

Abstract
Vascular calcification is caused by the deposition of calcium salts in the intimal or tunica media layer of the aorta, which increases the risk of cardiovascular events and all-cause mortality. However, the mechanisms underlying vascular calcification are not fully clarified. Recently it has been shown that transcription factor 21 (TCF21) is highly expressed in human and mouse atherosclerotic plaques. In this study we investigated the role of TCF21 in vascular calcification and the underlying mechanisms. In carotid artery atherosclerotic plaques collected from 6 patients, we found that TCF21 expression was upregulated in calcific areas. We further demonstrated TCF21 expression was increased in an in vitro vascular smooth muscle cell (VSMC) osteogenesis model. TCF21 overexpression promoted osteogenic differentiation of VSMC, whereas TCF21 knockdown in VSMC attenuated the calcification. Similar results were observed in ex vivo mouse thoracic aorta rings. Previous reports showed that TCF21 bound to myocardin (MYOCD) to inhibit the transcriptional activity of serum response factor (SRF)-MYOCD complex. We found that SRF overexpression significantly attenuated TCF21-induced VSMC and aortic ring calcification. Overexpression of SRF, but not MYOCD, reversed TCF21-inhibited expression of contractile genes SMA and SM22. More importantly, under high inorganic phosphate (3 mM) condition, SRF overexpression reduced TCF21-induced expression of calcification-related genes (BMP2 and RUNX2) as well as vascular calcification. Moreover, TCF21 overexpression enhanced IL-6 expression and downstream STAT3 activation to facilitate vascular calcification. Both LPS and STAT3 could induce TCF21 expression, suggesting that the inflammation and TCF21 might form a positive feedback loop to amplify the activation of IL-6/STAT3 signaling pathway. On the other hand, TCF21 induced production of inflammatory cytokines IL-1β and IL-6 in endothelial cells (ECs) to promote VSMC osteogenesis. In EC-specific TCF21 knockout (TCF21ECKO) mice, VD3 and nicotine-induced vascular calcification was significantly reduced. Our results suggest that TCF21 aggravates vascular calcification by activating IL-6/STAT3 signaling and interplay between VSMC and EC, which provides new insights into the pathogenesis of vascular calcification.
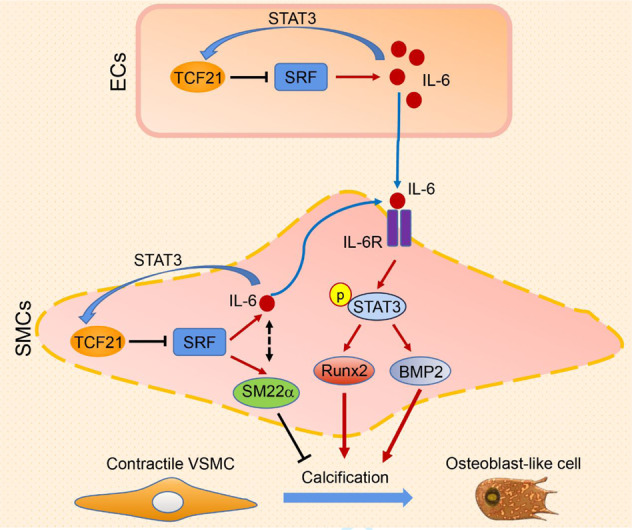
TCF21 enhances vascular calcification by activating the IL-6-STAT3 signaling pathway. TCF21 inhibition may be a new potential therapeutic strategy for the prevention and treatment of vascular calcification.
Keywords: TCF21, vascular calcification, SRF, IL-6/STAT3 signaling, inflammatory cytokines, vascular smooth muscle cell, endothelial cell
Introduction
The presence of vascular calcification increases the risk of acute cardiovascular events and all-cause mortality. The highly tunable and active process of vascular calcification is caused by the deposition of calcium salts in the intimal or tunica media layer of the aorta [1, 2], which is known as intimal (atherosclerotic) calcification or media calcification. Media calcification is accompanied by chronic kidney disease (CKD), diabetes mellitus, and aging, which can reduce vascular compliance and ultimately leads to heart failure or hypertension [3]. Moreover, intimal calcification often exists in advanced atherosclerotic plaques, and develops with the development of atherosclerotic lesions. Calcified nodules and spotty calcification at plaques will further increase the risk of plaque rupture, which is the main event leading to thrombosis and stroke [4]. However, no FDA-approved medications have been demonstrated to attenuate or reverse vascular calcification.
Vascular smooth muscle cells (VSMCs) are pluripotent cell types in blood vessels that physiologically exhibit a contractile phenotype. VSMCs are an essential component of the vessel wall, providing structural support, regulating vascular tone, and allowing vascular remodeling [5]. VSMCs can dedifferentiate into osteoblast-like cells, which is considered to be the initiation stage of vascular calcification [6]. VSMC-derived osteoblasts can secrete alkaline phosphatase (ALP), which is loaded into extracellular vesicles to promote the release of free phosphate, providing the basis for tissue and cell mineralization [7]. However, the underlying mechanisms of vascular calcification are not fully understood. Increasing evidence shows that vascular calcification develops when the tissue loses calcification inhibitory factors while accumulating calcification promoting factors. In addition, the interaction between endothelial cells (ECs) and VSMCs contributes to VSMC osteogenesis [8]. EC-derived inflammatory cytokines, chemokines, adhesion molecules and other types of molecules can promote VSMC phenotypic transition and calcification [9, 10]. Osteoblast-like cells derived from ECs are widely detected in calcified arterial media and atherosclerotic lesions [11].
Transcription factor 21 (TCF21), a member of the basic helix-loop-helix transcription factor family, is essential for the development of multiple cell types in the heart, lung, kidney and spleen during embryogenesis [12–15]. Recently, TCF21 was shown to be highly expressed in fibromyocytes, a collagen-synthesis cell type derived from VSMCs, and human and mouse atherosclerotic plaques [16]. Nagao M et al. demonstrated that TCF21 can directly bind to myocardin (MYOCD) to inhibit the transcriptional activity of the serum response factor (SRF)-MYOCD complex. TCF21 also inhibits SRF and MYOCD expression by binding to the promoter regions of these genes [17]. During heart development, TCF21-expressing epicardial cells are committed to the cardiac fibroblast lineage prior to the initiation of epicardial epithelial-to-mesenchymal transition, which is essential for cardiac fibroblast development [12]. Moreover, it has been demonstrated that TCF21-positve cells are distributed throughout early lesions, and accumulate in the fibrous cap and the subcapsular layer in advanced lesions, implying their important role during the development of atherosclerotic disease [18]. However, its role in vascular calcification remains to be investigated.
In the current study, we investigated the role and underlying mechanisms of TCF21 in vascular calcification. We found that TCF21 was increased in the calcification area of atherosclerotic lesions. Mechanistically, we elucidated that TCF21 facilitated VSMC osteogenesis in vivo and in vitro by promoting the interleukin 6 (IL-6)-signal transducer and activator of transcription 3 (STAT3) signaling pathway. These findings highlight the critical role of TCF21 in vascular calcification and suggest that TCF21 may act as a novel potential therapeutic target for vascular calcification.
Materials and methods
Reagents
Rabbit anti-TCF21, smooth muscle protein 22 alpha (SM22α), STAT3, phospho-STAT3 and GAPDH polyclonal antibodies were purchased from Affinity Biosciences (Cincinnati, OH, United States). Rabbit anti-Runt-related transcription factor 2 (RUNX2), bone morphogenetic protein 2 (BMP2), interleukin 1beta (IL-1β) and IL-6 polyclonal antibodies were purchased from ABclonal (Wuhan, China). Rabbit anti-osteopontin (OPN) and heat shock protein 90 (HSP90) polyclonal antibodies and mouse anti-actin alpha 2, smooth muscle (SMA) monoclonal antibodies were purchased from Proteintech Group (Chicago, IL, United States). TCF21 siRNA and control siRNA were purchased from RiboBio (Guangzhou, China). AceQ qPCR SYBR Green Master Mix, Lipofectamine RNAIMAX and Lipofectamine 2000 were purchased from Thermo Fisher Scientific (Waltham, MA, United States). Alizarin Red S reagent was purchased from Sinopharm Chemical Reagent (Shanghai, China). HA tag Nanoab or control magnetic beads were purchased from NuoyiBio (Shanghai, China). The dual-luciferase assay kit was purchased from Promega (Madison, WI, USA).
In vivo studies with animals
The in vivo study with mice was approved by the Ethics Committee of Hefei University of Technology and was consistent with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Animal studies are reported in compliance with the ARRIVE guidelines [19].
B6/JGpt-Tcf21em1Cflox/Gpt (TCF21flox) mice were purchased from GemPharmatech Co., Ltd. (Jiangsu, Nanjing, China). B6.FVB-Tg(Cdh5-Cre)7Mlia/J (Cdh5-Cre) mice were kindly provided by Dr. Suo-wen Xu from the University of Science and Technology of China. TCF21flox/floxCdh5-Cre (TCF21ECKO) mice were obtained by breeding male Cdh5-Cre mice with female TCF21flox mice.
To evaluate the effect of endothelial cell-specific TCF21 knockdown on acute vascular calcification, 8-week-old male TCF21flox/flox and TCF21ECKO mice (n = 6) were subcutaneously injected with 5.5 × 105 IU/kg vitamin D3 (VD3) at 09:00 on the first day and then given the first dose of nicotine (25 mg/kg) by intragastric administration. The mice were given nicotine again after a 12 h interval. Mice in both groups were injected with VD3 once per day for 3 d. After the last dose of VD3, all mice were maintained on a normal diet for 2 weeks. Then, the mice were anesthetized and euthanized in a CO2 chamber, and blood and whole aorta samples were collected.
Blood samples were kept at room temperature for 2 h. The serum was collected by centrifuging the blood for 20 min at 2000 ×g. Serum Ca2+ concentrations in mice were measured by a calcium colorimetric kit (Beyotime, China). Serum IL-6 levels were determined by an ELISA kit purchased from CUSABIO (Wuhan, China).
Cell culture
Human aortic smooth muscle cells (HASMCs) and human umbilical vein endothelial cells (HUVECs) were purchased from ATCC. HASMCs were cultured in DMEM containing 10% fetal bovine serum (FBS) and 50 μg/ml streptomycin and penicillin. HUVECs were cultured in Vasculife basal medium (EC medium) containing the VEGF Lifefactors kit (complete EC medium, Lifeline Cell Technology, Frederick, MD, USA). The cells were cultured in a 5% CO2 humidified incubator at 37 °C. Cells less than 10 passages were used for experiments.
The calcification medium was prepared by dissolving Na2HPO4 and NaH2PO4 (1:2) in DMEM containing 2% FBS and 50 μg/ml penicillin/streptomycin. The final concentration of PO43− was 3.0 mM. Cells at ~80% density were switched to calcification medium, which was replaced with new calcification medium every 2 days.
Determination of calcification
Vascular calcification in vivo or in vitro was determined by Alizarin Red S or Von Kossa staining [20]. Briefly, after fixation with 4% paraformaldehyde, the cells, aortic ring sections or aortic root sections were stained with Alizarin Red S (1%, pH = 4.0) for 30 min. Then, the excess dye was removed with acidic PBS. All images were photographed under a microscope. The stained cells were redissolved in 10% acetic acid solution, and the absorbance value was determined at 405 nm. The whole aorta was stained with Alizarin Red S (0.003% in 1% potassium hydroxide solution) for 48 h and then washed with 2% KOH solution twice.
For Von Kossa staining, the aortic ring sections were washed with PBS and then immersed in 2% silver nitrate solution under ultraviolet light for 45 min. After being washed with ddH2O for 5 min, the slides were washed with 5% sodium thiosulfate solution for 2 min. Then, the images were photographed under a microscope.
Immunofluorescent staining
The expression of BMP2, RUNX2, TCF21, SRF and IL-6 in the mouse aortic ring, aortic root and human carotid atherosclerotic plaques was determined by immunofluorescent staining as previously described [21].
Real-time quantitative RT-PCR (qPCR)
After treatment, total RNA from cells was extracted with RNApure reagent (Zomanbio, Beijing, China). Then, the expression of TCF21, RUNX2, BMP2, SM22α, OPN, IL-6 and IL-1β mRNA was determined by qPCR as described [22] using SYBR green PCR master mix from Thermo Fisher Scientific (Waltham, MA, United States) and the primers listed in Table S1. The mRNA levels were normalized to GAPDH mRNA in the corresponding samples.
Determination of protein expression by Western blotting
After treatment, total cell protein was extracted from HASMCs or HUVECs. The protein expression of TCF21, BMP2, RUNX2, OPN, SM22α, IL-6 and IL-1β was detected by Western blotting [23]. The density of each band was quantified by two technicians who were blinded to the treatments and each other’s results with ImageJ software (NIH, Bethesda, MD, USA).
Transfection of siRNA or overexpression vector
HASMCs or HUVECs in six-well plates (~40% density) were transfected with control siRNA (siControl) or TCF21 siRNA (siTCF21) using Lipofectamine RNAIMAX. After 24 h of transfection, the cells were switched to complete medium with or without high inorganic phosphate (3 mM, NaH2PO4/Na2HPO4) for 4 d. The overexpression vectors of TCF21 (pCMV-HA-TCF21, HA-TCF21), SRF (pCMV-HA-SRF, HA-SRF) and MYOCD (pCMV-HA-MYOCD, HA-MYOCD) were purchased from Tsingke Biotechnology Co., Ltd. (Beijing, China). HASMCs and HUVECs in six-well plates (~90% density) were transfected with the control vector (pCMV-HA, 2 μg/well), HA-TCF21, HA-SRF or HA-MYOCD plasmid with Lipofectamine 2000 for 24 h. After transfection, the cells were treated with or without calcification medium for 4 d.
Coimmunoprecipitation (Co-IP) assay
The interaction of STAT3 and TCF21 was determined by Co-IP as described previously [24]. Briefly, HASMCs were transfected with HA-TCF21 for 24 h in serum-free medium, after which the cells were switched to complete medium and cultured for another 24 h. The cell lysate was added to anti-HA tag or empty magnetic beads. After being washed five times, the proteins were pulled down with loading buffer, and the eluent and input cell lysates were used to analyze HA (representing TCF21) and STAT3 expression by Western blotting.
Preparation of plasmid DNA and determination of promoter activity
The human TCF21 promoter (from −1155 to −1) sequence was generated by PCR using HASMC genomic DNA as a template and the following primers: forward, 5′-AGCGGTACCCATTTTGCTCCAGCCCCTCAAC-3′, reverse, 5′-ACTGCTAGCCACCAAATTCCTCAGCGCTCG-3′. The PCR product was inserted into the luciferase reporter vector (pGL4.10). The constructed promoter was confirmed by sequencing and named pTCF21. Site-directed deletion of the predicted STAT3 binding sequence (from −498 to −490: TTCTAGGAA) was performed as described [25].
To determine the effect of STAT3 activation on TCF21 promoter activity, we transfected pTCF21 or pTCF21-delete, the internal reference plasmid (pGL4hRluc) and the STAT3 overexpression vector into HASMCs with Lipofectamine transfection reagent for 18 h. Then, the cells were cultured in complete medium for 24 h. The cell lysates were collected, and luciferase activity was measured using a dual luciferase reporter assay kit.
To determine the effect of TCF21 on STAT3 transcriptional activity, the STAT3 response reporter (pHHSTAT3-luc, purchased from HZBIO), the internal reference plasmid (pGL4hRluc) and the TCF21 overexpression vector were transfected into HASMCs with Lipofectamine transfection reagent for 18 h. Then, the cells were cultured in complete medium for 24 h. The cell lysates were collected, and the luciferase activity was measured using a dual luciferase reporter assay kit.
Collection and treatment of the EC-conditioned medium (EC-CM)
HUVECs in 100 mm dishes were transfected with control or TCF21 siRNA for 24 h. After being washed three times with PBS. The cells were cultured in 10 mL of complete EC medium for 24 h. The culture medium was centrifuged to remove cell debris and suspended cells, collected as EC-CM and stored frozen at −20 °C. IL-6 levels in EC-CM were determined by an ELISA kit purchased from CUSABIO (Wuhan, China). The EC-CM was mixed with fresh calcification medium (1:1) and used to treat HASMCs for 24 h.
Detection of SMA, TCF21 and RUNX2 expression in patient samples
All studies with human carotid atherosclerotic plaques were approved by the regional Ethical Committees and adhered strictly to the Declaration of Helsinki Principle 2008. All patients provided informed consent before sample collection. Human carotid atherosclerotic plaques were collected from 6 patients who underwent carotid endarterectomy at the First Affiliated Hospital of University of Science and Technology of China. Carotid atherosclerotic plaques were used to prepare 5 μm paraffin sections. The expression of SMA and TCF21 or SMA and RUNX2 in the lesion areas was determined by immunofluorescent staining [21].
Data analysis
All experiments were repeated at least three times, and representative results are shown. All values are displayed as the mean ± SD. Normal distribution was determined by the one-sample K-S of nonparametric test with SPSS 22 software, and the data were analyzed by parametric statistics (Student’s t test for two groups and one-way ANOVA for more than two groups). The difference was considered significant if P < 0.05.
Results
TCF21 expression was positively correlated with vascular calcification
To determine TCF21 expression in human atherosclerotic plaques, we collected lesions from 6 patients who underwent carotid endarterectomy (Table S2). Surprisingly, we found that TCF21 was highly expressed in patients with high RUNX2 expression and low SMA expression (Fig. 1a, b). TCF21 and RUNX2 exhibited similar expression patterns, suggesting that TCF21 may play a crucial role in vascular calcification. Then, we established an in vitro VSMC osteogenesis model using high levels of inorganic phosphate [26]. The calcification model was initially confirmed by Alizarin Red S staining (Fig. 1c). Moreover, corresponding to the increased calcification-related genes (RUNX2 and BMP2), the protein and mRNA levels of TCF21 were also significantly upregulated by inorganic phosphate (Fig. 1d, e). Taken together, the results in Fig. 1 indicate that the expression of TCF21 was increased during vascular calcification, suggesting that TCF21 may be involved in the development of vascular calcification.
Fig. 1. TCF21 was positively correlated with vascular calcification.

a, b Carotid artery atherosclerotic plaques were collected from 6 patients with carotid stenotic disease. Paraffin sections of plaques were prepared to determine SMA and TCF21 (a) or SMA and RUNX2 (b) expression by immunofluorescence staining. *P < 0.05 (n = 3). c–e HASMCs were cultured in control (Ctrl) or high phosphate medium (Pi, 3 mM) for 4 d. VSMC calcification was determined by Alizarin Red S staining (c). *P < 0.05 (n = 4). The protein and mRNA expression of TCF21, RUNX2 and BMP2 was detected by Western blotting (d) and qPCR (e), respectively. *P < 0.05 (n = 3).
TCF21 aggravates vascular calcification in vitro and ex vivo
To determine the effect of TCF21 on vascular calcification, we initially transfected HASMCs with the TCF21 overexpression vector. Overexpression of TCF21 aggravated inorganic phosphate-induced HASMC osteogenesis, as determined by Alizarin Red S staining and calcium quantitative assay (Fig. 2a, b). In addition to the increased calcification, the expression of BMP2 and RUNX2 mRNA and protein was increased in HASMCs overexpressing TCF21 (Fig. 2c, d). In contrast, knockdown of TCF21 by siRNA reduced phosphate-induced calcification and the expression of RUNX2 and BMP2 (Fig. 2e–g).
Fig. 2. TCF21 promotes HASMC osteogenesis.

HASMCs were transfected with the TCF21 overexpression vector, TCF21 siRNA or the corresponding control for 24 h. The cells were then switched to complete DMEM or complete medium containing high phosphate (Pi, 3 mM) for 3 d. At the end of the experiment, the cells were used for the following assays. a, e Calcium deposition was examined by Alizarin Red S staining. *P < 0.05 (n = 4). b, f Intracellular calcium levels were determined by a calcium detection kit, and the cell protein concentration was used for relative quantitative statistical analysis. *P < 0.05 (n = 4). c The mRNA expression of TCF21, RUNX2 and BMP2 in HASMC was analyzed by qPCR. *P < 0.05 (n = 3). d, g The protein expression of TCF21, RUNX2 and BMP2 was determined by Western blotting. *P < 0.05 (n = 3).
Next, we explored the role of TCF21 in an ex vivo calcification model. The rings of thoracic aortas (5 mm long) collected from C57BL/6J mice were transfected with the TCF21 overexpression vector or siRNA. The transfection efficiency was initially determined by immunofluorescence staining. As shown in Fig. S1, TCF21 overexpression increased TCF21 expression, while TCF21 siRNA inhibited TCF21 expression in the media layer of the aorta. Alizarin Red S and Von Kossa staining showed that phosphate significantly induced calcium deposition in the tunica media layer of aortic rings, and calcification was substantially enhanced by TCF21 overexpression (Fig. 3a, b). Compared to the control group, immunofluorescence staining showed that phosphate treatment significantly enhanced the expression of RUNX2, BMP2 and TCF21 (Figs. 3c, d, S1a). Transfection of the TCF21 overexpression vector further increased the expression of TCF21, BMP2 and RUNX2 (Figs. 3c, d, S1a). In contrast, TCF21 siRNA reduced phosphate-induced calcification and RUNX2, BMP2 and TCF21 expression (Figs. 3e–h, S1b).
Fig. 3. TCF21 enhances media calcification in the thoracic aorta ex vivo.

Thoracic aortas were collected from C57BL/6J mice and cut into 5-mm-long aortic rings. a–d The aortic rings were transfected with the TCF21 overexpression vector or empty vector (2 μg/well) for 24 h. e–h The aortic rings were transfected with TCF21 siRNA or control siRNA (20 nM) for 24 h. Then, the aortic rings were cultured in control or high phosphate medium for 14 d, during which the corresponding medium was changed another day. After treatment, the aortic rings were used to prepare 5-μm frozen sections. a, b, e, f The aortic calcification was determined by Alizarin Red S and Von Kossa staining. The Alizarin Red S positive area was quantitively analyzed. *P < 0.05 (n = 3). c, d, g, h The expression of BMP2 and RUNX2 in aortic rings was determined by immunofluorescence staining. The mean fluorescent intensity (MFI) was quantified. *P < 0.05 (n = 3).
SRF attenuates TCF21-induced vascular calcification
It has been reported that TCF21 induces VSMCs phenotypic transformation by binding to the SRF-MYOCD complex [27, 28]. We confirmed that overexpression of TCF21 reduced SM22α and SMA and increased OPN expression (Fig. 4a, b). SRF but not MYOCD reversed TCF21-mediated inhibition of SM22α and SMA expression (Figs. 4a, b, S2a, b). Interestingly, we determined that SRF mRNA expression was inhibited by TCF21 overexpression, while TCF21 was inhibited by SRF overexpression (Fig. 4a, b). These results suggest that TCF21 may act as an effective accelerator of vascular calcification through SRF-regulated VSMCs phenotype conversion.
Fig. 4. SRF abolishes TCF21-induced calcification.

a–d HASMCs were transfected with TCF21 or TCF21 and SRF vectors for 24 h. Then, the cells were cultured in high phosphate medium for 4 d. The protein expression of TCF21, SM22α, SMA, OPN, BMP2 and RUNX2 was determined by Western blotting (a, c). The mRNA expression of TCF21, SRF, SM22α and OPN was determined by qPCR (b). Osteogenesis was determined by Alizarin Red S staining (d). *P < 0.05 (n = 3). e–g Thoracic aortic rings collected from C57BL/6J mouse were transfected with TCF21 or the SRF overexpression vector for 24 h. After being cultured in control or high phosphate medium for 14 d, the aortic rings were used to prepare 5-µm cryosections. Aortic calcification was determined by Alizarin Red S and Von Kossa staining (e). The expression of TCF21 (f), BMP2 and RUNX2 (g) in aortic rings was determined by immunofluorescence staining. *P < 0.05 (n = 3).
To determine the effect of SRF on TCF21-induced calcification, HASMCs were cotransfected with TCF21 and SRF overexpression vectors under high phosphate conditions. As shown in Fig. 4c, overexpression of TCF21 significantly induced the expression of the osteogenic genes BMP2 and RUNX2, which was attenuated by SRF overexpression. Similarly, the Alizarin Red S staining showed that the TCF21-mediated increase in the deposition of calcium was inhibited by SRF overexpression in VSMCs (Fig. 4d). In addition, the results of Alizarin Red S and Von Kossa staining demonstrated that SRF overexpression abolished the TCF21-induced aortic ring calcification (Fig. 4e). To further investigate the molecular mechanism by which TCF21 affects vascular calcification via SRF, we examined the expression of TCF21, SRF, BMP2 and RUNX2 by immunofluorescent staining. SRF overexpression significantly reduced TCF21, BMP2 and RUNX2 expression in the aortic rings (Figs. 4f, g, S2c). Taken together, the results in Fig. 4 suggest that TCF21 induced VSMC osteogenesis by promoting VSMC phenotypic switching to an osteoblast-like phenotype.
TCF21 exacerbates inflammation by relieving the SRF-inhibited IL-6-STAT3 signaling pathway
Chronic inflammation appears to be a central factor in abnormal soft tissue calcification, including vascular calcification. Chronic inflammation in the vascular system has been shown to induce vascular calcification in mouse and human studies [29]. A study showed that TCF21 may activate proinflammatory gene expression in coronary artery smooth muscle cells by promoting aryl hydrocarbon receptor expression [30]. To investigate the mechanism of TCF21 in inflammation under vascular calcification conditions, we first overexpressed TCF21 in HASMCs with phosphate. IL-6 protein and mRNA levels were increased by TCF21 overexpression (Fig. 5a, b). STAT3 is the downstream effector of the IL-6 signaling pathway. Overexpression of TCF21 clearly enhanced the phosphorylation of STAT3 (p-STAT3) and its transcriptional activity (Figs. 5a, S3e). Further study indicated that TCF21 interacted with STAT3 (Fig. S3d), suggesting that the interaction of TCF21 with STAT3 may increase STAT3 activity. Conversely, knockdown of TCF21 reduced IL-6 expression (Fig. 5c, d). Interestingly, we determined that LPS induced TCF21 expression in HASMCs (Fig. S3a). Overexpression of STAT3 could also induce TCF21 expression (Fig. S3c). Sequence alignment analysis showed a putative STAT3 binding site in the TCF21 promoter region with the sequence TTCTAGGAA (from −498 to −490). To further determine whether TCF21 is a direct target of STAT3, we constructed a TCF21 promoter (pTCF21) and a promoter with a putative STAT3 binding site deletion (pTCF21-delete). STAT3 overexpression increased pTCF21 promoter activity but not that of the pTCF21-deleted promoter (Fig. S3f, g). Therefore, TCF21 increased IL-6-mediated STAT3 activation, which in turn enhanced TCF21 expression. We also determined that IL-6 levels in aortic rings were increased by TCF21 overexpression but reduced by TCF21 siRNA (Fig. 5e, f). Blocking IL-6 with a specific IL-6 antibody or inhibiting STAT3 with Stattic attenuated TCF21-induced HASMC calcification (Fig. 5g, h). Stattic was also able to block TCF21-induced aortic calcification ex vivo (Fig. 5i).
Fig. 5. TCF21 activates the IL-6-STAT3 signaling pathway.

a–d, j HASMCs were transfected with the TCF21 or SRF overexpression vector or Control siRNA/TCF21 siRNA for 24 h. The cells were then switched to high phosphate medium for 4 d. At the end of the experiment, total cell protein and total RNA were extracted. The expression of IL-6, p-STAT3 and STAT3 protein (a, c, j) and IL-6 mRNA (b, d) was determined by Western blotting and qPCR. *P < 0.05 (n = 3 for Western blotting, n = 4 for qPCR). e, f, k The aortic rings were transfected with the control vector/TCF21 or SRF overexpression vector or control siRNA/TCF21 siRNA for 24 h. Then, the aortic rings were cultured in control or high phosphate medium for 14 d. After treatment, the aortic rings were used to prepare 5 μm cryosections. The expression of IL-6 was determined by immunofluorescence staining. *P < 0.05 (n = 3). g, h HASMCs were transfected with the TCF21 overexpression vector for 24 h. The cells were then switched to high phosphate medium containing IL-6 antibodies (500 μg/ml) or Stattic (10 μM) for 4 d. HASMC calcification was determined by Alizarin Red S staining. *P < 0.05 (n = 3). i The aortic rings were transfected with the control vector or TCF21 overexpression vector for 24 h. Then, the aortic rings were cultured in control or high phosphate medium with or without Stattic (10 μM) for 14 d. After treatment, the aortic rings were used to prepare 5 μm frozen sections. Aortic calcification was determined by Alizarin Red S staining.
Considering that SRF can reduce TCF21-induced calcification, we determined whether SRF was involved in TCF21-induced IL-6 expression. The TCF21-mediated increase in IL-6 expression was significantly reversed by SRF overexpression in HASMCs and aortic rings (Fig. 5j, k). Consistently, SRF overexpression abolished TCF21-induced p-STAT3 level (Fig. 5j). Taken together, the data in Fig. 5 and S3 indicate that TCF21 increased the activation of the IL-6-STAT3 signaling pathway, which induced TCF21 expression through positive feedback. Therefore, the increase in TCF21 expression facilitated VSMC osteogenesis.
TCF21 promotes calcification through the interplay of ECs and VSMCs
ECs and VSMCs are adjacent cell types in the vasculature, and the interplay between ECs and VSMCs affects vascular calcification. We determined that TCF21 was also expressed in SMA-negative cells within human atherosclerotic lesions (Fig. 1a). Therefore, we hypothesized that TCF21 in EC may also be involved in the development of vascular calcification. To verify this hypothesis, we determined the changes in some key inflammatory molecules in ECs under TCF21 overexpression conditions. IL-1β and IL-6 were significantly increased by TCF21 (Fig. 6a, b). LPS also induced TCF21 expression in HUVECs (Fig. S3b). Next, we collected endothelial-conditioned medium (EC-CM) with or without TCF21 knockdown (Fig. 6c, d). IL-6 levels were significantly reduced in siTCF21-transfected EC-CM (Fig. 6e). HASMC calcification was induced using phosphate mixed in equal proportions with the EC-CM described above. Compared with EC-CM from control siRNA-transfected ECs, the addition of EC-CM from TCF21 knockdown ECs attenuated the expression of BMP2 and RUNX2 (Fig. 6f). To further determine the effect of EC TCF21 on vascular calcification, we generated TCF21flox/flox and EC-specific TCF21 knockout (TCF21ECKO) mice (Fig. 6g), and medial arterial calcification was induced by VD3 and nicotine-induced CKD. We determined that the expression of TCF21 was reduced in the aortic wall and was barely observed in the endothelial layer of TCF21ECKO mice (Fig. 6o). Calcium accumulation in the whole aorta and aortic root was significantly reduced in TCF21ECKO mice (Fig. 6h, i), indicating that medial calcification was attenuated by the genetic depletion of TCF21 in ECs. Interestingly, we determined that calcium levels in serum were also reduced in TCF21ECKO mice (Fig. 6j). Consistent with the reduction in calcification, the expression of the contractile gene SRF was increased, while that of the osteogenic gene RUNX2 was reduced in TCF21ECKO mice (Fig. 6k, l). The inflammatory cytokine IL-6 was reduced in the serum and aorta in TCF21ECKO mice (Fig. 6m, n). In conclusion, the results in Fig. 6 suggest that the interplay between ECs and VSMCs also contributes to TCF21-mediated enhancement of vascular calcification.
Fig. 6. TCF21 promotes calcification through ECs and VSMCs interactions.

a,b HUVECs were transfected with the control or TCF21 overexpression vector for 24 h and then cultured in complete DMEM for 1 day. The protein and mRNA expression of IL-6, IL-1β and TCF21 was determined by Western blotting and qPCR. *P < 0.05 (n = 3). c–e HUVECs were transfected with control or TCF21 siRNA for 24 h. After being washed with PBS, the cells were switched to serum-free DMEM for 48 h. Cellular RNA, protein and culture medium (defined as endothelial-conditioned medium, EC-CM) were collected separately. The protein and mRNA expression of TCF21 in the cells was determined by qPCR (c) and Western blotting (d). IL-6 levels in EC-CM were determined by ELISA (e). *P < 0.05 (n = 3). f HASMCs were cultured with a mixture of high-phosphate medium and EC-CM (1:1) for 4 d. After treatment, total cell protein was collected to determine the expression of BMP2 and RUNX2 by Western blotting. *P < 0.05 (n = 3). g–o After genotyping with the corresponding primers (g), TCF21flox/flox (TCF21f/f) and TCF21flox/floxCdh5-Cre (TCF21ECKO) mouse (6 mice/group) were injected daily with 100 μL of VD3 dissolved in peanut oil (5.5 × 105 U/kg) for 3 consecutive days. In addition, the mice were intragastrically administered nicotine (25 mg/kg) in the morning and evening on the first day. After 3 weeks, the mice were euthanized, and aorta and serum samples were collected. Vascular calcification in whole aorta (h) and aortic root (i) was determined by Alizarin Red S staining. *P < 0.05 (n = 6). Calcium (j) and IL-6 (m) levels in serum were determined by commercial kits. *P < 0.05 (n = 6). The expression of SRF (k), RUNX2 (l), IL-6 (n) and TCF21 (o) in the aortic root was determined by immunofluorescence staining. *P < 0.05 (n = 3). L: lumen; white arrow: endothelium layer.
Discussion
Vascular calcification is associated with cardiovascular morbidity and mortality in patients with metabolic syndrome, CKD or atherosclerosis [31]. Our study demonstrated increased TCF21 in calcified atherosclerotic plaques. TCF21 inhibition in VSMCs in vitro or endothelial TCF21 deficiency in vivo attenuated vascular calcification. Moreover, TCF21 is required for the interplay between VSMCs and ECs via inflammatory cytokines, especially IL-6, to activate STAT3 in VSMCs, thereby activating the osteogenesis of VSMCs. Thus, our findings demonstrate that TCF21 is a novel vascular calcification regulator and indicate the therapeutic potential of TCF21 inhibition.
Vascular calcification is an active cell-mediated regulatory process with similarities to bone formation [32, 33], which involves a phenotypic switch from contractile VSMCs to osteo/chondroblast-like cells [34, 35]. It has been demonstrated that TCF21 is highly expressed in unique fibroblast-like cells in human and mouse atherosclerotic plaques, which suggests that TCF21 may regulate VSMC phenotypic switching [17]. A subsequent study demonstrated that TCF21 increased VSMC proliferation by interacting with MYOCD to inhibit the SRF-MYOCD complex, thereby inhibiting the expression of contractile genes. TCF21 also inhibits the expression of SRF and MYOCD [17]. It is unclear whether TCF21 contributes to the progression of vascular calcification.
In the present study, we found that TCF21 expression was upregulated in calcific atherosclerotic plaques and osteoblast-like VSMCs. TCF21 overexpression promoted osteogenic differentiation in VSMCs. In contrast, TCF21 knockdown in VSMCs attenuated calcification. TCF21 disturbs the SRF-MYOCD complex, which regulates contractile genes [17] and affects vascular calcification. Thus, SRF but not MYOCD overexpression significantly attenuated TCF21-induced VSMC and aortic ring calcification. SRF is a MADS box transcription factor involved in the expression of contractile genes with low transcriptional activity [36]. However, it can bind multiple cofactors to enhance or block its activity [37]. Studies have shown that inactivation of SRF leads to increased arterial stiffness in mouse models [38]. Moreover, the roles of SRF in many cellular processes have been elucidated, including cell survival, contractility, synaptic activity, cell migration and inflammation [39]. We determined that SRF increased TCF21-mediated suppression of SMA and SM22α expression. More importantly, under high phosphate conditions, SRF reduced TCF21-induced BMP2 and RUNX2 expression and vascular calcification. Therefore, SRF-mediated contractile gene expression is involved in TCF21-mediated exacerbation of calcification.
Human and mouse studies indicate that sites of chronic inflammation in the vasculature are the sites of atherosclerotic or media calcification [29, 40, 41]. Inflammatory cytokines in the vasculature can enhance the expression of receptor activator of nuclear factor κB ligand (RANKL), a TNF superfamily member, thereby activating the expression of bone morphogenetic protein 4 (BMP4) and stimulating the osteogenic differentiation of VSMCs [42]. IL-6 and soluble IL-6 receptor (sIL-6R) induce the transformation of VSMCs into an osteoblast phenotype via STAT3-jumonji domain-containing protein 2B (JMJD2B)-regulated RUNX2 expression [43]. The binding of IL-29 to its receptor IL-28Ra enhances the activation of JAK2/STAT3 to upregulate BMP2 in VSMCs and promote their calcification and osteogenic transformation [44]. Moreover, STAT3 but not STAT1 mediates chronic inflammation-induced ectopic calcification [45]. We determined that TCF21 enhanced IL-6 and p-STAT3 expression, which further increased the expression of RUNX2. Blocking IL-6 with a specific antibody or inhibiting STAT3 with Stattic completely blocked TCF21-induced calcification, suggesting that TCF21 regulates vascular calcification via the IL-6-STAT3 signaling pathway. Interestingly, LPS or STAT3 overexpression can induce TCF21 expression. TCF21 and STAT3 can bind with each other to enhance STAT3 transcriptional activity. We also identified that TCF21 is a direct target of STAT3. These data suggest that inflammation and TCF21 can form a positive feedback loop to amplify IL-6-STAT3 signaling pathway activation. Moreover, SRF attenuated TCF21-induced IL-6 expression. SM22α and SRF can bind together to inhibit the expression of NF-κB inducing kinase (NIK) and downstream NF-κB signaling pathway [46]. However, we are not clear how TCF21 and SRF regulate IL-6 expression. SRF and TCF21 affect SM22α expression and may affect NF-κB expression, thereby affecting IL-6 expression. In addition, TCF21 and SRF may act directly on the IL-6 promoter, which requires further investigation.
ECs and SMCs are the main cell types in the vessel wall and act as semipermeable barriers. Oxidative stress and inflammation cause excessive endothelial permeability, which further leads to vascular hyperpermeability and abnormal metabolism [47]. Many studies have revealed that ECs can promote vascular calcification through endothelial-mesenchymal transition (EndoMT) and subsequent osteochondral differentiation [48, 49]. EndoMT is regulated by inflammatory cytokines, fibroblast growth factors (mechanical stress, hypoxia) and other conditions [50]. In addition, ECs and VSMCs crosstalk through paracrine mechanisms such as cytokines, extracellular vesicles containing pro-calcific mediators, and miRNAs, and myoendothelial gap junctions play critical roles in vascular calcification [20, 51, 52]. Interestingly, we determined that TCF21 was also expressed in SMA-negative cells within human atherosclerotic lesions, suggesting that TCF21 in non-VSMCs, especially in ECs, may be involved in vascular calcification. We found that TCF21 aggravated EC inflammatory cytokine production. Knockdown of TCF21 in ECs reduced IL-6 production. Moreover, conditioned medium collected from siTCF21-treated ECs attenuated phosphate-induced calcification in VSMCs compared with siCtrl-treated EC-conditioned medium. In vivo, EC-specific TCF21 deficiency reduced VD3- and nicotine-induced vascular calcification, suggesting that the interplay between ECs and VSMCs also plays an important role in TCF21-enhanced vascular calcification. However, future studies are still needed to elucidate the role of VSMC TCF21 in vascular calcification in vivo.
In conclusion, this study reveals a novel role of TCF21 in regulating vascular calcification by activating the IL-6-STAT3 signaling pathway. We also demonstrate that TCF21 can antagonize SRF, which affects the expression of contractile genes and IL-6-STAT3 signaling inactivation. TCF21 promotes the interplay between ECs and VSMCs via inflammatory cytokine production. Thus, we established TCF21 inhibition as a new potential therapeutic strategy for the prevention and treatment of vascular calcification.
Supplementary information
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) Grants U22A20272 to YLC and YJD, 82160094 to MXJ; Natural Science Foundation of Anhui province 2208085MH196 to BCZ.
Author contributions
Conceptualization, YLC and HH; methodology and investigation, XKZ, MMZ, SNW, TTZ, XNW, CYW, JZ, WYZ, XY, BCZ, and MXJ; resources: SWX; writing—original draft preparation, XKZ and YLC; writing—review and editing, YLC, SWX, YJD, JHH, QRM, and HH; supervision, YLC; funding acquisition, YLC, JHH, MXJ, and BCZ. All authors have read and agreed to the final version of the paper.
Data availability
All data shown are available in the paper and the supporting information.
Competing interests
The authors declare no competing interests.
Contributor Information
Hao Hu, Email: huhao1977@ustc.edu.cn.
Yuan-li Chen, Email: chenyuanli@hfut.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-023-01077-8.
References
- 1.Li Y, Pan Y, Wang L, Wang X, Chu H, Li Y, et al. 3-Arylcoumarin inhibits vascular calcification by inhibiting the generation of AGEs and anti-oxidative stress. J Enzym Inhib Med Chem. 2022;37:2147–57. doi: 10.1080/14756366.2022.2109024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Otsuka F, Sakakura K, Yahagi K, Joner M, Virmani R. Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler Thromb Vasc Biol. 2014;34:724–36. doi: 10.1161/ATVBAHA.113.302642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van den Bergh G, Opdebeeck B, D’Haese PC, Verhulst A. The vicious cycle of arterial stiffness and arterial media calcification. Trends Mol Med. 2019;25:1133–46. doi: 10.1016/j.molmed.2019.08.006. [DOI] [PubMed] [Google Scholar]
- 4.Kelly-Arnold A, Maldonado N, Laudier D, Aikawa E, Cardoso L, Weinbaum S. Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proc Natl Acad Sci USA. 2013;110:10741–6. doi: 10.1073/pnas.1308814110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chakraborty R, Saddouk FZ, Carrao AC, Krause DS, Greif DM, Martin KA. Promoters to study vascular smooth muscle. Arterioscler Thromb Vasc Biol. 2019;39:603–12. doi: 10.1161/ATVBAHA.119.312449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Rourke RA, Brundage BH, Froelicher VF, Greenland P, Grundy SM, Hachamovitch R, et al. American College of Cardiology/American Heart Association Expert Consensus document on electron-beam computed tomography for the diagnosis and prognosis of coronary artery disease. Circulation. 2000;102:126–40. doi: 10.1161/01.CIR.102.1.126. [DOI] [PubMed] [Google Scholar]
- 7.Goettsch C, Hutcheson JD, Aikawa M, Iwata H, Pham T, Nykjaer A, et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J Clin Invest. 2016;126:1323–36. doi: 10.1172/JCI80851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buendia P, Montes de Oca A, Madueno JA, Merino A, Martin-Malo A, Aljama P, et al. Endothelial microparticles mediate inflammation-induced vascular calcification. FASEB J. 2015;29:173–81. doi: 10.1096/fj.14-249706. [DOI] [PubMed] [Google Scholar]
- 9.Wang H, Xie Y, Salvador AM, Zhang Z, Chen K, Li G, et al. Exosomes: multifaceted messengers in atherosclerosis. Curr Atheroscler Rep. 2020;22:57. doi: 10.1007/s11883-020-00871-7. [DOI] [PubMed] [Google Scholar]
- 10.Xue C, Senchanthisai S, Sowden M, Pang J, White RJ, Berk BC. Endothelial-to-mesenchymal transition and inflammation play key roles in cyclophilin a-induced pulmonary arterial hypertension. Hypertension. 2020;76:1113–23. doi: 10.1161/HYPERTENSIONAHA.119.14013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med. 2010;16:1400–6. doi: 10.1038/nm.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Acharya A, Baek ST, Huang G, Eskiocak B, Goetsch S, Sung CY, et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development. 2012;139:2139–49. doi: 10.1242/dev.079970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hidai H, Bardales R, Goodwin R, Quertermous T, Quertermous EE. Cloning of capsulin, a basic helix-loop-helix factor expressed in progenitor cells of the pericardium and the coronary arteries. Mech Dev. 1998;73:33–43. doi: 10.1016/S0925-4773(98)00031-8. [DOI] [PubMed] [Google Scholar]
- 14.Quaggin SE, Schwartz L, Cui S, Igarashi P, Deimling J, Post M, et al. The basic-helix-loop-helix protein pod1 is critically important for kidney and lung organogenesis. Development. 1999;126:5771–83. doi: 10.1242/dev.126.24.5771. [DOI] [PubMed] [Google Scholar]
- 15.Lu J, Chang P, Richardson JA, Gan L, Weiler H, Olson EN. The basic helix-loop-helix transcription factor capsulin controls spleen organogenesis. Proc Natl Acad Sci USA. 2000;97:9525–30. doi: 10.1073/pnas.97.17.9525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL, et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med. 2019;25:1280–9. doi: 10.1038/s41591-019-0512-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagao M, Lyu Q, Zhao Q, Wirka RC, Bagga J, Nguyen T, et al. Coronary disease-associated gene TCF21 inhibits smooth muscle cell differentiation by blocking the myocardin-serum response factor pathway. Circ Res. 2020;126:517–29. doi: 10.1161/CIRCRESAHA.119.315968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nurnberg ST, Cheng K, Raiesdana A, Kundu R, Miller CL, Kim JB, et al. Coronary artery disease associated transcription factor TCF21 regulates smooth muscle precursor cells that contribute to the fibrous cap. Genom Data. 2015;5:36–7. doi: 10.1016/j.gdata.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. Br J Pharmacol. 2020;177:3617–24. doi: 10.1111/bph.15193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zeng P, Yang J, Liu L, Yang X, Yao Z, Ma C, et al. ERK1/2 inhibition reduces vascular calcification by activating miR-126-3p-DKK1/LRP6 pathway. Theranostics. 2021;11:1129–46. doi: 10.7150/thno.49771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu L, Zeng P, Yang X, Duan Y, Zhang W, Ma C, et al. Inhibition of vascular calcification: a new antiatherogenic mechanism of Topo II (DNA Topoisomerase II) inhibitors. Arterioscler Thromb Vasc Biol. 2018;38:2382–95. doi: 10.1161/ATVBAHA.118.311546. [DOI] [PubMed] [Google Scholar]
- 22.Liang Y, Han H, Liu L, Duan Y, Yang X, Ma C, et al. CD36 plays a critical role in proliferation, migration and tamoxifen-inhibited growth of ER-positive breast cancer cells. Oncogenesis. 2018;7:98. doi: 10.1038/s41389-018-0107-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y, Duan Y, Yang X, Sun L, Liu M, Wang Q, et al. Inhibition of ERK1/2 and activation of LXR synergistically reduce atherosclerotic lesions in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2015;35:948–59. doi: 10.1161/ATVBAHA.114.305116. [DOI] [PubMed] [Google Scholar]
- 24.Yang W, He R, Qu H, Lian W, Xue Y, Wang T, et al. FXYD3 enhances IL-17A signaling to promote psoriasis by competitively binding TRAF3 in keratinocytes. Cell Mol Immunol. 2023;20:292–304. doi: 10.1038/s41423-023-00973-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, Duan Y, Kang Y, Yang X, Jiang M, Zhang L, et al. Activation of liver X receptor induces macrophage interleukin-5 expression. J Biol Chem. 2012;287:43340–50. doi: 10.1074/jbc.M112.403394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hruska KA, Saab G, Mathew S, Lund R. Renal osteodystrophy, phosphate homeostasis, and vascular calcification. Semin Dial. 2007;20:309–15. doi: 10.1111/j.1525-139X.2007.00300.x. [DOI] [PubMed] [Google Scholar]
- 27.Sun Q, Chen G, Streb JW, Long X, Yang Y, Stoeckert CJ, Jr., et al. Defining the mammalian CArGome. Genome Res. 2006;16:197–207. doi: 10.1101/gr.4108706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie Y, Martin KA. TCF21: Flipping the phenotypic switch in SMC. Circ Res. 2020;126:530–2. doi: 10.1161/CIRCRESAHA.120.316533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, et al. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841–50. doi: 10.1161/CIRCULATIONAHA.107.732867. [DOI] [PubMed] [Google Scholar]
- 30.Kim JB, Pjanic M, Nguyen T, Miller CL, Iyer D, Liu B, et al. TCF21 and the environmental sensor aryl-hydrocarbon receptor cooperate to activate a pro-inflammatory gene expression program in coronary artery smooth muscle cells. PLoS Genet. 2017;13:e1006750. doi: 10.1371/journal.pgen.1006750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farrar DJ, Bond MG, Riley WA, Sawyer JK. Anatomic correlates of aortic pulse wave velocity and carotid artery elasticity during atherosclerosis progression and regression in monkeys. Circulation. 1991;83:1754–63. doi: 10.1161/01.CIR.83.5.1754. [DOI] [PubMed] [Google Scholar]
- 32.Johnson RC, Leopold JA, Loscalzo J. Vascular calcification: pathobiological mechanisms and clinical implications. Circ Res. 2006;99:1044–59. doi: 10.1161/01.RES.0000249379.55535.21. [DOI] [PubMed] [Google Scholar]
- 33.Lanzer P, Boehm M, Sorribas V, Thiriet M, Janzen J, Zeller T, et al. Medial vascular calcification revisited: review and perspectives. Eur Heart J. 2014;35:1515–25. doi: 10.1093/eurheartj/ehu163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. 2018;114:590–600. doi: 10.1093/cvr/cvy010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voelkl J, Luong TT, Tuffaha R, Musculus K, Auer T, Lian X, et al. SGK1 induces vascular smooth muscle cell calcification through NF-kappaB signaling. J Clin Invest. 2018;128:3024–40. doi: 10.1172/JCI96477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang G, Yu C, Xiang K, Gao M, Liu Z, Yang B, et al. Inhibition of ANXA2 regulated by SRF attenuates the development of severe acute pancreatitis by inhibiting the NF-kappaB signaling pathway. Inflamm Res. 2022;71:1067–78. doi: 10.1007/s00011-022-01609-8. [DOI] [PubMed] [Google Scholar]
- 37.Niu Z, Li A, Zhang SX, Schwartz RJ. Serum response factor micromanaging cardiogenesis. Curr Opin Cell Biol. 2007;19:618–27. doi: 10.1016/j.ceb.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galmiche G, Labat C, Mericskay M, Aissa KA, Blanc J, Retailleau K, et al. Inactivation of serum response factor contributes to decrease vascular muscular tone and arterial stiffness in mice. Circ Res. 2013;112:1035–45. doi: 10.1161/CIRCRESAHA.113.301076. [DOI] [PubMed] [Google Scholar]
- 39.Miano JM. Role of serum response factor in the pathogenesis of disease. Lab Invest. 2010;90:1274–84. doi: 10.1038/labinvest.2010.104. [DOI] [PubMed] [Google Scholar]
- 40.Abdelbaky A, Corsini E, Figueroa AL, Fontanez S, Subramanian S, Ferencik M, et al. Focal arterial inflammation precedes subsequent calcification in the same location: a longitudinal FDG-PET/CT study. Circ Cardiovasc Imaging. 2013;6:747–54. doi: 10.1161/CIRCIMAGING.113.000382. [DOI] [PubMed] [Google Scholar]
- 41.Shao JS, Cheng SL, Sadhu J, Towler DA. Inflammation and the osteogenic regulation of vascular calcification: a review and perspective. Hypertension. 2010;55:579–92. doi: 10.1161/HYPERTENSIONAHA.109.134205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Panizo S, Cardus A, Encinas M, Parisi E, Valcheva P, Lopez-Ongil S, et al. RANKL increases vascular smooth muscle cell calcification through a RANK-BMP4-dependent pathway. Circ Res. 2009;104:1041–8. doi: 10.1161/CIRCRESAHA.108.189001. [DOI] [PubMed] [Google Scholar]
- 43.Kurozumi A, Nakano K, Yamagata K, Okada Y, Nakayamada S, Tanaka Y. IL-6 and sIL-6R induces STAT3-dependent differentiation of human VSMCs into osteoblast-like cells through JMJD2B-mediated histone demethylation of RUNX2. Bone. 2019;124:53–61. doi: 10.1016/j.bone.2019.04.006. [DOI] [PubMed] [Google Scholar]
- 44.Hao N, Zhou Z, Zhang F, Li Y, Hu R, Zou J, et al. Interleukin-29 Accelerates vascular calcification via JAK2/STAT3/BMP2 signaling. J Am Heart Assoc. 2023;12:e027222. doi: 10.1161/JAHA.122.027222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fukuyo S, Yamaoka K, Sonomoto K, Oshita K, Okada Y, Saito K, et al. IL-6-accelerated calcification by induction of ROR2 in human adipose tissue-derived mesenchymal stem cells is STAT3 dependent. Rheumatology (Oxf) 2014;53:1282–90. doi: 10.1093/rheumatology/ket496. [DOI] [PubMed] [Google Scholar]
- 46.Dai X, Thiagarajan D, Fang J, Shen J, Annam NP, Yang Z, et al. SM22alpha suppresses cytokine-induced inflammation and the transcription of NF-kappaB inducing kinase (Nik) by modulating SRF transcriptional activity in vascular smooth muscle cells. PLoS One. 2017;12:e0190191. doi: 10.1371/journal.pone.0190191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lum H, Malik AB. Mechanisms of increased endothelial permeability. Can J Physiol Pharmacol. 1996;74:787–800. doi: 10.1139/y96-081. [DOI] [PubMed] [Google Scholar]
- 48.Yao Y, Jumabay M, Ly A, Radparvar M, Cubberly MR, Bostrom KI. A role for the endothelium in vascular calcification. Circ Res. 2013;113:495–504. doi: 10.1161/CIRCRESAHA.113.301792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yao J, Guihard PJ, Blazquez-Medela AM, Guo Y, Moon JH, Jumabay M, et al. Serine protease activation essential for endothelial-mesenchymal transition in vascular calcification. Circ Res. 2015;117:758–69. doi: 10.1161/CIRCRESAHA.115.306751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sanchez-Duffhues G, Garcia de Vinuesa A, Ten Dijke P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev Dyn. 2018;247:492–508. doi: 10.1002/dvdy.24589. [DOI] [PubMed] [Google Scholar]
- 51.Zhang YX, Tang RN, Wang LT, Liu BC. Role of crosstalk between endothelial cells and smooth muscle cells in vascular calcification in chronic kidney disease. Cell Prolif. 2021;54:e12980. doi: 10.1111/cpr.12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang H, Li L, Zhang L, Zang G, Sun Z, Wang Z. Role of endothelial cells in vascular calcification. Front Cardiovasc Med. 2022;9:895005. doi: 10.3389/fcvm.2022.895005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data shown are available in the paper and the supporting information.