
FIGURE 1.
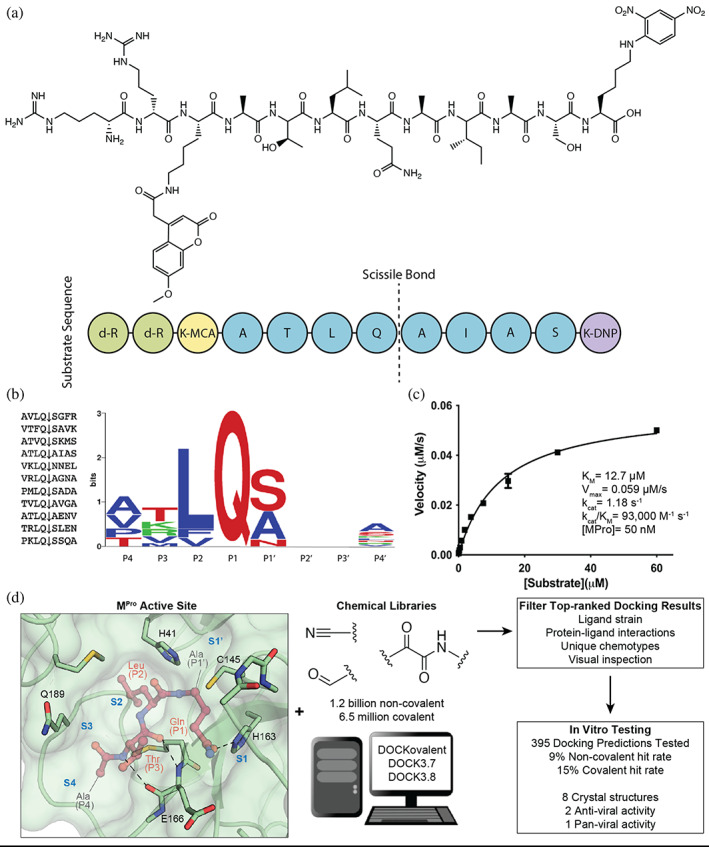
Substrate design and assay development allows structure‐based inhibitor discovery. (a) The chemical structure of the optimized NSP7 substrate shown as a schematic (top) of the substrate sequence highlights the role of each residue (bottom). The substrate contains the P4‐P4′ NSP7 extended substrate sequence (blue), the fluorophore (yellow), the fluorescent quencher (purple), and the residues for increasing solubility (green). (b) A list of the viral polypeptide NSP sequences (P4‐P4′) that are cleaved by MPro (left). The sequence logo highlighting the substrate specificity of MPro, yielding a P4‐P4′ consensus sequence: ATLQ(S/A)XXA (right). (c) The Michaelis‐Menten kinetics for the NSP7 substrate with MPro yield parameters indicative of an optimized, efficient substrate. (d) SARS‐CoV‐2 MPro active site (PDB 6Y2G) (Zhang, Lin, Sun, et al., 2020) (green; sub‐pockets S1′, S1, S2, S3, S4), shown here with substrate preferences (pink; P1′, P1, P2, P3, P4) (modeled after PDB 3SNE) (Zhu et al., 2011), was used to dock 1.2 billion non‐covalent molecules and 6.5 million electrophile molecules. Top‐ranked molecules were filtered and 395 were synthesized for in vitro testing. Some docking hits were prioritized for compound optimization, crystallography, pan‐viral enzymatic activity, and cell‐based antiviral activity. For C, experiments were performed in triplicate.