

Summary
Single-cell transcriptomics allows characterization of cerebrospinal fluid (CSF) cells at an unprecedented level. Here, we report a robust cryopreservation protocol adapted for the characterization of fragile CSF cells by single-cell RNA sequencing (RNA-seq) in moderate- to large-scale studies. Fresh CSF was collected from twenty-one participants at two independent sites. Each CSF sample was split into two fractions: one was processed fresh, while the second was cryopreserved for months and profiled after thawing. B and T cell receptor sequencing was also performed. Our comparison of fresh and cryopreserved data from the same individuals demonstrates highly efficient recovery of all known CSF cell types. We find no significant difference in cell type proportions and cellular transcriptomes between fresh and cryopreserved cells. Results were comparable at both sites and with different single-cell sequencing chemistries. Cryopreservation did not affect recovery of T and B cell clonotype diversity. Our CSF cell cryopreservation protocol provides an important alternative to fresh processing of fragile CSF cells.
Keywords: cryopreservation, cerebrospinal fluid, single-cell RNA-seq, immune cells, TCR sequencing, BCR sequencing
Graphical abstract
Highlights
-
•
We establish a robust protocol for cryopreservation of human cerebrospinal fluid cells
-
•
The method enables efficient scRNA-seq and TCR-BCR profiling of cryopreserved CSF cells
-
•
The method recovers all major CSF cell types with comparable transcriptomes
Motivation
Despite the considerable progress that the field of neuroimmunology has recently witnessed with the advent of single-cell transcriptomics, the characterization of cerebrospinal fluid (CSF) in clinical trials and large-scale translational studies remains largely limited to using fresh CSF samples. A robust CSF cell cryopreservation protocol that enables high-resolution single-cell (sc) transcriptomic data would facilitate the deployment of this important data generation modality in multi-center translational research studies and clinical trials, in which many sites do not have the expertise or resources to produce data from fresh samples. It would also serve to reduce technical variability in larger academic projects. Although previous studies have established a cryopreservation protocol, no study has systematically compared sc sequencing results from fresh and cryopreserved CSF cellular fractions from the same participants. Here, we evaluated one such protocol in detail to clearly document the differences between sc profiling of fresh and cryopreserved cells.
In this report, Touil et al. establish an efficient protocol to cryopreserve CSF cells, illustrating its utility using high-resolution single-cell analysis. Cryopreservation reduces technical variation by enabling batch processing and pooling of samples and will enable broad implementation of single-cell omics of CSF cells across many research sites.
Introduction
The composition of cerebrospinal fluid (CSF) provides insights into the physiological and pathological states of the central nervous system (CNS). Analysis of CSF represents an important element in the diagnosis and monitoring of neurological diseases; it can also play a critical role in clinical trials to demonstrate target engagement and uncover adverse events. The fluid component of CSF is sampled routinely in the diagnosis of CNS infections and a variety of neurological conditions for different biomarkers, including multiple sclerosis (MS) (oligoclonal bands),1 Alzheimer disease (AD) (Aβ/tau),2 neuromyelitis optica (NMO) (AQP4 antibody [Ab]),3 and autoimmune encephalitis (anti-GAD and anti-NMDA-R Abs).4,5 While proteins from the CSF supernatant are utilized for diagnostic purposes, currently, the evaluation of the CSF cellular component is often limited to measuring cell-type composition at low resolution (measuring the presence of different major immune cell subsets) given their fragile nature and low numbers in CSF. Cytology for malignant cells is performed clinically when malignancies are considered. Research assays typically require rigorous sample handling and larger CSF volumes; using flow cytometry,6 they have returned evidence of association between certain epitopes or cell subtypes and clinical outcomes. High-resolution single-cell analysis of fresh CSF samples has provided new insights into disorders such as MS7 and brain metastases.8
Reliable methods with which to cryopreserve CSF cells would significantly advance basic and translational research of neurological diseases. Here, we report an efficient protocol to cryopreserve CSF cells, illustrating its utility using high-resolution single-cell analysis. We show that the protocol is robust to batch effects, site of operation, and sequencing chemistries. The protocol has now been deployed in a large multi-center phase 4 MS clinical trial (https://clinicaltrials.gov/ct2/show/NCT03523858?term=Consonance&draw=2&rank=1). We provide a detailed protocol and video with step-by-step instructions as a valuable resource for the community for broader implementation in clinical and translational research.
Results
Highly efficient cryopreservation of CSF cells
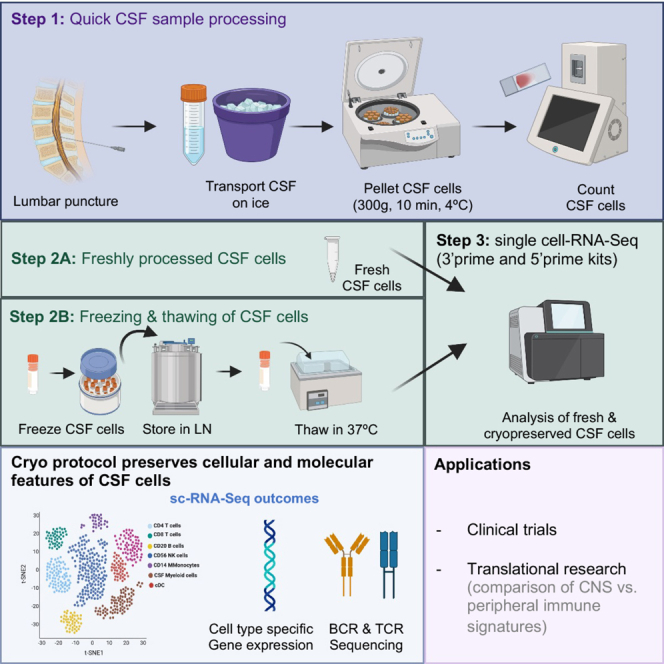
We assessed the performance of our cryopreservation protocol by processing CSF samples at two independent sites: six pairs of fresh and cryopreserved CSF samples using 3′ single-cell RNA sequencing (scRNA-seq) analysis at the Columbia University Irving Medical Center (CUIMC), USA, and fifteen pairs of fresh and cryopreserved CSF samples using both 3′ scRNA-seq (n = 7) and 5′ scRNA-seq (n = 8) at University Hospital Basel (UHB), Switzerland (Figure 1). Among the 6 Columbia samples, two were divided into three equal aliquots of CSF cells, and two of these aliquots were processed fresh to assess the extent of technical variability among fresh replicates, while the third aliquot was cryopreserved and processed at a later time.
Figure 1.

Schematic representation of the study design
CSF was collected via lumbar puncture and rapidly transported on ice for CSF cell processing. CSF cells were split into two fractions for fresh analysis or cryopreservation. CSF cells were analyzed using scRNA-seq.
3′ samples
Three out of thirteen fresh samples contained red blood cell (RBC) clusters compared with none in the cryopreserved samples (p = 0.18). The percentages of RBC contamination in three fresh samples were 1%, 2%, and 17%. The proportions of low-quality cells excluded during quality control (QC) (cells with <100 unique genes or a mitochondrial gene percentage >25%) were also not different between fresh and cryopreserved samples (p = 0.37) (Table S1). After removing RBCs, doublets, and low-quality cells (details in the STAR Methods), we retained 45,175 cells from 13 fresh-cryopreserved pairs and the two additional fresh technical replicates from Columbia for further analysis (Table 1). The number of cells passing QC was reduced but not significantly different between cryopreserved (median = 785 cells, interquartile range [IQR] = 1,656) and fresh (median = 1,163 cells, IQR = 2,088) samples (p = 0.24) (Figure 2A). Among sequencing QC metrics, the percentage of mitochondrial transcripts/cell was not significantly different in fresh (median = 2.6%, IQR = 1.6) versus cryopreserved (median = 3%, IQR = 2.1) (p = 0.68). The number of unique molecular identifiers (UMIs)/cell in fresh (median = 4,396, IQR = 1,215) versus cryopreserved (median = 3,368, IQR = 2,851) samples and the number of unique genes/cell in fresh (median = 1,532, IQR = 279) versus cryopreserved (median = 1,199, IQR = 772) samples were reduced in cryopreserved samples (p = 0.01 and 0.008 for log-transformed values, respectively). However, they remained within the acceptable range of parameters for downstream analysis (Figure S1A).
Table 1.
Patient demographics and sample description
| Research site | Sample ID | Age | Sex | Storage length (days) | Diagnosis | RNA-seq chemistry | Volume of CSF (mL) | Total fresh cell counts (cells) | Fresh cell counts | Cryopreserved cell counts | Viability after cell thawing (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Columbia University | PC050 | 26 | F | 7 | migraine | 3′ | 18 | 5,000 | 635 | 261 | 75 |
| PC051 | 42 | F | 7 | RRMS – active lesion | 3′ | 15 | 11,000 | 2,706 | 1,147 | 80 | |
| PC052 | 52 | F | 7 | RRMS – active lesion | 3′ | 15 | 16,000 | 3,729 | 4,120 | 85 | |
| PC056 | 29 | F | 30 | RRMS | 3′ | 15 | 13,000 | fresh 1: 3,085fresh 2: 2,576 | 1,869 | 100 | |
| PC057 | 31 | F | 30 | RRMS | 3′ | 12 | 9,500 | 2,177 | 2,181 | 88 | |
| PC058 | 70 | F | 14 | PML, BM transplant | 3′ | 13 | 8,000 | fresh 1: 1,163fresh 2: 1,031 | 717 | 94 | |
| University Hospital Basel | CR061 | 54 | M | 60 | idiopathic intracranial hypertension | 5′ | 3 | 6,000 | 1,332 | 1,320 | 90 |
| CR062 | 27 | F | 60 | idiopathic intracranial hypertension | 5′ | 8 | 15,000 | 2,370 | 2,369 | 95 | |
| CR063 | 34 | F | 60 | unknown | 5′ | 5 | 9,000 | 734 | 735 | 83 | |
| CR064 | 68 | F | 30 | cutaneous borreliosis | 5′ | 3 | 5,000 | 755 | 192 | 70 | |
| CR065 | 48 | M | 7 | migraine | 5′ | 3 | 6,000 | 896 | 165 | 75 | |
| CR066 | 39 | F | 30 | migraine | 3′ | 8 | 14,000 | 3,847 | 3,841 | 86 | |
| CR067 | 59 | M | 30 | low-grade thalamic glioma | 3′ | 8 | 3,000 | 741 | 785 | 93 | |
| CR068 | 32 | F | 14 | temporal lobe epilepsy | 3′ | 3 | 3,000 | 625 | 526 | 81 | |
| CR069 | 31 | F | 14 | hereditary spastic paresis | 3′ | 5.2 | 3,000 | 615 | 441 | 80 | |
| CR070 | 43 | F | 14 | bulbar lesion | 3′ | 4.5 | 2,000 | 427 | 275 | 78 | |
| CR071 | 44 | M | 7 | paravertebral increased muscle | 3′ | 4.5 | 3,000 | 468 | 533 | 89 | |
| CR072 | 27 | F | 7 | visual deficit | 3′ | 4.2 | 16,000 | 1,924 | 2,770 | 75 | |
| CR074 | 46 | M | 30 | brainstem encephalitis | 5′ | 5.2 | 70,000 | 5,899 | 5,414 | 92 | |
| CR075 | 24 | F | 14 | PRES | 5′ | 3.7 | 4,000 | 631 | 302 | 88 | |
| CR076 | 72 | M | 14 | Bell’s palsy | 5′ | 4.7 | 8,000 | 1,304 | 541 | 86 |
Metadata for fresh and cryopreserved samples used in 3′ and 5′ single-cell RNA sequencing. M, male; F, female: RRMS, relapsing-remitting multiple Ssclerosis: PML, progressive multifocal leukoencephalopathy; BM, bone marrow; PRES, posterior reversible encephalopathy syndrome. See also Table S1.
Figure 2.

Efficient CSF cell cryopreservation validated by 3′ single-cell transcriptomics
(A) Post-quality control (QC) CSF cell counts in the fresh and cryopreserved (Cryo) sample pairs (p = 0.24).
(B) Uniform manifold approximation and projection (UMAP) plot of 21 clusters/cell states color coded by their annotations.
(C) UMAP indicating good representation of fresh and Cryo cells in each cluster.
(D) Annotation of clusters using selected marker genes. Heatmap colors correspond to the proportion of cells in each cluster expressing cluster marker gene.
(E) Bar chart indicating similar cell-type proportions in thirteen fresh and Cryo sample pairs.
(F and G) Significant positive correlation (F) between the different cluster proportions from thirteen fresh and Cryo sample pairs (p = 0.93) and (G) between two fresh-fresh sample pairs (p = 0.98, 0.99). Each point represents a cluster color coded by its sample ID.
5′ samples
None of the samples contained RBCs, and there was no significant difference in the frequency of excluded low-quality cells between fresh and cryopreserved samples (p = 0.37) (Table S1). After excluding doublets and low-quality cells, we analyzed 24,989 cells from 16 samples (8 pairs of fresh and cryopreserved samples) (Table 1). The number of cells passing QC were reduced in cryopreserved samples: median = 1,099 cells and IQR = 825 in fresh versus median = 638 and IQR = 1,309 in cryopreserved samples (p = 0.03) (Figure 3A). However, all sequencing metrics were similar between fresh and cryopreserved samples: the percentage of mitochondrial transcripts/cell in fresh (median = 1.7%, IQR = 0.35) versus cryopreserved (median = 1.9%, IQR = 0.75) (p = 1); the number of UMIs/cell in fresh (median = 1,860, IQR = 757) versus cryopreserved (median = 1,629, IQR = 265) (p = 0.54); and the number of genes/cell in fresh (median = 795, IQR = 279) versus cryopreserved (median = 712, IQR = 64) (p = 0.54) (Figure S1B). Overall, the two sets of results are consistent: although there might be slight differences in recovered cells between fresh and cryopreserved samples and in sequencing metrics, cryopreserved CSF cells generate data that meet QC parameters for downstream analyses. In both 3′ and 5′ data, the number of cells was lower in the cryopreserved samples, and the difference was marginally significant in the 5′ data (before accounting for the testing of multiple hypotheses); in toto, we interpret these observed reductions as reproducible but of small effect. There is no large effect of cryopreservation on cell recovery. Importantly, the cryopreservation protocol is robust to site/batch effects and different single-cell chemistries (3′ versus 5′) in recovering cryopreserved CSF cells: we see the same patterns in the data at both sites and with both chemistries.
Figure 3.

Efficient CSF-cell cryopreservation validated by 5′ single-cell transcriptomics
(A) Post- quality control (QC) CSF cell counts in the fresh and cryopreserved (Cryo) sample pairs (p = 0.37).
(B) Uniform Manifold Approximation and Projection (UMAP) plot of 21 clusters color coded by their annotations.
(C) UMAP indicating good representation of fresh and Cryo cells in each cluster.
(D) Annotations of clusters using selected marker genes. Heatmap colors correspond to the proportion of cells in each cluster expressing cluster marker gene.
(E) Bar chart indicating similar CSF cell type proportions in eight fresh-Cryo sample pairs.
(F) Significant positive correlation between the different cluster proportions in eight fresh-Cryo sample pairs (p = 0.97).
(G) Clonal rank indicating similar distribution of the T and B cell clonotypes in the eight fresh and Cryo CSF samples.
Identification of all major CSF cell types
Normalization, integration, and clustering of cells was performed independently on the 3′ and 5′ samples. We identified 21 clusters of CSF cells in each dataset (Figures 2B and 3B). The top 10 marker genes for each cluster are described in Figure 2D and Tables S2 and S3 for 3′ and 5′ samples, respectively. Prior single-cell studies of CSF have reported CSF-specific microglia-like clusters.7,9,10 We refined our azimuth-based annotations for myeloid clusters using known marker genes for CSF-specific cells7,9,10 and annotated clusters 7 and 9 in the 3′ samples (Figure 2B; Table S1) and clusters 5, 6, and 17 in the 5′ samples (Figure 3B; Table S1) as “CSF-myeloid” cells. Figures 2D and 3D illustrate each cluster marker gene expression in the 3′ and 5′ samples, respectively. Sequencing QC metrics between fresh and cryopreserved cells showed similar patterns across individual clusters, suggesting that cryopreservation did not disproportionately affect RNA quality of any specific CSF cell type (Figures S1C–S1H). We identified all major cell types previously reported to be present in CSF.
Cryopreservation does not result in loss of CSF cell types
We assessed the sensitivity of CSF cell types to cryopreservation by comparing their proportion/frequency between fresh and cryopreserved samples. The counts and percentage of cells found in each cluster from the 3′ and 5′ samples are available in Figures 1A and 2A and Table S1. We observed a high correlation of cell-type frequencies between all fresh-cryopreserved sample pairs (median Spearman correlation = 0.93 for 3′ samples and 0.97 for 5′ samples) (Figures 2F, 3F, S2A, and S2B). Correlation was slightly higher between the two fresh-fresh replicate pairs from a subset of the 3′ samples (0.98 and 0.99) (Figure 2G). Principal-component analysis (PCA) of cluster frequencies further highlights that the fresh and cryopreserved cells from individual samples are more similar to each other than they are to their sample class (fresh or cryopreserved) (Figures S2C and S2D). However, both correlation and PCA suggest that cryopreserved samples with very low numbers of recovered cells (e.g., CR064FRZ and CR065FRZ, both <200 cells) show higher variability in their expected cluster frequencies. Hence, we recommend that researchers exercise extra caution when analyzing data from samples with lower-than-expected recovered cell counts.
We also compared differences in each cell cluster frequency between fresh and cryopreserved sample pairs using the Wilcoxon signed-rank test. We observed no significant difference in the frequencies of each broad cell type (e.g., when all CD4 subclusters are aggregated as one cell type) or individual cell clusters in 3′ (Figures 2E and S2E) and 5′ samples (Figures 3E and S2F) (all adjusted p > 0.05). These results suggest that our cryopreservation method does not result in significant loss of any specific CSF cell type.
Cryopreservation does not affect gene expression of CSF cells
We next assessed the impact of cryopreservation on gene expression in CSF cell types.
We observed a high correlation in gene expression between fresh and cryopreserved cells across all 21 cell clusters in 3′ samples (median correlation = 0.99, IQR = 0.005; Figure S3; Table S2) and in 5′ samples (median correlation = 0.99, IQR = 0.01; Figure S3; Table S2). We observed a low number of differentially expressed genes between fresh and cryopreserved cells both in the 3′ samples (between 0 and 10 genes with fold change >1.5 across 21 clusters; Figure S3; Table S3) and in the 5′ samples (between 0 and 4 genes with fold change >1.5 across all 21 clusters; Figure S3; Table S3).
Among differentially expressed genes, hemoglobin beta gene (HBB), and the mitochondrial MT-ND4L gene, mitochondrial-like paralogs MTRNR2L8 and MTRNR2L12, long non-coding RNA (lncRNA) genes AL138963.4 and SNHG25, histone gene HIST1H1E, and the PABPC1 and MYH9 genes showed reduced expression, while the ribosomal gene RPS20 and the mitochondrial gene MT-ATP8 showed increased expression in more than one cell type in either the 3′ or 5′ cryopreserved samples (Figures S4A and S4B), suggesting that cryopreservation affects these genes in a non-cell-type-specific manner. HBB and MTRNR2L8 were the only 2 genes that were differentially expressed in more than 1 cluster with >2-fold change. MTRNR2L8 is implicated as an antiapoptotic factor, and its decreased expression might be related to the biological effects of cryopreservation on cell survival or transcriptome. On the other hand, HBB, which encodes hemoglobin subunit β and is highly expressed in RBCs, was mostly found in RBC-contaminated fresh samples and probably represents contamination with ambient RNA in these samples (Figures S4C and S4D). These results suggest that our cryopreservation method maintains the overall gene expression profile of CSF cell types except for very few genes.
CSF clonotypes are conserved after cryopreservation
Lastly, we assessed the effect of cryopreservation on the frequency of T and B cell clonotypes in 5′ scRNA-seq data. The number of cells identified from each clonotype and their corresponding CDR3 amino acid sequences are available in Table S4. 77% of the cells in the T cell clusters and 87% of the cells in B or plasma cell clusters had an identified CDR3 sequence. There was no difference in the frequency of cells with an identified CDR3 sequence between fresh and cryopreserved samples in the T cell or B cell components (p = 0.44 and 0.2, respectively). Of all identified unique CDR3 sequences, 25% were commonly found in fresh and cryopreserved samples. The non-shared clones were equally distributed between fresh and cryopreserved samples, and all had lower frequencies compared with shared clones (Figure S2G). The frequency of the shared clones was highly correlated between paired fresh and cryopreserved samples (Spearman’s ρ = 0.94, p < 0.001; Figure S2H). Within each patient, T and B cell clonotypes were identified by their unique CDR3 sequence and ranked using their occurrence frequency (i.e., clonal expansion). Figure 3G is a clonal rank plot showing that both expanded clones (rank 1 to 10) and unexpanded clones (rank 0) remain unaffected by cryopreservation (overlapping “triangles” and “crosses” of the same color). Altogether, these findings suggest that cryopreservation has no effect on the diversity (or lack thereof) of the adaptive immune receptor repertoire.
Discussion
High-quality peripheral blood and CSF leukocyte samples are imperative for elucidating the cellular and molecular cascades orchestrating neuroinflammatory and neurodegenerative disorders. Recent scRNA-seq investigations in fresh CSF samples have allowed comprehensive fine-grained mapping of CSF cell types, transient cell states, and disease-associated cell-type-specific signatures,10,11,12 observations that cannot be achieved using conventional flow cytometry in CSF. Cryopreservation of CSF cells using a robust protocol that preserves cellular and molecular phenotypes would significantly advance basic and clinical research. Recently, Oh and colleagues13 published a CSF cryopreservation protocol; however, no direct comparison of fresh and cryopreserved CSF samples was conducted.
We developed a simple yet rigorous and cost-efficient protocol to cryopreserve CSF cells that showed highly reproducible results at two independent sites when data from fresh and cryopreserved CSF cells are compared. We reported in an earlier study14 in human peripheral blood mononuclear cells (PBMCs) that our cryopreservation method performs better than other cryopreservation media in the recovery of immune cells with minimal impact on gene expression profile. The CSF protocol enabled us to analyze between 165 and 5,414 cryopreserved cells, depending on the volume of the starting sample, which was as low as 3 mL, and on the participant’s diagnosis. We recovered >70% of cryopreserved cells post-thawing using our protocol at both sites. We validated the efficiency of our protocol in recovering good-quality cells using two scRNA-seq chemistries, the 3′ scRNA-seq and the 5′ scRNA-seq, which are known to have different sensitivities to detect genes. We identified all known CSF cell types including microglia-like cells reported by others, which we report as CSF-myeloid cells given the lack of clarity as to their origin. Our cryopreservation method did not alter the composition of CSF cells; the frequency and gene expression profiles of cell types (even minor cell subsets like B cells) were preserved. We noted a higher hemoglobin gene expression in the fresh samples despite the exclusion of RBCs from the analyses. However, no RBCs were found in our cryopreserved pairs for RBC-contaminated fresh samples, suggesting that the cryopreservation protocol might be beneficial in removing RBC contamination and hemoglobin ambient RNA, when present.
Our findings highlight comparable fresh and cryopreserved CSF cell profiles despite a slightly reduced number of QC-passed cells post-cryopreservation. CSF cell clusters and T/B cell clonotypes in fresh versus cryopreserved samples are preserved and comparable. Given the practical challenges of fresh CSF characterization in large multi-center settings with different levels of technological sophistication and considering cost savings, adopting the proposed method will enable large-scale and multi-center investigations—crucial for clinical trials.
Limitations of the study
While we present a robust protocol for cryopreservation of CSF cells that can be broadly implemented in clinical trial and research settings for single-cell transcriptomic characterization, there are still limitations that will require further research. First, we evaluated the efficacy of our protocol on CSF cells stored for only up to 2 months. 2 months might not be a sufficient storage period for certain large multi-center, worldwide clinical trial settings. Future work is needed to test the efficacy of our protocol on CSF cells stored for periods longer than 2 months, ideally 6 months to 1 year. Second, our study has evaluated the largest number of CSF samples/donors published to date and is powered to detect large changes in cell composition, as observed in diseased conditions.7,13,15 Nonetheless, larger sample sizes would be needed to evaluate the impact of cryopreservation on smaller changes in cell composition. Finally, due to ethical limitations, which exist at many clinical sites in relation to obtaining larger CSF volumes from patients, and the known variability in the yield/recovery of input cells using the 10× Genomics platform, we compared relatively low numbers of high-quality CSF cells between fresh and cryopreserved samples. It is possible that some rare and functionally relevant immune subtypes may not be recovered after cryopreservation, a possibility that should be explored in future studies using multi-modal assays such as cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq).16 Users may need to perform additional optimizations if they plan to implement the protocol using smaller CSF volumes for single-cell transcriptomics or additional complementary downstream applications such as flow cytometry.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Neuroinflammatory patient’s CSF cells | Columbia University University Hospital Basel |
IRB#AAAR5606 IRB#2027-00198 and 00908 |
| MS patient’s CSF cells | Columbia University | IRB#AAAR5606 |
| Chemicals, peptides, and recombinant proteins | ||
| Dimethyl Sulfoxide (DMSO) | Sigma-Aldrich | Cat#472301-1L; CAS: 67-68-5 |
| Fetal Bovine Serum (FBS) | ThermoFischer Scientific | Cat#10-438-026 |
| RPMI-1640 | ThermoFischer Scientific | Cat#11-875-093 |
| Critical commercial assays | ||
| 3′ sc-RNA-seq kit (V3.1 GEM kit) | 10x Genomics | N/A |
| Chromium Next GEM Chip G Single Cell Kit | 10x Genomics | Cat#PN-1000120 |
| Chromium Next GEM Single Cell 3′ Kit v3.1 | 10x Genomics | Cat#PN-1000268 |
| Dual Index Kit TT Set A | 10x Genomics | Cat#PN-1000215 |
| 5′ sc-RNA-seq kit (V1.1 and v2.0 V(D)J Kits) | 10x Genomics | N/A |
| V1.1 V(D)J Kit | 10x Genomics | N/A |
| Chromium Next GEM Chip G Single Cell Kit | 10x Genomics | Cat#PN-1000120 |
| Chromium Single Cell V(D)J Reagent Kits, v1.1 | 10x Genomics | Cat#PN-1000165 |
| Single Index Kit T Set A | 10x Genomics | Cat#PN-1000213 |
| V2 V(D)J Kit | 10x Genomics | N/A |
| Chromium Next GEM Chip K Single Cell Kit | 10x Genomics | Cat#PN-1000286 |
| Chromium Next GEM Single Cell 5ʹ Kit v2 | 10x Genomics | Cat#PN-1000263 |
| Dual Index Kit TT Set A | 10x Genomics | Cat#PN-1000215 |
| Deposited data | ||
| Human CSF sc-RNA-seq | This paper | GEO: GSE234069 |
| Software and algorithms | ||
| Cell Ranger v3.1.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/installation |
| Cell Ranger v6.0.2 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/installation |
| Scrublet | Wolock et al.17 | https://github.com/swolock/scrublet |
| Seurat v4.0.1 | Hao et al.18 | https://github.com/satijalab/seurat |
| R | https://cran.r-project.org/ | |
| Harmony | Korsunsky et al.19 | https://github.com/immunogenomics/harmony |
| Azimuth Human PBMC Atlas | Hao et al.18 | https://app.azimuth.hubmapconsortium.org/app/human-pbmc |
| Shiny interactive browser | This paper |
https://malhotralab.shinyapps.io/csf_cryo_3prime/and https://malhotralab.shinyapps.io/csf_cryo_5prime/ |
| Other | ||
| Sprotte spinal needle | 4MDMECICAL | Cat#CF4301C |
| Ice Bucket | ThermoFischer Scientific | Cat#07-210-128 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Philip De Jager, pld2115@cumc.columbia.edu.
Material availability
This study did not generate new unique reagents.
Experimental model and study participant details
Human cerebrospinal fluid (CSF) samples
Diagnostic lumbar punctures (LPs) were performed on twenty-one patients who presented with suspected neuroinflammatory disorders to the Multiple Sclerosis Center at either Columbia University Irving Medical Center, in patient hospital unit or University Hospital Basel. Six patients recruited at the Columbia University Medical Center and seven patients at the University Hospital Basel were analyzed using droplet-based 3′ single-cell RNA sequencing (3′ sc-RNA-Seq) on the 10x Genomics Chromium platform. Eight patients recruited at the University Hospital Basel were analyzed using 5′ sc-RNA-seq on the same platform. All participants provided written informed consent forms as part of a protocol approved by Columbia and Basel university’s institutional review board (Columbia University IRB # AAAR5606 and Basel University IRB #2017-00198 and 2021-00908, respectively). All LPs were performed in sterile conditions. CSF samples were collected in sterile tubes, using the Sprotte spinal needle and immediately transported to the laboratory in an ice bucket to maintain a stable temperature of 0°C to 4°C. The demographic characteristics of the sample cohort including age, sex, diagnosis, and the cell counts from each sample used for the analysis are described in (Table 1). Samples were collected from both male and female patients. Paired analysis was performed between fresh and cryopreserved CSF sample pairs.
Demographic characteristics including sex were not expected to influence the results, and hence their effects were not studied.
Method details
Sample processing of CSF cells
The CSF samples were immediately processed upon arrival to the laboratory without delay. The CSF samples were processed using 15mL or 50mL Falcon tubes depending on the volume of collected CSF sample, centrifuged at 300g for 10 min at 4°C and the supernatant was aliquoted into 1mL aliquots and stored in the −80°C freezers. The CSF-cell pellet was resuspended in 70μL of autologous CSF. Using 10 μL of the CSF cell suspension, cell viability and counts were assessed using automated cell counters at Nexcelom (Nexcelom, Bioscience) with acridine orange and propidium iodide (AOPI) viability dye (Nexcelom, Bioscience).The volume and cell-count of CSF samples varied between 3 and 18mL, 427–5899 cells, respectively.
The CSF-cell suspension was then split into two aliquots: the first aliquot was used to analyze the transcriptomic profile of fresh CSF-cells using single-cell RNA sequencing (3′ and 5’sc-RNA-seq), while the second CSF aliquot was immediately cryopreserved and stored in the liquid nitrogen (LN), as described below. For samples PC056 and PC058, we prepared three CSF-cell aliquots: two fresh (technical replicates) and the third aliquot was cryopreserved for up to 2 months.
Cryopreservation of CSF cells
For cryopreservation, the CSF-cell suspension was diluted into 750μL of RPMI-1640 supplemented with 40% FBS in a gentle manner. We further diluted the CSF-cell suspension dropwise by adding 750uL of freezing medium (RPMI-1640 supplemented with 40% FBS and 20% DMSO) pre-chilled at 4°C. The CSF sample was immediately placed inside a freezing container (Mister Frosty, Thermo Fisher) and stored in the −80°C freezer overnight, before it was moved to the LN for 7 days to 2 months.
Thawing of CSF cells
After the storage period elapsed, the cryopreserved cells were retrieved from the LN, and thawed at 37°C for 1–2 min, until a tiny ice crystal remained in the cryotube. The thawed cells were subsequently diluted 1:4 using pre-warmed RPMI-1640 supplemented with 10% FBS and centrifuged at 300g for 10 min at room temperature. The supernatant was discarded and the cells were resuspended in fresh 60μL RPMI-1640 media, we used 10μL to count the cells and processed for 3′ or 5′-single cell RNA-seq as in (Figure 1).
Single-cell RNA and TCR-BCR sequencing
Fresh and cryopreserved CSF-cells from 44 individual samples (14 from Columbia university and 30 from university hospital Basel) were loaded into the 10x Genomics Chromium Controller for droplet-encapsulation. cDNA libraries were prepared using either the Chromium Next GEM Single Cell 3ʹ v3.1 or Chromium Next GEM Single Cell V(D)J v1.1 and v2.0 kits (10x Genomics) according to the manufacturer’s instructions. When the latter was used, TCR- and BCR-enriched libraries were prepared for each sample using Chromium Single Cell V(D)J Enrichment Kit (Human T cell) and Chromium Single Cell V(D)J Enrichment Kit (Human B Cell) respectively. All libraries were sequenced using NovaSeq 6000 (Illumina) and NovaSeq 6000 S2 Reagent Kit v1.5 (100 cycles) (Illumina) to get a sequencing depth of 50K reads/cell (whole transcriptome libraries) or 10K reads/cell (TCR and BCR enriched libraries).
Single-cell RNA-sequencing data processing
scRNA sequenced reads were aligned and quantified using Cell Ranger v3.1 to ref. 3.1.0 (Ensembl 93) transcriptome for 3′ samples, and using Cell Ranger v6.0.2 multi pipeline to ref. 2020-A (Ensembl 98) transcriptome and VDJ ref. 5.0.0 (Ensembl 94) for 5′ whole transcriptome and TCR and BCR sequencing data.
The rest of the pre-processing and analysis were performed using Seurat library (v 4.0.1).18 We did not exclude any sample from the study. Data analysis was not performed in a blinded manner. However, all data from paired fresh and cryopreserved samples underwent the same quality control process with the same criteria. For each sample, we performed quality control and filtered out cells: (1) that were likely doublets using Scrublet,17 (2) with less than 100 genes, (3) with more than 25% mitochondrial transcripts, (4) Red Blood Cell (RBC) clusters (defined as clusters that showed high expression of hemoglobin genes).
Data from 3′ samples (13 fresh and cryopreserved pairs, and 2 additional fresh replicates) and 5′ samples (8 fresh and cryopreserved pairs) were then independently merged using harmony integration, resulting in two separate datasets for subsequent analyses. The sample size was not pre-determined except for the paired froup comparison in each dataset which was comparable to the largest CSF sc-RNA-sq studies published today.7,13 The top 1000 variable genes were identified for each sample, aggregated over all samples in each dataset, and used for dimensionality reduction using principal component analysis (PCA). Integration was performed using the top 50 PCs using Harmony https://doi.org/10.1038/s41592-019-0619-0). UMAP representation and clustering were performed using default Seurat parameters. Each cluster’s marker cells were identified using Seurat FindAllMarkers function for genes that were expressed in at least 25% of the cells from that cluster.
We annotated cells using a three-pronged approach to improve the annotation accuracy. (1) We performed differential gene expression (DE) of clusters using Seurat FindAllMarkers function to identify each cluster marker gene (gene expressed in at least 25% of the cells with fold change >1.28 at false discovery rate <0.05 between clusters). (2) We also annotated our cells by mapping our data to the Azimuth18 human peripheral blood mononuclear cells CITE-seq atlas, developed as part of the NIH Human Biomolecular Atlas Project, using supervised PCA (reference-based annotation).18 Finally, we refined our annotations by manual inspection of expression of marker genes from published CSF single-cell RNA-seq studies.7,9,10
Differential gene expression analysis
We performed differential expression analyses between fresh and paired cryopreserved samples within each cluster using Seurat FindMarkers function with MAST20 test, fitting generalized linear models that are adapted for zero-inflated single-cell gene expression data. Patient ID was considered as a latent variable in order to account for the paired data structure. Genes detected in at least 10% of cells in either of the fresh or cryopreserved groups were included in the analyses. Groups with <5 cells were excluded from gene differential expression analysis. Genes with Bonferroni-adjusted p value <0.05 and absolute log2 fold change >0.58 (fold change >1.5) were considered significant. P-value and log fold change for all significant genes are available in Tables S2 and S3.
Quantification and statistical analysis
Wilcoxon signed-rank test was used to compare recovered cell counts, RNA-sequencing quality control metrics, cluster cell frequencies, and frequencies of RBCs and low-quality cells between fresh and cryopreserved samples. Spearman’s ρ was used to assess correlation between variables. Both tests are nonparametric and require no assumptions about the shape of the population distribution. Number of samples in each group comparison, median and interquartile range for each group, and p values for each statistical test are reported in the main text. Reported p values are all two-sided. Adjustment for multiple comparisons was performed using the Benjamini and Hochberg false discovery rate method, unless otherwise specified. Enrichment of clonotypes with ≥2 cells in each cluster were assessed using one-sided Fisher’s exact test.
Additional resources
The proposed protocol has now been deployed in a large Phase 4 MS clinical trial (ROCHE Consonance Trial), description available at: https://clinicaltrials.gov/ct2/show/NCT03523858?term=Consonance&draw=2&rank=1.
The implementation of the proposed protocol is further videotaped and can be found at:https://we.tl/t-OLoUj5O10N.
Acknowledgments
We thank all patients who generously offered CSF and blood samples. We are grateful to the single-cell sequencing core of Columbia University and FGCZ at the University of Zurich. We thank Imran Fanaswala for helping with TCR BCR analysis and Neelang Parghi from the P.LD.J. lab for helping with GEO data upload. We are grateful to the entire Roche Consonance Team for their help with the implementation of the CSF cryopreservation protocol and sharing the video. This work was funded, in part, by a sponsored research agreement from Roche. Video link: https://we.tl/t-uismrf1Ph3. This work was funded by F. Hoffmann-La Roche. H.T. was funded by the Canadian MS Society and the FRQS (Fonds de recherche du Québec en santé) for a post-doctoral fellowship. K.T.T. was supported by an NIH grant. The work was also supported, in part, by CS-02018-191971, P30 AG066462, K23NS105935-01, and CZF2019-002456.
Author contributions
D.M., P.L.D.J. and H.T. designed the study. H.T. and D.C. carried out experiments. T.R. analyzed data. H.T., T.R., and D.M. drafted the manuscript. D.C., S.E., I.C., T.D., and K.O. performed the lumbar punctures and coordinated sample acquisition. C.R. and L.C. contributed to the study design, preparation of protocol video, and interpretation of results. V.M., D.M., J.B., D.C., P.L.D.J., T.R., and H.T. contributed to the analysis plan and interpretation of results. K.T.T. helped with sample collection and provided clinical data of patients. D.M. and P.L.D.J. conceived and supervised the overall project and edited the manuscript.
Declaration of interests
D.M., D.C., C.R., L.C., and J.B. are full-time employees of F. Hoffmann-La Roche. D.M. was employed by F. Hoffmann-La Roche when the study was completed and the manuscript submitted. D.M. moved to Biogen while the manuscript was under review.
Published: July 14, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2023.100533.
Contributor Information
Philip L. De Jager, Email: pld2115@cumc.columbia.edu.
Dheeraj Malhotra, Email: dheeraj21malhotra@gmail.com.
Supplemental information
Data and code availability
-
•
Single-cell RNAseq NGS data from Basel samples have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. The gene expression data from Basel are available via an interactive browser. Link to the browser is listed in the key resources table. Gene expression and metadata from cells included in the alaysis from Basel sampes are available as Supplementary data file 1 and 2. The gene expression data from 3′ and 5′ sc-RNA-seq samples are available via the interactive browser “Shiny app’’ at: https://malhotralab.shinyapps.io/csf_cryo_3prime/and https://malhotralab.shinyapps.io/csf_cryo_5prime/, respectively. The Columbia University single cell data will be made available by the lead contact upon reasonable request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Thompson A.J., Banwell B.L., Barkhof F., Carroll W.M., Coetzee T., Comi G., Correale J., Fazekas F., Filippi M., Freedman M.S., et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17:162–173. doi: 10.1016/S1474-4422(17)30470-2. [DOI] [PubMed] [Google Scholar]
- 2.Lee J.C., Kim S.J., Hong S., Kim Y. Diagnosis of Alzheimer's disease utilizing amyloid and tau as fluid biomarkers. Exp. Mol. Med. 2019;51:1–10. doi: 10.1038/s12276-019-0250-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wingerchuk D.M., Banwell B., Bennett J.L., Cabre P., Carroll W., Chitnis T., de Seze J., Fujihara K., Greenberg B., Jacob A., et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85:177–189. doi: 10.1212/WNL.0000000000001729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graus F., Saiz A., Dalmau J. GAD antibodies in neurological disorders - insights and challenges. Nat. Rev. Neurol. 2020;16:353–365. doi: 10.1038/s41582-020-0359-x. [DOI] [PubMed] [Google Scholar]
- 5.Graus F., Titulaer M.J., Balu R., Benseler S., Bien C.G., Cellucci T., Cortese I., Dale R.C., Gelfand J.M., Geschwind M., et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15:391–404. doi: 10.1016/S1474-4422(15)00401-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han S., Lin Y.C., Wu T., Salgado A.D., Mexhitaj I., Wuest S.C., Romm E., Ohayon J., Goldbach-Mansky R., Vanderver A., et al. Comprehensive immunophenotyping of cerebrospinal fluid cells in patients with neuroimmunological diseases. J. Immunol. 2014;192:2551–2563. doi: 10.4049/jimmunol.1302884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schafflick D., Xu C.A., Hartlehnert M., Cole M., Schulte-Mecklenbeck A., Lautwein T., Wolbert J., Heming M., Meuth S.G., Kuhlmann T., et al. Integrated single cell analysis of blood and cerebrospinal fluid leukocytes in multiple sclerosis. Nat. Commun. 2020;11:247. doi: 10.1038/s41467-019-14118-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rubio-Perez C., Planas-Rigol E., Trincado J.L., Bonfill-Teixidor E., Arias A., Marchese D., Moutinho C., Serna G., Pedrosa L., Iurlaro R., et al. Immune cell profiling of the cerebrospinal fluid enables the characterization of the brain metastasis microenvironment. Nat. Commun. 2021;12:1503. doi: 10.1038/s41467-021-21789-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esaulova E., Das S., Singh D.K., Choreño-Parra J.A., Swain A., Arthur L., Rangel-Moreno J., Ahmed M., Singh B., Gupta A., et al. The immune landscape in tuberculosis reveals populations linked to disease and latency. Cell Host Microbe. 2021;29:165–178.e8. doi: 10.1016/j.chom.2020.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farhadian S.F., Mehta S.S., Zografou C., Robertson K., Price R.W., Pappalardo J., Chiarella J., Hafler D.A., Spudich S.S. Single-cell RNA sequencing reveals microglia-like cells in cerebrospinal fluid during virologically suppressed HIV. JCI Insight. 2018;3 doi: 10.1172/jci.insight.121718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pappalardo J.L., Zhang L., Pecsok M.K., Perlman K., Zografou C., Raddassi K., Abulaban A., Krishnaswamy S., Antel J., van Dijk D., Hafler D.A. Transcriptomic and clonal characterization of T cells in the human central nervous system. Sci. Immunol. 2020;5 doi: 10.1126/sciimmunol.abb8786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramesh A., Schubert R.D., Greenfield A.L., Dandekar R., Loudermilk R., Sabatino J.J., Jr., Koelzer M.T., Tran E.B., Koshal K., Kim K., et al. A pathogenic and clonally expanded B cell transcriptome in active multiple sclerosis. Proc. Natl. Acad. Sci. USA. 2020;117:22932–22943. doi: 10.1073/pnas.2008523117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oh H., Leventhal O., Channappa D., Henderson V.W., Wyss-Coray T., Lehallier B., Gate D. Methods to investigate intrathecal adaptive immunity in neurodegeneration. Mol. Neurodegener. 2021;16:3. doi: 10.1186/s13024-021-00423-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lütge A., Zyprych-Walczak J., Brykczynska Kunzmann U., Crowell H.L., Calini D., Malhotra D., Soneson C., Robinson M.D. CellMixS: quantifying and visualizing batch effects in single-cell RNA-seq data. Life Sci. Alliance. 2021;4 doi: 10.26508/lsa.202001004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heming M., Li X., Räuber S., Mausberg A.K., Börsch A.L., Hartlehnert M., Singhal A., Lu I.N., Fleischer M., Szepanowski F., et al. Neurological Manifestations of COVID-19 Feature T Cell Exhaustion and Dedifferentiated Monocytes in Cerebrospinal Fluid. Immunity. 2021;54:164–175.e6. doi: 10.1016/j.immuni.2020.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stoeckius M., Hafemeister C., Stephenson W., Houck-Loomis B., Chattopadhyay P.K., Swerdlow H., Satija R., Smibert P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods. 2017;14:865–868. doi: 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolock S.L., Lopez R., Klein A.M. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 2019;8:281–291.e9. doi: 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.-R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finak G., McDavid A., Yajima M., Deng J., Gersuk V., Shalek A.K., Slichter C.K., Miller H.W., McElrath M.J., Prlic M., et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Single-cell RNAseq NGS data from Basel samples have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. The gene expression data from Basel are available via an interactive browser. Link to the browser is listed in the key resources table. Gene expression and metadata from cells included in the alaysis from Basel sampes are available as Supplementary data file 1 and 2. The gene expression data from 3′ and 5′ sc-RNA-seq samples are available via the interactive browser “Shiny app’’ at: https://malhotralab.shinyapps.io/csf_cryo_3prime/and https://malhotralab.shinyapps.io/csf_cryo_5prime/, respectively. The Columbia University single cell data will be made available by the lead contact upon reasonable request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.