steady-state (i.e. after the 7th dose). The simulations were based on N=100 simulated PK and PD parameters in all cases.
Western blotting for LCK and pLCK
Human T-ALL cells were harvested and washed once with ice-cold phosphate-buffered saline (PBS: Gibco, #10010–023) and then lysed with RIPA Lysis and Extraction Buffer (Thermo Scientific, #89901) supplemented with protease and phosphatase inhibitor cocktail (Thermo Scientific, #78440). Protein lysates were incubated on ice with gentle shaking for 15 minutes before being centrifuged at 4°C, 15 000 rpm for 15 minutes. Supernatants were transferred into new centrifuge tubes, and an equal volume of 2x Laemmli sample buffer (Bio-Rad, #1610737) supplemented with 2-mercaptoethanol (Bio-Rad, #1610710) was added. Protein samples were heated at >95°C before being stored at −20°C or western blotting. Equal amounts of protein samples were separated by precast 4–15% Tris-glycine Mini-PROTEAN TGX gels (Bio-Rad, # 4561086). Resolved proteins were transferred onto Immobilon-FL PVDF membranes (Millipore, #IPFL00010). The membranes were blocked with Intercept® (TBS) blocking buffer (LI-COR, #927–60001) for 2 hrs at room temperature. The membranes were then probed with primary antibodies at an optimal concentration in the same blocking buffer supplemented with 0.2% Tween 20 (Fisher BioReagents, #BP337–500) overnight at 4°C: Antibodies against LCK (Cell Signaling Technology, #2657; 1:2 000), pLCK (Cell Signaling Technology, #6943; 1:2 000), and GAPDH (Cell Signaling Technology, #2118; 1:5 000) for loading control. The membranes were then washed with TBS-T three times (10 minutes each time on a shaker) and incubated with the IRDye® 800CW goat anti-rabbit and 680CW goat anti-mouse IgG secondary antibodies (LI-COR, #926–32211 and #926–32210; 1:5 000) at room temperature for 2 hrs. Excessive antibodies were washed out with TBS-T, and the membranes were exposed in LI-COR Odyssey imaging system. Fluorescent signal intensities were quantified and analyzed with Image Studio software (lite version 5.2). The fluorescent signals of LCK and pLCK were respectively normalized to that of GAPDH, and then pLCK/LCK were calculated for each sample, followed by the normalization to control (0 hr as 100%).
Statistical analysis
For the experiments involving human T-ALL patient samples, sample size was strictly based on the availability of specimen and data without other filtering. Therefore, samples size was not determined in advance. The associations of LC50 values between dasatinib, ponatinib, and saracatinib were evaluated by Pearson correlation and Wilcoxon matched-pairs signed rank test. In the ponatinib efficacy studies in vivo, human leukemic burden in NSG mice and their survivals were assessed using Wilcoxon matched-pairs signed rank tests and log-rank tests, respectively. P-values were considered significant if <0.05. All statistical tests were two-sided. All analyses were performed with GraphPad Prism (version 9.3.1).
Data availability statement
Whole genome sequencing data is deposited with the European Genome-Phenome Archive (accession number: EGAD00001006434). To request reagents included in this work, please contact Dr. Jun J. Yang (jun.yang@stjude.org).
Details of other experiments (e.g., ex vivo drug sensitivity assay for dasatinib, ponatinib and saracatinib, LC-MS for dasatinib and ponatinib) are provided in the Supplemental Methods.
Results
Cytotoxicity profile of dasatinib and ponatinib in T-ALL
Using primary human T-ALL samples, we first determined leukemia sensitivity to ponatinib and dasatinib ex vivo, quantified as the concentration at which 50% of leukemia cells were killed (LC50). In a total of 52 T-ALL cases, the distribution of LC50 for ponatinib was bi-modal with a median of 432.6nM (Supplemental Fig. 1A, Supplemental Table 1). Based on achievable drug concentration estimated from PK studies in humans(30, 36–38), we chose 90nM as the cutoff to define ponatinib sensitivity. 32.7% (n=17) of samples were classified as sensitive to ponatinib, representing 33.6% and 18.2% of children and adults with T-ALL, respectively (Fig. 1A). Similarly, dasatinib sensitivity (LC50 <80nM) was observed in 47.5% and 9.1% of pediatric and adult T-ALL, respectively, consistent with our previous report(16). Analyzing 51 samples with both drugs tested, we observed a highly significant correlation between their LC50 values (Fig. 1B, r=0.735, P <0.0001, by Pearson correlation test); and ponatinib LC50 was significantly lower than that of dasatinib (P =0.0038, by Wilcoxon matched-pairs signed rank test). Measuring phosphorylation of LCK, CD247, and ZAP70, we confirmed that ponatinib efficiently inhibited the preTCR-LCK signaling in a dose-dependent manner (Supplemental Fig 2A, B), as we previously described with dasatinib(16). Furthermore, we also tested this cohort of T-ALL samples for sensitivity to a selective LCK inhibitor, namely saracatinib(39). As shown in Fig. 1C and 1D, saracatinib LC50 was significantly correlated with that of dasatinib (r=0.891, P <0.0001) and ponatinib (r=0.826, P <0.0001), suggesting LCK as the common driver of these drug response phenotypes.
Using xenograft model of three representative ponatinib-sensitive T-ALL cases, we evaluated anti-leukemic efficacy in vivo. Ponatinib given at 30 mg/kg once daily significantly inhibited leukemia growth in mice compared to those receiving vehicle control across three cases, resulting in prolonged survival ranging from 122% to 216% (Fig. 1E, 1F).
PK and PD evaluation of dasatinib and ponatinib in mice
Next we sought to determine plasma concentration of orally administered ponatinib or dasatinib in NSG mice, from which we then performed PK modeling for each drug across dosages. After a single oral dose of dasatinib, plasma concentration rose quickly in a dose-dependent manner: Cmax reached 102 ng/mL at 20 mg/kg and 366.36 ng/mL at 40 mg/kg, both with 1.17 hrs of Tmax, and AUC0–24hrs were 495.2 and 1940.0 ng h/mL, respectively (Fig. 2A, Table 1). Drug concentration in the plasma then decreased, with t1/2 of 0.35 and 0.34 hr. By comparison, ponatinib orally given at 15 and 30 mg/kg resulted in Cmax of 181.36 and 355.84 ng/mL and AUC0–24hrs of 1533.2 and 2996.5 ng h/mL, respectively. Ponatinib apparent clearance was slower than dasatinib, with a Tmax of 3.04 and 3.08 hrs, and t1/2 of 1.92 hrs for both dosages (Fig. 2B, Table 1). Other PK parameters are described in Table 1.
Figure 2. PK and PD profiles of dasatinib and ponatinib in mice.
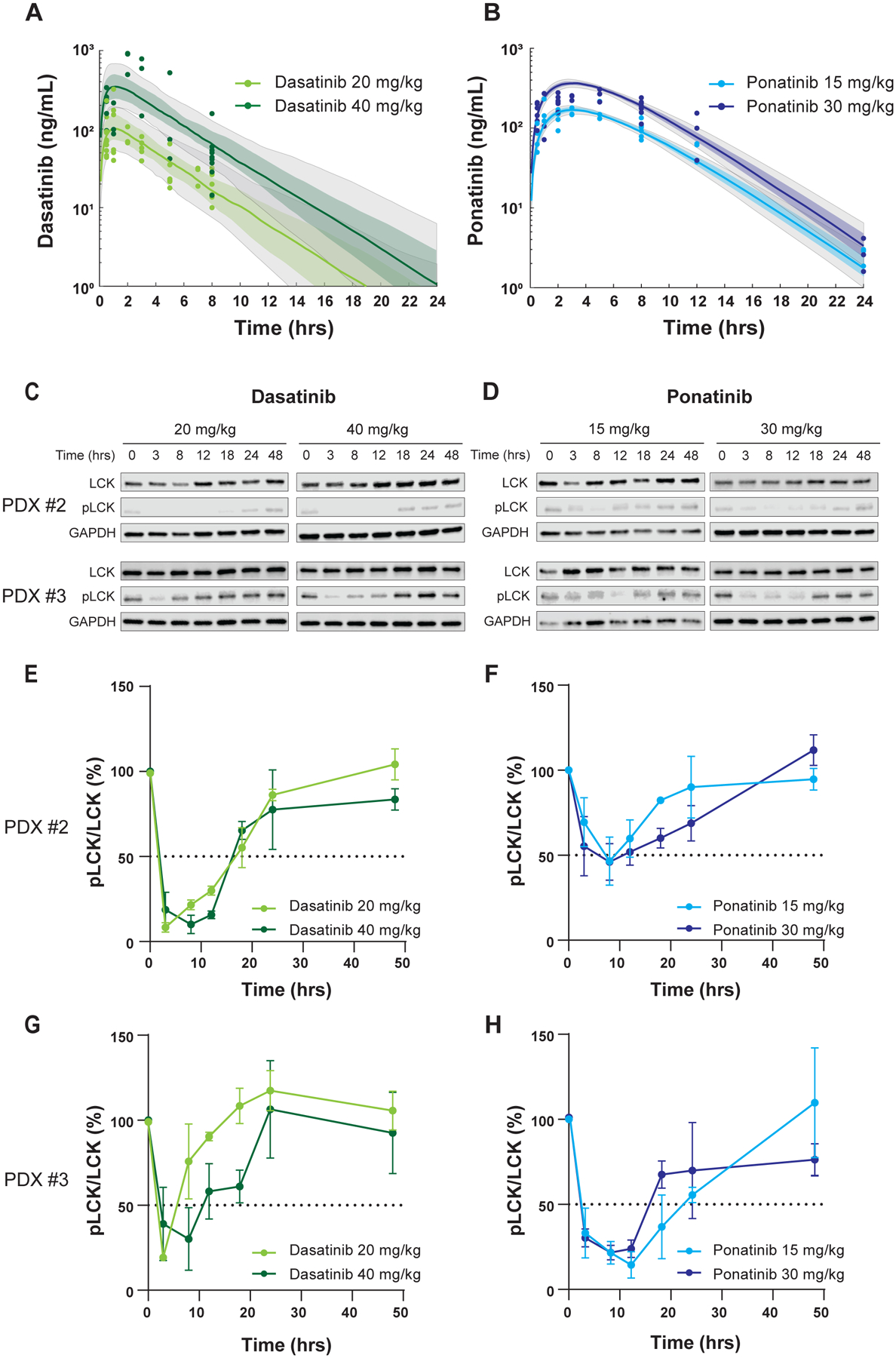
A, B, Time-dependent change in plasma drug concentration after single dose administered orally. For dasatinib, observed plasma concentrations are plotted in light green (20 mg/kg) and dark green (40 mg/kg) (A), while those are shown in light blue (15 mg/kg) and dark blue (30 mg/kg) for ponatinib (B). The corresponding curves and shaded regions are the median and 25th-75th percentile drug concentrations predicted by a one-compartment PK model with first-order absorption and linear elimination. The grey shaded regions are the 10th-90th percentile model predicted drug concentrations. 100 ng/ml is equivalent to 204.9nM for dasatinib, and 187.8nM for ponatinib. C, D, LCK phosphorylation over time after dasatinib and ponatinib treatment was used as the PD endpoint in the two T-ALL PDX models. pLCK was quantified by Western blotting using near-infrared fluorescence detection. Human T-ALL blasts were collected from mouse bone marrow (derived from PDXs #2 and #3 in Fig. 1E, F) at various time points after a single dose of each drug. Representative results are shown. E-H, The y axes indicate relative phosphorylation levels normalized to mice not receiving drug (0 hr). Each plot is a mean from three mice shown with S.D. as an error bar.
Table 1.
PK parameters of dasatinib and ponatinib in orally-dosed mice
| Dasatinib | Ponatinib | ||
|---|---|---|---|
| Median (5th-95th percentile) | Median (5th-95th percentile) | ||
| AUC0–24hrs 20 mg (ng hr/mL) | 495.21 (253.15–1170.58) | AUC0–24hrs 15 mg (ng hr/mL) | 1524.53 (1363.64–1718.81) |
| AUC0–24hrs 40 mg (ng hr/mL) | 1940.07 (907.47–4024.05) | AUC0–24hrs 30 mg (ng hr/mL) | 2996.56 (2707.46–3351.17) |
| AUC0-infinity 20 mg (ng hr/mL) | 499.51 (253.33–1173.71) | AUC0-infinity 15 mg (ng hr/mL) | 1533.22 (1367.25–1727.10) |
| AUC0-infinity 40 mg (ng hr/mL) | 1952.88 (908.72–4027.31) | AUC0-infinity 30 mg (ng hr/mL) | 3010.21 (2716.52–3365.01) |
| Cmax 20 mg (ng/mL) | 102.00 (37.81–230.62) | Cmax 15 mg (ng/mL) | 181.36 (153.95–210.12) |
| Cmax 40 mg (ng/mL) | 366.36 (166.52–847.16) | Cmax 30 mg (ng/mL) | 355.84 (308.83–412.35) |
| Tmax 20 mg (hrs) | 1.17 (0.83–1.42) | Tmax 15 mg (hrs) | 3.04 (2.54–3.58) |
| Tmax 40 mg (hrs) | 1.17 (0.92–1.42) | Tmax 30 mg (hrs) | 3.08 (2.63–3.58) |
| t1/2 20 mg (hrs) | 0.35 (0.27–0.45) | t1/2 15 mg (hrs) | 1.92 (1.32–2.57) |
| t1/2 40 mg (hrs) | 0.34 (0.27–0.34) | t1/2 30 mg (hrs) | 1.92 (1.42–2.51) |
| CL/f 20 mg (mL/hr/kg) | 667.41 (284.01–1315.89) | CL/f 15 mg (mL/hr/kg) | 9.78 (8.69–10.97) |
| CL/f 40 mg (mL/hr/kg) | 341.38 (165.82–733.74) | CL/f 30 mg (mL/hr/kg) | 9.97 (8.92–11.04) |
AUC, area under the curve; CL/f, apparent clearance; PK, pharmacokinetic.
We then evaluated the PD of each drug in two T-ALL PDX models in which we had confirmed dasatinib and ponatinib efficacies in vivo (Fig. 1E, 1F)(16). The doses and dosing schedule were the same as those used in the PK studies noted above. pLCK inhibition in human T-ALL blasts from mouse bone marrow was examined as the PD endpoint by Western blotting and quantified using near-infrared fluorescence detection (Fig. 2C, 2D). With both drugs, a higher dosage resulted in better pLCK inhibition and slower recovery (Fig. 2E–H).
Using the observed pLCK/LCK ratios, we performed PK/PD modeling to explore exposure-to-response relationships for each drug. The model fit estimated a peak pLCK inhibition by dasatinib of 88% (CV%: 8%), while ponatinib produced a peak pLCK inhibition of 72% (CV%: 16%) (Fig. 3A, 3B). Other PD parameters are summarized in Table 2. With these models, we first simulated the steady-state drug concentration and pLCK inhibitory effect for dasatinib and ponatinib with daily dosing schedule for a 7-day treatment course in mice. For dasatinib at 20 mg/kg or 40 mg/kg, drug levels oscillated with each 24-hr period, with concomitant changes in pLCK inhibition, consistently throughout the 7-day treatment. The median duration of pLCK inhibition (at least 50%) by dasatinib was simulated to be 8.2 hrs per day ([4.0, 13.6], 10th and 90th percentile) at 20 mg/kg and 14.5 hrs per day ([9.7, 21.8], 10th and 90th percentile) at 40 mg/kg (Supplemental Fig. 3A, 3B). Simulations of ponatinib data yielded a similar pattern of time-dependent change in drug plasma concentration and pLCK inhibition. The median duration of pLCK inhibition (>50%) by ponatinib was 8.5 hrs per day ([0, 13.3], 10th and 90th percentile) at 15 mg/kg and 12.0 hrs per day ([0, 16.6], 10th and 90th percentile) at 30 mg/kg (Supplemental Fig. 3C, 3D).
Figure 3. Simulation of PK-PD after repetitive dasatinib and ponatinib dosing in human T-ALL.
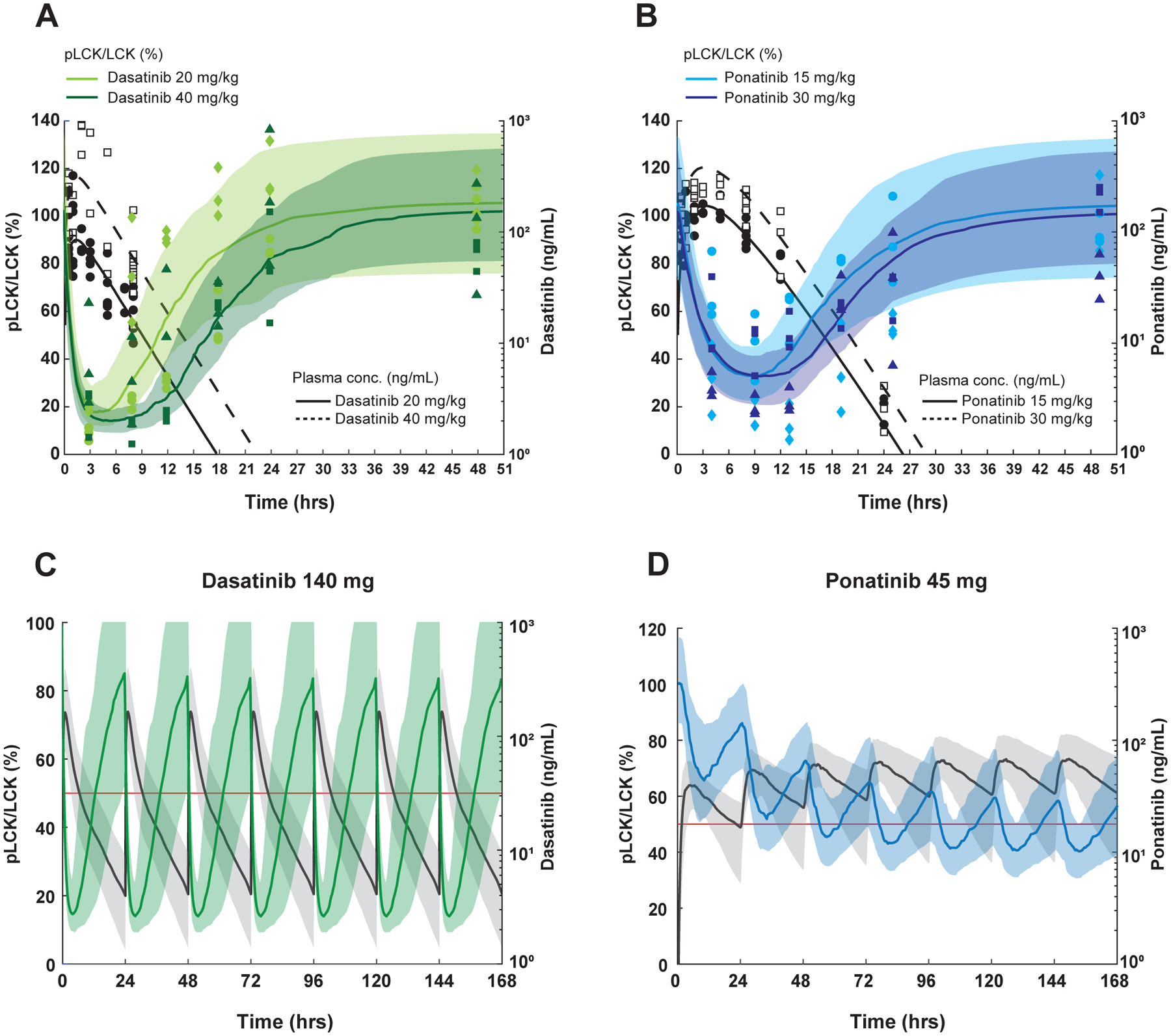
A, B, PK/PD modeling in mice after receiving a single dose of dasatinib (A) or ponatinib (B). For pLCK inhibition (left y axis, normalized to untreated animals), data are plotted as light green (20 mg/kg) and dark green symbols for dasatinib (40 mg/kg), while they are plotted as light blue (15 mg/kg) and dark blue symbols (30 mg/kg) for ponatinib Data from PDX #2 are shown as circles and squares, and those from PDX #3 are in rhombi and triangles. The median, 10th-90th percentile pLCK levels were predicted and are respectively shown in the curves and shaded regions in the same colors. For plasma drug concentrations (right y axis) The black circles (lower doses) and black squares (higher doses) show measured plasma drug concentrations and the black solid (lower doses) and black dashed (higher dose) curves indicate the median model estimated drug concentrations. 100 ng/ml is equivalent to 204.9nM for dasatinib, and 187.8nM for ponatinib. C, D, PK and PD simulations at FDA-approved dosages of dasatinib and ponatinib in humans. The steady-state after seven doses of dasatinib 140 mg (C) and ponatinib 45 mg (D) given daily are simulated (N=100). In the dasatinib simulation, the median, and 10th-90th percentile model estimated pLCK levels are shown by the green curve and shaded regions, respectively. For ponatinib, the blue curve and shaded regions indicate the median, 10th-90th percentile model estimated pLCK levels. In both simulations, the black solid curve and shaded regions indicate the median, 10th-90th percentile model estimated drug concentrations. The right y axes, plasma drug concentrations; the left y axes, pLCK levels normalized to the untreated mice. The red lines indicate model estimated pLCK levels equal to 50%.
Table 2.
Estimated PD parameters of dasatinib and ponatinib in orally-dosed mice
| Dasatinib | Ponatinib | |
|---|---|---|
| Emax (ng/ml) (CV%) | 7.3 (67%) | 2.6 (56%) |
| EC50 (ng/ml) (CV%) | 32.0 (69%) | 50.2 (47%) |
| EC90 (ng/ml) (CV%) | 33.0 (14%) | 63.0 (13%) |
| Maximum pLCK inhibition (%) (CV%) | 88.0 (8%) | 72.0 (16%) |
| Median duration of pLCK/LCK <50% (hrs/day) (10th-90th percentile) | 20 mg/kg 10.0 (4.7, 19.7) | 15 mg/kg 10.5 (0, 24.0) |
| 40 mg/kg 15.5 (8.9, 24.0) | 30 mg/kg 14.8 (0, 24.0) |
PD, pharmacodynamic.
PK/PD-based simulations of steady-state effects of dasatinib or ponatinib in humans
To estimate the dosage and dosing schedule required for T-ALL treatment in humans, we then sought to simulate pLCK inhibition in human T-ALL with FDA-approved adult doses of dasatinib (140 mg daily) and ponatinib (45 mg daily) for BCR::ABL1 ALL (12, 21, 23, 34, 40–42). We used previously reported human PK data for these agents to estimate systemic exposure(30–32). During the 7-day course of dasatinib treatment, pLCK inhibition oscillated significantly for each 24-hr period, although this still produced >50% pLCK inhibition for 13.0 hrs per day ([5.7, 23.3], 10th and 90th percentile) given 140 mg once daily (Fig. 3C). By contrast, simulations of ponatinib in humans showed a progressively increasing plasma drug concentrations over the first three daily doses along with a gradual increase in pLCK inhibition until steady-state was reached after the third dose. Ponatinib achieved >50% inhibition for 13.9 hrs per day ([0, 24.0], 10th and 90th percentile) given 45 mg once daily (Fig. 3D). Lower doses of both drugs also showed sustained pLCK inhibition: dasatinib 100 mg would result in >50% pLCK inhibition for 11.0 hrs per day ([4.6, 18.4], 10th and 90th percentile), ponatinib 40 mg would suppress pLCK to the same degree for 9.6 hrs per day ([0, 24.0], 10th and 90th percentile).
Ponatinib can overcome dasatinib resistance in T-ALL in vitro
Given that dasatinib resistance caused by mutations in ABL1 in BCR::ABL1 leukemia can be treated with ponatinib(13, 26), we explored whether this is also true in LCK-activated T-ALL. First, we developed a dasatinib-resistant cell model by continuously treating a dasatinib-sensitive T-ALL cell line, KOPT-K1, with dasatinib. After five weeks, cells (hereinafter referred to as KOPT-K1-R) acquired resistance to dasatinib (Fig. 4A, LC50=1717.5nM). Whole genome sequencing showed that the acquired resistance in KOPT-K1-R might come from LCK mutation at T316I, which was later confirmed by Sanger sequencing (Supplemental Fig. 4). Interestingly, these cells retained substantial sensitivity to ponatinib with an LC50 of 87.4nM (Fig. 4B), which is an achievable drug level based on human PK data of this drug(30, 36–38). Western blotting confirmed that dasatinib no longer inhibited LCK in KOPT-K1-R, whereas ponatinib blocked LCK phosphorylation to some extent, around 50% at 100nM (Fig. 4C, 4D).
Figure 4. Dasatinib-resistant KOPT-K1 retains its sensitivity to ponatinib.
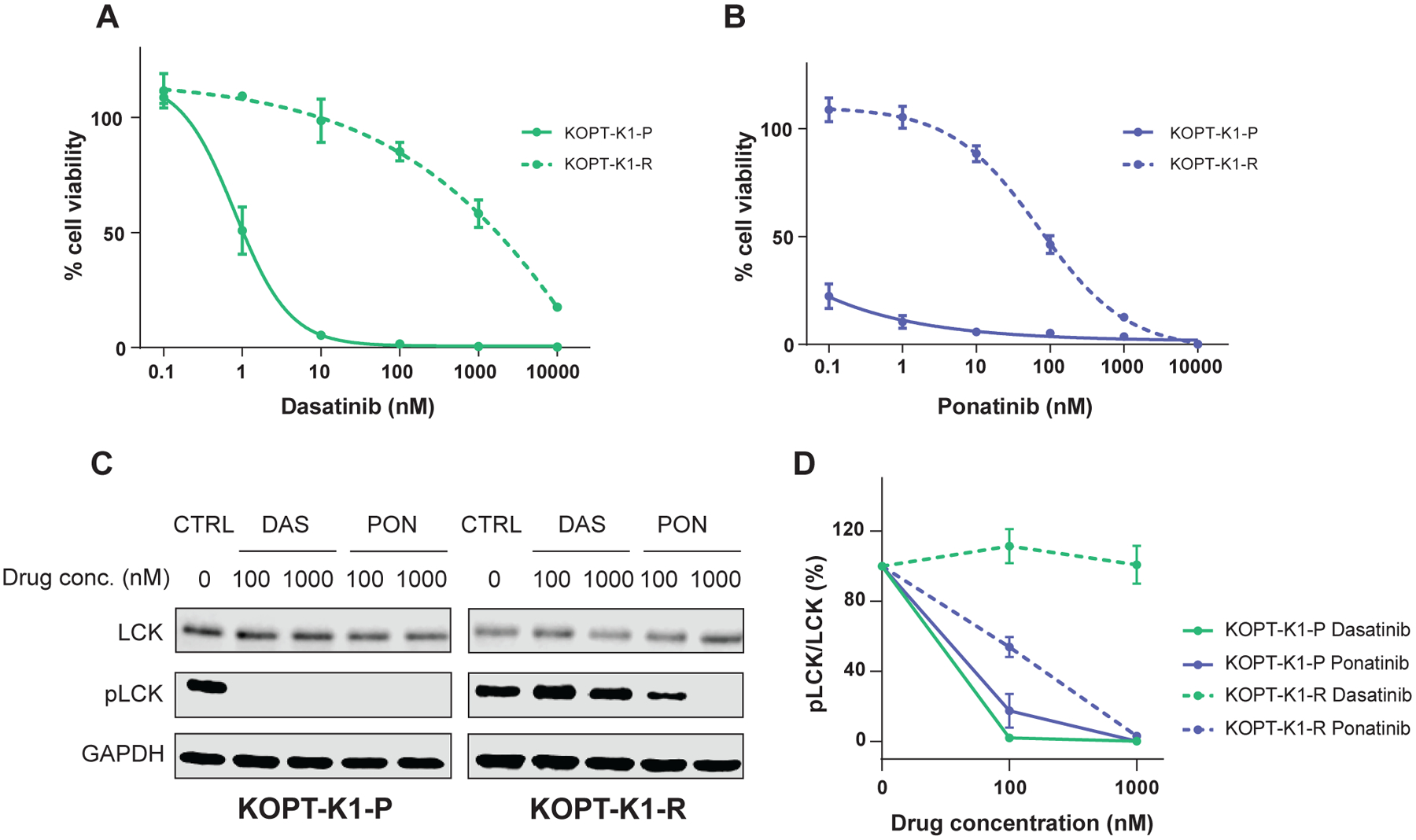
A, B, The resistant cells (KOPT-K1-R, dashed line) as well as parental cells (KOPT-K1-P, solid line) were subject to either dasatinib (A) or ponatinib (B) treatment for 72 hrs at various concentrations, followed by a CTG assay to evaluate cell viability. The y axes indicate % cell viability compared to the respective untreated cells. C, D, LCK phosphorylation in dasatinib- or ponatinib-treated cells were quantified by Western blotting. The cells were treated at indicated concentrations for 3 hrs. The experiments were repeated three times and a representative image is shown (C). CTRL, no-treatment control; DAS, dasatinib; PON, ponatinib. D, The y axes indicate relative LCK phosphorylation normalized to the level in untreated cells. Each plot is a mean from independent experiments (N=3) shown with S.D. as an error bar.
Discussion
For reasons that are still not fully understood, T-ALL is associated with inferior prognosis relative to B-ALL, in the context of conventional cytotoxic chemotherapy(1, 3, 5–8). The discovery of LCK as a therapeutic target in a substantial subset of T-ALL is potentially exciting because 1) there are a number of FDA-approved TKIs that can act as LCK inhibitors(16–18, 43); 2) TKIs such as dasatinib and ponatinib have been integrated into BCR::ABL1 ALL chemotherapy with documented safety(13, 21, 22, 34, 40). To aid the development of LCK-targeting therapy for T-ALL, we herein performed comprehensive preclinical PK and PD studies of two potent LCK inhibitors, namely dasatinib and ponatinib, to establish the exposure-to-response relationship in T-ALL PDX models and then simulated dose and dosing schedules appropriate for treatment in patients with T-ALL.
As multi-target kinase inhibitors, dasatinib and ponatinib exhibit similar potency against LCK vs ABL based on binding affinity and enzymatic kinetics (26, 44, 45). Moreover, our group previously reported that cytotoxicity as measured by LC50 was also comparable for dasatinib and ponatinib in LCK-activated T-ALL vs BCR::ABL1 B-ALL(16). Therefore, we reason that pharmacodynamic profile of dasatinib and ponatinib in BCR::ABL1 leukemias can be used as a benchmark for these agents in LCK-activated T-ALL. In chronic myeloid leukemia (CML) patients, dasatinib exposure was considered as sufficient to elicit a clinical response if the time above 50% pCRKL inhibition is 12.8 hrs per day(46). This is in line with our estimated duration of pLCK inhibition in human T-ALL by dasatinib at 140 mg and ponatinib at 45 mg once daily in human T-ALL. For this reason, our results are informative of the design of future trials of dasatinib or ponatinib in LCK-activated T-ALL. However, it should be noted that there is significant variability in T-ALL sensitivity to dasatinib or ponatinib ex vivo (16), plausibly linked to the degree of LCK activation, suggesting that the level of pLCK inhibition required for anti-leukemic efficacy may vary from patient to patient. Future studies are warranted to include additional T-ALL PDX models representing the entire range of LCK activation in order to model exposure-to-response relationships more precisely.
A potential limitation of our study is that we did not consider the effects of age on dasatinib or ponatinib PK. As an ABL inhibitor, dasatinib is currently approved to be given at a fixed dose of 140 mg in adults with accelerated or blast phase CML or BCR::ABL1 ALL(21, 40, 47), though many trials use daily dosing at 100mg in combination with chemotherapy(23, 41). By contrast, it is dosed based on body-surface area, ranging from 60 to 80 mg/m2 in children (22, 48, 49). In pediatric BCR::ABL1 ALL, there is also emerging data that suggest therapy with dasatinib at 80 mg/m2 is associated with improved survival (22); among adults, dasatinib at higher dose (140 mg, equivalent to 80mg/m2 in children) also showed increased central nervous system penetration as compared to 100 mg(50). Because the PK profile of dasatinib is similar between adults and children(51), it might be reasonable to start with dasatinib at 80 mg/m2 for pediatric T-ALL patients. However, ponatinib human PK data in children is scarce though an ongoing Phase I/II trial (NCT04501614) for pediatric BCR::ABL1 leukemia patients will provide critical information. It remains unclear what the optimal dose of ponatinib would be in the context of LCK inhibition in T-ALL and whether that differs between children and adults. We are conducting a Phase II trial of ponatinib in patients with T-ALL (NCT05268003), which hopefully will shed light in this regard in the near future. Finally, ponatinib is associated with notable toxicity, especially with higher doses (45 mg)(13, 52, 53). Therefore, it is possible that ponatinib dose should be gradually reduced as leukemic burden decreases over time to limit the exposure while maintaining efficacy against residual disease(12, 34, 54).
A common cause of treatment failure with TKI therapy is the acquisition of drug resistance mutations, and multiple lines of agents are often needed to maintain remission. For instance, acquired resistance to dasatinib including mutation of ABL1 T315I in BCR::ABL1 leukemia can be addressed by switching to ponatinib(13, 26). It is unknown how often LCK mutation may arise during either dasatinib or ponatinib therapy, or different types of mutation can be associated with each agent in patients with T-ALL. Our in vitro data suggests that LCK T316I directly causes dasatinib resistance in T-ALL cell line, but this does not seem to completely abrogate ponatinib’s interaction with LCK, and therefore its cytotoxic effects. However, it should be noted that ponatinib activity was significantly weakened in T-ALL with mutant LCK, whereas its effects were largely unaffected by ABL mutations(26). Even though T316 in LCK and T315 in ABL are both gatekeeper residues for the kinase domain(55), they may have differential impact on ponatinib binding. These results are potentially important and may inform the choice of LCK inhibitor in T-ALL, but their relevance in human trials need to be examined carefully.
In conclusion, we characterized PK and PD profiles of dasatinib and ponatinib in preclinical T-ALL models and our results established potential dosages for future human trials of LCK inhibitors in T-ALL.
Supplementary Material
Acknowledgements
We thank the Hartwell Center for Biotechnology, the Flow Cytometry and Cell Sorting Core, and the Animal Research Center at St. Jude Children’s Research Hospital for their technical assistance. We also thank all the patients and families for donating research specimens, as well as the clinicians and research staff for assistance in sample collection. This work was in part supported by the National Institutes of Health (P30CA21765, R01CA264837, and U01CA264610), American Lebanese Syrian Associated Charities, the Translational Research Program at the Leukemia & Lymphoma Society (6665-23), and the Takeda Pharmaceutical Company.
Competing interests
This work was partly supported by Takeda Pharmaceutical Company. J.H. is a current employee of Amgen Inc.
References
- 1.Jeha S, Pei D, Choi J, Cheng C, Sandlund JT, Coustan-Smith E, et al. Improved CNS Control of Childhood Acute Lymphoblastic Leukemia Without Cranial Irradiation: St Jude Total Therapy Study 16. J Clin Oncol. 2019;37(35):3377–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moricke A, Zimmermann M, Valsecchi MG, Stanulla M, Biondi A, Mann G, et al. Dexamethasone vs prednisone in induction treatment of pediatric ALL: results of the randomized trial AIEOP-BFM ALL 2000. Blood. 2016;127(17):2101–12. [DOI] [PubMed] [Google Scholar]
- 3.Pieters R, de Groot-Kruseman H, Van der Velden V, Fiocco M, van den Berg H, de Bont E, et al. Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J Clin Oncol. 2016;34(22):2591–601. [DOI] [PubMed] [Google Scholar]
- 4.Place AE, Stevenson KE, Vrooman LM, Harris MH, Hunt SK, O’Brien JE, et al. Intravenous pegylated asparaginase versus intramuscular native Escherichia coli L-asparaginase in newly diagnosed childhood acute lymphoblastic leukaemia (DFCI 05–001): a randomised, open-label phase 3 trial. Lancet Oncol. 2015;16(16):1677–90. [DOI] [PubMed] [Google Scholar]
- 5.Maloney KW, Devidas M, Wang C, Mattano LA, Friedmann AM, Buckley P, et al. Outcome in Children With Standard-Risk B-Cell Acute Lymphoblastic Leukemia: Results of Children’s Oncology Group Trial AALL0331. J Clin Oncol. 2020;38(6):602–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conter V, Bartram CR, Valsecchi MG, Schrauder A, Panzer-Grumayer R, Moricke A, et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood. 2010;115(16):3206–14. [DOI] [PubMed] [Google Scholar]
- 7.Winter SS, Dunsmore KP, Devidas M, Wood BL, Esiashvili N, Chen Z, et al. Improved Survival for Children and Young Adults With T-Lineage Acute Lymphoblastic Leukemia: Results From the Children’s Oncology Group AALL0434 Methotrexate Randomization. J Clin Oncol. 2018;36(29):2926–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schrappe M, Valsecchi MG, Bartram CR, Schrauder A, Panzer-Grumayer R, Moricke A, et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood. 2011;118(8):2077–84. [DOI] [PubMed] [Google Scholar]
- 9.Pieters R, Kaspers GJ, van Wering ER, Huismans DR, Loonen AH, Hahlen K, et al. Cellular drug resistance profiles that might explain the prognostic value of immunophenotype and age in childhood acute lymphoblastic leukemia. Leukemia. 1993;7(3):392–7. [PubMed] [Google Scholar]
- 10.Teachey DT, Pui CH. Comparative features and outcomes between paediatric T-cell and B-cell acute lymphoblastic leukaemia. Lancet Oncol. 2019;20(3):e142–e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanasi I, Ba I, Sirvent N, Braun T, Cuccuini W, Ballerini P, et al. Efficacy of tyrosine kinase inhibitors in Ph-like acute lymphoblastic leukemia harboring ABL-class rearrangements. Blood. 2019;134(16):1351–5. [DOI] [PubMed] [Google Scholar]
- 12.Jabbour E, Short NJ, Ravandi F, Huang X, Daver N, DiNardo CD, et al. Combination of hyper-CVAD with ponatinib as first-line therapy for patients with Philadelphia chromosome-positive acute lymphoblastic leukaemia: long-term follow-up of a single-centre, phase 2 study. Lancet Haematol. 2018;5(12):e618–e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre P, Paquette R, Chuah C, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369(19):1783–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kantarjian H, Stein A, Gokbuget N, Fielding AK, Schuh AC, Ribera JM, et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N Engl J Med. 2017;376(9):836–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gocho Y, Liu J, Hu J, Yang W, Dharia NV, Zhang J, et al. Network-based systems pharmacology reveals heterogeneity in LCK and BCL2 signaling and therapeutic sensitivity of T-cell acute lymphoblastic leukemia. Nat Cancer. 2021;2(3):284–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frismantas V, Dobay MP, Rinaldi A, Tchinda J, Dunn SH, Kunz J, et al. Ex vivo drug response profiling detects recurrent sensitivity patterns in drug-resistant acute lymphoblastic leukemia. Blood. 2017;129(11):e26–e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laukkanen S, Gronroos T, Polonen P, Kuusanmaki H, Mehtonen J, Cloos J, et al. In silico and preclinical drug screening identifies dasatinib as a targeted therapy for T-ALL. Blood Cancer J. 2017;7(9):e604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu J, Jarusiewicz J, Du G, Nishiguchi G, Yoshimura S, Panetta JC, et al. Preclinical evaluation of proteolytic targeting of LCK as a therapeutic approach in T cell acute lymphoblastic leukemia. Sci Transl Med. 2022;14(659):eabo5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, et al. Dasatinib in imatinibresistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354(24):2531–41. [DOI] [PubMed] [Google Scholar]
- 21.Lilly MB, Ottmann OG, Shah NP, Larson RA, Reiffers JJ, Ehninger G, et al. Dasatinib 140 mg once daily versus 70 mg twice daily in patients with Ph-positive acute lymphoblastic leukemia who failed imatinib: Results from a phase 3 study. Am J Hematol. 2010;85(3):164–70. [DOI] [PubMed] [Google Scholar]
- 22.Shen S, Chen X, Cai J, Yu J, Gao J, Hu S, et al. Effect of Dasatinib vs Imatinib in the Treatment of Pediatric Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA Oncol. 2020;6(3):358–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rousselot P, Coude MM, Gokbuget N, Gambacorti Passerini C, Hayette S, Cayuela JM, et al. Dasatinib and low-intensity chemotherapy in elderly patients with Philadelphia chromosome-positive ALL. Blood. 2016;128(6):774–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serafin V, Capuzzo G, Milani G, Minuzzo SA, Pinazza M, Bortolozzi R, et al. Glucocorticoid resistance is reverted by LCK inhibition in pediatric T-cell acute lymphoblastic leukemia. Blood. 2017;130(25):2750–61. [DOI] [PubMed] [Google Scholar]
- 25.He Y, Zhang J, Zhang Y, Hu Z, Wang P, Gan W, et al. Dasatinib-therapy induced sustained remission in a child with refractory TCF7-SPI1 T-cell acute lymphoblastic leukemia. Pediatr Blood Cancer. 2022;69(8):e29724. [DOI] [PubMed] [Google Scholar]
- 26.O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutationbased resistance. Cancer Cell. 2009;16(5):401–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pophali PA, Patnaik MM. The Role of New Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia. Cancer J. 2016;22(1):40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Rix U, Fang B, Bai Y, Edwards A, Colinge J, et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat Chem Biol. 2010;6(4):291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gozgit JM, Wong MJ, Wardwell S, Tyner JW, Loriaux MM, Mohemmad QK, et al. Potent activity of ponatinib (AP24534) in models of FLT3-driven acute myeloid leukemia and other hematologic malignancies. Mol Cancer Ther. 2011;10(6):1028–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanley MJ, Diderichsen PM, Narasimhan N, Srivastava S, Gupta N, Venkatakrishnan K. Population Pharmacokinetics of Ponatinib in Healthy Adult Volunteers and Patients With Hematologic Malignancies and Model-Informed Dose Selection for Pediatric Development. J Clin Pharmacol. 2022;62(4):555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Broniscer A, Baker SD, Wetmore C, Pai Panandiker AS, Huang J, Davidoff AM, et al. Phase I trial, pharmacokinetics, and pharmacodynamics of vandetanib and dasatinib in children with newly diagnosed diffuse intrinsic pontine glioma. Clin Cancer Res. 2013;19(11):3050–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Argiris A, Feinstein TM, Wang L, Yang T, Agrawal S, Appleman LJ, et al. Phase I and pharmacokinetic study of dasatinib and cetuximab in patients with advanced solid malignancies. Invest New Drugs. 2012;30(4):1575–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Montani D, Bergot E, Gunther S, Savale L, Bergeron A, Bourdin A, et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation. 2012;125(17):2128–37. [DOI] [PubMed] [Google Scholar]
- 34.Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre PD, Paquette R, Chuah C, et al. Ponatinib efficacy and safety in Philadelphia chromosome-positive leukemia: final 5-year results of the phase 2 PACE trial. Blood. 2018;132(4):393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goutelle S, Maurin M, Rougier F, Barbaut X, Bourguignon L, Ducher M, et al. The Hill equation: a review of its capabilities in pharmacological modelling. Fundam Clin Pharmacol. 2008;22(6):633–48. [DOI] [PubMed] [Google Scholar]
- 36.Ye YE, Woodward CN, Narasimhan NI. Absorption, metabolism, and excretion of [(14)C]ponatinib after a single oral dose in humans. Cancer Chemother Pharmacol. 2017;79(3):507–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Narasimhan NI, Dorer DJ, Niland K, Haluska F, Sonnichsen D. Effects of food on the pharmacokinetics of ponatinib in healthy subjects. J Clin Pharm Ther. 2013;38(6):440–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cortes JE, Kantarjian H, Shah NP, Bixby D, Mauro MJ, Flinn I, et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N Engl J Med. 2012;367(22):2075–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buffiere A, Accogli T, Saint-Paul L, Lucchi G, Uzan B, Ballerini P, et al. Saracatinib impairs maintenance of human T-ALL by targeting the LCK tyrosine kinase in cells displaying high level of lipid rafts. Leukemia. 2018;32(9):2062–5. [DOI] [PubMed] [Google Scholar]
- 40.Kantarjian H, Cortes J, Kim DW, Dorlhiac-Llacer P, Pasquini R, DiPersio J, et al. Phase 3 study of dasatinib 140 mg once daily versus 70 mg twice daily in patients with chronic myeloid leukemia in accelerated phase resistant or intolerant to imatinib: 15-month median follow-up. Blood. 2009;113(25):6322–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoon JH, Yhim HY, Kwak JY, Ahn JS, Yang DH, Lee JJ, et al. Minimal residual disease-based effect and long-term outcome of first-line dasatinib combined with chemotherapy for adult Philadelphia chromosome-positive acute lymphoblastic leukemia. Ann Oncol. 2016;27(6):1081–8. [DOI] [PubMed] [Google Scholar]
- 42.Ribera JM, Garcia-Calduch O, Ribera J, Montesinos P, Cano-Ferri I, Martinez P, et al. Ponatinib, chemotherapy, and transplant in adults with Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood Adv. 2022;6(18):5395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uitdehaag JC, de Roos JA, van Doornmalen AM, Prinsen MB, de Man J, Tanizawa Y, et al. Comparison of the cancer gene targeting and biochemical selectivities of all targeted kinase inhibitors approved for clinical use. PLoS One. 2014;9(3):e92146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26(1):127–32. [DOI] [PubMed] [Google Scholar]
- 45.Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig PA, et al. The target landscape of clinical kinase drugs. Science. 2017;358(6367). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ishida Y, Murai K, Yamaguchi K, Miyagishima T, Shindo M, Ogawa K, et al. Pharmacokinetics and pharmacodynamics of dasatinib in the chronic phase of newly diagnosed chronic myeloid leukemia. Eur J Clin Pharmacol. 2016;72(2):185–93. [DOI] [PubMed] [Google Scholar]
- 47.Abaza Y, Kantarjian H, Alwash Y, Borthakur G, Champlin R, Kadia T, et al. Phase I/II study of dasatinib in combination with decitabine in patients with accelerated or blast phase chronic myeloid leukemia. Am J Hematol. 2020;95(11):1288–95. [DOI] [PubMed] [Google Scholar]
- 48.Jeha S, Coustan-Smith E, Pei D, Sandlund JT, Rubnitz JE, Howard SC, et al. Impact of tyrosine kinase inhibitors on minimal residual disease and outcome in childhood Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer. 2014;120(10):1514–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zwaan CM, Rizzari C, Mechinaud F, Lancaster DL, Lehrnbecher T, van der Velden VH, et al. Dasatinib in children and adolescents with relapsed or refractory leukemia: results of the CA180–018 phase I dose-escalation study of the Innovative Therapies for Children with Cancer Consortium. J Clin Oncol. 2013;31(19):2460–8. [DOI] [PubMed] [Google Scholar]
- 50.Gong X, Li L, Wei H, Liu B, Zhou C, Zhang G, et al. A Higher Dose of Dasatinib May Increase the Possibility of Crossing the Blood-brain Barrier in the Treatment of Patients With Philadelphia Chromosome-positive Acute Lymphoblastic Leukemia. Clin Ther. 2021;43(7):1265–71 e1. [DOI] [PubMed] [Google Scholar]
- 51.Aplenc R, Blaney SM, Strauss LC, Balis FM, Shusterman S, Ingle AM, et al. Pediatric phase I trial and pharmacokinetic study of dasatinib: a report from the children’s oncology group phase I consortium. J Clin Oncol. 2011;29(7):839–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dorer DJ, Knickerbocker RK, Baccarani M, Cortes JE, Hochhaus A, Talpaz M, et al. Impact of dose intensity of ponatinib on selected adverse events: Multivariate analyses from a pooled population of clinical trial patients. Leuk Res. 2016;48:84–91. [DOI] [PubMed] [Google Scholar]
- 53.Lipton JH, Chuah C, Guerci-Bresler A, Rosti G, Simpson D, Assouline S, et al. Ponatinib versus imatinib for newly diagnosed chronic myeloid leukaemia: an international, randomised, open-label, phase 3 trial. Lancet Oncol. 2016;17(5):612–21. [DOI] [PubMed] [Google Scholar]
- 54.Jabbour E, Short NJ, Jain N, Huang X, Montalban-Bravo G, Banerjee P, et al. Ponatinib and blinatumomab for Philadelphia chromosome-positive acute lymphoblastic leukaemia: a US, single-centre, single-arm, phase 2 trial. Lancet Haematol. 2023;10(1):e24–e34. [DOI] [PubMed] [Google Scholar]
- 55.Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol. 2008;15(10):1109–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole genome sequencing data is deposited with the European Genome-Phenome Archive (accession number: EGAD00001006434). To request reagents included in this work, please contact Dr. Jun J. Yang (jun.yang@stjude.org).
Details of other experiments (e.g., ex vivo drug sensitivity assay for dasatinib, ponatinib and saracatinib, LC-MS for dasatinib and ponatinib) are provided in the Supplemental Methods.