

Abstract
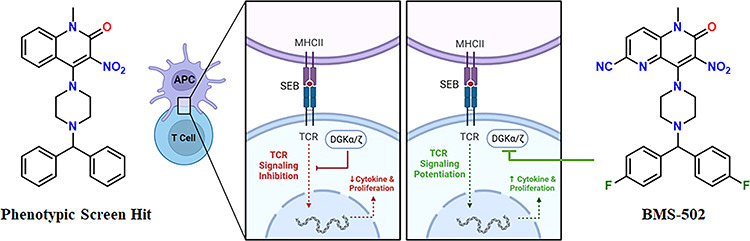
We describe a phenotypic screening and optimization strategy to discover compounds that block intracellular checkpoint signaling in T-cells. We identified dual DGKα and ζ inhibitors notwithstanding the modest similarity between α and ζ relative to other DGK isoforms. Optimized compounds produced cytokine release and T-cell proliferation consistent with DGK inhibition and potentiated an immune response in human and mouse T-cells. Additionally, lead inhibitor BMS-502 demonstrated dose-dependent immune stimulation in the mouse OT-1 model, setting the stage for a drug discovery program.
Keywords: diacylglycerol, phenotypic, DGK, checkpoint inhibitor
Diacylglycerol kinases (DGKs) phosphorylate diacylglycerols (DAGs), generating phosphatidic acids (PAs) (Figure 1A).1 DAG is produced upon T-cell receptor (TcR) stimulation, promoting proliferation and release of interleukin 2 (IL2) and interferon γ (IFNγ).2 DAG phosphorylation is an intracellular checkpoint that modulates this pathway to suppress the immune response,3,4 analogous to the extracellular checkpoints CTLA4 and PD-1.5 Extracellular checkpoint inhibition can effectively treat certain cancers where there is an insufficient immune response.6,7 Because the DGK isoforms α and ζ predominate in T-cells,8,9 it has been hypothesized that α and/or ζ inhibition could overcome immune system suppression in a similar fashion, possibly in combination with extracellular checkpoint inhibitors.10,11 Our discovery and optimization of DGK inhibitors was unanticipated and indirect because it was driven exclusively by phenotypic assays.12,13
Figure 1.

(A) Diacylglycerol phosphorylation. (B) TcR engagement leads to IL2 production. DGK interrupts this pathway by depletion of DAG.
A phenotype-driven approach finds molecules acting through unanticipated mechanisms and captures interactions with biomolecules in a native cellular environment, which is challenging to replicate using target-based biochemical assays. A compound’s impact on protein–protein interactions, post-translational modifications, or protein localization into different cellular compartments can result in the phenotype deviating from the expected outcome suggested by a biochemical assay.13,14 Conversely, phenotypic screening’s holistic nature provides chemical matter that has surmounted hurdles that often confound translation from biochemical to whole-cell activity. Given these attributes, a strategy was designed to identify compounds acting upon intracellular checkpoints, agnostic of specific molecular mechanisms of action.12 Authentic leads were desired to initiate a drug discovery program without bias toward specific targets. To accomplish this objective, over one million compounds were evaluated in primary human CD4+ T-cells stimulated with agonist antibodies for the TcR. Compounds that amplified proliferation in this high-throughput assay were optimized using a two-cell assay wherein CD4+ T-cells were stimulated by B-cells (Raji) presenting staphylococcus enterotoxin B (SEB) on the B-cell major histocompatibility complex (MHCII) (Figure 2.). SEB induces interaction between cells through the TcR and the MHCII, leading to secretion of IL2. When Raji cells expressed PD-L1, active compounds overcame the negative regulation of the PD-1/PD-L1 pathway (Figure 2A). A counter-screen with Raji cells lacking PD-L1 (Figure 2B) selected for compounds that overcame, but acted independently of, the PD-1/PD-L1 pathway.
Figure 2.

(A) Assay format using the SEB-mediated T-cell interaction with Raji cells to stimulate IL2 production. (B) Counter-screen without PD-L1 presentation. .
This screening strategy identified quinolone 1 (BMS-684, Table 1) with EC50 values of 2–4 μM and structural attributes that make it a synthetically tractable starting point for a medicinal chemistry program. Retrosynthetically, the compound could be divided to apply parallel synthesis methodology, allowing for rapid exploration of the structure–activity relationships (SARs). BMS-684 demonstrated favorable metabolic stability in human and mouse liver microsomal preparations (Table 1).15,16 It exhibited promising membrane permeability of 670 nm/sec at pH 5 and 952 nm/sec at pH 7, as measured in a parallel artificial membrane permeability assay. A kinase panel, designed to identify hinge binders, revealed no noteworthy binding to the 327 protein kinases tested at concentrations of 15 μM.17 At a concentration of 30 μM, BMS-684 exhibited no activity in a safety screening panel consisting of 11 ion channels, 3 transporters, 5 enzymes, 4 nuclear hormone receptors, and 19 G-protein-coupled receptors, including serotonin. When these assays were conducted, we were unaware that DGK was the biochemical target and did not fully appreciate the significance of our selectivity versus the serotonin receptors. Ritanserin,18 an extensively studied DGKα inhibitor, is a potent 5-HT2A and 5-HT2c receptor antagonist. The profile of BMS-684 suggested that it was not a pan-assay interference compound,19 and we set out to explore the SAR with four goals in mind. (1) We aimed to identify compounds with enhanced potency while maintaining metabolic stability, which would support in vivo characterization of the phenotype. (2) We wanted to demonstrate rational optimization using the cellular assay, considering the uncertainty surrounding the timeline for target identification and the reliance on cell-based SAR. (3) We sought potent photoaffinity probes that would facilitate target tagging and enrichment using established techniques. (4) Concerned about the potential formation of reactive metabolites and the associated risk of idiosyncratic toxicity,20−22 we desired alternatives to the nitro group. However, nitro group replacement was not considered crucial for fully characterizing the phenotype and identifying the biochemical target.
Table 1. Potency and Metabolic Stability Dataa.

| Compound |
RAJI EC50 μM (SD, n) |
Met. stab. % remaining |
|||
|---|---|---|---|---|---|
| R1 | PD-L1 positive | PD-L1 negative | MLMs | HLMs | |
| 1 (BMS-684) | 3.2 (4.31, 164) | 2.4 (1.84, 94) | 82 | 93 | |
| 2 | 1.5 (0.81, 3) | 3.7 (1.28, 3) | 84 | 86 | |
| 3 | 1.5 (0.41, 3) | 2.5 (0.80, 3) | 8 | 43 | |
| 4 | >100 | >100 | 6 | 28 | |
| 5 | >100 | >100 | |||
| 6 | 6.39 (3.05, 2) | 9.28 (3.07, 5) | 4 | 25 | |
| 7 | –H | >5 | >16 | 63 | 93 |
| 8 | –C(O)CF3 | 7.1 (5.34, 2) | 5.8 (5.16, 2) | 63 | 75 |
| 9 | –CO2Et | 4.1 (1.3, 2) | 20 (18.66, 2) | 52 | 49 |
| 10 | –CN | 1.37 (0.07, 3) | 1.24 (0.94, 3) | ||
| 11 | –C(O)CH3 | 8.5 (0.16, 2) | 13 (2.84, 2) | ||
| 12 | –CO2H | >100 | >100 | ||
| 13 | >33 | >100 | |||
| 14 | >100 | >100 | |||
SD: standard deviation, n: number of test occasions. MLMs: mouse liver microsomes, HLMs: human liver microsomes.
Initial modifications focused on the benzhydryl moiety (Table 1). Polar groups, particularly hydrogen-bond donors and acceptors, were poorly tolerated, providing no IL2 production up to 100 μM concentrations (not shown). Interestingly, benzyl compound 3 had similar potency to BMS-684 but exhibited poor metabolic stability. The addition of a methylene spacer, 4, or the replacement of the benzhydryl moiety with an ethyl substituent, 5, resulted in the loss of all activity. Replacing one of the phenyl rings with a methyl group, 6, resulted in a slight loss of targeted activity but provided poor metabolic stability. Phenyl ring fluorination resulted in the identification of 2 with similar activity to BMS-684. As the program progressed, this motif was retained in many subsequent analogues, as it consistently conferred improved metabolic stability compared to the unsubstituted prototype. With these results in hand, we chose to maintain the benzhydryl piperazine and explore the quinolone ring SARs.
Nitro group (R1) replacements and modifications of the N-1 substituent were investigated, as presented in Table 1. Replacing the 3-nitro substituent with a hydrogen atom, 7, led to a complete loss of activity. Partial activity was preserved when the nitro group was replaced with electron-withdrawing substituents, as observed in compounds 8–11.
The 3-nitrile compound 10 had similar activity to 2. In contrast, activity was lost with the carboxylic acid 12, despite its potential to directly mimic the nitro group. Anticipating that further examination of nitro group replacements could be conducted in an intrinsically more potent structural background, the SAR survey shifted focus to N-1 substituents. It was found that this position exhibited high sensitivity toward elaboration, as unsubstituted 13 and ethyl 14 compounds were inactive. The ensuing modifications examined substitution at positions 5–8 (Table 2). This endeavor delivered the first notable potency improvement, with 6-fluoro 15 and 6-cyano 18. Additionally, an improvement was observed for the 6-methoxy 24 and 6-dimethylamino 25 derivatives. However, larger alkoxy or amino groups at the 6-position resulted in diminished activity and poor metabolic stability. Functionalizing the remaining carbocyclic positions, such as 7-fluoro derivative 16, did not improve activity.
Table 2. SARs for Modifying Positions 5–8a.

| Compound | RAJI EC50 μM (SD, n) |
Met. stab. % remaining |
||
|---|---|---|---|---|
| PD-L1 positive | PD-L1 negative | MLMs | HLMs | |
| 15 | 0.34 (0.70, 13) | 0.49 (1.02, 15) | 77 | 89 |
| 16 | 2.6 (1.25, 2) | 1.8 (0.64, 2) | ||
| 17 | 0.61 (0.26, 7) | 1.1 (0.67, 7) | 84 | |
| 18 | 0.13 (0.05, 11) | 0.12 (0.07, 11) | 93 | 97 |
| 19 | 0.56 (0.20, 4) | 0.84 (0.32, 4) | 63 | 100 |
| 20 | 7.7 (NA, 1b) | 13 (NA, 1b) | ||
| 21 | 0.29 (0.10, 5) | 0.31 (0.20, 5) | 88 | 92 |
| 22 (BMS-502) | 0.037 (0.03, 13) | 0.037 (0.05, 12) | 99 | 90 |
| 23 | 0.19 (0.27, 5) | 0.067 (0.03, 4) | 80 | 92 |
| 24 | 0.63 (0.17, 4) | 1.2 (0.66, 4) | 8 | 79 |
| 25 | 0.67 (NA, 1b) | 0.76 (NA, 1b) | 5 | 69 |
| 26 | 0.39 (NA, 1b) | 0.40 (NA, 1b) | 43 | 100 |
| 27 | 0.42 (0.32, 5) | 0.37 (0.17, 5) | <1 | 10 |
SD: standard deviation, n: number of test occasions. MLMs: mouse liver microsomes, HLMs: human liver microsomes.
N = 1 data were used when equivalent activity was observed in the PD-L1 positive and negative formats and the compounds were not pursued due to poor activity or metabolic stability.
Considering that the most potent analogues feature an electron withdrawing group on the quinolone ring, we endeavored to imitate this effect using the naphthyridinones 19–21. The most potent isomer was 21, exhibiting an 8-fold improvement over BMS-684. Ultimately, the merging of an electron withdrawing substituent at C-6 with a napthyridinone ring resulted in 22 (BMS-502), which had an EC50 value less than 100 nM and excellent metabolic stability. With these improvements, only a minor potency loss occurred when cyano replaced nitro, as exemplified by comparing naphthyridinones 23 and BMS-502. In this potent structural backdrop, we introduced a photolabile diazirine and a terminal alkyne, 27, with satisfactory potency to be used for photolabeling and enrichment of the biological target. However, photoaffinity probes failed to significantly enrich likely candidates. Instead, DGKα and ζ were identified through a lipid-probe chemoproteomics strategy, confirmed by knockout studies, enzyme inhibition, cellular thermal shift, and a novel finding that compounds induce DGKα and ζ translocation to the plasma membrane.12
At this stage, the overall progression from lead BMS-684 to BMS-502 represented an 80-fold enhancement of potency, along with improved in vitro metabolic stability. This optimization process took place prior to establishing the connection between cellular activity and DGK inhibition. A retrospective analysis of DGK inhibition revealed that BMS-684 exhibited selectivity for DGKα, whereas BMS-502 demonstrated substantial inhibitory activity against DGKs α, ζ and ι (Table 3).
Table 3. BMS-502 and BMS-684 Compared to Published DGK Inhibitorsa.

| Compound | Diacylglycerol kinase
IC50 (μM) |
Human
whole-blood EC50 (μM) |
CD8+ proliferation EC50 (μM) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| α | β | γ | κ | ζ | ι | IFNγ | pERK | ||
| BMS-684 | 0.015 | 2.5 | 1.9 | >10 | >10 | >10 | >20 | >20 | |
| BMS-502 | 0.0046 | 1.0 | 0.68 | 4.6 | 0.0021 | 0.0026 | 0.28 | 0.52 | 0.065 |
| 18 | 0.034 | 10 | 6.2 | ||||||
| 21 | 0.053 | 2.7 | 1.0 | ||||||
| ritanserin | >5 | >10 | >10 | >10 | >10 | >20 | >10 | ||
| R59949 | 4.3 | >10 | 3.4 | >10 | >10 | >10 | >10 | ||
| CU-3 | 0.6 | ||||||||
The 6-nitrile 18 and naphthyridinone 21 were tested to reveal the contributions of these molecular edits to ζ and ι inhibition. The results presented in Table 3 indicate that the change from carbon to nitrogen in 21 had a more significant impact on the enhanced inhibition of ζ and ι. Furthermore, the combination of these structural changes exhibited an unexpected synergy, resulting in the low nanomolar inhibition displayed by BMS-502.
At this stage, we had successfully optimized the chemotype in a rational manner by employing combinations of preferred groups. In Table 3, the in vitro characterization data for BMS-684 and BMS-502 are compared with published DGKα inhibitors. Among the 10 known DGK isoforms, our compounds exhibited significant selectivity for DGKs α, ζ, and ι with limited inhibitory activity toward the β, γ, and κ isoforms. No significant inhibitory activity observed for the δ, η, ε, and θ isoforms. Consequently, the cellular-driven optimization process had incorporated DGK ζ and ι inhibition into the chemotype. DGKs α and ζ are expressed in T-cells, while ι is not, consistent with knockout studies revealing that the cellular activity of BMS-502 depends on α and ζ.12 Based on these findings, we concluded that the dual α and ζ inhibition was driving the phenotypic response.
We further profiled compounds in human whole-blood (hWB) measuring IFNγ production or phosphorylation of extracellular signal-regulated kinase (pERK, Figure 1B). It was expected that pERK levels should increase with increasing DGK inhibition.4BMS-502 demonstrated significant activity in both of these assays, while ritanserin and R59949 were inactive at 20 μM. Also, an EC50 value of 340 nM for BMS-502 was measured in mouse cytotoxic T-cell IFNγ assay (mCTC) that compares well with the hWB IFNγ EC50 of 280 nM. Separately, proliferation of human effector CD8+ T-cells was measured as a marker of immune stimulation. Again, while BMS-502 exhibited an EC50 of 65 nM in the proliferation assay, ritanserin and R59949 had negligible activity at 10 μM. These results were consistent with our compounds inhibiting DGKα, and ζ mediated DAG depletion to potentiate an immune response initiated by TcR activation. With this promising in vitro data in hand, we were encouraged to assess BMS-502’s potential in vivo.
The mouse pharmacokinetic parameters for BMS-502 are presented in Figure 3. The compound exhibited favorable oral bioavailability, low clearance, and a long half-life. The measured plasma concentration for the 5 mg/kg dose remained above the mCTC for 72 h, indicating that potentially efficacious exposures could be maintained after a single oral dose. An OT-1 mouse model was chosen to determine the increase in activated effector T-cells in response to oral dosing. The OT-1 mouse responds to SIINFEKL peptide antigen with an increase in activated effector T-cells, the percent of CD8+ T-cells that are also CD69+ in the spleen. Without the SIINFEKL antigen, a 10 mpk dose of BMS-502 caused no significant increase in activated effector T-cells (Figure 3B). However, when the SIINFEKL antigen and BMS-502 were administered simultaneously, increasing doses of BMS-502 provided a dose-dependent increase in effector T-cells (Figure 3B).
Figure 3.

(A) BMS-502 pharmacokinetic profile in C57 black mice. (B) BMS-502 dose response in the OT1 model. %CD69+ T-cells = 100(CD69+ CD8s)/total CD8s in the spleen. (C) Pharmacokinetic parameters for BMS-502.
We have described the optimization of a phenotypic screening hit using whole-cell assays to provide potent DGKα and ζ inhibitors and connected the cellular effects to DGK inhibition as reported elsewhere.12 The resulting compounds demonstrated potency in an in vivo model of immune stimulation. Surprisingly, using only the CD4+/Raji cellular format and beginning with a selective type 1 DGK inhibitor (α, β, and γ) we built in type 4 DGK inhibition (ζ and ι). This result is unexpected in light of the usual DGK isoform classification into five types according to the presence of additional functional domains, substrate specificity, and the observation that they show lower sequence identity within shared domains outside their respective groups (Figure 4).1,23−25
Figure 4.
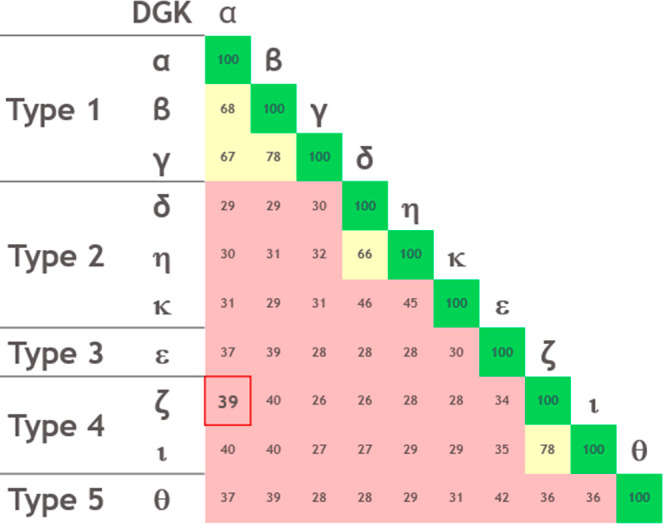
Percent identity of DGK isoforms in catalytic and accessory domains grouped by type.
DGKs α, β, and γ share approximately 67% sequence identity in the catalytic domain and are considered closely related. Conversely, α and ζ share 39% sequence identity in the catalytic domain. Accordingly, it seems unlikely that given an α-selective inhibitor, one would expect to design in ζ inhibition. To the best of our knowledge, these compounds are the first inhibitors targeting both DGKα and ζ.26−31 Due to the lack of structural information, we are unable to speculate on the mechanism of dual inhibition.32,33 Regardless, we have successfully selected for compounds with a unique and desirable profile that represents significant progress in the field of DGK inhibition.
In conclusion, our study has uncovered potent and selective inhibitors of DGKα and ζ through the utilization of a phenotypic screening and optimization strategy. We have successfully showcased the capability of a cellular assay to drive the evolution of lead compounds in valuable and unforeseen directions. Despite the modest similarity between these enzymes, our cellular assay identified dual DGK α and ζ inhibitors, highlighting its effectiveness in identifying compounds with unique profiles. These inhibitors exhibited cellular effects consistent with DGK inhibition and potentiated an in vitro immune response. Additionally, our selection process prioritized excellent microsomal stability, which translated into a long half-life for BMS-502 in vivo. The exposures achieved exceeded the mCTC EC50 for 72 h and elicited an immune response in the mouse OT-1 model. Importantly, BMS-502 exhibited little or no effect in the absence of SIINFEKL peptide antigen. This indicates that BMS-502 does not directly stimulate T-cells; instead, it amplifies inadequate stimulation provided by suboptimal SIINFEKL antigen concentrations.
DGKs have been implicated in tumor initiation, progression, and metastasis, with DGKα and DGKζ shown to promote tumor cell growth in hepatocellular carcinoma, osteosarcoma, melanoma, and glioblastoma.34−38 Furthermore, DAG levels orchestrate T cell function and perform a key role in tumor immunosurveillance.39 Therefore, DGK inhibitors hold the potential to significantly impact immune checkpoint modulation, adoptive T-cell-based immunotherapy, and direct inhibition of tumor growth. The discovery of these compounds and the accompanying data represent a substantial advancement in the field of DGK inhibition, opening new avenues for studying DGKs as promising therapeutic targets.
Acknowledgments
The table of contents graphic was created with BioRender.com.
Glossary
Abbreviations
- DGK
diacylglycerol kinase
- DAG
diacylglycerol
- PA
phosphatidic acid
- IL2
interleukin 2
- IFNγ
interferon γ
- SEB
staphylococcus enterotoxin B
- MHC
major histocompatibility complex
- SAR
structure activity relationship
- MLM
mouse liver microsome
- HLM
human liver microsome
- ERK
extracellular signal-regulated kinase
- pERK
phosphorylated extracellular signal-regulated kinase
- mCTC
mouse cytotoxic T-cell assay
- hWB
human whole-blood
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00063.
Compound synthesis, characterization, and assay protocols (PDF)
Author Contributions
This manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was funded by Bristol Myers Squibb.
The authors declare no competing financial interest.
Supplementary Material
References
- Shulga Y. V.; Topham M. K.; Epand R. M. Regulation and functions of diacylglycerol kinases. Chem. Rev. 2011, 111 (10), 6186–208. 10.1021/cr1004106. [DOI] [PubMed] [Google Scholar]
- Sim J. A.; Kim J.; Yang D. Beyond lipid signaling: pleiotropic effects of diacylglycerol kinases in cellular signaling. Int. J. Mol. Sci. 2020, 21 (18), 6861. 10.3390/ijms21186861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco S.; Merida I. Diacylglycerol-dependent binding recruits PKCθ and RasGRP1 C1 domains to specific subcellular localizations in living T lymphocytes. Mol. Biol. Cell 2004, 15 (6), 2932–2942. 10.1091/mbc.e03-11-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riese M. J.; Grewal J.; Das J.; Zou T.; Patil V.; Chakraborty A. K.; Koretzky G. A. Decreased Diacylglycerol Metabolism Enhances ERK Activation and Augments CD8+ T Cell Functional Responses. J. Biol. Chem. 2011, 286 (7), 5254–5265. 10.1074/jbc.M110.171884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledzinska A.; Menger L.; Bergerhoff K.; Peggs K. S.; Quezada S. A. Negative immune checkpoints on T lymphocytes and their relevance to cancer immunotherapy. Mol. Oncol. 2015, 9 (10), 1936–1965. 10.1016/j.molonc.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J.; Chehrazi-Raffle A.; Reddi S.; Salgia R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J. Immunother Cancer 2018, 6 (1), 8. 10.1186/s40425-018-0316-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehdizadeh S.; Bayatipoor H.; Pashangzadeh S.; Jafarpour R.; Shojaei Z.; Motallebnezhad M. Immune checkpoints and cancer development: Therapeutic implications and future directions. Pathol., Res. Pract. 2021, 223, 153485. 10.1016/j.prp.2021.153485. [DOI] [PubMed] [Google Scholar]
- Gu J.; Wang C.; Cao C.; Huang J.; Holzhauer S.; Desilva H.; Wesley E. M.; Evans D. B.; Benci J.; Wichroski M.; Wee S.; Riese M. J. DGKζ exerts greater control than DGKα over CD8+ T cell activity and tumor inhibition. OncoImmunology 2021, 10 (1), 1941566. 10.1080/2162402X.2021.1941566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna S.; Zhong X.-P. Role of diacylglycerol kinases in T cell development and function. Crit. Rev. Immunol. 2013, 33 (2), 97–118. 10.1615/CritRevImmunol.2013006696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitaram P.; Uyemura B.; Malarkannan S.; Riese M. J. Beyond the cell surface: targeting intracellular negative regulators to enhance T cell anti-tumor activity. Int. J. Mol. Sci. 2019, 20 (23), 5821. 10.3390/ijms20235821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao S.; Akiyama R.; Sakane F. Combined inhibition/silencing of diacylglycerol kinase alpha and zeta simultaneously and synergistically enhances interleukin-2 production in T cells and induces cell death of melanoma cells. J. Cell. Biochem. 2021, 122 (5), 494–506. 10.1002/jcb.29876. [DOI] [PubMed] [Google Scholar]
- Wichroski M.; Benci J.; Liu S.; Chupak L.; Fang J.; Cao C.; Wang C.; Russo J.; Qiu H.; Shan Y.; Banas D.; Powles R.; Locke G.; Witt A.; Stromko C.; Zheng X.; Martin S.; Ding M.; Gentles R.; Meanwell N. A.; Velaparthi U.; Olson R.; Wee S.; Tenney D.; Parker C. G.; Cravatt B. F.; Lawrence M.; Borzilleri R.; Lees E.. Discovery Of Novel DGKa/z Inhibitors That Combine With PD-1 Checkpoint Therapy To Induce Robust T Cell-Mediated Anti-Tumor Immunity. Science Translational Medicine 2023, submitted. [DOI] [PubMed] [Google Scholar]
- Vincent F.; Nueda A.; Lee J.; Schenone M.; Prunotto M.; Mercola M. Phenotypic drug discovery: recent successes, lessons learned and new directions. Nat. Rev. Drug Discovery 2022, 21 (12), 899–914. 10.1038/s41573-022-00472-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinney D. C. Phenotypic vs. Target-Based Drug Discovery for First-in-Class Medicines. Clin. Pharmacol. Ther. (N. Y., NY, U. S.) 2013, 93 (4), 299–301. 10.1038/clpt.2012.236. [DOI] [PubMed] [Google Scholar]
- Kieltyka K.; Zhang J.; Li S.; Vath M.; Baglieri C.; Ferraro C.; Zvyaga T. A.; Drexler D. M.; Weller H. N.; Shou W. Z. A high-throughput bioanalytical platform using automated infusion for tandem mass spectrometric method optimization and its application in a metabolic stability screen. Rapid Commun. Mass Spectrom. 2009, 23 (11), 1579–1591. 10.1002/rcm.4037. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Vath M.; Ferraro C.; Li Y.; Murphy K.; Zvyaga T.; Weller H.; Shou W. A high-speed liquid chromatography/tandem mass spectrometry platform using multiplexed multiple-injection chromatography controlled by single software and its application in discovery ADME screening. Rapid Commun. Mass Spectrom. 2013, 27 (7), 731–737. 10.1002/rcm.6514. [DOI] [PubMed] [Google Scholar]
- PubChem. Bioassay Record for AID 1258054. Source: ChEMBL. 2023.
- Boroda S.; Niccum M.; Raje V.; Purow B. W.; Harris T. E. Dual activities of ritanserin and R59022 as DGKα inhibitors and serotonin receptor antagonists. Biochem. Pharmacol. (Amsterdam, Neth.) 2017, 123, 29–39. 10.1016/j.bcp.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B.; Nissink J. W. M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations. ACS Chem. Biol. 2018, 13 (1), 36–44. 10.1021/acschembio.7b00903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsayed M. S. A.; Su Y.; Wang P.; Sethi T.; Agama K.; Ravji A.; Redon C. E.; Kiselev E.; Horzmann K. A.; Freeman J. L.; Pommier Y.; Cushman M. Design and Synthesis of Chlorinated and Fluorinated 7-Azaindenoisoquinolines as Potent Cytotoxic Anticancer Agents That Inhibit Topoisomerase I. J. Med. Chem. 2017, 60 (13), 5364–5376. 10.1021/acs.jmedchem.6b01870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostinski Y.; Heynen G. J. J. E.; Lopez-Alberca M. P.; Paul J.; Miksche S.; Radetzki S.; Schaller D.; Shanina E.; Seyffarth C.; Kolomeets Y.; Ziebart N.; de Schryver J.; Oestreich S.; Neuenschwander M.; Roske Y.; Heinemann U.; Rademacher C.; Volkamer A.; von Kries J. P.; Birchmeier W.; Nazare M. From Pyrazolones to Azaindoles: Evolution of Active-Site SHP2 Inhibitors Based on Scaffold Hopping and Bioisosteric Replacement. J. Med. Chem. 2020, 63 (23), 14780–14804. 10.1021/acs.jmedchem.0c01265. [DOI] [PubMed] [Google Scholar]
- Tseng C.-C.; Baillie G.; Donvito G.; Mustafa M. A.; Juola S. E.; Zanato C.; Massarenti C.; Dall’Angelo S.; Harrison W. T. A.; Lichtman A. H.; Ross R. A.; Zanda M.; Greig I. R. The Trifluoromethyl Group as a Bioisosteric Replacement of the Aliphatic Nitro Group in CB1 Receptor Positive Allosteric Modulators. J. Med. Chem. 2019, 62 (10), 5049–5062. 10.1021/acs.jmedchem.9b00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakubchyk Y.; Abramovici H.; Maillet J. C.; Daher E.; Obagi C.; Parks R. J.; Topham M. K.; Gee S. H. Regulation of neurite outgrowth in N1E-115 cells through PDZ-mediated recruitment of diacylglycerol kinase zeta. Mol. Cell. Biol. 2005, 25 (16), 7289–302. 10.1128/MCB.25.16.7289-7302.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai Y.; Ikeda M.; Saito N. Regulation of diacylglycerol kinase by phosphorylation. Adv. Biol. Regul 2012, 52 (1), 239–47. 10.1016/j.advenzreg.2011.09.004. [DOI] [PubMed] [Google Scholar]
- Ware T. B.; Franks C. E.; Granade M. E.; Zhang M.; Kim K.-B.; Park K.-S.; Gahlmann A.; Harris T. E.; Hsu K.-L. Reprogramming fatty acyl specificity of lipid kinases via C1 domain engineering. Nat. Chem. Biol. 2020, 16 (2), 170–178. 10.1038/s41589-019-0445-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granade M. E.; Lemke M. C.; Manigat L. C.; Purow B. W.; Harris T. E. Identification of ritanserin analogs that display DGK isoform specificity. Biochem. Pharmacol. 2022, 197, 114908. 10.1016/j.bcp.2022.114908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velnati S.; Massarotti A.; Antona A.; Talmon M.; Fresu L. G.; Galetto A. S.; Capello D.; Bertoni A.; Mercalli V.; Graziani A.; Tron G. C.; Baldanzi G. Structure activity relationship studies on Amb639752: toward the identification of a common pharmacophoric structure for DGKα inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35 (1), 96–108. 10.1080/14756366.2019.1684911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casarez A. D.; Coburn C. A.; Kellar T. A.; Buzard D. J.; Arora N.. Preparation of oxime compounds useful as T-cell activators and inhibitors of diacylglycerol kinases for the treatment and prevention of diseases. WO2021258010A1, 2021.
- Velnati S.; Ruffo E.; Massarotti A.; Talmon M.; Varma K. S. S.; Gesu A.; Fresu L. G.; Snow A. L.; Bertoni A.; Capello D.; Tron G. C.; Graziani A.; Baldanzi G. Identification of a novel DGKα inhibitor for XLP-1 therapy by virtual screening. Eur. J. Med. Chem. 2019, 164, 378–390. 10.1016/j.ejmech.2018.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chupak L. S.; Olson R. E.; Velaparthi U.; Zheng X.; Gentles R. G.; Ding M.; Warrier J. S.. Substituted quinolinonyl piperazine compounds useful as T-cell activators. WO2021133748, 2021.
- Chupak L. S.; Ding M.; Gentles R. G.; Huang Y.; Martin S. W.; McDonald I. M.; Mercer S. E.; Olson R. E.; Velaparthi U.; Wichroski M.; Zheng X.. Preparation of naphthyridinone compounds useful as T cell activators. WO2020006016, 2020.
- Takahashi D.; Suzuki K.; Sakamoto T.; Iwamoto T.; Murata T.; Sakane F. Crystal structure and calcium-induced conformational changes of diacylglycerol kinase α EF-hand domains. Protein Sci. 2019, 28 (4), 694–706. 10.1002/pro.3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q.; Srinivasan L.; Gabelli S. B.; Raben D. M. Elusive structure of mammalian DGKs. Adv. Biol. Regul 2022, 83, 100847. 10.1016/j.jbior.2021.100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X.; Wan G.; Chen N.; Li J.; Chen B.; Tang Y.; Gu W.; Jin C.; Meng J.; Zhang P.; Liu L.; Yang Z.; Lu C. DGKζ plays crucial roles in the proliferation and tumorigenicity of human glioblastoma. Int. J. Biol. Sci. 2019, 15 (9), 1872–1881. 10.7150/ijbs.35193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K.; Xue B.; Bai G.; Zhang W. Downregulation of Diacylglycerol kinase zeta (DGKZ) suppresses tumorigenesis and progression of cervical cancer by facilitating cell apoptosis and cell cycle arrest. Bioengineered 2021, 12 (1), 1517–1529. 10.1080/21655979.2021.1918505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeishi K.; Taketomi A.; Shirabe K.; Toshima T.; Motomura T.; Ikegami T.; Yoshizumi T.; Sakane F.; Maehara Y. Diacylglycerol kinase alpha enhances hepatocellular carcinoma progression by activation of Ras-Raf-MEK-ERK pathway. J. Hepatol. 2012, 57 (1), 77–83. 10.1016/j.jhep.2012.02.026. [DOI] [PubMed] [Google Scholar]
- Yamaki A.; Akiyama R.; Murakami C.; Takao S.; Murakami Y.; Mizuno S.; Takahashi D.; Kado S.; Taketomi A.; Shirai Y.; Goto K.; Sakane F. Diacylglycerol kinase α-selective inhibitors induce apoptosis and reduce viability of melanoma and several other cancer cell lines. J. Cell. Biochem. 2019, 120 (6), 10043–10056. 10.1002/jcb.28288. [DOI] [PubMed] [Google Scholar]
- Yu W.; Tang L.; Lin F.; Yao Y.; Shen Z. DGKZ Acts as a Potential Oncogene in Osteosarcoma Proliferation Through Its Possible Interaction With ERK1/2 and MYC Pathway. Front Oncol 2019, 8, 655. 10.3389/fonc.2018.00655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke M.; Kazanietz M. G. Overarching roles of diacylglycerol signaling in cancer development and antitumor immunity. Sci. Signaling 2022, 15 (729), eabo0264 10.1126/scisignal.abo0264. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.