
FIGURE 11.
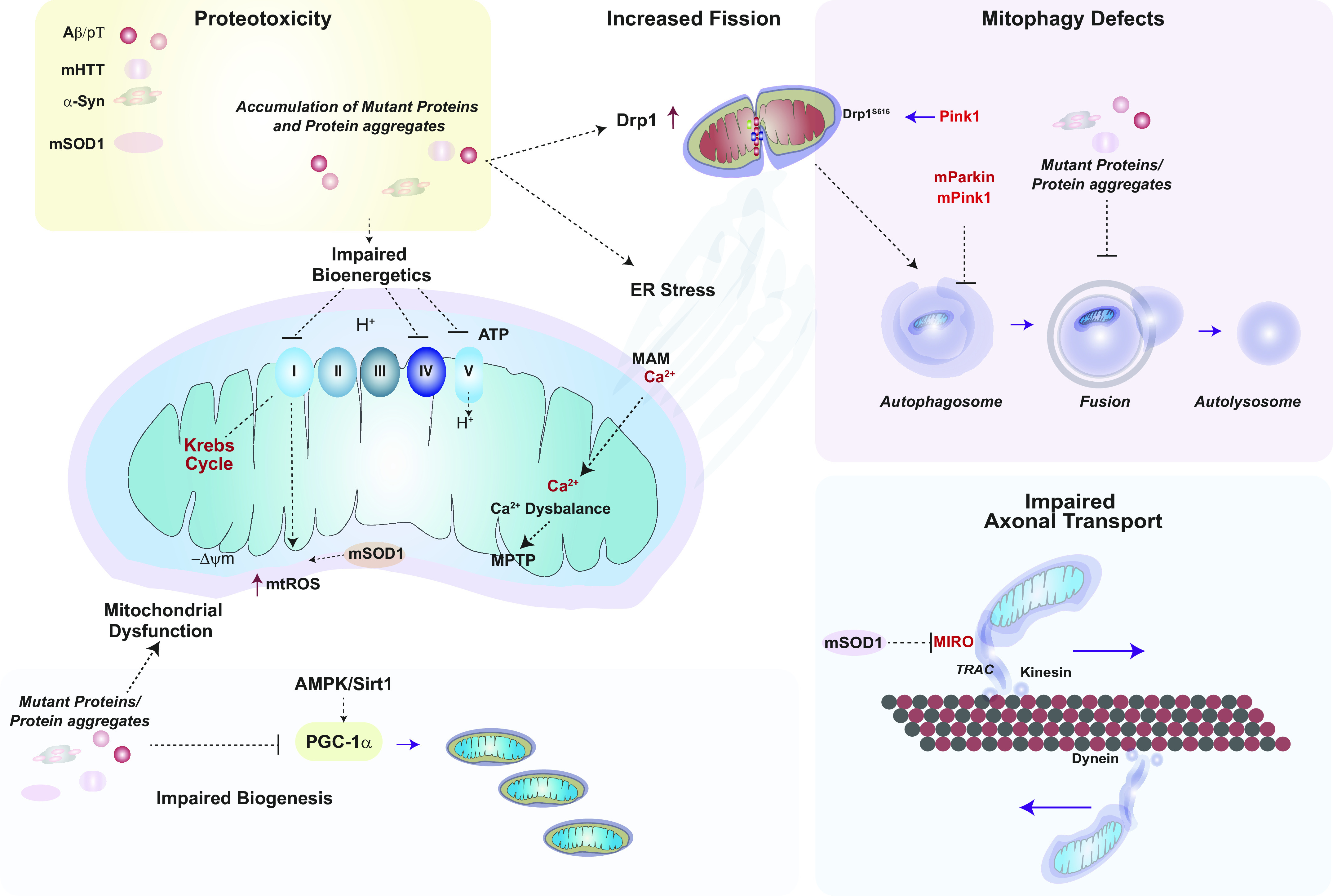
Mitochondrial dysfunction in neurodegenerative disease. Mitochondrial dysfunction is a hallmark of neurodegenerative diseases. Mutant proteins and their aggregates have been implicated in the pathogenesis of various neurodegenerative diseases, including AD, HD, PD, and ALS, by disrupting proteostasis and mitochondrial function. Mutant Aβ and hyperphosphorylated Tau (p-T), mutant Huntingtin (mHTT), and α-synuclein (α-Syn) aggregates can block electron transport chain (ETC) activities at various complexes. α-Syn and mHTT may also trigger mitochondrial dysfunction and mitochondrial reactive oxygen species (mtROS) production. mHTT, p-T, and mutant Aβ can interfere with the Sirt1-PGC-1α axis and its initiation of mitochondrial biogenesis. mHTT, p-T, and mutant Aβ can promote mitochondrial fragmentation via interactions with dynamin-related protein-1 (Drp1). Mutant forms of PTEN-induced kinase 1 (PINK1) can also interfere with PINK1-dependent Drp1 S616 phosphorylation. Autophagy and mitophagy processes may be disrupted by mutant proteins: mHTT can interfere with autophagosome formation, whereas p-T and mutant Aβ block autophagosome-lysosome fusion. Mutant PINK1 (mPINK1) and Parkin (mParkin) may cause mitophagy dysfunction. Superoxide dismutase 1 (SOD1) catalyzes the dismutation of superoxide generated by mitochondria, whereas in ALS the mutant form (mSOD1) is defective at removing superoxide, which favors peroxynitrite formation. mSOD1 can also interfere with mitochondrial axonal transport via inhibiting the transport regulator protein mitochondrial Rho GTPase (MIRO) and by promoting the autophagosome-dependent degradation of MIRO. −ΔΨm, mitochondrial membrane potential (loss); ER, endoplasmic reticulum; MAM, mitochondria-associated membrane; p62, SQSTM1 autophagy cargo adaptor protein. See glossary for other abbreviations.