
Figure 2.
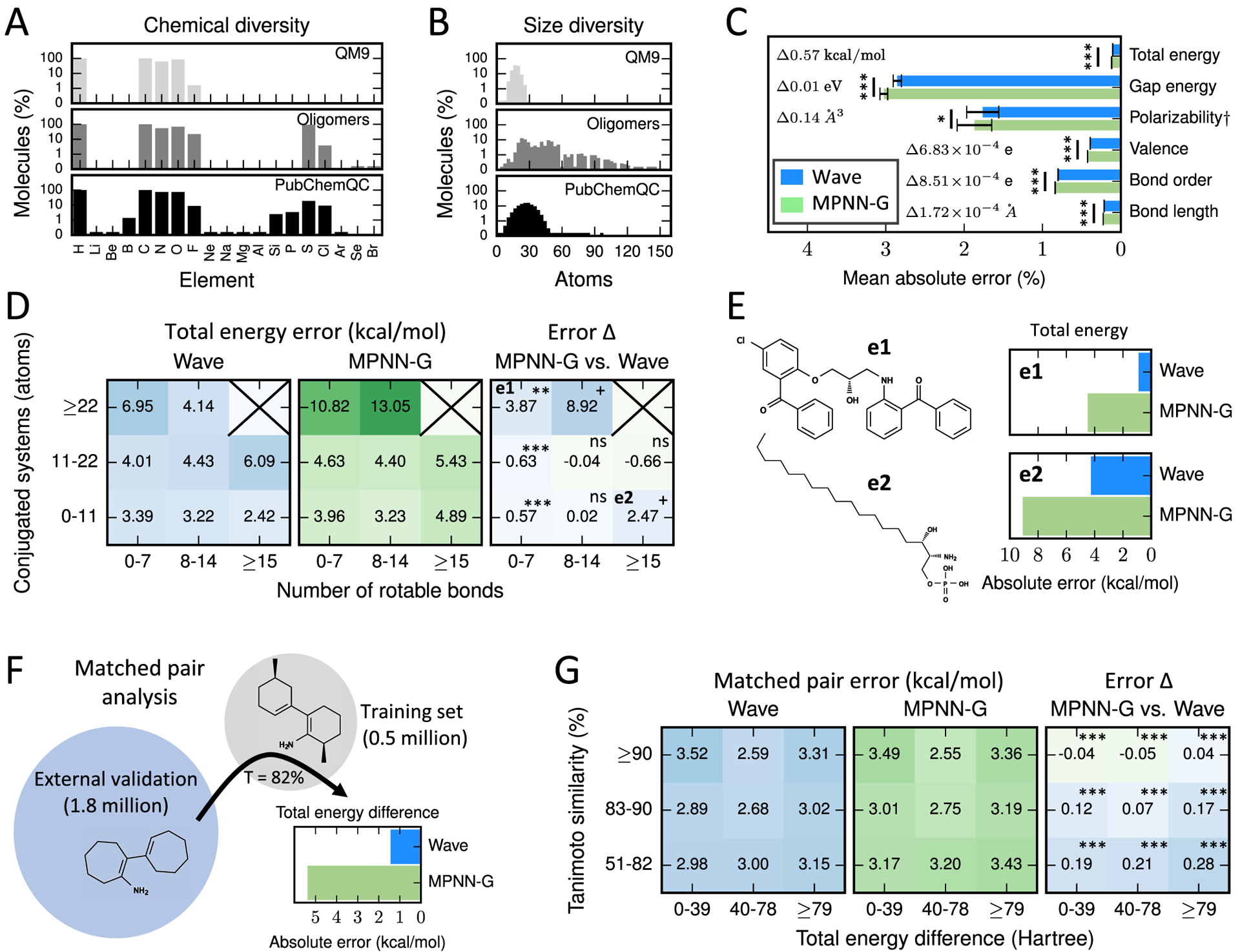
Wave enables more accurate coordinate-free calculation of QC properties across diverse molecules. (A) PubChemQC and Oligomer datasets used in this study cover a substantially larger number of atom types and (B) include substantially larger molecules than QM9. (C) Wave was slightly more accurate than MPNN-G on the six QC properties included in this study. (D) Wave exhibits substantially lower error compared to MPNN-G when calculating total energy for molecules with large conjugated systems and also outperformed MPNN-G on large, flexible molecules. (E) Example molecules on which Wave achieves lower absolute error on total energy (kcal/mol) compared to MPNN-G with (e1) many atoms in conjugated systems and (e2) many rotable bonds. (F) Matched pairs were selected by choosing topologically similar molecules from a large external validation set. (G) Coordinate-free methods exhibit a small increase in error when computing the difference in total energy between matched pairs of molecules. Wave slightly outperformed MPNN-G on this task. Statistical tests were performed by paired t-test. +: p < 0.1, *: p < 0.05, **: p < 0.01, and ***: p < 0.001.