

Abstract
Kidney repair after injury involves the cross-talk of injured kidney tubules with interstitial fibroblasts and immune cells. Although tubular cells produce multiple cytokines, the role and regulation of specific cytokines in kidney repair are largely undefined. In this study, we detected the induction of fibroblast growth factor 2 (FGF2) in mouse kidneys after repeated low-dose cisplatin (RLDC) treatment and in RLDC-treated renal proximal tubule cells in vitro. We further detected FGF2 in the culture medium of RLDC-treated renal tubular cells but not in the medium of control cells, indicating that RLDC induces FGF2 expression and secretion. Compared with the medium of control cells, the medium of RLDC-treated renal tubular cells was twice as effective in promoting fibroblast proliferation. Remarkably, the proliferative effect of the RLDC-treated cell medium was diminished by FGF2-neutralizing antibodies. In addition, the RLDC-treated cell medium induced the expression of fibrosis-related proteins, which was partially suppressed by FGF2-neutralizing antibodies. In mice, FGF2 deficiency partially prevented RLDC-induced decline in kidney function, loss of kidney weight, renal fibrosis, and inflammation. Together, these results indicate that FGF2 is produced by renal tubular cells after kidney injury and acts as an important paracrine factor in maladaptive kidney repair and disease progression.
Keywords: fibroblast growth factor 2, cisplatin, renal fibrosis, maladaptive kidney repair
Introduction
Cisplatin, a potent chemotherapy drug for cancers, can cause serious side effects in normal tissues and organs, especially in the kidneys. Damage to the kidney by cisplatin may induce acute kidney injury (AKI) within a few days or chronic kidney problems in months to years. AKI or acute nephrotoxicity of cisplatin is characterized by abrupt damage to renal tubules and rapid loss of renal function. Cisplatin-induced chronic kidney problems have some of the key features of chronic kidney disease (CKD), including tubular atrophy, atubular glomeruli, tubulointerstitial fibrosis, and a gradual decline in renal function.1–9
After AKI, kidney tubules have the capacity for repair. Specifically, the surviving tubular cells may dedifferentiate and then proliferate to fill the wound. If the repair is complete, it will reconstruct an intact tubule that is fully functional. However, when the initial injury is severe or repeated, tubular repair becomes incomplete or maladaptive. Under this situation, tubular cells may change to a secretory phenotype to release various profibrotic and proinflammatory factors for interstitial fibroblast activation and proliferation, leading to renal interstitial fibrosis and progression toward CKD.10–13
The extent of renal fibrosis after cisplatin exposure depends on experimental models. For example, very limited renal fibrosis developed in mice after 2 relatively high doses (15 mg/kg) of cisplatin treatment administered 2 weeks apart2,3; however, considerable renal interstitial fibrosis was induced by 4 weekly injections of 7 to 9 mg/kg cisplatin, a model of repeated low-dose cisplatin (RLDC) treatment.1,5,6,8 Our recent work further analyzed the changes in transcriptional profile at the single cell level during the development of CKD in this RLDC model. We identified a cell population of failed-repair proximal tubules, which was proinflammatory owing to high expressions of Krt20, Vcam1, Dcdc2a, Sema5a, and Ccl2 genes. Moreover, we demonstrated increased transcription of several profibrotic growth factors, including transforming growth factor-β, connective tissue growth factor (also named CCN2), and platelet-derived growth factor,4 in proximal tubule cells after RLDC treatment. These observations support the scenario that injured or partially repaired proximal tubules may produce paracrine factors that activate fibroblasts in the interstitium for fibrogenesis. However, the key paracrine factor(s) involved in this pathologic process remains to be identified.
Fibroblast growth factor 2 (FGF2) is a member of the fibroblast growth factor (FGF) family of proteins, which is a group of heparin-binding growth factors involved in many biological processes, such as embryonic development, angiogenesis, cell proliferation, differentiation, cell survival, and tumorigenesis.14 FGF2 has many isoforms generated using alternative translational start sites. Among them, the isoform with the lowest molecular weight, called LMW FGF2, is the only secreted form that usually functions via binding to high-affinity FGF receptors. Once bound to FGFs, FGF receptors undergo conformational changes, leading to their tyrosine kinase activation and subsequent intracellular signaling events in an autocrine or paracrine fashion.15 FGF2 expression was elevated in patients with renal interstitial fibrosis, suggesting its possible involvement in fibrogenesis.16
The present study was designed to analyze changes in FGF2 and its function during the development of CKD after RLDC treatment. We found that FGF2 expression was induced by RLDC in mouse kidneys. FGF2 deficiency in mice alleviated renal fibrosis, decreased renal inflammation, and improved renal function after RLDC treatment. In cultured renal proximal tubule cells, RLDC increased FGF2 expression and secretion, which could stimulate the proliferation of fibroblasts and their production of fibrotic proteins. These results indicate that FGF2, produced by proximal tubule cells, is an important paracrine factor for fibroblast activation and fibrogenesis after cisplatin treatment.
Materials and Methods
Animals and RLDC Treatment
C57BL/6 mice and Fgf2 knockout (KO) mouse line (Fgf2tm1Doe/J, stock number 003256) were originally purchased from the Jackson Laboratory and bred in-house for this study. Mice were housed in a pathogen-free animal facility at Charlie Norwood VA Medical Center, with free access to food and water under a 12/12-hour light/dark cycle. For Fgf2 wild-type (WT) and KO mice genotyping, primers 5′- CAA AGA ACT TAT AGC CCC CC-3′ and 5′- TAG CGA TGA TGA ACC AGG-3′ were used to detect the mutant band of approximately 200 base pairs, and primers 5′-CGA GAA GAG CGA CCC ACA C-3′ and 5′-CCA GTT CGG GGA CCC TAT T-3′ were used to detect the WT band of approximately 185 base pairs. Littermate male mice of 11 to 12 weeks old were used for the experiments. Mice were administered 4 consecutive weekly injections of 8 mg/kg of cisplatin for the RLDC treatment. Kidney tissue samples were collected 4 weeks after the first injection for renal function and renal pathology analysis. Blood urea nitrogen (BUN) levels and estimated glomerular filtration rates (GFRs) were determined to indicate renal function. BUN was measured using a commercial kit from EKF Diagnostics USA (Stanbio Laboratory).
Transcutaneous Measurement of GFR
Estimated GFR was measured in mice by transcutaneously monitoring the clearance of fluorescein isothiocyanate (FITC)-labeled sinistrin using the transdermal GFR measurement system from MediBeacon as previously described.4 Briefly, mice were anesthetized with isoflurane inhalation, shaved, and depilated 1 day before the measurement. The transdermal GFR monitors were adhered to the skin using a double-sided adhesive patch and mounted on the mouse using silk tape. FITC-sinistrin (15 mg/mL dissolved in 0.9% sterile saline) was injected via a tail vein. The estimated GFR was monitored for 1 hour, and data were analyzed using the elimination kinetics curve of FITC-sinistrin.
Immunoblot Analysis
Samples of the kidney cortex and outer medulla were collected from the C57BL/6 and FGF2 mice and lysed in 2% sodium dodecyl sulfate (SDS) buffer with a protease inhibitor cocktail (Sigma) and Benzonase nuclease (EMD Millipore). The kidney lysate was used for immunoblot analysis. Protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Equal amounts of protein were loaded into each lane for SDS-polyacrylamide gel electrophoresis. After being transferred onto polyvinylidene difluoride membranes, the blots were incubated sequentially with a blocking buffer, primary antibodies at 4 °C overnight, and secondaryantibodies for 1 hour at room temperature. Antigens on the blots were revealed using an enhanced chemiluminescence kit (Thermo Fisher Scientific). Primary antibodies were obtained from the following sources: anti-FGF2 (ab106245), anti-Vcam1 (ab134047), antie–α-SMA (ab5694), antifibronectin (ab2413), and anti–cyclophilin B (ab16045) from Abcam; anti-FGF2 (05–118) from Millipore; anti-Kim1 (AF1817) from R&D System; anti–collagen I (NBP1–30054) from Novus Biologicals; GAPDH (#2118) and antivimentin (#3932) from Cell Signaling; and anti–β-actin (A5441) from Sigma. Secondary antibodies for immunoblot analysis were obtained from Jackson ImmunoResearch Laboratories.
RNA Extraction and Real-Time Polymerase Chain Reaction
Kidney cortical and outer medulla samples were dissected, freshly frozen in liquid nitrogen, and stored at −80 °C until use. Total RNA was extracted with a mirVana miRNA Isolation Kit (Life Technologies, Thermo Fisher Scientific). For Fgf2 messenger RNA (mRNA) detection, reverse transcription was performed using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) and polymerase chain reaction (PCR) was performed using the TaqMan Universal PCR Master Mix (Thermo Fisher Scientific). The Fgf2 and Gapdh quantitative PCR probe assays were purchased from Integrated DNA Technologies, Inc. For other real-time quantitative PCR, 1 mg of RNA was reverse transcribed using a cDNA Transcription Kit (Bio-Rad), and real-time quantitative PCR was performed using the SYBR Green PCR Master Mix (Bio-Rad). GAPDH was used for normalization. The sequences of the primers used for quantitative PCR are listed in the Table.
Table.
Polymerase chain reaction primers
| Primer | Sequence |
|---|---|
| IL-7 primer 1 | 5′-TTGCCCGAATAATGAACCAAAC-3′ |
| IL-7 primer 2 | 5′-TGCGAGCAGCACGATTTA-3′ |
| Ccl2 primer 1 | 5′-CATCCACGTGTTGGCTCA-3′ |
| Ccl2 primer 2 | 5′-GATCATCTTGCTGGTGAATGAGT-3′ |
| Gapdh primer 1 | 5′-AATGGTGAAGGTCGGTGTG-3′ |
| Gapdh primer 2 | 5′-GTGGAGTCATACTGGAACATGTAG-3′ |
Ccl2, chemokine (C-C motif) ligand 2. Gapdh, glyceraldehyde-3-phosphate dehydrogenase; IL, interleukin.
RLDC Treatment in Boston University mouse proximal tubular cell line Cells
Boston University mouse proximal tubular cell line (BUMPT) cells were seeded in 35-mm dishes at a density of 0.2 × 106 cells/dish. After overnight growth, the cells were subjected to 4 days of RLDC treatment, which consisted of 7 hours of 2-μM cisplatin incubation, followed by 17 hours of recovery in a cisplatin-free normal medium. After the treatment, the medium and/or cell lysate was collected for analysis.
Tubular Cell Medium Collection, Concentration, and Treatment of Renal Fibroblasts
The BUMPT cells were seeded in 60-mm dishes at a density of 0.5 × 106 cells/dish. After overnight growth, the cells were subjected to 3 days of RLDC treatment, whereas control cells were stored in a control medium without cisplatin for 3 days. Then, the culture medium was discarded, and the cells were washed twice with phosphate-buffered saline. Finally, the cells were incubated with a fresh serum-free medium with or without 2-M cisplatin for another day. The final culture medium was collected for concentration using Amicon Ultra-4 Centrifugal Filter Units (Millipore, UFC8003) with a molecular weight cut-off of 3 kDa. Briefly, 4 ml of culture medium from each 60-mm dish was collected and centrifuged at 1,000g for 5 minutes at room temperature to remove cell debris. The supernatant was then transferred to a prerinsed (Milli-Q water, Millipore) Amicon centrifugal filter unit and centrifuged at 7,500g on a fixed-angle rotor for 1 hour at 4 °C to recover approximately 100 μL of concentrated solution. To ensure optimal activity, the concentrated medium was immediately used for treatment or stored at −80 °C for no more than 3 days before use. Repeated freeze/thaw cycles were avoided at all times.
To treat fibroblasts with tubular cell medium, normal rat kidney-49F (NRK-49F) fibroblasts were seeded in a 12-well plate at a density of 0.15 × 106 cells/well to reach approximately 80% confluence by the next day. After overnight starvation in a serum-free medium, subconfluent NRK-49F fibroblasts were incubated with the tubular cell medium (100 μL concentrated medium diluted in 1 mL of serum-free Dulbecco’s Modified Eagle medium) for 2 days. Cells were monitored morphologically, trypsinized for counting using a Bio-Rad TC20 automated cell counter, and then lysed for protein analysis. In the experiments testing the effect of the FGF2-neutralizing antibody, the antibody or mouse immunoglobulin G (IgG) was preincubated with a tubular cell medium for approximately 1.5 hours at room temperature before adding to NRK-49F fibroblasts.
Enzyme-linked Immunosorbent Assay of FGF2
To measure the release or secretion of FGF2 from tubular cells, the culture media of the control or RLDC-treated BUMPT cells were collected and concentrated as described earlier in the article. Enzyme-linked Immunosorbent Assay (ELISA) of FGF2 was performed using a mouse FGF2 ELISA Kit (ELISAGenie, 894 MOFI00666). In brief, a series of standard dilutions was freshly prepared as instructed, and 100 μL of each standard was added into appropriate wells. Meanwhile, samples were diluted 2-fold (50-μL samples mixed with 50 μL of 1× diluent) to make up a volume of 100 μL for each well. After overnight incubation at 4 °C, the wells were carefully washed 4 times. The wells were then sequentially incubated at room temperature with 100 μL/well of biotinylated FGF2 detection antibody for 1 hour and 100 μL/well of streptavidin horseradish peroxidase (HRP) for 45 minutes. A complete wash was performed after each incubation. The color was developed by incubating with 100 μL/well of tetramethylbenzidine substrate for 30 minutes at room temperature. After adding 50 μL/well of stop solution, the absorbance at 450 nm was immediately measured on an Infinite 200 PRO reader (Tecan Life Sciences), the concentrations of released FGF2 were calculated on the basis of standard curves.
Histologic Analysis
Kidney tissue samples were fixed with 4% paraformaldehyde in phosphate-buffered saline, embedded in paraffin, and cut into 5-μM sections. For immunohistochemistry staining, tissue sections were heated in an antigen retrieval buffer. Then, the sections were incubated with 3% hydrogen peroxide, followed by a blocking buffer. The slides were then exposed to the primary antibody at 4 °C overnight. After washing, the slides were incubated with ImmPRESS HRP Horse Anti-Rabbit IgG (MP-7401; Vector Laboratories) or ImmPRESS-AP Horse Anti-Goat IgG (MP-5405; Vector Laboratories) for 1 hour at room temperature. After washing, color was developed with an ImmPACT DAB Substrate Kit, Peroxidase (HRP) (SK-4105; Vector Laboratories) and ImmPACT Vector Red Substrate Kit, Alkaline Phosphatase (AP) (SK-5105; Vector Laboratories). The primary antibodies for immunostaining included antimacrophage (ab22506, Abcam) and antie-α-SMA (ab5694, Abcam). Sirius red staining was performed according to a standard protocol from the manufacturer (Chondrex Inc).
Statistical Analysis and Software
Quantitative data were expressed as means ± SEM. Statistical analysis was conducted using the GraphPad Prism software 9.2.0(332) (GraphPad Software). Statistical differences were determined using a 2-tailed unpaired Student’s t test or 1-way analysis of variance. A P value of <.05 was considered significantly different.
Results
RLDC Induces Fibrotic Changes in Mouse Kidneys and in BUMPT Cells
Renal interstitial fibrosis is a key pathologic feature of CKD. Consistent with our previous studies,4 the expression of fibrosis marker proteins, including fibronectin (FN), collagen I, α-SMA, and vimentin, was increased in RLDC-treated kidneys compared with that in untreated controls (Fig.1A). Correspondingly, RLDC increased the deposition of collagen in kidney tissues, as shown by Sirius red staining (Fig. 1B). We further examined the in vitro expression of fibrosis marker proteins in RLDC-treated BUMPT mice. As shown in Figure 1C, RLDC treatment increased the expression of FN, collagen I, and vimentin protein, indicating the induction of fibrotic changes in this model.
Figure 1.
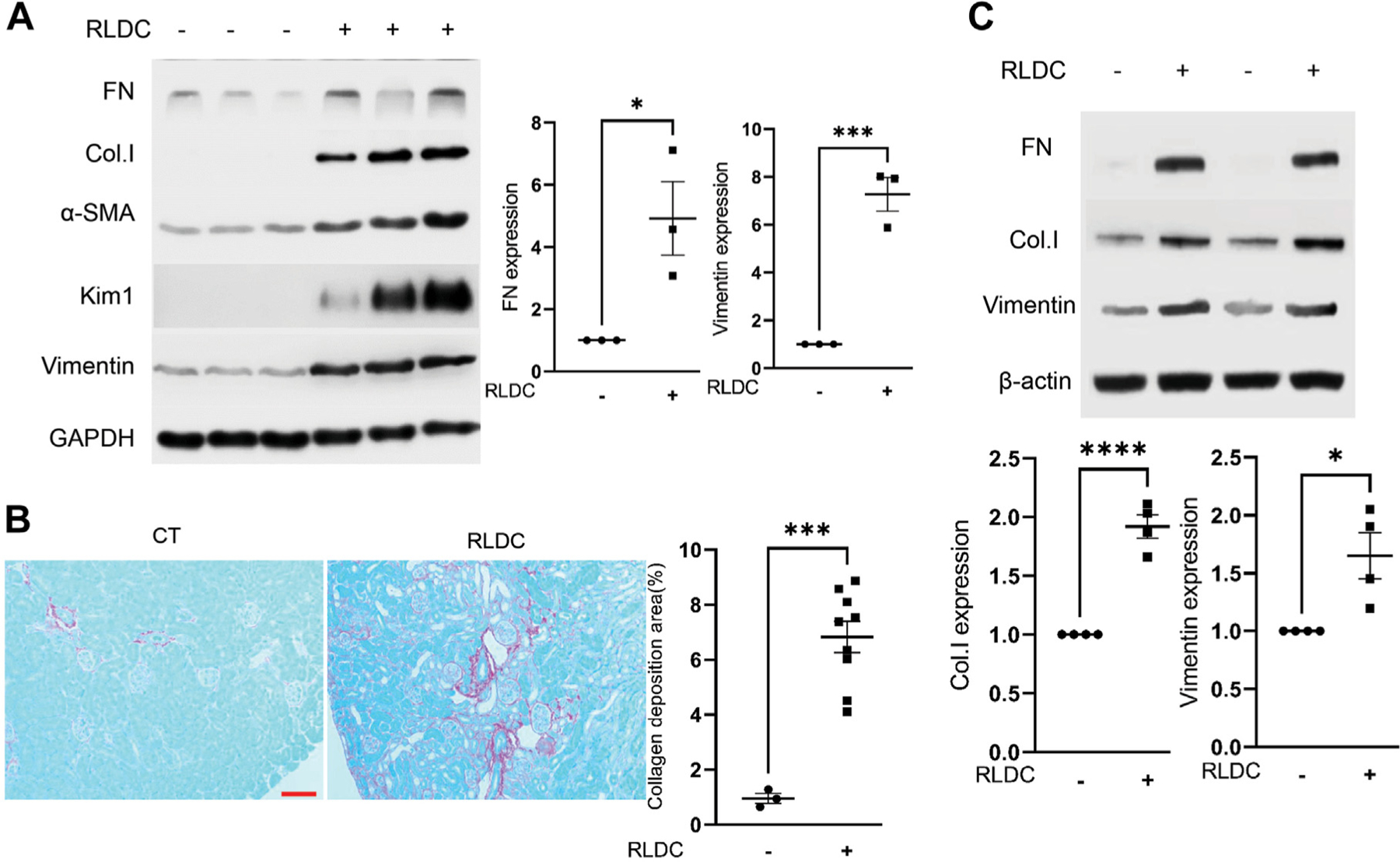
Repeated low-dose cisplatin (RLDC) induces fibrotic changes in mouse kidneys and in Boston University mouse proximal tubular cell line cells. (A, B) Male C57BL/6 mice were administered 4 consecutive weekly injections of 8 mg/kg cisplatin (RLDC) or saline as control (CT) to collect kidney cortex and outer medulla samples for analysis. (A) Representative immunoblots of fibronectin (FN), collagen I (Col.I), α-SMA, Kim1, vimentin, and GAPDH (loading control). (B) Sirius red and fast green staining images showing increased collagen deposition in post-RLDC kidneys. Scale bar = 0.05 mm. (C) BUMPT cells were subjected to RLDC treatment (7 hours of cisplatin incubation, followed by 17 hours of recovery for 4 days) or control incubation to collect cell lysate for immunoblot analysis of FN, Col.I, vimentin, and β-actin (loading control). Data were expressed as mean ± SEM. *P < .05. ***P < .001, ****P < .0001.
RLDC Induces FGF2 Expression in Mouse Kidneys and BUMPT Cells
The interaction or cross-talk among tubular cells, interstitial fibroblasts, immune cells, and others plays a pivotal role in kidney repair. In this regard, it is generally understood that after AKI, incompletely repaired proximal tubules may produce various profibrotic and proinflammatory factors that stimulate fibrosis and chronic inflammation.10–13 FGF2 is one of these factors that is increased during renal fibrogenesis in humans and experimental models.14,16,17 To determine the role of FGF2 in RLDC-induced chronic kidney injury and renal fibrosis, we initially analyzed the expression of FGF2 after RLDC treatment in mice. Immunoblot analysis detected a remarkably higher FGF2 expression in the kidney tissues of RLDC-treated mice than in controls (Fig. 2A). Quantitative PCR also detected a significant increase in FGF2 mRNA in the kidneys of RLDC-treated mice (Fig. 2B). Consistently, both protein and mRNA levels of FGF2 were increased in RLDC-treated BUMPT cells compared with control cells (Fig. 2C, D). Thus, FGF2 was induced in both in vivo and in vitro models of RLDC treatment.
Figure 2.

Repeated low-dose cisplatin (RLDC) induces fibroblast growth factor 2 (FGF2) expression in mouse kidneys and Boston University mouse proximal tubular cell line cells. (A, B) Male C57BL/6 mice were administered 4 consecutive weekly injections of 8 mg/kg cisplatin (RLDC) or saline as control (CT) to collect kidney cortex and outer medulla samples for analysis. (A) Representative immunoblots of FGF2 and cyclophilin B (Cyclo.B) (loading control). (B) Quantitative polymerase chain reaction (PCR) analysis showing the upregulation of FGF2 messenger (mRNA) expression in RLDC-treated mice kidneys. (C, D) BUMPT cells were treated with or without RLDC for 4 days to collect cell lysate for immunoblot and quantitative PCR analyses. (C) Representative immunoblots of FGF2 and Cyclo.B (loading control). (D) Quantitative PCR analysis of FGF2 mRNA induction by RLDC. Quantitative data were expressed as means ± SEM. *P < .05. **P < .01. L, long exposure; S, short exposure.
RLDC Increases the Secretion of FGF2 in Renal Tubular Cells
FGF2 has multiple isoforms, among which the LMW FGF2 is primarily located in the cytoplasm and secreted from the cell via a nonclassical secretory pathway.18,19 Therefore, we hypothesized that this FGF2 isoform is produced and secreted from renal tubular cells during RLDC treatment and then acts as a paracrine factor to stimulate interstitial fibroblasts for fibrogenesis. To test this possibility, we first examined the effect of RLDC on FGF2 secretion from BUMPT cells into a culture medium. FGF2 was undetectable in the medium of control cells during immunoblot analysis; however, it was detected in the medium collected from the RLDC-treated cells at the expected size of approximately 18 kDa (Fig. 3A). We further confirmed this result via ELISA analysis. As shown in Figure 3B, the FGF2 level increased from 45.6 pg/mL in the control cell medium to 233.3 pg/mL in the medium of the RLDC-treated cells, indicating a 5-fold increase in FGF2 secretion. The RLDC-treated cells also released more collagen I into the culture medium than the control cells (Fig. 3C). These results, together with those in Figure 2, suggest that RLDC treatment may induce the production and secretion of FGF2 in renal tubular cells for renal fibrosis.
Figure 3.

Repeated low-dose cisplatin (RLDC) increases the secretion of fibroblast growth factor 2 (FGF2) in renal tubular cells. Boston University mouse proximal tubular cell line cells were subjected to 4 days of RLDC treatment (RLDC) or kept untreated as control (CT) to collect the final culture media for immunoblot and enzyme-linked immunosorbent assay analyses. (A) Representative immunoblot showing FGF2 in the medium of RLDC-treated cells but not in that of control cells. (B) ELISA quantification of FGF2 secretion by RLDC-treated cells in culture medium. Data were expressed as mean ± SEM (n = 3). **P < .01. (C) Representative immunoblot of collagen I (Col.I) in the medium of RLDC-treated cells.
Increased Secretion of FGF2 by RLDC Promotes the Proliferation and Fibrotic Protein Production in Fibroblasts
To investigate the paracrine effect of FGF2 secreted from the RLDC-treated tubular cells in renal fibrosis, we incubated NRK-49F (a renal fibroblast cell line) cells with the medium collected from the control or RLDC-treated cells. As shown in Figure 4A, the number of fibroblasts incubated with the RLDC cell medium was almost twice as high as that incubated with the control cell medium, confirming that the medium from the RLDC-treated cells was much more effective in stimulating fibroblast proliferation. To clarify the specific role of FGF2 in the RLDC medium, we tested the effect of FGF2-neutralizing antibodies. As shown in Figure 4A, the proliferation-stimulating effect of the RLDC cell medium was dramatically reduced by pretreatment of the medium with 20 or 40 μg of FGF2-neutralizing antibodies. Furthermore, the RLDC medium induced a higher expression of FN and vimentin in fibroblasts than the control cell medium (Fig. 4B), whereas a-SMA expression was indifferent. The inductive effect of the RLDC medium was reduced by the FGF2-neutralizing antibodies (Fig. 4C). These results suggest that FGF2 secreted from RLDC-treated tubular cells may have an important role in renal fibrosis via its paracrine function on fibroblasts.
Figure 4.
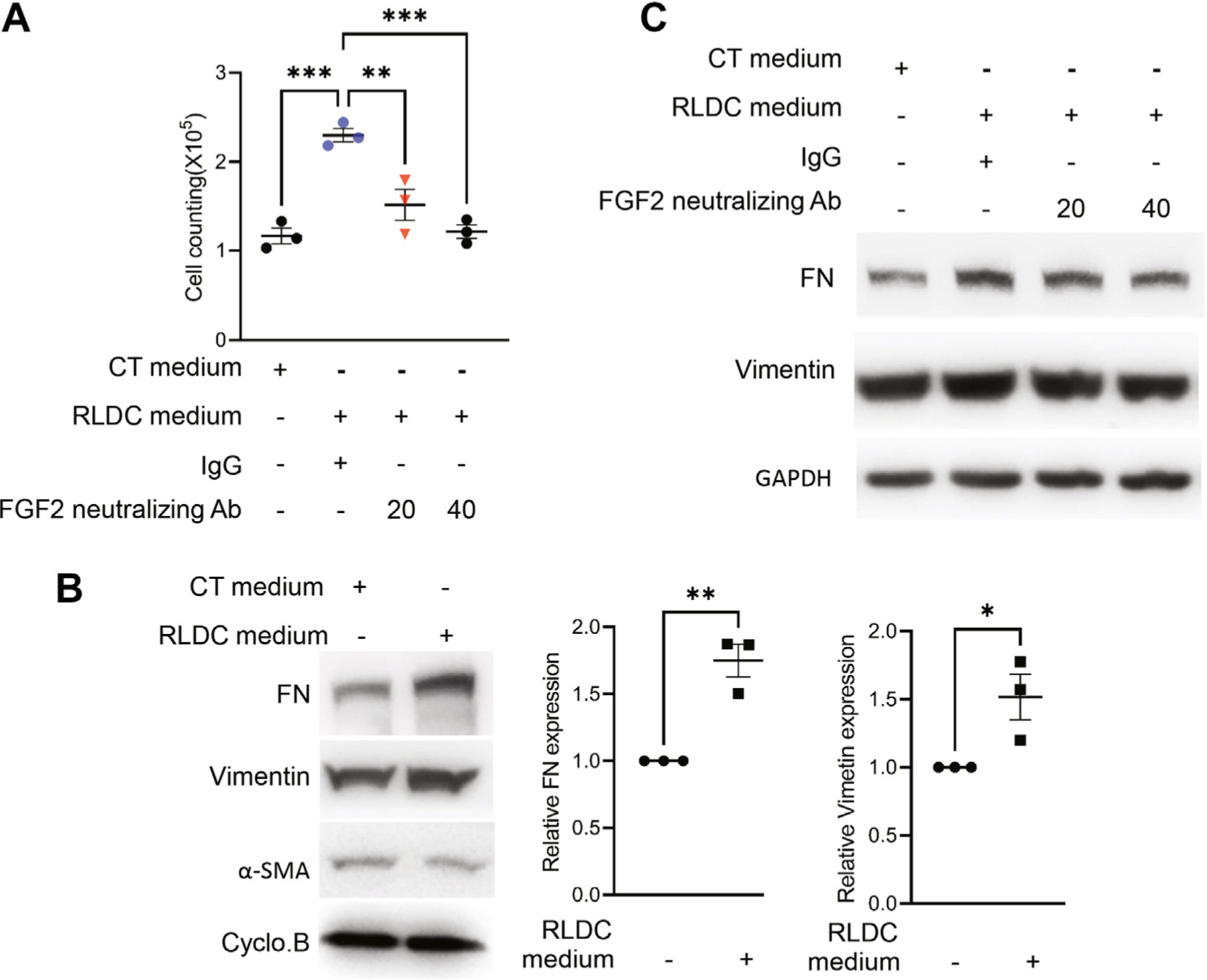
Increased secretion of fibroblast growth factor 2 (FGF2) by repeated low-dose cisplatin (RLDC) promotes proliferation and fibrotic protein production in fibroblasts. Boston University mouse proximal tubular cell line cells were subjected to 4 days of RLDC treatment (RLDC) or kept untreated as control (CT) to collect the final culture media. The media were preincubated with 20 or 40 μg FGF2-neutralizing antibodies (Abs) or nonimmune immunoglobulin G (IgG) and then added to normal rat kidney-49F fibroblasts. After 48 hours of culture, the number of fibroblasts was counted, and cell lysates were collected for immunoblot analysis. (A) Cell counting showing the proliferative effect of RLCD-treated cell medium on fibroblasts that was suppressed by FGF2-neutralizing antibodies. Data were expressed as mean ± SEM (n = 3). **P < .01. ***P < .001. (B) Representative immunoblots showing the induction of fibronectin (FN) and vimentin (but not α-SMA) in fibroblasts by the RLCD-treated cell medium. Data were expressed as mean ± SEM (n = 3). *P < .05. **P < .01. (C) Representative immunoblots showing that FGF2-neutralizing antibodies partially suppressed the induction of fibrosis proteins by RLCD-treated cell medium. Cyclo.B, cyclophilin B.
RLDC-Induced Chronic Kidney Injury Is Ameliorated in Fgf2-Deficient Mice
To determine the role of FGF2 in RLDC-induced CKD in vivo, we compared Fgf2-deficient (Fgf2−/−) mice with their WT (Fgf2+/+) littermates. Genotypes of the mice were analyzed via PCR-based genotyping (Fig. 5A). We further verified that FGF2 was markedly induced by RLDC treatment in the kidneys of WT mice but not in Fgf2-deficient mice (Fig. 5B). The RLDC treatment caused a significant decline in renal function, as shown by the estimated GFR in WT mice, which was partially but significantly prevented in Fgf2-deficient mice (Fig. 5C). Consistently, RLDC treatment led to a decrease in kidney weight, as shown by the kidney weight-to-body weight ratio in WT mice, which was also partially prevented in the Fgf2-deficient mice (Fig. 5D). In terms of kidney injury, the Fgf2-deficient mice showed significantly lower BUN levels after RLDC treatment when compared with the WT mice (Fig. 5E). These results indicate that FGF2 plays a pathogenic role in developing chronic kidney problems after RLDC treatment, especially in renal tubular degeneration and decline of renal function.
Figure 5.

Repeated low-dose cisplatin (RLDC)-induced chronic kidney injury was ameliorated in the Fgf2-deficient mice. Male Fgf2-deficient (Fgf2−/−) mice and their wild-type (WT) (Fgf2+/+) littermates were administered 4 consecutive weekly injections of 8 mg/kg cisplatin. Blood and kidney samples were collected for analysis 5 weeks after the first cisplatin injection. (A) Representative genotyping results of fibroblast growth factor 2 (FGF2) mice. (B) Quantitative polymerase chain reaction results showing that the induction of FGF2 by RLDC was inhibited by the Fgf2-deficient mice. (C) Estimated glomerular filtration rate (eGFR) values showing that RLDC-induced kidney function decline was partially prevented in the Fgf2-deficient mice. (D) The ratio of kidney weight (KW) to body weight (BW) indicating the partial preservation of KW in the Fgf2-deficient mice. (E) Blood urea nitrogen (BUN) values indicating better renal function in the Fgf2-deficient mice after RLDC treatment than in the WT mice. Quantitative data were expressed as mean ± SEM. *P < .05. **P < .01. ****P < .0001. bp, base pair; CT, control; KO, knockout; Mu, mutant; ns, not significant; WT, wild type.
RLDC-Induced Renal Fibrosis Is Alleviated in Fgf2-Deficient Mice
For renal fibrosis, the WT mice showed remarkable increases in fibrosis marker proteins, including FN, α-SMA, and collagen I, after RLDC treatment, which were significantly attenuated in the Fgf2-deficient (KO) mice (Fig. 6A). Correspondingly, Sirius red staining detected less collagen deposition in the Fgf2-deficient mice than in the WT mice (Fig. 6B). To further verify the role of FGF2 in fibroblast transition to myofibroblast, α-SMA, a myofibroblast activation marker, was analyzed via immunostaining. As shown in Figure 6C, the positive α-SMA signal was significantly elevated in the kidney tissues of the WT mice after RLDC treatment compared with that in untreated controls, and this α-SMA induction was remarkably reduced in the Fgf2-deficient mice. These results support a critical role of FGF2 in fibroblast activation and renal fibrosis in RLDC-treated mice.
Figure 6.

Repeated low-dose cisplatin (RLDC)-induced renal fibrosis was alleviated in the Fgf2-deficient mice. Male Fgf2-deficient (Fgf2−/−) mice and their wild-type (Fgf2+/+) littermates were administered 4 consecutive weekly injections of 8 mg/kg cisplatin. Kidney samples were collected 5 weeks after the first cisplatin injection for analysis. (A) Representative immunoblots showing the suppressive effect of fibroblast growth factor 2 deficiency on RLDC-induced expression of fibrosis-related proteins. Control (CT) +/+ group, n = 5; CT−/− group, n = 5; RLDC+/+ group (n = 6); RLDC−/−group, n = 6. (B) Sirius red and fast green staining showing decreased collagen deposition in post-RLDC kidneys in the Fgf2-deficient mice compared with the wild-type mice. (C) Immunostaining images showing α-SMA induction by RLDC, which was reduced in the Fgf2-deficient mice. Scale bar = 0.05 mm. Quantitative data are expressed as mean ± SEM. *P < .05. **P < .01. ****P < .0001. Col.I, collagen I; CT+/+, wild type CT mice; CT−/−, knockout CT mice; Cyclo.B, cyclophilin B; FN, fibronectin; ns, not significant; RLDC+/+, wild type mice with RLDC; RLDC−/−, knockout mice with RLDC.
FGF2 Knockout Decreases Renal Inflammation in RLDC-Treated Mice
Inflammation is crucially involved in kidney injury and post-injury fibrosis.20,21 The inflammatory response has also been demonstrated in post-RLDC kidneys.6,7,9,22,23 FGF2 has been reported to play a pivotal role in inflammation regulation in a variety of diseases, including HIV-associated chronic kidney injuries, lipopolysaccharide (LPS)-induced kidney injury, atherosclerosis due to a high-fat diet,24–27 obesity, asthma, and autoimmune arthritis.28 To explore the role of Fgf2 deficiency in inflammatory infiltration after RLDC treatment, we analyzed macrophages via immunohistochemistry staining. Compared with the untreated control, RLDC treatment induced macrophages in kidney tissues in both the WT and Fgf2-deficient mice; however, the number of macrophages was significantly lower in the Fgf2-deficient mice than in the WT mice (Fig. 7A), suggesting a role of FGF2 in macrophage infiltration. We further analyzed chemokine (C-C motif) ligand 2 (Ccl2) and interleukin 7 (IL-7), 2 important chemokines/cytokines that were shown to be significantly induced by RLDC in the kidneys.4 As shown in Figure 7B, Ccl2 and IL-7 mRNA expression was significantly induced by RLDC in the kidneys of the WT mice compared with the untreated controls, and this induction was suppressed in the Fgf2-deficient mice. These results indicate the involvement of FGF2 in chronic renal inflammation during the maladaptive repair of RLDC-injured kidneys.
Figure 7.
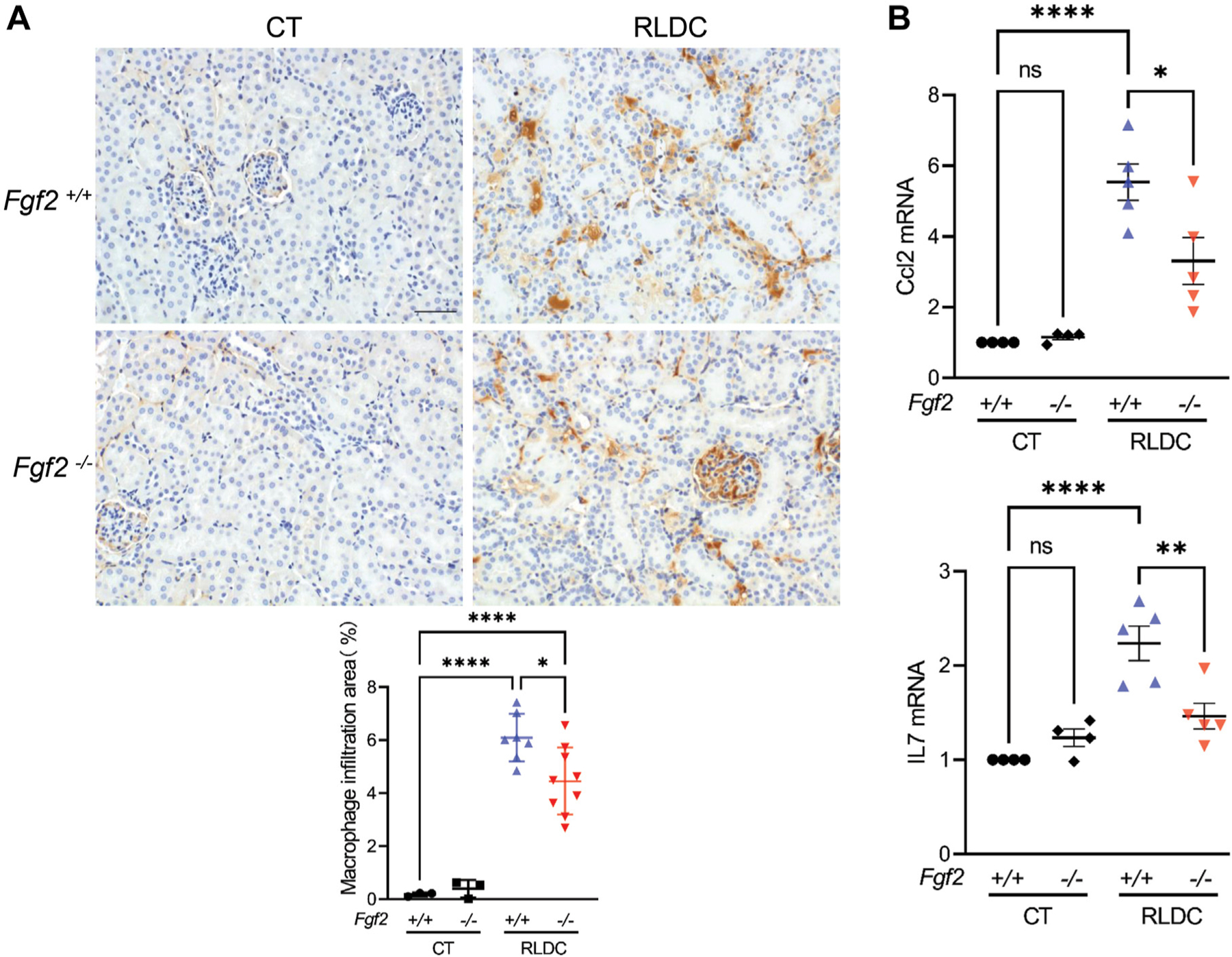
Fibroblast growth factor 2 (FGF2) knockout decreased renal inflammation in Repeated low-dose cisplatin (RLDC)-treated mice. Male Fgf2-deficient (Fgf2−/−) mice and their wildtype (Fgf2+/+) littermates were administered 4 consecutive weekly injections of 8 mg/kg cisplatin. Blood and kidney samples were collected 5 weeks after the first cisplatin injection for analysis. (A) Immunostaining images showing that RLDC-induced macrophage infiltration in kidney tissues was reduced in the Fgf2-deficient mice compared with the wild-type mice. (B) Quantitative polymerase chain reaction analysis showing the induction of chemokine (C-C motif) ligand 2 and interleukin 7 (IL7) by RLDC, which was partially reduced in Fgf2-deficient mice. Quantitative data are expressed as mean ± SEM. *P < .05. **P < .01. ****P < .0001. CT, control; mRNA, messenger RNA; ns, not significant.
Discussion
It is generally understood that during maladaptive repair, kidney tubule cells may produce various profibrotic and proinflammatory factors, which stimulate fibroblasts and immune cells in the interstitium in a paracrine fashion for fibrogenesis or scar formation. However, the roles of specific tubule-secreted factors in this process remain unclear. In the present study, we have shown that FGF2 was notably upregulated and secreted from kidney tubule cells after RLDC injury in mice and BUMPT cells. Functionally, we have demonstrated that chronic kidney damage and functional decline after RLDC treatment were alleviated in the Fgf2-deficient mice. Mechanistically, we have provided evidence that FGF2 released from tubular cells contributed significantly to fibroblast proliferation and chronic inflammation during post-RLDC maladaptive kidney repair. Together, these findings support a pathogenic role of FGF2 in maladaptive kidney repair and consequent development of chronic kidney problems after RLDC treatment.
Normally, FGF2 is produced at low levels, and its expression has been shown to be confined to glomerular, vascular, and a few tubular and interstitial fibroblast-like cells in the kidneys. However, FGF2 was dramatically induced under pathologic conditions, especially in renal fibrosis. In renal fibrosis, FGF2 expression was increased, specifically in interstitial and injured kidney tubule cells.14 Subsequently, emerging studies have suggest that damaged proximal tubular cells produce and secrete profibrotic cytokines that activate interstitial fibroblasts, leading to CKD.11 Consistently, our data show that RLDC induced both FGF2 mRNA and protein in cultured BUMPT proximal tubular cells and post-RLDC kidneys (Fig. 2A–D). Moreover, our immunoblot and ELISA analysis detected the secretion of FGF2 into the culture medium of RLDC-treated BUMPT cells (Fig. 3A, B). Therefore, the proximal tubule cells may serve as the main source of profibrotic cytokines or paracrine factors, such as FGF2, for renal fibrosis during maladaptive kidney repair.
FGF2 has been implicated in kidney injury and repair; however, its role remains controversial. On the one hand, FGF2 has been shown to protect against renal ischemia-reperfusion injury by suppressing mitochondrial damage and emergency room stress.29–33 Xu et al29 further showed that cisplatin-induced nephrotoxic AKI was ameliorated in kidney tubule-FGF receptor 2 (FGFR2)-ablated mice. On the other hand, overexpression of FGF2 induced a strong apoptotic response in kidney cells and was suggested to mediate Middle East respiratory syndrome coronavirus–induced kidney failure.34 In addition, excessive FGF2 in the kidney accelerates LPS-induced kidney damage,27 and higher circulating FGF2 levels may increase the risk of HIV nephropathy in children by promoting the adhesion of endotoxins or viruses to endothelial cells.35,36 For kidney repair, Villanueva et al32 reported that recombinant FGF2 promoted renal regeneration via re-expression of nephrogenic proteins, which was impaired by bFGFR2 antisense oligonucleotide in early ischemic injury. They further verified the repair function of FGF2 in rats subjected to the 5/6 nephrectomy (NPX).33 In the current study, we tested the Fgf2-deficient mice with the global knockout of the Fgf2 gene. After RLDC treatment, the Fgf2-deficient mice showed better renal function, as demonstrated by higher estimated GFR levels and lower BUN levels. Notably, these mice also had better kidney weight and less renal fibrosis after RLDC treatment. These results suggest that Fgf2 may play a critical pathologic role in kidney injury and/or repair in this model.
Specifically, our results indicate that FGF2 is a key factor produced and secreted by renal tubule cells for renal fibrosis. This is illustrated by the findings that FGF2 deficiency alleviated renal fibroblast activation and interstitial fibrosis in post-RLDC kidneys (Fig. 6) and partially prevented kidney shrinkage or the loss of kidney weight (Fig. 5). Such a profibrotic function of FGF2 is also supported by other studies using different models. For example, a remarkable upregulation of FGF2 was observed within the tubulointerstitium in diabetic mice, and diabetic nephropathy was alleviated by FGF2 inhibition.37 FGF2 knockout or inhibition also reduced renal fibrosis in obstructive nephropathy.38 In addition, ablation of fibroblast FGFR2 suppressed renal fibrogenesis after renal ischemia-reperfusion injury in mice.30 Notably, FGF2 expression was increased in fibrotic human kidneys.16 All these findings point to a role of FGF2 in renal fibrosis under pathologic or renal disease conditions.
One mechanism of the profibrotic action of FGF2 is through its paracrine effect on fibroblasts. This was shown in our study by incubating fibroblasts with the media collected from RLDC-treated or untreated cells. Compared with the control cell medium, the RLDC-treated cell medium dramatically increased the proliferation of fibroblasts. Remarkably, this proliferative effect was largely attenuated by FGF2-neutralizing antibodies (Fig. 4A), indicating a critical role of FGF2 in stimulating fibroblast proliferation. The RLDC cell medium also induced the expression of fibrosis marker proteins, such as FN and vimentin, which was also suppressed by FGF2-neutralizing antibodies, albeit at a partial or low efficacy (Fig. 4C). Interestingly, the RLDC cell medium did not significantly induce α-SMA in fibroblasts. It is possible that the level of FGF2 secreted by RLDC-treated cells in the culture medium was too low for inducing the transition of fibroblast to myofibroblast, although it was sufficient to stimulate fibroblast proliferation and production of fibrosis marker proteins. If true, this result indicates the different thresholds of FGF2 for fibroblast proliferation and its transition to myofibroblasts.
FGF2 may also contribute to renal fibrosis and maladaptive kidney repair by modulating renal inflammation. In the present study, the Fgf2-deficient mice had less macrophage infiltration and less chemokine Ccl2 production in the kidneys after RLDC treatment (Fig. 7). These mice also had a lower level of IL-7 than that in the WT mice. Consistent with our results, the development of CKD was delayed by the depletion of macrophages or the proinflammatory protein galectin-3 released by macrophages39 and the blockade of the associated receptors CCR1 and CCR2,40–42 implying the role of macrophages in CKD progression. Growing evidence supports the immunoregulatory effect of FGF2. For instance, FGF2 may induce renal damage by increasing the adhesion of inflammatory cells to the endothelium.43 Furthermore, FGF2 can not only stimulate the attachment of macrophages to HIV-infected tubular epithelial cells but also promote recruitment and adhesion of inflammatory cells to the perivascular and tubulointerstitial areas in mice.36 Mattison et al27 have also shown that FGF2 increases the recruitment of F4/80-positive macrophages in LPS-treated kidneys; although this increase did not reach statistical significance. In light of these studies, our results support a proinflammatory function of FGF2 in maladaptive kidney repair by recruiting macrophages and inducing proinflammatory factors.
Therapeutically, for kidney protection, it is important to consider the effects of cisplatin chemotherapy on tumors. For example, a kidney protective strategy would not be clinically applicable if it also protects cancer cells and diminishes the chemotherapy effect of cisplatin in tumors.44,45 In this regard, there is abundant evidence to suggest that FGF2 is a protumor factor.46,47 High expression of FGF2 was observed in the primary tumor cells of renal cell carcinoma and head and neck squamous cell carcinoma.46,47 Moreover, overexpression of FGF2 has led to cisplatin resistance in tumor cells,47–49 whereas FGF2 inhibition by small interfering RNA enhanced cisplatin-induced apoptosis in A549 lung cancer cells.48 In a previous study, Fgf2-deficient mice had significantly less liver tumor burden, probably because of increased T-cell recruitment for tumor regression50. Consistently, other studies revealed that liver metastases were steadily reduced by anti-FGF2 antibody in mice.51,52 Based on the evidence in these studies, blocking FGF2 may not only protect the kidneys, as suggested by our current study, but also enhance the anticancer effect in tumors during cisplatin chemotherapy.
The use of global knockout of the Fgf2 gene is a limitation in the present study. It has been shown that FGF2 is expressed in various renal cells, such as tubular cells, fibroblasts, and glomerular cells, and contributes to renal fibrosis14 For instance, fibroblast-secreted FGF2 promotes the proliferation and activation of fibroblasts, and the release of FGF2 from glomerular sources augments podocyte injury in membranous nephropathy.53 In this regard, it is difficult to distinguish the effect of FGF2 in renal tubules from that of other renal cells in our study using global FGF2 knockout mice. Thus, a mouse model with renal tubular cell–specific Fgf2 deletion should be established in future studies to explore the specific role of FGF2 produced by renal tubular cells in kidney diseases.
Funding
The work was supported in part by the National Institutes of Health of the United States of America (5R01DK087843 and 5R01DK058831) and the Department of Veterans Affairs of the United States of America (I01 BX000319). Zheng Dong is a recipient of the Senior Research Career Scientist award (1TK6BX005236) from the Department of Veterans Affairs of the United States of America.
Footnotes
Declaration of Competing Interest
The authors declare no competing interests.
Data Availability
Data reported in this study are available from the corresponding author upon reasonable request.
References
- 1.Sharp CN, Doll MA, Dupre TV, et al. Repeated administration of low-dose cisplatin in mice induces fibrosis. Am J Physiol Renal Physiol 2016;310(6): F560–F568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torres R, Velazquez H, Chang JJ, et al. Three-dimensional morphology by multiphoton microscopy with clearing in a model of cisplatin-induced CKD. J Am Soc Nephrol 2016;27(4):1102–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Landau SI, Guo X, Velazquez H, et al. Regulated necrosis and failed repair in cisplatin-induced chronic kidney disease. Kidney Int 2019;95(4):797–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma Z, Hu X, Ding HF, Zhang M, Huo Y, Dong Z. Single-nucleus transcriptional profiling of chronic kidney disease after cisplatin nephrotoxicity. Am J Pathol 2022;192(4):613–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharp CN, Siskind LJ. Developing better mouse models to study cisplatin-induced kidney injury. Am J Physiol Renal Physiol 2017;313(4):F835–F841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharp CN, Doll MA, Megyesi J, Oropilla GB, Beverly LJ, Siskind LJ. Subclinical kidney injury induced by repeated cisplatin administration results in progressive chronic kidney disease. Am J Physiol Renal Physiol 2018;315(1): F161–F172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Black LM, Lever JM, Traylor AM, et al. Divergent effects of AKI to CKD models on inflammation and fibrosis. Am J Physiol Renal Physiol 2018;315(4): F1107–F1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu Y, Cai J, Li F, et al. Chronic effects of repeated low-dose cisplatin treatment in mouse kidneys and renal tubular cells. Am J Physiol Renal Physiol 2019;317(6):F1582–F1592. [DOI] [PubMed] [Google Scholar]
- 9.Sears S, Siskind L. Potential therapeutic targets for cisplatin-induced kidney injury: lessons from other models of AKI and fibrosis. J Am Soc Nephrol 2021;32(7):1559–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Humphreys BD, Cantaluppi V, Portilla D, et al. Targeting endogenous repair pathways after AKI. J Am Soc Nephrol 2016;27(4):990–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol 2015;26(8):1765–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gewin L, Zent R, Pozzi A. Progression of chronic kidney disease: too much cellular talk causes damage. Kidney Int 2017;91(3):552–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu BC, Tang TT, Lv LL, Lan HY. Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int 2018;93(3):568–579. [DOI] [PubMed] [Google Scholar]
- 14.Strutz F The role of FGF-2 in renal fibrogenesis. Front Biosci (Schol Ed) 2009;1(1):125–131. [DOI] [PubMed] [Google Scholar]
- 15.Okada-Ban M, Thiery JP, Jouanneau J. Fibroblast growth factor-2. Int J Biochem Cell Biol 2000;32(3):263–267. [DOI] [PubMed] [Google Scholar]
- 16.Li XY, Zhang F, Qu LL, et al. Identification of YAP1 as a novel downstream effector of the FGF2/STAT3 pathway in the pathogenesis of renal tubulointerstitial fibrosis. J Cell Physiol 2021;236(11):7655–7671. [DOI] [PubMed] [Google Scholar]
- 17.Livingston MJ, Ding HF, Huang S, Hill JA, Yin XM, Dong Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 2016;12(6):976–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.La Venuta G, Zeitler M, Steringer JP, Müller HM, Nickel W. The startling properties of fibroblast growth factor 2: how to exit mammalian cells without a signal peptide at hand. J Biol Chem 2015;290(45):27015–27020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brough D, Pelegrin P, Nickel W. An emerging case for membrane pore formation as a common mechanism for the unconventional secretion of FGF2 and IL-1beta. J Cell Sci 2017;130(19):3197–3202. [DOI] [PubMed] [Google Scholar]
- 20.He L, Wei Q, Liu J, et al. AKI on CKD: heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int 2017;92(5):1071–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo X, Xu L, Velazquez H, et al. Kidney-targeted renalase agonist prevents cisplatin-induced chronic kidney disease by inhibiting regulated necrosis and inflammation. J Am Soc Nephrol 2022;33(2):342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ozkok A, Edelstein CL. Pathophysiology of cisplatin-induced acute kidney injury. BioMed Res Int 2014;2014:967826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharp CN, Doll M, Dupre TV, Beverly LJ, Siskind LJ. Moderate aging does not exacerbate cisplatin-induced kidney injury or fibrosis despite altered inflammatory cytokine expression and immune cell infiltration. Am J Physiol Renal Physiol 2019;316(1):F162–F172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Das JR, Jerebtsova M, Tang P, Li J, Yu J, Ray PE. Circulating fibroblast growth factor-2 precipitates HIV nephropathy in mice. Dis Model Mech 2021;14(7): dmm048980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.ZhuGe DL, Javaid HMA, Sahar NE, Zhao YZ, Huh JY. Fibroblast growth factor 2 exacerbates inflammation in adipocytes through NLRP3 inflammasome activation. Arch Pharm Res 2020;43(12):1311–1324. [DOI] [PubMed] [Google Scholar]
- 26.Liang W, Wang Q, Ma H, Yan W, Yang J. Knockout of low molecular weight FGF2 attenuates atherosclerosis by reducing macrophage infiltration and oxidative stress in mice. Cell Physiol Biochem 2018;45(4):1434–1443. [DOI] [PubMed] [Google Scholar]
- 27.Mattison PC, Soler-García AA, Das JR, et al. Role of circulating fibroblast growth factor-2 in lipopolysaccharide-induced acute kidney injury in mice. Pediatr Nephrol 2012;27(3):469–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shao X, Chen S, Yang D, et al. FGF2 cooperates with IL-17 to promote autoimmune inflammation. Sci Rep 2017;7(1):7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu Z, Zhu X, Wang M, Lu Y, Dai C. FGF/FGFR2 protects against tubular cell death and acute kidney injury involving Erk1/2 signaling activation. Kidney Dis (Basel) 2020;6(3):181–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu Z, Dai C . Ablation of FGFR2 in fibroblasts ameliorates kidney fibrosis after ischemia/reperfusion injury in mice. Kidney Dis (Basel) 2017;3(4):160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan XH, Zheng XM, Yu LX, et al. Fibroblast growth factor 2 protects against renal ischaemia/reperfusion injury by attenuating mitochondrial damage and proinflammatory signalling. J Cell Mol Med 2017;21(11):2909–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Villanueva S, Cespedes C, Gonzalez AA, Roessler E, Vio CP. Inhibition of bFGF-receptor type 2 increases kidney damage and suppresses nephrogenic protein expression after ischemic acute renal failure. Am J Physiol Regul Integr Comp Physiol 2008;294(3):R819–R828. [DOI] [PubMed] [Google Scholar]
- 33.Villanueva S, Cespedes C, Gonzalez A, Vio CP. bFGF induces an earlier expression of nephrogenic proteins after ischemic acute renal failure. Am J Physiol Regul Integr Comp Physiol 2006;291(6):R1677–R1687. [DOI] [PubMed] [Google Scholar]
- 34.Yeung ML, Yao Y, Jia L, et al. MERS coronavirus induces apoptosis in kidney and lung by upregulating Smad7 and FGF2. Nat Microbiol 2016;1:16004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ray PE, Li J, Das JR, Yu J. Association of circulating fibroblast growth factor-2 with progression of HIV-chronic kidney diseases in children. Pediatr Nephrol 2021;36(12):3933–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang P, Jerebtsova M, Przygodzki R, Ray PE. Fibroblast growth factor-2 increases the renal recruitment and attachment of HIV-infected mononuclear cells to renal tubular epithelial cells. Pediatr Nephrol 2005;20(12): 1708–1716. [DOI] [PubMed] [Google Scholar]
- 37.Dong QL, Zhao XH, Wang Q, et al. Anti-aging gene Klotho ameliorates diabetic nephropathy in mice by inhibiting FGF2 signaling pathway. J Biol Regul Homeost Agents 2020;34(4):1369–1377. [DOI] [PubMed] [Google Scholar]
- 38.Guan X, Nie L, He T, et al. Klotho suppresses renal tubulo-interstitial fibrosis by controlling basic fibroblast growth factor-2 signalling. J Pathol 2014;234(4):560–572. [DOI] [PubMed] [Google Scholar]
- 39.Ko GJ, Boo CS, Jo SK, Cho WY, Kim HK. Macrophages contribute to the development of renal fibrosis following ischaemia/reperfusion-induced acute kidney injury. Nephrol Dial Transplant 2008;23(3):842–852. [DOI] [PubMed] [Google Scholar]
- 40.Henderson NC, Mackinnon AC, Farnworth SL, et al. Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am J Pathol 2008;172(2):288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kitagawa K, Wada T, Furuichi K, et al. Blockade of CCR2 ameliorates progressive fibrosis in kidney. Am J Pathol 2004;165(1):237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eis V, Luckow B, Vielhauer V, et al. Chemokine receptor CCR1 but not CCR5 mediates leukocyte recruitment and subsequent renal fibrosis after unilateral ureteral obstruction. J Am Soc Nephrol 2004;15(2):337–347. [DOI] [PubMed] [Google Scholar]
- 43.Zittermann SI, Issekutz AC. Basic fibroblast growth factor (bFGF, FGF-2) potentiates leukocyte recruitment to inflammation by enhancing endothelial adhesion molecule expression. Am J Pathol 2006;168(3):835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 2008;73(9):994–1007. [DOI] [PubMed] [Google Scholar]
- 45.Pabla N, Dong G, Jiang M, et al. Inhibition of PKCdelta reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J Clin Invest 2011;121(7):2709–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Volkova M, Tsimafeyeu I, Olshanskaya A, et al. Immunochemical expression of fibroblast growth factor and its receptors in primary tumor cells of renal cell carcinoma. Am J Clin Exp Urol 2021;9(1):65–72. [PMC free article] [PubMed] [Google Scholar]
- 47.McDermott SC, Rodriguez-Ramirez C, McDermott SP, Wicha MS, Nör JE. FGFR signaling regulates resistance of head and neck cancer stem cells to cisplatin. Oncotarget 2018;9(38):25148–25165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He L, Meng Y, Zhang Z, Liu Y, Wang X. Downregulation of basic fibroblast growth factor increases cisplatin sensitivity in A549 non-small cell lung cancer cells. J Cancer Res Ther 2018;14(7):1519–1524. [DOI] [PubMed] [Google Scholar]
- 49.Miyake H, Hara I, Gohji K, Yoshimura K, Arakawa S, Kamidono S. Expression of basic fibroblast growth factor is associated with resistance to cisplatin in a human bladder cancer cell line. Cancer Lett 1998;123(2): 121–126. [DOI] [PubMed] [Google Scholar]
- 50.Im JH, Buzzelli JN, Jones K, et al. FGF2 alters macrophage polarization, tumour immunity and growth and can be targeted during radiotherapy. Nat Commun 2020;11(1):4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang L, Park H, Chhim S, et al. A novel monoclonal antibody to fibroblast growth factor 2 effectively inhibits growth of hepatocellular carcinoma xenografts. Mol Cancer Ther 2012;11(4):864–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gordon-Weeks AN, Lim SY, Yuzhalin AE, et al. Neutrophils promote hepatic metastasis growth through fibroblast growth factor 2-dependent angiogenesis in mice. Hepatology 2017;65(6):1920–1935. [DOI] [PubMed] [Google Scholar]
- 53.Floege J, Kriz W, Schulze M, et al. Basic fibroblast growth factor augments podocyte injury and induces glomerulosclerosis in rats with experimental membranous nephropathy. J Clin Invest 1995;96(6):2809–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data reported in this study are available from the corresponding author upon reasonable request.