

Abstract

High-precision nanomaterials to entrap DNA-binding molecules are sought after for applications such as controlled drug delivery and scaffold-assisted structural biology. Here, we engineered protein–DNA cocrystals to serve as scaffolds for DNA-binding molecules. The designed cocrystals, isoreticular cocrystals, contain DNA-binding protein and cognate DNA blocks where the DNA–DNA junctions stack end-to-end. Furthermore, the crystal symmetry allows topology preserving (isoreticular) expansion of the DNA stack without breaking protein–protein contacts, hence providing larger solvent channels for guest diffusion. Experimentally, the resulting designed isoreticular cocrystal adopted an interpenetrating I222 lattice, a phenomenon previously observed in metal–organic frameworks (MOFs). The interpenetrating lattice crystallized dependably in the same space group despite myriad modifications at the DNA–DNA junctions. Assembly was modular with respect to the DNA inserted for expansion, providing an interchangeable DNA sequence for guest-specified scaffolding. Also, the DNA–DNA junctions were tunable, accommodating varied sticky base overhang lengths and terminal phosphorylation. As a proof of concept, we used the interpenetrating scaffold crystals to separately entrap three distinct guest molecules during crystallization. Isoreticular cocrystal design offers a route to a programmable scaffold for DNA-binding molecules, and the design principles may be applied to existing cocrystals to develop scaffolding materials.
Keywords: cocrystal, design, DNA-binding protein, scaffold, X-ray crystallography
An alluring promise of nanotechnology is the ability to precisely direct the assembly of functional domains within a highly organized material (e.g., a molecular pegboard). DNA assemblies have been a popular strategy to create nanostructured materials for many reasons. DNA is relatively inexpensive and quick to synthesize, making it possible to test numerous designed crystal variants. Furthermore, under the right conditions, the structure of B-DNA and the pairing of canonical bases is predictable.1 Therefore, the final designed structure is highly likely to assemble from the sequence of DNA building blocks. Covalent attachment of DNA to proteins is one promising route to control protein assembly into high-order materials including crystals.2 In this case, a major design parameter is the number and spatial distribution of the DNA attachment sites.3 For example, it is possible to control fiber assembly4 or to direct single crystal formation5 with a single surface attached DNA. At the other extreme, it is possible to attach numerous DNA linkers to a protein allowing the protein to serve as a node akin to a colloidal crystal nanoparticle.6,7 However, it has proven difficult to obtain designed materials that simultaneously [1] attain ultrahigh precision (e.g., high-resolution X-ray diffracting single crystals), [2] offer significant free space for the installation of guest domains, and [3] adopt a truly modular target structure which can offer any desired DNA sequence as an installation site for guest molecules. Arguably, the reported materials that most closely match this vision of a high-precision modular scaffold crystal are the tensegrity triangle family of designed DNA crystals pioneered by Seeman and colleagues.8−11 These DNA crystals offer significant sequence flexibility in the component DNA tiles as well as significant porosity and guest capacity when the tiles are large but have limited nanostructure precision, as assessed via X-ray diffraction resolution.
We hypothesized that we could create a family of porous scaffold crystals that combine the programmability advantages of DNA crystals with the high-resolution diffraction that has been previously observed for protein–DNA cocrystals. We further hypothesized that incorporating protein components into the designed crystal lattice would remove both the necessity of incorporating Holliday junctions into the DNA and the necessity of close packing DNA. Instead, our target lattice would consist of contiguous DNA double helices “insulated” by scaffold proteins. Additionally, the protein portion of the designed porous lattice would offer additional guest installation sites (e.g., via accessible thiols, fusion to the N- or C-termini, or insertion into loops) and greater control over the physicochemical composition of the scaffold crystal interior (given the larger variety of amino acids compared to nucleotides). Meanwhile, the DNA component offers the compelling advantage of using sequence specific DNA-binding events for both construction and guest incorporation.12−15
A suitably engineered scaffold crystal will self-assemble consistently and independently of the target biomolecule. In principle, guest molecules that adopt a uniform conformation with respect to the crystal lattice can also be revealed by X-ray diffraction (XRD). Scaffold-assisted X-ray crystallography is a potentially high-throughput structural biology strategy that decouples the crystallization problem from the target biomolecule, reviewed previously.16 Limited examples of scaffolds using DNA-binding for host–guest structural biology include cryo-EM goniometers17 and protein–DNA host crystals for small-molecule drugs.18 Here, we pursue a scaffold lattice that can be optimized independently of the guest.
We present a designed scaffold protein and DNA lattice suitable for site-specific guest molecule installation via programmed DNA-binding (Figure 1). The crystal DNA–DNA junctions are designable with modular sequences for varied guest targets. We demonstrate the concept with a target crystal lattice that contains two symmetry distinct protein–protein interfaces that should be maintained during 2D DNA expansion. Engineered cocrystal variants consistently grew, adopting the target interfaces despite a variety of modifications at the DNA–DNA junction, albeit in an interpenetrating, less porous habit. The interpenetrating cocrystals were found to host three diverse representative guest molecules (DNA, protein, small molecule), all installed via site-specific DNA binding during crystallization. Our crystals therefore represent a significant step toward a full porous programmable assembly scaffold crystal that can be assembled prior to site-specific guest installation. Our general strategy of isoreticular cocrystal expansion will ultimately enable the design and synthesis of high-precision, modular protein–DNA crystals with a variety of solvent pore sizes, topologies, and guest molecule installation schemes.
Figure 1.

An isoreticular cocrystal (A) is built of a scaffold protein (gray sphere) and DNA (green rod). Inserted DNA (dark green rod) might be added at the DNA–DNA junctions, and insertion lengths that respect the DNA helical twist may allow junctions to be stabilized by coding sticky overhangs. (B) The simplest example of crystal expansion would occur in a 1D expansion with parallel DNA stacks throughout the crystal. (C) 2D expansion may occur when DNA–DNA junctions mediate growth in two dimensions. (D) The inserted DNA provides site-specific scaffolding for the DNA-binding guest of interest.
Results and Discussion
The Isoreticular Cocrystal Design
Our designed protein–DNA cocrystals consist of a scaffold DNA-binding protein and cognate DNA sequence (Figure 1). Insertion of DNA at the junctions results in a periodic, isoreticular (topology preserving) expansion of existing cocrystals. To design a porous isoreticular cocrystal (ICC), we first sought a DNA–protein cocrystal lattice where (1) coaxial dsDNA:dsDNA contacts are the primary contacts enabling growth of the crystal in at least one dimension, (2) these DNA:DNA junctions align with the crystallographic symmetry so that expansion of the DNA blocks could occur without altering other important crystallographic contacts, and (3) any important protein–protein contacts are orthogonal to the DNA–DNA contacts and might therefore be preserved during assembly of the resulting ICC.
We identified candidate cocrystal building blocks by filtering Protein Data Bank (PDB) entries (as of 01/2017) derived from X-ray crystallography. To this end, we used the PDB advanced search and custom Python scripts. We excluded entries that were very large (>1 Mb) or lacked sufficient DNA content (less than 20 O5′ atoms) or lacked sufficient protein content (less than 19 amino acids). We also excluded candidates with significant RNA content (more than 2 O2′ atoms) or candidates with unknown atoms (UNK). After these content filters, we proceeded to filter the list using geometry, rejecting candidates that lacked stacked duplex DNA junctions between DNA in the asymmetric unit and symmetry copy DNA through inspection of pairwise distances between the ribose C1′ atoms in terminal nucleotides. Candidate crystals were discarded unless there was a terminal C1′ in the asymmetric unit and a nearby (3–7 Å) terminal C1′ in a symmetry copy. Next, a more sophisticated script was used to identify DNA–DNA junctions spanning crystallographic interfaces in which both sides feature base paired dsDNA. First, we determined if the junctions were DNA stacks (end-to-end). We generated symmetry copies of the C1′ positions, calculated distances between asymmetric unit C1′ and the symmetry copy C1′ and flagged possible stacking C1′ pairs as those between 3 and 7 Å. Further, we analyzed termini base pair status. Base paired termini included structures in which C1′ distances between terminal bases within the same asymmetric unit were within 9–12 Å. The last part of this script computes the step rotations at the termini to further assess the junctions (Figure S1 and Python scripts on Zenodo19). Finally, we collected the structures in which there were stacked junctions containing duplexes that were base paired at these junctions.
The resulting list of 609 PDB entries was visually assessed. Specifically, we used PyMOL and the Supercell script20 to inspect the lattice contacts for each candidate, and to flag candidates where expansion of the DNA stacks might occur without breaking protein–protein contacts. Most of the candidates that appeared to be suitable for isoreticular expansion were cases in which all of the DNA blocks in the crystal were aligned in the same direction (1D expansion candidates). A minority of the candidates had symmetry such that DNA block insertions might expand the crystal in two dimensions, thereby rapidly increasing the diameter of the crystal solvent channels. We did not encounter a candidate that appeared to be expandable in three dimensions.
Cocrystal 1 Expansion
Here, we highlight the 2D expansion system composed of RepE54 replication initiation protein and cognate DNA sequence (21-mer).21 For convenience, we name this system cocrystal 1 (CC1). We recently reported an updated PDB model for CC1 with improved resolution of 1.9 Å (PDB code: 7rva), while optimizing a protocol to dramatically stabilize such crystals using chemical ligation.22 The asymmetric unit for the original, nonexpanded CC1 system contains only one copy of the protein bound to cognate dsDNA (Figure 2). In accordance with the C121 space group, both ends of the dsDNA form DNA–DNA junctions that span crystallographic interfaces between the asymmetric unit and symmetry copies (Figure 2F). Notably, the blunt end DNA–DNA junctions stack closely, resembling contiguous DNA. One important difference from contiguous DNA is that the best nonexpanded CC1 structure (PDB code 7rva) has dangling 3′ thymine residues. The same crystal lattice grew with blunt ended DNA–DNA junctions (PDB codes 7sgc and 7sdp) if we removed the dangling T bases (albeit with slightly lower X-ray diffraction resolution). There are two distinct protein–protein interfaces in the CC1 unit cell (Figure 2C). During expansion of the DNA–DNA junctions, we aimed to preserve existing protein–protein contacts.
Figure 2.

Asymmetric unit of cocrystal 1 (A) is the complex of RepE54 replication initiator protein and a cognate 21-mer binding sequence (based on PDB code 7rva). The CC1 lattice (B) is in a C121 space group, and there are two protein–protein interfaces (C) conserved throughout the lattice. Upon expansion of the DNA with 10 additional base pairs (D) the lattice is highly porous (E) and the DNA–DNA junctions (G) are accessible for DNA-binding guest targets and are not sequence constrained by scaffold interactions.
To achieve an isoreticular expanded cocrystal and insert another twist in the DNA at the junction, we truncated the dangling Ts that serve to anchor the strands on the sides of the DNA, and we added DNA bases. Per x3DNA,23 the geometric parameters of the blunt ended junction with terminal phosphates are in Table 1. Explicit modeling of possible DNA inserts (Protocol S1, custom Python scripts are available on Zenodo19) suggested than a 10 base pair insertion would approximate a contiguous dsDNA geometry at the DNA:DNA junction (CC1+10). The target crystal lattice would be quite porous with a solvent fraction of 80% (Figure 2E, Protocol S2) and solvent channels large enough to permit transport of guests with diameter of 5 nm (per MAPCHANNELS24).
Table 1. DNA–DNA Junction Geometry Parameters in Different Versions of the CC1 Native Crystala.
| Junction parameters from x3dna | CC1 (original) | B-DNAb | CC1–5′P (blunt-ended) | CC1–3′P (blunt-ended) |
|---|---|---|---|---|
| PDB code | 7rva | 7sgc | 7sdp | |
| Base pair step parameters | GC/CG | GC/CG | GC/CG | |
| Shift (Å) | –0.03 | 0.0 ± 0.51 | 0.10 | 0.05 |
| Slide (Å) | –0.81 | 0.35 ± 0.78 | –1.00 | –2.03c |
| Rise (Å) | 3.49 | 3.32 ± 0.19 | 3.78c | 3.56 |
| Tilt (deg) | 1.87 | 0.0 ± 3.4 | 2.51 | 0.72 |
| Roll (deg) | 1.25 | 1.4 ± 5.1 | 1.94 | 4.03 |
| Twist (deg) | 36.64 | 35.4 ± 6.3 | 39.10 | 28.18 |
The original CC1 contains 3′ terminal dangling thymines. For expansion, these thymines were truncated for two bluntly ended versions: CC1-5′p and CC1-3′p.
Base pair step parameters from Olson et al. 1998.25 C5′ to O3′ distance from x3dna idealized B-DNA.
Values that differ from B-DNA by more than 2 standard deviations.
We obtained crystals which featured the designed DNA–DNA junctions while maintaining the two original protein–protein interactions. This was in keeping with our goal to obtain “isoreticular” crystals with expanded DNA but maintaining the same lattice topology. However, under the crystallization conditions tested to date, we did not obtain a porous crystal. Instead of the original space group of C121, our data was best explained using a space group of I222 in which a second copy of the target lattice is found within the body of the crystal, rotated by 180° (Figure 3). In other words, when we expanded the DNA struts which hold together the CC1 lattice in two dimensions, we obtained two (interpenetrating) copies of the target lattice rather than a single porous lattice. Interpenetrating crystal growth is an interesting phenomenon which has been repeatedly observed in the context of metal organic frameworks (MOFs), particularly when researchers attempt to grow isoreticular MOFs with larger pores.26−28 Despite being a less porous system, interpenetrating CC1 shows modularity and utility for hosting DNA-binding molecules. In the future, additional building block design or assembly process engineering may allow us to grow only one copy of the target lattice.
Figure 3.

Target porous lattice (A), shown here in the I121 setting, was similar to (B) half of the contents of the I222 experimental structure. (C) The other half of the I222 experimental structure is obtained via a 180° rotation about the x-axis (vertical in this diagram). (D) The full I222 structure requires close packing of DNA duplexes and involves additional 2-fold symmetry axes perpendicular to the y-axis (which is itself perpendicular to the page).
The designed lattice and the experimentally obtained lattice were quite similar (Figure 3). The obtained I222 unit cell (75.26 Å, 132.07 Å, 134.99 Å, 90.00°, 90.00°, 90.00° in the best resolution CC1+10bp data set (PDB code: 7u6k)) differed from the C121 target model (161.43 Å, 121.86 Å, 73.89 Å, 90.00°, 121.53°, 90.00°). To reveal the similarity (Figure 3), we select an alternate I121 setting of the design model (135.99 Å, 120.57 Å, 73.30 Å, 90.00°, 95.63°, 90.00°) and rotate the axes for the experimentally obtained orthorhombic I222 crystal (so that the unit cell vectors are 134.99 Å, 132.07 Å, 75.26 Å).
The obtained protein structure closely matches the structure in the parent structure (PDB 7rva) and thus also the protein in the designed expanded model (Figure S2). Specifically, when superimposing the 210 alpha carbons found in both models, the RMSDCα = 0.49 Å. Given this alignment, the positions of the two most significant protein symmetry neighbors (both of which are rotated 180° about the y-axis) closely match those of the design model. The neighbor with the larger deviation (RMSDCα = 1.42 Å) is adjoined via a protein–protein interface formed between the C-terminal beta-sheets. The neighbor with the smaller deviation (RMSDCα = 1.23 Å) is connected via a protein–protein interface that surrounds a 2-fold symmetry axis that passes between Gly91, Pro113, and Ile116 and the symmetry copies thereof. The slightly more precise conservation of position for this neighbor may be due to the larger interface (1465 Å2 SASA buried) compared to the C-terminal β sheet interface (775 Å2), per calculations with MSMS.29 Considering the positions of both protein neighbors, the small deviations are consistent with minor changes in the size of the shortest unit cell vector (73.3 Å versus 75.3 Å) and the unit cell vector (c⃗) that is aligned with the direction of the protein stack in the crystal (Figure 3A-B).
The other two critical symmetry neighbors are connected via pure
translational displacement and DNA:DNA stacking. If a⃗, b⃗, and c⃗ represent
the unit cell edge vectors (not just the scalar edge lengths), the
DNA:DNA stacking neighbors are found at 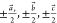 . To quantify the displacement of the DNA
in one of these symmetry neighbors, we compute RMSDC1′ = 8.60 Å, comparing the 62 C1′ atoms in the obtained
crystal to the same atoms in the design model. We attribute the larger
displacement of the neighboring DNA, compared to neighbor proteins,
to a “lever arm” movement of the expanded DNA.
. To quantify the displacement of the DNA
in one of these symmetry neighbors, we compute RMSDC1′ = 8.60 Å, comparing the 62 C1′ atoms in the obtained
crystal to the same atoms in the design model. We attribute the larger
displacement of the neighboring DNA, compared to neighbor proteins,
to a “lever arm” movement of the expanded DNA.
Whereas the RMSD numbers above refer to entire protein or DNA domains, it is also interesting to quantify the deviation between the target crystal design and the obtained crystal at the key interfaces. For example, if we select 28 key alpha and beta carbon positions across the C-terminal β sheet protein–protein interface, from (I180, S182, S225, I227, K229, V239, S241) in each symmetry partner, we can superpose all 28 carbon atoms with RMSDCαβ = 0.75 Å. Essentially, as shown in Figure S3, the details of the obtained interface strongly match the parent structure (PDB entry 7rva) and the expanded design model built from it. We can similarly select 26 residues at the other protein–protein interface, 13 residues from each symmetry partner (A90, G91, E93, E110, S111, F112, P113, I116, K117, P118, N132, P133, Y134). We can superpose all 50 alpha and beta carbons with RMSDCαβ = 0.56 Å. In keeping with the results presented above, this second protein–protein interface more precisely recapitulates the interface in the parent structure (Figure S3).
Cocrystal 1 Modularity
We demonstrated that assembly of the target expanded lattice (31-mer rather than 21-mer) was modular by growing crystals using three different DNA expansion strategies (Figure 4). Symmetric expansion (Figure 4B) used a single DNA block with 5 additional base pairs flanking the scaffold protein binding segment, thereby converting a 21-mer to a 31-mer. Asymmetric expansion (Figure 4C) used a single DNA block with 10 bp added to one side of the scaffold protein binding site. Lastly, the two-part scaffold-insert expansion strategy (Figure 4D) used two separate DNA blocks, one scaffold duplex that consisted of the scaffold protein binding site (21-mer) and one insert duplex (10-mer). All three strategies led to the growth of crystals with the same long, tapered prismatic habit and the same I222 interpenetrating lattices (Figure 5).
Figure 4.

Isoreticular cocrystal design schemes vary the original, nonexpanded duplex (A). The symmetrical expansion (B) fused 5 base pairs to each side of the original duplex. The asymmetrical expansion (C) fused 10 base pairs to a single side of the original duplex. The scaffold-insert expansion (D) maintained a 21 base pair scaffold strand and added an independent 10 base pair insert strand with matching sticky overhangs to the scaffold strand. In each expansion scheme, the DNA–DNA junctions were tunable with varied sticky base overhang lengths and terminal phosphorylation (no phosphate, 3′P, or 5′P).
Figure 5.

Interpenetrating CC1+10bp crystal growth for various DNA blocks cocrystallized with RepE54 Transcription Factor. Symmetric expansion crystals were grown with (A) G-C rich or (B) T-A rich addition. (C) Asymmetric expansion crystals were grown with a G-C rich addition. Scaffold-insert expansion crystals were grown with a (D) G-C rich insert strand or (E) T-A rich insert strand. (F–J) Symmetric expansion crystals grew with varied sticky base overhang lengths: blunt end, 1-nt, 2-nt, 3-nt, and 4-nt. Note: panel H intentionally duplicates panel A. Matching crystal growth conditions are found in Table S1. PDB codes for A–E are in the upper right corner. Scale bars are 100 μm.
For isoreticular cocrystals to realize their potential as a general-purpose scaffold crystal, it is crucial to reliably assemble into the intended lattice while making changes to the sequence of the inserted DNA segment (Figure S4). To demonstrate reliable assembly regardless of the inserted DNA sequence, we grew crystals using both the symmetric and scaffold-insert strategies with two dramatically different expansion sequences: a G-C rich sequence (5′-GACGGCCCGG) and a T-A rich sequence (5′-GACGGTAATT) (Table S2). Crystals consistently grew into the interpenetrating lattice. There were slight differences in favorable crystal growth conditions and crystal habit (Figure 5, Table S1), although we cannot definitively attribute these to the sequence difference (as opposed to other variables such as oligo purity and concentration). X-ray diffraction revealed that the nanostructure was essentially identical for both expansion sequences. For example, in symmetric expanded crystals, both sequences resulted in the same space group I222 (Unit Cell dimensions Symmetric G-C rich: 75.261 132.066 134.986 90.000 90.000 90.000; Unit cell dimensions Symmetric T-A rich: 72.141 129.124 131.290 90.000 90.000 90.000).
To understand how these crystals were affected by changes at the DNA–DNA junction, we grew crystals with varied sticky base overhang lengths and terminal phosphorylation status. Using the symmetric expansion strategy and a G-C rich expansion sequence, we grew a variety of crystals featuring blunt ends as well as crystals with 1, 2, 3, or 4 sticky base overhangs encoding the desired assembly (Figure 5F–J). While crystals with differing sticky overhang lengths did grow with differences in size and optimum growth conditions, we nonetheless observed crystal growth for all such systems within one family of growth conditions (300–500 mM MgCl2, 25–35% PEG 400, and 100 mM tris HCl pH 8.0). We have similarly verified crystal growth (with blunt ends or 1, 2, or 3 nt-overhangs) when using DNA oligos that were modified to include either 3′ or 5′ phosphorylation. Crystal growth was reliable despite these changes. The nucleation and growth phenomenon that underly crystal formation are very sensitive to small differences in the initial conditions. For this reason, it is challenging to unambiguously attribute changes in crystal growth to one variable (e.g., sticky overhangs or phosphorylation). Our results to date, consistent with the work of others,30 suggest that crystal growth might be tuned by changing the DNA–DNA junctions, but future work will be needed to carefully quantify such effects using identical growth conditions.
Guest Protein Entrapment
To demonstrate the potential utility of the designed expanded crystals as molecular scaffolds, we embedded within the DNA insert a binding site (5′-TAATTA) for the engrailed homeodomain protein (EnH) (Figure 6 and Figure S5).31 Modeling suggests that EnH binding is incompatible with the presence of a symmetry neighbor from the interpenetrating lattice (Figure S6). Therefore, we hypothesized that EnH binding in the crystallization experiment might compete with formation of the interpenetrating lattice. To test this hypothesis, we first fused EnH at the genetic level to an enhanced green fluorescent protein (eGFP) (Figure S5 and Protocol S3). In principle, EnH-eGFP fluorescence might allow visualization and quantification of guest capture within the cocrystals via fluorescence or confocal microscopy.
Figure 6.

Symmetric expanded CC1+10bp was designed with the Engrailed homeodomain (EnH) binding site (A). After cocrystallization with EnH-eGFP fusion protein (B), the crystal fluoresced when exposed to 488 nm with confocal microscopy, indicating guest EnH-eGFP entrapment (C–D). The control crystal, grown without EnH-eGFP, did not fluoresce. Scale bar: 100 μm.
Our first EnH installation experiment used the symmetric expansion strategy. In this case, the full EnH binding sequence was divided between neighboring blocks (Figure 6). Our hypothesis was that EnH-eGFP would bind during crystal growth as the DNA–DNA sticky base overhangs hybridize and thus reconstitute the full EnH binding site. We further hypothesized that EnH-eGFP binding would stabilize labile DNA–DNA junctions during crystal growth. We cocrystallized scaffold symmetric expanded CC1+10bp with a moderate stoichiometric excess of EnH-eGFP (1:1.2:1.2 RepE54 scaffold protein, DNA, and guest EnH-eGFP). Consistent with the presence of EnH-eGFP in solution, the initial crystallization solution appeared green by eye. After crystal growth, the solution was transparent, but the crystal had not become bright green by eye. However, when illuminated with a blue laser (420 nm) using a confocal microscope, the crystals grown in the presence of EnH-eGFP were bright green (Figure 6). A control crystal, grown with the same scaffold protein and scaffold DNA in the absence of EnH-eGFP was not fluorescent (though, since reflections can occur at crystal facets due to refractive index changes, the EnH-eGFP doped crystal appears to illuminate part of the nearby control crystal).
The presence of a full interpenetrating lattice would be expected to block the EnH binding site (and would leave little room for the full EnH-eGFP fusion protein) (Figure S6). Therefore, we were unsurprised to find that we could not observe bound EnH in crystal structures where EnH-eGFP was present during crystallization (X-ray diffraction data sets for CC1+10bp crystals grown with EnH-eGFP present are published on Zenodo13). Instead, we were surprised to find, using z-stack confocal microscopy, that EnH-eGFP nonetheless appeared to be uniformly incorporated within the cocrystals and that strong incorporation required the cognate EnH DNA binding site. Specifically, control crystals lacking the EnH DNA binding site had much lower evident doping of the EnH-eGFP (Figure S7). Significant doping of EnH-eGFP might correspond to isolated defects in an otherwise ideal interpenetrating double lattice. Alternately, the crystal may have sectors that have only one of the two interpenetrating lattices (Figure S8). These regions of the crystal could be enriched with EnH-eGFP guest domains.
Guest DNA Cocrystallization
Using ICC modularity to generate scaffold crystals with specific protein binding sites is only one application for this class of tunable nanoscaffolds. Capturing functional cargo nucleic acids represents an intriguing alternative. For example, guest DNA might encode information or a guest RNA might have a functional therapeutic role. In both cases, the crystal might serve as a tough protective matrix.22 The scaffold-insert strategy described above provides a starting point if the insert DNA is viewed as a guest molecule. To track incorporation of guest DNA we grew crystals with a stoichiometric mixture of scaffold DNA (21-mer), scaffold protein, and insert “guest” DNA (10-mer). One strand of the insert duplex was modified with the red fluorescent dye TAMRA (conjugated to the C7 atom in thymine 2) (Figure 7). The insert DNA was varied to be G-C rich or T-A rich, both with a strand containing a TAMRA-T2. When control crystals were set up without the insert strand, crystals did not grow (Figure S9). We therefore expect that the quantity of guest DNA incorporated within the crystal is stoichiometrically matched to the quantity of scaffold DNA and scaffold protein.
Figure 7.

(A) The scaffold-insert expanded complex shows the scaffold DNA in cyan and the insert strand in dark blue. The TAMRA-labeled thymine 2 (magenta) is conjugated on the C7 atom (B). Cocrystals grew with the TAMRA-T2 in the interpenetrating I222 space group. Crystals consistently grew for both a G-C rich insert sequence (C) and a T-A rich insert sequence (D). Substituents R1 and R2 are the 5′ and 3′ neighboring nucleobase, respectively.
Postcrystallization, the guest-loaded crystals were magenta by eye (Figure 7C–D). The X-ray structures again adopted the intended expanded lattice twice within the same space via interpenetration. The thymine containing the TAMRA-T2 (Figure 7) had prominent electron density at the C7 atom (PDB codes 7uv6 and 7uv7) in both crystals, G-C rich and T-A rich inserts. The lack of electron density for the TAMRA ring system suggested that the fluorescent dye was not rigid in the crystal.
Guest Small Molecule Cocrystallization
Yet another use of a scaffold crystal is the entrapment of DNA-binding small molecules. We cocrystallized CC1+10bp with two different small molecule drugs, netropsin and doxorubicin. Netropsin, a well-studied minor groove DNA-binding drug, has been cocrystallized with DNA previously, and was shown to prefer T-A rich binding sequences, especially 5′-AATT.18 Here, we grew CC1+10bp cocrystals in the presence of a slight stoichiometric excess of netropsin (1:1.2:1.2 protein to DNA to netropsin). As with the previous guest protein and DNA, the presence of a DNA-binding guest did not prevent the assembly of the interpenetrating lattice. Post cocrystallization, the netropsin was visible in the X-ray diffraction pattern (Figure 8). Consistent with previous research on netropsin DNA binding specificity, the small molecule bound in the minor groove at the 5′-AATT sequence, which is located in the middle of the RepE54 protein recognition sequence (Figure 8A). RepE54 directly interacts with the two flanking major grooves. We did not find any direct protein to netropsin interaction. Each netropsin amide was hydrogen bonded to DNA (Figure S10) resembling the quintessential Class I netropsin-DNA binding which includes bifurcated hydrogen bonds between the amides and surrounding A-T bases.18 The ends of netropsin were ordered, with the amidinium atom hydrogen bonded to complementary A and T and the guanidinium end hydrogen bonded to neighboring adenines (Figure S10 panel B). Future experiments might relocate the netropsin binding site. Possibly, netropsin binding at the DNA–DNA junction could serve to tune crystal growth kinetics or to disfavor formation of the interpenetrating lattice.
Figure 8.

X-ray diffraction highlight of netropsin bound to CC1+10bp. (A) The electron density indicated that netropsin, shown in magenta, was bound at the 5′-AATT site. The cocrystal resolution was 3.1 Å. (B) The Polder map (an omit map for netropsin that excludes bulk solvent generated with PHENIX Polder Maps32) in gray mesh shows the electron density around netropsin in the binding pocket. The DNA backbone of the binding site is highlighted in orange. (C) The Polder map of netropsin (contour level 3 rmsd).
Doxorubicin is an antitumor compound that inhibits topoisomerase II by dsDNA intercalation.33 Doxorubicin is proposed to interact with dsDNA by either intercalation between G-C base pairs or electrostatics-driven minor groove binding at A-T rich areas.34 The drug was previously cocrystallized with 6-mer DNA between two G–C base pairs.35 As doxorubicin is bright red in solution, we anticipated a colorimetric difference in the crystals with the guest. We grew CC1+10bp cocrystals in the presence of a slight stoichiometric excess of doxorubicin (1:1.2:1.2 protein to DNA to doxorubicin). In solution, doxorubicin was bright pink, and the resulting cocrystals were lightly pink (Figure S12). Upon X-ray diffraction analysis, the scaffold crystal diffraction was maintained (3.1 Å), but doxorubicin was not visible in the electron density (Table S7, Zenodo19). In future studies, it may be feasible to use the DNA sequence modularity of CC1+10bp, or other crystals designed using the same principles, to probe the sequence specificity of DNA intercalating small molecules like doxorubicin.
Conclusions
The designed host cocrystals we present here are modular and consistent scaffolds that offer a route to arrange or entrap DNA-binding molecules within a high-precision material. In the interpenetrating crystal habit, the DNA designs repeatably obtained the same crystal symmetry (I222) despite changes in the expansion scheme, sequence, sticky base overhang lengths, and terminal phosphorylation. Within the expanded, interpenetrating cocrystals, we installed three diverse guest molecules via DNA-binding: a protein, a discrete DNA insert, and a small molecule. Therefore, we conclude that the crystal structure is robust and tunable for DNA-binding guests of interest. Our design principles may be used to expand the repertoire of protein–DNA scaffolds, as we have shown just one example among numerous alternative starting point cocrystals that already exist in the PDB.
A limiting feature of the interpenetrating habit cocrystals is the low porosity. Ideally, one would host guest proteins post-crystallization in a cocrystal with large solvent pores whereas the current interpenetrating form only allowed entrapment of guests during crystallization. We hypothesize the interpenetrating crystal form is preferred due to the high magnesium content in the growth solution, with divalent cations allowing the polyanionic DNA strands to pack tightly (Figure 3). Optimization of the growth solutions may produce a porous lattice and isoreticular expanded crystals for guest installation via DNA-binding. Alternatively, cocrystallization with a fully intact guest binding site and excess guest protein may force the crystal to grow only the intended porous lattice. From a different perspective, the less porous interpenetrating crystal form may be an advantageous material for controlled cargo release.
With a few exceptions, biomolecular crystals adopt adventitious lattices. Here we show that isoreticular expansion, a concept from metal organic framework engineering, can be applied to re-engineer protein–DNA cocrystals for the modular insertion of designed DNA blocks. The resulting scaffolds may have favorable biomaterial properties. For example, subjecting such an interpenetrating network to chemical ligation may result in robust biomaterials22 with myriad potential applications in sensing, delivery, or structural biology.
Materials and Methods
Protein Cloning, Expression, and Purification
The cloning and expression of the scaffold protein, RepE54 Replication Initiator, was described by Komori et al.21 The CC1 RepE54 was produced at the Histone Source at Colorado State University exactly as described in our previous chemical ligation study.22
The guest protein, Engrailed Homeodomain-eGFP Fusion (EnH-eGFP), was cloned into the PetDuet plasmid by using Gibson cloning (Protocol S3). EnH-eGFP protein was expressed with a T7 promoter in E. coli BL21(DE3) cells. Upon the addition of 0.5 mM IPTG, the cells were outgrown at 25 °C for 20 h. The cell pellets were sonicated in lysis buffer ((100 mM tris HCl, 200 mM NaCl, 10% glycerol, 10 mM imidazole, pH 8.0) and applied to HisTrap (HisPur Ni-NTA Resin) equilibrated with HisTrap buffer (100 mM tris HCl, 200 mM NaCl, 10% glycerol, 10 mM imidazole, pH 8.0). The protein was eluted with 100 mM imidazole in a HisTrap buffer. The EnH-eGFP protein was purified further with Nuvia cPrimeTM Hydrophobic Cation Exchange Media, equilibrated with cation exchange buffer (100 mM NaCl, 100 mM tris HCl, 10% glycerol, pH 6.8), and eluted with 400 mM NaCl in cation exchange buffer. Following cation exchange, the samples were purified via HiLoad Superdex 200 PG column (Cytiva) at the Histone Source at Colorado State University. The fractions containing EnH-eGFP protein were pooled, concentrated using an Amicon Ultra-15 10 kDa MWCO centrifugal filter unit (EMD Millipore), and dialyzed with EnH-eGFP storage buffer (200 mM NaCl, 100 mM tris HCl, 10% glycerol, pH 8.0). EnH-eGFP protein was collected, concentrated to 42 mg/mL, and stored at −80 °C after being flash-frozen with liquid nitrogen.
Protein purification steps were analyzed with SDS-PAGE (NuPAGE 4–12% Bis-Tris Gel) with MES SDS running buffer and stained with Imperial Protein stain. Bradford Assay using Coomassie Plus Protein Assay Reagent was used to determine final protein concentrations.
DNA Duplex Annealing
The DNA oligomer sequences used for cocrystallization are listed in Table S2. Each oligomer contains the 19-bp iteron sequence for RepE54 Protein–DNA binding, but the flanking DNA sequences vary depending on the expansion scheme and sequence. Individual oligomers were synthesized and HPLC purified by Integrated DNA Technologies and annealed in Snow Lab. The oligomers were resuspended in CC1 oligo buffer (50 mM tris HCl, 100 mM KCl pH 7.0) and combined in an equal molar ratio (1:1) with the complementary strand. The strands were annealed by heating to 94 °C for 2 min and slowly cooling to room temperature over approximately 60 min. The final concentration of all of the CC1 duplexes was 4 mM.
Scaffold Protein–DNA Complex Cocrystallization
The scaffold protein and DNA (1:1.2) were incubated on ice 30 min prior to crystallization via sitting drop vapor diffusion. Crystallization conditions for the interpenetrating lattice (CC1+10bp) were 300–500 mM MgCl2, 20–35% PEG 400 and 80–100 mM tris HCl pH 8.0. A detailed list of individual crystals, PDB codes, and corresponding growth conditions are in Table S1. Crystals grew to a span of 50–300 μm3 in a range of 24 h to 30 days. Crystal pictures were taken with a Moticam 3.0 MP camera attached to a Motic SMZ-168 stereozoom microscope.
Cocrystallization with Guest Protein, DNA and Small Molecule
Crystallization conditions for all guest entrapped cocrystals are in Table S1. Scaffold-guest cocrystallization were grown via sitting-drop vapor diffusion. The guest fusion protein (EnH-eGFP) was incubated with scaffold protein and symmetrical expanded DNA (1:1.2:1.2 scaffold protein to DNA to EnH-eGFP) on ice 30 min prior to the crystallization setup. The fluorescently labeled DNA guest was cocrystallized in CC1+10bp using the scaffold-insert scheme. The insert duplex was a 10-mer with 2 sticky base overhangs and a G-C rich sequence. The complementary strand contained the thymine-2 with a TAMRA. The oligomers making up the insert were synthesized and HPLC purified by Integrated DNA Technologies and the insert duplex was annealed in the Snow Lab. Cocrystallization proceeded with a modest excess of each of the scaffold and insert duplexes (1:1.2:1.2 scaffold protein to scaffold DNA to insert DNA). Both small molecule guests, netropsin dihydrochloride (VWR) and doxorubicin hydrochloride (UbpBio) were resuspended in molecular biology grade water and added to the G-C rich symmetric CC1+10bp complex prior to crystallization (1:1.2:1.2 scaffold protein to DNA to small molecule).
Confocal Microscopy
Fluorescence microscopy was performed on a Nikon Eclipse Ti spinning-disk confocal microscope with an AndoriXon Ultra 897U EMCCD camera. EnH-eGFP protein entrapment was analyzed under visible light (DIC-N1) and laser exposure (488 nm excitation).
X-ray Diffraction Data Collection and Refinement
Crystals were flash frozen in liquid nitrogen using the growth conditions as the cryo-protectant. Single-crystal X-ray diffraction data was collected at ALS beamline 4.2.2 using a CMOS detector from 0 to 180° with an omega delta of 0.2° and an exposure time of 0.5 s and data was processed with XDS.36 The doxorubicin cocrystal data set was collected in the Biomolecular Crystallography Facility in the Vanderbilt University Center for Structural Biology on a D8 Venture (Bruker AXS, Madison, WI) system, and the data set was reduced using Proteum3 software (Bruker AXS, Madison, WI). The best resolution expanded structure (CC1+10bp Symmetric G-C rich; PDB code: 7u6k; 2.38 Å) was solved by molecular replacement with the updated original RepE54 structure (PDB code: 7rva; 1.89 Å) and refined (PHENIX32 and COOT37). The later expanded structures were solved with molecular replacement using the best resolution expanded structure, and the same Rfree flags were applied. A flowchart of X-ray diffraction refinement for all expanded structures is given in Figure S11. Tables S3–S7 list X-ray diffraction and refinement statistics for all structures.
Guest Small Molecule Refinement
X-ray diffraction data was obtained from the cocrystal grown with netropsin. Molecular replacement was performed with the starting model as the best resolution expanded structure (CC1+10bp Symmetric G-C rich; PDB code: 7u6k; 2.38 Å). We first refined the structure without the netropsin ligand and produced a discovery map. To add netropsin to the structure, we used the previous structure (PDB code: 1ztt) with the same binding site (5′-AATT) and aligned the DNA binding sequences in PyMOL.31 The resulting orientation of netropsin was added to the discovery map structure, and this CC1+10bp-netropsin model was used for further refinement in PHENIX (and unselecting the automatic setting “link ligand to protein”). We produced Polder maps from the structure with the ligand with PHENIX Polder Maps32 and visualized the Polder maps in PyMOL.38 The final structure was analyzed for hydrogen bonds in PyMOL and LigPlot+ v.2.2,39 a ligand-protein interaction diagram package.
Acknowledgments
We thank H. Scherman, Director of the Histone Source at Colorado State University, for the expression and purification of the RepE54 Initiation Protein. We thank J. Nix, at the ALS Beamline 4.2.2, for extensive support of the XRD data collection. We thank P.S. Ho for crystallography advice and guest DNA-binding molecule suggestions. We thank T. Huber for cloning expertise and PSB3 plasmid. We thank J. Stuart for advice on confocal microscopy and TAMRA-labelled oligomer handling. We thank J. Harp, at the Biomolecular Crystallography Facility in the Vanderbilt University Center for Structural Biology, for training and support. We thank M. Zintel and C. Loewecke for proofreading.
Data Availability Statement
The code and data presented in this study are openly available in Zenodo at DOI: 10.5281/zenodo.7900381.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.2c07282.
Supporting cocrystal design and experimental evidence are in supplemental figures S1–S12. Protein–DNA crystallization conditions and X-ray diffraction data are in supplemental tables S1–S7. Co-crystal modeling details, cocrystal solvent calculations, and protein sequences are in supplemental protocols S1–S3 (PDF)
Author Present Address
⊥ A.R.O.: Department of Medicine, Division of Diabetes, Endocrinology and Metabolism, Vanderbilt University Medical Center, Nashville, Tennessee 37232, United States
Author Present Address
¶ A.V.: Department of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, Georgia 30332, United States
This material is based upon work supported by the National Science Foundation under Grant No. NSF DMR 2003748 and NSF DMR 1506219. The team also gratefully acknowledges support for undergraduate researchers from the Nelson Family Faculty Excellence Award.
The authors declare no competing financial interest.
Supplementary Material
References
- Hays F. A.; Teegarden A.; Jones Z. J. R.; Harms M.; Raup D.; Watson J.; Cavaliere E.; Ho P. S. How Sequence Defines Structure: A Crystallographic Map of DNA Structure and Conformation. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (20), 7157–7162. 10.1073/pnas.0409455102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan J. R.; Hayes O. G.; Winegar P. H.; Mirkin C. A. Protein Materials Engineering with DNA. Acc. Chem. Res. 2019, 52 (7), 1939–1948. 10.1021/acs.accounts.9b00165. [DOI] [PubMed] [Google Scholar]
- Hayes O. G.; Partridge B. E.; Mirkin C. A. Encoding Hierarchical Assembly Pathways of Proteins with DNA. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (40), e2106808118. 10.1073/pnas.2106808118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan J. R.; Hayes O. G.; Remis J. P.; Mirkin C. A. Programming Protein Polymerization with DNA. J. Am. Chem. Soc. 2018, 140 (46), 15950–15956. 10.1021/jacs.8b10011. [DOI] [PubMed] [Google Scholar]
- Winegar P. H.; Hayes O. G.; McMillan J. R.; Figg C. A.; Focia P. J.; Mirkin C. A. DNA-Directed Protein Packing within Single Crystals. Chem. 2020, 6, 1007. 10.1016/j.chempr.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodin J. D.; Auyeung E.; Mirkin C. A. DNA-Mediated Engineering of Multicomponent Enzyme Crystals. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (15), 4564–4569. 10.1073/pnas.1503533112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan J. R.; Brodin J. D.; Millan J. A.; Lee B.; Olvera de la Cruz M.; Mirkin C. A. Modulating Nanoparticle Superlattice Structure Using Proteins with Tunable Bond Distributions. J. Am. Chem. Soc. 2017, 139 (5), 1754–1757. 10.1021/jacs.6b11893. [DOI] [PubMed] [Google Scholar]
- Seeman N. C. Nucleic Acid Junctions and Lattices. J. Theor. Biol. 1982, 99 (2), 237–247. 10.1016/0022-5193(82)90002-9. [DOI] [PubMed] [Google Scholar]
- Paukstelis P. J.; Nowakowski J.; Birktoft J. J.; Seeman N. C. Crystal Structure of a Continuous Three-Dimensional DNA Lattice. Chem. Biol. 2004, 11 (8), 1119–1126. 10.1016/j.chembiol.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Birktoft J. J.; Chen Y.; Wang T.; Sha R.; Constantinou P. E.; Ginell S. L.; Mao C.; Seeman N. C. From Molecular to Macroscopic via the Rational Design of a Self-Assembled 3D DNA Crystal. Nature 2009, 461 (7260), 74–77. 10.1038/nature08274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons C. R.; Zhang F.; Birktoft J. J.; Qi X.; Han D.; Liu Y.; Sha R.; Abdallah H. O.; Hernandez C.; Ohayon Y. P.; Seeman N. C.; Yan H. Construction and Structure Determination of a Three-Dimensional DNA Crystal. J. Am. Chem. Soc. 2016, 138 (31), 10047–10054. 10.1021/jacs.6b06508. [DOI] [PubMed] [Google Scholar]
- Mou Y.; Yu J.-Y.; Wannier T. M.; Guo C.-L.; Mayo S. L. Computational Design of Co-Assembling Protein-DNA Nanowires. Nature 2015, 525 (7568), 230–233. 10.1038/nature14874. [DOI] [PubMed] [Google Scholar]
- Praetorius F.; Dietz H. Self-Assembly of Genetically Encoded DNA-Protein Hybrid Nanoscale Shapes. Science 2017, 355 (6331), eaam5488. 10.1126/science.aam5488. [DOI] [PubMed] [Google Scholar]
- Pippig D. A.; Baumann F.; Strackharn M.; Aschenbrenner D.; Gaub H. E. Protein–DNA Chimeras for Nano Assembly. ACS Nano 2014, 8, 6551. 10.1021/nn501644w. [DOI] [PubMed] [Google Scholar]
- McCluskey J. B.; Clark D. S.; Glover D. J. Functional Applications of Nucleic Acid–Protein Hybrid Nanostructures. Trends Biotechnol. 2020, 38, 976. 10.1016/j.tibtech.2020.02.007. [DOI] [PubMed] [Google Scholar]
- Ward A. R.; Snow C. D. Porous Crystals as Scaffolds for Structural Biology. Curr. Opin. Struct. Biol. 2020, 60, 85–92. 10.1016/j.sbi.2019.12.008. [DOI] [PubMed] [Google Scholar]
- Aksel T.; Yu Z.; Cheng Y.; Douglas S. M.. Molecular Goniometers for Single-Particle Cryo-EM of DNA-Binding Proteins. bioRxiv, Feb. 28, 2020, 2020.02.27.968883. 10.1101/2020.02.27.968883. [DOI] [PMC free article] [PubMed]
- Goodwin K. D.; Long E. C.; Georgiadis M. M. A Host–Guest Approach for Determining Drug–DNA Interactions: An Example Using Netropsin. Nucleic Acids Res. 2005, 33 (13), 4106–4116. 10.1093/nar/gki717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orun A. R.; Snow C. D.. Scripts for Designing Expanded, Isoreticular Co-Crystals and Crystal Datasets with Guest Protein; Zenodo, 2022. 10.5281/zenodo.6646957. [DOI]
- Holder T.Crystallographic Symmetry Related Commands; PyMOLWiki, 2012. [Google Scholar]
- Komori H.; Matsunaga F.; Higuchi Y.; Ishiai M.; Wada C.; Miki K. Crystal Structure of a Prokaryotic Replication Initiator Protein Bound to DNA at 2.6 A Resolution. EMBO J. 1999, 18 (17), 4597–4607. 10.1093/emboj/18.17.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orun A.; Dmytriw S.; Vajapayajula A.; Snow C. Stabilizing DNA–Protein Co-Crystals via Intra-Crystal Chemical Ligation of the DNA. Crystals 2022, 12 (1), 49. 10.3390/cryst12010049. [DOI] [Google Scholar]
- Lu X.-J.; Olson W. K. 3DNA: A Versatile, Integrated Software System for the Analysis, Rebuilding and Visualization of Three-Dimensional Nucleic-Acid Structures. Nat. Protoc. 2008, 3 (7), 1213–1227. 10.1038/nprot.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juers D. H.; Ruffin J. MAP_CHANNELS: A Computation Tool to Aid in the Visualization and Characterization of Solvent Channels in Macromolecular Crystals. J. Appl. Crystallogr. 2014, 47 (6), 2105–2108. 10.1107/S160057671402281X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson W. K.; Gorin A. A.; Lu X.-J.; Hock L. M.; Zhurkin V. B. DNA Sequence-Dependent Deformability Deduced from Protein–DNA Crystal Complexes. Proc. Natl. Acad. Sci. U. S. A. 1998, 95 (19), 11163–11168. 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasell T.; Culshaw J. L.; Chong S. Y.; Schmidtmann M.; Little M. A.; Jelfs K. E.; Pyzer-Knapp E. O.; Shepherd H.; Adams D. J.; Day G. M.; Cooper A. I. Controlling the Crystallization of Porous Organic Cages: Molecular Analogs of Isoreticular Frameworks Using Shape-Specific Directing Solvents. J. Am. Chem. Soc. 2014, 136 (4), 1438–1448. 10.1021/ja409594s. [DOI] [PubMed] [Google Scholar]
- Safarifard V.; Rodríguez-Hermida S.; Guillerm V.; Imaz I.; Bigdeli M.; Azhdari Tehrani A.; Juanhuix J.; Morsali A.; Casco M. E.; Silvestre-Albero J.; Ramos-Fernandez E. V.; Maspoch D. Influence of the Amide Groups in the CO2/N2 Selectivity of a Series of Isoreticular, Interpenetrated Metal–Organic Frameworks. Cryst. Growth Des. 2016, 16 (10), 6016–6023. 10.1021/acs.cgd.6b01054. [DOI] [Google Scholar]
- Tranchemontagne D. J.; Hunt J. R.; Yaghi O. M. Room Temperature Synthesis of Metal-Organic Frameworks: MOF-5, MOF-74, MOF-177, MOF-199, and IRMOF-0. Tetrahedron 2008, 64 (36), 8553–8557. 10.1016/j.tet.2008.06.036. [DOI] [Google Scholar]
- Sanner M. F.; Olson A. J.; Spehner J.-C. Reduced Surface: An Efficient Way to Compute Molecular Surfaces. Biopolymers 1996, 38 (3), 305–320. . [DOI] [PubMed] [Google Scholar]
- Li Z.; Zheng M.; Liu L.; Seeman N. C.; Mao C. 5′-Phosphorylation Strengthens Sticky-End Cohesions. J. Am. Chem. Soc. 2021, 143 (37), 14987–14991. 10.1021/jacs.1c07279. [DOI] [PubMed] [Google Scholar]
- Kissinger C. R.; Liu B. S.; Martin-Blanco E.; Kornberg T. B.; Pabo C. O. Crystal Structure of an Engrailed Homeodomain-DNA Complex at 2.8 A Resolution: A Framework for Understanding Homeodomain-DNA Interactions. Cell 1990, 63 (3), 579–590. 10.1016/0092-8674(90)90453-L. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX : A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 (2), 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutts S. M.; Swift L. P.; Rephaeli A.; Nudelman A.; Phillips D. R. Sequence Specificity of Adriamycin-DNA Adducts in Human Tumor Cells1. Mol. Cancer Ther. 2003, 2 (7), 661–670. [PubMed] [Google Scholar]
- Pérez-Arnaiz C.; Busto N.; Leal J. M.; García B. New Insights into the Mechanism of the DNA/Doxorubicin Interaction. J. Phys. Chem. B 2014, 118 (5), 1288–1295. 10.1021/jp411429g. [DOI] [PubMed] [Google Scholar]
- Frederick C. A.; Williams L. D.; Ughetto G.; Van der Marel G. A.; Van Boom J. H.; Rich A.; Wang A. H. J. Structural Comparison of Anticancer Drug-DNA Complexes: Adriamycin and Daunomycin. Biochemistry 1990, 29 (10), 2538–2549. 10.1021/bi00462a016. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 (2), 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60 (12), 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Delano W.The PyMOL Molecular Graphics System; 2002.
- Laskowski R. A.; Swindells M. B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51 (10), 2778–2786. 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code and data presented in this study are openly available in Zenodo at DOI: 10.5281/zenodo.7900381.