

Abstract
Murine norovirus (MNV) is a positive-sense, plus-stranded RNA virus in the Caliciviridae family. Viruses in this family replicate in the intestine and are transmitted by the fecal-oral route. MNV is related to the human noroviruses, which cause the majority of nonbacterial gastroenteritis worldwide. Given the technical challenges in studying human norovirus, MNV is often used to study mechanisms in norovirus biology since it combines the availability of a cell culture and reverse genetics system with the ability to study infection in the native host. Adding to our previous protocol collection (Hwang et al., 2014), we herein describe additional techniques that have since been developed to study MNV biology.
Basic Protocol 1: Indirect method for measuring cell cytotoxicity and antiviral activity
Basic Protocol 2: Measuring MNV genome titers by RT-qPCR
Support protocol 1: Preparation of standard
Basic Protocol 3: Generation of recombinant murine norovirus with minimal passaging
Basic Protocol 4: Generation of recombinant murine norovirus via circular polymerase extension reaction (CPER)
Basic Protocol 5: Expression of norovirus NS1–2 in insect cell suspension cultures using a recombinant baculovirus
Support protocol 2: Isotope-labeling of the norovirus NS1–2 in insect cells
Support protocol 3 : Purification of the norovirus NS1–2 protein
Support protocol 4: Expression of norovirus NS1–2 in mammalian cells by transduction with a recombinant baculovirus
Basic Protocol 6: Infection of enteroids in transwellⓇ inserts with MNV
Support protocol 5: Preparation of conditioned media for enteroids culture
Support protocol 6: Isolation of crypts for enteroids generation
Support protocol 7: Enteroid culture passaging and maintenance
Basic Protocol 7: Quantification of murine norovirus-induced diarrhea using neonatal mouse infections
Alternate Protocol 1: Intragastric inoculation of neonatal mice
Alternate protocol 2: Scoring colon contents
Keywords: Murine norovirus, circular polymerase extension reaction, baculovirus expression, tuft cell-enriched murine intestinal enteroid culture, neonatal infections
INTRODUCTION:
Murine norovirus (MNV) is a small non-enveloped virus with a plus-sense RNA genome ~7.5 kb in length that infects laboratory and wild mice (Farkas et al., 2012; Karst & Wobus, 2015; Smith et al., 2012; Thackray et al., 2007). MNV infections can be acute or persistent depending on the virus strain. The first MNV strain, MNV-1, to be isolated causes an acute infection in immunocompetent mice that is characterized by infection of immune cells starting in the small intestine, minimal fecal shedding, and viral clearance from tissues within 1 to 2 weeks (Gonzalez-Hernandez et al., 2014; Grau et al., 2017; Karst et al., 2003). The persistent MNV strains (e.g., MNV-CR6, MNV-CR3) initiate infection in the large intestine and replicate in tuft cells from which they are shed into the stool for weeks to months (Gonzalez-Hernandez et al., 2014; Nice et al., 2013; Wilen et al., 2018). All MNV strains belong to genogroup V in the norovirus genus in the Caliciviridae family (Wobus, 2020). These viruses are related to human noroviruses and share a number of biological characteristics, including fecal-oral spread and intestinal infections. Research on human noroviruses has been technically challenging given the historic lack of cell culture models (Duizer et al., 2004). The discovery of MNV in mice, development of cell culture and reverse genetics systems provided the field with a versatile system to study norovirus biology (Karst et al., 2003; Ward et al., 2007; Wobus et al., 2004). Although significant progress has been made in the development of laboratory cultivation systems for human norovirus (Estes et al., 2019; Ettayebi et al., 2016; Van Dycke et al., 2019), significant challenges remain, including the inability to generate a cell culture-derived virus stock, for their molecular study. Thus, MNV continues to provide a powerful tool for the study of norovirus-host interactions and pathogenesis.
The following protocols present an update to our 2014 protocol collection (Hwang et al., 2014) and describe updated and recently developed methods to study various aspects of MNV biology. The protocols begin with a description of how to measure antiviral efficacy of compounds using an indirect measure of cell viability (Basic Protocol 1). We then provide an updated protocol to quantify MNV genome copies by RT-qPCR (Basic Protocol 2 and Support Protocol 1). This is followed by a recently developed method for the rapid recovery of MNV directly from virus stocks without the need for bacterial cloning of the viral genome, namely circular polymerase extension reaction (CPER) (Basic Protocol 3). Much remains to be learned about the function of individual viral proteins. Thus, the next set of protocols describes the expression, labeling, purification of norovirus NS1–2 using a recombinant baculovirus (Basic Protocol 4, Support Protocols 2–4) and provides a blueprint for studying other viral proteins. While the ability of MNV to infect immune cells was the basis for the first cell culture system (Wobus et al., 2004), the discovery of a tropism of persistent MNV strains for intestinal tuft cells is relatively recent (Wilen et al., 2018). To study the interaction of MNV with tuft cells, Basic Protocol 5 and Support Protocols 5–7 describe the generation of intestinal enteroids and tuft-cell enriched enteroids cultures for MNV infection experiments. The unit ends with the MNV diarrhea model and a description of how to infect neonatal mice and quantify diarrhea ( Basic Protocol 6, Alternate Protocols 1–2).
CAUTION: Murine norovirus is a Biosafety Level 2 (BSL-2) pathogen in some countries (e.g., USA). Follow all appropriate guidelines and regulations for the use and handling of pathogenic microorganisms.
NOTE:All solutions and equipment coming into contact with living cells must be sterile, and aseptic techniques should be used accordingly.
NOTE:All culture incubations should be performed in a humidified 37°C, 5% CO2 incubator unless otherwise specified.
BASIC PROTOCOL 1: Indirect method for measuring cell cytotoxicity and antiviral activity.
Introduction.
The assessment of antiviral activity for putative antiviral drugs in the development pipeline facilitates the discovery of new antiviral compounds. Screening of compound libraries for both CC50 and EC50 is an important step to ascertain the effectiveness of compounds and to assess the medicinal chemistry path being followed. This process is facilitated by a rapid first screen of antiviral activity that can then be followed up with more expensive or intensive methods that directly quantify the virus, such as qRT-PCR (see Basic Protocol 2) or TCID50 (Basic Protocol 5 in (Hwang et al., 2014)). Several methods have been developed to calculate the TCID50 and have been summarized recently (Lei et al., 2021), where the authors also provide a spread-sheet calculator as an electronic supplementary material.
Here, we present a method for indirectly assessing cell viability in the presence of MNV and antiviral compounds using CellTiter-Glo (Promega, WI, USA). The regent lyses cells to release ATP which is then converted to a stable luminescent signal proportional to the health of the cells at the time of harvest. The sensitivity and linear range of CellTiter-Glo allow for the detection of subtle changes in the cells or virus in response to an antiviral, that may go undetected with other reagents. The reagent does not require fixing or washing of cells, thereby significantly limiting loss of cells that lift off the plate during infection. This protocol generates both CC50 and EC50 values in technical triplicate on one 96-well plate. This is achieved by seeding plates with an MNV permissible cell line (Haga et al., 2016; Lingemann, 2020; Orchard et al., 2016; Wobus et al., 2004), treating with a dilution series of the antiviral, and infecting with MNV. The plate is incubated for 24 hours, until 90% of virally infected control cells are dead, following this CellTiter-Glo reagent is added. We also illustrate the utility of indirect measurements of virus replication to observe unexpected effects that compound carriers such as DMSO can have on norovirus infection.
Materials:
DMEM, high glucose, GlutaMAX (Thermo Fisher Scientific, cat. no. 10566–016)
Fetal bovine serum
CellTiter-Glo® Luminescent Cell Viability Assay (Promega, cat. no. G8461)
MNV-1 virus stock
RAW 264.7 cells (ATCC no. TIB-71)
Huh7mCD300lf cells (a gift from Prof S. Taube, University Lübeck) (Lingemann, 2020)
2’-C-methylcytidine (Sigma-Aldrich, cat. no. M4949) (reference antiviral)
Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, cat. no. D5879)
White flat bottom 96-well plate (Greiner Bio-One, cat. no. 655083)
Clear flat bottom 96-well plate
Multi-channel pipette (capable of volumes up to 100 μl)
Plate reader capable of reading luminescent signals in a 96-well plate format
Filtered pipette tips
Plate rocker
Protocol steps:
Preparation of cells in the test plate
-
1
Take a 75-cm2 flask with RAW 264.7 cells that have reached 70–80% confluence.
-
2
Scrape, count and collect the cells.
-
3
Seed the test plate (white flat-bottom 96-well) with 2×104 RAW264.7 cells/well in 100 μl of DMEM-10 (DMEM supplemented with 10% FBS).
Set up a clear 96-well plate, to visually monitor the cells throughout the experiment. Include antiviral-treated and infected wells in parallel to the test plate. This parallel plate is useful for observing the cell culture directly but can be omitted to reduce time or conserve compound and reagents.
Huh7 cells expressing the CD300lf receptor (Huh7mCD300lf) can be used as an alternate cell line and are seeded at 1×104 cells/well.
-
4
Gently rock the plate to ensure an even monolayer of cells.
-
5
Incubate the plates overnight at 37°C, 5% CO2.
Setting up the dilution plate (see Figure 1A)
Figure 1:
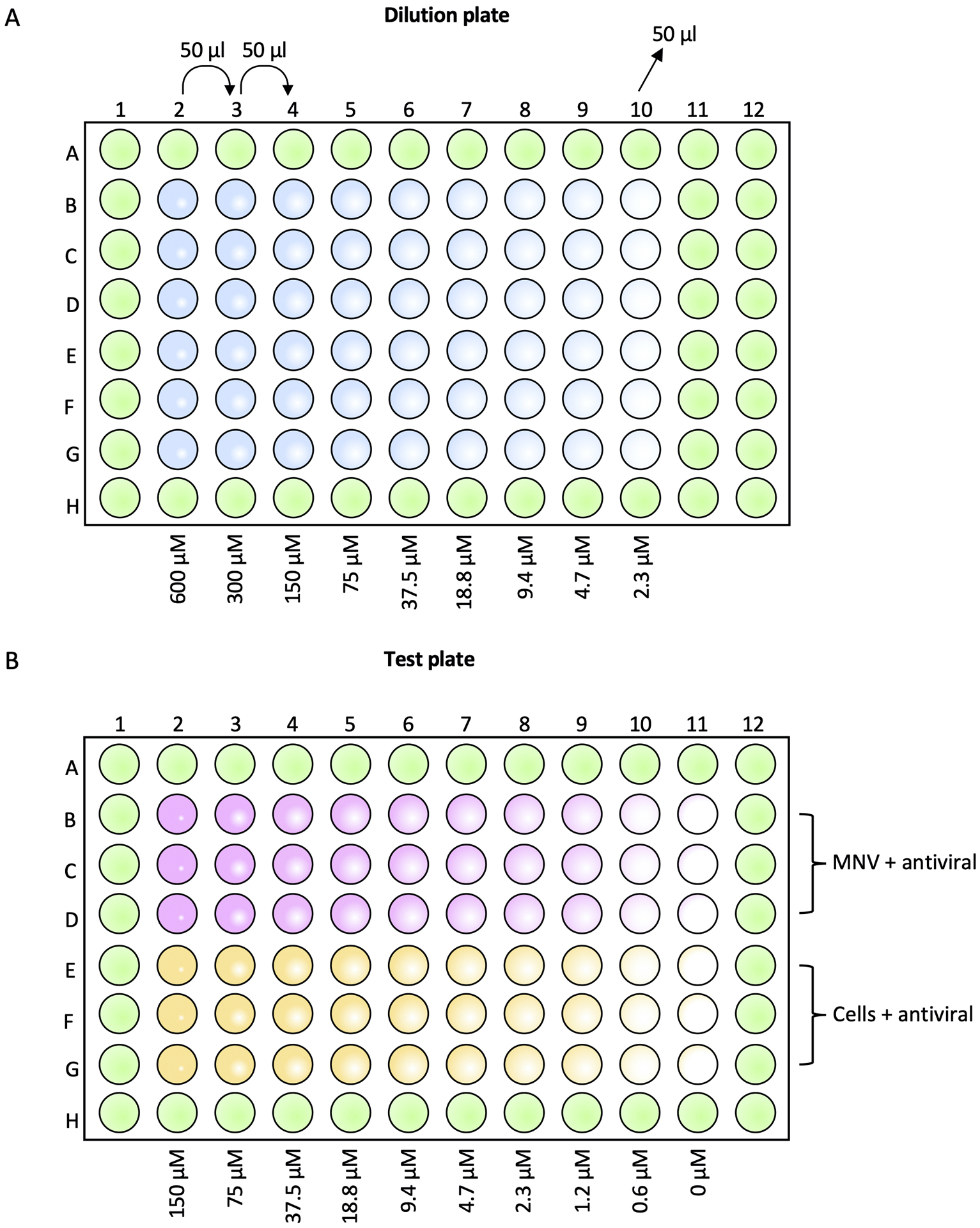
Preparation of the inhibitor dilution (A) and test (B) plates. Wells in green contain only DMEM-10. (A) Wells in blue contain dilutions of the inhibitor at the indicated concentrations. (B) Pink indicates wells containing RAW 264.7 cells, MNV and a 2-fold dilution series of inhibitor, for measuring antiviral effects. Yellow indicates wells containing RAW 264.7 cells and a 2-fold serial dilution of inhibitor, for measuring cell cytotoxicity. Inhibitor concentrations range from 150 μM to 0.6 μM with a 0 μM cells only control and virus only control.
-
6
Prepare inhibitor dilutions in a round-bottom 96-well plate at 4X the final concentration.
For the reference data, 150 μM of 2’-C-methylcytidine (2-CMC) on the test plate would be equivalent to 600 μM (4X) on the dilution plate.
-
7
Add 50 μl of DMEM-10 to all wells of the dilution plate, except column 2 rows B-G.
-
8
Add 100 μl of 2-CMC at 600 μM to column 2 rows B-G.
-
9
Using a multichannel pipette, perform a 2-fold serial dilution of the inhibitor, transferring 50 μl from column 2 into column 3.
Other serial dilutions can be used (e.g., 3-fold) as appropriate.
Use of an electronic multichannel pipette is ideal as it speeds up the task.
The plate set-up can be modified for screening of antivirals at a single concentration to identify lead compounds for further validation.
-
10
Mix gently up and down three times and repeat the dilution series down to column 10, mix and discard 50 μl from the last column.
Addition of antiviral and MNV to the test plate (see Figure 1B)
-
11
Remove 50 μl from wells in columns 2–11 rows B-D of the test plate.
-
12
Remove 25 μl from wells in columns 2–11 rows E-G of the test plate.
-
13
Using a multichannel pipette transfer 25 μl of inhibitor from the dilution plate into the respective wells of the test plate.
-
14
Infect cells with MNV at a final MOI of 0.1 by adding 25 μl of virus to columns 2–11 rows B-D of the test plate.
Make 1 ml of virus stock at 8×104 PFU/ml in DMEM-10.
The CellTiter-Glo assay has shown that MNV infections in RAW264.7 cells are sensitive to DMSO at concentrations > 0.07% (Figure 2B). In the presence of MNV (MOI 0.1), increased concentrations of DMSO caused an increase in percent cell viability. This is indicative of a decrease in MNV-induced killing of cells. In comparison, cell viability in the absence of MNV is not impaired until ≥ 2% DMSO (Figure 2A). Wells currently designated for DMEM-10 only can be utilized as paired DMSO + virus controls if DMSO will exceed 0.07%.
For Huh7mCD300lf cells infect at an MOI of 0.01 and incubate for 48 hours. We have observed that replication of MNV in Huh7 CD300lf cells does not show the same sensitivity to DMSO as RAW264.7 cells.
-
15
Incubate the plate at 37°C, 5% CO2 for ~24 hours until 90–95% cell death (5–10% cell viability) is observed in the virus only control wells (Figure 2B).
Figure 2:
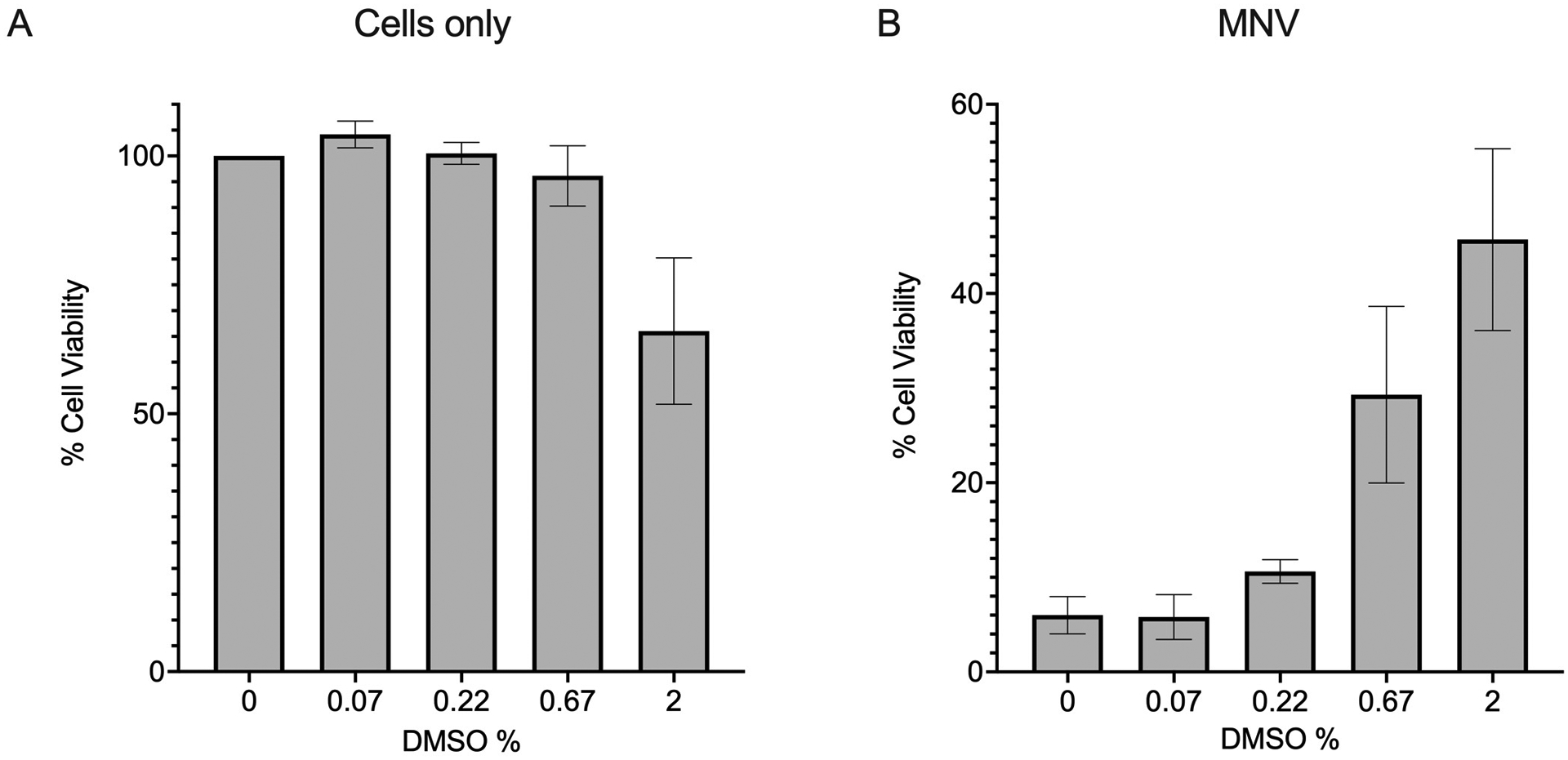
Effect of DMSO on cell viability. RAW 264.7 cells were treated with DMSO at various final percentages and either left uninfected (cells only) (A) or infected with MNV at an MOI of 0.1 (B). Cells were incubated for 24 hours, and viability measured with CellTiter-Glo. (A) Effect of DMSO alone upon RAW264.7 cells. Luminescence signals were normalised to those of the 0% DMSO control to determine the % cell viability. (B) Luminescence signals for MNV infected samples were normalised against those of the 0% DMSO cells only control, representing 100% viable cells.
Indirect measurement of cell viability using CellTiter-Glo
-
16
Thaw CellTiter-Glo reagent and equilibrate the cell plate to RT for 20 minutes.
-
17
Using a multichannel pipette, add 100 μl of CellTiter-Glo reagent to each well.
Change tips between samples to avoid ATP cross-contamination.
-
18
Incubate plates on orbital rotator at 200 RPM for 2 minutes to lyse cells.
-
19
Incubate the plate in the dark for 10 minutes to allow luminescent signal to develop.
-
20
Measure the luminescence on a plate reader.
-
21
Calculate the % cell viability, virus only sample represents 0% cell viability and the cells only sample represents 100% cell viability. These values are then graphed to determine the EC50 and CC50 values for the chosen inhibitor.
Example EC50 and CC50 curves are shown in Figure 3.
Figure 3:
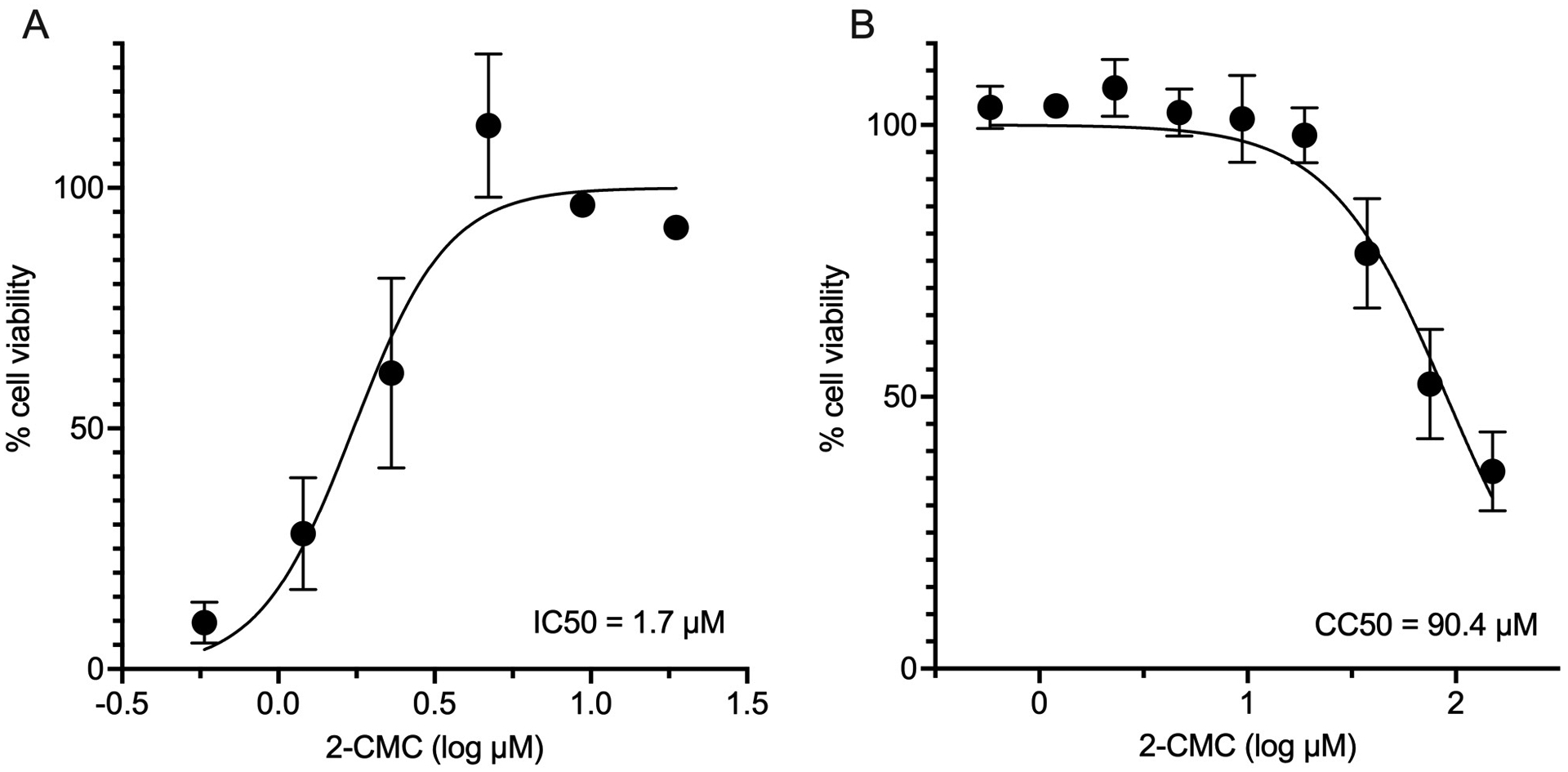
Reference EC50 and CC50 data with 2-CMC. Luminescence data obtained with CellTiter-Glo was normalized, with virus only and cells only representing 0% and 100% cell viability respectively. Graphs were generated using GraphPad Prism and represent the mean and standard deviation of three technical repeats. (A) Determination of 2-CMC EC50 with MNV. Concentrations of 2-CMC that are cytotoxic to cells were excluded. (B) A CC50 graph was generated for cells in the presence of 2-CMC (150 μM – 0.6 μM). Curves were generated with a non-linear regression model, log (inhibitor) vs. normalized response – variable slope.
BASIC PROTOCOL 2: MEASURING MNV GENOME TITERS BY RT-qPCR
Introduction.
MNV levels can be assessed through reverse transcription quantitative polymerase chain reaction (RT-qPCR). This technique involves purifying viral RNA, reverse transcribing the RNA into cDNA (complementary DNA) using a reverse primer, and then exponentially amplifying the cDNA. RT-qPCR can be conducted as either a single reaction or in combination with qPCR (quantitative RealTime PCR). Ct values obtained from the qPCR values will correlate with the cDNA levels. Comparing the Ct values to a known MNV standard can be used to determine genome or cDNA equivalents.
Here, we describe an updated protocol based on primers and probe previously described for the quantification of murine norovirus genome (Taube et al., 2009). In this protocol, we use guanidinium thiocyanate-phenol-chloroform extraction, also known as TRIzol™ extraction because it quickly inhibits RNase activity and inactivates infectious virus. Coupled with Zymo-Spin™ columns this is an extremely fast and streamlined method to extract MNV genomes from a variety of starting materials, including but not limited to cell culture lysates, homogenized tissues, or stools samples. Alternatively, silica-column based RNA extraction methods are also very reliable and described previously (Alternate Protocol 4 in (Hwang et al., 2014)). The RNA extraction should involve a DNase I treatment step to remove DNA contamination. After RNA extraction, a One-Step RT-qPCR is performed to titrate the MNV cDNA. Linearized plasmid DNA and in vitro transcribed RNA are widely used as a standard and their respective advantages and disadvantages have been described previously (Hwang et al., 2014). The commercial availability of double-stranded gene fragments of sufficient length has now become an easy and highly reproducible alternative to linearized plasmid DNA.
Materials:
Two sets of pipettors (one designated for handling RNA and master-mix(es), one designated for handling DNA)
Filter tips 1000 μL, 100 μL, 10 μL, nuclease-free
Nuclease-free 1.5 ml micro-centrifuge tubes
(Designated for RNA or DNA/plasmid work)
96 Well PCR-plate, white, 100 μl volume, (Sarstedt, Cat. No. 72.1977.23)
qPCR-Film, DNase-/RNase-free, highly transparent (Sarstedt, Cat. No. 95.1999)
RNase decontamination solution
Nuclease-free water
Nuclease-free 100 % ethanol
Nuclease-free 70 % ethanol
Direct-zol™ RNA Miniprep kit with TRI reagent (Zymo Research, Cat. No. R2051)
iTaq™ Universal Probes One-Step Kit (BioRad, Cat. No. 1725140)
Forward Primer (5’ → 3’): GTGCGCAACACAGAGAAACG (desalted and/or HPLC purified)
Reverse Primer (5’ → 3’): CGGGCTGAGCTTCCTGC (desalted and/or HPLC purified)
Probe (5’→ 3’): [6-FAM]-CTAGTGTCTCCTTTGGAGCACCTA-[BHQ1] (HPLC purified), modified at the 5′-end with a 6-carboxyfluorescein (FAM), and at the 3′-end with a quencher, i.e. Black Hole Quencher 1 (BHQ1)
MNV-1 gBlock (shown is the sequence of the top strand 5’ → 3’, generate as double-stranded DNA): GTGCGCAACACAGAGAAACGCAAAAACAAGAAGGCTTCGTCTAAAGCTAGTGTCTCCTTTGGAGCACCTAGCCCCCTCTCTTCGGAGAGCGAAGACGAAATTAATTACATGACCCCTCCTGAGCAGGAAGCTCAGCCCG
RealTime qPCR machine
Viral RNA isolation
-
1
Prepare an RNA-designated workspace cleaned with RNase decontamination solution (15 % bleach, 0.05 % SDS, 1 % NaOH), incubate for at least 5 min, then rinse with nuclease-free water and nuclease-free 70 % ethanol.
Note: Use of a clean designated “RNA only” workspace with its own set of pipettors, tips, and microcentrifuge tubes is highly advisable. Treat your gloved hands with RNase decontamination and thoroughly clean your equipment including pipettors, tip-boxes etc. before use to minimize the presence of nucleases in the work area. Alternative commercial decontamination solutions may also be used such as RNase Away® (Sigma, cat. no. 83931).
-
2
Extract viral RNA from your sample (i.e., cell culture lysate or homogenized tissue) following the manufacturer’s recommendations.
Note: For TRIzol™ extraction it is highly recommended to use the silica membrane-based columns (i.e., Zymo-Spin™ IICR Column), to remove any phenol contaminants. Treat your sample with TRI-reagent (TRIzol™ or TRIzol LS™). For cell suspensions use 3 – 4 volumes of TRI-reagent. For direct lysis in a cell culture dish use 100 μl TRI-reagent per square-centimeter surface. Follow the manufacturer’s instructions and include the recommended DNase I digest.
For any “open-tube” spin-column extraction, such as the QIAamp Viral RNA kit or RNeasy kit an RNA-only microcentrifuge is recommended. A vacuum manifold is not recommended because cross-contamination with plasmid DNA cannot easily be excluded.
-
3
Elute or resuspend the RNA in 50 μl of DNase/RNase-Free water. Use an RNase-free microcentrifuge tube. The eluted RNA can be used immediately or stored aliquoted at ‑ 80 °C.
Note: This protocol works well without the use of RNase inhibitors. If commercial RNase inhibitors are used, they need to be tested because there is a chance they interfere with the RT-qPCR protocol. The use of spectrophotometry to estimate the purity/integrity of RNA is discouraged, as it will only reliably measure cellular RNA or carrier RNA present in some commercial RNA kits.
RT-qPCR
Note: The RT-qPCR is performed as previously described (See Alternate Protocol 4 in (Hwang et al., 2014)) using the iTaq™ Universal Probes One-Step Kit. Be aware that the optics of the qPCR machine need to be compatible with your qPCR master-mix and the fluorophore used for the probe.
-
4
Plan the plate arrangement according to the number of samples, including standard dilutions, a no-template control (NTC), that serves as a general control for extraneous nucleic acid contamination, and a no transcriptase control (NRT), that assesses the amount of DNA contamination present in an RNA preparation. Each sample and control should be run in duplicate, but preferably in triplicate, to observe for pipetting error.
-
5
Thaw reaction components on ice, mix them thoroughly and briefly centrifuge them to collect the content at the bottom of the tubes.
Note: Light-sensitive components should be protected from light (i.e., probe).
-
6
Set-up the RT-qPCR reaction by calculating the amount of each component needed (see Table 1). A reaction volume of 20 μl is sufficient.
-
7
Prepare a master mix in a 1.5 mL nuclease-free tube, containing each component except for the template. The NRT control-mix can be removed from the master-mix before adding the reverse transcriptase. This step can be carried out at room temperature, as the polymerase used in commercial master-mixes usually requires heat-activation (hot-start) and the components are stable at room temperature. Alternatively, this step can also be carried out on ice.
-
8
Mix thoroughly and pipet 15 μl into the required wells of a 96-well plate. Components of the master-mix should be pipetted in the RNA working area while the standard should be pipetted in the DNA working area using a designated set of pipettors.
-
9
Pipet 5 μl of each sample RNA into the required well. Nuclease-free water should be added to NTC and NRT to match the total volume of the samples.
-
10
The standard should contain at least six 10-fold dilutions of a control dsDNA and should be added last (see support protocol 1 on preparing the standard). The standard should be pipetted on a designated DNA working area with its own set of pipettors.
-
11
Cover the plate with the optical adhesive sheet and then briefly centrifuge the plate. If no plate-centrifuge is available, tap the plate to remove bubbles and collect all the volume at the bottom of the well.
-
12
Load the plate into the instrument and run the RT-qPCR reaction after programming the thermal cycler settings as follows:
Reverse transcription for 25 min, 50°C
Activation and initial denaturation for 5 min, 95°C
40 cycles of denaturation for 10 sec, 94°C, and annealing and elongation for 30 sec, 60°C
-
13
Perform data analysis according to the instrument’s specific instructions.
Table 1:
Pipetting scheme for the RT-qPCR master-mix1
| Component | Initial Concentration | Final Concentration 1 reaction |
Volume 1 reaction |
MasterMix 100 reaction |
|---|---|---|---|---|
| Master Mix | 2 x | 1 x | 10 μl | 1000 μl |
| For primer | 25 μM | 0.3 μM | 0.24 μl | 24 μl |
| Rev primer | 25 μM | 0.3 μM | 0.24 μl | 24 μl |
| Probe | 25 μM | 0.125 μM | 0.1 μl | 10 μl |
| RNA template | – | – | 5 μl | – |
| iScript RT | 40 x | – | 0.5 μl | 50 μl |
| Nuclease-free H 2 O | – | – | 3.92 μl | 392 μl |
| Total | – | – | 20 μl | 1500 μl |
| Each | 15 μl |
The volumes and components resemble the recommended reaction setup from BioRad One-Step RT-qPCR kit (cat. no. 1725140, 1725141 and 12013250). To account for pipetting error, a 100-reaction master-mix is recommended for a complete 96-well plate.
Support protocol 1: Preparation of standard
Introduction.
Depending on your application, you can use a standard derived from in vitro transcribed RNA, or dsDNA (double-stranded DNA). Both have their advantages and disadvantages. In vitro transcribed RNA needs to be generated, purified, and aliquots must be stored at ‑ 80 °C. On the other hand, dsDNA standards are easily obtained and highly reproducible but lack the control-step for reverse transcription. For dsDNA standards you can use plasmid DNA or commercial custom dsDNA fragments, such as gBlocks™ from Integrated DNA Technologies (IDTA) that comprise the complete amplicon of the qPCR. If you use plasmid DNA, a linearization step followed by column-based purification method is highly recommended, as circular DNA is not as efficiently amplified during cycling because primer/probe binding may be impaired for super-coiled DNA. Copy numbers can be derived from the measured mass of the ssRNA (single-stranded RNA) or dsDNA molecule.
Determine the concentration of the linearized plasmid by spectrophotometry and convert the readout to g per liter. Accurate measurements are key here to obtain reproducible results.
-
Calculate the molecular weight (MW) by multiplying the DNA size (in base-pairs) with 662 grams / mol, which is the average weight of a nucleotide pair. Alternatively, online calculators can be used to calculate the precise MW of the DNA based on sequence (MW needs to be multiplicated by 2 to account for dsDNA).
Example: a gBlock™ corresponding to the size of the PCR amplicon of MNV-1 (139 bp) has a MW of 85,758 Da or 85.76 kDa.
-
Calculate the molar concentration (M) of your linearized plasmid:Example: a gBlock™ of MNV-1 (139 bp) usually has a concentration of 3,000 fM
- The copy number (cp) can be calculated based on Avogadro’s constant:
- 1 M is about copies:
The dsDNA is now adjusted to 2 × 108 / μl (in this example 1.8 × 1012 cp are resuspended in 9 ml nuclease-free water)
The PCR pipetting scheme requires 5 μl template/standard. Froma 2 × 108 / μl stock this equals 1×109 copies per PCR well.
-
To generate a standard curve, a series of at least six 10-fold serial dilution is performed, changing tips between each dilution-step. A suitable dilution range for a standard set is from 107 to 102 copies per well. Each dilution is stored as a single-use aliquot at ‑ 80 °C. 8-strip PCR tubes have proven useful for a set of standards.
Make sure that your standard curve has a correlation coefficient (R2) higher than 0.98 and shows a slope close to −3.3, which ensures that your primers have close to 100 % efficiency for amplification (you have double the original amount of template as each cycle progresses). The most dilute standard within the linear range of the standard curve can be as little as 2 genome copies, thus, the reliable limit of detection for this particular protocol of MNV-1 amplification is 2 genome copies per PCR well.
If the entire well was used in the RNA extraction, you can calculate the genome copies per well. For this you need to consider that only 1/10th of your RNA extraction (5 μl out of 50 μl elution volume) was analyzed. To obtain the genome-equivalents per well (GE / well) multiply your GE reading from the qPCR machine with your dilution factor, in this case 10.
Alternatively, if you have used a specific volume for the RNA extraction, you can calculate genome copies per ml. For this you need to again consider that only 1/10th of your RNA extraction (5 μl out of 50 μl elution volume) was analyzed. Additionally, you need to take the sample volume into account used in the lysis reaction, e.g., if you used 500 μl of a virus stock together with the TRI-reagent, you need to multiply your copy numbers also by 2 to bring it up to 1 ml. In total for this example, you need to multiply by 20 to obtain GE / ml.
Basic Protocol 3: GENERATION OF RECOMBINANT MURINE NOROVIRUS WITH MINIMAL PASSAGING
Introduction.
Reverse genetics provides an invaluable technique for exploring norovirus biology, allowing for the introduction of specific mutations into a cDNA clone of the viral genome via site-directed mutagenesis (Liu & Naismith, 2008) and subsequent recovery of the recombinant virus through transfection of genomic cDNA or in vitro transcribed RNA into permissive cells. The original DNA-based reverse genetics protocol is based on a publication by Ward et al. (Ward et al., 2007). An RNA-based protocol was later developed in the laboratory of Ian Goodfellow (Arias et al., 2012), which allowed minimal passaging by directly transfecting capped RNA transcripts into BV-2 cells using the specialized Neon (Life Technologies) electroporation system. An account of the available DNA and RNA based reverse genetics systems has previously been summarized (see Basic Protocol 3 and 4 in (Hwang et al., 2014)).
Generation of infectious MNV from cDNA is technically challenging in naturally susceptible cell lines such as RAW264.7 cells or BV-2, due to difficulties efficiently transfecting these cells.
The identification of mCD300lf as an entry receptor for MNV (Orchard et al., 2016) has now enabled the generation of susceptible cell lines from any permissive cell line, thus overcoming the limitation in the choice of easily transfectable cell lines. Huh7 human hepatoma cells are easily transfectable with a variety of transfection reagents and have been transduced to stably express mCD300lf generating Huh7mCD300lf (Lingemann, 2020). Lentiviruses are the tool of choice for fast and efficient generation of stable homogeneous cell lines for gene expression (Zufferey et al., 1998). A protocol for the lentivirus vector generation (Zufferey et al., 1998) is available from the Trono-Lab website (Lentivector Toolbox1). Here, we provide an updated RNA-based reverse genetics protocol for MNV (based on Arias et al. 2012) using Huh7mCD300lf cells with minimal passaging. However, the protocol can also be easily applied to cDNA from the Ward et al system. Transfection of Huh7mCD300lf with MNV.CW1 wild type in vitro transcribed RNA typically yields between 1×105 – 1×107 TCID50/ml within 48 to 96 hours post transfection while DNA transfection typically yields about a log less.
Materials
MNV cDNA (pT7: MNV 3’Rz) (a gift from Prof I. Goodfellow, University of Cambridge)
Huh7mCD300lf cells (a gift from Prof S. Taube, University Lübeck) (Lingemann, 2020)
NheI Restriction Enzyme (NEB R0131S)
MAXIscript™ T7 Transcription Kit (ThermoFisher AM1314M)
DNase I (RNAse free) (NEB M0303S)
ScriptCap™ m7G Capping System (Epicentre Biotechnologies SCCE0610)
Or Faustovirus Capping Enzyme (NEB M2081L)
Opti-MEM™ I (ThermoFisher 31985070)
Lipofectamine™ MessengerMAX™ (ThermoFisher LMRNA003) for RNA-based transfection
Fugene HD Transfection reagent (Promega, E2311) for DNA-based transfection
Protocols Steps
Synthesis of infectious capped MNV transcripts
-
Digest 10 – 20 μg of the plasmid containing the wild-type MNV cDNA (pT7:MNV3’Rz) with NheI to obtain linear DNA. Table 2 shows a typical restriction setup for NheI digestion.
NheI recognizes a unique restriction site after the 3′-end poly(A) tail of the MNV genome; thus, it defines the 3′ end of transcription with a tolerated addition of a single guanosine to the 3′ end of the genome.
Incubate at 37°C for 2 h. Keep at least 2 μl from the linearized DNA for later electrophoresis analysis.
Purify the linearized DNA by phenol/chloroform extraction, ensuring RNase-free conditions. Determine DNA concentration measuring the absorbance at 260 nm by spectrometry (i.e. NanoDrop)..
Ensure complete linearization and condition of purified DNA by gel electrophoresis.
Perform the in vitro transcription from the purified, linearized DNA using T7 RNA polymerase. Multiple kits are available from various manufacturers such as the MAXIscript™ T7 Transcription Kit (Invitrogen™). Ensure all centrifuge tubes and filter tips are certified as RNase-free. Table 3 shows a typical reaction for the MAXIscript™ T7 Transcription Kit.
Incubate at 37 °C for 2 hrs. Digest the DNA with 1 μl TURBO DNase I (MAXIscript™ T7 Transcription Kit, 2 U/μl) at 37 °C for 30 min to ensure complete DNA digestion. Keep back at least 2 μl from the in vitro transcription for electrophoresis analysis.
Purify the RNA to remove unincorporated nucleotides, enzymes and other impurities. This can be done using TRI reagent (Direct-zol RNA Miniprep, Zymo Research) for RNA extraction (see also Viral RNA Isolation in Basic Protocol 2). Elute the RNA with 69 μl RNase-free-ddH2O by centrifugation as before. Determine RNA concentration and purity. Keep back at least 2 μl RNA for electrophoresis analysis.
-
Capping can be performed with the ScriptCap™ m7G Capping System (CellScript). Alternatively, the Faustovirus Capping Enzyme (New England Biolabs) can also be used. Cap the purified RNA by first denaturing at 65 °C for 5 – 10 min and immediately store on ice. A typical ScriptCap™ reaction is shown in Table 4.
Note: the official ScriptCap™ protocol states to use 4 μl capping enzyme but 1 μl has been tested and works sufficiently if needed.
Incubate at 37 °C for 1.5 hrs. Reserve 2 μl for electrophoresis analysis (Figure 4). Capped RNA can be used straight away or stored at −80 °C.
-
Run the in vitro transcribed, purified and capped RNA samples together by gel electrophoresis (1 % Agarose, 1x TAE) (Figure 4). Run the electrophoresis at 120 V for only 10 min to avoid RNA degradation. Bands should be of equal size with decreasing intensity as the RNA becomes more dilute. Smears or multiple bands may indicate degradation at different steps but can be a result of the gel electrophoresis.
Note: Ensure the electrophoresis chamber is as RNase-free as possible to avoid RNA degradation. An occasional clean with a solution containing NaOH should be sufficient, but repeated use will corrode the components.
One day prior to transfection, trypsinize a monolayer of Huh7mCD300lf cells and seed 2×105 cells per well of a 6-well plate, in DMEM (supplemented with 10 % FCS, 1 % Penicillin/Streptomycin, 1 % L-glutamine, 1 % non-essential amino acids). Cells should be approximately 70 % confluent at the time of transfection.
-
Prepare an RNA- and a transfection-mix. For the transfection-mix add 3.75 μl Lipofectamine MessengerMAX to 100 μl Opti-MEM and incubate at RT for 10 min. For the RNA-mix add 2.5 μg RNA to 100 μl Opti-MEM (always thaw and store RNA on ice to avoid degradation).
For a DNA based reverse genetics system the RNA can be substituted with MNV cDNA from plasmid (Ward system). We recommend the Fugene HD-based transfection protocol (Hwang et al., 2014) with a plasmid to Fugene HD ration of 1:4. However simple DNA transfection protocols, such as calcium phosphate (Kwon & Firestein, 2013) also work.
Combine the RNA (or DNA) and transfection mix with a slight flick and incubate at room temperature for ca. 5 min (but do not exceed 30 min). Other transfection reagents may have different incubation times, so adjust accordingly if needed.
Meanwhile, remove the DMEM from the seeded 6-well and replace with 800 μl Opti-MEM.
Add the ~200 μl transfection mix to the cells, drop-wise (carefully allowing the transfection mix to drop directly onto the cells instead of the more common addition of into the well wall).
Incubate at 37 °C, 5 % CO2, 95 % RH for 3 – 4 hr.
Replace the Opti-MEM transfection with 2 ml DMEM (supplemented with 10 % FCS, 1 % Penicillin/Streptomycin, 1 % L-glutamine, 1 % non-essential amino acids).
Freeze both medium and cells at 2 days post-transfection at −80°C. Use cell lysate for titration by end-point dilution assay or plaques assay.
Passage on standard BV-2 cells or RAW246.7 cells to expand the viral stock if necessary.
Table 2:
Reaction Setup for NheI Linearization of MNV cDNA
| Components | Final Concentration | Volume (μl) |
|---|---|---|
| 10x CutSmart Buffer (NEB) | 1x | 20 |
| NheI-HF 20 U/μl (NEB) | 0.3 U/μl | 3 |
| cDNA (pT7:MNV3’Rz) | 100 ng/μl | Variable |
| ddH2O | -- | add to 200 |
Table 3:
Reaction set up for in vitro transcription of linearized MNV cDNA
| Reagent | Final Concentration | Volume (μl) |
|---|---|---|
| 10x Buffer | 1x | 4.0 |
| rNTPs (10 mM each) | 0.5 mM each | (2.0 each) 8.0 |
| T7 Enzyme (15 U/μl) | 0.75 U/μl | 2.0 |
| Linear DNA | 100 ng/μl | (3–4 μg) ~10–15 |
| ddH2ORNase-free | -- | Up to 40 |
Table 4:
Reaction set up for capping of in vitro transcribed MNV RNA
| Reagent | Per 100 μl Reaction | Notes |
|---|---|---|
| Denatured IVT RNA | (50 – 60 μg) 69.0 μl | |
| 10x Capping Buffer | 10.0 μl | Dissolve at 37°C, thaw at RT,
vortex before use |
| 10 mM GTP | 10.0 μl | |
| 2 mM SAM | 5.0 μl | Dilute fresh from 20 mM stock |
| RNase Inhibitor | 2.5 μl | ScriptGuard RNase Inhibitor |
| Capping Enzyme | 4.0 μl | As little as 1 μl can also be used |
Figure 4:
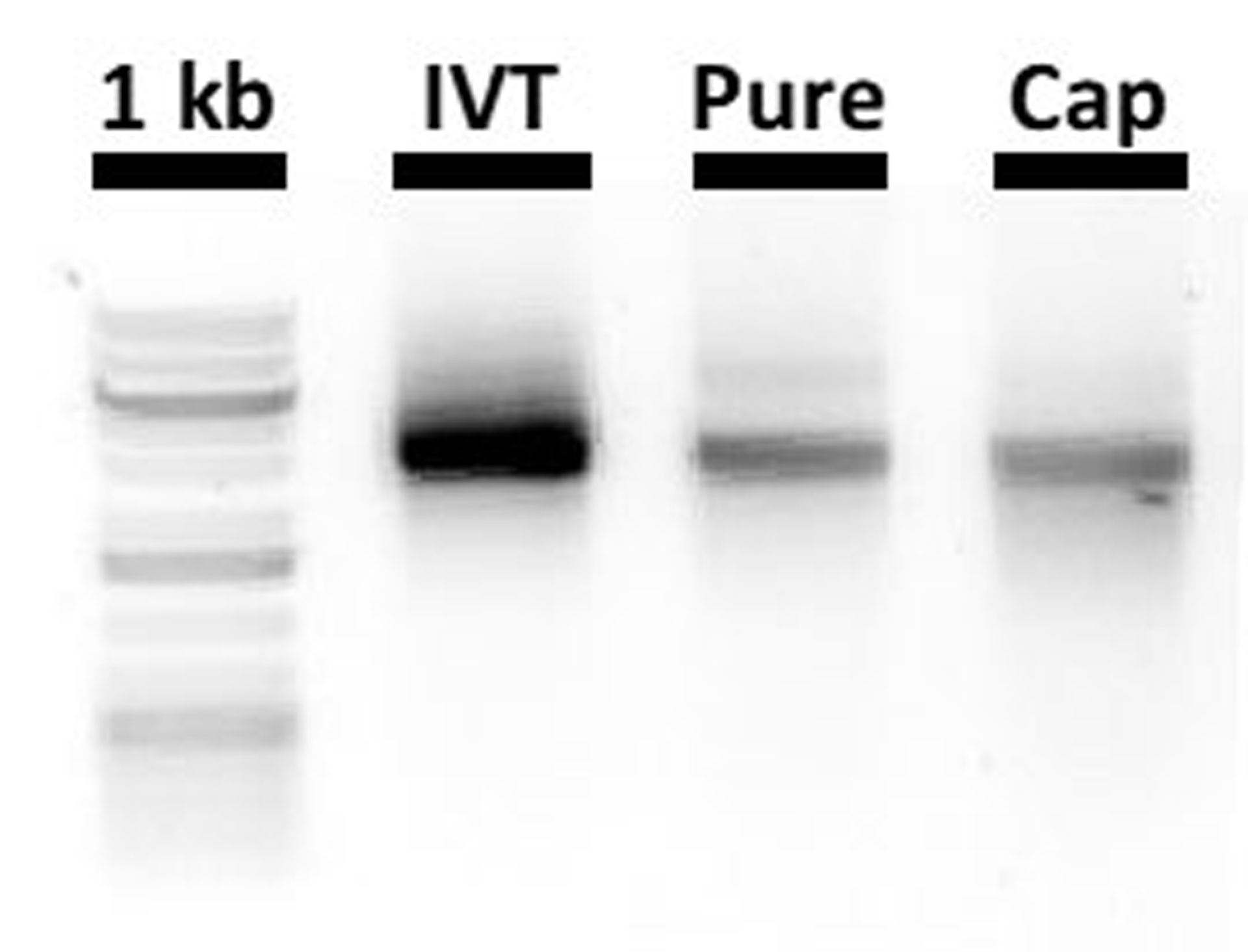
Analysis of MNV RNA transcript integrity by nondenaturing gel electrophoresis. In vitro transcribed RNA (IVT), purified RNA (Pure), and capped RNA (Cap) products are run at 120 V for 10 min on a nondenaturing 1% agarose gel (TAE buffer) in parallel to a GeneRuler™ 1 kb Plus DNA Ladder (1 kb, lane 1, not representative of RNA sizes).
Basic Protocol 4: GENERATION OF RECOMBINANT MURINE NOROVIRUS VIA CIRCULAR POLYMERASE EXTENSION REACTION (CPER)
Introduction.
The use and utility of recombinant virus infectious clones and replicons has revolutionised our understanding of the molecular events of the intracellular replication of many (+)-sense RNA viruses. In addition, it has informed some attributes of protein function and guided the development of antiviral compounds and therapies. Many reverse genetics systems have been employed to derive authentic full-length copies of the MNV genome in addition to mutants and to some extent the development of reporter viruses (see also Basic Protocol 4 and Alternate Protocol 2 in (Hwang et al., 2014)). The protocol described within is founded on the publication by Amarilla et al. (Amarilla et al., 2021). Essentially, we describe the assembly of PCR fragments encompassing the entire genome that are circularised with a linker containing a poly-A tail, Hepatitis D virus ribozyme and a CMV promoter (see Figure 5). This assembly is then transfected into NIH3T3 cells whereupon the full-length viral genome is transcribed by the host RNA pol II enzyme before translation and replication in the cytoplasm. In NIH3T3 cells this usually yields about 103 pfu/mL of MNV which is subsequently transferred and amplified in RAW264.7 cells to generate ~108 pfu/mL. The advantages of this approach are: (i) rapid recovery of virus, (ii) no requirement for cloning or transformation in bacterial cultures, and (iii) gene blocks can be synthesised from sequence alone to generate the fragments required for the CPER assembly.
Figure 5:
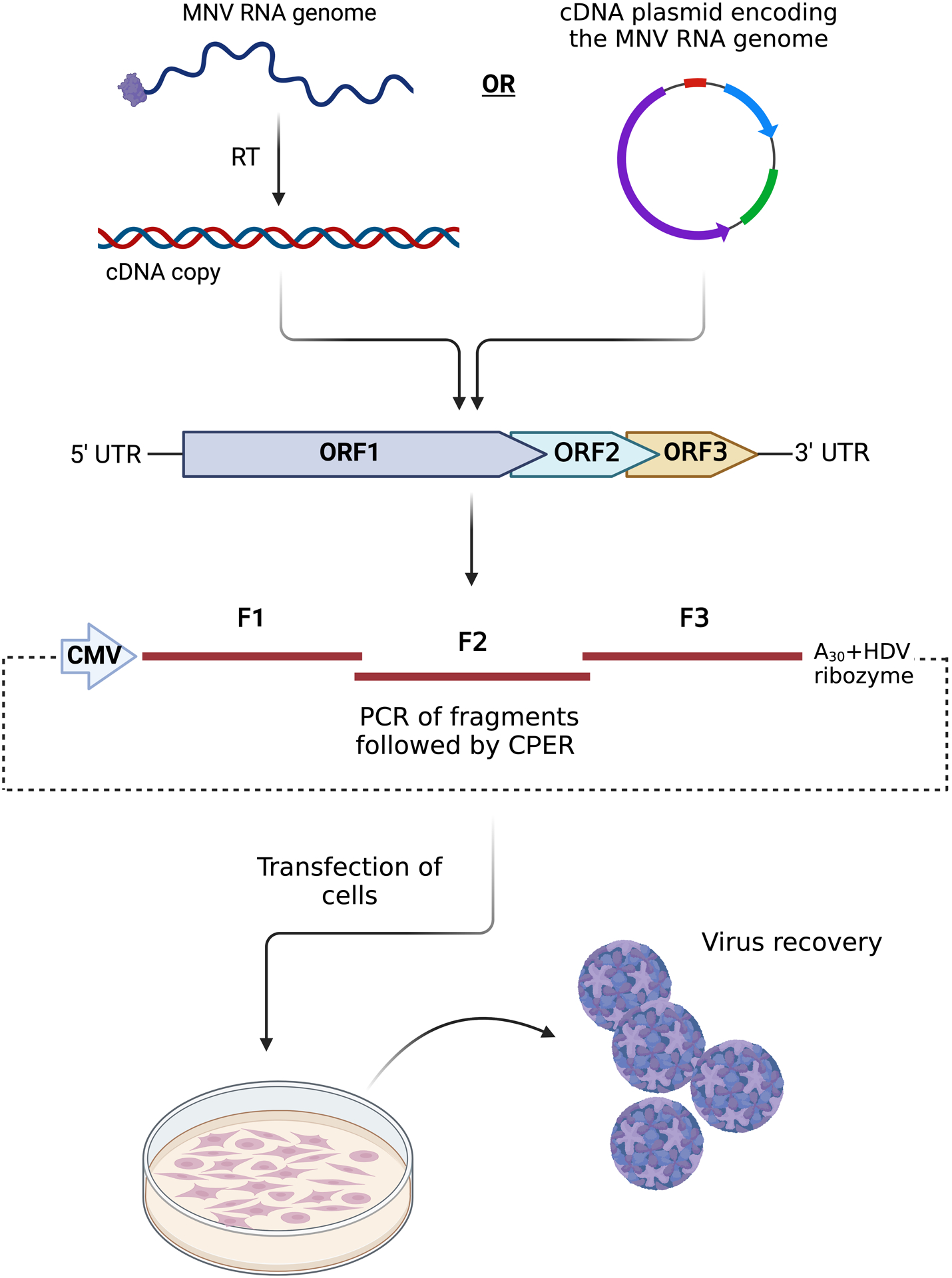
Schematic showing the workflow and conceptual design of the CPER approach to generate MNV. The PCR fragments can be produced from isolated viral RNA that has been reverse-transcribed (RT) or from a cDNA plasmid entire MNV genome. The amplified PCR fragment are combined and ligated with the linker fragment for assembly. The reaction is subsequently transfected into cells and infectious virus is recovered.
Materials
Full-length MNV-containing plasmid (pSPORT-T7-MNV1; was received via a materials transfer agreement from Dr. Herbert Virgin)
Plasmid containing MNV linker sequence and HDV ribozyme (pUC19-MNV-CMV_Linker) (available from Dr. Jason Mackenzie)
Dulbecco modified Eagle medium (MEM; Gibco) supplemented with 10% FCS (Gibco), 1% GlutaMAX (Gibco), and 1% sodium pyruvate (100 mM; Gibco)
Serum Free Dulbecco modified Eagle medium (DMEM; Gibco) supplemented with 1% GlutaMAX (Gibco), and 1% sodium pyruvate (100 mM; Gibco)
Dulbecco modified Eagle medium (DMEM; Gibco) supplemented with 4% FCS (Gibco), 1% GlutaMAX (Gibco), and 1% sodium pyruvate (100 mM; Gibco)
NIH3T3 (ATCC CRL-1658) or HEK 293T (ATCC CRL-3216) cells growing in culture (Note: low passage number works better)
RAW 264.7 cells (ATCC no. TIB-71)
High-fidelity PrimeSTAR GXL DNA Polymerase (Takara Bio, Cat# R050B)
5X PrimeSTAR GXL Buffer (Takara Bio, Cat# R050B)
10 mM dNTP mix (Takara Bio, Cat# R050B)
- Primers (all listed as sequence 5’−3’):
Fragment 1 MNV_Frag1.F GTGAAATGAGGATGGCAACGCCATCTTC MNV_Frag1.R CTGACACAGTTGACAAGTGTTTTGGGC Fragment 2 MNV_Frag2.F GCCCAAAACACTTGTCAACTGTGTCAG MNV_Frag2.R CACTCATCCTCATTCACAAAGACTGCTGAG Fragment 3 MNV_Frag3.F CTCAGCAGTCTTTGTGAATGAGGATGAGTG MNV_Frag3.R GCAGTAAGCAGAAATCATTTTCACAAAAGGTTTC - Linker fragment (Full sequence of the linker described in Appendix)
MNV_Linker.F GAAACCTTTTGTGAAAATGATTTCTGCTTACTG MNV_Linker.R GAAGATGGCGTTGCCATCCTCATTTCAC Nuclease-free H2O (purchased, e.g., from Invitrogen)
Zymoclean Gel DNA Recovery Kit (Zymo Research, Cat# D4007)
Transfection reagent (Lipofectamine 3000; Invitrogen, Cat# L3000015)
Opti-MEM™ (Gibco, Cat# 11058021)
6-well cell culture plate
100 mm cell culture dish
15-ml conical centrifuge tubes (e.g., BD Falcon)
1.5-ml microfuge tubes (e.g. Eppendorf)
PCR tubes
NanoDrop™ 2000 Spectrophotometer
-
Sequencing facility to confirm the MNV fragments (if necessary)
NOTE: All steps are carried out at room temperature. Media should be warmed to 37ºC or equilibrated to room temperature.
CPER reaction
NOTE: All primers are designed with a 27–33bp overlap and approximately equal melting temperature of 60 TmºC. High-fidelity PrimeSTAR GXL DNA Polymerase (Takara Bio) is used for all PCR amplifications.
-
PCR amplify CPER fragments using primers listed above under the following conditions
Example:CR amplification of MNV Fragment#15X PrimeSTAR GXL Buffer 10μL 10 mM dNTP mix 4μL MNV_Frag1.F (10 μM) 1μL MNV_Frag1.R (10 μM) 1μL pSPORT-T7-MNV1 (1μg/μL) 1μL PrimeSTAR GXL DNA Polymerase 1μL Nuclease-free H2O 32μL Total 50μL Amplification cycles:- 98 °C for 30s,
- 98°C for 10s, 55°C for 15s, for 30 cycles
- 68°C for 3min,
- and a final extension at 68°C for 10 min.
Purify PCR reactions by agarose gel electrophoresis and recover DNA using Zymoclean™ Gel DNA Recovery Kit (Zymo Research) following the manufacturer’s instructions.
-
Determine DNA concentration of each fragment using NanoDrop 2000 spectrophotometer.
NOTE: where necessary these DNA fragments can be sequenced using the primers listed above.
-
Each CPER reaction requires 0.1 picomole of each respective cDNA fragment and the corresponding linker fragment. Use the online calculator to calculate the amount of DNA (ng) given the size (bp) of each fragment that equates to 0.1 picomole http://endmemo.com/bio/dnacopynum.php. Using this value, calculate the volume of each fragment required for the CPER reaction.
Fragment Size (bp) MNV-Linker 1156 MNV_Fragment #1 2398 MNV_Fragment #2 2697 MNV_Fragment #3 2312 Example: Calculate the amount of MNV Fragment#1 required for CPER reaction. Assuming recovered DNA is 109.5 ng/μL and the size of the fragment is 2398 bp, 0.1 picomole equates to 155.87 ng of DNA (at 2398 bp).155.87.1 ng divided by 109.5 ng/μL = 1.4μL of Fragment#1 required for CPER reaction. 155.87.2 -
Prepare the CPER reaction by combining 0.1 picomole of each fragment and linker with PrimeSTAR GXL DNA polymerase (Takara Bio) to a total reaction volume of 50 μL.
Example: CPER reaction for MNV. Assuming recovered DNA for each fragment is: Fragment#1 109.5 ng/μL; Fragment#2 86.9 ng/μL; Fragment#3 111.4 ng/μL; Linker Fragment 72.7 ng/μL.MNV Fragment#1 1.4μL MNV Fragment#2 2μL MNV Fragment #3 1.4μL MNV_Linker Fragment 1μL 5X PrimeSTAR GXL Buffer 10μL 10 mM dNTP mix 4μL PrimeSTAR GXL DNA Polymerase 2μL Nuclease-free H2O 28.2 μL Total 50μL Cycling conditions:- 98°C for 30 s,
- 98°C for 10s, 55°C for 30s, for 12 cycles
- 68°C for 10min,
- and a final extension at 68°C for 10 min.
Transfect NIH3T3 cells with MNV CPER reaction and passaging on RAW264.7 cells.
NOTE: Prepare NIH3T3 cells the day before by seeding cells into 6-well plate so that they are approximately 70–80% confluent at the time of transfection. It is important to be gentle with NIH3T3 cells as they are easily dislodged from the flask/plate. It is also possible to use HEK 293T cells instead of NIH3T3.
In one 1.5 mL microfuge tube dilute 5 μL Lipofectamine 3000 in 120 μL Opti-MEM, vortex briefly and allow to sit for 5 minutes.
In a second 1.5 mL microfuge tube combine the entire CPER reaction (50 mL DNA) with 70 μL Opti-MEM and 5 μL of the P3000 reagent provided with the Lipofectamine 3000 to enhance transfection. Mix well.
Add all 125 μL of Lipofectamine 3000 mixture to the 1.5 mL microfuge tube containing CPER DNA and mix well.
Incubate solutions for 15 mins at room temperature while preparing cells.
To prepare cells, gently remove cell culture media and replace with 1000 μL of serum free DMEM.
Carefully tilt 6-well plate containing NIH3T3 cells and add the transfection mix (from step 3) to the cell culture media (from step 5). To ensure even distribution of the cDNA, gently rock the plates before placing them at 37°C with 5% CO2.
After 3 to 5 hours add 800 μL of DMEM supplemented with 4% FCS.
-
For the recovery of MNV the transfected cell supernatant is collected at 72 hours post-transfection (h.p.t.)
NOTE: Prepare RAW264.7 cells the day before by seeding into 100 mm cell culture dish with 10 mls complete media so that they are ~80% confluent at the time of infection.
The collected supernatant is then clarified from cell debris by centrifugation at 500 x g for 5 min. 100 uL of clarified supernatant is stored at −80 °C (for subsequent virus quantitation by plaque assay) while the remaining supernatant (~1900 μL) is immediately passaged onto a 100 mm culture dish seeded to 80% confluency with RAW264.7 cells.
After a 1-hour inoculation of RAW264.7 cells with transfection supernatant, add an additional 10 mL of DMEM supplemented with 2% FCS to each dish.
Monitor cells daily for cytopathic effect and harvest when CPE is observed (~3 days).
Cell supernatant from the passaged RAW264.7 cell cultures was collected at 3 days post infection.
-
Clarify from cell debris by centrifugation at 500 x g for 5 min and store at −80 °C.
Use plaque assay (see Alternate Protocol 3) or TCID50 (see Basic Protocol 5) to confirm the presence of viruses both in the cleared lysates from NIH3T3 and RAW264.7 cells.
Basic Protocol 5: EXPRESSION OF NOROVIRUS NS1–2 IN INSECT CELL SUSPENSION CULTURES USING A RECOMBINANT BACULOVIRUS
Introduction.
The expression of mammalian viral proteins in Escherichia coli is commonly undertaken. However, many viral proteins are difficult to express in E. coli and the issues of solubility and lack of post-translational modification remain. Recombinant baculovirus infection in insect cells is an effective, robust, and widely used alternative. This eukaryotic expression system has been used for norovirus virus-like particle production and is available in a range of commercial options, including the bacmid system and homologous recombination systems for recombinant virus construction. The benefits of expressing proteins through baculovirus includes the availability of the host eukaryote post-translational modification systems, such as phosphorylation, and the ability to transduce mammalian cells at high efficiency in the absence of viral replication. The Baculovirus Expression Vector System (BEVS) is therefore a relevant system to express the potentially phosphorylated HuNV NS1–2 protein for recombinant protein production in insect cells, mammalian cells and to generate isotopically labelled proteins for NMR analysis. Herein, we describe the expression of the NS1–2 protein in insect cell suspension cultures in serum-free medium. Using this system, we and others have also expressed NS5 (VPg), NS6 (Pro), NS7 (Pol), NS6/7 (ProPol) and VP1 from GI, GII and GV noroviruses. The generation of the recombinant virus can be performed using any commercially available baculovirus system, for this protocol the FlashBac Ultra system from Oxford Expression Technologies (Oxford, UK) was utilised.
Note: The NS1–2 coding sequence used in this protocol for recombinant protein production and purification represents amino acids 3–249 of the NS1–2 protein and is therefore devoid of the C-terminal transmembrane region. The construct includes an N-terminal histidine and/or STREP-II tag for protein purification and with or without an NT* fusion protein (Kronqvist et al., 2017) containing an enterokinase cleavage site for removal. Typically, we express and purify 4–6 mg of NS1–2 per 50 ml of cell culture.
Materials
ESF921 insect cell medium (Expression Systems, cat. no. 96–001-01)
Trichoplusia ni (T.ni) insect cells (Expression Systems, cat. no. 94–002F)
0.1% Trypan Blue in PBS pH6.2
Titred NS1–2 expressing recombinant baculovirus. See Current Protocols Unit 5.5 (Irons et al., 2018)
250 ml Schott bottle
Haemocytometer
Innova 42R Incubator shaker, 1.9 cm orbit (New Brunswick Scientific)
50 ml polypropylene tube
Seed T.ni cells into 50 ml of ESF921 medium at a final concentration of 8×105 cells/ml in a 250 ml Schott bottle.
Other types of cell culture vessel can be used. Schott bottles are an easily reusable vessel that provides excellent cell viability and can be scaled down to smaller bottles at a culture volume of 20% of the bottle volume.
Incubate at 27°C overnight with orbital shaking at 125 rpm.
Check the cell count and viability of the cells by mixing 10 μl of cell suspension with 10 μl of 0.1% Trypan Blue and loading onto a haemocytometer.
We recommend a cell density of 1× 106 cells/ml or greater and a cell viability of >98%.
Infect the 50 ml cell suspension culture by pipetting the recombinant baculovirus containing the norovirus NS1–2 gene directly into the cell suspension at a multiplicity of infection (MOI) of 1.0.
Viral titre can be generated by plaque assay or dilution series.
Incubate the infected culture at 27°C for 2–4 days.
Monitor the culture each day and harvest at 50–80% cell viability as determined by trypan blue staining. Optimisation of MOI and harvest time may be required for some proteins.
Collect the cells from the infected culture by centrifugation at 500 g for 5 mins.
The cell pellets can be stored at −80°C for extended periods (years) prior to protein purification.
SUPPORT PROTOCOL 2: Isotope-labeling of the norovirus NS1–2 in insect cells.
This protocol is to facilitate isotopic labelling of recombinant NS1–2 protein. The analysis of proteins by NMR or metabolomic mass spectrometry is benefited from the use of isotopic labels for the protein of interest. Isotopic labelling is well-established for E. coli but less so for the BEVS. This protocol is based upon the method of Sitarska et al. (Sitarska et al., 2015) for Spodoptera frugiperda cells and has been adapted to T.ni cells for labelling with either 15N or 15N/13C. On average, 73% of 14N is replaced with 15N for the NS1–2 protein of human GII.4 norovirus after isotope labelling.
Materials
Basal ΔESF921 (All Amino Acid Deficient modified medium, Expression Systems, cat. no. 96–276-01)
ISOGRO-15N Growth Medium, (Merck, cat. no. 606871)
D-Glucose
L-Tryptophan-15N (10 mg/ml) (Merck, cat. no. 609064)
15N-NH4Cl (300 mg/ml) (Merck, cat. no. 299251)
L-Cycloserine (5 mg/ml) (Merck, cat.no. C1159)
Nalgene 0.22 μM bottle-top filter (Thermofisher Scientific, cat. no. 596–4520)
- 250 ml Schott bottle
- To make 15N-ISOGRO medium, take 200 ml of basal ΔESF921 and add:
- 2 g of 15N-ISOGRO
- 2 g of Glucose
- then stir for 15 mins at room temperature.
-
Add:
- 400 μl of 10 mg/ml 15N-tryptophan
- 167 μl of 300 mg/ml 15N-NH4Cl
- 200 μl of 5 mg/ml L-cycloserine
- then stir for 1 min at room temperature.
15N-NH4Cl is included as a nitrogen source, unlabelled D-Glucose is included as a carbon source.Addition of L-cycloserine inhibits unlabelled-alanine synthesized from the unlabelled-glucose source thus maximizing the incorporation of labelled-alanine from the algal extract. Addition of L-cycloserine is preferential during 13C incorporation but is included in the 15N-labelling protocol. -
Filter sterilise the 15N-ISOGRO medium using a bottle-top 0.22 μM filter into a 250 ml Schott bottle and store at 4°C.The medium is now ready for use in step 10 below.
-
Seed a 50 ml T.ni culture as in Basic Protocol 5 in ESF921 medium.Note that the starting point is to use normal ESF921 medium (or equivalent) to grow the cells.
- Incubate at 27°C for 24 hours shaking at 125 rpm.
- Infect the 50 ml culture with the recombinant baculovirus at an MOI of 2.0 and incubate at 27°C for 16 hours.
-
Check the cell count and viability by haemocytometer as described in the basic protocol.Make sure the cell viability remains above 80% before proceeding. It may be necessary to adjust the MOI for different proteins / recombinant baculoviruses.
- Collect the infected cells by centrifugation at 500 g for 5 mins.
- Remove the supernatant and wash the cells by gently resuspending the cell pellet in 10 ml of basal ΔESF921 medium and recollecting the cells by centrifugation at 500 g for 5 mins.
-
Remove the basal ΔESF921 medium and gently resuspend the cell pellet in 10 ml of 15N-ISOGRO medium.This is the labelled medium made in steps 1–3.
-
Add the resuspended cells to 40 ml of 15N-ISOGRO medium in a 250 ml Schott bottle.It is recommended to check the cells again for viability at this stage, proceed if the viability remains above 90%.
- Incubate the infected culture in 15N-ISOGRO medium at 27°C at 125 rpm and monitor daily until the cell viability drops below 80%, as described in Basic Protocol 5.
- Harvest the infected cell pellet by centrifugation as described in Basic Protocol 5 and store at −80°C.
SUPPORT PROTOCOL 3: PURIFICATION OF THE NOROVIRUS NS1–2 PROTEIN
Introduction.
This protocol describes a small-scale batch purification of the norovirus NS1–2 protein from a 50 ml T. ni suspension culture as described in Basic Protocol 4. While many laboratories will have chromatographic purification systems and these are often the methods of choice, the use of small-scale batch purifications using epitope affinity tags can be used effectively for the extraction of the NS1–2 protein from insect cells.
Materials
Lysis Buffer: 50 mM NaH2PO4.2H2O pH8, 300 mM NaCl, 10% glycerol, 1% Triton X-100.
Wash Buffer I: 50 mM NaH2PO4.2H2O pH8, 300 mM NaCl, 10% glycerol, 0.05% Triton X-100.
Wash Buffer II: 50 mM NaH2PO4.2H2O pH8, 300 mM NaCl, 10% glycerol.
Elution Buffer: 50 mM NaH2PO4.2H2O pH8, 300 mM NaCl, 50 mM biotin.
Complete, EDTA-free Protease Inhibitor Cocktail (Merck, cat. no. 11873580001)
RNAse (10 mg/ml)
DNAse I (10 mg/ml)
Strep-Tactin XT 4Flow high-capacity resin (IBA Lifesciences, cat.no. 2–5030-002)
20 G needle and 5 ml disposable syringe
0.8 μM filter
- 50 ml polypropylene tube
-
Thaw and resuspend a 50 ml T.ni cell pellet from Basic Protocol 4 in lysis buffer containing 1x protease inhibitor (Roche) at approximately 5× 106-1× 107 cells/ml.Note that for some recombinant proteins, such as proteases, the protease inhibitor is omitted.
- Add RNAse and DNase I at a final concentration of 10 μg/ml each and incubate on ice for 10 mins.
- Shear the sample by passing it through a 20 G needle five times.
- Centrifuge the lysate at 14,000 g for 20 mins.
- Collect the supernatant and filter through a 0.8 μM filter.
- Add Strep-Tactin resin (0.5 ml per 20 ml) to the clarified supernatant and incubate for 1 hour at 4°C, ensuring the resin remains in suspension.
- Load the lysate plus Strep-Tactin onto a gravity flow purification column (Pierce) at room temperature to collect the resin.
- Wash the resin with 20 ml of Wash Buffer I, followed by 20 ml of Wash Buffer II.
- Add 0.5 ml of Elution Buffer and collect the eluant.
-
Repeat the elution seven times to collect eight 0.5 ml elution fractions.The eluted protein can be stored at −80°C but avoid repeated freeze-thaw cycles.
-
Analyse the fractions by SDS-PAGE and western blot analysis to confirm expression and purification.A polishing step of size exclusion chromatography can be used if higher purity is desired.
-
SUPPORT PROTOCOL 4: EXPRESSION OF NOROVIRUS NS1–2 IN MAMMALIAN CELLS BY TRANSDUCTION WITH A RECOMBINANT BACULOVIRUS
Introduction.
The baculovirus, Autographa californica nucleopolyhedrovirus, is unable to replicate in mammalian cells but can transduce a wide variety of cell types. Commercial vectors for the use of baculovirus in mammalian cell transduction are available. This protocol is to use a recombinant baculovirus expressing the GII.4 NS1–2 protein under the control of a constitutive CMV mammalian cell promoter to express the NS1–2 protein in HEK293T cells, thereby facilitating the analysis of this protein in a mammalian cell.
Materials
HEK293T mammalian cells (ATCC, cat. no. CRL-3216)
DMEM-10, DMEM Glutamax medium (Thermofisher Scientific, cat. no. 10566–024) supplemented with 10% FBS
Dulbecco’s A (Calcium free) PBS pH7.4
Recombinant baculovirus stock
Sodium Butyrate
25 cm2 tissue culture flask
- Cell Scrapers
- Seed a 25 cm2 flask with 3× 106 HEK293T cells in 5 ml DMEM-10 and incubate overnight at 37°C, 5% CO2.
- Remove medium and wash the cell monolayer with 2 ml of Dulbecco’s A (Calcium free) PBS pH7.4.
-
Dilute recombinant baculovirus inoculum by adding 600 μl of virus to 2.4 ml of Dulbecco’s A (Calcium free) PBS pH 7.4 (1:4 ratio) to dilute the insect cell medium.The baculovirus titre should be typically 108 pfu/ml or higher. The baculovirus can be concentrated by centrifugation to generate a high titre stock if desired. The above method describes an alternative to centrifugation, which involves diluting the insect cell culture medium, which can reduce transduction efficiency (Hsu et al., 2004). Typically, a 1:4 dilution in PBS is sufficient for direct use on mammalian cells without a loss in transduction efficiency.Freshly prepared baculovirus stock is recommended for this protocol. See Current Protocols Unit 5.5 (Irons et al., 2018).
- Add 3 ml of the diluted inoculum to the HEK293T cells.
- Incubate at room temperature for 8 hr to allow the baculovirus to transduce the cells.
- Remove the inoculum and wash the cells once with 2 ml of Dulbecco’s A PBS pH 7.4.
-
Add fresh DMEM-10 containing 5 mM sodium butyrate to the cells and incubate at 37°C for 48 hours.Sodium butyrate has been shown to enhance expression from the baculovirus system in mammalian cells (Condreay et al., 1999; Fung et al., 2016).
- Wash the cells with 5 ml of PBS then detach into 5 ml of fresh PBS using a cell scraper.
- Collect the cells by centrifugation at 300 g for 5 mins, remove the PBS and use immediately or store the pellet at −80°C.
BASIC PROTOCOL 6: INFECTION OF ENTEROIDS IN TRANSWELLⓇ INSERTS WITH MNV
Introduction.
This procedure describes the process of infecting mouse small intestinal enteroids with MNV. This protocol offers an ex vivo infection system to interrogate MNV infection in primary epithelial cells, without any immune, stromal, or mesenchymal cellular contributors. Intestinal tuft cells are the exclusive epithelial cell type which expresses the MNV receptor CD300lf. However, CD300lf is expressed apically on tuft cells, while enteroids grown in 3D culture grow with their apical surface facing inward. When enteroids are polarized on a transwell filter, their apical surface becomes exposed to enable viral entry (Strine et al., 2022). Tuft cells can be robustly infected by both MNVCW3 and MNVCR6 in enteroids in vitro (Strine et al., 2022).
Materials
MNV virus stock
TranswellⓇ inserts (Costar, 6.5 mm insert, 0.4 μm pore polyester membrane, cat. no. 3470)
50% L-WRN Conditioned media (see Supporting protocol 1)
Y27632 dihydrochloride (ROCK inhibitor) (Tocris, cat. no. 1254)
MatrigelⓇ (Corning Basement Membrane Matrix, cat. no. 354234 or 356231)
MatrigelⓇ needs to be thawed on ice and aliquoted for use in small volumes. Aliquots should be stored at −20°C.
1X Phosphate buffered saline (PBS−/−), pH 7.2 (Gibco, cat. no. 20012027)
CostarⓇ 24-well clear TC-treated plates (Corning, cat. no. 3524)
0.05% Trypsin-EDTA (Gibco, cat. no. 25300054)
EDTA, 0.5M solution, pH 8 (AmericanBio, cat. no. AB00502)
Dulbecco’s Modified Eagle Medium (DMEM, Gibco, cat. no. 11965092)
Fetal Bovine Serum (FBS, Corning, cat. no. 35–010-CV)
IntestiCult™ Organoid Growth Medium (Mouse, STEMCELL Technologies, cat. no. 06005)
Recombinant murine IL-4 (Peprotech, cat. no. 214–14)
37°C water bath
37°C/5% CO2 incubator
Table-top centrifuge
15 mL conical centrifuge tube (e.g., Falcon)
Pre-coat the TranswellⓇ inserts
-
1
Place one TranswellⓇ insert per one well of a 24-well plate.
-
2
Add 100 μl of 10% MatrigelⓇ solution in sterile 1X PBS to each TranswellⓇ insert
Collagen IV (Sigma, cat. no. C5533, stock solution: 1 mg/ml) can also be used as a coating reagent at a final concentration of 33 μg/ml diluted in sterile 1X PBS. Incubate at 37°C for at least 90 minutes or overnight at 37°C. Cultrex® RDF Basement Membrane Extract Type 2 (R&D system, cat. no: 3533–005-02) is another alternative and can be used at the same concentration as Matrigel®.
-
3
Incubate at 37°C for at least 20 min.
Dissociate the 3D enteroid cultures
-
4
Dissociate 3D enteroids gently in Matrigel® bubbles (see Support Protocol 7, steps 1–6).
Plan to use 2–3 wells of enteroids per each TranswellⓇ insert in a 24-well plate format.
Usually, mouse enteroids are ready to be used around ~5 days of growth.
-
5
Manually disrupt the enteroids by pipetting up and down to produce a single-cell suspension.
Generating a single-cell suspension is critical for growth and differentiation on the TranswellⓇ insert. Gentle pipetting ~100 times without generating bubbles may be necessary.
-
6
Neutralize the dissociation reaction with approximately 10 mL DMEM-10 (see recipe).
-
7
Pellet the cells at 500 x g for 5 minutes and aspirate the supernatant without disturbing the pellet.
-
8
Resuspend the cells in 50% L-WRN conditioned media with 10 mM Y27632 (see recipe; see Supporting Protocol 1).
Resuspension volume will depend on how many TranswellⓇ inserts will be seeded. Each insert will require 100 μL of cells. Counting the cells at this stage is not necessary but be sure to use 2–3 densely grown wells per/100 μL to ensure proper monolayer formation.
Plate the single-cell suspension on the TranswellⓇinserts
-
9
Aspirate MatrigelⓇ/PBS solution from the pre-incubated TranswellⓇ inserts.
-
10
Add 100 μL of resuspended cells into the upper (top) TranswellⓇ chamber and add 600 μL of 50% L-WRN conditioned media with 10 mM Y27362 to the lower compartment beneath the insert.
-
11
The next day, replace the media in the upper and lower compartments with fresh mouse IntestiCult™ Organoid Growth Medium supplemented with 50 ng/mL rIL-4 (STEMCELL).
Use 100 μL for the upper compartment and 600 μL for the lower compartment. This step is essential to achieve tuft cell differentiation and subsequent MNV infection. Tuft cell differentiation is supported by supplementation with recombinant murine IL-4 at 50 ng/ml in the differentiation media added to the apical and lower chambers.
Differentiating the enteroids on the TranswellⓇinserts
-
12
Replace the media every 1–2 days.
Dense monolayers may result in media becoming yellow/orange but be sure to change the media consistently every 1–2 days even if the media is not yellow/orange yet. Monolayer establishment can also be tracked using an epithelium volt/ohmmeter by measuring transepithelial resistance (TER) (Moon et al., 2014).
-
13
Maintain the culture for 14 days before infection.
Culturing for <14 days is not sufficient for effective tuft cell differentiation or MNV infection.
Infect the enteroids with MNV
-
14
On day 14, infect the enteroids apically (i.e., adding virus to the upper chamber) by adding 100 μL L-WRN conditioned media containing MNVCR6 or MNVCW3 at the desired multiplicity of infection (MOI).
Each TranswellⓇ insert will contain approximately 106 cells/mL at this stage, and MOI can be estimated using this value.
-
15
Incubate the virus on the enteroids for 1 hour at 37°C to facilitate viral binding.
-
16
After 1 hour, wash off any unbound virus using 100 μL pre-warmed 1X PBS(−/−) in the apical chamber.
3 washes of 100 μL may be necessary to remove unbound virus, as it may stick to the plastic of the TranswellⓇ inserts. Washing the basolateral chamber is not necessary. If infections proceed for >1 day, do not change the media during this time.
-
17
After three washes, add fresh L-WRN conditioned media to the apical chamber.
-
18
Harvest the virus by collecting supernatant from the upper chamber of the TranswellⓇ insert at desired timepoints.
Samples can also be harvested from the lower chamber if desired, but maximal viral shedding will be detected from the apical supernatant. Cells can also be collected for qPCR or other analyses via trypsin or TRIzol treatment.
SUPPORT PROTOCOL 5: PREPARATION OF CONDITIONED MEDIA FOR ENTEROID CULTURE
Materials:
L-WRN cells (available from ATCC, CRL-3276)
L-cell medium (see recipe)
G418 (Gibco, cat. no. 10131035)
Hygromycin (InvivoGen, cat. no. ant-hg-5)
Primary culture medium (see recipe)
0.05% Trypsin-EDTA (Gibco, cat. no. 25300054)
1X Phosphate buffered saline (PBS), pH 7.2 (Gibco, cat. no. 20012027)
0.22 μM filter system (Millipore Stericup)
50 ml conical centrifuge tube (e.g., Falcon)
150 cm2 tissue culture treated vented flasks (e.g., Corning)
Table-top centrifuge
37°C/5% CO2 incubator
- Note: This protocol is for small scale preparation and has been adapted from (Miyoshi & Stappenbeck, 2013).
- Grow L-WRN cells in L-cell media (see recipe) at 37°C in a 150 cm2 flask.
- When confluent, trypsinize the cells using 0.05% Trypsin-EDTA and incubate at 37°C until they detach (approx. ~1–2 min).
-
Once detached, neutralize with L-cell medium and re-plate them at approximately 1–2 × 106/flask in 150 cm2 flasks containing 500 μg/mL G418 and 2 mM hygromycin in L-cell medium. Incubate at 37°C.For a small-scale preparation, make ten 150 cm2 flasks from 1 confluent 150 cm2 flask (e.g., split 1:10).
-
When cells are overly confluent (approx. 2–3 days), wash the flasks twice with 20 mL 1X PBS and once with 10 mL primary culture medium (see recipe).Be sure to use primary culture medium here and not the L-cell media used in step 1.
- Add 25 mL fresh primary culture medium to each flask and incubate at 37°C.
-
Every 24 hours, collect the culture medium (“L-WRN conditioned media”). Store these fractions at 4°C.In total, change and collect conditioned media 4 times.
- Pool all of the L-WRN conditioned media fractions and clarify them by centrifugation at 500 x g for 5 minutes. Collect the media, leaving the pelleted cellular debris behind.
- Filter the L-WRN conditioned media through a 0.22 μM filter.
- Dilute the L-WRN conditioned media with an equal volume of fresh primary culture medium, generating 50% L-WRN conditioned medium.
-
Aliquot the 50% L-WRN conditioned medium stocks in 50 ml falcon tubes and store at −80°C until further use.Medium can be thawed in a water bath at 37°C before use. If thawed, aliquots should be used within ~2 weeks and stored at 4°C.
SUPPORT PROTOCOL 6: ISOLATION OF CRYPTS FOR ENTEROID GENERATION
Materials:
Mouse of desired genotype
1X Phosphate buffered saline (PBS−/−), pH 7.2 (Gibco, cat. no. 20012027)
Surgical scissors
Forceps
Wide bore syringe
Crypt culture medium (see recipe)
Collagenase type I (Invitrogen, cat. no. 17100–017)
Gentamicin (Sigma, cat. no. G1397)
MatrigelⓇ (Corning Basement Membrane Matrix, cat. no. 354234 or 356231)
Y27632 dihydrochloride (ROCK inhibitor) (Tocris, cat. no. 1254)
35 mm tissue culture treated dish (e.g., Corning)
100 mm sterile petri dish (e.g., Corning)
24-well plate (e.g., Corning)
40 μM cell strainer (e.g., Fisher)
L-WRN Conditioned media (see recipe)
50 mL conical tube (e.g., Falcon)
15 mL conical tube (e.g., Falcon)
37°C water bath
37°C/5% CO2 incubator
- Table-top centrifuge
- Sacrifice the mouse according to the Institutional Animal Care and Use Committee protocol. Cut open the mouse abdominal cavity and harvest the distal small intestine (~10 cm), gently clearing it of fat and connective tissue (Figure 6).
- Using a wide bore blunt syringe or P1000 pipet tip, flush the tissue with cold 1X PBS to remove any contents.
- Cut the tissue longitudinally and vortex 15 seconds in cold 1X PBS.
-
Mince the tissue and transfer it in a 15 ml conical tube in 1–2 mL crypt culture medium (see recipe) supplemented with 2 mg/ml collagenase type I and 50 μM gentamicin. Incubate at 37°C for 30 minutes.Tissue pieces should be small enough to freely pipet using a wide bore P1000 tip.Pipetting up and down every ~10 minutes can help with crypt dissociation.In subsequent steps, do not supplement the crypt culture medium with collagenase or gentamicin.
- After 30 minutes, filter the isolated crypts through a 40 μM cell strainer into a 50 mL conical tube. Add 10 mL crypt culture medium and centrifuge at 500 x g for 5 minutes.
-
Aspirate the supernatant without disturbing the pellet and add another 10 mL crypt culture medium. Gently resuspend the pellet, transfer to a 15 mL conical tube, and centrifuge at 500 x g for 5 minutes.This second wash step is critical to minimize contamination with mesenchymal cells.
-
Aspirate the supernatant and resuspend the purified crypts in one drop of 30 μL of MatrigelⓇ per well of a 24-well tissue plate.A good isolation of crypts in steps 4–5 from 10 cm of small intestine should yield approx. 6 MatrigelⓇ bubbles worth of purified crypts. Poor isolation will be evidenced by a very small pellet in steps 6–7. Continued trouble after this stage (e.g., crypts are too sparse and do not grow well) can be overcome by tweaking collagenase treatment time to improve crypt release and viability.
- Invert the plate and incubate it for ∼10 min at 37°C to polymerize the MatrigelⓇ.
- Once the MatrigelⓇ is solidified, return the plate to its correct orientation, and add 600 μL 50% L-WRN conditioned media with 10 mM Y27632 per well of a 24-well plate.
Figure 6:
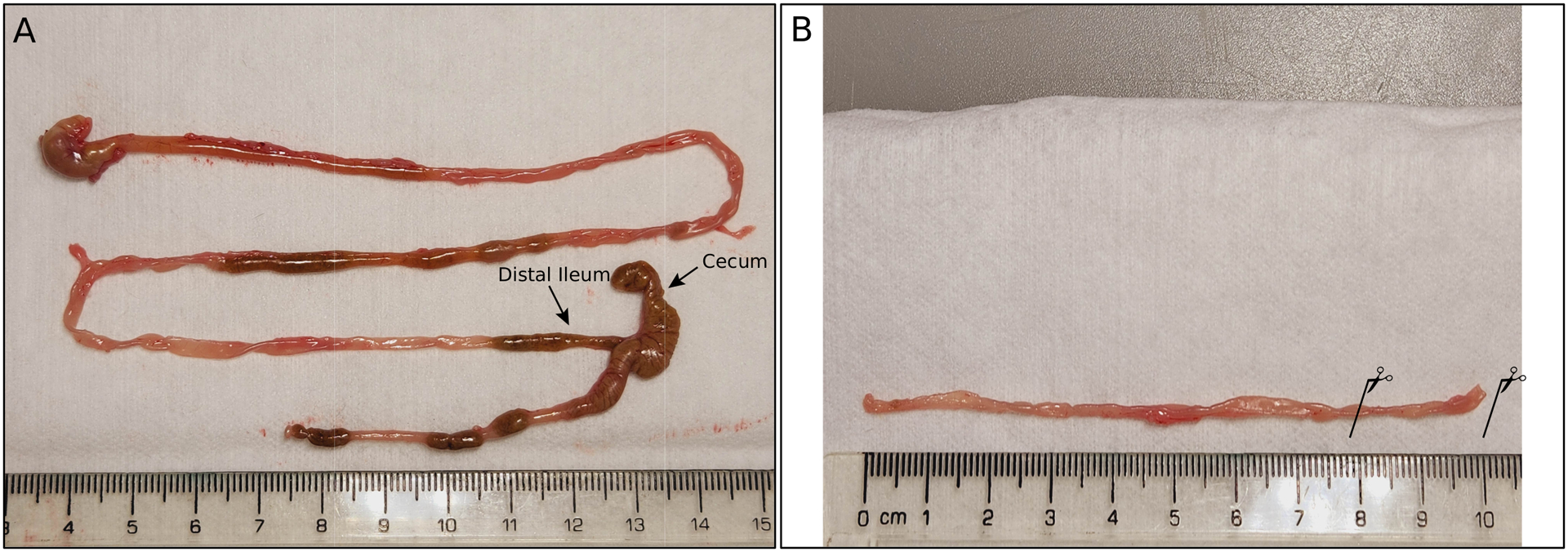
dissection process for crypt isolation. (A) The whole gastrointestinal tract was removed from the animal and displayed to identify the distal ileum. (B) The ileum was cleaned by removing its contents, and the part to be used for crypt isolation was identified (see solid lines with scissors).
SUPPORT PROTOCOL 7: ENTEROID CULTURE PASSAGING AND MAINTENANCE
Materials:
1X Phosphate buffered saline (PBS−/−), pH 7.2 (Gibco, cat. no. 20012027)
PBS-EDTA (see recipe)
0.05% Trypsin-EDTA (Gibco, cat. no. 25300054)
MatrigelⓇ (Corning Basement Membrane Matrix, cat. no. 354234 or 356231)
50% L-WRN Conditioned media (see Supporting Protocol 1Supporting Protocol 1)
DMEM-10 (see recipe)
24-well plate (e.g., Corning)
- 15 ml conical centrifuge tubes (e.g., Falcon)
- Aspirate the culture media from each well.
- Dissociate 3D enteroids gently in MatrigelⓇ bubbles by adding 500 μL PBS-EDTA to each well of the 24-well plate containing MatrigelⓇ enteroid bubbles.
-
Manually scratch and triturate the enteroid bubbles gently by pipetting up and down approximately 5 times.A single cell suspension is not critical at this stage, and pipetting too much at this step may result in cell death and lower viability.
- Pool the enteroids and transfer them to a single 15 mL conical tube and centrifuge at 500 x g for 5 minutes.
-
Aspirate the PBS-EDTA without disturbing the pellet and add 200 μl of pre-warmed (37°C) 0.05% Trypsin-EDTA per 8 dissociated bubbles (e.g., from 8 wells with 30 μL enteroids in one Matrigel bubble in each one).The amount of trypsin will depend on the number of bubbles. Usually, 200 μl trypsin is enough to trypsinize 8 bubbles. The pellet may be loose after centrifugation.
- Incubate the cells in a 37°C water bath for 90 seconds.
-
Manually dissociate the enteroids by gently pipetting up and down.times is typically sufficient for the mouse distal small intestine.
- Neutralize the dissociation reaction with 10 mL DMEM-10.
- Pellet the cells at 500 x g for 5 minutes and aspirate the supernatant without disturbing the pellet.
-
Resuspend the cells in one drop of 30 μL MatrigelⓇ per well of a 24-well plate and grown in 50% L-WRN conditioned media.MatrigelⓇ exists in a viscous liquid form at 4°C and is solid at 37°C. MatrigelⓇ should be placed on ice before use in plating enteroids.Repeat steps 1–10 any time the enteroids need to be passaged. When the enteroids within the growth medium grow too large, they will eventually become clouded with intracellular debris and die. Enteroids processed from the mouse distal small intestine typically require a passage every 3–5 days.
Basic Protocol 7: QUANTIFICATION OF MURINE NOROVIRUS-INDUCED DIARRHEA USING NEONATAL MOUSE INFECTIONS
Introduction.
This procedure outlines the quantification of MNV-induced diarrhea using the neonatal mouse model. We describe the inoculation methodology and disease scoring system. This protocol can be used with neonatal mice 3 – 6 days post birth (P2-P5) with no significant difference in intestinal disease outcomes, but diarrhea severity is reduced in mice infected at P7 or later (Peiper et al., 2023). The oral gavage method has been validated using BALB/c mice (Roth et al., 2020) while the intragastric inoculation method has been validated in both BALB/c (Roth et al., 2020) and C57BL/6J neonatal mice (Peiper et al., 2023). Both inoculation methods can deliver up to 40 μL of volume to the pups. Disease outcomes can be observed 24–72 hours post-infection (hpi). Depending on the application, infection doses of 107 to 108 are useful. Fecal scoring can be performed at multiple time points using the same neonatal mouse, but the mice do not defecate at every timepoint, representing a limitation of this readout. Colon content scores can be used to overcome this limitation, although the mouse must be euthanized to acquire a colon content score (Helm et al., 2022). This model is useful for a wide range of applications including disease kinetics of MNV strains (Peiper et al., 2023; Roth et al., 2020), identification of virulence determinants and host determinants of protection (Helm et al., 2022), and efficacy of small molecules and therapeutics.
Materials
P0-P5 neonatal mice (BALB/c or C57BL/6J)
Phosphate-buffered saline (PBS), pH 7.4, sterile
MNV stock (107 to 108 TCID50 units/mouse recommended)
1.5–2 mL Eppendorf tubes
1 mL insulin syringes (Fisher Scientific, Cat #14955462)
30-G (0.3mm × 25mm) needles (BD, Cat #305128)
22-G × 25 mm plastic feeding tubes (Instech, Cat #FTP-22–25-50)
Oral gavage inoculation of neonatal mice
Note: It is recommended that only BALB/c pups are used with this method since the relatively small size of C57BL/6J mice make them vulnerable to gavage-induced trauma. In our experience, approximately 20% of C57BL/6J pups inoculated at P3-P5 succumb to oral gavage independent of infection, a confounding variable not observed in BALB/c pups.
-
1
Make the desired virus inoculum in Eppendorf tubes in a volume of 10–40 μL per mouse. Additional volume should be made for loss of inoculum in the tip of the syringe.
-
2
Attach 22-G plastic feeding tube to the end of syringe and remove any air bubbles. The same tube can be used between each mouse as long as the tube filter remains clear.
-
3
Insert feeding tube into the mouth of each pup and thread down to the esophagus. Make sure to insert slowly to avoid punctures in the esophagus.
-
4
Deliver up to 40 μL to each mouse.
-
5
Carefully withdraw the tube.
-
6
Observe the mouse for 5 minutes to monitor for any signs of distress before returning it to the cage. If signs of distress such as difficulty breathing or lethargy are observed, euthanize the mouse per IACUC protocol.
Scoring intestinal disease in fecal samples
-
7
At desired time points, palpate the abdomen of each mouse to induce defecation.
-
8
Assess feces based on color and consistency according to a 6-point scale (Figure 7): 0, no defecation; 1, firm, orange, does not smear; 2, pasty, orange, or mixed color, does not smear; 3, orange or yellow, semi-liquid and smears; 4, yellow, liquid, and smears; 5, clear, liquid, and smears.
-
9
Disease severity is reported as the average fecal score per condition, excluding any scores of 0. Disease incidence is reported at the percentage of mice per condition with a fecal score of 3 or higher. For statistical purposes and to control for inter-litter heterogeneity, at least 10 neonates from 3 separate litters should be used per condition tested.
Figure 7:

Scoring scale for fecal samples. Color and consistency are scored on a 6-point scale with 0, no defecation or solid; 1, firm, orange, does not smear; 2, pasty, orange, or mixed color, does not smear; 3, orange or yellow, semi-liquid and smears; 4, yellow, liquid, and smears; 5, clear, liquid, and smears. (Figure reproduced from: (Roth et al., 2020)).
Alternate Protocol 1: Intragastric inoculation of neonatal mice
Note: This method can be used in BALB/c and C57BL/6J mice up to P7. After P7, the milk spot disappears and intragastric delivery is no longer possible.
Make desired inoculum in Eppendorf tubes in a volume of 10–40 μL per mouse. Additional volume should be made for loss of inoculum in the tip of the syringe.
Attach 30-G needle to the end of syringe and remove any air bubbles. The same needle can be used between each mouse if no blood is present on the tip of the needle.
Restrain the neonatal mouse so the milk spot becomes clear and close to the surface. The milk spot in the stomach is visible through the skin on the abdominal wall. It should appear in the upper right quadrant of the abdomen. It is recommended to do inoculations when the milk spot is clearly visible to ensure accurate injections (Figure 8).
Insert the needle parallel to the stomach so that the needle pierces the stomach lengthwise. If the needle is inserted properly, the stomach wall will indent and then bounce back when the stomach is pierced (Figure 8).
Deliver up to 40 μL to each mouse.
Carefully withdraw the needle. Minimal flowback is expected.
Observe the neonate for 5 minutes to monitor for any signs of distress before returning it to the cage. If signs of distress are observed, euthanize the mouse per IACUC protocol.
Figure 8:
Intragastric inoculation of neonatal mice. (A) Restrain neonatal mouse so milk spot becomes visible on abdominal surface in right upper quadrant. (B) Insert needle parallel to stomach so that the needle enters stomach lengthwise.
Alternate protocol 2: Scoring colon contents
At the desired timepoint, euthanize the mouse according to the guidelines of the home institution and IACUC protocol.
Briefly dip the mouse carcass into 70% ethanol. Using forceps and scissors, cut both the skin and peritoneum longitudinally along the midline of the abdomen being careful not to cut the intestines.
Locate the junction of the anus and colon and cut the colon as close to the anus as possible.
Carefully remove the colon without tearing it until you locate the cecum and then cut at the cecum.
Using the scissors in a closed position, squeeze out the contents of the colon by running the closed scissors across the colon.
-
Score the contents according to the scale in Figure 7.
Calculations for disease severity and incidence are calculated in the same manner as fecal scoring (see Basic protocol 7). For statistical purposes and to control for inter-litter heterogeneity, at least 10 neonates from 3 separate litters should be used per condition tested.
REAGENTS AND SOLUTIONS:
15N-ISOGRO medium (in Support Protocol 2):
Basal ΔESF921 (All Amino Acid Deficient modified medium, Expression Systems, cat. no. 96–276-01)
1% (w/v) 15N-ISOGRO Growth medium, (Merck, cat. no. 606871)
56mM D-Glucose
100μM L-Tryptophan-15N (10 mg/ml) (Merck, cat. no. 609064)
5mM 15N-NH4Cl (300 mg/ml) (Merck, cat. no. 299251)
-
50μM L-Cycloserine (5 mg/ml) (Merck, cat.no. C1159)
Filter sterilize using a Nalgene 0.22 μM bottle-top filter (Thermofisher Scientific, cat. no. 596–4520) and store at 4°C.
15N-NH4Cl (in Support Protocol 2):
250mg in 833μl in MQH2O (300 mg/ml stock). Filter sterilize and store at −20°C.
Cell culture medium (in Basic protocol 3):
DMEM Low glucose (Gibco, cat. no. 10567022)
1M HEPES (Gibco, cat. no. 15630080)
100X GlutaMAX (Gibco, cat. no. 35050061)
Foetal bovine serum (Bovogen, cat. no. SFBS)
1% Penicillin/Streptomycin (produced in-house by the Microbiology Preparation Unit)
Crypt culture medium:
DMEM/F12 (Gibco, cat. no. 11320033)
15 mM HEPES (Gibco, cat. no. 15630080)
1X L-Glutamine (Gibco, 25030081)
1X Penicillin/Streptomycin (Gibco, cat. no. 15070063)
10% heat inactivated FBS (Corning, cat. no. 35–010-CV)
DMEM-10:
DMEM (Gibco, cat. no. 11965092)
10% FBS heat inactivated (Corning, cat. no. 35–010-CV)
DNAse I:
10 mg/ml in 20 mM Tris pH7.5, 1 mM MgCl2, 50% glycerol. Store at −20°C.
Dulbecco’s A (Calcium-free) PBS pH7.4:
1x PBS tablet
100ml MQH2O
Filter sterilize and store aliquots at −20°C.
L-cell medium:
High glucose DMEM (Gibco, cat. no. 11965092)
1% Penicillin/Streptomycin (Gibco, cat. no. 15070063)
10% FBS heat inactivated (Corning, cat. no. 35–010-CV)
L-Cycloserine:
10mg in 2ml in MQH2O (5 mg/ml stock). Filter sterilise and store at −20°C.
L-Tryptophan-15N:
20 mg in 2ml in MQH2O (10mg/ml stock). Filter sterilise and store in aliquots at −80°C.
NS1–2 purification buffers:
Base Buffer:
50 mM NaH2PO4.2H2O pH8
300 mM NaCl
Make up to 1L and autoclave.
To make Lysis Buffer add:
Base Buffer
10% glycerol
1% Triton X-100.
To make Wash Buffer I add:
Base Buffer
10% glycerol
0.05% Triton X-100.
To make Wash Buffer II add:
Base Buffer
10% glycerol
To make Elution Buffer add:
Base Buffer
50 mM biotin
-
pH to 8.0 with 10M NaOH
Filter sterilise all solutions and store at 4°C before use. The Elution Buffer can be aliquoted and stored at −20°C.
PBS pH6.2:
1mM Na2HPO4
10.5mM KH2PO4
140mM NaCl
40mM KCl
PBS-EDTA:
1X Phosphate buffered saline (PBS), pH 7.2 (Gibco, cat. no. 20012027)
5 mM EDTA (AmericanBio, cat. no. AB00502)
Primary culture medium:
Advanced DMEM/F-12 (Gibco, cat. no. 12634010)
100X L-glutamine (Gibco, 25030081)
1% Penicillin/Streptomycin (Gibco, cat. no. 15070063)
4% FBS heat inactivated (Corning, cat. no. 35–010-CV)
RNAse:
10 mg/ml in 20 mM Tris pH7.5, 1 mM MgCl2, 50% glycerol. Store at −20°C.
Sodium Butyrate:
5mM Sodium Butyrate
DMEM Glutamax medium (Thermofisher Scientific, cat. no. 10566–024)
10% FBS
Filter sterilise and use immediately.
COMMENTARY:
MNV antiviral efficacy determination:
Large scale screening of putative antivirals against human norovirus remains challenging. Herein, we use MNV as a model to illustrate a laboratory scale system utilizing the measurement of ATP to determine cell health, and the utility of indirect measurements of virus replication to observe unexpected effects that compound carriers such as DMSO can have on norovirus infection. Specifically, we present a method for indirectly assessing cell viability in the presence of MNV and antiviral compounds using CellTiter-Glo (Promega, WI, USA). The regent lyses cells to release ATP which is then converted to a stable luminescent signal proportional to the health of the cells at the time of harvest. The sensitivity and linear range of CellTiter-Glo allow for the detection of subtle changes in the cells or virus in response to an antiviral, that may go undetected with other reagents. The reagent does not require fixing or washing of cells, thereby significantly limiting loss of cells that lift off the plate during infection. This protocol generates both CC50 and EC50 values in technical triplicate on one 96-well plate. This is achieved by seeding plates with an MNV permissible cell line (Haga et al., 2016; Lingemann, 2020; Orchard et al., 2016; Wobus et al., 2004), treating with a dilution series of the antiviral, and infecting with MNV. The plate is incubated for 24 hours, until 90% of virally infected control cells are dead, following this CellTiter-Glo reagent is added. Limitations of using CellTiter-Glo include the potential cost compared to other indirect cell viability reagents. This limitation can be mitigated as the “one-solution” method is simple and quick, includes non-adherent cells, and due to the reagent’s sensitivity could be adapted to a 1536 well plate assay. Another potential limitation of CellTiter-Glo, is that the reagent measures ATP as an indicator of cell metabolism and therefore health. Activation of immune cells and potentially viral infection also have the ability to increase ATP levels within cells. Therefore, before antiviral screening, a viral infection comparing the TCID50 generated using CellTitre-Glo to the titer calculated by plaque assay would need to be performed to confirm there is no to minimal effect between assays. Finally, because the values generated using this method are an indirect measure of viral replication, and potentially some antivirals tested may interact with the reagent itself, positive results should be confirmed using a direct measure of replication.
Baculovirus expression of NS1–2:
Despite progress in the development of cell culture systems for human norovirus (Ettayebi et al., 2016; Ettayebi et al., 2021; Jones et al., 2015; Van Dycke et al., 2019), much is yet to be discovered regarding the function of individual proteins during viral replication. Some norovirus proteins remain challenging to express and analyse, therefore a potent eukaryotic system is needed to further our understanding of virus-host interactions and to advance antiviral drug discovery. Norovirus NS1–2, the N-terminal non-structural protein present in ORF1, contains both an inherently disordered region (IDR) and a transmembrane domain. Much is still to be determined regarding the function of this protein, posttranslational modifications, and its potential cleavage products. Recombinant baculovirus infection in insect cells is an effective, robust, and widely used eukaryotic expression system that has been used for norovirus virus-like particle production and is available in a range of commercial options. The benefits of expressing proteins through baculovirus includes the availability of the host eukaryote post-translational modification systems, such as phosphorylation, and the ability to transduce mammalian cells at high efficiency in the absence of viral replication.
MNV infection of intestinal enteroids:
MNV was initially shown to infect immune cells in cell culture (Wobus et al., 2004). While the acute MNV-1 strain (and MNV-1 clone MNV.CW3) also infects immune cells in vivo (Grau et al., 2017; Van Winkle et al., 2018), persistent MNV strains, including MNV.CR6, infect intestinal tuft cells, a feature critical for virus persistence (Strine et al., 2022; Wilen et al., 2018). Tuft cells are rare epithelial cells located in the gut and lung that exhibit functions akin to those of immune cells. They mediate important interkingdom interactions between the host and helminths, protists, viruses, and bacteria and have diverse and critical functions in immunity, inflammation, and tumorigenesis (Strine & Wilen, 2022). To study the interaction of MNV with tuft cells, we have turned to intestinal enteroids, non-transformed biopsy-derived small intestinal organoids. Murine enteroids recapitulate the mouse intestinal epithelium but lack hematopoietic and stromal compartments. To generate an in vitro enteroid culture system that supports tuft cell development, intestinal stem cells are isolated from the small intestinal epithelium of adult mice, propagated, and differentiated in the presence of recombinant interleukin-4 to stimulate tuft cell expansion. Since the viral receptor CD300lf is expressed on the apical surface of tuft cells, which are facing inward in the three-dimensional (3D) enteroid structure, the receptor is made accessible by plating enteroids-derived cells in 2D and polarizing the monolayer in transwells. This in vitro system supports MNV infection of tuft cells from the apical surface. Contrary to in vivo infections, both acute and persistent strains of MNV similarly infect tuft cells in this culture model (Strine et al., 2022). This model enables the study of MNV interaction with murine tuft cells.
Neonatal infections with MNV:
Human noroviruses are a leading cause of severe childhood diarrhea worldwide. However, very little is known about the pathogenic mechanisms underlying norovirus diarrhea, principally because of the lack of tractable small animal models. Mice are a versatile small animal model for MNV and the MNV model has greatly facilitated progress in understanding host-norovirus interactions and norovirus strain variability (Karst & Wobus, 2015; Wobus et al., 2006). However, the model remains limited because immunocompetent adult mice do not develop diarrhea upon MNV infection, and mice lack the emetic reflex. While diarrhea was observed in MNV-infected interferon-deficient adult mice, these hosts also develop systemic pathology and lethality, confounding translatability of findings to human norovirus disease. Thus, the mechanistic basis of norovirus diarrhea has remained unclear. Recently, we developed a norovirus small animal disease model in wild-type mice by demonstrating that MNV-1 causes self-resolving diarrhea in neonatal C57Bl6 mice (Roth et al., 2020). We have now extended these studies to Balb/c mice and another MNV strain, WU23, that was isolated from a diarrhetic mouse (Peiper et al., 2023). These models mirror key features of human norovirus disease, such as acute self-resolving diarrhea and weight loss, and thus enable studies to understand the mechanism of norovirus diarrhea.
Critical Parameters:
MNV antiviral efficacy determination:
The health and quality of the cells (passage number below 20 for RAW264.7 cells) plated into the 96-well plate will determine the reproducibility of results.
It is important to know the DMSO sensitivity of the cells. Different cell lines have differing sensitivities and this affects the interpretation of results.
Be careful to avoid ATP contamination.
MNV control infection should show >90% death.
MNV qRT-PCR protocol:
Follow the critical parameters previously outlined (Hwang et al., 2014), additionally:
All plastic consumables including tips and mixing tubes should be RNase-DNase-free for all steps of this protocol.
Using commercial DNAse and RNase free water dedicated for primer and probe resuspension is highly recommended.
Setting up separate, dedicated areas for different processes in the qPCR workflow, e.g., RNA extraction, standard preparation, master-mix preparation with its own set of pipettors, tips, and microcentrifuge tubes helps to avoid cross-contamination.
Never use autoclaved materials, such as filter-tips or tubes, as the autoclave can be a source for nucleic acid contamination.
The DNA and RNA PCR standard should be aliquoted in single-use aliquots and stored accordingly.
When pipetting the qPCR plates, it is recommended to pipette the samples first, then the standard (from the lowest concentration to the highest) and finally the negative controls.
MNV reverse genetics protocol:
Cells used should be in a healthy growing state and with a reasonable passage number.
Work swiftly if possible and keep the RNA on ice at all times.
To verify successful transfection, the RNA concentration should be determined. However, this cannot be readily be measured by NanoDrop spectrophotometer after the capping reaction without an extra purification step. It is therefore recommended to determine the RNA concentration after purification and estimate the RNA concentration after capping based on the RNA-control gel.
If the RNA control gel shows a smear due to partial digestion it is better to start over as it will have great impact on viral yields
MNV CPER protocol:
-
The design and quality of the primers are one of the key issues for the CPER reaction.
Ensure that there is at least a 25–35 bp overlap with the primers
that the annealing temperatures are close to equal
Ensure a high-fidelity DNA polymerase is used
We found that the CMV promoter worked in the linker and the use of EF1a did not result in the recovery of infectious virus
All cells used for transfection should be highly transfectable (i.e., NIH3T3 or HEK 293T) and in a healthy growing state
We have found that for virus amplification after transfer of the transfection supernatant that the RAW264.7 cells should be a of a low passage number i.e., no greater than 20 passages.
Baculovirus expression of NS1–2:
-
The overall quality of the T.ni insect cells will determine the success of the baculovirus infection and the production of high-quality protein.
Routinely count cells and check viability during cell maintenance
Keep cell viability above 90%, discard cells if they drop below this
To maintain exponential growth of T.ni cells and to prevent clumping when they reach a high density, split cells every 2 days at 8 ×105 cells/ml. Cells can be split every 3 days at 5 ×105 cells/ml but this can increase the likelihood of cell aggregation.
-
To generate good quality protein, a time course needs to be performed prior to expression to determine the best time to harvest for:
when the most protein is expressed
before the state of the infected cells begins to affect protein quality
To reduce viscosity of the cell lysate, include RNAse and DNAse, shear with a 20G needle and filter through a 0.8 μM filter, this will prevent the solution from blocking on the gravity flow column.
MNV infection of intestinal enteroids:
Generating a single cell suspension prior to plating on the transwell is essential for an optimal monolayer.
A second wash step during crypt isolation is important to remove mesenchymal cells.
Differentiation with 50 ng/ml rIL-4 is critical for tuft cell differentiation and infection.
General tips:
-Pre-wet (using media or FBS) pipette tips before manipulating intestinal pieces or crypts to prevent them from sticking to the wall of the pipettes.
-Make sure to remove the mesentery/membrane around the intestine before processing as it may be difficult to spin it out later in the process.
-Use gentle dissociation reagents to avoid too much damage to the intestine.
- When incubating with the dissociation media to generate the single cell suspension, the next steps need to be done as quickly as possible, as the cells will start to clump together.
-Usually, monolayers (transwell) can reach 100% confluency within ~3 days, but if the attachment is poor, this may take longer. Monolayers with good attachments can last at least 3 weeks, with proper maintenance (i.e., change of media every 2 days).
Neonatal infections with MNV:
One of the most important factors for observing intestinal disease is the age of the mice at initial infection. MNV intestinal disease is highly age restricted with ~80% incidence of disease at 48hpi when mice are infected at P3-P5 with incidence dropping to ~30% by P7 (Peiper et al., 2023). The infection dose is another major determinant in the development of intestinal disease; 80% mice infected with 1×107 TCID50 units of MNV develop intestinal disease at 48 hpi, but only 40% of mice develop disease when infected with a one log lower dose (Roth et al., 2020).
Time Considerations:
MNV antiviral efficacy determination:
The whole assay will take two days. Allow ~1 ½ hours for setting up inhibitor dilutions and addition of virus. Harvesting of the plate takes ~30 minutes with another 30 minutes to analyse the data.
MNV qRT-PCR protocol:
Time for the RNA extraction depends on the number of samples and the method used. For a TRIzol™ extraction using the column-based spin protocol, this can be done within 30 min for a set ca. 10 samples. Preparation of the standard dilutions and aliquoting takes about 1 hour and lasts for many qRT-PCR runs. Pipetting the qRT-PCR requires about 30 min for a 96-well plate if the workspace is setup. The qRT-PCR run takes about 1 hour 15 min depending on the cycler.
MNV reverse genetics protocol:
Plasmid linearization and purification takes approximately 3 hours. This can be prepared in advance and stored at −20 °C (day 1). The generation of in vitro transcribed RNA, purification, and capping takes ca. 5 hours (day 2), storage at −80° C. The initial transfection of Huh7mCD300lf cells takes about one hour but requires a medium change after 4 hours (day 3). Amplification of MNV from transfection of Huh7mCD300lf cells takes another 48 hours for recombinant wild type and maybe 96 hours for viruses with mutations leading to reduced fitness.
MNV CPER protocol:
The PCR amplification of the linker and fragments will take ~2 ½ hours and an additional 1 hour for purification and gel analysis. The CPER reaction itself takes ~90 mins and the transfection ~30 mins. The initial transfection of NIH3T3 cells occurs for 3 days and the amplification of MNV in RAW264.7 cells takes another 48–72 hours.
Baculovirus expression of NS1–2:
The baculovirus protein expression in insect cells will generally take two to three days, determined by a prior time-course. The harvest and subsequent protein purification can take ~3 hours.
MNV infection of intestinal enteroids:
Crypt isolation takes several hours depending on how many mice/genetic backgrounds are being used. Enteroid passaging requires several hours every 3–5 days depending on how dense the cells are seeded and the growth rate. Virus infection itself is less than one hour.
Neonatal infections with MNV:
The time commitment for infecting neonatal mice depends on the number of pups to be infected but takes ~1 hour. The total time for the experiment depends on the experimental design but is typically 3 days. The kinetics of intestinal disease are determined by the viral dose at initial infection. With an infection of 1×107 TCID50 units of MNV, disease starts to develop by 24 hpi. Peak intestinal disease is observed at 48 hpi with a disease plateauing at 72 hpi and fully resolving within 96 hpi.
Troubleshooting:
Table 5:
MNV antiviral efficacy determination
| Problem | Possible Cause | Solution |
|---|---|---|
| High luminescent reading for all wells. | Bacterial contamination of CellTiter Glo. | Use a fresh aliquot stored at −20°C. |
| Luminescence signal of MNV infected and cells-only controls are similar. | MNV infection of cells has not progressed enough. | Extend the timecourse of infection. |
Table 6:
MNV reverse genetics protocol
| Problem | Possible Cause | Solution |
|---|---|---|
| Purified linearized DNA shows lower band. | DNA degradation, possibly due to high temperature and/or poor handling | If only very faint, typically can be ignored as this should not affect IVT. If prominent, perform linearization and purification again working faster and ensuring DNA is kept cool. |
| RNA gel shows smears in bands. | RNA degradation | If capped product and/or others are smeared, clean the electrophoresis chamber with a solution containing NaOH and run another gel. If capped end product is as expected, continue with transfection. |
Table 7:
MNV CPER protocol
| Problem | Possible Cause | Solution |
|---|---|---|
| No infectious virus is recovered. | Primers do not amplify fragments. | Check that the overlapping areas of the primers have the same melting temperature. We suggest ~55°C. |
| No infectious virus is recovered. | Purity of DNA fragments is low. | Re-purify the fragments after gel extraction until 260/280 and 260/230 ratios are optimal for DNA. |
| No infectious virus is recovered. | CPER ligation did not occur. | Check the CPER ligation reaction by gel electrophoresis. You should see the complete ligated product. |
| No infectious virus is recovered. | CPER ligation did not occur. | Transfect immediately after the CPER reaction is complete, i.e. do not freeze CPER reaction (it is ok to freeze the fragments though). |
| No CPE after 3 days. | Tranfection efficiency is low. | Keep monitoring the transfected cells for several additional days (up to 7 days) to allow for virus to spread through culture. |
Table 8:
Baculovirus expression of NS1–2
| Problem | Possible Cause | Solution |
|---|---|---|
| No protein expressed. | Titer of baculovirus too low. | Plaque assay inoculum to determine
titer. Re-amplify the inoculum through Sf21 insect cells. |
| Multiple NS1–2 protein bands detected after purification. | Baculovirus infection allowed to progress for too long or too high an m.o.i. was used. | Optimise the point of harvest by time
course. Determine cell viability before harvest, optimum viability at harvest should be approximately 50–70%. |
Table 9:
MNV infection of intestinal enteroids
| Problem | Possible cause | Solution |
|---|---|---|
| Poor recovery of enteroids from mice | Sub-optimal crypt dissociation. If cells are not treated long enough with collagenase, the crypts will not dissociate, and if cells are treated too long, cell viability will be poor. | Examine the cells under the microscope every 10 minutes during the 30 min dissociation period to ensure optimal dissociation. Adjust incubation time accordingly. |
| Inadequate monolayer on transwells | Transwell was not properly coated or plated with single-cell suspension | Pre-coat entire transwell surface evenly; pre-wet pipette tips, ensure cells are in a single cell suspension before adding to transwell; use lobular rather than spheroidal enteroids to create single cell suspension |
Table 10:
Neonatal infections with MNV
| Problem | Possible Cause | Solution |
|---|---|---|
| The neonatal mice do not defecate when abdomen is palpated. | The neonatal mice are not eating as they are too ill to eat. | Check the weights of the mice. If the weights are decreasing, virus dose likely needs to be decreased. |
| The neonatal mice do not defecate when abdomen is palpated. | The neonatal mouse has defecated recently, and colon is empty or the stool is too proximal in the colon. | Each mouse can be palpated up to 3 times in order to induce defecation. After 3 attempts, the mouse needs to be returned to the dam, and palpation can be repeated in 1–2 hours. |
ACKNOWLEDGMENTS:
C.E.W. was supported by NIH R21 AI 154647, S.M.K. by NIH R01AI162970 and R01AI141478, C.B.W. by NIH R01 AI148467, A.M.P. by NIH T32AI007110, and E.W.H. by NIH F30AI154834. Research in the lab of V.W. was funded by the New Zealand Ministry for Business Innovation and Employment, grant number UOOX1904. J.M.M. was funded by the National Health and Medical Research Council of Australia, grant number 1123135, and the Miller Foundation.
CONFLICT OF INTEREST STATEMENT:
The funding sources of all authors had no conflicting interests with the protocols and research described within, nor did they have a role in protocol design and the collection, analysis, and interpretation of data.
Appendix
Sequence of MNV Linker fragment
GACATTGATTATTGACTAGTTATTAATAGTAATCAATTACGGGGTCATTAGTTCATAGCCCATATATGGAGTTCCGCGTTACATAACTTACGGTAAATGGCCCGCCTGGCTGACCGCCCAACGACCCCCGCCCATTGACGTCAATAATGACGTATGTTCCCATAGTAACGCCAATAGGGACTTTCCATTGACGTCAATGGGTGGAGTATTTACGGTAAACTGCCCACTTGGCAGTACATCAAGTGTATCATATGCCAAGTCCGCCCCCTATTGACGTCAATGACGGTAAATGGCCCGCCTGGCATTATGCCCAGTACATGACCTTACGGGACTTTCCTACTTGGCAGTACATCTACGTATTAGTCATCGCTATTACCATGGTGATGCGGTTTTGGCAGTACACCAATGGGCGTGGATAGCGGTTTGACTCACGGGGATTTCCAAGTCTCCACCCCATTGACGTCAATGGGAGTTTGTTTTGGCACCAAAATCAACGGGACTTTCCAAAATGTCGTAACAACTGCGATCGCCCGCCCCGTTGACGCAAATGGGCGGTAGGCGTGTACGGTGGGAGGTCTATATAAGCAGAGCTCGTTTAGTGAACCGGTGAAATGAGGATGGCAACGCCATCTTCgaaaccttttgtgaaaatgatttctgcttactgctttcttttctttgtggtagttagatgcattttAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGGGTCGGCATGGCATCTCCACCTCCTCGCGGTCCGACCTGGGCATCCGAAGGAGGACGTCGTCCACTCGGATGGCTAAGGGAGAGCCACTTTTCTCTCGATTCTCTATCGGAATCTAGGGAGCTCGGATCCAGACATGATAAGATACATTGATGAGTTTGGACAAACCACAACTAGAATGCAGTGAAAAAAATGCTTTATTTGTGAAATTTGTGATGCTATTGCTTTATTTGTAACCATTATAAGCTGCAATAAACAAGTTAACAACAACAATTGCTCGAGGGGGGGCCCGGTACCTTGAAGCTGTCCCTGATGGTCGTCATCTACCTGCCTGGACAGCATGGCCTGCAACGCGGGCATCCCGATGCCGCCGGAAGCGAGAAGAATCATAATGGGGAAGGCCATCCAGCCTCGCGTCGGCGCTTAA
LITERATURE CITED:
- Amarilla AA, Sng JDJ, Parry R, Deerain JM, Potter JR, Setoh YX, Rawle DJ, Le TT, Modhiran N, Wang X, Peng NYG, Torres FJ, Pyke A, Harrison JJ, Freney ME, Liang B, McMillan CLD, Cheung STM, Guevara D, Hardy JM, Bettington M, Muller DA, Coulibaly F, Moore F, Hall RA, Young PR, Mackenzie JM, Hobson-Peters J, Suhrbier A, Watterson D, & Khromykh AA (2021). A versatile reverse genetics platform for SARS-CoV-2 and other positive-strand RNA viruses. Nat Commun, 12(1), 3431. 10.1038/s41467-021-23779-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias A, Urena L, Thorne L, Yunus MA, & Goodfellow I (2012). Reverse genetics mediated recovery of infectious murine norovirus. J Vis Exp(64). 10.3791/4145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condreay JP, Witherspoon SM, Clay WC, & Kost TA (1999). Transient and stable gene expression in mammalian cells transduced with a recombinant baculovirus vector. Proc Natl Acad Sci U S A, 96(1), 127–132. 10.1073/pnas.96.1.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duizer E, Schwab KJ, Neill FH, Atmar RL, Koopmans MP, & Estes MK (2004). Laboratory efforts to cultivate noroviruses. J Gen Virol, 85(Pt 1), 79–87. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=14718622 [DOI] [PubMed] [Google Scholar]
- Estes MK, Ettayebi K, Tenge VR, Murakami K, Karandikar U, Lin SC, Ayyar BV, Cortes-Penfield NW, Haga K, Neill FH, Opekun AR, Broughman JR, Zeng XL, Blutt SE, Crawford SE, Ramani S, Graham DY, & Atmar RL (2019). Human Norovirus Cultivation in Nontransformed Stem Cell-Derived Human Intestinal Enteroid Cultures: Success and Challenges. Viruses, 11(7). 10.3390/v11070638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettayebi K, Crawford SE, Murakami K, Broughman JR, Karandikar U, Tenge VR, Neill FH, Blutt SE, Zeng XL, Qu L, Kou B, Opekun AR, Burrin D, Graham DY, Ramani S, Atmar RL, & Estes MK (2016). Replication of human noroviruses in stem cell-derived human enteroids. Science, 353(6306), 1387–1393. 10.1126/science.aaf5211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettayebi K, Tenge VR, Cortes-Penfield NW, Crawford SE, Neill FH, Zeng XL, Yu X, Ayyar BV, Burrin D, Ramani S, Atmar RL, & Estes MK (2021). New Insights and Enhanced Human Norovirus Cultivation in Human Intestinal Enteroids. mSphere, 6(1). 10.1128/mSphere.01136-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas T, Fey B, Keller G, Martella V, & Egyed L (2012). Molecular detection of murine noroviruses in laboratory and wild mice. Vet Microbiol, 160(3–4), 463–467. 10.1016/j.vetmic.2012.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung KL, Kapoor K, Pixley JN, Talbert DJ, Kwit AD, Ambudkar SV, & Gottesman MM (2016). Using the BacMam Baculovirus System to Study Expression and Function of Recombinant Efflux Drug Transporters in Polarized Epithelial Cell Monolayers. Drug Metab Dispos, 44(2), 180–188. 10.1124/dmd.115.066506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Hernandez MB, Liu T, Payne HC, Stencel-Baerenwald JE, Ikizler M, Yagita H, Dermody TS, Williams IR, & Wobus CE (2014). Efficient norovirus and reovirus replication in the mouse intestine requires microfold (M) cells. J Virol, 88(12), 6934–6943. 10.1128/JVI.00204-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau KR, Roth AN, Zhu S, Hernandez A, Colliou N, DiVita BB, Philip DT, Riffe C, Giasson B, Wallet SM, Mohamadzadeh M, & Karst SM (2017). The major targets of acute norovirus infection are immune cells in the gut-associated lymphoid tissue. Nat Microbiol, 2(12), 1586–1591. 10.1038/s41564-017-0057-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga K, Fujimoto A, Takai-Todaka R, Miki M, Doan YH, Murakami K, Yokoyama M, Murata K, Nakanishi A, & Katayama K (2016). Functional receptor molecules CD300lf and CD300ld within the CD300 family enable murine noroviruses to infect cells. Proc Natl Acad Sci U S A, 113(41), E6248–E6255. 10.1073/pnas.1605575113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm EW, Peiper AM, Phillips M, Williams CG, Sherman MB, Kelley T, Smith HQ, Jacobs SO, Shah D, Tatum SM, Iyer N, Grodzki M, Morales Aparicio JC, Kennedy EA, Manzi MS, Baldridge MT, Smith TJ, & Karst SM (2022). Environmentally-triggered contraction of the norovirus virion determines diarrheagenic potential. Front Immunol, 13, 1043746. 10.3389/fimmu.2022.1043746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CS, Ho YC, Wang KC, & Hu YC (2004). Investigation of optimal transduction conditions for baculovirus-mediated gene delivery into mammalian cells. Biotechnol Bioeng, 88(1), 42–51. 10.1002/bit.20213 [DOI] [PubMed] [Google Scholar]
- Hwang S, Alhatlani B, Arias A, Caddy SL, Christodoulou C, Cunha JB, Emmott E, Gonzalez-Hernandez M, Kolawole A, Lu J, Rippinger C, Sorgeloos F, Thorne L, Vashist S, Goodfellow I, & Wobus CE (2014). Murine norovirus: propagation, quantification, and genetic manipulation. Curr Protoc Microbiol, 33, 15K 12 11–61. 10.1002/9780471729259.mc15k02s33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irons SL, Chambers AC, Lissina O, King LA, & Possee RD (2018). Protein Production Using the Baculovirus Expression System. Curr Protoc Protein Sci, 91, 5 5 1–5 5 22. 10.1002/cpps.45 [DOI] [PubMed] [Google Scholar]
- Jones MK, Grau KR, Costantini V, Kolawole AO, de Graaf M, Freiden P, Graves CL, Koopmans M, Wallet SM, Tibbetts SA, Schultz-Cherry S, Wobus CE, Vinje J, & Karst SM (2015). Human norovirus culture in B cells. Nat Protoc, 10(12), 1939–1947. 10.1038/nprot.2015.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst SM, & Wobus CE (2015). Viruses in Rodent Colonies: Lessons Learned from Murine Noroviruses. Annu Rev Virol, 2(1), 525–548. 10.1146/annurev-virology-100114-055204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst SM, Wobus CE, Lay M, Davidson J, & Virgin IV HW (2003). STAT1-dependent innate immunity to a Norwalk-like virus. Science, 299(5612), 1575–1578. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12624267 [DOI] [PubMed] [Google Scholar]
- Kronqvist N, Sarr M, Lindqvist A, Nordling K, Otikovs M, Venturi L, Pioselli B, Purhonen P, Landreh M, Biverstal H, Toleikis Z, Sjoberg L, Robinson CV, Pelizzi N, Jornvall H, Hebert H, Jaudzems K, Curstedt T, Rising A, & Johansson J (2017). Efficient protein production inspired by how spiders make silk. Nat Commun, 8, 15504. 10.1038/ncomms15504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon M, & Firestein BL (2013). DNA transfection: calcium phosphate method. Methods Mol Biol, 1018, 107–110. 10.1007/978-1-62703-444-9_10 [DOI] [PubMed] [Google Scholar]
- Lei C, Yang J, Hu J, & Sun X (2021). On the Calculation of TCID(50) for Quantitation of Virus Infectivity. Virol Sin, 36(1), 141–144. 10.1007/s12250-020-00230-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingemann M (2020). New Tools to Study Murine Norovirus-Host Interactions. University of Lubeck; ]. Lubeck, Germany. [Google Scholar]
- Liu H, & Naismith JH (2008). An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol, 8, 91. 10.1186/1472-6750-8-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, & Stappenbeck TS (2013). In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat Protoc, 8(12), 2471–2482. 10.1038/nprot.2013.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon C, VanDussen K, Miyoshi H, & Stappenbeck T (2014). Development of a primary mouse intestinal epithelial cell monolayer culture system to evaluate factors that modulate IgA transcytosis. Mucosal Immunology, 7(4). 10.1038/mi.2013.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nice TJ, Strong DW, McCune BT, Pohl CS, & Virgin HW (2013). A single-amino-acid change in murine norovirus NS1/2 is sufficient for colonic tropism and persistence. J Virol, 87(1), 327–334. 10.1128/JVI.01864-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard RC, Wilen CB, Doench JG, Baldridge MT, McCune BT, Lee YC, Lee S, Pruett-Miller SM, Nelson CA, Fremont DH, & Virgin HW (2016). Discovery of a proteinaceous cellular receptor for a norovirus. Science, 353(6302), 933–936. 10.1126/science.aaf1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiper AM, Helm EW, Nguyen Q, Phillips M, Williams CG, Shah D, Tatum S, Iyer N, Grodzki M, Eurell LB, Nasir A, Baldridge MT, & Karst SM (2023). Infection of neonatal mice with the murine norovirus strain WU23 is a robust model to study norovirus pathogenesis. Lab Anim (NY), 52(6), 119–129. 10.1038/s41684-023-01166-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth AN, Helm EW, Mirabelli C, Kirsche E, Smith JC, Eurell LB, Ghosh S, Altan-Bonnet N, Wobus CE, & Karst SM (2020). Norovirus infection causes acute self-resolving diarrhea in wild-type neonatal mice. Nat Commun, 11(1), 2968. 10.1038/s41467-020-16798-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitarska A, Skora L, Klopp J, Roest S, Fernandez C, Shrestha B, & Gossert AD (2015). Affordable uniform isotope labeling with (2)H, (13)C and (15)N in insect cells. J Biomol NMR, 62(2), 191–197. 10.1007/s10858-015-9935-6 [DOI] [PubMed] [Google Scholar]
- Smith DB, McFadden N, Blundell RJ, Meredith A, & Simmonds P (2012). Diversity of murine norovirus in wild-rodent populations: species-specific associations suggest an ancient divergence. J Gen Virol, 93(Pt 2), 259–266. 10.1099/vir.0.036392-0 [DOI] [PubMed] [Google Scholar]
- Strine MS, Alfajaro MM, Graziano VR, Song J, Hsieh LL, Hill R, Guo J, VanDussen KL, Orchard RC, Baldridge MT, Lee S, & Wilen CB (2022). Tuft-cell-intrinsic and -extrinsic mediators of norovirus tropism regulate viral immunity. Cell Rep, 41(6), 111593. 10.1016/j.celrep.2022.111593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strine MS, & Wilen CB (2022). Tuft cells are key mediators of interkingdom interactions at mucosal barrier surfaces. PLoS Pathog, 18(3), e1010318. 10.1371/journal.ppat.1010318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube S, Perry JW, Yetming K, Patel SP, Auble H, Shu L, Nawar HF, Lee CH, Connell TD, Shayman JA, & Wobus CE (2009). Ganglioside-linked terminal sialic acid moieties on murine macrophages function as attachment receptors for murine noroviruses [10.1128/JVI.02245–08]. J Virol, 83(9), 4092–4101. 10.1128/JVI.02245-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackray LB, Wobus CE, Chachu KA, Liu B, Alegre ER, Henderson KS, Kelley ST, & Virgin IV HW (2007). Murine noroviruses comprising a single genogroup exhibit biological diversity despite limited sequence divergence. J Virol, 81(19), 10460–10473. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17652401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dycke J, Ny A, Conceicao-Neto N, Maes J, Hosmillo M, Cuvry A, Goodfellow I, Nogueira TC, Verbeken E, Matthijnssens J, de Witte P, Neyts J, & Rocha-Pereira J (2019). A robust human norovirus replication model in zebrafish larvae. PLoS Pathog, 15(9), e1008009. 10.1371/journal.ppat.1008009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Winkle JA, Robinson BA, Peters AM, Li L, Nouboussi RV, Mack M, & Nice TJ (2018). Persistence of Systemic Murine Norovirus Is Maintained by Inflammatory Recruitment of Susceptible Myeloid Cells. Cell Host Microbe, 24(5), 665–676 e664. 10.1016/j.chom.2018.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward VK, McCormick CJ, Clarke IN, Salim O, Wobus CE, Thackray LB, Virgin H. W. t., & Lambden PR (2007). Recovery of infectious murine norovirus using pol II-driven expression of full-length cDNA. Proc Natl Acad Sci U S A, 104(26), 11050–11055. 10.1073/pnas.0700336104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilen CB, Lee S, Hsieh LL, Orchard RC, Desai C, Hykes BL Jr., McAllaster MR, Balce DR, Feehley T, Brestoff JR, Hickey CA, Yokoyama CC, Wang YT, MacDuff DA, Kreamalmayer D, Howitt MR, Neil JA, Cadwell K, Allen PM, Handley SA, van Lookeren Campagne M, Baldridge MT, & Virgin HW (2018). Tropism for tuft cells determines immune promotion of norovirus pathogenesis. Science, 360(6385), 204–208. 10.1126/science.aar3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wobus CE, Green KY (2020). Caliciviridae: The Viruses and Their Replication. In Howley D. M. K. Peter M., Whelan Sean (Ed.), Fields Virology: Emerging Viruses (7 ed., Vol. 1, pp. 129–169). Wolters Kluwer. [Google Scholar]
- Wobus CE, Karst SM, Thackray LB, Chang KO, Sosnovtsev SV, Belliot G, Krug A, Mackenzie JM, Green KY, & Virgin HW (2004). Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol, 2(12), e432. 10.1371/journal.pbio.0020432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wobus CE, Thackray LB, & Virgin H. W. t. (2006). Murine norovirus: a model system to study norovirus biology and pathogenesis. J Virol, 80(11), 5104–5112. 10.1128/JVI.02346-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L, & Trono D (1998). Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol, 72(12), 9873–9880. 10.1128/JVI.72.12.9873-9880.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]