

Abstract
Multiple mechanisms restrain inflammation in neonates, most likely to prevent tissue damage caused by overly robust immune responses against newly encountered pathogens. Here, we identify a population of pulmonary dendritic cells (DCs) that express intermediate levels of CD103 (CD103int) and appear in the lungs and lung–draining lymph nodes of mice between birth and two weeks of age. CD103int DCs express XCR1 and CD205, and require expression of the transcription factor BATF3 for development, suggesting that they belong to the cDC1 lineage. Additionally, CD103int DCs express CCR7 constitutively and spontaneously migrate to the lung–draining lymph node, where they promote stromal cell maturation and lymph node expansion. CD103int DCs mature independently of microbial exposure and TRIF– or MyD88–dependent signaling, and are transcriptionally related to efferocytic and tolerogenic DCs as well as mature, regulatory DCs. Correlating with this, CD103int DCs show limited ability to stimulate proliferation and IFNγ production by CD8+ T cells. Moreover, CD103int DCs acquire apoptotic cells efficiently, in a process that is dependent on the expression of the TAM receptor, Mertk, and which drives their homeostatic maturation. The appearance of CD103int DCs coincides with a temporal wave of apoptosis in developing lungs and explains, in part, dampened pulmonary immunity in neonatal mice. Taken together, these data suggest a mechanism by which dendritic cells sense apoptotic cells at sites of non-inflammatory tissue remodeling, such as tumors or the developing lungs, and limit local T cell responses.
Keywords: dendritic cell, lymph node stroma, neonatal immunity, pulmonary immunity, Mertk
One-sentence summary:
Mertk promotes maturation of regulatory dendritic cells in neonatal lungs.
INTRODUCTION.
Dendritic cells (DCs) are key antigen–presenting cells (1). In the lung, conventional DCs (cDCs) are divided into XCR1+CD103+ cDC1 cells and SIRPα+CD103− cDC2 cells that reside in different locations, acquire different antigens and prime different kinds of T cells (2, 3). For example, cDC1 cells reside beneath the respiratory epithelium, where they collect inhaled antigens as well as apoptotic cell–associated antigens, and potently cross–prime CD8+ T cells (4–6). In contrast, cDC2 cells reside in the lung parenchyma, but are biased towards priming CD4+ T cells (3, 7). Although both cDC1 and cDC2 cells are derived from common dendritic cell precursors (8), the development of cDC1 cells requires BATF3 and IRF8 (9–11), whereas the development of cDC2 cells requires the transcription factor IRF4 (11, 12).
Under homeostatic conditions, most DCs in the lung are immature and non–migratory (13). However, TLR signaling or exposure to inflammatory cytokines causes DC maturation and increases their expression of CCR7, which promotes migration to the lung–draining mediastinal lymph node (mLN), where they present captured and processed antigens to T cells (13–15). Migratory DCs also promote LN expansion by activating fibroblastic reticular cells (FRCs)(16, 17) and maturing high endothelial venules (HEVs)(18–20). This process enhances the recruitment of T cells from the blood through an expanded network of HEVs and provides space for T cell clonal expansion and differentiation (16, 19).
Although DCs activated by TLRs and inflammatory cytokines are typically associated with productive immunity, DCs activated by alternative mechanisms may be tolerogenic. For example, mechanical disruption of E-cadherin-mediated cell:cell contacts and β-catenin signaling promote sterile, non-inflammatory activation of DCs that help maintain physiological non–responsiveness (21, 22). Similarly, a population of homeostatically–activated, mature DCs in the thymus is thought to promote T cell tolerance rather than activation (23). Moreover, DCs in the intestinal lamina propia can be activated following efferocytosis of dying intestinal epithelial cells, leading to the upregulation of CCR7 and CD40, but also PD–L1, Aldh1a2 and TGFβ, which help maintain tolerance in intestinal CD8+ T cells (24, 25).
Similar types of DCs are found in tumors and are referred to as mature, regulatory DCs (mregDCs) or DC3 cells (26–29). Although the exact relationship between mregDCs and DC3 cells remains unclear, both cell types are activated (mature), and express high amounts of CCR7, IL–12b and CD40, in addition to immune regulatory molecules like PD–L1. Although some data suggest that mregDCs dampen CD8+ T cell responses and promote the differentiation of FoxP3–expressing Tregs (26), other data suggest that they help maintain CD8+ T cells in tumors via the production of CXCL16 and the trans–presentation of IL–15 (30) or that they promote the differentiation of CD103+ tissue–resident memory CD8+ T cells (28). The origins of mregDCs and DC3 cells are also unclear, as they seem to have mixed characteristics of both cDC1 and cDC2 cells (26, 30). In lung tumors, the accumulation of mregDCs is linked with signaling through Axl (26), a member of the TAM family of receptors that facilitates the clearance of apoptotic cells (31), suggesting that the process of efferocytosis may control the maturation of those cells. However, it is unclear whether mregDCs are normally found in the lung or how they might function.
Here we describe a novel population of DCs that appear in the lungs and lung–draining LNs of mice between birth and two weeks of age and express intermediate levels of CD103 (CD103int). CD103int cDCs require BATF3, suggesting that they are derived from bona fide cDC1 cells, but that they are constitutively activated and express high amounts of CCR7, CD40, IL–12b and PD–L1. Transcriptional analysis reveals that CD103int DCs closely resemble tolerogenic DCs (23), efferocytic DCs (24), and mregDCs (26). Maturation of CD103int DCs occurs independently of microbial exposure, IL–33, Trif– or MyD88–dependent signaling. However, CD103int DCs spontaneously migrate to the lung–draining mLN, where they promote the expansion of the FRC network and increase the number and size of HEVs. CD103int DCs express Mertk and Axl and robustly engulf apoptotic lung epithelial cells, which are abundant in the still-developing lungs of neonatal mice. Genetic deletion of Mertk impaired the accumulation of lung CD103int cDCs, reduced their expression of CCR7, CD40 and PD–L1, and improved the differentiation of IFNγ+ CD8 T cells responding to pulmonary antigens. Thus, the appearance of CD103int regulatory DCs coincided with a temporal wave of apoptosis as the lungs undergo alveolarization in the neonatal period. These data suggest that the process of lung remodeling activates a distinct maturation pathway in cDC1 cells and contributes to impaired pulmonary immunity in neonates.
RESULTS
CD103int DCs accumulate in the lungs and draining LNs of neonatal mice.
To determine whether populations of pulmonary DCs were altered in the post–natal period, we characterized DCs in the lungs of mice at various times after birth. After gating on B220−CD3− (Lineage)−CD64−CD11c+MHCII+ cells (gating strategies are in fig. S1), we enumerated DCs based on expression of CD11b, SIRPα and CD103. We found that the lungs of neonatal mice had CD11b+SIRPα+CD103− cDC2 cells as well as CD11b−SIRPα−CD103hi cDC1 cells (Fig. 1A). However, we also found a third subset of CD11b−SIRPα−CD103intermediate (CD103int) DCs (Fig. 1A). By frequency and numbers, the CD103int DCs were the most dominant population of DCs in the lung during the first week after birth, but declined thereafter, leaving a majority of SIRPα+ cDC2 cells and a smaller population of cDC1 cells (Fig. 1B–C). We also analyzed Lin−CD64−MHC+CD11c+ migratory DCs in the mLN and observed a similar expansion of CD103int DCs during the first two weeks after birth (Fig. 1D–F).
Figure 1. CD103int DCs accumulate in the lungs and mLNs of neonatal mice.
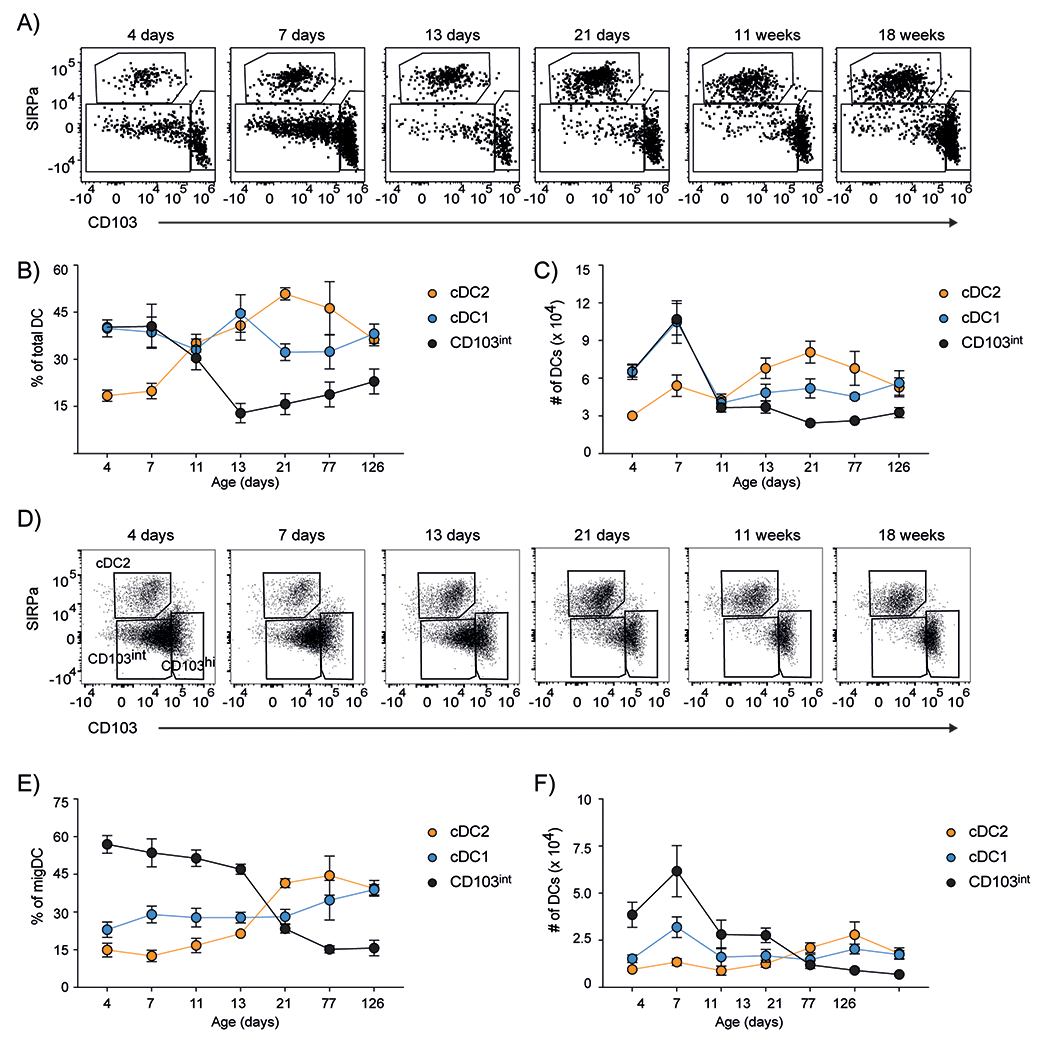
A. Representative plots (gated on Lin−CD64−MHCII+CD11c+ cells) of DCs in the lungs of mice at various times after birth. B. The frequencies of DC subsets, expressed as a proportion of total DCs determined by gating on Lin−CD64−MHCII+CD11c+ cells, in the lungs. C. Numbers of DC subsets in the lungs. D. Representative plots (gated on Lin−CD64−MHCII+CD11c+ cells) of DCs in the mLNs of mice at various times after birth. E. The frequencies of migratory DC (migDC) subsets in the mLN, expressed as a proportion of total migDCs. F. Numbers of DC subsets in the mLNs. Data are plotted as mean ± SD. This experiment was performed 5 times with 5 mice/timepoint on days 4, 7, 11 and 4 mice/timepoint on days 13, 21, 77 and 126.
CD103int DCs are related to DC1 cells.
To evaluate whether CD103int DCs were transcriptionally distinct from cDC1 and cDC2 cells, we sorted CD11b+SIRPα+CD103− cDC2 cells, CD11b−SIRPα−CD103hi cDC1 cells and CCR7+CD11b−SIRPα−CD103int CD103int DCs from the lungs of 7-day old mice (Fig. 2A) and performed RNAseq. As shown by principal component analysis, (Fig. 2B), CD103int DCs had a distinct pattern of gene expression distinct from either cDC1 or cDC2 cells. As expected, cDC1 cells had high expression of markers like CD103 (Itgae), Irf8 and Xcr1 (Fig. 2C middle panel), whereas cDC2 cells expressed markers like Irf4, CD11b (Itgam) and Sirpa (Fig. 2C top panel). In contrast, CD103int DCs expressed a combination of activation markers like Ccr7, PD-L1 (Cd274) and Cd40, that was not detected in the other DC subsets (Fig. 2C second panel).
Figure 2. CD103int DCs express markers of maturation.
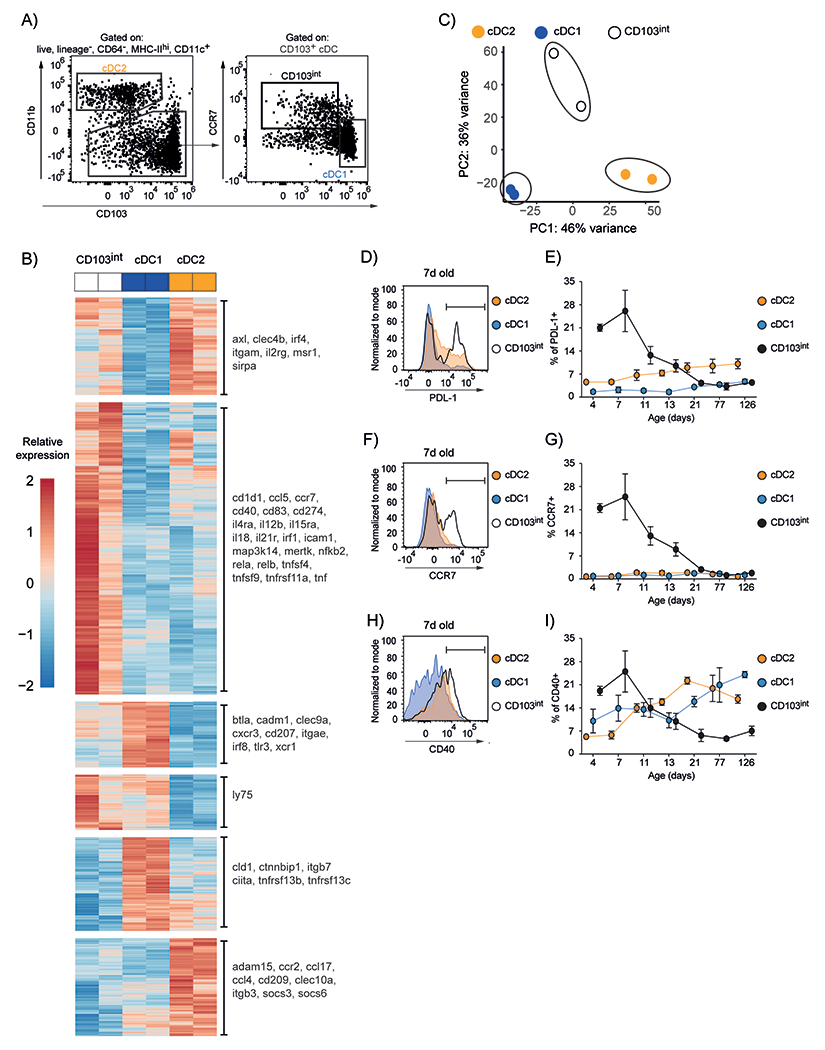
A. Gating strategy for sorting DC subsets from the lungs of 7–day old mice. The plot on the left was gated on live, singlet lymphocyte, Lin−CD64−MHCII+CD11c+ cells, according to the gating strategy in fig. S1. B. mRNA was obtained from DCs sorted from the lungs of eight 7–day old C57BL/6 mice in 2 independent experiments. Principal component analysis of gene expression among DC subsets. C. Heatmap visualization of differentially expressed genes (DEG) among DC subsets. Scale is row z-score. D. Representative histogram of PD–L1 expression on DC subsets at day 7. The gate representing PD–L1+ cells is indicated. E. Frequency of PD-L1-expressing cells within the total population of each subset. F. Representative histogram of CCR7 expression on DC subsets at day 7. The gate representing CCR7+ cells is indicated G. Frequency of CCR7-expressing cells within the total population of each subset. H. Representative histogram of CD40 expression on DC subsets at day 7. The gate representing CD40+ cells is indicated I. Frequency of CD40-expressing cells within the total population of each subset. Graphs are plotted as mean ± SD. This experiment was performed 5 times with 5 mice/timepoint on days 4, 7, 11 and 4 mice/timepoint on days 13, 21, 77 and 126.
To test whether these activation markers were also temporally regulated in CD103int DCs, we analyzed their expression on cDC1, cDC2 and CD103int DCs between birth and 18 weeks. We found that PD–L1 was highly expressed on CD103int DCs for the first week after birth, but was minimally expressed on cDC1 and cDC2 cells at any time (Fig. 2D–E, fig. S2A). Similarly, CCR7 was highly expressed on CD103int DCs for the first two weeks after birth, but was not expressed on either cDC1 or cDC2 cells at any time (Fig. 2F–G, fig. S2B). In contrast, CD40 was highly expressed on CD103int DCs for the first week after birth and declined thereafter, whereas its expression on cDC1 and cDC2 cells continued to increase over time and exceeded that on CD103int DCs after 2 weeks (Fig. 2H–I, fig. S2C). As predicted by our RNAseq data, we also found that CD103int DCs in day-7 lungs spontaneously produced IL-12p40, but not IL-12p70 (fig. S3A–C). These data confirmed our RNAseq analysis and defined a temporal window in the lung during which activated, CD103int DCs appear.
Our RNAseq data suggested that CD103int DCs expressed multiple cDC1–defining genes, including Ly75 (CD205), Irf8 and Xcr1. To confirm these data, we quantified the protein expression of CD205, XCR1, and IRF8 on DC subsets from adult and neonatal lungs using flow cytometry. As expected, we found that CD205 was poorly expressed by cDC2 cells, but strongly expressed by cDC1 cells and CD103int DCs, particularly in neonates (Fig. 3A–B). Similarly, we found that XCR1 was not expressed by cDC2 cells, was expressed at modest levels in CD103int DCs and was highly expressed on DC1 cells, particularly those from adults (Fig. 3C–D). We also found that IRF8 was poorly expressed by cDC2 cells, but was clearly expressed by cDC1 cells and CD103int DCs, particularly in neonates (Fig. 3E–F).
Figure 3. CD103int DCs are related to Batf3-dependent cDC1 cells.
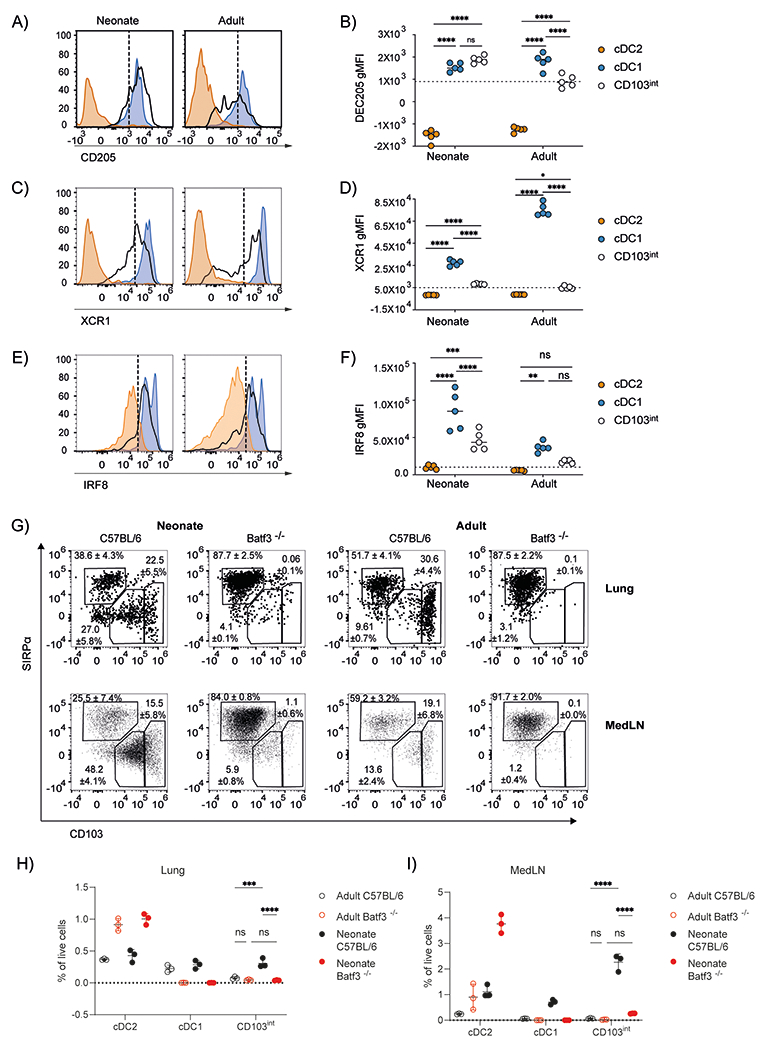
A–F. CD11b−SIRPα−CD103hi cDC1 cells, CD11b+SIRPα+CD103− cDC2 cells, and CD11b−SIRPα−CD103int CD103int DCs were isolated from the lungs of 7–day old neonatal and 8–week old adult mice. Representative histograms of CD205 (A), XCR1 (C), and IRF8 (E) expression in each subset are shown. Dashed lines divide marker+ from marker− cells. The geometric mean fluorescent intensity (gMFI) of CD205 (B), XCR1 (D), and IRF8 (F) expression on DC subsets is plotted as mean ± SD. This experiment was performed 3 times with 5 mice/timepoint. Significance was tested by using ordinary one-way ANOVA and Tukey post-test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. G. Representative plots and frequencies (mean ± SD) of DC subsets from the lungs and mLNs of 7–day old neonatal and 8–week old adult C57BL/6 and Batf3−/− mice. H. Frequency of DC subsets in the total cell population from the lungs of 7–day old neonatal and 8-week old adult C57BL/6 and Batf3−/− mice. I. Frequency of migratory DC subsets in the total cell population of mediastinal LN in 7-day old neonatal and 8-week old adult C57BL/6 and Batf3−/− mice. Bar and whiskers represent mean ± SD. This experiment was performed 3 times with 3–5 mice/timepoint. Significance was tested by using ordinary one-way ANOVA and Tukey post-test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Given that the gene expression profile of CD103int DCs did not conclusively assign them to the cDC1 lineage, we next tested whether they were dependent on the transcription factor, BATF3, which is essential for the development of cDC1 cells (10). We found that both DC1 cells and CD103int DCs were nearly absent from the lungs and mLNs of Batf3−/− mice (Fig. 3G–I). These data confirm that CD103int DCs in the neonatal lung belong to the DC1 lineage, albeit with a distinct activation signature.
CD103int DCs promote stromal cell maturation and lymph node expansion.
The activated phenotype of CD103int DCs and their expression of CCR7 suggested that they should spontaneously migrate to the draining LN. To test this possibility, we exposed 3–day old KikGR mice to violet laser light through the chest wall (the pups are hairless) to photoconvert cells in the lung from green fluorescence to red fluorescence. We found that only about 10% of the total DCs in the lung were photoconverted (fig. S4A–C). In addition, although all three DC subsets were present in the lung at the time of photoconversion (fig S4D–E), photoconverted cells consisted primarily of cDC2 cells (fig. S4F–G). We characterized the red fluorescent cells that migrated to the mLN over the next 24 hours. We found that neonatal mice had very few red DCs in the mLN prior to exposure or immediately following exposure (Fig. 4A), confirming that we were not directly photoconverting cells in the mLN. At this time, the DCs in the LN consisted primarily of CD103int DCs and smaller populations of cDC1 and cDC2 cells (Fig. 4B–C). After gating on red fluorescent DCs, we also found that most had a CD103int phenotype (Fig. 4D–E). Thus, despite inefficient labeling of CD103int DCs in the lung, most cells that migrated to the LN had a CD103int phenotype.
Figure 4. Migratory CD103int DCs promote LN expansion and stromal cell maturation.
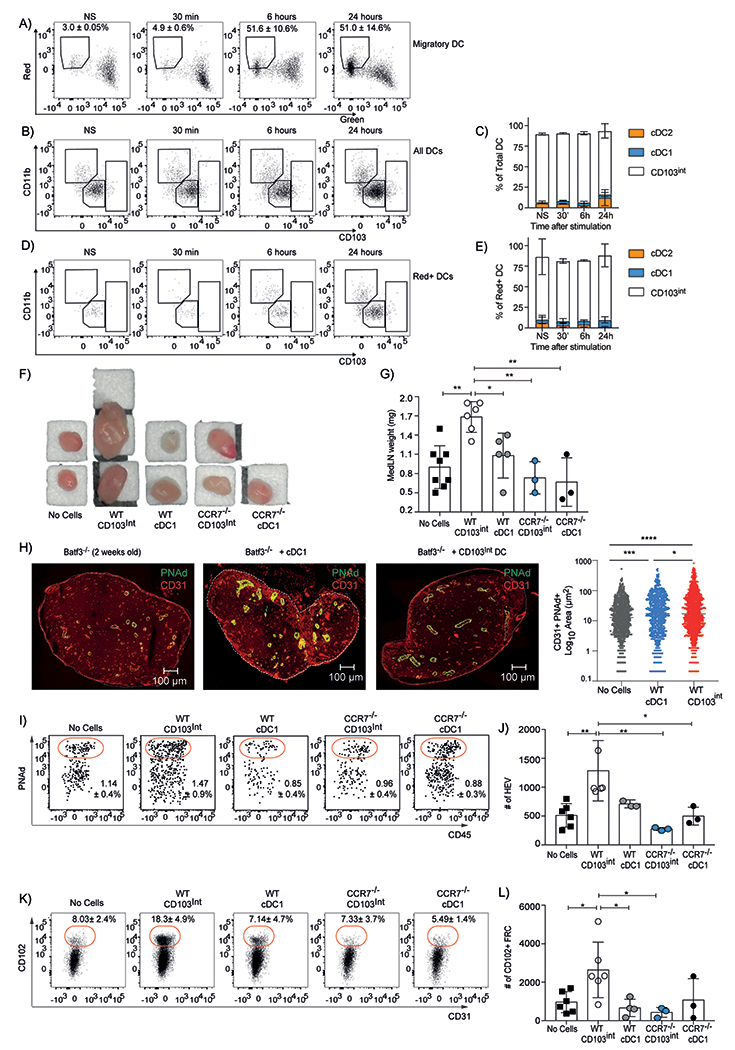
A. Representative plots show the frequency (mean ± SD) of photo–converted migratory DCs (gated on Lin−CD64−CD11c+MHCII+ cells) in the mLNs. B. Representative plots of total DCs in the mLN after photoconversion. C. The relative proportions (mean ± SD) of DC subsets in mLN after photoconversion. D. Representative plots of photoconverted (red) DCs in the mLN. E. The relative proportions (mean ± SD) of photoconverted DC subsets in mLN. This experiment was performed twice with 3 mice/timepoint. F-G. CD103int and cDC1 cells were sorted from the lungs of 7–day old C57BL/6 and Ccr7−/− mice, intranasally administered to 14–day old Batf3−/− mice and the size (F) and mass (G) of the mLNs of recipient mice were determined 2 days later. Data in G are combined from 2 independent experiments. Significance was tested using ordinary one-way ANOVA and Tukey test. *p<0.05, **p<0.01. H. Representative images of PNAd and CD31 expression in mLNs of recipient Batf3−/− mice 2 days after intranasal DC transfer. The area of PNAd+CD31+ HEVs was quantified using 3 sections/mLN from 2 independent experiments with 3 mice per group. Significance was tested using ordinary one-way ANOVA and Tukey test. *p<0.05, ***p<0.01, ****p<0.0001. I. Representative plots of HEV cells (gated on CD31+PDPN− cells) in mLNs of recipient Batf3−/− mice 2 days after DC transfer. J. The numbers (mean ± SD) of CD45−CD31+PNAd+ HEV cells per mLN. K. Representative plots of FRC cells (gated on CD45−PDPN+CD31− cells) in mLNs of recipient Batf3−/− mice 2 days after DC transfer. L. The numbers (mean ± SD) of CD45−CD31−PDPN+CD102+ FRC cells per mLN. Data are representative of 2 experiments with at least 3 mice/group. Significance was determined using 2–way ANOVA. Bar and whiskers represent the mean ± SD. *p<0.05, **p<0.01.
In adult mice, the homeostatic migration of DCs to LNs helps maintain FRCs and HEVs that support the lymphoid architecture of the lymph node (16, 18, 19). Since the stromal architecture of LNs is not yet fully matured in neonatal mice (32), an early wave of DC migration may enhance this process. To test whether the migration of CD103int DCs to the mLN might promote stromal cell maturation, we sorted cDC1 cells as well as CD103int DCs from the lungs of 7–day old mice, transferred them intranasally into 14–day old Batf3−/− mice and evaluated mLN expansion. We found that mLNs from Batf3−/− mice that received CD103int DCs expanded dramatically compared to those that received either no DCs or cDC1 cells (Fig. 4F–G). We performed a similar experiment in which we transferred CD103int DCs or cDC1 cells from Ccr7−/− mice and found that the mLNs did not expand in either group (Fig. 4F–G). We also found that cDC2 cells transferred from neonatal lungs to Batf3−/− recipients failed to expand the mLN (fig. S5A–B).
We next used fluorescent microscopy to evaluate the architecture of mLNs from Batf3−/− mice that received no cells, CD103int DCs or cDC1 cells. We found that the mLNs of Batf3−/− that received CD103int DCs had larger and more abundant CD31+PNAd+ HEVs than mice receiving no cells or cDC1 cells (Fig. 4H). We used flow cytometry to quantify our findings by enumerating CD45−CD31+PNAd+ HEV cells and CD45−PDPN+CD102+ FRC cells in mLN cell suspensions. Again, we found that Batf3−/− mice receiving CD103int DCs had increased numbers of HEV cells (Fig. 4I–J) and FRC cells (Fig. 4K–L) than those receiving no cells, cDC1 cells or Ccr7−/− CD103int DCs. Finally, we found that cDC2 cells transferred from neonatal lungs to Batf3−/− recipients failed to increase the numbers of CD45−CD31+PNAd+ HEV cells or CD45−PDPN+CD102+ FRC cells (fig. S5C–D). Thus, our data show that the CCR7–mediated migration of CD103int DCs promotes the maturation of HEVs and FRCs in the lung draining mLNs of neonatal mice.
CD103int DCs are activated independently of microbiota and TLR signaling
DCs are typically activated by inflammatory or microbial signals (33). To test whether microbial colonization or TLR–mediated sensing of microbial molecules promoted the maturation of CD103int DCs in neonatal lungs, we first enumerated DC subsets in the lungs of 7–day old conventional mice and germ–free (GF) mice. Although the frequency of cDC2 cells was slightly reduced and the frequency of cDC1 cells was slightly increased in GF mice compared to conventional mice, we observed no change in the frequency of CD103int DCs (Fig. 5A). Moreover, the expression of CCR7, a marker of DC activation, was slightly increased on CD103int DCs and cDC1 cells in the lungs of neonatal GF mice (Fig. 5B). To test whether pathways downstream of TLR signaling were involved in the generation or activation of CD103int DCs, we characterized DC subsets in the lungs of 7–day old Myd88−/− or Trif−/− mice. Although we observed minor changes in the frequencies of cDC1 and cDC2 populations in Myd88−/− mice, we saw no change in the frequency of CD103int DCs (Fig. 5C). Moreover, CD103int DCs expressed high levels of CCR7 regardless of genotype (Fig. 5D). We obtained similar results when we examined DC subsets in Trif−/− mice (Fig. 5E–F), suggesting that microbial recognition through TLR–dependent pathways does not play a role in the generation or activation of CD103int DCs.
Figure 5. Microbiota and TLR signaling are dispensable for CD103int DCs.
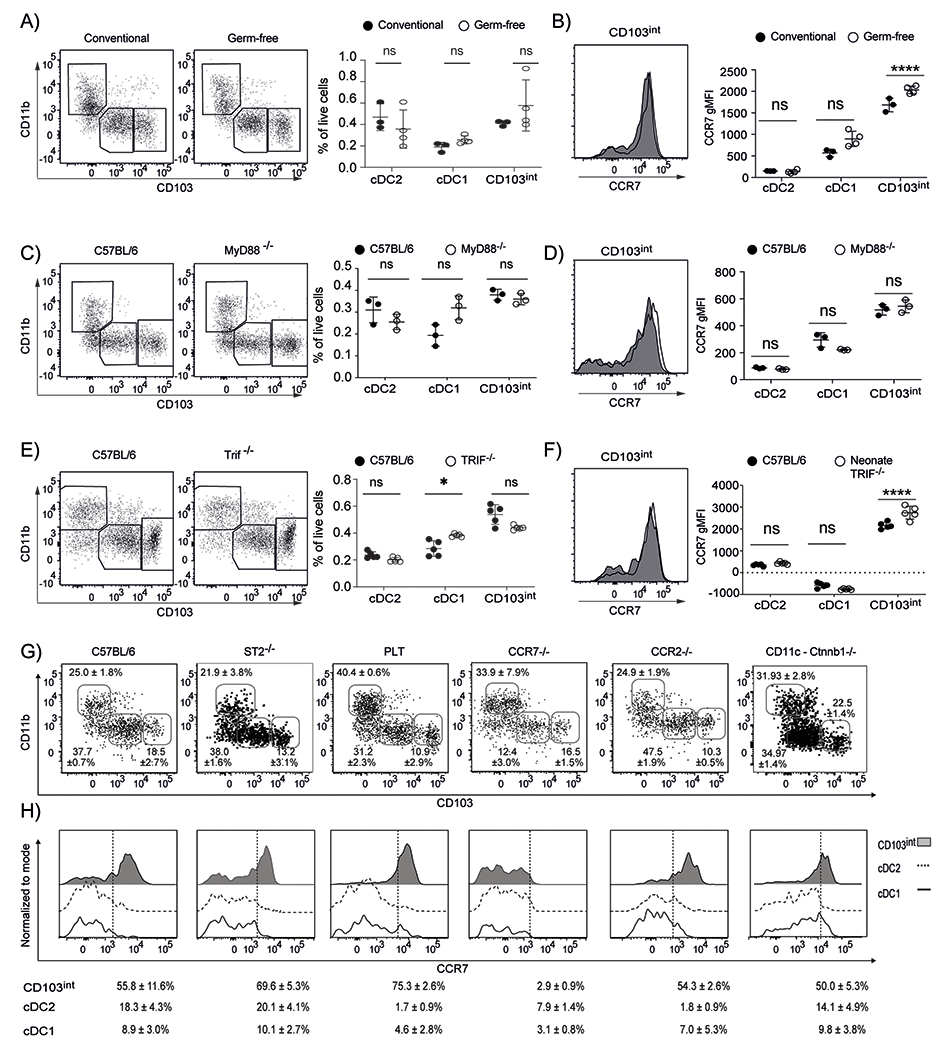
A. Representative plots (gated on Lin−CD64−CD11c+MHCII+ cells) and frequencies (mean ± SD) of DC subsets in total live cells in the lungs of 7–day old C57BL/6 mice housed in conventional or GF conditions. B. Representative histograms of CCR7 expression on CD103int DCs and graphs of gMFI of CCR7 on DC subsets from the lungs of 7–day old C57BL/6 mice housed in conventional or GF conditions. C. Representative plots (gated on Lin−CD64−CD11c+MHCII+ cells) and frequencies (mean ± SD) of DC subsets in total live cells in the lungs of 7–day old C57BL/6 and Myd88−/− mice. D. Representative histograms of CCR7 expression on CD103int DCs and graphs of gMFI of CCR7 on DC subsets from the lungs of 7–day old C57BL/6 and Myd88−/− mice. E. Representative plots (gated on Lin−CD64−CD11c+MHCII+ cells) and frequencies (mean ± SD) of DC subsets in total live cells in the lungs of 7–day old C57BL/6 and Trif−/− mice. F. Representative histograms of CCR7 expression on CD103int DCs and graphs of gMFI of CCR7 on DC subsets from the lungs of 7–day old C57BL/6 and Trif−/− mice. These experiments were performed 2 times for each comparison with 3 mice (A–B), 3 mice (C–D) and 5 mice (E–F) per group. Significance was determined using 2-way ANOVA with Sidak multiple comparision test. *p<0.05, ***p<0.001, ****p<0.0001. G. Representative plots (gated on Lin−CD64−CD11c+MHCII+ cells) and frequencies (mean ± SD) of DC subsets in the lungs of 7–day old C57BL/6, ST2−/−, plt/plt, Ccr7−/−, Ccr2−/− mice and CD11c-Ctnnb1−/− mice. H. Representative histograms of CCR7 expression and frequency (mean ± SD) of CCR7+ cells in DC subsets. Dashed lines divide marker+ from marker− cells. These experiments were performed at least 2 times with 3 C57BL/7, 4 ST2−/−, 3 plt/plt, 3 Ccr7−/−, 3 Ccr2−/− mice and 6 CD11c-Ctnnb1−/− mice.
IL–33 is highly expressed by neonatal lung epithelial cells and promotes cDC2 activation (34, 35). However, we found that the proportions of DC subsets were similar in the lungs of 7–day old C57BL/6 mice and Il1rl1−/− mice that lack ST2, the IL-33 receptor (Fig. 5G), and that the ST2-deficient CD103int DCs retained their activated phenotype, including high expression of CCR7 (Fig. 5H). These data therefore, suggest that IL–33 was not involved in the generation or activation of CD103int DCs.
In addition to promoting chemotaxis, receptors like CCR7 contribute to DC survival and maturation (36, 37). To test whether CCR7 signaling contributed to the activated phenotype of CD103int DCs, we characterized DCs subsets in the lungs of 7–day old Ccr7−/− mice as well as plt/plt mice, which lack the CCR7 ligands, CCL19 and CCL21–Ser (38). Interestingly, although the proportion of CD103int DCs in the lungs of neonatal plt/plt mice was similar to that in neonatal C57BL/6 mice, the frequency of CD103int DCs was reduced in Ccr7−/− mice (Fig. 5G). Nevertheless, the CD103int DCs maintained their activated phenotype in plt/plt mice, with high expression of CCR7 (Fig. 5H). These data suggest that signaling through CCR7 is important for the maintenance of CD103int mregDC in neonatal lungs, most likely through CCL21–Leu, which is expressed normally in plt/plt mice (39). Finally, given the role of β-catenin signaling in the sterile activation of DCs (21, 22), we evaluated DC subsets in the lungs of neonatal CD11c-Ctnnb−/− mice, which lack β-catenin expression in dendritic cells. We observed no difference in the formation of CD103int DCs (Fig. 5G) or their activation and expression of CCR7 in response to β-catenin-deficiency (Fig. 5H).
CD103int DCs are a distinct subset of mature DCs
To better understand how CD103int DCs were related to other DC subsets, we performed gene set enrichment analysis (GSEA) comparing DEGs in CD103int DCs and cDC1 cells to previously identified gene sets in published data (23, 24, 26). We found that genes expressed in mature DCs (Mat ON)(23) were enriched in CD103int DCs relative to cDC1 cells (Fig. 6A), whereas genes expressed in immature DCs (MAT OFF)(23) were negatively enriched in cDC1 cells relative to CD103int DCs (fig. S6A). Moreover, we found that genes expressed by tolerogenic DCs (23) were enriched in CD103int DCs relative to cDC1 cells (Fig. 6B), but that genes expressed by immunogenic DCs (23) were not enriched in either CD103int DCs or cDC1 cells (fig. S6B).
Figure 6. CD103int DCs have a regulatory signature.
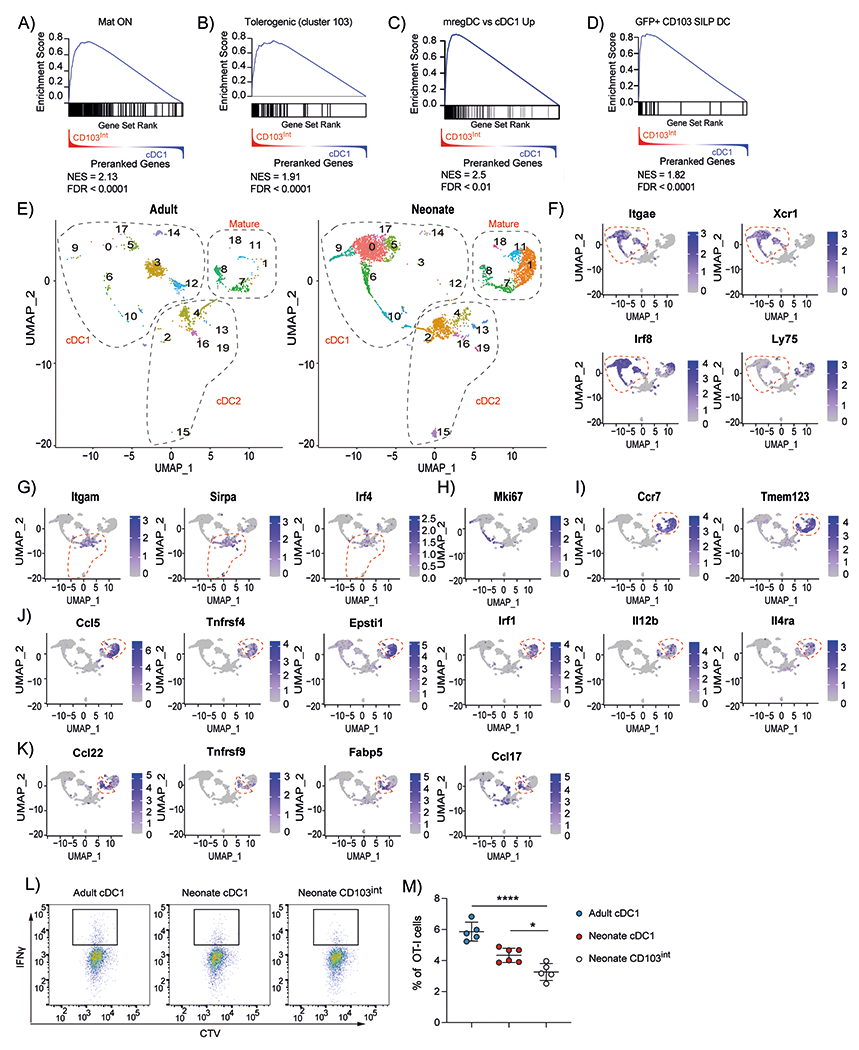
A ranked list of genes differentially expressed in CD103int over cDC1 cells was compared with genes enriched in mature DCs from the thymus (A), genes enriched in tolerogenic DCs from the thymus (B), genes enriched in tumor–derived mregDCs (C), genes enriched in efferocytic CD103+ DCs from small intestine (D). Normalized enrichment scores (NES) and false discovery rates (FDR) are listed in each panel. E. UMAP representation of single cell RNAseq of live, CD45.2+Zbtb46–YFP+ cells sorted from adult or neonatal lungs. Dashed outlines indicate clusters of cDC1, cDC2 and mature cDC1 cells. F. Expression of cDC1–defining genes. Dashed outline indicates clusters of cDC1 cells. G. Expression of cDC2–defining genes. Dashed outline indicates clusters of cDC2 cells. H. Expression of Mki67. I. Expression of maturation genes. Dashed outline indicates clusters of mature cDC1 cells. J. Expression of genes enriched in CD103int DCs. Dashed outline indicates clusters of CD103int cells. K. Expression of genes enriched in homeostatically mature DCs. Dashed outline indicates clusters of homeostatically mature DCs. Data are combined from two independent experiments. L-M. Naïve OT-I cells were cultured with sorted DC subsets loaded with OVA257–264 peptide and IFNγ expression was determined on day 4. This experiment was performed twice with 5 replicates for adult cDC1, 6 replicates for neonatal cDC1 and 5 replicates for neonatal CD103int DCs. Data are shown as mean ± SD. Significance was determined using 1-way ANOVA. *p<0.05, ****p<<0.0001.
Recent studies investigating mregDCs in tumors (26) suggest that they may have a phenotype similar to the CD103int DCs that we identified in neonatal lungs. We found that genes highly expressed in mregDCs from lung tumors (26) were enriched in CD103int DCs relative to cDC1 cells (Fig. 6C), whereas genes poorly expressed in mregDCs from lung tumors (26) were negatively enriched in CD103int DCs (fig. S6C). Interestingly, the phenotype of mregDCs is controlled in part by the tyrosine kinase, Axl (26), suggesting that the uptake of apoptotic cells by CD103int DCs may control their phenotype. To test this possibility in another context, we next compared genes expressed in CD103int DCs with those previously identified by comparing gene expression in CD103+ DCs in the small intestine lamina propria (SILP DCs) either before or after engulfing apoptotic, GFP-expressing epithelial cells. We found that genes enriched in SILP DCs that had engulfed GFP-expressing epithelial cells (GFP+SILP DC) (24) were strongly enriched in CD103int DCs (Fig. 6D), whereas genes expressed by CD103+ DCs in the intestinal lamina propria that had not engulfed apoptotic epithelial cells (GFP−SILP DC) (24) were not enriched in CD103int DCs (fig. S6D).
To understand whether the maturation of CD103int DCs differed from homeostatic maturation of DCs in adult lungs, we generated Zbtb46-YFP reporter mice (Zbtb46-cre mice crossed to Rosa-YFPfl/fl mice), sorted CD45.2+YFP+ cells from the lungs of adult and neonatal mice, and performed single cell RNA-seq analysis. Dimensional reduction of the transcriptional data allowed us to display the results as 20 distinct clusters and to determine that many of the clusters differed widely in their abundance between neonatal and adult lungs (Fig. 6E). Clusters 3, 4, 12 and 14 were over-represented in adult mice, whereas clusters 0, 1, 2, 15, and 17 were over-represented in neonatal mice (Fig. 6E, fig. S6E).
To identify clusters that corresponded to cDC1 cells, we analyzed the expression of CD103 (Itgae), Xcr1, Irf8 and CD205 (Ly75) and found that these genes were enriched in clusters 0, 3 5, 6, 9, 10, 12 and 14 (Fig. 6E–F). In contrast, cDC2-defining genes, CD11b (Itgam), Sirpa and Irf4, were enriched in clusters 2, 4, 13, 15 and 16 (Fig. 6E, G). Proliferating cells, identified by expression of KI-67 (Mki67), were observed in clusters 6, 9, 10 and 2 (Fig. 6E, H). Mature DCs were identified by Ccr7 and Tmem123 expression and were found in clusters 1, 7, 8, 11 and 18 (Fig. 6E, I).
To characterize the clusters that represented mature DCs, we used GSEA to compare gene expression in cells from each cluster with a gene set preferentially expressed by mature cDC1 cells (MAT ON)(23). We found that cells in clusters 1, 7, 8, and 11 most closely resembled mature DCs (fig. S6F). Similarly, genes preferentially expressed by CD103int DCs identified in our bulk RNA-seq analysis were predominantly enriched in cluster 1 (fig. S6F), which was almost exclusively found in neonatal rather than adult lungs (Fig. 6E). Genes expressed by CD103int DCs were also enriched to a lesser extent in clusters 7, 8 and 11, consistent with their mature status (fig. S6F). Interestingly, clusters 7 and 8 were similarly abundant in neonatal and adult lungs (Fig. 6E), suggesting that these cells underwent homeostatic maturation by similar mechanisms in neonates and adults, whereas the CD103int DCs represented in cluster 1 were matured by mechanisms specific to neonatal lungs. Some of the genes preferentially expressed by cells in cluster 1 (CD103int DCs), included Il12b and Il4ra (Fig. 6J), both of which were identified in mregDCs (26), as well as Ccr5, Tnfrsf4, Epsti1 and Irf1. In contrast, the cells in clusters 7 and 8, representing homeostatically matured DCs, preferentially expressed Ccl22, Tnfrsf9, Fabp5 and Ccl17 (Fig. 6K). Thus, it seems that CD103int DCs are distinct from typical homeostatically mature DCs.
CD103int DCs dampen CD8+ T cell responses.
Given that CD103int DCs were transcriptionally similar to mregDCs and GFP+SILP DCs, which have regulatory functions, we next assessed the ability of CD103int DCs to induce CD8+ T cell activation. For this we sorted cDC1 cells from adult lungs as well as cDC1 cells and CD103int DCs from neonatal lungs, loaded them with the Ovalbumin (OVA)-derived peptide antigen, OVA257-264, and cultured them for 4 days with naïve, CTV-labeled, OVA257-264-specific CD8+ OT-I T cells. Although T cells in each culture proliferated similarly, T cells cultured with neonatal CD103int DCs had the lowest capacity to induce IFNγ production (Fig. 6L–M). We repeated the experiment using DC subsets loaded with OVA protein instead of OVA peptide and obtained similar results (fig. S6G). Since other groups reported that mregDCs promote Treg differentiation, we cultured naïve OVA-antigen specific CD4+ OTII cells with cDC1 cells, cDC2 cells and CD103int DCs sorted from neonatal lungs that were loaded with OVA protein. We found that a higher frequency of OTII cells stimulated with cDC2 cells expressed FOXP3 than those stimulated with cDC1 cells or CD103int DCs (fig. S6H–I), suggesting that cDC2 cells rather than CD103int DCs preferentially stimulated Treg differentiation. Consistent with this observation, Helios−FOXP3+ inducible Tregs were observed at a very low frequency and numbers in the lungs and mLNs for the first 2 weeks after birth and increased thereafter (fig. S6J). This increase in Tregs coincided with the decline of CD103int DCs and an increase in cDC2 cells in the lungs and mLNs (Fig. 1).
CD103int DCs from neonates are efferocytic and are dependent on Mertk signaling
Our data showing that CD103int DCs had a gene expression program similar to that of efferocytic DCs in the intestine (Fig. 6D) suggested that CD103int DCs in neonatal lungs might engulf apoptotic cells, causing their maturation, in a non–inflammatory, tolerogenic manner. To test whether CD103int DCs engulfed apoptotic cells in neonatal lungs, we crossed CC10–CreERT mice with ROSA–YFPfl/fl mice to produce offspring (CC10–YFP) that, after an injection of tamoxifen 4 days prior to the analysis, expressed YFP in airway epithelial cells. Using these mice, we tested what types of DCs from the lungs of 7–day old neonatal or 8–week old adult CC10–YFP mice, became YFP+ cells, indicating that they had engulfed epithelial cells. We found that CD103int DCs from neonates, but not from adults, took-up YFP+ epithelial cells and that cDC1 and cDC2 cells engulfed YFP+ cells poorly in either neonatal or adult mice (Fig. 7A–C).
Figure 7. The generation and activation of CD103int DCs depends on Mertk.
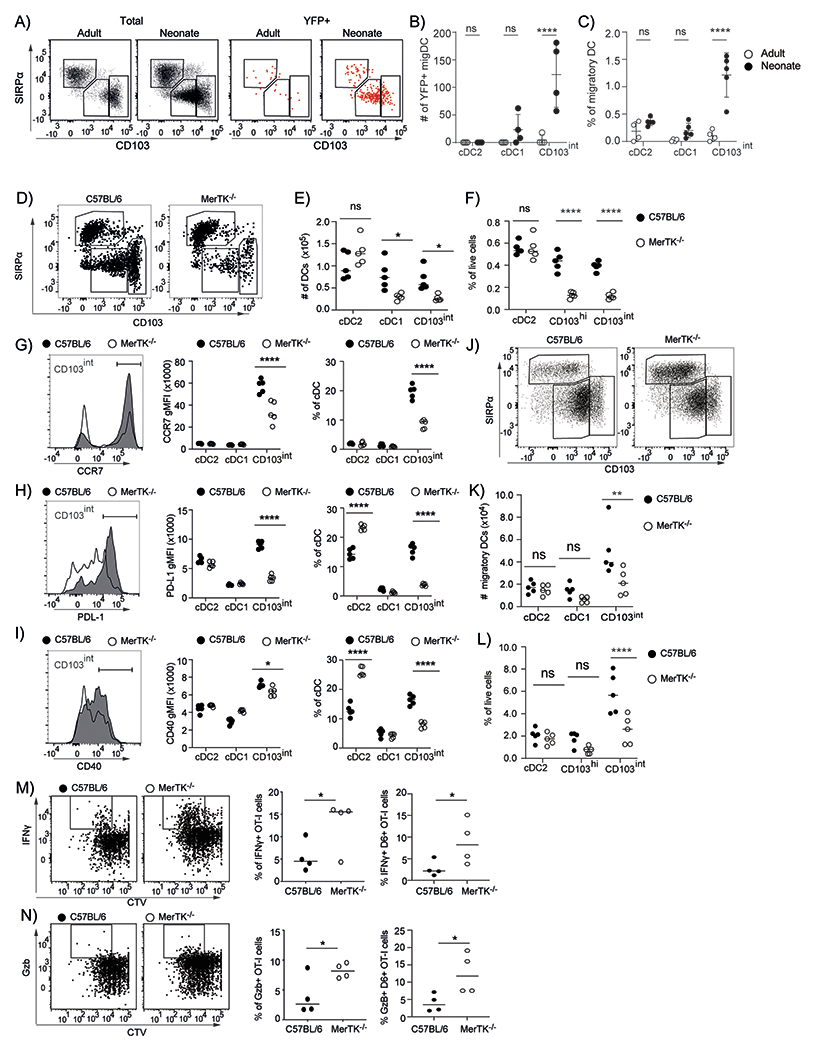
A. Representative plots of total DCs (gated on Lin−CD64−CD11c+MHCII+ cells) and YFP+ DCs from adult and neonatal lungs. B. The number of YFP+ DCs in each subset in adult and neonatal lungs. C. The frequency of YFP+ DCs within each subset in adult and neonatal lungs. This experiment was performed 2 times with 4-5 mice/group. Data are shown as mean ± SD. Significance was determined using 2-way ANOVA and Tukey test for multiple comparisons. *** p < 0.0002. D. Representative plots of DCs (gated on Lin−CD64−CD11c+MHCII+ cells) from the lungs of neonatal C57BL/6 and Mertk−/− mice. E-F. Numbers (E) and frequencies (F) of DC subsets within total live cells in the lungs of neonatal C57BL/6 and Mertk−/− mice. G-I. Representative histograms of CCR7 (G), PD–L1 (H) and CD40 (I) on CD103int DCs from the lungs of neonatal C57BL/6 (shaded) and Mertk−/− (open) mice. Graphs show gMFI and frequency of positive cells within each DC subset. J. Representative plots of DCs (gated on Lin−CD64−CD11c+MHCII+ cells) from the mLNs of neonatal C57BL/6 and Mertk−/− mice. K-L. Numbers (K) and frequencies (L) of DC subsets within the total live cell population in the mLNs of neonatal C57BL/6 and Mertk−/− mice. The experiments in D-L were performed 3 times using 4-5 mice/group. Graphs show individual points and a bar indicating the mean. Significance was tested using 2-way ANOVA and Sidak test for multiple comparisons. * p < 0.0332, ** p < 0.0021, p < 0.0001. M-N. CTV-labeled OTI T cells were transferred to neonatal C57BL/6 or Mertk−/− mice followed by intranasal delivery of 30 μg of OVA. Representative plots (gated on CD45.1+CD8+ cells) show CTV dilution and IFNγ expression (M) or granzyme B (GZB) expression (N). Graphs show frequencies of OTI cells that express IFNγ+ or GZB+ or the frequencies of OTI cells that express IFNγ+ or GZB+ OTI cells and have divided 5 times or more. Bars represent the mean. This experiment was performed 3 times using 4 mice/group. Significance was tested using unpaired t test analysis. * p < 0.05.
The process of efferocytosis involves signaling through TAM receptors like Tyro3, Axl and Mertk (31). To determine what TAM receptors might be expressed by CD103int DCs, we examined our RNA-seq data and found that both Mertk and Axl were expressed by CD103int DCs. We next examined the proportions of DC subsets in the lungs of neonatal Mertk−/− mice. We found that neonatal Mertk−/− mice had reduced frequencies of both cDC1 cells and CD103int DCs (Fig. 7D–F). We also found that the remaining CD103int DCs from neonatal Mertk−/− mice expressed lower levels of CCR7, PD–L1 and CD40 (Fig. 7G–I), whereas the cDC1 and cDC2 cells in WT and Mertk−/− mice expressed similar levels of these molecules. We observed a similar reduction in the frequency and number of CD103int DCs in the mLNs of neonatal mice (Fig. 7J–L). These data show that Mertk signaling is an important event in the maturation of CD103int DCs in neonatal lungs.
Given the role of Mertk in the activation of CD103int DCs, we next tested whether T cell activation in response to pulmonary antigens differed between WT and Mertk−/− mice. To test this possibility, we transferred CTV–labeled, CD8+ OTI T cells to neonatal WT and Mertk−/− mice and administered OVA intranasally 24 hours later. After 4 days, we recovered OT-I cells from the mLN. We found that OT-I cells stimulated in Mertk−/− mice proliferated more and produced more IFNγ (Fig. 7M) and more granzyme B (Fig. 7N), than OT–I cells activated in WT mice.
The appearance of CD103int DCs coincided with apoptosis in the developing lungs.
One possibility why CD103int DCs appear in the lungs of neonatal mice is that the lungs are not yet fully developed at birth, and the process of alveolarization and lung remodeling continues in the post-natal period (40–42). To test whether the alveolarization process is accompanied by a high frequency of apoptotic cells, we performed TUNEL staining on neonatal and adult lungs. We found numerous TUNEL+ cells clustered around the periphery of neonatal lungs, but only observed a few, scattered TUNEL+ cells in adult lungs (Fig. 8A–B). To quantify this difference, we determined the number of TUNEL stained (apoptotic) cells per area of tissue (mm2) and found that the relative abundance of apoptotic cells was much higher in neonatal lungs than in adult lungs (Fig. 8C). In addition, we enumerated cells with activated caspases–3 and 7 using Magic Red–conjugated (DEVD)2, a peptide that binds specifically to activated caspase–3/7. We found that the frequency of caspase–3/7+ cells decreased substantially during the first 3 weeks after birth (Fig. 8D–E) as measured by the area occupied by apoptotic cells. Thus, we conclude there was a temporal wave of apoptosis in the developing lungs in the first few weeks after birth that coincides with the appearance of CD103int DCs.
Figure 8. Extensive apoptosis in neonatal lungs.
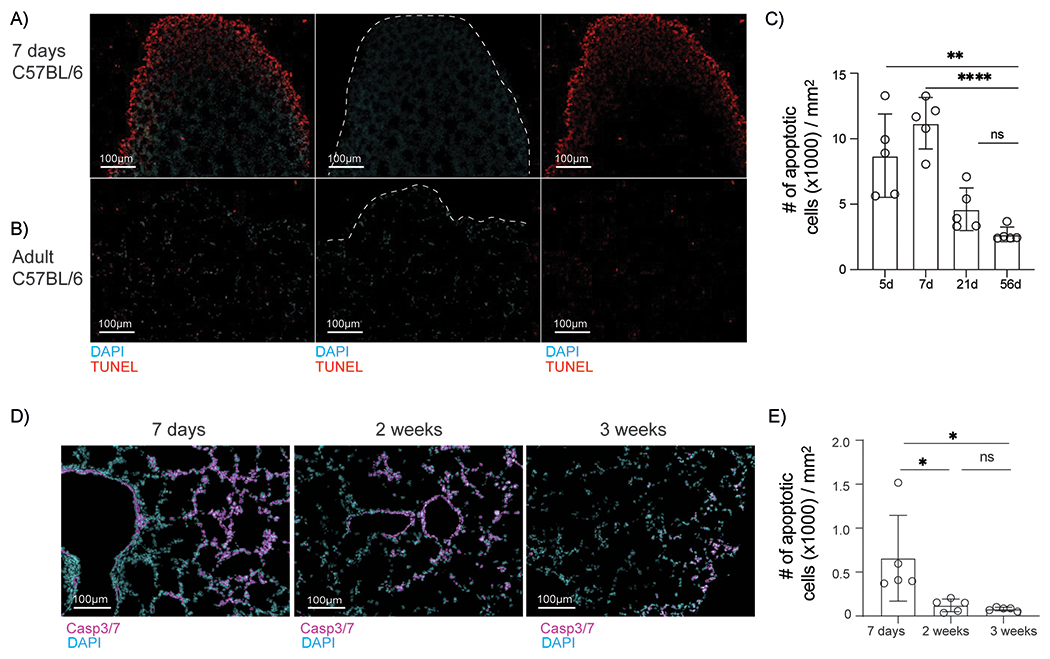
A-B. Representative images of TUNEL-stained apoptotic cells in sections from the lungs of neonatal (A) and adult (B) mice. C. Density of apoptotic cells (mean ± SD) in lung sections from mice at the indicated ages. Data are obtained from 4-5 sections per group. Significance was determined by one-way ANOVA and Tukey test for multiple comparisons. * p < 0.0332, ** p < 0.0021 D. Representative images of lung sections stained for activated caspase-3/7. Data were obtained from 5 sections per group. E. Density of apoptotic cells (mean ± SD) in lung sections from mice at the indicated ages. Significance was determined by one-way ANOVA and Tukey test for multiple comparisons.
DISCUSSION
Here we describe a distinct population of mature cDC1 cells that express intermediate levels of CD103 and accumulate in the lungs and mLNs of neonatal mice during the first two weeks of life. CD103int DCs require the transcription factor, BATF3, for their development, and express IRF8, XCR1, and CD205, suggesting that they are part of the cDC1 lineage. However, CD103int DCs are constitutively activated and express maturation markers, including CCR7, CD40 and IL–12b, as well as regulatory molecules like PD–L1, similar to the mregDCs and DC3 cells described in tumors. Maturation of CD103int DCs occurs in the absence of microbiota, TLR signaling, IL-33 or β-catenin and promotes spontaneous migration to the draining LN, where they trigger the maturation of HEVs and FRCs and increase LN size and cellularity. CD103int DCs also express the TAM receptor, Mertk, and efficiently engulf apoptotic lung epithelial cells. Genetic deletion of Mertk reduces the frequency of CD103int DCs and lowers their expression of CCR7, PD–L1, and CD40. As a result, CD8+ T cells are more efficiently primed by respiratory antigens in neonatal Mertk−/− mice than in WT mice. Finally, we identified a temporal wave of apoptosis in the post–natal lung that likely drives the activation of CD103int DCs via efferocytosis. These data suggest that CD103int DCs are a distinct maturation state of cDC1 cells that accumulate spontaneously in the neonatal lung.
The neonatal period is notable for Th2-biased immune responses that promote allergies and asthma, in part due to poor expression of IL-12 by DCs (43). This effect is partly explained by the increased expression of IL-33 in the lung after birth (34, 35), which promotes Th2 responses by activating ILC2 cells and inhibiting the expression of IL-12a (p35) in DCs. Consistent with this idea, we found that CD103int DCs made abundant IL-12b (p40), but failed to make the functional IL-12 p40p35 heterodimer. However, the formation and phenotype of CD103int DCs were unaffected by the loss of IL-33 signaling. Similarly, de novo exposure to microbiota programs cDC2 cells in neonatal lungs to express PD-L1 and promotes the differentiation of inducible Tregs that dampen responses to pulmonary allergens (44). However, we found that CD103int DCs appeared normal in frequency and phenotype in the lungs of GF mice and in mice lacking TLR signaling pathways, suggesting that they are not activated by microbial exposure.
Notably, the lungs of mice continue to develop in the neonatal period, with alveolarization occurring 5-25 days after birth (40). This process requires Wnt/β-catenin signaling (45), which is also associated with sterile activation of DCs and the acquisition of a regulatory phenotype (21, 22). However, we found that the numbers and phenotype of CD103int DCs were normal in CD11c-Ctnnb−/− mice. Interestingly, the process of alveolarization involves extensive pulmonary remodeling that involves cell proliferation as well as apoptosis (42). Consistent with this idea, we found substantial numbers of TUNEL+ apoptotic cells in neonatal lungs. Given the preferential uptake of apoptotic epithelial cells by CD103int DCs and their requirement for Mertk activation, it seems likely that the temporal restriction of CD103int DCs to the neonatal period is due to the remodeling of the lung during alveolarization and the abundance of apoptotic cells during that period.
The similarity between CD103int DCs in neonatal lungs and mregDCs in tumors (26) may be explained by their exposure to apoptotic cells. CD103int DCs in neonatal lungs acquire apoptotic lung epithelial cells and mregDCs in tumors acquire apoptotic tumor cells. In both cases, the regulatory phenotype of DCs is controlled by TAM receptors – Mertk in CD103int DCs and Axl in mregDCs – suggesting that both Mertk and Axl facilitate the acquisition of apoptotic debris and program DC1 cells with a regulatory phenotype. A similar phenomenon occurs in the small intestine, in which CD103+ DCs acquire apoptotic intestinal epithelial cells and express a regulatory transcriptional program similar to that in both CD103int DCs and mregDCs (24). Thus, the acquisition of apoptotic cells by DCs via TAM receptors leads to a regulatory phenotype in multiple locations. In fact, DCs with a similar mature, regulatory phenotype are found in a variety of lymphoid and non-lymphoid tissues (27–30, 46), albeit at very low frequencies, likely because of the paucity of apoptotic cells under homeostatic conditions. However, our data also suggest that regulatory DCs should be formed at any time in the lung or other tissues that are being remodeled or repaired in a way that generates substantial numbers of apoptotic cells without overt inflammation.
The acquisition and transport of apoptotic cells from the lung to the LNs in adults is limited to BATF3-dependent cDC1 cells (5). This finding is consistent with our observation that apoptotic lung epithelial cells are primarily captured by CD103int DCs and that CD103int DCs in neonatal lungs are entirely dependent on BATF3 and express a high amount of CD205, XCR1 and IRF8, suggesting that they are part of the cDC1 lineage. In tumors however, DCs with a CCR7+PD-L1+ regulatory phenotype are a mixed population; some with characteristics of cDC1 cells and others with characteristics of cDC2 cells. We find that although some cDC2 cells in neonatal lungs express PD-L1, only about 1% express CCR7, suggesting that they may be regulatory, but are not mature migratory DCs. This is an important distinction, as CCR7 is essential for the migration of CD103int DCs to the draining LN and the promotion of stromal cell maturation (16, 18–20). Interestingly, most reports examining DC–mediated stromal cell maturation either do not distinguish between DC subsets or focus on attributes of cDCs that are linked to cDC2 cells rather than cDC1 cells. For example, SIRPα–expressing cDC2 cells are important for maintaining FRCs in the spleen (17). Similarly, CLEC–2, which is expressed by cDC2 cells and not cDC1 cells, helps expand the FRC network following immunization (16). These studies contrast with our results showing that cDC2 cells from neonatal lungs do not promote LN expansion and stromal cell maturation. These results imply that the stroma-activating functions of cDC2 cells likely require DC maturation triggered by microbial or inflammatory signals, which causes CCR7 expression and migration to the LN. In the absence of these signals (i.e. in naïve mice), the cDC2 cells in the neonatal lung are not involved in stromal cell maturation.
An interesting result from our single cell RNAseq experiment is that homeostatically mature DCs (clusters 7 and 8) appear in both neonatal and adult lungs, whereas CD103int DCs (cluster 1) appear almost exclusively in neonates. These data suggest that distinct mechanisms may govern homeostatic maturation and the maturation driven by efferocytosis. In addition, the developing lungs of neonates or tumors may provide specific signals, in addition to Mertk- and Axl-mediated efferocytosis, that help shape local DC maturation.
CD103int DCs, mregDCs, efferocytic CD103+ DCs in the small intestine and tolerogenic DCs in the thymus all express a variety of molecules, including PD-L1, CD200, CD95 and Aldh1a2, that imply a regulatory or tolerogenic function of these cells (23, 24, 26, 30). Consistent with this idea, multiple groups report that these DCs prime CD4+FoxP3+ Tregs in vitro (24, 26). In contrast, we found that cDC2 cells from neonatal lungs could prime CD4+FoxP3+ Tregs, but that cDC1 cells and CD103int DCs from neonatal lungs could not. Our result is consistent with the idea that pulmonary cDC2 cells are specialized to prime CD4+ T cells, including Tregs, whereas pulmonary DC1 cells are specialized to prime and cross-prime CD8+ T cells (47, 48). One distinction between our findings and the results of others is that the mregDCs from tumors are a mix of cDC1 and cDC2 cells, whereas the CD103int DCs in neonatal lungs belonged exclusively to the cDC1 lineage.
Given that mregDCs, efferocytic CD103+ DCs in the intestine and CD103int DCs acquire apoptotic cells and rely on TAM receptor signals, rather than microbial or inflammatory stimuli for their activation (24, 26), it seems unlikely that they are involved in the initial priming of T cells in response to infection/inflammation. Instead, given their apparent role in sustaining ongoing T cell responses in tumors or promoting tissue-resident memory T cells, they are more likely to be the “clean-up crew” that help to clear apoptotic debris after the acute response, that dampen the proliferation and effector functions of activated T cells and that promote memory formation, thereby resolving inflammation and preventing ongoing tissue damage. In specialized cases, such as the neonatal lung, the intestinal epithelium or in tumors, each of which have numerous apoptotic cells but lack an acute inflammatory response, the regulatory DCs dominate the DC compartment and, as we see in the neonatal lung, dampen, but do not entirely prevent CD8+ T cell responses.
In summary, our data show that a novel population of CD103int cDCs is activated in neonatal lungs by Mertk signaling and the acquisition of apoptotic bodies. CD103int DCs actively migrate to the mLN and promote stromal cell maturation, but dampen CD8+ T cell responses to pulmonary antigens. These data help explain the qualitative differences between pulmonary immune responses in neonates and adults and provide insight into how immune responses in the lung and elsewhere may be impacted by tissue remodeling that involves extensive apoptosis in the absence of overt inflammation.
MATERIALS and METHODS
Study design.
This study was designed to determine the phenotype, origin and function of DCs in the lungs of neonatal mice. The goal of the study was to define how pulmonary DCs in neonatal mice differed from their adult counterparts and the mechanisms underlying this difference. Comparisons were made between pulmonary DCs in neonates and those in adults, and between pulmonary DCs in C57BL/6 (wild type) mice and mice with mutations that impact DC development or activity. In each case, we compared the number or frequency of pulmonary DC subsets, the activation phenotype of pulmonary DC subsets and the functional activity of DC subsets. Most measurements were performed using flow cytometry. Mice were not randomized as we compared cells from mice of different genotypes or ages. No mice or samples were excluded from analysis. Personnel were not blinded to the groups being studied. The number of times each experiment was performed and the number of mice per group are included in the figure legends.
Mice, intranasal administration and cell transfer.
C57BL/6J (Jax #000664), B6;129–Mertktm1Grl/J (Mertk−/− Jax #011122), C57BL/6J–Ticam1Lps2/J (Trif−/− Jax #005037), B6.129S4–Ccr2tm1Ifc/J (Ccr2−/− Jax #017586), B6.Tg(CAG–KikGR)33Hadj/J (KikGR Jax #013753), B6.129S(C)–Batf3tm1Kmm/J (Batf3−/− Jax #013755), B6.129P2(SJL)–Myd88tm1.1Defr/J (Myd88−/− Jax #009088), B6.SJL–PtprcaPepcb/BoyJ (CD45.1 Jax #002014), B6.129P2(C)–Ccr7tm1Rfor/J (Ccr7−/− Jax #006621), C57BL/6–Tg(TcraTcrb)1100Mjb/j (OT–I Jax #003831), B6;129(Cg)-Scgb1a1tm1(cre/ERT)Blh/J (CC10–CreERT Jax #016225), B6.129-Ctnnb1tm2Kem/KnwJ (Ctnnbfl/fl Jax #022775) B6.Cg-Tg(Itgax-cre)1–1Reiz/J (CD11c-cre Jax #008068), B6.Cg-Zbtb46tm3.1(cre)Mnz/J (zDC-cre Jax #028538) and B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J (ROSA–YFPfl/fl Jax #006148) mice were purchased from Jackson Laboratories. CD45.1 mice were crossed to OTI mice to produce CD45.1.OTI mice. CC10-CreERT mice were crossed to ROSA-YFPfl/fl mice to produce CC10–YFP mice. CC10-YFP mice were treated with 200 μg tamoxifen in corn oil delivered intraperitoneally 4 days prior to analysis. CD11c-cre mice were crossed with Ctnnbfl/fl mice to produce CD11c-Ctnnb−/− mice. Germ–free (GF) C57BL/6J mice were obtained from the University of Alabama at Birmingham (UAB) gnotobiotic core facility. B6.Il1rl1−/− (ST2−/−) mice were obtained from Dr. Andrew MacKenzie (MRC Laboratory of Molecular Biology, Cambridge, UK). B6.plt/plt mice were obtained from Dr. Hideki Nakano (NIEHS, Durham, NC). All mice were bred and maintained in the UAB vivarium under specific pathogen–free conditions, except for GF mice, which were bred and maintained in the UAB gnotobiotic facility.
Anesthesia for mice up to 2 weeks–old was performed in a chamber containing 3.5% isofluorane in 1.5 L/min of oxygen. Endofit Ovalbumin (InvivoGen), and sorted cDCs were resuspended in sterile, endotoxin–free PBS and administered intranasally in a final volume of 15 μL (neonates) or 30 μL (2–weeks old mice).
Male and female mice of each genotype were used. All procedures involving mice were approved by the UAB Institutional Animal Care and Use Committee under protocol 09710.
Cell preparation and flow cytometry
Lungs were cut into small pieces and then incubated for 30 minutes in PBS with Collagenase (1.25 mg/mL, Sigma) and DNase (150 U/mL, Sigma). After incubation, suspensions were pressed through a 70 μm filter with a 3 mL sterile syringe plunger. Cell suspensions from mLN were obtained by pressing through a 70 μm filter with a 3 mL sterile syringe plunger. Stromal cells from mLN were obtained as described (49), using a mix of collagenase (0.2 mg/mL), DNase (0.1 mg/mL), dispase (10 U/mL, Corning). For staining, cells were resuspended in PBS with 2% Bovine Calf Serum (BCS – GE Healthcare Life Sciences), incubated with anti-CD16/32 (BioXcell) for 15 minutes at 4°C, and stained with antibody cocktail in PBS with 2% BCS. A live–dead cell discriminator (Live/Dead fixable, Thermo–Fisher) was added at this point. For intracellular staining, we incubated the cells with brefeldin A (BFA, Sigma–Aldrich, 12.5 μg/mL) without restimulation for the last 4 h of culture at 37°C. Cell surface staining was performed as indicated above prior to fixation (BD cytofix), and permeablization (BD cytoperm). Intracellular staining was performed during permeabilization by mixing BD cytoperm 1X with the anti–cytokine antibody mix. Activated caspase–9 was detected using sulforhodamine (SR) –FLICA Caspase 9 kit (Bio-Rad). In brief, lungs were harvested and finely chopped in a well of 24-well plate containing 800 μL of PBS. Immediately after chopping, 14 μL of 60X FLICA solution was added to the wells and were incubated for 30 minutes at 37° C. Lung tissues were then washed with 1X apoptosis wash buffer and passed through a 70 μm cell strainer. The cell suspension was then prepared for flow cytometry as described above. A list of antibodies used in these studies is provided in Table S1. Gating strategies for all cell types discussed are shown in fig. S1.
Cell purification and transfer
OTI cells were purified from the spleens of CD45.1+ OTI mice by incubating single cell suspensions with anti-CD16/32 (BioXcell) and anti–CD8 magnetic beads (Miltenyi Biotec), for 45 minutes at 4°C. After washing, CD8+ cells were enriched by magnetic separation using LS–columns (Miltenyi Biotec) and stained with cell trace violet (CTV, Thermo–Fisher) according to instructions. Cells were adjusted to 6 x 105 cells/mL in sterile PBS and 50 μL of the cell suspension were transferred intraperitoneally. For DC enrichment, cell suspensions from dissociated lungs were incubated with anti–CD11c magnetic beads followed by magnetic separation using LS columns (Miltenyi Biotec). After enrichment, DCs were purified by first gating on Lin−CD64−MHCII+CD11c+ cells and sorted based on expression of CD103, CD11b, SIRPα. Post–sorting purity was >95%. All sorting was performed using a FACSAriaII provided by the Flow Cytometry and Single Cell services Core at UAB.
Cell culture.
CD8+ T cells were purified from splenocytes of naive OTI mice as described above. Sorted dendritic cells were first incubated with OVA257-264 peptide (InVivoGen) at a final concentration of 15 uM. After washing the unbound peptide, 3 x 103 DCs were plated with CTV-labeled OT-I cells at a 1:10 ratio. Intracellular cytokine production by the OTI T cells was measured after 4-days of incubation. CD4+ cells were purified from splenocytes of OTII mice using anti–CD4 magnetic beads (Miltenyi Biotec) and CTV labeled (Thermo-Fisher) as indicated by manufacturer. OTII cells were cultured with 1 x 103 sorted DCs in a 1:5 ratio in the presence of 1 μg of OVA protein (Endofit OVA, InvivoGen) in supplemented RPMI media (10% FBS, 1% HEPES, 1% Sodium pyruvate, 2% Penicillin / streptomycin, 2% L-Glutamine, 0.1% 2-Mercaptoethanol). After 4.5 days in culture, OT-II cells were stained for detection of intracellular FoxP3 using the eBioscience™ Mouse Regulatory T Cell Staining Kit according to the provided instructions. Sorted DCs were cultured at 2.5 – 5.0 ×104 cells per mL in supplemented RPMI media. After 12-hours of incubation, supernatants were collected for further analysis of IL-12p40 and IL-12p70 concentration by ELISA.
RNA isolation from sorted dendritic cells
Sorted DCs were pelleted, resuspended in 500 μL Trizol and incubated for 5 minutes at room temperature before adding 100 μL chloroform. After centrifuging, the aqueous phase was transferred to a clean tube and an equal volume of isopropanol was added. The resulting solution was transferred to a MinElute spin column (Qiagen) and the RNA extraction was continued as recommended by the RNeasy Plus Micro kit (Qiagen). RNA was eluted in 10 μL of RNAse free water and quality assessed by electrophoresis in a Bio–Rad Experion analyzer (Bio–Rad). All samples had a RIN > 8.0. Next generation sequencing was performed at Genewiz using Illumina HiSEq system with a minimum of 120 million reads per sample.
RNAseq analysis
Sequencing data was analyzed for quality control using FastQC and aligned to the mm10 genome using STAR aligner version 2.5.2a. The count matrix was analyzed using the R package DESeq2 (version 1.20.0)(50), and genes with counts higher than 20 were deemed expressed. For differential expression analysis, DESeq function was implemented on the raw counts, and an additive model (~ subset + batch) was applied to account for differences between batches. Genes with an adjusted P–value < 0.1 and an absolute value of log2 fold change ≥ 1 were defined as differentially expressed genes (DEGs). For visualization purposes, the removeBatchEffect function in the limma package (51) was used to adjust batch effect. The regularized–logarithm transformation was then conducted using rlog function, and heatmap was generated using the R package pheatmap.
For Gene Set Enrichment Analysis (GSEA)(52), all detected genes were ranked by the −log10 P–value with the sign, and the ranked list was used as input for the GSEA PreRanked analysis. For comparison with reference (23), we retrieved ranked gene lists labeled “immunogenic cluster 49, tolerogenic cluster 103, MAT OFF and MAT ON” and compared them to ranked gene lists obtained from our studies. For comparison with reference (24), we retrieved from GEO2R lists of DEGs obtained from CD103+ DCs from the small intestine lamina propria (SILP) that had either engulfed apoptotic GFP–expressing epithelial cells (GFP+SILP DC) or had not engulfed apoptotic epithelial cells (GFP−SILP DCs). The DEGs were separated into ranked lists of upregulated and downregulated genes and were compared to ranked gene lists obtained from our studies. For comparison with reference (26), the count matrix was analyzed using the Seurat package (53), and DEGs between mregDC and cDC1 populations were obtained using FindMarkers function. Again, ranked lists of upregulated and downregulated genes and were compared to ranked gene lists from our studies.
Single-cell RNAseq data analysis.
Live CD45+YFP+ cells were sorted from the lungs of two 7–day–old neonatal or two 8–week–old adult zDC-cre x ROSA–YFPfl/fl mice. We obtained 5074 and 4985 cells from the neonatal lungs and 2416 and 2235 cells from adult lungs. An aggregate count matrix was generated using Cell Ranger (version 6.1.2, 10x Genomics). Briefly, cell barcodes were demultiplexed, and reads were aligned to the mm10 genome using the cellranger multi pipeline. The cell ranger aggr was then used to normalize counts across samples. We next processed the aggregated matrix using the R package Seurat (54)(version 4.2.0). Cells with a total number of genes (nFeature_RNA) between 200 and 3500 and a gene count (nCount_RNA) lower than 15,000 were kept (1943 and 1706 for neonatal cells and 631 and 528 for adult cells). We further excluded cells whose mitochondrial gene counts accounted for more than 10% of total counts. Highly variable genes were detected using FindVariableDeatures function with the nFeatures parameter set to 1500. PCA was first implemented, and the top 20 PCs that reached statistical significance were used for UMAP transformation and cluster analysis. Using the expression of lineage genes (Ms4a1, Cd19, Ighd, Igha, Sdc1, Mzb1, Cd3e, Cd3g, Lck, Cd8a, Cd8b1, Cd4, Tcrg-C1, Trdc, Klrb1b, Klrb1c, Klrc1, Klrd1, Klrg1, Klra7, Nkg7, Lyz2, Cd68, Fcgr1, Ccr2, Ly6g, Zbtb46, Il12b, Siglech, Siglecf), we excluded non-DC contaminants from downstream analyses. We performed the normalization, detection of variable genes, and scaling on the resulting DC-specific subset. Cluster analysis identified 20 clusters, and the assignment of cells from neonatal and adult animals to clusters was determined. To examine the expression of gene modules associated with mregDCs, signature gene sets were compiled as in the bulk RNAseq analysis, and the module scores were calculated using AddModuleScore function. The module scores were further scaled across all the cells for visualization.
Photo–labeling of lung cells
Three–day old KIK–green mice were anesthetized in a mix of Oxygen–Isofluorane for 60 seconds then placed on their backs on a warm pad and exposed for 15 seconds to a focusable 405–nm laser (Laserpointerpro, 150 mW). Laser exposure was performed only on top of the rib cage above the lung left lobe avoiding as much as possible the sternum. After exposure mice were returned to their cage and euthanized at the indicated time points.
Histology
LNs from 2–week old mice were embedded in OCT (Scigen) and frozen at −80°C. LN sections were cut at a thickness of 8 μm using a Leica CM1850 cryostat (Leica) at −25°C and then fixed on a glass slides with cold acetone for 10 minutes. Dried slides were blocked with anti-CD16/32 and stained with antibody mix for 12 h at 4°C. Slides were washed 4 times in PBS before mounting with Prolong Gold Antifade Mountant (Invitrogen). Lungs were inflated with OCT (80% in PBS) and embedded in OCT (Scigen) and frozen at −80°C. Lung sections were cut at 8 μm thickness, then fixed with 4% PFA for 30 minutes at room temperature. TUNEL staining was performed with One-step TUNEL apoptosis kit – AF555 (Elabscience) following manufacturer’s instructions. Images were captured on a Nikon Ti–eclipse microscope using a 20X objective and analyzed with NIS AR software. The area of PNAd+CD31+ HEVs was quantified by first generating a PNAd+ mask and a CD31+ mask based on the fluorescence intensity of FITC and PE respectively. Then we generated a third mask composed of the intersection between the PNAd+ and CD31+ masks, which was used to quantify the HEV area. The proportion of TUNEL+ cells in neonatal and adult lungs was determined by calculating the density of AF555 stained cells per tissue area. Activated caspase 3/7 were detected using Magic Red (MR) Caspase 3/7 kit (Bio-Rad). Lung tissues were sectioned as described above, and the area of apoptotic cells was determined by calculating the density of MR+ cells per tissue area.
Statistical analysis
Statistical analyses were performed using GraphPad Prism versions 7.0 and 8.0. Comparison between two groups were analyzed with Student’s t–test. Comparisons with more than two groups were analyzed using one–way or two–way ANOVA followed by Tukey or Sidak post–test.
Supplementary Material
Data file S1. Raw data files.
Fig. S1 Gating strategies for flow cytometry analysis.
Fig. S2 Maturation markers expressed by lung dendritic cells.
Fig. S3. CD103int DCs produce IL-12p40 but not IL-12p70.
Fig. S4. Green-to-red photoconversion in neonatal lungs.
Fig. S5. cDC2 cells from neonatal lungs do not promote stromal cell maturation in mLNs.
Fig. S6. CD103int DCs have a regulatory signature.
Table S1. List of antibodies.
ACKNOWLEDGEMENTS.
We would like to dedicate this manuscript to Dr. Leopoldo Flores–Romo, who died on March 23, 2020. We thank Uma Mudunuru, Scott Simpler and Rebecca Burnham for their expertise in animal husbandry and Enid Keyser and Sagar Hanumanthu for assistance with cell sorting and Shanrun Liu for assistance with single cell RNAseq.
FUNDING.
The Flow Cytometry and Single Cell services Core is supported by the O’Neal Comprehensive Cancer Center CA013148 and the Center for AIDS Research AI027767. This work was supported by NIH grants AI100127 to T.D.R, AI125180 to F.E.L, and CA237742 to E.U.
Footnotes
COMPETING INTERESTS. The authors declare that they have no competing interests.
MATERIALS AND DATA AVAILABILITY STATEMENT.
The bulk RNAseq data is available under GEO accession number GSE112270. The single cell RNAseq data is available under GEO accession number GSE224178. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES.
- 1.Merad M, Sathe P, Helft J, Miller J, Mortha A, The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 31, 563–604 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guilliams M, Lambrecht BN, Hammad H, Division of labor between lung dendritic cells and macrophages in the defense against pulmonary infections. Mucosal Immunol 6, 464–473 (2013). [DOI] [PubMed] [Google Scholar]
- 3.del Rio ML, Rodriguez-Barbosa JI, Kremmer E, Forster R, CD103− and CD103+ bronchial lymph node dendritic cells are specialized in presenting and cross-presenting innocuous antigen to CD4+ and CD8+ T cells. J Immunol 178, 6861–6866 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Sung SS, Fu SM, Rose CE Jr., Gaskin F, Ju ST, Beaty SR, A major lung CD103 (alphaE)-beta7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J Immunol 176, 2161–2172 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Desch AN, Randolph GJ, Murphy K, Gautier EL, Kedl RM, Lahoud MH, Caminschi I, Shortman K, Henson PM, Jakubzick CV, CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell-associated antigen. J Exp Med 208, 1789–1797 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.del Rio ML, Bernhardt G, Rodriguez-Barbosa JI, Forster R, Development and functional specialization of CD103+ dendritic cells. Immunol Rev 234, 268–281 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco-Madeira F, Toussaint W, Vanhoutte L, Neyt K, Killeen N, Malissen B, Hammad H, Lambrecht BN, Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity 38, 322–335 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Sichien D, Lambrecht BN, Guilliams M, Scott CL, Development of conventional dendritic cells: from common bone marrow progenitors to multiple subsets in peripheral tissues. Mucosal Immunol 10, 831–844 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Ginhoux F, Liu K, Helft J, Bogunovic M, Greter M, Hashimoto D, Price J, Yin N, Bromberg J, Lira SA, Stanley ER, Nussenzweig M, Merad M, The origin and development of nonlymphoid tissue CD103+ DCs. J Exp Med 206, 3115–3130 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM, Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bajana S, Turner S, Paul J, Ainsua-Enrich E, Kovats S, IRF4 and IRF8 Act in CD11c+ Cells To Regulate Terminal Differentiation of Lung Tissue Dendritic Cells. J Immunol 196, 1666–1677 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, Ho AW, See P, Shin A, Wasan PS, Hoeffel G, Malleret B, Heiseke A, Chew S, Jardine L, Purvis HA, Hilkens CM, Tam J, Poidinger M, Stanley ER, Krug AB, Renia L, Sivasankar B, Ng LG, Collin M, Ricciardi-Castagnoli P, Honda K, Haniffa M, Ginhoux F, IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity 38, 970–983 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hammad H, Lambrecht BN, Lung dendritic cell migration. Adv Immunol 93, 265–278 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Randolph GJ, Ochando J, Partida-Sanchez S, Migration of dendritic cell subsets and their precursors. Annu Rev Immunol 26, 293–316 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Forster R, Davalos-Misslitz AC, Rot A, CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol 8, 362–371 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Astarita JL, Cremasco V, Fu J, Darnell MC, Peck JR, Nieves-Bonilla JM, Song K, Kondo Y, Woodruff MC, Gogineni A, Onder L, Ludewig B, Weimer RM, Carroll MC, Mooney DJ, Xia L, Turley SJ, The CLEC-2-podoplanin axis controls the contractility of fibroblastic reticular cells and lymph node microarchitecture. Nat Immunol 16, 75–84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saito Y, Respatika D, Komori S, Washio K, Nishimura T, Kotani T, Murata Y, Okazawa H, Ohnishi H, Kaneko Y, Yui K, Yasutomo K, Nishigori C, Nojima Y, Matozaki T, SIRPalpha(+) dendritic cells regulate homeostasis of fibroblastic reticular cells via TNF receptor ligands in the adult spleen. Proc Natl Acad Sci U S A 114, E10151–E10160 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Z, Li J, Zheng W, Zhao G, Zhang H, Wang X, Guo Y, Qin C, Shi Y, Peripheral Lymphoid Volume Expansion and Maintenance Are Controlled by Gut Microbiota via RALDH+ Dendritic Cells. Immunity 44, 330–342 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moussion C, Girard JP, Dendritic cells control lymphocyte entry to lymph nodes through high endothelial venules. Nature 479, 542–546 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Wendland M, Willenzon S, Kocks J, Davalos-Misslitz AC, Hammerschmidt SI, Schumann K, Kremmer E, Sixt M, Hoffmeyer A, Pabst O, Forster R, Lymph node T cell homeostasis relies on steady state homing of dendritic cells. Immunity 35, 945–957 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Jiang A, Bloom O, Ono S, Cui W, Unternaehrer J, Jiang S, Whitney JA, Connolly J, Banchereau J, Mellman I, Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity 27, 610–624 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manicassamy S, Reizis B, Ravindran R, Nakaya H, Salazar-Gonzalez RM, Wang YC, Pulendran B, Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science 329, 849–853 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ardouin L, Luche H, Chelbi R, Carpentier S, Shawket A, Montanana Sanchis F, Santa Maria C, Grenot P, Alexandre Y, Gregoire C, Fries A, Vu Manh TP, Tamoutounour S, Crozat K, Tomasello E, Jorquera A, Fossum E, Bogen B, Azukizawa H, Bajenoff M, Henri S, Dalod M, Malissen B, Broad and Largely Concordant Molecular Changes Characterize Tolerogenic and Immunogenic Dendritic Cell Maturation in Thymus and Periphery. Immunity 45, 305–318 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Cummings RJ, Barbet G, Bongers G, Hartmann BM, Gettler K, Muniz L, Furtado GC, Cho J, Lira SA, Blander JM, Different tissue phagocytes sample apoptotic cells to direct distinct homeostasis programs. Nature 539, 565–569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joeris T, Gomez-Casado C, Holmkvist P, Tavernier SJ, Silva-Sanchez A, Klotz L, Randall TD, Mowat AM, Kotarsky K, Malissen B, Agace WW, Intestinal cDC1 drive cross-tolerance to epithelial-derived antigen via induction of FoxP3(+)CD8(+) Tregs. Sci Immunol 6, (2021). [DOI] [PubMed] [Google Scholar]
- 26.Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, Chudnovskiy A, Maskey S, Walker L, Finnigan JP, Kirkling ME, Reizis B, Ghosh S, D’Amore NR, Bhardwaj N, Rothlin CV, Wolf A, Flores R, Marron T, Rahman AH, Kenigsberg E, Brown BD, Merad M, A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580, 257–262 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, Krishnan I, Maroni G, Meyerovitz CV, Kerwin CM, Choi S, Richards WG, De Rienzo A, Tenen DG, Bueno R, Levantini E, Pittet MJ, Klein AM, Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 50, 1317–1334 e1310 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bourdely P, Anselmi G, Vaivode K, Ramos RN, Missolo-Koussou Y, Hidalgo S, Tosselo J, Nunez N, Richer W, Vincent-Salomon A, Saxena A, Wood K, Lladser A, Piaggio E, Helft J, Guermonprez P, Transcriptional and Functional Analysis of CD1c(+) Human Dendritic Cells Identifies a CD163(+) Subset Priming CD8(+)CD103(+) T Cells. Immunity 53, 335–352 e338 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerhard GM, Bill R, Messemaker M, Klein AM, Pittet MJ, Tumor-infiltrating dendritic cell states are conserved across solid human cancers. J Exp Med 218, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Pilato M, Kfuri-Rubens R, Pruessmann JN, Ozga AJ, Messemaker M, Cadilha BL, Sivakumar R, Cianciaruso C, Warner RD, Marangoni F, Carrizosa E, Lesch S, Billingsley J, Perez-Ramos D, Zavala F, Rheinbay E, Luster AD, Gerner MY, Kobold S, Pittet MJ, Mempel TR, CXCR6 positions cytotoxic T cells to receive critical survival signals in the tumor microenvironment. Cell 184, 4512–4530 e4522 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lemke G, Biology of the TAM receptors. Cold Spring Harb Perspect Biol 5, a009076 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cupedo T, Lund FE, Ngo VN, Randall TD, Jansen W, Greuter MJ, de Waal-Malefyt R, Kraal G, Cyster JG, Mebius RE, Initiation of cellular organization in lymph nodes is regulated by non-B cell-derived signals and is not dependent on CXC chemokine ligand 13. J Immunol 173, 4889–4896 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Iwasaki A, Medzhitov R, Toll-like receptor control of the adaptive immune responses. Nat Immunol 5, 987–995 (2004). [DOI] [PubMed] [Google Scholar]
- 34.de Kleer IM, Kool M, de Bruijn MJ, Willart M, van Moorleghem J, Schuijs MJ, Plantinga M, Beyaert R, Hams E, Fallon PG, Hammad H, Hendriks RW, Lambrecht BN, Perinatal Activation of the Interleukin-33 Pathway Promotes Type 2 Immunity in the Developing Lung. Immunity 45, 1285–1298 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Saluzzo S, Gorki AD, Rana BMJ, Martins R, Scanlon S, Starkl P, Lakovits K, Hladik A, Korosec A, Sharif O, Warszawska JM, Jolin H, Mesteri I, McKenzie ANJ, Knapp S, First-Breath-Induced Type 2 Pathways Shape the Lung Immune Environment. Cell Rep 18, 1893–1905 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanchez-Sanchez N, Riol-Blanco L, de la Rosa G, Puig-Kroger A, Garcia-Bordas J, Martin D, Longo N, Cuadrado A, Cabanas C, Corbi AL, Sanchez-Mateos P, Rodriguez-Fernandez JL, Chemokine receptor CCR7 induces intracellular signaling that inhibits apoptosis of mature dendritic cells. Blood 104, 619–625 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Marsland BJ, Battig P, Bauer M, Ruedl C, Lassing U, Beerli RR, Dietmeier K, Ivanova L, Pfister T, Vogt L, Nakano H, Nembrini C, Saudan P, Kopf M, Bachmann MF, CCL19 and CCL21 induce a potent proinflammatory differentiation program in licensed dendritic cells. Immunity 22, 493–505 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Mori S, Nakano H, Aritomi K, Wang CR, Gunn MD, Kakiuchi T, Mice lacking expression of the chemokines CCL21-ser and CCL19 (plt mice) demonstrate delayed but enhanced T cell immune responses. J Exp Med 193, 207–218 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luther SA, Tang HL, Hyman PL, Farr AG, Cyster JG, Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc Natl Acad Sci U S A 97, 12694–12699 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warburton D, El-Hashash A, Carraro G, Tiozzo C, Sala F, Rogers O, De Langhe S, Kemp PJ, Riccardi D, Torday J, Bellusci S, Shi W, Lubkin SR, Jesudason E, Lung organogenesis. Curr Top Dev Biol 90, 73–158 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Narayanan M, Owers-Bradley J, Beardsmore CS, Mada M, Ball I, Garipov R, Panesar KS, Kuehni CE, Spycher BD, Williams SE, Silverman M, Alveolarization continues during childhood and adolescence: new evidence from helium-3 magnetic resonance. Am J Respir Crit Care Med 185, 186–191 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bruce MC, Honaker CE, Cross RJ, Lung fibroblasts undergo apoptosis following alveolarization. Am J Respir Cell Mol Biol 20, 228–236 (1999). [DOI] [PubMed] [Google Scholar]
- 43.Lee HH, Hoeman CM, Hardaway JC, Guloglu FB, Ellis JS, Jain R, Divekar R, Tartar DM, Haymaker CL, Zaghouani H, Delayed maturation of an IL-12-producing dendritic cell subset explains the early Th2 bias in neonatal immunity. J Exp Med 205, 2269–2280 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gollwitzer ES, Saglani S, Trompette A, Yadava K, Sherburn R, McCoy KD, Nicod LP, Lloyd CM, Marsland BJ, Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat Med 20, 642–647 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Pongracz JE, Stockley RA, Wnt signalling in lung development and diseases. Respir Res 7, 15 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Domingo-Gonzalez R, Zanini F, Che X, Liu M, Jones RC, Swift MA, Quake SR, Cornfield DN, Alvira CM, Diverse homeostatic and immunomodulatory roles of immune cells in the developing mouse lung at single cell resolution. Elife 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, Cheong C, Liu K, Lee HW, Park CG, Steinman RM, Nussenzweig MC, Differential antigen processing by dendritic cell subsets in vivo. Science 315, 107–111 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Vander Lugt B, Khan AA, Hackney JA, Agrawal S, Lesch J, Zhou M, Lee WP, Park S, Xu M, DeVoss J, Spooner CJ, Chalouni C, Delamarre L, Mellman I, Singh H, Transcriptional programming of dendritic cells for enhanced MHC class II antigen presentation. Nat Immunol 15, 161–167 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Fletcher AL, Malhotra D, Acton SE, Lukacs-Kornek V, Bellemare-Pelletier A, Curry M, Armant M, Turley SJ, Reproducible isolation of lymph node stromal cells reveals site-dependent differences in fibroblastic reticular cells. Front Immunol 2, 35 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Satija R, Farrell JA, Gennert D, Schier AF, Regev A, Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33, 495–502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data file S1. Raw data files.
Fig. S1 Gating strategies for flow cytometry analysis.
Fig. S2 Maturation markers expressed by lung dendritic cells.
Fig. S3. CD103int DCs produce IL-12p40 but not IL-12p70.
Fig. S4. Green-to-red photoconversion in neonatal lungs.
Fig. S5. cDC2 cells from neonatal lungs do not promote stromal cell maturation in mLNs.
Fig. S6. CD103int DCs have a regulatory signature.
Table S1. List of antibodies.
Data Availability Statement
The bulk RNAseq data is available under GEO accession number GSE112270. The single cell RNAseq data is available under GEO accession number GSE224178. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.