
Fig. 2.
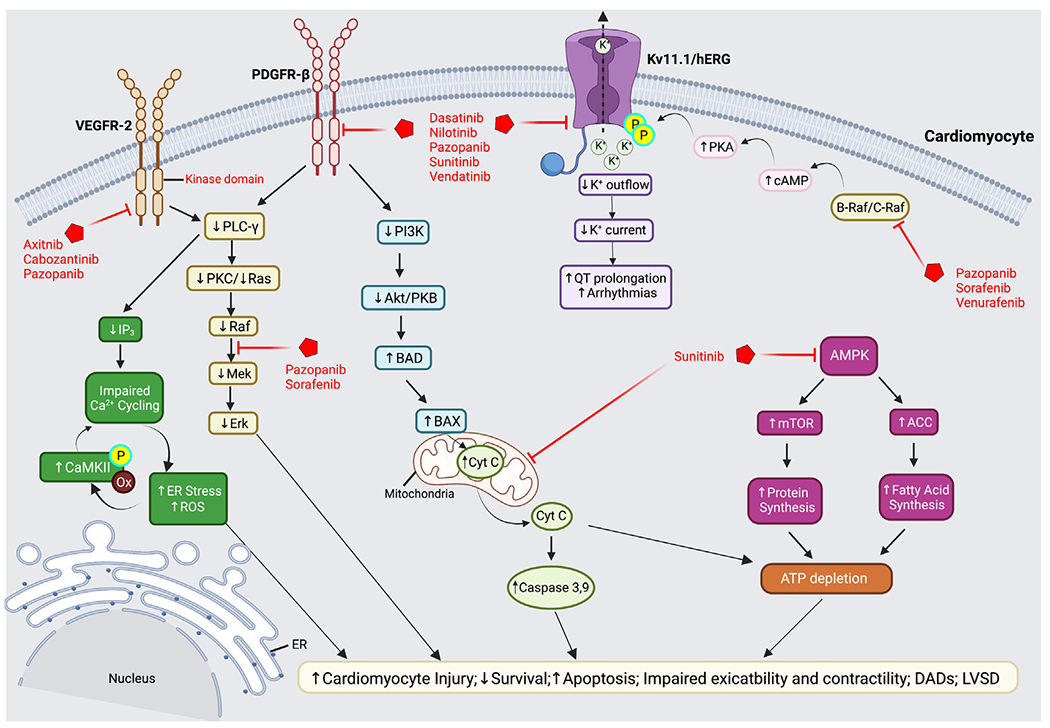
Intracellular signaling pathways mediating tyrosine kinase inhibitor cardiotoxicity. TKI-mediated inhibition of VEGFR-2 results in the downregulation of PLC-γ, which alters IP3 and Ras/Raf/Mek/Erk signal transduction cascades. Reduced IP3 contributes LVSD and reduced myocardial contractility, which results from impaired calcium cycling and increased ER stress and ROS. Increased ROS is proposed to activate and increase CaMKII phosphorylation and further dysregulation in Ca2+ homeostasis. Reduced Ras/Raf/Mek/Erk signaling is also directly affected by PDGFR-β inhibition resulting in decreased cardiomyocyte survival, increased apoptosis, and LVSD. Inhibition of PDGFR-β reduces PI3K/Akt signaling and upregulates the proapoptotic proteins, BAD and BAX, leading to mitochondrial dysfunction, release of Cyt C, activation of caspase 3 and 9, ATP depletion, and cell death. AMPK inhibition by sunitinib also depletes ATP due to increased energy sink from mTOR and ACC-mediated protein and fatty acid synthesis, respectively. Loss of ATP contributes to cardiomyocyte injury and death. TKIs also directly inhibit myocyte Kv11.1/hERG channels disrupting K+ currents. Inhibition of cytoplasmic C-RAF/B-RAF enzymes increases cAMP promoting PKA phosphorylation and inhibition of hERG channels, leading to QT prolongation and the development of arrhythmias. ACC, acetyl-coenzyme A carboxylase; Akt, protein kinase B; AMPK, adenosine 5′-monophosphate-activated protein kinase; BAD, BCL2-antagonist of cell death; BAX, BCL2-associated X protein; CaMKII, Ca2+/calmodulin-dependent protein kinase II; cAMP, cyclic adenosine monophosphate; Cyt C, cytochrome c; hERG, human ether-à-go-go; IP3, inositol-trisphosphate-3 kinase; LVSD, left ventricular systolic dysfunction; mTOR, mammalian target of rapamycin; PI3K, phosphatidyl inositol-3 kinase; PLC- γ, phospholipase C gamma; PKA/C, protein kinase A/C; PDGFR-β, platelet-derived growth factor receptor; Raf-1, rapidly accelerated fibrosarcoma-1; ROS, reactive oxygen species. (Created with BioRender.com.)