

Abstract
Antimicrobial peptides (AMPs) are short peptides that play crucial roles in diverse biological processes and have various functional activities against target organisms. Due to the abuse of chemical antibiotics and microbial pathogens’ increasing resistance to antibiotics, AMPs have the potential to be alternatives to antibiotics. As such, the identification of AMPs has become a widely discussed topic. A variety of computational approaches have been developed to identify AMPs based on machine learning algorithms. However, most of them are not capable of predicting the functional activities of AMPs, and those predictors that can specify activities only focus on a few of them. In this study, we first surveyed 10 predictors that can identify AMPs and their functional activities in terms of the features they employed and the algorithms they utilized. Then, we constructed comprehensive AMP datasets and proposed a new deep learning-based framework, iAMPCN (identification of AMPs based on CNNs), to identify AMPs and their related 22 functional activities. Our experiments demonstrate that iAMPCN significantly improved the prediction performance of AMPs and their corresponding functional activities based on four types of sequence features. Benchmarking experiments on the independent test datasets showed that iAMPCN outperformed a number of state-of-the-art approaches for predicting AMPs and their functional activities. Furthermore, we analyzed the amino acid preferences of different AMP activities and evaluated the model on datasets of varying sequence redundancy thresholds. To facilitate the community-wide identification of AMPs and their corresponding functional types, we have made the source codes of iAMPCN publicly available at https://github.com/joy50706/iAMPCN/tree/master. We anticipate that iAMPCN can be explored as a valuable tool for identifying potential AMPs with specific functional activities for further experimental validation.
Keywords: antimicrobial peptides, bioinformatics, sequence analysis, machine learning, deep learning, functional activities
INTRODUCTION
Due to the microbial pathogens’ increasing resistance to chemical antibiotics, it is urgent to develop novel infectious therapeutics [1, 2]. Over the past decade, there have been several developments in utilizing antimicrobial peptides (AMPs) as potential alternatives to treat infections since most natural AMPs are particular polypeptide substances in living organisms and are critical components of the innate immune system which protects the host against invading pathogens [3]. AMPs are generally small-molecule polypeptides and have diverse functional activities against target organisms such as bacteria, yeasts, fungi, viruses and cancer cells. Compared with traditional chemical antibiotics, AMPs have higher antibacterial activities, broader antibacterial spectrums and fewer possibilities resulting in target strains’ resistance mutation [4]. Therefore, AMPs have a wide range of application prospects in the pharmaceutical industry and have become a hotspot in biomedical research [4].
Based on existing research data about AMPs, many efforts have been made to construct various databases containing experimentally validated AMPs. To date, a number of databases have been developed to provide comprehensive experimentally verified annotations of AMPs. For example, the antimicrobial peptide database (APD3) [5] includes various AMPs with different functional activities such as antibacterial, antifungal, antiviral and anticancer. It also provides a user-friendly web page for peptide classification, search and prediction. dbAMP [6, 7] is another comprehensive AMP database specifically focusing on AMPs’ functional and physicochemical characteristics in high-throughput transcriptome and proteome data. It also provides relevant annotations of AMP–protein interactions and targeting species of AMPs. DRAMP [8–10] offers access to minimum inhibitory concentration values and structure information of AMPs, and LAMP [11, 12] crosslinks existing AMP databases into account and provides the related information. In addition to comprehensive databases, some disease-specific databases integrate AMPs with specific functional activities, such as AntiTbPdb Field [13], a database for antitubercular (anti-TB) peptides. With the continuous expansion and development of AMP databases, identifying AMPs and their functional types accurately by computational methods has become increasingly important for AMP research, due to the time-consuming, expensive and laborious wet-laboratory experiments.
In the last decade, a plethora of computational predictors have been developed for identifying AMPs [13–16]; a few attempts have also been made to review, benchmark and evaluate these approaches [17, 18]. However, the majority of these predictors only focus on identifying AMPs. As such, they cannot be used to predict the functional activities of particular interest to biomedical researchers. Several predictors have been proposed to predict AMPs with one specific functional activity, such as DeepAVP for predicting antiviral peptides [19], Deep-AFPpred for predicting antiviral peptides [20], and StaBle-ABPpred [21] and Deep-ABPpred [22] for predicting antibacterial peptides. However, these predictors have limited predictive capability and cannot provide comprehensive functional activity annotations of AMPs. Furthermore, only a few predictors can predict multiple functional activities of AMPs, given the significance of AMP functional activities and the fact that none of the work systematically summarized and evaluated these computational approaches for predicting AMPs and their functional activities. Herein, we reviewed these computational approaches comprehensively, including the involved functional activities, benchmark datasets, machine learning algorithms, feature selection algorithms, and performance evaluation strategies and metrics. Then, we developed a predictive framework named iAMPCN (identification of AMPs and their functional activities based on convolutional neural networks), which is composed of multiple single-class models. We evaluated its ability to identify different kinds of functional activities of AMPs. The performance evaluation results demonstrated that iAMPCN achieved superior performances in identifying AMPs and their functional types compared with available predictive tools. We anticipate that iAMPCN can serve as a prominent tool for identifying potential AMPs and their specific functions that can be experimentally validated.
MATERIALS AND METHODS
Existing approaches for predicting AMPs and their functional types
In the present study, we comprehensively reviewed 10 computational predictors for AMPs and their activities regarding the predicted functional activities, data sources, algorithms and performance evaluation strategies in Table 1 and Figure 1. We also provided a summary of the key features used in these approaches in Table 2. We grouped these tools into traditional machine learning (ML)-based and deep learning (DL)-based according to the algorithms they applied.
Table 1.
A comprehensive summary of the reviewed approaches for AMP prediction
| Category | Tool | Year | Prediction of functional activities | Positive data source | CD-HIT cut-off | Algorithm | Sampling strategy | Feature selection | Evaluation strategy | Evaluation metrics | Webserver/software availability |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Machine learning based | iAMP-2L | 2013 | Antibacterial, anticancer, antiviral, antifungal, anti-HIV | APD2 | 1st: 40%; 2nd: 40% | FKNN | N.A. | N.A. | Jack-knife validation test, independent test | Acc, Sp, Sn, MCC, macro Pr, macro Sn, macro Acc, SA, HL | Yes |
| MLAMP | 2016 | Antibacterial, anticancer, antifungal, anti-HIV, antiviral | APD2 | 1st: 40%; 2nd: 40% | RF, ECC | SMOTE | N.A. | Jack-knife validation test, independent test | Acc, Sp, Sn, MCC, macro Pr, macro Sn, macro Acc, SA, HL | Yes | |
| AMAP | 2019 | Antibacterial, anticancer, antifungal, anti-HIV, antiviral, antibiofilm, antiparasitic, chemotactic, insecticidal, antimalarial, antioxidant, spermicidal, anti-protist | APD3 | 1st: N.A.; 2nd: N.A. | SVM, XGB, one-vs-rest classifier fusion | N.A. | N.A. | LOCO, 5-fold CV, independent test | AUC, AUPR, MCC | Yes | |
| AMPfun | 2019 | Antiparasitic, anticancer, antifungal, antiviral, targeting mammals, targeting Gram-positive bacteria, targeting Gram-positive bacteria | APD3, ADAM, ParaPep, AVPdb, CancerPPD, MLACP, AntiCP, AntiFP, DRAMP | 1st: 50%; 2nd: N.A. | DT, RF, SVM | N.A. | SFS | 10-fold CV, independent test | Acc, Sp, Sn, MCC, AUC | Yes | |
| Zhang et al.’ s work | 2020 | Anti-MRSA, antibacterial, antiviral, antifungal, anticancer, anti-HIV, antiparasitic | APD3, APD2 | 1st: 40%; 2nd: 40% | GBDT, ET, ECC | ADASYN | Lasso | 10-fold CV, independent test | Acc, Sp, Sn, MCC, macro Pr, macro Sn, macro Acc, HL, AP | No | |
| iAMP-RAAC | 2021 | Antiparasitic, anticancer, antifungal, antiviral, targeting mammals, targeting Gram-positive bacteria, targeting Gram-positive bacteria | APD3, ADAM, ParaPep, AVPdb, CancerPPD, MLACP, AntiCP, AntiFP, DRAMP | 1st: 50%; 2nd: N.A. | SVM | N.A. | ANOVA, IFS | 10-fold CV,independent test | Acc, Sp, Sn, MCC | Yes | |
| dbAMP 2.0 | 2021 | Anticancer, antifungal, antiviral, targeting mammals, targeting Gram-positive bacteria, targeting Gram-positive bacteria | dbAMP 2.0, APD2, CAMP | 1st: 50%; 2nd: N.A. | GBDT | N.A. | N.A. | 5-fold CV | Sn, Sp, balanced Acc | Yes | |
| Deep learning based | iAMP-CA2L | 2021 | Antibacterial, anticancer, antifungal, anti-HIV, antiviral, antibiofilm, antiparasitic, chemotactic, anti-MRSA, antiendotoxin | APD3, AMPer, ADAM | 1st: 90%; 2nd: 90% | DL | N.A. | N.A. | Jack-knife validation, independent test | Acc, Pr, Sp, Sn, MCC, F1, SA, AP, coverage, HL, RL, OE, macro Pr, macro Sn, macro F1, micro Pr, micro Sn, micro F1 | Yes |
| Multi-AMP | 2021 | Anti-MRSA, antibacterial, antiviral, antifungal, anticancer, anti-HIV, antiparasitic | APD3, APD2 | 1st: 40%; 2nd: 40% | DL | N.A. | N.A. | 5-fold CV, independent test | Acc, Sp, Sn, MCC, F1, macro Sp, macro Sn, macro Acc, HL, macro MCC, macro F1 | No | |
| AMPDiscover | 2021 | Antibacterial, antifungal, antiviral, antiparasitic | starPepDB | 1st: 50%; 2nd: N.A. | RF, DL | N.A. | Correlation subset, SE, Relif-F, IG, gain ratio, symmetrical uncertainty | 5-fold CV, 10-fold CV, 15-fold CV, 20-fold CV, 25-fold CV, independent test | Acc, Sp, Sn, MCC, AUC | Yes |
Figure 1.
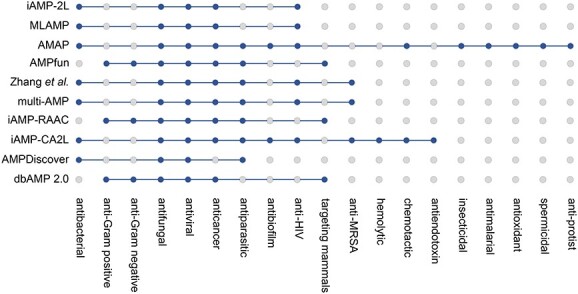
A summary of current computational approaches for predicting AMPs and their functional activities. The blue dots indicate the functional activities that the predictors can predict.
Table 2.
Different types of features employed by the reviewed approaches for AMP prediction
| Feature type | Feature | Tools |
|---|---|---|
| Composition features | Amino acid composition (AAC) | AMAP, AMPfun, dbAMP 2.0 |
| Dipeptide composition (DPC) | dbAMP 2.0 | |
| N-gram composition found by counting (NCC) | AMPfun | |
| N-gram composition found by t-test (NTC) | AMPfun | |
| Motifs composition (MC) | AMPfun | |
| Position features | N-gram binary profiling of position found by counting (NCB) | AMPfun |
| N-gram binary profiling of position found by t-test (NTB) | AMPfun | |
| Motifs binary profiling of position (MB) | AMPfun | |
| Structural and physicochemical properties | Physicochemical properties | dbAMP 2.0 |
| Composition/transition/distribution (CTD) | AMPfun | |
| Pseudo amino acid composition (PseAAC) | iAMP-2L, MLAMP, AMPfun, dbAMP 2.0 | |
| Conjoint triad (CTriad) | AMAP | |
| Reduced amino acid clusters (RAAC) | iAMP-RAAC | |
| Protein Descriptors Calculation (ProtDCal) | AMPDiscover | |
| Evolutionary information | Cellular automata images (CAIs) | iAMP-CA2L |
Machine learning-based approaches
To our best knowledge, iAMP-2L [23] is the first AMP predictor, which utilizes the fuzzy K-nearest neighbor (FKNN) algorithm to identify both AMPs and their functional activities, including antibacterial, anticancer, antiviral, antifungal and anti-human immunodeficiency virus (anti-HIV). The first-level prediction of iAMP-2L is to determine whether a peptide is an AMP, for which the FKNN [24] algorithm was utilized. The second-level prediction of iAMP-2L was designed to characterize the functional activity types of the queryAMP, for which the multi-label fuzzy K-nearest neighbor (ML-FKNN) classifier was applied. Both levels of prediction utilized the pseudo amino acid composition (PseAAC) [25] to represent sequences. In another work, Lin et al. proposed MLAMP [26] by mainly focusing on addressing the unbalanced labeled problem in the two-level AMP prediction. ML-SMOTE was developed by modifying the synthetic minority oversampling technique (SMOTE) [27] based on the multi-label classification. In addition, MLAMP converted the PseAAC [25] into new vectors as sequence representations with the grey model [28]. Another approach utilizing oversampling technique was proposed by Zhang et al. [29], which employed adaptive synthetic sampling (ADASYN) [30] to handle the data imbalance issue. Both MLAMP and Zhang et al.’s work applied the ensemble classifier chain (ECC) [31] to classify the AMP functional activities where the relationship between labels was considered. The only difference is that Zhang et al. utilized the gradient boosting decision tree (GBDT) [32] as the classifier for the first stage of prediction and the extra tree (ET) [33] for the second stage. In contrast, both stages of MLAMP applied the random forest (RF) as the classifier. Besides, Zhang et al. adopted Lasso [34] for feature selection to further improve the performance. AMAP [35] utilized the support vector machine (SVM) [36] and extreme gradient boosting (XGBoost) [37] as the classifiers, trained with amino acid composition (AAC) [38] and physicochemical properties to identify AMPs. Compared with other predictors, AMAP can identify the most functional types. AMPfun [39] is also based on RF but was built on a more comprehensive training dataset by integrating multiple AMP databases. AMPfun employed abundant sequence-based features, including compositional information, physicochemical descriptions and binary profiles, for model training. It can classify a range of functional activities, including antiparasitic, antiviral, anticancer, antifungal and anti-mammalian cells, anti-Gram positive and anti-Gram negative. iAMP-RAAC [40] is also a two-stage AMP predictor based on SVM and reduced amino acid cluster (RAAC) [41] features. iAMP-RAAC was trained on the same datasets as AMPfun and can predict the same functional activities as AMPfun. In addition, dbAMP 2.0 [6], an AMP database, has a user interface developed based on the GBDT algorithm for identifying AMPs and their functional types.
Deep learning-based approaches
With the advances and applications of DL techniques in bioinformatics, several DL-based predictors have been recently developed. Among them, iAMP-CA2L [42] is the first DL-based predictor for AMPs and their functional activities. iAMP-CA2L used the ANTIALIAS [43] technology to extract features from cellular automata images (CAIs) [44] of sequences and convolutional neural networks (CNNs) [45] and long short-term memory (LSTM) [46] to further extract features. Then, two SVMs were trained with these features to distinguish AMPs from non-AMPs and annotate 10 different functional activities, including antibacterial, antiviral, antifungal, antibiofilm, antiparasitic, anti-HIV, anticancer, chemotactic, anti-Methicillin-resistant Staphylococcus aureus (anti-MRSA) and antiendotoxin. Another recent approach, multi-AMP [47], has also been developed based on DL. However, different from iAMP-CA2L, multi-AMP considered the AMP identification as a multi-task problem. Accordingly, the final output layers of multi-AMP are used to address two tasks: AMP identification and their functional activity prediction. The models for these tasks shared identical parameters in the front layers (CNNs) for feature extraction but were structured with different dense layers for the final prediction. Multi-AMP used the position-specific scoring matrix [48] to represent peptide sequences. AMPDiscover [49] is another recently developed webserver, which provides two predictive models for AMPs: RF models with six different feature selection strategies, and the RNNs (recurrent neural networks) [50].
Sequence similarity threshold
To remove the redundant sequences from the training datasets to avoid the overfitting issue during training machine learning models, most studies employed CD-HIT [51–53] with a relatively stringent sequence similarity threshold to reduce the sequence redundancy. For example, iAMP-2L [23] and Zhang et al.’s work [29] set the sequence identity threshold at 40%. AMPfun [39], dbAMP 2.0 [6] and AMPDiscover [49] set the sequence identity threshold at 50%. However, in the case of functional activity prediction of AMPs, most studies set a relatively loosened threshold or did not remove redundant sequences, which is probably because of the rather limited annotations of functional activities. For example, iAMP-CA2L [42] set the sequence identity threshold at 90%, while AMAP [35], AMPfun [39], dbAMP 2.0 [6] and AMPDiscover [49] did not remove the sequence redundancy.
Model development
Although several approaches have been proposed to characterize AMPs and their functional activities, they have some drawbacks or limitations. First, the majority of the existing approaches only focused on limited functional activities. Although other approaches can cover more functional types, they were developed based on relatively out-of-date databases with fewer AMPs, resulting in the inaccurate annotation of the functional activities. AMAP can predict several activities that other methods cannot, such as antibiofilm, insecticidal, antimalarial, antioxidant, spermicidal and anti-protist. However, its models for identifying these functional activities were trained with less than 50 positive samples, which was particularly the case that only four positive samples were used to identify the ‘anti-protist’ activity of AMPs. Accordingly, we assume that the predictive results of these models could be less reliable, given the fact that many publicly available AMP databases have been updated recently. Second, different AMP databases may have some differences in AMPs that they include. Nevertheless, most existing AMP predictors were developed based on the data collected from one or only a few databases. As a result, the training datasets on which these predictors were built could be biased and poorly annotated. In view of these shortcomings, it is necessary to curate a comprehensive training dataset by integrating the available databases and developing more accurate predictors for identifying AMPs and their functional activities.
In this study, we introduce a two-stage computational framework, termed identification of AMPs and their functional activities based on Convolutional Neural networks (iAMPCN), to address the aforementioned shortcomings and improve the predictive performance of AMPs and their functional activities. Specifically, iAMPCN employs the one-hot encoding scheme to encode the peptide sequences and a two-stage framework coupled with CNN to train the model and conduct the prediction.
Dataset construction
iAMPCN is a two-stage computational framework for identifying AMPs and their functional types. Accordingly, we constructed comprehensive benchmark training and independent test datasets for both tasks. Our detailed procedures are described below.
In the first stage, for the positive dataset, we collected the AMPs with functional activity annotations from several databases, including APD3 [5], dbAMP [6, 7], DRAMP [8–10], dbaasp [54–56], CAMP [57, 58], LAMP [11, 12], ParaPep [59], phytAMP [60], AVPdb [61], CancerPPD [62], AntiTbPdb [63], Hemolytik [64] and milkAMP [65], as well as the training datasets of 24 AMP classifiers, including ADAM [66], iAMP-2L [23], AMPfun [39], iAMP-CA2L [42], MLACP [67], AntiCP 2.0 [68], ACPred [69], ACPred-FL [70], ACPred-Fuse [71], ACP-DL [72], iACP-DRLF [73], mACPpred [74], AntiFP [75], AVPpred [76], AVPIden [77], AntiTbPred [78], AtbPpred [78], BIOFIN [79], BIPEP [80], dPABBs [81], HemoPI [82], HLPpred-Fuse [83], iAntiTB [84], AnOxPePred [85] and HAPPENN [86]. Then, we eliminated those sequences with non-standard residues such as ‘B’, ‘J’, ‘O’, ‘U’, ‘X’ or ‘Z’. Finally, 49 115 experimentally validated AMP sequences were extracted from these resources. We performed the following procedures to generate the negative dataset: (i) peptide sequences were downloaded from UniProt [87–90] (http://www.uniprot.org), and all entries containing the keyword ‘antimicrobial’ and related keywords (e.g. ‘antibacterial’, ‘antifungal’, ‘anticancer’, ‘antiviral’, ‘antiparasitic’, ‘anticancer’, ‘antibiotic’, ‘antibiofilm’ or ‘effector’) were removed; (ii) the sequences with the length greater than 200 or less than 10 amino acid residues and with non-standard residues ‘B’, ‘J’, ‘O’, ‘U’, ‘X’ or ‘Z’ were discarded; (iii) the CD-HIT program [51–53] was applied to remove the sequences that had 40% pairwise sequence identity to those in the positive dataset. Finally, 195 525 negative samples were obtained.
Here, we applied the same strategy as AMPfun [39] to construct the training and independent test datasets for each functional type. We collected positive and negative samples from 49 115 experimentally validated AMP sequences for each functional activity. The authors of AMPfun [39] and iAMP-CA2L [42] also constructed a relatively large AMP dataset from several different databases. As mentioned in AMPfun [39], a peptide with a specific functional activity would be used as a positive sample of this activity; otherwise, it would be treated as a negative sample. We integrated the positive and negative datasets for 22 different functional types in the same way. It is known that the dataset containing instances with missing labels is an unavoidable problem. Even though all AMP sequences are obtained from only one database, it is very likely that these sequences need to be completely annotated since different AMP databases have been updated several times. In addition, there are a large number of overlapping AMPs between different AMP databases, and the linking antimicrobial peptides (LAMP) [11, 12] database was constructed to reflect the crosslink between several AMP databases [11, 12]. Therefore, combining peptides from multiple databases can make the activity annotations more complete. Irrespective of the combinations or not, instances with missing labels must exist. Because of the large number of overlapping AMPs, combining peptides from multiple datasets will be better than using peptides from only one dataset.
A detailed summary of positive and negative datasets for the first and second stages is shown in Table 3. For training and evaluating predictive models for AMP and functional activity predictions, we randomly selected 20% samples from positive and negative datasets to construct test datasets. We also used the remaining 80% of the samples as training datasets. We then filtered those sequences used in the training datasets of other predictive tools and split the remaining datasets to construct the independent datasets and training datasets. Detailed information on training, test and independent test datasets is shown in Supplementary Tables S1 and S2.
Table 3.
The numbers of positive and negative samples for predicting AMP and their functional activities
| Positive samples | Negative samples | |
|---|---|---|
| AMPs | 49 115 | 195 525 |
| Functional activity | ||
| Antibacterial | 16 058 | 13 551 |
| Antibiofilm | 372 | 29 202 |
| Anticancer | 4698 | 24 876 |
| Anticandidal | 669 | 28 905 |
| Antifungal | 5955 | 23 619 |
| Anti-Gram negative | 8537 | 21 044 |
| Anti-Gram positive | 8053 | 21 539 |
| Anti-HIV | 812 | 28 762 |
| Antimalarial | 73 | 29 501 |
| Anti-MRSA | 267 | 29 307 |
| Antiparasitic | 457 | 29 117 |
| Antiplasmodial | 63 | 29 511 |
| Antiprotozoal | 53 | 29 521 |
| Anti-TB | 275 | 29 299 |
| Antiviral | 6495 | 23 093 |
| Anti-mammalian cells | 4345 | 25 229 |
| Anuran defense | 779 | 28 795 |
| Chemotactic | 85 | 29 489 |
| Cytotoxic | 180 | 29 394 |
| Endotoxin | 82 | 29 492 |
| Hemolytic | 2383 | 27 191 |
| Insecticidal | 415 | 29 159 |
Model construction
An overview of the architecture of iAMPCN is illustrated in Figures 2 and 3. Here, we utilized four different types of amino acid information to represent peptide sequences, including one-hot encoding, BLOSUM62 encoding, AAIndex [91, 92] encoding and PAAC encoding. When using the one-hot encoding scheme, a peptide sequence is converted into a numerical matrix with the dimension of 200*21, where 200 represents the length of a peptide sequence and a 21-dimensional binary vector represents each amino acid type. For example, the one-hot encoding of A is [1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0], C is [0,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0] and the one-hot encoding of the non-standard amino acids is [0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0, 0,0,0,1]. The model with one-hot vector encoding with 21 bits and the model with one-hot vector encoding with 20 bits are the same when training and testing the sequences with usual amino acids (a detailed analysis is provided in the Supplementary Methods). We padded the vectors with ‘0’ for those sequences shorter than 200 and removed parts of sequences for those longer than 200 to obtain fixed dimensional matrices. For BLOSUM62 encoding, a peptide sequence is converted into a numerical matrix with the dimension of 200*23, where 200 again represents the length of a peptide sequence and a 23-dimensional vector represents each amino acid type. This encoding reflects the evolutionary information of amino acid residues. For the AAIndex encoding, a peptide sequence is converted into a numerical matrix with the dimension of 200*531. Each amino acid type is represented by a 531-dimensional vector that presents various physicochemical and biochemical properties of amino acids. Lastly, for the PAAC encoding, a peptide sequence is converted into a numerical matrix with a dimension of 200*3. Each amino acid type is represented by a three-dimensional vector that indicates the original hydrophobicity, hydrophilicity and side-chain masses of amino acid residues. Recently, Otović et al. [93] proposed a model based on the recursive neural network (RNN) to predict therapeutic peptides. This model also used physicochemical encodings to represent sequences. Without using the AAIndex [91, 92], it used the ‘AAdata’ from the R package ‘Peptides’ [94]. Each amino acid of ‘AAdata’ has 94 physicochemical properties that reflect hydrophobicity, electronegativity, alpha and turn propensities, etc. Most of these physicochemical properties were calculated using some formulas, such as principal components and Z-scale. Compared with ‘AAdata’, ‘AAIndex’ contains more enriched and detailed physicochemical properties. For example, ‘AAIndex’ includes the ‘short and medium range non-bonded energy per residue’, which are not included in ‘AAdata’. In addition, ‘AAIndex’ also provides other original, informative physicochemical properties.
Figure 2.

The architecture of iAMPCN. Four kinds of sequence representations were utilized, and different CNN models with different filter lengths ranging from two to six were utilized to extract feature information of varying sequence lengths from each type of encodings.
Figure 3.

Sequence analysis of AMPs collected for this study. (A) Amino acid distributions of AMP sequences versus non-AMP sequences. (B) An UpSet plot demonstrating the statistics of AMPs with overlapping and/or unique functional activities. (C) A Venn diagram illustrating the distributions of AMPs with five main functional activities, including antibacterial, anticancer, antifungal, antiviral and anti-mammalian cells. (D) Amino acid distributions of the AMPs with the five major functional activities. (E) Amino acid distributions of anti-Gram-positive and anti-Gram-negative AMPs.
The one-dimensional CNNs (i.e. Conv1d) with different filter lengths ranging from two to six were utilized to extract feature information of varying sequence lengths from each type of encoding. First, the features extracted by each CNN were normalized and inputted into a pooling layer. Then, the extracted features from the same encoding type were combined and inputted into a pooling layer to obtain the final features. After combining these features, a final dense layer was used to give the final predictive output. The parameters of this framework are described in detail in the Supplementary Methods.
In this study, we utilized the transfer learning strategy—specifically, we applied the model for predicting AMPs and non-AMPs trained at the first stage of AMP prediction, as the pretrained model to initialize all parameters’ weights of the models for predicting functional activities. The learning rates of model training for both the AMP prediction and AMP functional activity prediction were set as the same, i.e. 0.0001. Due to the imbalanced datasets of certain functional activities, the focal loss [95] was applied as follows:
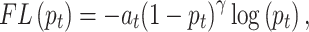 |
where 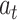 controls the weights of the samples belonging to positive and negative classes, and
controls the weights of the samples belonging to positive and negative classes, and 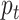 is an estimated probability by the model,
is an estimated probability by the model, 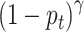 is considered to be a modulating factor that controls the effects of easy-classified samples. Here,
is considered to be a modulating factor that controls the effects of easy-classified samples. Here, 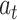 and
and 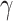 were set to 1 and 2, respectively. In addition, the early stopping strategy was applied during the 10-fold stratified cross-validation test to avoid overfitting.
were set to 1 and 2, respectively. In addition, the early stopping strategy was applied during the 10-fold stratified cross-validation test to avoid overfitting.
Performance evaluation strategies and metrics
Performance evaluation strategies, including the K-fold stratified cross-validation test, jack-knife validation test and independent test, are commonly used to evaluate predictors and optimize the parameters of models. In this study, we employed the 10-fold stratified cross-validation test on training data sets for model selection and optimization. In addition, we used the independent test to compare the predictive performance of different tools. To objectively assess and compare the model’s performance with different tools and web servers, six performance metrics were used, including sensitivity, specificity, precision, accuracy, Matthew’s correlation coefficient (MCC) [96] and area under the ROC curve (AUROC or AUC), which are defined as follows:
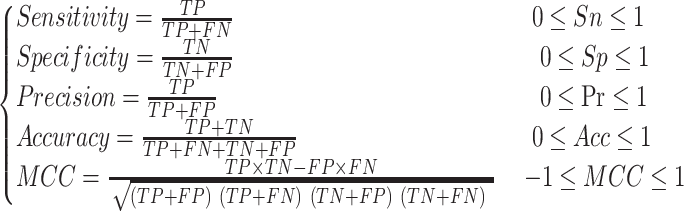 |
where TP and TN represent the numbers of correctly predicted positive and negative samples, respectively, while FP and FN represent the numbers of incorrectly predicted positive and negative samples, respectively.
RESULTS AND DISCUSSION
Sequence analysis of collected AMPs
The amino acid distributions of AMPs and non-AMPs in the constructed datasets are shown in Figure 3A. Compared with non-AMPs, cysteine (C), lysine (K) and arginine (R) residues appeared to be abundant in AMP sequences. This observation is consistent with previous studies, which show that most active AMP sequences are abundant in hydrophilic or positively charged amino acids [3, 97]. An overview of the extracted functional activities of the AMP sequences is shown in Figure 3B–C. We compared the amino acid distributions of AMPs in terms of five main functional activities (Figure 3D). We found that the AMPs with the activity of targeting mammalian cells have more lysine (K) and leucine (L) residues, and the AMPs with antifungal activity have more cysteine residues. We also compared the amino acid distributions of AMPs with anti-Gram-negative and anti-Gram-positive activity, given that most AMPs with anti-Gram-negative activity can also usually perform the anti-Gram-positive activity (Figure 3B). We found that the amino acids of these two types of AMPs have similar compositions (Figure 3E). The detailed amino acid distributions of the other activities are shown in Supplementary Figure S1. In addition, we compared the length distributions of AMPs and non-AMPs (Supplementary Figure 2), as well as the length distributions of positive samples and negative samples of different functional activity datasets (Supplementary Figure 3). As a result, we noticed no significant length differences between the AMPs with various activities.
Predictive performance for AMP identification
Most studies usually utilized sequence cluster programs, such as CD-HIT, to remove highly similar sequences from the training dataset to reduce homology bias and redundancy. However, some studies indicated that this step would affect the predictive performance of the trained model. Therefore, we first evaluated the predictive performance of the models using training datasets with different sequence identity cut-offs ranging from 40 to 100%. Figure 4A shows the numbers of positive and negative samples of the training datasets with varying thresholds of identity, and the performance evaluation results based on 10-fold stratified cross-validation tests on these training datasets are shown in Figure 4B–C and Table 4. The performance comparison results indicate that the AUC and accuracy of the model trained on a dataset pre-processed with a higher identity cut-off are better than that with a lower identity cut-off value. In addition, we compared our model with an identity cut-off of 40% with several available webserver/tools, including AMAP [35], AMPDiscover [49], AMPfun [39], AMPlify [98], AMPscanner [15], APIN [16], iAMP-CA2L [42], iAMP-RAAC [40] and dbAMP [6], using the independent test dataset. Furthermore, we compared the performance of our model trained by training datasets with different identity cut-off thresholds ranging from 40 to 100% on the independent test dataset and provided the results in Figure 4D–E. iAMPCN trained on the dataset with an identity cut-off of 40% achieved the AUC value of 0.9973, outperforming other approaches. Along with the identity thresholds increasing, the AUC values increased slightly. We also evaluated the predictive probability distributions and performance metrics of different computational approaches on the independent validation dataset and demonstrated the results in Figure 5 and Table 5. These distributions also suggest that the iAMPCN has much better prediction performance. Moreover, we selected those AMPs with lengths ranging from 5 to 30 amino acids from the independent test dataset to construct a new test dataset to further evaluate the performance of iAMPCN in identifying short AMPs. The performance comparison results in terms of the accuracy of iAMPCN and two state-of-the-art approaches for predicting short AMPs, RF-AMPEP30 [99] and sAMPEP-PFPDeep [100], are provided in Figure 4F. All these performance evaluation results demonstrated that iAMPCN achieved a highly competitive predictive performance and outperformed state-of-the-art predictors for AMP identification.
Figure 4.
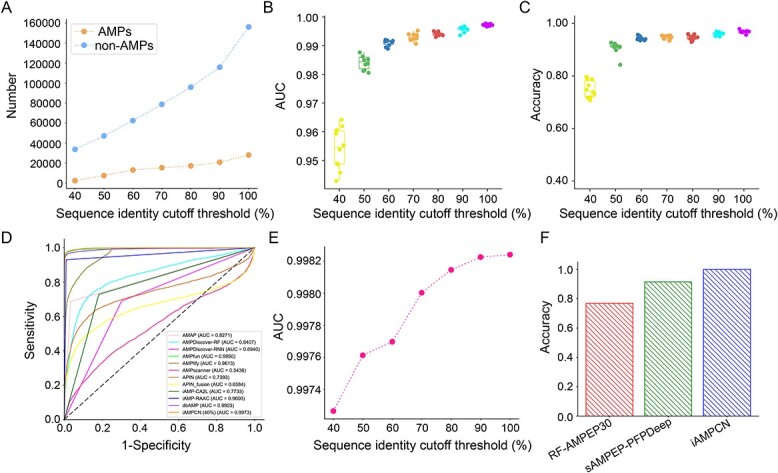
Performances of AMP prediction. (A) The numbers of AMPs and non-AMPs in the training datasets with different sequence identity cut-off thresholds. (B–C) AUCs and accuracy values of iAMPCN built on the training datasets with different sequence identity cut-off thresholds based on the 10-fold stratified cross-validation test. (D) ROC curves and the corresponding AUC values of various modes using the independent test dataset. (E) AUCs of iAMPCN built on the training datasets with different identity thresholds via the independent test dataset. (F) The accuracy values of identifying short AMPs (5–30 AAs) on the independent test dataset.
Table 4.
Performances of 10-fold cross-validation test based on the AMP training dataset
| Sequence identity | Sensitivity | Specificity | Accuracy | MCC | AUC |
|---|---|---|---|---|---|
| 40% | 0.5065 (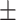 0.0648) 0.0648) |
0.9929 (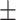 0.0032) 0.0032) |
0.7497 (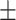 0.0309) 0.0309) |
0.6312 (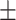 0.0276) 0.0276) |
0.9541 (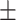 0.0070) 0.0070) |
| 50% | 0.8265 (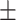 0.0493) 0.0493) |
0.9874 (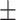 0.0048) 0.0048) |
0.9070 (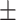 0.0227) 0.0227) |
0.8492 (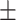 0.0191) 0.0191) |
0.9841 (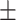 0.0023) 0.0023) |
| 60% | 0.8960 (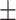 0.0145) 0.0145) |
0.9890 (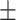 0.0031) 0.0031) |
0.9425 (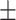 0.0065) 0.0065) |
0.9040 (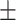 0.0085) 0.0085) |
0.9910 (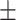 0.0011) 0.0011) |
| 70% | 0.9034 (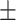 0.0158) 0.0158) |
0.9914 (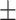 0.0026) 0.0026) |
0.9474 (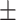 0.0068) 0.0068) |
0.9147 (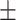 0.0053) 0.0053) |
0.9930 ( 0.0012) 0.0012) |
| 80% | 0.8972 ( 0.0207) 0.0207) |
0.9942 ( 0.0022) 0.0022) |
0.9457 ( 0.0094) 0.0094) |
0.9187 (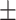 0.0071) 0.0071) |
0.9941 (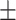 0.0006) 0.0006) |
| 90% | 0.9270 (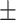 0.0122) 0.0122) |
0.9942 (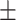 0.0011) 0.0011) |
0.9606 (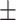 0.0060) 0.0060) |
0.9370 (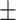 0.0075) 0.0075) |
0.9958 (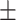 0.0009) 0.0009) |
| 100% | 0.9409 (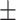 0.0124) 0.0124) |
0.9961 (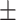 0.0021) 0.0021) |
0.9685 (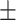 0.0053) 0.0053) |
0.9518 (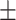 0.0032) 0.0032) |
0.9972 (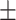 0.0003) 0.0003) |
Figure 5.

Predictive probability distributions of different computational approaches based on the independent test dataset. These approaches include (A) AMAP, (B) AMPDiscover-RF, (C) AMPDiscover-RNN, (D) AMPfun, (E) AMPlify, (F) AMPscanner, (G) APIN, (H) APIN-fusion, (I) iAMP-CA2L, (J) iAMP-RAAC, (K) dbAMP, (L) iAMPCN (CD-HIT 40%), (M) iAMPCN (CD-HIT 50%), (N) iAMPCN (CD-HIT 60%), (O) iAMPCN (CD-HIT 70%), (P) iAMPCN (CD-HIT 80%), (Q) iAMPCN (CD-HIT 90%) and (R) iAMPCN (CD-HIT 100%).
Table 5.
Performances of different predictors based on the AMP independent test dataset
| Tool | Precision | Sensitivity | Specificity | Accuracy | F1 | MCC | AUC |
|---|---|---|---|---|---|---|---|
| AMAP | 0.8908 | 0.6875 | 0.9668 | 0.8271 | 0.776 | 0.7134 | 0.8271 |
| AMPDiscover-RF | 0.3671 | 0.6882 | 0.5326 | 0.6104 | 0.4788 | 0.1992 | 0.8407 |
| AMPDiscover-RNN | 0.4740 | 0.6894 | 0.6986 | 0.6940 | 0.5617 | 0.3551 | 0.6940 |
| AMPfun | 0.9784 | 0.9687 | 0.9916 | 0.9801 | 0.9735 | 0.9632 | 0.9950 |
| AMPlify | 0.9611 | 0.6162 | 0.9902 | 0.8032 | 0.7510 | 0.7089 | 0.9613 |
| AMPscanner | 0.3045 | 0.5885 | 0.4707 | 0.5296 | 0.4014 | 0.0535 | 0.5436 |
| APIN | 0.6618 | 0.5868 | 0.8819 | 0.7343 | 0.6220 | 0.4870 | 0.7393 |
| APIN_fusion | 0.5229 | 0.5585 | 0.7993 | 0.6789 | 0.5401 | 0.3509 | 0.6584 |
| iAMP-CA2L | 0.6123 | 0.7283 | 0.8184 | 0.7733 | 0.6653 | 0.5211 | 0.7733 |
| iAMP-RAAC | 0.9736 | 0.9299 | 0.9901 | 0.9600 | 0.9513 | 0.9332 | 0.9600 |
| dbAMP | 0.9835 | 0.9473 | 0.9937 | 0.9705 | 0.9650 | 0.9519 | 0.9923 |
| iAMPCN (40%) | 0.9809 | 1.0000 | 0.9923 | 0.9962 | 0.9904 | 0.9866 | 0.9973 |
| iAMPCN (50%) | 0.9869 | 1.0000 | 0.9948 | 0.9974 | 0.9934 | 0.9908 | 0.9976 |
| iAMPCN (60%) | 0.9892 | 1.0000 | 0.9957 | 0.9978 | 0.9946 | 0.9924 | 0.9977 |
| iAMPCN (70%) | 0.9922 | 1.0000 | 0.9969 | 0.9985 | 0.9961 | 0.9946 | 0.9980 |
| iAMPCN (80%) | 0.9957 | 1.0000 | 0.9983 | 0.9992 | 0.9979 | 0.9970 | 0.9981 |
| iAMPCN (90%) | 0.9943 | 1.0000 | 0.9978 | 0.9989 | 0.9972 | 0.9960 | 0.9982 |
| iAMPCN (100%) | 0.9969 | 1.0000 | 0.9988 | 0.9994 | 0.9985 | 0.9979 | 0.9982 |
Performance on identifying functional activities of AMPs
To preliminarily assess the performance of iAMPCN using the extracted features, we evaluated the predictive performances of identifying functional activities based on three existing datasets constructed by previous studies [23, 39, 42] and provided the performance comparison results in Supplementary Tables S3–S5. The results indicate that the models built on the combination of four types of sequence features usually outperformed those models based on one type of sequence representation. Furthermore, on the iAMP-2L benchmark dataset, iAMPCN achieved the best performance in terms of all performance measures except hamming loss, which might be caused by the small dataset. Besides, iAMPCN performed best on the iAMP-CA2L benchmark dataset in terms of accuracy. These results indicate that iAMPCN is a competitive predictor for AMPs and their functional activities.
To explore the effects of different sequence identity thresholds in predicting functional activities, we also evaluated the predictive models trained on the benchmark datasets with different CD-HIT thresholds ranging from 40 to 100% (Supplementary Tables S6–S27, Supplementary Figure S4). For most functional activities, the higher the threshold of sequence identity, the higher the accuracy and robustness of the models achieved. Therefore, we utilized the trained models based on the training datasets with a CD-HIT threshold of 100% for the rest of the analysis.
Performance comparison of AMP function prediction on the independent test datasets
We first compared the performance of iAMPCN with that of several state-of-the-art approaches for predicting AMP functional types, including AMAP [35], iAMP-CA2L [42], AMPfun [39], iAMP-RAAC [40] and AMPDiscover [49], based on both balanced and imbalanced independent test datasets, respectively. A statistical summary of the balanced and imbalanced independent test datasets is provided in Supplementary Table S2. Figure 6 shows the ROC curves of the compared methods. Other performance evaluation metrics of these methods on the balanced independent test datasets are provided in Table 6. The corresponding pairwise comparisons (based on the Wilcoxon signed-rank test) of the accuracy, F1, MCC and AUC for measuring the performance differences between these methods are provided in Table 7. The ROC curves and other performance evaluation metrics on the imbalanced independent test datasets are shown in Supplementary Figure S5 and Table S28, respectively. Altogether, we conclude that except for ‘endotoxin activity’, iAMPCN outperforms other compared methods for predicting all the functional activities with the highest accuracy and AUC values. As can be seen from Table 6, several methods including AMAP, AMPDiscover-RF, AMPDiscover-RNN, AMPfun, dbAMP and iAMP-RAAC achieved low precision and specificity values, which indicates that the numbers of their predicted false positive (FP) samples were much higher than those of the predicted true negative (TN) samples since the number of tested negative samples was fixed (Supplementary Table S2). In addition, iAMP-CA2L also achieved low specificity values, which means that the numbers of its predicted false negative (FN) samples were higher than those of the predicted true positive (TP) samples since the number of tested positive samples was fixed (Supplementary Table S2). Higher FN or FP values often result in lower MCC values. When both FN and FP values were very high, the MCC value would tend to be very low, which is the case for iAMP-CA2L for predicting the antibacterial activity (Table 6). Compared with these methods, iAMPCN achieved lower FN or FP values for predicting most functional activities.
Figure 6.
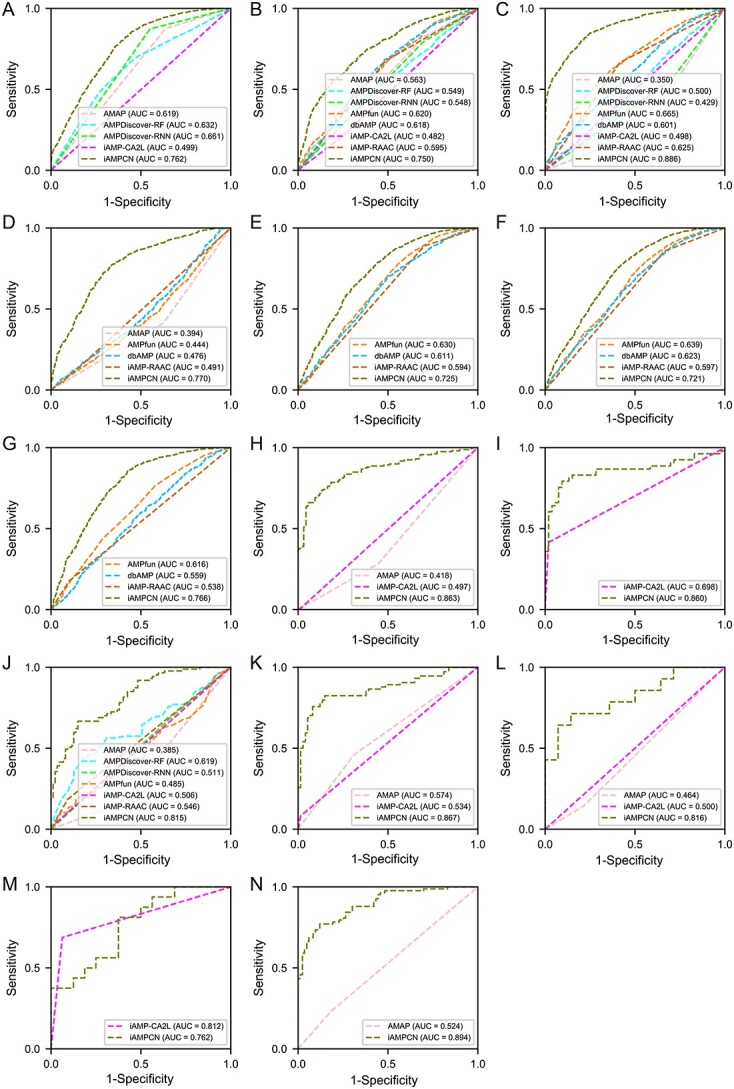
ROC curves and the corresponding AUC values of different tools for predicting 14 AMP functional activities on independent test datasets. These functional activities are (A) antibacterial, (B) antifungal, (C) antiviral, (D) anticancer, (E) anti-Gram positive, (F) anti-Gram negative, (G) anti-mammalian cells, (H) anti-HIV, (I) anti-MRSA, (J) antiparasitic, (K) antibiofilm, (L) chemotactic, (M) endotoxin and (N) insecticidal.
Table 6.
Performances of different computational approaches for predicting AMP functional activities based on balanced independent test datasets
| Activity | Method | Precision | Sensitivity | Specificity | Accuracy | F1 | MCC | AUC |
|---|---|---|---|---|---|---|---|---|
| Antibacterial | AMAP | 0.5788 | 0.8750 | 0.3633 | 0.6192 | 0.6967 | 0.2774 | 0.6192 |
| AMPDiscover-RF | 0.6162 | 0.8803 | 0.4518 | 0.6660 | 0.7250 | 0.3675 | 0.6322 | |
| AMPDiscover-RNN | 0.6132 | 0.8727 | 0.4495 | 0.6611 | 0.7203 | 0.3557 | 0.6611 | |
| iAMP-CA2L | 0.4990 | 0.6826 | 0.3148 | 0.4987 | 0.5766 | -0.0028 | 0.4987 | |
| iAMPCN | 0.6709 | 0.7914 | 0.6118 | 0.7016 | 0.7262 | 0.4099 | 0.7621 | |
| Antifungal | AMAP | 0.5354 | 0.8228 | 0.2860 | 0.5544 | 0.6487 | 0.1289 | 0.5544 |
| AMPDiscover-RF | 0.5385 | 0.7851 | 0.3272 | 0.5561 | 0.6388 | 0.1263 | 0.5398 | |
| AMPDiscover-RNN | 0.5316 | 0.7895 | 0.3044 | 0.5469 | 0.6354 | 0.1073 | 0.5469 | |
| AMPfun | 0.5901 | 0.5746 | 0.6009 | 0.5877 | 0.5822 | 0.1755 | 0.6256 | |
| dbAMP | 0.5524 | 0.8044 | 0.3482 | 0.5763 | 0.6550 | 0.1715 | 0.6082 | |
| iAMP-CA2L | 0.5000 | 0.2140 | 0.7860 | 0.5000 | 0.2998 | 0.0000 | 0.5000 | |
| iAMP-RAAC | 0.5850 | 0.6763 | 0.5202 | 0.5982 | 0.6273 | 0.1989 | 0.5982 | |
| iAMPCN | 0.7347 | 0.5368 | 0.8061 | 0.6715 | 0.6204 | 0.3561 | 0.7483 | |
| Antiviral | AMAP | 0.3042 | 0.2336 | 0.4656 | 0.3496 | 0.2643 | -0.3092 | 0.3496 |
| AMPDiscover-RF | 0.4283 | 0.5432 | 0.2749 | 0.4091 | 0.4790 | -0.1888 | 0.4999 | |
| AMPDiscover-RNN | 0.4447 | 0.5683 | 0.2902 | 0.4293 | 0.4989 | -0.1473 | 0.4293 | |
| AMPfun | 0.5862 | 0.7502 | 0.4705 | 0.6103 | 0.6582 | 0.2299 | 0.6653 | |
| dbAMP | 0.5392 | 0.8618 | 0.2635 | 0.5627 | 0.6633 | 0.1564 | 0.6006 | |
| iAMP-CA2L | 0.4615 | 0.0243 | 0.9717 | 0.4980 | 0.0461 | -0.0126 | 0.4980 | |
| iAMP-RAAC | 0.6156 | 0.6653 | 0.5845 | 0.6249 | 0.6395 | 0.2506 | 0.6249 | |
| iAMPCN | 0.8248 | 0.7308 | 0.8448 | 0.7878 | 0.7750 | 0.5794 | 0.8865 | |
| Anticancer | AMAP | 0.3973 | 0.4118 | 0.3753 | 0.3935 | 0.4044 | -0.2130 | 0.3935 |
| AMPfun | 0.4450 | 0.4172 | 0.4796 | 0.4484 | 0.4306 | -0.1034 | 0.4444 | |
| dbAMP | 0.4828 | 0.6344 | 0.3204 | 0.4774 | 0.5483 | -0.0476 | 0.4759 | |
| iAMP-RAAC | 0.4658 | 0.1172 | 0.8656 | 0.4914 | 0.1873 | -0.0259 | 0.4914 | |
| iAMPCN | 0.6830 | 0.7667 | 0.6441 | 0.7054 | 0.7224 | 0.4139 | 0.7698 | |
| Anti-Gram positive | AMPfun | 0.5607 | 0.8865 | 0.3054 | 0.5960 | 0.6869 | 0.2359 | 0.6304 |
| dbAMP | 0.5513 | 0.8265 | 0.3272 | 0.5768 | 0.6614 | 0.1774 | 0.6107 | |
| iAMP-RAAC | 0.5590 | 0.8879 | 0.2995 | 0.5937 | 0.6860 | 0.2317 | 0.5937 | |
| iAMPCN | 0.6372 | 0.7995 | 0.5449 | 0.6722 | 0.7092 | 0.3561 | 0.7246 | |
| Anti-Gram negative | AMPfun | 0.5683 | 0.8868 | 0.3262 | 0.6065 | 0.6927 | 0.2572 | 0.6390 |
| dbAMP | 0.5603 | 0.8515 | 0.3317 | 0.5916 | 0.6758 | 0.2144 | 0.6232 | |
| iAMP-RAAC | 0.5635 | 0.8582 | 0.3354 | 0.5968 | 0.6803 | 0.2271 | 0.5968 | |
| iAMPCN | 0.6335 | 0.8058 | 0.5338 | 0.6698 | 0.7093 | 0.3529 | 0.7212 | |
| Anti-mammalian cells | AMPfun | 0.6024 | 0.3574 | 0.7641 | 0.5608 | 0.4486 | 0.1330 | 0.6163 |
| dbAMP | 0.5374 | 0.6486 | 0.4416 | 0.5451 | 0.5878 | 0.0923 | 0.5585 | |
| iAMP-RAAC | 0.6364 | 0.1769 | 0.8989 | 0.5379 | 0.2768 | 0.1096 | 0.5379 | |
| iAMPCN | 0.6748 | 0.7942 | 0.6173 | 0.7058 | 0.7297 | 0.4181 | 0.7665 | |
| Anti-HIV | AMAP | 0.3879 | 0.2830 | 0.5535 | 0.4182 | 0.3273 | -0.1699 | 0.4182 |
| iAMP-CA2L | 0.3333 | 0.0063 | 0.9874 | 0.4969 | 0.0123 | -0.0325 | 0.4969 | |
| iAMPCN | 0.8489 | 0.7421 | 0.8679 | 0.8050 | 0.7919 | 0.6149 | 0.8633 | |
| Anti-MRSA | iAMP-CA2L | 0.9565 | 0.4151 | 0.9811 | 0.6981 | 0.5789 | 0.4806 | 0.6981 |
| iAMPCN | 0.8600 | 0.8113 | 0.8679 | 0.8396 | 0.8350 | 0.6803 | 0.8597 | |
| Antiparasitic | AMAP | 0.2692 | 0.1609 | 0.5632 | 0.3621 | 0.2014 | -0.3013 | 0.3621 |
| AMPDiscover-RF | 0.5000 | 0.7241 | 0.2759 | 0.5000 | 0.5915 | 0.0000 | 0.6092 | |
| AMPDiscover-RNN | 0.4841 | 0.7011 | 0.2529 | 0.4770 | 0.5728 | -0.0514 | 0.4770 | |
| AMPfun | 0.4750 | 0.4368 | 0.5172 | 0.4770 | 0.4551 | -0.0461 | 0.4970 | |
| iAMP-CA2L | 0.4000 | 0.0230 | 0.9655 | 0.4943 | 0.0435 | -0.0344 | 0.4943 | |
| iAMP-RAAC | 0.5517 | 0.1839 | 0.8506 | 0.5172 | 0.2759 | 0.0463 | 0.5172 | |
| iAMPCN | 0.7534 | 0.6322 | 0.7931 | 0.7126 | 0.6875 | 0.4309 | 0.7861 | |
| Antibiofilm | AMAP | 0.5965 | 0.4595 | 0.6892 | 0.5743 | 0.5191 | 0.1527 | 0.5743 |
| iAMP-CA2L | 0.8571 | 0.0811 | 0.9865 | 0.5338 | 0.1481 | 0.1592 | 0.5338 | |
| iAMPCN | 0.8243 | 0.8243 | 0.8243 | 0.8243 | 0.8243 | 0.6486 | 0.8667 | |
| Chemotactic | AMAP | 0.4000 | 0.1429 | 0.7857 | 0.4643 | 0.2105 | -0.0933 | 0.4643 |
| iAMP-CA2L | 0.0000 | 0.0000 | 1.0000 | 0.5000 | 0.0000 | 0.0000 | 0.5000 | |
| iAMPCN | 1.0000 | 0.5000 | 1.0000 | 0.7500 | 0.6667 | 0.5774 | 0.8163 | |
| Endotoxin | iAMP-CA2L | 0.9167 | 0.6875 | 0.9375 | 0.8125 | 0.7857 | 0.6455 | 0.8125 |
| iAMPCN | 0.7059 | 0.7500 | 0.6875 | 0.7188 | 0.7273 | 0.4384 | 0.7617 | |
| Insecticidal | AMAP | 0.5588 | 0.2289 | 0.8193 | 0.5241 | 0.3248 | 0.0597 | 0.5241 |
| iAMPCN | 0.8514 | 0.7590 | 0.8675 | 0.8133 | 0.8025 | 0.6302 | 0.8936 |
Table 7.
The Wilcoxon signed-rank test for measuring the statistical significance of the performance between different approaches in terms of accuracy, F1, MCC and AUC
| Accuracy | |||||||
|---|---|---|---|---|---|---|---|
| Wilcoxon P-value | AMAP | AMPDiscover-RF | AMPDiscover-RNN | AMPfun | iAMP-CA2L | iAMP-RAAC | dbAMP |
| AMPDiscover-RF | – | ||||||
| AMPDiscover-RNN | – | – | |||||
| AMPfun | – | – | – | ||||
| iAMP-CA2L | 0.008362 | – | – | – | |||
| iAMP-RAAC | – | – | – | 0.006714 | – | ||
| dbAMP | – | – | – | 0.030151 | – | 0.035339 | |
| iAMPCN | 6.10E−05 | – | – | 6.10E−05 | 0.000122 | 6.10E−05 | 6.10E−05 |
| F1 | |||||||
| Wilcoxon P-value | AMAP | AMPDiscover-RF | AMPDiscover-RNN | AMPfun | iAMP-CA2L | iAMP-RAAC | dbAMP |
| AMPDiscover-RF | – | ||||||
| AMPDiscover-RNN | – | – | |||||
| AMPfun | – | – | – | ||||
| iAMP-CA2L | 0.187622 | – | – | – | |||
| iAMP-RAAC | – | – | – | 0.106995 | – | ||
| dbAMP | – | – | – | 0.002014 | – | 0.00061 | |
| iAMPCN | 0.000122 | – | – | 6.10E−05 | 0.000122 | 0.000122 | 0.000305 |
| MCC | |||||||
| Wilcoxon P-value | AMAP | AMPDiscover-RF | AMPDiscover-RNN | AMPfun | iAMP-CA2L | iAMP-RAAC | dbAMP |
| AMPDiscover-RF | – | ||||||
| AMPDiscover-RNN | – | – | |||||
| AMPfun | – | – | – | ||||
| iAMP-CA2L | 0.008362 | – | – | – | |||
| iAMP-RAAC | – | – | – | 0.008362 | – | ||
| dbAMP | – | – | – | 0.035339 | – | 0.047913 | |
| iAMPCN | 6.10E−05 | 6.10E−05 | 6.10E−05 | 6.10E−05 | 0.000183 | 6.10E−05 | 6.10E−05 |
| AUC | |||||||
| Wilcoxon P-value | AMAP | AMPDiscover-RF | AMPDiscover-RNN | AMPfun | iAMP-CA2L | iAMP-RAAC | dbAMP |
| AMPDiscover-RF | – | ||||||
| AMPDiscover-RNN | – | – | |||||
| AMPfun | – | – | – | ||||
| iAMP-CA2L | 0.008362 | – | – | – | |||
| iAMP-RAAC | – | – | – | 0.072998 | – | ||
| dbAMP | – | – | – | 0.035339 | – | 0.00116 | |
| iAMPCN | 6.10E−05 | – | – | 6.10E−05 | 0.000122 | 6.10E−05 | 6.10E−05 |
‘–’ means that the number of functional activities that both tools can predict is less than 5.
Nevertheless, it is challenging to improve the performance of iAMPCN in predicting some specific functional activities, including antibacterial, antifungal, anticancer, antiparasitic, endotoxin and anti-mammalian cell activities, as indicated by the low MCC values in Table 6. Previous studies using traditional machine-learning algorithms also demonstrated a similar challenge (as illustrated using the MCC values) in predicting such functional activities based on the constructed datasets of these studies [39, 40]. As such, we would like to argue that predicting these functional activities is difficult, regardless of machine-learning models, feature engineering strategies applied or the training data. Here, we explain the possible reasons: First, the predictions for those functional activities with low MCC values may be related to the killing targets and the mechanism of action. The killing targets of the AMPs with some functional activities are not cells, such as antiviral AMPs, but the targets of the AMPs with antibacterial, antifungal, anticancer, antiparasitic, endotoxin and anti-mammalian cell activities are typically cells, and as such, in order to kill the cells, the cell membrane structure needs to be destroyed. To achieve this, the AMPs tend to possess similar physicochemical properties (e.g. positively charged), structures (e.g. α-helical) and mechanisms of action [101, 102]. Due to their similar physicochemical properties, the AMPs with such functional activities may have high sequence similarities [101, 102]. Our analysis has indeed demonstrated that the amino acid distributions of the positive and negative data of the antibacterial, antifungal, anticancer, antiparasitic, endotoxin and anti-mammalian cell activities are similar (Supplementary Figure 1), thereby making it difficult to distinguish specific functional activities from each other. Second, the dataset imbalance of some activities could also negatively affect the predictive performance. Although we tried several strategies to reduce the impact (e.g. focal loss) and improve the TP and TF values, such improvement was limited. Therefore, some advanced strategies to better handle the imbalanced data need to be developed in the future work. Third, we also utilized the PHOENIX (https://phoenix.arize.com/) tool to explore the embedding performance of representing each peptide sequence. In particular, the embeddings were extracted from the nodes preceding the final dense layer. The training and test datasets of specific functional activities (e.g. antibacterial, antifungal, anticancer and anti-mammalian cells) were utilized as the inputs to the PHOENIX tool. As a result, we observed that some clusters of specific functional activities only contained the points representing the peptides from the test datasets, indicating that the representations of the training data could not provide sufficient information for distinguishing these peptide sequences (Supplementary Figures 6–9). Furthermore, we tried the t-distributed stochastic neighbor embedding (t-SNE) [103] algorithm to visualize the embedding representations of the test data. For example, although the peptides with antibacterial activities could be roughly clustered into two groups, some peptides still could not be clearly clustered (Supplementary Figure 10). Taken together, the experiments indicate that (i) the sequences between the positive and negative datasets have similarities to a certain extent, (ii) the samples from the training datasets cannot provide enough information for distinguishing the samples of test datasets and (iii) the sequence embedding module (i.e. CNNs and peptide encodings) of iAMPCN needs to be optimized in order to improve the peptide embedding representations in the future work. Lastly, the peptide structure also influences its functional activity. For example, peptides with an α-helical structure are more likely to have antimicrobial activities [101, 102]. In the present study, we only incorporated the physicochemical properties of peptides to encode the sequences to improve the predictive performance, but did not explicitly explore the structural information of peptides. In the future work, we will explore effective strategies to integrate their structural information to further improve the predictive performance of those specific functional activities with low MCC values.
Adaptability and stability analysis
To further assess the adaptability in AMP identification, we compared the predictive performance of iAMPCN with that of other existing predictors. We employed the datasets on which these existing predictors were trained and evaluated to evaluate the performance. These predictors include García-Jacas et al.’s work [104], BERT-based [105], ACEP [106], APIN-fusion [16], APIN [16], AMPScannerV2 [15], DeepAVP [19], UnidLSTM [19], MultiLSTM [19], DynEvo [19], StaEvo [19], Deep-AmPEP30 [99], Deep-ABPpred [22], AniAMPpred [13], Deep-AFPpred [20], Deep-AVPpred [107] and StaBle-ABPpred [21]. Based on the datasets from Veltri et al. [15], Nishant et al. [76], Li et al. [19], Yan et al. [99], Sharma et al. [13, 20, 22, 107] and Singh et al. [21], the performance evaluation metrics of iAMPCN and these predictors were calculated and provided in Supplementary Tables S29 and S30. It is shown that iAMPCN achieved the highest MCC values on 5 out of 11 test datasets, including the Nishant et al. AVP dataset [76], Nishant et al. AVP* dataset [76], Sharma et al. AMP dataset [13], Sharma et al. AFP dataset [20], Singh et al. evaluation dataset [21] and Singh et al. test dataset [21]. Moreover, iAMPCN also achieved the second-highest MCC values on 2 of 11 test datasets, including Li et al. [19] and Sharma et al. AVP [107]. These results show that iAMPCN is a very competitive predictor for identifying AMPs and AMPs with one specific functional activity. To further evaluate the performance, iAMPCN was compared with some latest state-of-the-art predictors, including the consensus model [14] and García-Jacas et al.’s model [108], based on the Pinacho-Castellanos et al.’s general AMP datasets [49] (Supplementary Table S31). The consensus model [14] is an ensemble model combining several predictive results from RNN and BERT [14, 109, 110] models. As a result, iAMPCN achieved an accuracy which was only 0.004 lower than that of the consensus model when being evaluated on the external test dataset, while iAMPCN also achieved a higher accuracy on the test dataset. All these results indicate that iAMPCN has the outstanding adaptability for identifying AMPs or AMPs with one specific functional activity.
As the predictive performance of different models might be affected by the selection of negative data sampling strategies [98], we applied the same datasets and evaluation strategy provided by Sidorczuk et al. [98] to further measure the effect caused by the selection of negative data. The corresponding AUC values are listed in Supplementary Table S32. We can see that iAMPCN achieved a much better performance than the other compared methods, with only one average AUC of <0.8. In addition, all the AUC’s standard deviations of iAMPCN were relatively low. In summary, these results suggest that iAMPCN has excellent stability.
Performance comparison of alternative machine-learning algorithms
To further analyze and evaluate the feature extraction capacity of iAMPCN and the prediction capacity of the final dense layer of iAMPCN, we extracted the feature representation used as inputs into the final dense layer to conduct the comparison. We then replaced the final dense layer with other state-of-the-art machine-learning algorithms, including random forest (RF), adaptive boosting (AdaBoost), XGBoost, logistic regression (LR) and GBDT. In addition, we utilized two oversampling strategies, SMOTE [27] and ADASYN [30], to generate balance datasets. Finally, we compared the AUC values of these algorithms on different functional activity training datasets by 10-fold stratified cross-validation tests and plotted the ROC curves on test datasets. The corresponding performance comparison results are provided in Supplementary Figures S11 and S12, respectively. These results illustrated no significant differences in terms of predictive performance by these machine-learning algorithms. From another perspective, these results also indicate that the feature extraction part by CNN modules of iAMPCN can already learn appropriate feature representations from AMP sequences, leading to outstanding and robust performance, regardless of any machine-learning algorithm in the final dense layer. Furthermore, we conclude that the entire deep-learning framework of iAMPCN outperformed other machine-learning algorithms for the prediction of most functional activities and that the last part of iAMPCN has an excellent predictive capability for the AMP functional activities and accordingly, the trained models have the potential to be applied as effective feature extraction modules in future studies.
Apart from the performance comparison between iAMPCN with several different classic algorithms, we also modified the final dense layers of iAMPCN to evaluate the effect of dense layers on the predictive performance. In particular, we selected several different hidden dense layers, including three hidden layers with the node numbers [128, 64, 32], two hidden layers with the node numbers [128, 64], [64, 32], and one hidden layer with the node number [128], [64] and [32], to compare the performance of iAMPCN which connected all the extracted feature representations to a single final output dense layer without any hidden dense layers by performing 10-fold stratified cross-validation tests. Five datasets were selected for performance evaluation, including the AMP, antibacterial, antifungal, antiviral and antiparasitic datasets. Among these datasets, the antibacterial dataset is nearly balanced for positive and negative samples, while the antiviral and antifungal datasets are imbalanced, and the antiparasitic dataset is very imbalanced. Thus, these selected datasets for the performance comparison are representative. The corresponding AUCs are calculated and listed in Supplementary Table S33. It can be seen that the deep learning structures with more straightforward dense layers achieved higher AUCs and performed better when being tested on the AMP, antibacterial, antifungal and antiviral datasets, while the deep learning structures with different dense layers achieved similar AUC values when being tested on the antiparasitic dataset. Considering that training more complex structures is more time consuming and computationally expensive, the deep learning structure of iAMPCN without the hidden dense layers is more suitable for AMP identification.
Performance comparison with the unsupervised pretrained model
In addition, we also compared the predictive performance of our proposed iAMPCN method with an unsupervised pretrained model. Particularly, we applied the Evolutionary Scale Modeling (ESM-2) [111] model to extract the sequence features and utilized the RF and XGBoost models to predict AMPs and their functional activities. To make a fair comparison, the same training datasets of iAMPCN were utilized. The predictive performance is shown in Supplementary Tables S34 and S35. As a result, we can see that compared with the other two models, iAMPCN achieved the best MCC value for AMP identification (Supplementary Table S34) and also achieved the best MCC values in predicting 12 out of 14 functional activities (Supplementary Table S35). In addition, we also performed the pairwise comparisons (based on the Wilcoxon signed-rank test), and the results showed that the accuracies and MCCs between our method and the other two methods were significantly different (Supplementary Tables S36), with the only exception for the AUC score between iAMPCN and XGBoost, which was statistically insignificant with the P-value of 0.3028.
Model interpretability analysis
Model interpretability is critically important for researchers to better understand machine learning-based models and make informed decisions regarding the discovery of new potential AMPs. Here, we analyzed the AMPs with the six most common functional activities (i.e. anti-Gram positive, anti-Gram negative, antifungal, antiviral, anti-mammalian cells and anticancer) and applied the ‘GradientExplainer’ method from the SHapley Additive exPlanations (SHAP) algorithm to conduct the interpretability analysis. In particular, we selected three AMPs for each functional activity, and displayed the amino acids’ relative importance for each AMP sequence in Figure 7. We can see that lysine and arginine residues play a relatively important role in antimicrobial activities, presumably due to their positively charged characteristics (Figure 7). This observation is consistent with many previous studies [112, 113]. In addition, leucine, tryptophan and phenylalanine also contribute to antimicrobial activities because of their hydrophobicity (Figure 7) [114, 115]. Furthermore, the aspartic acid was also identified to be significant for anticancer activity (Figure 7), which is also suggested in previous studies [116, 117].
Figure 7.
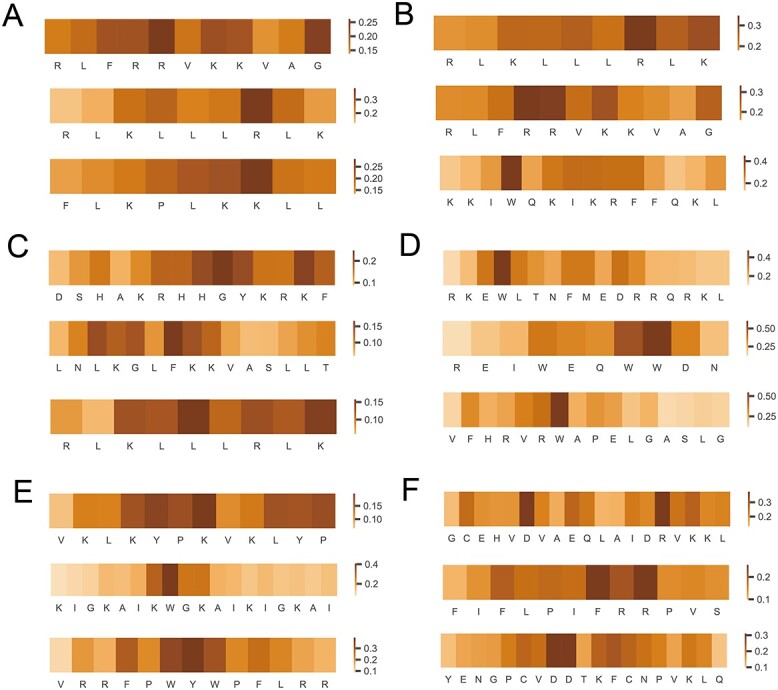
Model interpretability analysis using SHAP values for selected AMPs. (A) Anti-Gram negative, (B) anti-Gram positive, (C) antifungal, (D) antiviral, (E) anti-mammalian cells and (F) anticancer. The value on the color scale of each panel indicates the relative importance. The higher the value, the more important the corresponding amino acid residue for the functional activity of AMPs.
CONCLUSIONS
Accurate identification of AMPs and their functional activities is critical for functional peptide design and antimicrobial therapy development. In this study, based on a systematic community-wide assessment of computational methods for AMP and their function prediction, we constructed a comprehensive AMP benchmark dataset by integrating a number of public databases and developed a novel deep learning-based framework, iAMPCN, to accurately identify AMPs and their 22 functional activities. Extensive stratified cross-validation tests based on the training datasets and independent test results demonstrated that iAMPCN achieved superior performance for predicting AMPs and most AMP functional types. In addition, the model structure analysis indicates that iAMPCN can be utilized as a feature extraction tool for AMP prediction. The superior predictive performance of iAMPCN can be attributed to three major factors: (i) the reliable dataset curation of the up-to-date annotations of AMPs and their functional activity to provide the most comprehensive training data; (ii) the highly accurate deep-learning framework of iAMPCN learns from the effective feature representations to build robust predictive power for AMP and functional activity prediction; (iii) the selection of appropriate sequence identity thresholds for AMP and function prediction, respectively. Our analysis indicates that a threshold of 40% is sufficient for identifying AMPs, while it is suggested that a higher threshold should be chosen for predicting AMP functional activities. Taken together, we anticipate iAMPCN will be a practical approach for identifying AMPs and their functional activities. In our future work, we will focus on tackling the label imbalance problem to improve model performance in predicting AMP functional activities.
Key Points
We provided a comprehensive summary of existing tools for predicting antimicrobial peptides (AMPs) and their functional activities.
We constructed comprehensive benchmark datasets of AMPs and 22 functional activities and accordingly developed a two-stage deep learning-based framework, termed iAMPCN, for the identification of AMPs and their functional activities based on four different types of sequence encodings.
Performance benchmarking based on the independent test datasets suggested that iAMPCN outperformed other available state-of-the-art tools for the prediction of AMPs and the majority of functional activities.
The source codes of iAMPCN are publicly available at https://github.com/joy50706/iAMPCN/tree/master for the wider research community to use.
Supplementary Material
Author Biographies
Jing Xu is currently a PhD student in the Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Australia. Her research interests are bioinformatics, computational biology, machine learning and deep learning.
Fuyi Li received his PhD in Bioinformatics from Monash University, Australia. He is currently a Professor at the College of Information Engineering, Northwest A&F University, China. His research interests are bioinformatics, computational biology, machine learning and deep learning.
Chen Li is a research fellow in the Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University. His research interests include systems proteomics, immunopeptidomics, personalized medicine and experimental bioinformatics.
Xudong Guo received his MEng degree from Ningxia University, China. He is currently a research assistant at the College of Information Engineering, Northwest A&F University. His research interests are bioinformatics and data mining.
Cornelia Landersdorfer is an associate professor at the Monash Institute of Pharmaceutical Sciences, Monash University, Australia. Her research interests include antibiotic mono- and combination therapy to achieve synergistic bacterial killing and prevent resistance, mechanism-based mathematical modeling, population pharmacokinetics, pharmacodynamics, and optimized dosage regimens of anti-infectives and other agents.
Hsin-Hui Shen is an NHMRC Career Development Fellow at Monash University. She completed her PhD in Physical Chemistry at Oxford University in 2008. Her research interests include interaction of polymyxins with bacterial membranes and synergistic effects of antibiotics against Gram-positive bacteria.
Anton Y. Peleg is a professor of Infectious Diseases and Microbiology and Director of the Department of Infectious Diseases at the Alfred Hospital and Monash University. He is also a research group leader in the Department of Microbiology, Monash University. His research spans clinical to basic research, with a focus on hospital-acquired infections, antimicrobial resistance, infections in immunocompromised hosts and understanding mechanisms of disease caused by hospital pathogens.
Jian Li is a professor and an NHMRC Principal Research Fellow in the Biomedicine Discovery Institute and Department of Microbiology, Monash University, Australia. His research interests include the pharmacology of polymyxins and the discovery of novel, safer polymyxins.
Seiya Imoto is professor and director of the Human Genome Center, Institute of Medical Science, The University of Tokyo, Japan. His research interests include genome sequence data analysis, metagenome data analysis, immunogenomics, big data analysis, disease risk prediction and supercomputing in bioinformatics and computational biology.
Jianhua Yao is an expert fellow and head of the Medical Algorithm Group in Tencent AI Lab, China. He received his PhD degree from John Hopkins University. His research interests include bioinformatics, computational biology, medical imaging and pattern recognition.
Tatsuya Akutsu received his DEng degree in Information Engineering in 1989 from the University of Tokyo, Japan. Since 2001, he has been a professor in the Bioinformatics Centre, Institute for Chemical Research, Kyoto University, Japan. His research interests include bioinformatics and discrete algorithms.
Jiangning Song is currently an associate professor and group leader in the Monash Biomedicine Discovery Institute, Monash University. He is also a member of the Monash Data Futures Institute. His research interests include bioinformatics, computational biology, machine learning, medical imaging and pattern recognition.
Contributor Information
Jing Xu, Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC 3800, Australia; Monash Data Futures Institute, Monash University, Melbourne, VIC 3800, Australia.
Fuyi Li, Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC 3800, Australia; College of Information Engineering, Northwest A&F University, Shaanxi 712100, China; The Peter Doherty Institute for Infection and Immunity, The University of Melbourne, Melbourne, VIC 3800, Australia.
Chen Li, Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC 3800, Australia; Monash Data Futures Institute, Monash University, Melbourne, VIC 3800, Australia.
Xudong Guo, College of Information Engineering, Northwest A&F University, Shaanxi 712100, China.
Cornelia Landersdorfer, Monash Institute of Pharmaceutical Sciences, Monash University, Melbourne, VIC 3800, Australia.
Hsin-Hui Shen, Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC 3800, Australia; Department of Materials Science and Engineering, Faculty of Engineering, Monash University, Clayton, VIC, 3800, Australia.
Anton Y Peleg, Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC 3800, Australia; Department of Infectious Diseases, Alfred Hospital, Alfred Health, Melbourne, Victoria, Australia.
Jian Li, Monash Biomedicine Discovery Institute and Department of Microbiology, Monash University, Melbourne, VIC 3800, Australia.
Seiya Imoto, Division of Health Medical Intelligence, Human Genome Center, Institute of Medical Science, The University of Tokyo, Minato-ku, Tokyo, Japan; Collaborative Research Institute for Innovative Microbiology, The University of Tokyo, Bunkyo-ku, Tokyo, Japan.
Jianhua Yao, Tencent AI Lab, Tencent, Shenzhen, China.
Tatsuya Akutsu, Bioinformatics Center, Institute for Chemical Research, Kyoto University, Uji 611-0011, Japan.
Jiangning Song, Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC 3800, Australia; Monash Data Futures Institute, Monash University, Melbourne, VIC 3800, Australia; Bioinformatics Center, Institute for Chemical Research, Kyoto University, Uji 611-0011, Japan.
FUNDING
National Health and Medical Research Council of Australia (NHMRC) (APP1127948, APP1144652), Australian Research Council (ARC) (LP110200333, DP120104460), National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI111965), Major and Seed Inter-Disciplinary Research (IDR) projects awarded by Monash University. This work was also partially supported by the International Collaborative Research Program of Institute for Chemical Research, Kyoto University and a Grant from the International Joint Usage/Research Center, Institute of Medical Science, The University of Tokyo (K23-2074).
DATA AVAILABILITY
The source codes and data are available at https://github.com/joy50706/iAMPCN/tree/master.
REFERENCES
- 1. D’Costa VM, King CE, Kalan L, et al. Antibiotic resistance is ancient. Nature 2011;477(7365):457–61. [DOI] [PubMed] [Google Scholar]
- 2. Blair JMA, Webber MA, Baylay AJ, et al. Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol 2015;13:42–51. [DOI] [PubMed] [Google Scholar]
- 3. Fjell CD, Hiss JA, Hancock REW, et al. Designing antimicrobial peptides: form follows function. Nat Rev Drug Discov 2012;11:37–51. [DOI] [PubMed] [Google Scholar]
- 4. Magana M, Pushpanathan M, Santos AL, et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect Dis 2020;20:e216–30. [DOI] [PubMed] [Google Scholar]
- 5. Wang G, Li X, Wang Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res 2015;44:D1087–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jhong J-H, Yao L, Pang Y, et al. dbAMP 2.0: updated resource for antimicrobial peptides with an enhanced scanning method for genomic and proteomic data. Nucleic Acids Res 2021;50:D460–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jhong J-H, Chi Y-H, Li W-C, et al. dbAMP: an integrated resource for exploring antimicrobial peptides with functional activities and physicochemical properties on transcriptome and proteome data. Nucleic Acids Res 2018;47:D285–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kang X, Dong F, Shi C, et al. DRAMP 2.0, an updated data repository of antimicrobial peptides. Scientific Data 2019;6:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shi G, Kang X, Dong F, et al. DRAMP 3.0: an enhanced comprehensive data repository of antimicrobial peptides. Nucleic Acids Res 2021;50:D488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fan L, Sun J, Zhou M, et al. DRAMP: a comprehensive data repository of antimicrobial peptides. Sci Rep 2016;6:24482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ye G, Wu H, Huang J, et al. LAMP2: a major update of the database linking antimicrobial peptides. Database 2020;2020:baaa061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao X, Wu H, Lu H, et al. LAMP: a database linking antimicrobial peptides. PloS One 2013;8:e66557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sharma R, Shrivastava S, Kumar Singh S, et al. AniAMPpred: artificial intelligence guided discovery of novel antimicrobial peptides in animal kingdom. Brief Bioinform 2021;22:bbab242. [DOI] [PubMed] [Google Scholar]
- 14. Ma Y, Guo Z, Xia B, et al. Identification of antimicrobial peptides from the human gut microbiome using deep learning. Nat Biotechnol 2022;40:921–31. [DOI] [PubMed] [Google Scholar]
- 15. Veltri D, Kamath U, Shehu A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 2018;34:2740–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Su X, Xu J, Yin Y, et al. Antimicrobial peptide identification using multi-scale convolutional network. BMC Bioinformatics 2019;20:730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu J, Li F, Leier A, et al. Comprehensive assessment of machine learning-based methods for predicting antimicrobial peptides. Brief Bioinform 2021;22:bbab083. [DOI] [PubMed] [Google Scholar]
- 18. Gabere MN, Noble WS. Empirical comparison of web-based antimicrobial peptide prediction tools. Bioinformatics 2017;33:1921–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li J, Pu Y, Tang J, et al. DeepAVP: a Dual-Channel deep neural network for identifying variable-length antiviral peptides. IEEE J Biomed Health Inform 2020;24:3012–9. [DOI] [PubMed] [Google Scholar]
- 20. Sharma R, Shrivastava S, Kumar Singh S, et al. Deep-AFPpred: identifying novel antifungal peptides using pretrained embeddings from seq2vec with 1DCNN-BiLSTM. Brief Bioinform 2022;23:bbab422. [DOI] [PubMed] [Google Scholar]
- 21. Singh V, Shrivastava S, Kumar Singh S, et al. StaBle-ABPpred: a stacked ensemble predictor based on biLSTM and attention mechanism for accelerated discovery of antibacterial peptides. Brief Bioinform 2022;23(1):bbab439. [DOI] [PubMed] [Google Scholar]
- 22. Sharma R, Shrivastava S, Kumar Singh S, et al. Deep-ABPpred: identifying antibacterial peptides in protein sequences using bidirectional LSTM with word2vec. Brief Bioinform 2021;22(5):bbab065. [DOI] [PubMed] [Google Scholar]
- 23. Xiao X, Wang P, Lin W-Z, et al. iAMP-2L: a two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal Biochem 2013;436:168–77. [DOI] [PubMed] [Google Scholar]
- 24. Keller JM, Gray MR, Givens JA. A fuzzy K-nearest neighbor algorithm. IEEE Trans Syst Man Cybern 1985;SMC-15:580–5. [Google Scholar]
- 25. Shen H-B, Chou K-C. PseAAC: a flexible web server for generating various kinds of protein pseudo amino acid composition. Anal Biochem 2008;373:386–8. [DOI] [PubMed] [Google Scholar]
- 26. Lin W, Xu D. Imbalanced multi-label learning for identifying antimicrobial peptides and their functional types. Bioinformatics 2016;32:3745–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chawla NV, Bowyer KW, Hall LO, et al. SMOTE: synthetic minority over-sampling technique. Journal of Artificial Intelligence Research 2002;16:321–57. [Google Scholar]
- 28. Liu S, Lin Y. Introduction to Grey Systems Theory. Grey Systems: Theory and Applications. Berlin, Heidelberg: Springer Berlin Heidelberg, 2011, 1–18. [Google Scholar]
- 29. Zhang J, Li S, Ding Y et al. An two-layer predictive model of ensemble classifier chain for detecting antimicrobial peptides. In: 2020 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). Seoul, Korea (South), 2020, p. 56–61.
- 30. Haibo H, Yang B, Garcia EA et al. ADASYN: adaptive synthetic sampling approach for imbalanced learning. In: 2008 IEEE International Joint Conference on Neural Networks (IEEE World Congress on Computational Intelligence). Hong Kong, 2008, p. 1322–8.
- 31. Read J, Pfahringer B, Holmes G, et al. Classifier chains for multi-label classification. Machine Learning 2011;85:333. [Google Scholar]
- 32. Ye J, Chow J-H, Chen J et al. Stochastic gradient boosted distributed decision trees. In: Proceedings of the 18th ACM Conference on Information and Knowledge Management. Hong Kong, China: Association for Computing Machinery, 2009, p. 2061–4. [Google Scholar]
- 33. Geurts P, Ernst D, Wehenkel L. Extremely randomized trees. Machine Learning 2006;63:3–42. [Google Scholar]
- 34. Tibshirani R. Regression shrinkage and selection via the lasso: a retrospective. J R Stat Soc Series B Stat Methodology 2011;73:273–82. [Google Scholar]
- 35. Gull S, Shamim N, Minhas F. AMAP: hierarchical multi-label prediction of biologically active and antimicrobial peptides. Comput Biol Med 2019;107:172–81. [DOI] [PubMed] [Google Scholar]
- 36. Chang C-C, Lin C-J. LIBSVM: a library for support vector machines. ACM Trans Intell Syst Technol 2011;2:Article 27. [Google Scholar]
- 37. Chen T, Guestrin C. XGBoost: a scalable tree boosting system. In: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. San Francisco, California, USA: Association for Computing Machinery, 2016, p. 785–94. [Google Scholar]
- 38. Fountoulakis M, Lahm H-W. Hydrolysis and amino acid composition analysis of proteins. J Chromatogr A 1998;826:109–34. [DOI] [PubMed] [Google Scholar]
- 39. Chung C-R, Kuo T-R, Wu L-C, et al. Characterization and identification of antimicrobial peptides with different functional activities. Brief Bioinform 2019;21:1098–114. [DOI] [PubMed] [Google Scholar]
- 40. Dong G-F, Zheng L, Huang S-H, et al. Amino acid reduction can help to improve the identification of antimicrobial peptides and their functional activities. Front Genet 2021;12:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zheng L, Liu D, Yang W, et al. RaacLogo: a new sequence logo generator by using reduced amino acid clusters. Brief Bioinform 2021;22(3):bbaa096. [DOI] [PubMed] [Google Scholar]
- 42. Xiao X, Shao Y-T, Cheng X, Stamatovic B. iAMP-CA2L: a new CNN-BiLSTM-SVM classifier based on cellular automata image for identifying antimicrobial peptides and their functional types. Brief Bioinform 2021;22(6):bbab209. [DOI] [PubMed] [Google Scholar]
- 43. Wang Q, Yuan Y. Learning to resize image. Neurocomputing 2014;131:357–67. [Google Scholar]
- 44. Xiao X, Wang P, Chou K-C. Cellular automata and its applications in protein bioinformatics. Current Protein and Peptide Science 2011;12:508–19. [DOI] [PubMed] [Google Scholar]
- 45. Kim Y. Convolutional Neural Networks for Sentence Classification. In: Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP). Doha, Qatar, 2014, p. 1746–51. Association for Computational Linguistics. [Google Scholar]
- 46. Hochreiter S, Schmidhuber J. Long short-term memory. Neural Comput 1997;9:1735–80. [DOI] [PubMed] [Google Scholar]
- 47. Qiaozhen M, Tang J, Guo F. Multi-AMP: detecting the antimicrobial peptides and their activities using the multi-task learning. In: 2021 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). Houston, TX, USA, 2021, p. 710–3.
- 48. Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. Edited by G. Von Heijne. J Mol Biol 1999;292:195–202. [DOI] [PubMed] [Google Scholar]
- 49. Pinacho-Castellanos SA, García-Jacas CR, Gilson MK, et al. Alignment-free antimicrobial peptide predictors: improving performance by a thorough analysis of the largest available data set. J Chem Inf Model 2021;61:3141–57. [DOI] [PubMed] [Google Scholar]
- 50. Graves A, Mohamed A, Hinton G. Speech recognition with deep recurrent neural networks. In: 2013 IEEE International Conference on Acoustics, Speech and Signal Processing. Vancouver, BC, Canada, 2013, p. 6645–9.
- 51. Huang Y, Niu B, Gao Y, et al. CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics 2010;26:680–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006;22:1658–9. [DOI] [PubMed] [Google Scholar]
- 53. Fu L, Niu B, Zhu Z, et al. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 2012;28:3150–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pirtskhalava M, Amstrong AA, Grigolava M, et al. DBAASP v3: database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res 2020;49:D288–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pirtskhalava M, Gabrielian A, Cruz P, et al. DBAASP v.2: an enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res 2015;44:D1104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gogoladze G, Grigolava M, Vishnepolsky B, et al. Dbaasp: database of antimicrobial activity and structure of peptides. FEMS Microbiol Lett 2014;357:63–8. [DOI] [PubMed] [Google Scholar]
- 57. Thomas S, Karnik S, Barai RS, et al. CAMP: a useful resource for research on antimicrobial peptides. Nucleic Acids Res 2009;38:D774–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Waghu FH, Gopi L, Barai RS, et al. CAMP: collection of sequences and structures of antimicrobial peptides. Nucleic Acids Res 2013;42:D1154–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mehta D, Anand P, Kumar V, et al. ParaPep: a web resource for experimentally validated antiparasitic peptide sequences and their structures. Database 2014;2014:bau051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hammami R, Ben Hamida J, Vergoten G, et al. PhytAMP: a database dedicated to antimicrobial plant peptides. Nucleic Acids Res 2008;37:D963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Qureshi A, Thakur N, Tandon H, et al. AVPdb: a database of experimentally validated antiviral peptides targeting medically important viruses. Nucleic Acids Res 2013;42:D1147–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tyagi A, Tuknait A, Anand P, et al. CancerPPD: a database of anticancer peptides and proteins. Nucleic Acids Res 2014;43:D837–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Usmani SS, Kumar R, Kumar V, et al. AntiTbPdb: a knowledgebase of anti-tubercular peptides. Database 2018;2018:bay025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gautam A, Chaudhary K, Singh S, et al. Hemolytik: a database of experimentally determined hemolytic and non-hemolytic peptides. Nucleic Acids Res 2013;42:D444–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Théolier J, Fliss I, Jean J, et al. MilkAMP: a comprehensive database of antimicrobial peptides of dairy origin. Dairy Sci Technol 2014;94:181–93. [Google Scholar]
- 66. Lee H-T, Lee C-C, Yang J-R, et al. A large-scale structural classification of antimicrobial peptides. Biomed Res Int 2015;2015:475062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Manavalan B, Basith S, Hwan Shin T, et al. MLACP: machine-learning-based prediction of anticancer peptides. Oncotarget 2017;8(44):77121–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Agrawal P, Bhagat D, Mahalwal M, et al. AntiCP 2.0: an updated model for predicting anticancer peptides. Brief Bioinform 2020;22(3):22. [DOI] [PubMed] [Google Scholar]
- 69. Schaduangrat N, Nantasenamat C, Prachayasittikul V, et al. ACPred: a computational tool for the prediction and analysis of anticancer peptides. Molecules 2019;24(10):1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wei L, Zhou C, Chen H, et al. ACPred-FL: a sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 2018;34:4007–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rao B, Zhou C, Zhang G, et al. ACPred-fuse: fusing multi-view information improves the prediction of anticancer peptides. Brief Bioinform 2019;21:1846–55. [DOI] [PubMed] [Google Scholar]
- 72. Yi H-C, You Z-H, Zhou X, et al. ACP-DL: a deep learning long short-term memory model to predict anticancer peptides using high-efficiency feature representation. Molecular Therapy - Nucleic Acids 2019;17:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lv Z, Cui F, Zou Q, et al. Anticancer peptides prediction with deep representation learning features. Brief Bioinform 2021;22(5):bbab008. [DOI] [PubMed] [Google Scholar]
- 74. Boopathi V, Subramaniyam S, Malik A, et al. mACPpred: a support vector machine-based meta-predictor for identification of anticancer peptides. International Journal of Molecular Sciences 2019;20:1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Agrawal P, Bhalla S, Chaudhary K, et al. In silico approach for prediction of antifungal peptides. Front Microbiol 2018;9:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Thakur N, Qureshi A, Kumar M. AVPpred: collection and prediction of highly effective antiviral peptides. Nucleic Acids Res 2012;40:W199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Pang Y, Yao L, Jhong J-H, et al. AVPIden: a new scheme for identification and functional prediction of antiviral peptides based on machine learning approaches. Brief Bioinform 2021;22(6):bbab263. [DOI] [PubMed] [Google Scholar]
- 78. Manavalan B, Basith S, Shin TH, et al. AtbPpred: a robust sequence-based prediction of anti-tubercular peptides using extremely randomized trees. Comput Struct Biotechnol J 2019;17:972–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gupta S, Sharma AK, Jaiswal SK, et al. Prediction of biofilm inhibiting peptides: an in silico approach. Front Microbiol 2016;7:00949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Fallah Atanaki F, Behrouzi S, Ariaeenejad S, et al. BIPEP: sequence-based prediction of biofilm inhibitory peptides using a combination of NMR and physicochemical descriptors. ACS Omega 2020;5:7290–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sharma A, Gupta P, Kumar R, et al. dPABBs: a novel in silico approach for predicting and designing anti-biofilm peptides. Sci Rep 2016;6:21839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chaudhary K, Kumar R, Singh S, et al. A web server and mobile app for computing hemolytic potency of peptides. Sci Rep 2016;6:22843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hasan MM, Schaduangrat N, Basith S, et al. HLPpred-fuse: improved and robust prediction of hemolytic peptide and its activity by fusing multiple feature representation. Bioinformatics 2020;36:3350–6. [DOI] [PubMed] [Google Scholar]
- 84. Khatun S, Hasan M, Kurata H. Efficient computational model for identification of antitubercular peptides by integrating amino acid patterns and properties. FEBS Lett 2019;593:3029–39. [DOI] [PubMed] [Google Scholar]
- 85. Olsen TH, Yesiltas B, Marin FI, et al. AnOxPePred: using deep learning for the prediction of antioxidative properties of peptides. Sci Rep 2020;10:21471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Timmons PB, Hewage CM. HAPPENN is a novel tool for hemolytic activity prediction for therapeutic peptides which employs neural networks. Sci Rep 2020;10:10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Consortium TU . The universal protein resource (UniProt). Nucleic Acids Res 2007;36:D190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Consortium TU . UniProt: a hub for protein information. Nucleic Acids Res 2014;43:D204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Consortium TU . UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 2018;47:D506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Consortium TU . UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res 2023;51:D523–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kawashima S, Pokarowski P, Pokarowska M, et al. AAindex: amino acid index database, progress report 2008. Nucleic Acids Res 2007;36:D202–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kawashima S, Kanehisa M. AAindex: amino acid index database. Nucleic Acids Res 2000;28:374–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Otović E, Njirjak M, Kalafatovic D, et al. Sequential properties representation scheme for recurrent neural network-based prediction of therapeutic peptides. J Chem Inf Model 2022;62:2961–72. [DOI] [PubMed] [Google Scholar]
- 94. Daniel O, Paola R-VR, Torres. Peptides: a package for data mining of antimicrobial peptides. The R Journal 2015;7:4–14. [Google Scholar]
- 95. Lin T-Y, Goyal P, Girshick R et al. Focal loss for dense object detection. In: 2017 Proceedings of the IEEE international conference on computer vision. Venice, Italy, 2017, p. 2980–8.
- 96. Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochimica et Biophysica Acta (BBA) - Protein Structure 1975;405:442–51. [DOI] [PubMed] [Google Scholar]
- 97. Wang P, Bang J-K, Kim HJ, et al. Antimicrobial specificity and mechanism of action of disulfide-removed linear analogs of the plant-derived Cys-rich antimicrobial peptide Ib-AMP1. Peptides 2009;30:2144–9. [DOI] [PubMed] [Google Scholar]
- 98. Sidorczuk K, Gagat P, Pietluch F, et al. Benchmarks in antimicrobial peptide prediction are biased due to the selection of negative data. Brief Bioinform 2022;23:bbac343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Yan J, Bhadra P, Li A, et al. Deep-AmPEP30: improve short antimicrobial peptides prediction with deep learning. Molecular Therapy - Nucleic Acids 2020;20:882–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Hussain W. sAMP-PFPDeep: improving accuracy of short antimicrobial peptides prediction using three different sequence encodings and deep neural networks. Brief Bioinform 2022;23(1):bbab487. [DOI] [PubMed] [Google Scholar]
- 101. Zhang Q-Y, Yan Z-B, Meng Y-M, et al. Antimicrobial peptides: mechanism of action, activity and clinical potential. Mil Med Res 2021;8(1):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Mookherjee N, Anderson MA, Haagsman HP, et al. Antimicrobial host defence peptides: functions and clinical potential. Nat Rev Drug Discov 2020;19:311–32. [DOI] [PubMed] [Google Scholar]
- 103. Van der Maaten L, Hinton G. Visualizing data using t-SNE. JMLR 2008;9:2579–605. [Google Scholar]
- 104. García-Jacas CR, Pinacho-Castellanos SA, García-González LA, Brizuela CA. Do deep learning models make a difference in the identification of antimicrobial peptides? Brief Bioinform 2022;23(3):bbac094. [DOI] [PubMed] [Google Scholar]
- 105. Zhang Y, Lin J, Zhao L, et al. A novel antibacterial peptide recognition algorithm based on BERT. Brief Bioinform 2021;22(6):bbab200. [DOI] [PubMed] [Google Scholar]
- 106. Fu H, Cao Z, Li M, Wang S. ACEP: improving antimicrobial peptides recognition through automatic feature fusion and amino acid embedding. BMC Genomics 2020;21:597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Sharma R, Shrivastava S, Singh SK, et al. Deep-AVPpred: artificial intelligence driven discovery of peptide drugs for viral infections. IEEE J Biomed Health Inform 2022;26:5067–74. [DOI] [PubMed] [Google Scholar]
- 108. García-Jacas CR, García-González LA, Martinez-Rios F, et al. Handcrafted versus non-handcrafted (self-supervised) features for the classification of antimicrobial peptides: complementary or redundant? Brief Bioinform 2022;23(6):bbac428. [DOI] [PubMed] [Google Scholar]
- 109. Rives A, Meier J, Sercu T, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc Natl Acad Sci 2021;118:e2016239118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Devlin J, Chang M-W, Lee K, et al. Bert: pre-training of deep bidirectional transformers for language understanding. arXiv preprint arXiv:181004805 2018; preprint: not peer reviewed. [Google Scholar]
- 111. Lin Z, Akin H, Rao R, et al. Evolutionary-scale prediction of atomic level protein structure with a language model. Science 2023;379(6637):1123–30. [DOI] [PubMed] [Google Scholar]
- 112. Chan DI, Prenner EJ, Vogel HJ. Tryptophan- and arginine-rich antimicrobial peptides: structures and mechanisms of action. Biochimica et Biophysica Acta (BBA) - Biomembranes 2006;1758:1184–202. [DOI] [PubMed] [Google Scholar]
- 113. Cutrona KJ, Kaufman BA, Figueroa DM, et al. Role of arginine and lysine in the antimicrobial mechanism of histone-derived antimicrobial peptides. FEBS Lett 2015;589:3915–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hong J, Oren Z, Shai Y. Structure and organization of hemolytic and nonhemolytic diastereomers of antimicrobial peptides in membranes. Biochemistry 1999;38:16963–73. [DOI] [PubMed] [Google Scholar]
- 115. Shahmiri M, Cornell B, Mechler A. Phenylalanine residues act as membrane anchors in the antimicrobial action of Aurein 1.2. Biointerphases 2017;12:05G605. [DOI] [PubMed] [Google Scholar]
- 116. Yokoyama M, Okano T, Sakurai Y, et al. Toxicity and antitumor activity against solid tumors of micelle-forming polymeric anticancer drug and its extremely long circulation in blood. Cancer Res 1991;51:3229–36. [PubMed] [Google Scholar]
- 117. Norouzi P, Mirmohammadi M, Houshdar Tehrani MH. Anticancer peptides mechanisms, simple and complex. Chem Biol Interact 2022;368:110194. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source codes and data are available at https://github.com/joy50706/iAMPCN/tree/master.